

Abstract
β-hCG expression in breast cancer is highly controversial with reports supporting both protective and tumorigenic effects. It has also been reported that risk of breast cancer at an early age is increased with full-term pregnancies if a woman is a BRCA1 mutation carrier. We have already demonstrated that BRCA1-defective cells express high levels of β-hCG and that when BRCA1 is restored, β-hCG level is reduced. Also, BRCA1 can bind to the promoter and reduce the levels of β-hCG. β-hCG induces tumorigenicity in BRCA1-defective cells by directly binding to TGFBRII and induces TGFBRII-mediated cell proliferation. In this study, we analyzed the mechanism of action of β-hCG on BRCA1 expression and its influence on drug sensitivity in breast cancer cells. We demonstrate that β-hCG induces mutant BRCA1 protein expression in BRCA1 mutant cells; however, in BRCA1 wild-type cells, β-hCG reduced wild-type BRCA1 protein expression. Transcriptionally, β-hCG could induce Slug/LSD1-mediated repression of wild-type and mutant BRCA1 messenger RNA levels. However, β-hCG induces HSP90-mediated stabilization of mutant BRCA1 and hence the overexpression of mutant BRCA1 protein, resulting in partial restoration of homologous recombination repair of damaged DNA. This contributes to drug resistance to HSP90 inhibitor 17AAG in BRCA1-defective cancer cells. A combination of HSP90 inhibitor and TGFBRII inhibitor has shown to sensitize β-hCG expressing BRCA1-defective breast cancers to cell death. Targeting the β-hCG–HSP90–TGFBRII axis could prove an effective treatment strategy for BRCA1-mutated breast tumors.
We demonstrate that β-hCG induces mutant BRCA1 protein expression which is stabilized by HSP90. This results tumor progression and drug resistance in BRCA1 mutated but not in BRCA1 wild type breast cancer cells.
Introduction
Women who have had multiple pregnancies have been documented to be protected against breast cancer (1,2). However, it has been shown that the protective effect is limited to ER/PR positive but not ER/PR negative breast cancers (2,3). Jernström et al. have shown that pregnancy is associated with the increased risk of breast cancer in BRCA1/2 mutation carriers (4). It has also been reported that among BRCA1 mutation carriers, the risk of breast cancer was inversely related to the women’s age at first child birth (5,6). Also the risk of breast cancer by age 40 years in BRCA1/2 mutation carriers increased with full-term pregnancies (4,5).
As majority of BRCA1-related cancers are triple negative in nature, the contribution of pregnancy-associated hormones, estrogen, progesterone and epidermal growth factor to the tumorigenesis of BRCA1-mutated cancers is questionable. However, one of the other major hormones produced during pregnancy, which is critically important for the development and differentiation of the breast tissue, is human chorionic gonadotropin (hCG). We hypothesize that exposure to β-hCG could be the reason for increased breast cancer risk due to pregnancy in BRCA1 mutation carriers whereas the same could be protective for individuals possessing wild-type BRCA1. With regard to cancer, β-hCG expression is highly controversial as it is known to possess both protective and tumorigenic effect (7–11). We have already demonstrated that wild-type BRCA1 inhibits β-hCG expression and that β-hCG can induce tumor progression in BRCA1-defective cells through activation of TGFBRII receptor (12). In this study, we assessed the mechanistic effect of β-hCG supplementation on BRCA1-defective and wild-type cancer cells, which has not been analyzed till date. Here, we demonstrate that β-hCG differentially regulates the expression of wild-type and mutant BRCA1 protein. This results in tumor progression and drug resistance in BRCA1-defective breast cancer cells.
Methods
Cell lines
HCC1937 and HCC1937/wt BRCA1 were obtained as gift from Dr Grant McArthur, Peter MacCallum Cancer Centre, VIC, Australia. Both HCC1937 (BRCA1 mutant) and HCC1937/wt BRCA1 (BRCA1 wild type) were grown in RPMI 1640 media. MDAMB-231 (BRCA1 wild type) and MCF7 (BRCA1 wild type) were grown in Dulbecco’s modified Eagle’s medium. SUM149 (BRCA1 mutant) was grown in Hams F-12 media supplemented with hydrocortisone and insulin. All cell lines were maintained at 37°C in a CO2 incubator supplemented with 10% fetal bovine serum. Short Tandem Repeat (STR) profiling has been conducted to authenticate the cell lines that are predominantly used in this study. Stable clones of β-hCG overexpressing cells were generated by transfecting HCC1937, HCC1937/wt BRCA1, MDAMB-231 with β-hCG plasmids and the cells were selected with G418 for 45 days. Stable clones of BRCA1 knockdown cells were generated by transfecting MDAMB-231 with sh BRCA1 plasmid (sc-29219) and the cells were selected with puromycin for 45 days. Stable clones of BRCA1 knockdown cells, β-hCG overexpressing MDAMB-231 cells, were selected with both G418 and puromycin.
Real-time PCR, semiquantitative PCR and Chromatin Immunoprecipitation (ChIP)
Real-time PCR was carried out using cDNA synthesized from 1000 ng of total RNA isolated by High Pure RNA Isolation Kit (11828665001, Roche, Switzerland) as per manufacturer’s protocol. Real time PCR was carried out using SYBR Green (KM4101, Kappa Biosciences, Norway) as per the manufacturer’s protocol. Green GoTaq polymerase system (M791B, Promega, Madison, WI, USA) was used for semiquantitative PCR as per manufacturer’s protocol. The sonicated chromatin fragments were immunoprecipitated with anti-LSD1 or IgG antibody (sc-2025, SantaCruz Biotechnology, Dallas, TX, USA). Specific primers were used to amplify the DNA fragments using real-time PCR.
Immunofluorescence, immunoblotting and immunoprecipitation
Immunofluorescence and immunoblotting were carried out as described earlier (12). For immunoprecipitation, 1 mg of protein sample was immunoprecipitated with anti-BRCA1 or IgG antibody and the immunoblotting was carried out using anti-HSP90 antibody.
Antibodies and primers
Antibodies used for all the experiments were as follows:
For immunofluorescence, BRCA1 (9010S; Cell Signaling Technology, Danvers, MA, USA), γH2AX (2577S; Cell Signaling Technology) and β-catenin (8480P; Cell Signaling Technology) primary antibodies were followed by secondary antibodies conjugated with FITC (35552, Cell Signaling Technology). For immunoblotting, BRCA1 (sc-642, Santa Cruz Biotechnology), β-actin (sc-47778, Santa Cruz Biotechnology), β-hCG (ab9582, Abcam, Cambridge, United Kingdom), HSP90 (4877S, Cell Signaling Technology), β-catenin (8480P, Cell Signaling Technology), Slug (9585S, Cell Signaling Technology), Ubiquitin (3933S, Cell Signaling Technology), ABCG2 (sc-69988, Santa Cruz Biotechnology) and LSD1 (2184S, Cell Signaling Technology) were used. For immunoprecipitation BRCA1 (A301-377A, Bethyl Laboratories, Montgomery, TX, USA), HSP90 (4877S, Cell Signaling Technology) and normal IgG (sc-2025, Santa Cruz Biotechnology) were used. For ChIP, BRCA1 (A301-377A, Bethyl Laboratories) and LSD1 (2184S, Cell Signaling Technology) were used.
The primers used for all the experiments were listed as follows:
GAPDH forward 5′-CAACTACATGGTTTACATGTTC-3′
GAPDH reverse 5′-GCCAGTGGACTCCACGAC-3′
BRCA1 forward 5′’-AAGGTTGTTGATGTGGAGGAG-3′
BRCA1 reverse 5′-CAGAGGTTGAAGATGGTATGTTG-3′
ChIP: BRCA1 0.4kb up forward 5′-TTCCCTCCACCCCCCCAACAATC-3′
ChIP: BRCA1 0.4 kb up reverse 5′-CCCAATCCCCCACTCTTTCCGCC-3′
Cyclin D1 forward 5′-GGAACTGCTTCTGGTGAACAAG-3′
Cyclin D1 Reverse 5′-TGGAGGGTGGGTTGGAAATG-3′
Cyclin D2 forward 5′-GACTCCTAAGACCCATCTTCAG-3′
Cyclin D2 Reverse 5′-GCGAAGGATGTGCTCAATG -3′
Cyclin D3 forward 5′-GCCCCTGACTATTGAGAAGC-3′
Cyclin D3 Reverse 5′-GGTGTAATCTGTAGCACAGAGG-3′
Homologous recombination assay
Cells were supplemented with β-hCG and/or treated with 17AAG for 24 h, and homologous recombination (HR) assay procedure was followed as per manufacturer’s protocol (35600, Norgen Biotek Corporation, Thorold, ON, Canada). Two plasmids dl-1 and dl-2 were co-transfected into the cells supplemented with β-hCG and/or treated with 17AAG and plasmid isolation was carried out after 48 h. Semiquantitative and real-time PCR was carried out using universal primer that amplifies backbone dl-1 (546 bp) and/or dl-2 (258 bp) plasmids, and assay primers that amplifies HR product (420 bp).
Exogenous β-hCG, full-length native hCG and drug treatment
Purified β-hCG (ab126653, Abcam) and full-length native hCG (ab126654, Abcam) were reconstituted with defined volumes of nuclease-free water. Cells were allowed to adhere for 16–24 h in 10% fetal-bovine-serum-supplemented media. Later the media were replaced with serum-free media containing 5 IU/ml of β-hCG or full-length hCG for additional 24 h. The drugs, 17-N-allylamino-17-demethoxygeldanamycin (17AAG; D5193-1MG, Sigma Aldrich, St. Louis, MO, USA) and cycloheximide (C1988-1G, Sigma Aldrich), were procured from Sigma. Cycloheximide 100 μg/ml was used for all experiments. Different concentrations of 17AAG (0–1000 nM) were used in experiments. For treatments, cells were incubated with or without β-hCG for 24 h, followed by treating with or without cycloheximide or treating with or without 250-nM 17AAG, and the cells were either fixed for immunofluorescence or protein was isolated at different time points from 0 to 24 h.
Transgenic mouse model and xenograft tumor
The generation of transgenic mouse model was carried out as per the protocol mentioned earlier (13). Protein was isolated and immunoblot was conducted from the tumor sample excised from WAP and MMTV Cre: BRCA1ko/ko transgenic mouse models. Female SCID mice (6-week old) were used for the study. All the protocols were approved by Institute Animal Ethical Committee and performed in accordance with the guidelines and regulations of Institute Animal Ethical Committee of Rajiv Gandhi Centre for Biotechnology (RGCB), Thiruvananthapuram, Kerala, India. Xenograft tumor was developed by injecting 3 × 106 β-hCG overexpressing MDAMB-231 or control MDAMB-231 cells. After 15 days, the animals were killed and the tumor was collected as per the protocol. RNA was isolated as per manufacturers protocol (11828665001, Roche).
MTS assay, colony formation assay
For MTS Tetrazolium assay, the cells seeded at a density of 6000 per well in 96-well plate were supplemented with or without β-hCG for 24 h followed by treating with 17AAG for 96 h or ABT888 for 48 h. Similarly, β-hCG-silenced cells were treated with 17AAG for 96 h or ABT888 for 48 h or combination of 17AAG and ABT888 for 72 h.
For colony formation assay, the cells supplemented with or without β-hCG for 24 h were treated with 17AAG for 72 h. Thousand cells per well were seeded in 6-well plate and 14 days later, the colonies were fixed with methanol to acetic acid (3:1) and stained with crystal violet. Colonies with a minimum of 50 cells were counted manually.
Statistical analysis
One-way analysis of variance (Student’s t-test) was employed for statistical analysis. Experiments were performed in technical replicates and the data were shown in standard deviation. P < 0.05 was considered statistically significant.
Results
β-hCG induces proliferation in BRCA1 mutant but not in BRCA1 wild-type cells
We used HCC1937 cell line that has homozygous BRCA1 5382insC mutation and reconstituted HCC1937 (HCC1937/wt BRCA1) where BRCA1 has been reconstituted and codes for a functional wild-type BRCA1 protein. Also HCC1937 is a triple negative cell line, which has acquired TP53 mutation and PTEN mutation and causes aggressive basal breast cancer. Here, we analyzed the influence of β-hCG on cell proliferation, and stable β-hCG overexpressing HCC1937 (HCC1937 β) and HCC1937/wt BRCA1 (HCC1937/wt BRCA1 β) were analyzed for colony-forming ability (Figure 1A). The colony-forming ability was induced in HCC1937 β whereas it was reduced in HCC1937/wt BRCA1 β. In addition, no significant change on cell cycle progression marker, Cyclin D1, was observed in HCC1937 β whereas a significant reduction in Cyclin D1 was observed in HCC1937/wt BRCA1 β (Figure 1B). Further, to confirm the influence of β-hCG in BRCA1 wild-type cells, β-hCG overexpressing stable clones were developed in non-isogenic MDAMB-231 and the cell proliferation was analyzed. Cell proliferation was severely inhibited upon stable overexpression of β-hCG in MDAMB-231 as analyzed by cell proliferation and colony formation assays (Figure 1C and D). As cell proliferation is induced in BRCA1 mutant condition in the presence of β-hCG, we analyzed whether knockdown of BRCA1 in β-hCG overexpressing MDAMB-231 would induce cell proliferation. Stable knockdown of BRCA1 in MDAMB-231 impaired cell proliferation (Figure 1E and F), as BRCA1 was essential for cell cycle progression (14,15). Stable overexpression of β-hCG further impaired cell proliferation, indicating the tumorigenic effect of β-hCG occurs in the presence of BRCA1 mutant protein but not at suboptimal levels of wild-type BRCA1 expression (Figure 1E and F). In addition, xenograft tumors have been induced using β-hCG overexpressing MDAMB-231 cells and the levels of Cyclin D1/D2/D3 were analyzed. Cyclin D1 and D2 are significantly downregulated in β-hCG overexpressing MDAMB-231 in comparison to MDAMB-231 control tumor (Figure 1G), which clearly indicates the inhibition of proliferation by β-hCG in BRCA1 wild-type cells.
Figure 1.

β-hCG induces proliferation in BRCA1-mutated but reduces proliferation in BRCA1 wild-type breast cancer cells. (A) Analysis of cell proliferation by colony formation assay in β-hCG overexpressing HCC1937 (HCC1937 β) and HCC1937/wt BRCA1 (HCC1937/wt BRCA1 β). (B) RT-qPCR analysis of Cyclin D1 from β-hCG overexpressing HCC1937 (HCC1937 β) and HCC1937/wt BRCA1 (HCC1937/wt BRCA1 β). Cell proliferation assay of stable β-hCG overexpressing MDAMB-231 cells using (C) MTS and (D) colony formation assay. Cell proliferation assay of stable BRCA1 silenced and/or BRCA1 silenced with β-hCG overexpressing MDAMB-231 using (E) MTS assay and (F) colony formation assay. (G) Quantitative Real Time PCR (qRT-PCR) analysis of Cyclin D1, Cyclin D2 and Cyclin D3 from xenograft tumor induced by Control MDAMB-231 and/or β-hCG overexpressing MDAMB-231 cells. All the experiments are conducted in triplicates. All error bars in the graphs represent SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Further, we analyzed the effect of exogenous supplementation of full-length hCG, which has both α and β-hCG subunits, mimicking various physiological conditions such as (i) normal (<5 mIU), b) pregnancy (5 mIU to 288 IU) and (iii) gestational trophoblastic disease (>300 IU) to compare whether hCG has any effect on the proliferation of BRCA1 mutant and wild-type breast cancer cells. A concentration-dependent cell death was observed in BRCA1 wild-type cells whereas cell proliferation was induced in BRCA1 mutant cells (Supplementary Figure 1A, available at Carcinogenesis Online). All these observations clearly show that β-hCG or full-length hCG induces proliferation in BRCA1 mutant breast cancers whereas it inhibits cell proliferation in BRCA1 wild-type breast cancers.
β-hCG transcriptionally represses the expression of BRCA1 via LSD1/Slug in wild-type TNBC cells
Next, we analyzed whether β-hCG alters the expression of BRCA1 protein as proliferation was differentially affected with respect to BRCA1 status. Interestingly, we found that, in HCC1937, the expression of mutant BRCA1 was increased whereas the expression of wild-type BRCA1 protein was decreased in HCC1937/wt BRCA1 upon supplementation of 5IU of β-hCG for 24 h (Figure 2A). Similarly, BRCA1 wild-type protein expression is decreased upon supplementation of increasing concentrations of full-length hCG as well (Supplementary Figure 1B and C, available at Carcinogenesis Online). The increase in the mutant BRCA1 expression was significant only at the protein level because a decrease in BRCA1 expression was observed at messenger RNA (mRNA) level, in both BRCA1 wild-type and mutant cell lines (Figure 2B). However, in HCC1937/wt BRCA1 cells, the level of BRCA1 mRNA was decreased significantly upon 5 IU of β-hCG supplementation concomitant with wild-type BRCA1 protein reduction (Figure 2B). Similarly, a reduction in the expression of BRCA1 mRNA was observed in other breast cancer cell line, SUM149 (BRCA1 mutated) and benign breast transformed cell line, MCF10A (BRCA1 wild type; Figure 2C)
Figure 2.

β-hCG induces mutant but not wild-type BRCA1. (A) Western blot analysis of BRCA1 upon supplementation of 5 IU of β-hCG for 24 h in HCC1937 and HCC1937/wt BRCA1 cells. Quantification of the western blot on the right panel. β-actin was used as an endogenous control. (B) Quantitative Real Time PCR (qRT-PCR) analysis of BRCA1 expression upon supplementation of 5 IU of β-hCG for 24 h in HCC1937 and HCC1937/wt BRCA1. (C) qRT-PCR analysis of BRCA1 expression upon supplementation of 5 IU of β-hCG for 24 h in SUM149 and MCF10A. (D) Western blot analysis of β-catenin and Slug upon supplementation of 5 IU of β-hCG for 24 h in HCC1937 and HCC1937/wt BRCA1 cells. Quantitation was given on right panel. (E) Western blot analysis of LSD1 upon supplementation of 5 IU of β-hCG for 24 h in HCC1937 and HCC1937/wt BRCA1. Quantitation was given on right panel. (F) TCGA dataset analysis of LSD1 in BRCA1 wild-type and BRCA1 mutant human breast cancer tissue samples (Waddell’s dataset) using Oncomine. (G) qRT-PCR amplification of BRCA1 promoter immunoprecipitated with anti-LSD1 antibody from HCC1937 and HCC1937/wt BRCA1 supplemented with and/or without β-hCG. Normal chromatin fragments were used as input. (H) Western blot confirmation of silencing of LSD1 and Slug in HCC1937. (I) RT-qPCR analysis of BRCA1 in control, LSD1 and Slug-silenced HCC1937 in the presence and/or absence of β-hCG. Full blots of cropped images are included in supplementary information. *P < 0.05, **P < 0.01, ***P < 0.001.
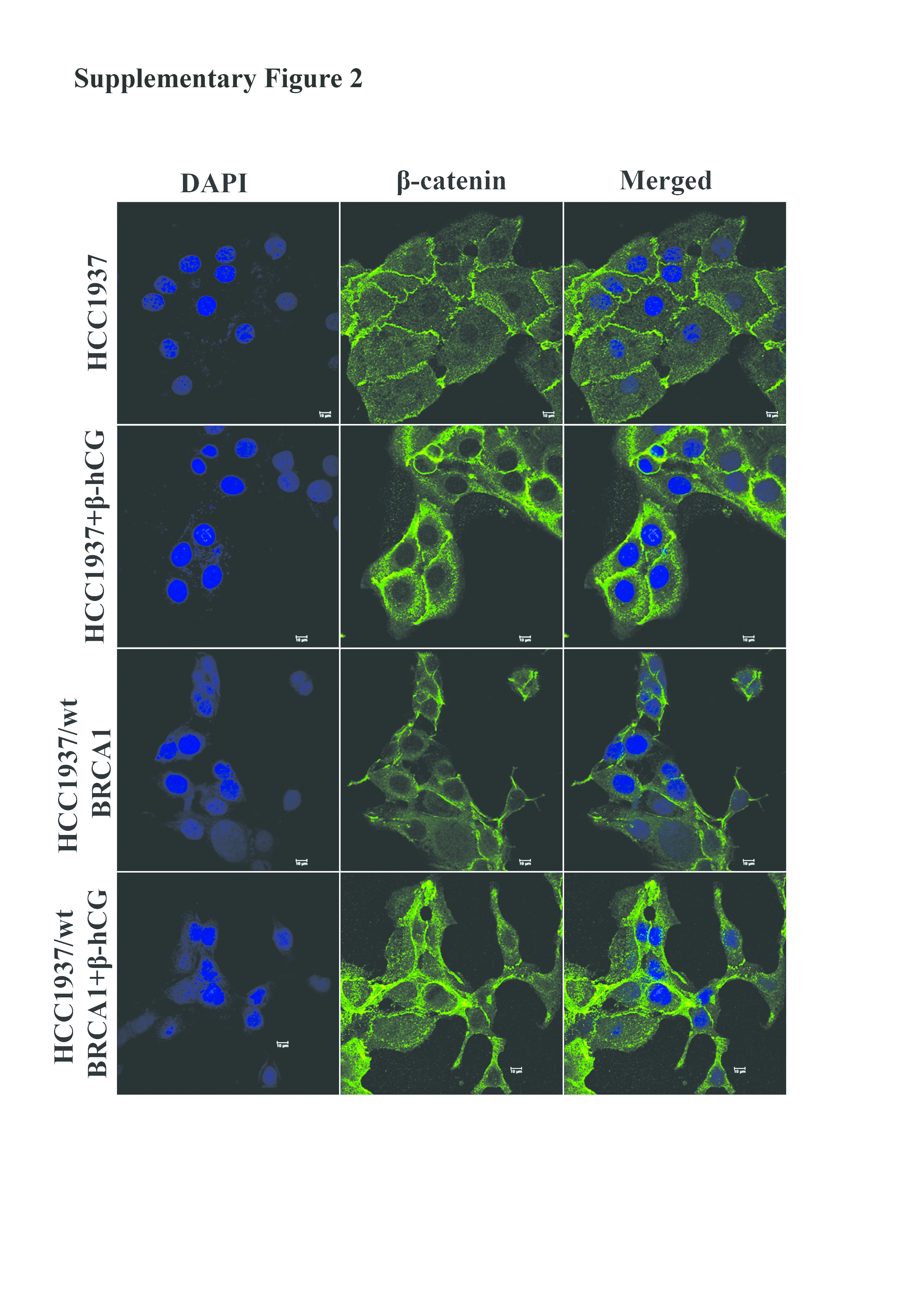
To identify the mechanism behind the reduced expression of BRCA1 mRNA in the presence of β-hCG, we looked for β-catenin–Slug–LSD1 axis that has been already reported to transcriptionally repress BRCA1 (16). It is observed that the expression of β-catenin and Slug was increased in HCC1937/wt BRCA1 but not in HCC1937 upon β-hCG supplementation (Figure 2D). Further, the activation of β-catenin is marked by the nuclear translocation upon supplementation of β-hCG in HCC1937/wt BRCA1 (Supplementary Figure 2, available at Carcinogenesis Online). Expression of Slug was correspondingly reversed when β-hCG is inhibited by siRNA in HCC1937 and HCC1937/wt BRCA1 (Supplementary Figure 3A, available at Carcinogenesis Online). Reduced expression of BRCA1 observed in HCC1937/wt BRCA1 could be due to Slug-mediated repression of BRCA1. On the other hand, expression of LSD1 was found to be high in HCC1937 but unaltered in HCC1937/wt BRCA1 upon β-hCG supplementation (Figure 2E). In accordance, LSD1 expression was higher in BRCA1-mutated than BRCA1 wild-type human breast cancer TNBC tissue samples in Waddell’s dataset (Reporter: ILMN_1813840) as analyzed by Oncomine (Figure 2F and Supplementary Figure 3B, available at Carcinogenesis Online). Further, the recruitment of LSD1 to the promoter of BRCA1 was found to be induced in HCC1937 but reduced in HCC1937/wt BRCA1 upon β-hCG supplementation (Figure 2G). In order to confirm the involvement of LSD1 and slug in reducing BRCA1 mRNA levels in HCC1937, we have analyzed the BRCA1 mRNA levels by silencing LSD1 and Slug in HCC1937. Silencing of LSD1 and Slug has been confirmed by western blot (Figure 2H). LSD1 silencing has significantly induced BRCA1 mRNA levels in HCC1937; however, supplementation of β-hCG in LSD1 silenced HCC1937 reduced the levels of BRCA1 mRNA as LSD1 is found to be induced upon β-hCG supplementation. Slug silencing has significantly induced BRCA1 mRNA levels; however, supplementation of β-hCG in Slug-silenced HCC1937 further increased the levels of BRCA1 mRNA as slug is found to be induced upon β-hCG supplementation (Figure 2I). Altogether, a net reduction in the mRNA levels of mutant and wild-type BRCA1 was observed (Supplementary Figure 3C, available at Carcinogenesis Online).
β-hCG induces HSP90-mediated stabilization of mutant BRCA1 protein
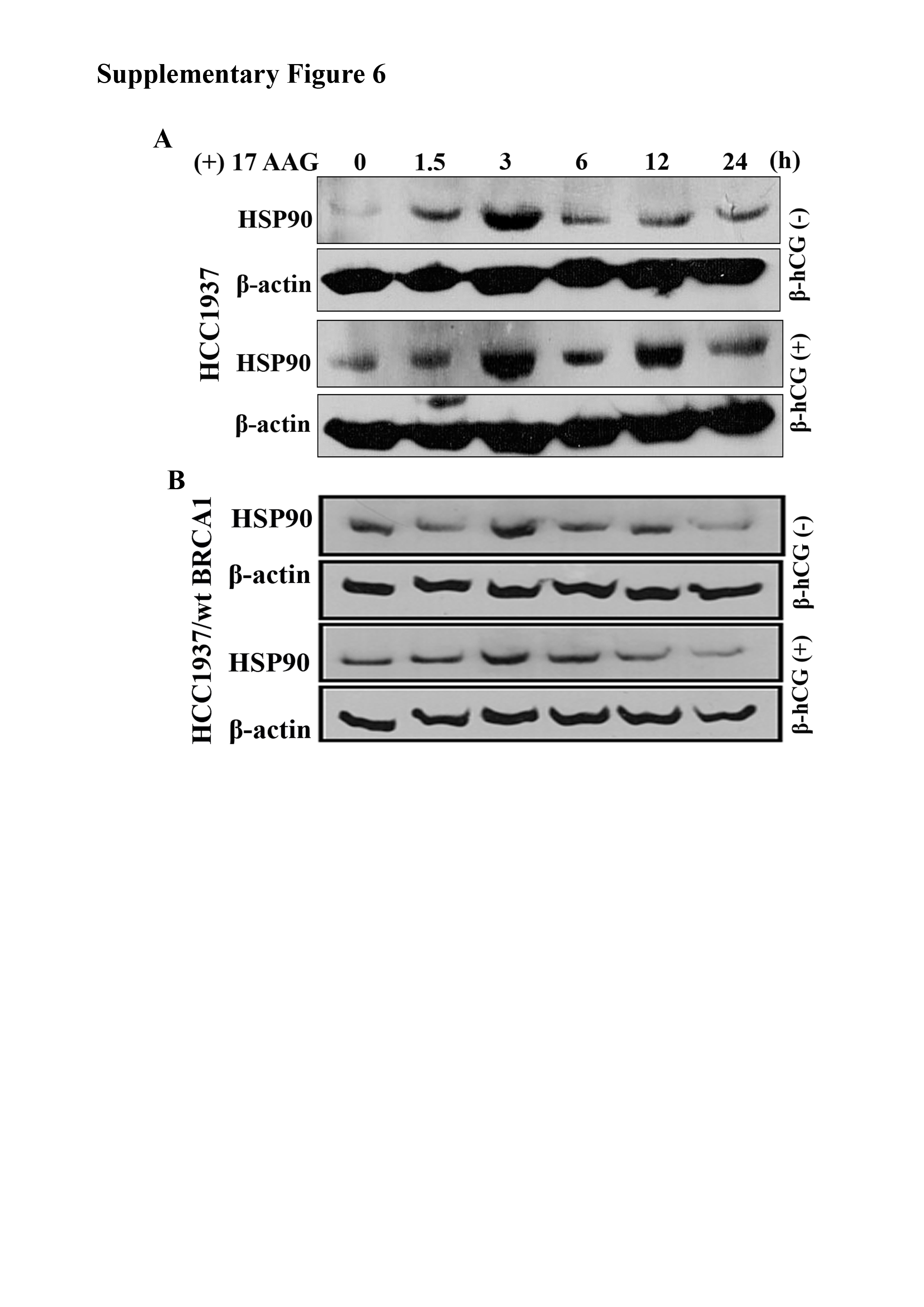
As transcriptional inhibition of mutant BRCA1 mRNA was observed, we determined whether the increase in mutant BRCA1 in HCC1937 upon β-hCG supplementation could be due to posttranslational modifications. At first, we analyzed the stability of mutant and wild-type BRCA1 with the help of cycloheximide, a protein synthesis inhibitor. As expected, mutant BRCA1 is highly unstable; however, upon supplementation of β-hCG, mutant BRCA1 protein was stable even up to 24 h in HCC1937. In contrast, wild-type BRCA1 is highly stable; however, in cells having wild-type BRCA1, wild-type BRCA1 protein stability was reduced upon β-hCG supplementation even at 6 h (Figure 3A and B). In addition, a high level of ubiquitinated BRCA1 was observed upon supplementation of β-hCG in HCC1937/wt BRCA1 whereas such effect was absent in HCC1937 upon blocking proteasome-mediated degradation of BRCA1 by MG132 (Figure 3C). All these observations clearly indicate that the stability of mutant BRCA1 has been induced upon β-hCG supplementation. Next, we looked for the mechanism underlining the stability of mutant BRCA1. By proteomic approach, using mass spectrometry, we identified HSP90AA1 and HSP90AB1 to be induced upon supplementation of β-hCG in BRCA1 mutant cells (data not shown). HSP90AA1 (HSP90α) and HSP90AB1 (HSP90β) share sequence and functional similarities and hence total HSP90 was analyzed in this study. HSP90 is known to stabilize many mutant proteins, including mutant BRCA1 (17). So, we analyzed the stability of BRCA1 with respect to HSP90 in the presence or in the absence of β-hCG. The level of HSP90 protein is increased in HCC1937 upon β-hCG supplementation and decreased upon endogenous silencing of β-hCG in these cells (Supplementary Figure 4A and B, available at Carcinogenesis Online). Also, HSP90 protein expression was higher in BRCA1-mutated than BRCA1 wild-type human breast cancer tissue samples (Figure 4A and B). In our previous report, we have developed BRCA1 conditional knockout mouse models (WAP-Cre; BRCA1KO/CO and MMTV-Cre; BRCA1KO/CO) and found that WAP-Cre; BRCA1KO/CO, which specifically induces tumor in breast tissue expresses high expression of β-hCG than MMTV-Cre; BRCA1KO/CO (12). Further, HSP90 was found to be higher in WAP-Cre; BRCA1KO/CO where β-hCG expression was higher than in MMTV-Cre; BRCA1KO/CO (Figure 4C). Further, a direct interaction between mutant BRCA1 and HSP90 was observed, which was increased upon supplementation of β-hCG (Figure 4D and E). To confirm the HSP90-mediated stabilization of mutant BRCA1, HSP90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin (17AAG) was used. The expression of HSP90 was not significantly altered by 17AAG in HCC1937 and HCC1937/wt BRCA1 supplemented with or without β-hCG, as 17AAG is reported to inhibit the ATPase activity of HSP90, not the expression levels (Supplementary Figure 5A and B, available at Carcinogenesis Online). Mutant BRCA1, but not wild-type BRCA1 protein expression, was significantly reduced upon 17AAG treatment. However, supplementation of β-hCG could retain mutant but not wild-type BRCA1 protein expression even in the presence of 17AAG, indicating that the presence of β-hCG could induce HSP90-mediated stabilization of mutant BRCA1 but not wild-type BRCA1 (Figure 4F). Similar phenomenon was observed in β-hCG overexpressing stable clones of HCC1937 and HCC1937/wt BRCA1 (Supplementary Figure 6, available at Carcinogenesis Online).
Figure 3.

β-hCG stabilizes mutant but not wild-type BRCA1. (A) Expression BRCA1 upon treating with 100 μM of cycloheximide (Cyclohex) over a time-period of up to 24 h in the presence (+) or in the absence (−) of β-hCG in HCC1937. (B) Similar experiment was performed to analyze expression of BRCA1 in HCC1937/wt BRCA1. Scale bars, 20 μm. Blue color in the immunofluorescence images represents 4′,6-diamidino-2-phenylindole, which stains the nucleus, whereas the green color indicates the staining for BRCA1 protein. (C) Western blot analysis of ubiquitin from β-hCG-supplemented HCC1937 and HCC1937/wt BRCA1 cells immunoprecipitated with anti-BRCA1 antibody. Proteasome-mediated degradation of protein from HCC1937 and HCC1937/wt BRCA1 was blocked using MG132. Full blots of cropped images are included in supplementary information.
Figure 4.

Presence of β-hCG results in the stabilization of BRCA1 even in the presence of HSP90 inhibitor. (A) TCGA dataset analysis of HSP90AB1 and HSP90AA1 in BRCA1 wild-type and BRCA1 mutant human breast cancer tissue samples (Waddell’s dataset) using Oncomine. (B) Immunohistochemical analysis of HSP90 in BRCA1 wild-type and BRCA1 mutant (5382insC and 185delA) human breast cancer tissues. (C) Western blot analysis of HSP90 from WAP-Cre; BRCA1KO/CO and MMTV-Cre; BRCA1KO/CO mouse tumor samples. Quantitation of the western blot is given on the right panel. (D) Western blot analysis of HSP90 immunoprecipitated with BRCA1 or IgG antibody in HCC1937, HCC1937/wt BRCA1 and MCF-7. (E) Western blot analysis of HSP90 immunoprecipitated with BRCA1 or IgG antibody in HCC1937 and MCF-7 supplemented with β-hCG. (F) Immunofluorescence for BRCA1 upon treating with 17AAG for a period of 24 h in the presence (+) or in the absence (−) of β-hCG in HCC1937 and HCC1937/wt BRCA1. Scale bars, 20 μm. Blue color in the immunofluorescence images represents 4′,6-diamidino-2-phenylindole, which stains the nucleus, whereas the green color indicates the staining for BRCA1 protein. Full blots of cropped images are included in supplementary information.
β-hCG rescues HR and induces drug resistance in BRCA1 mutant cells
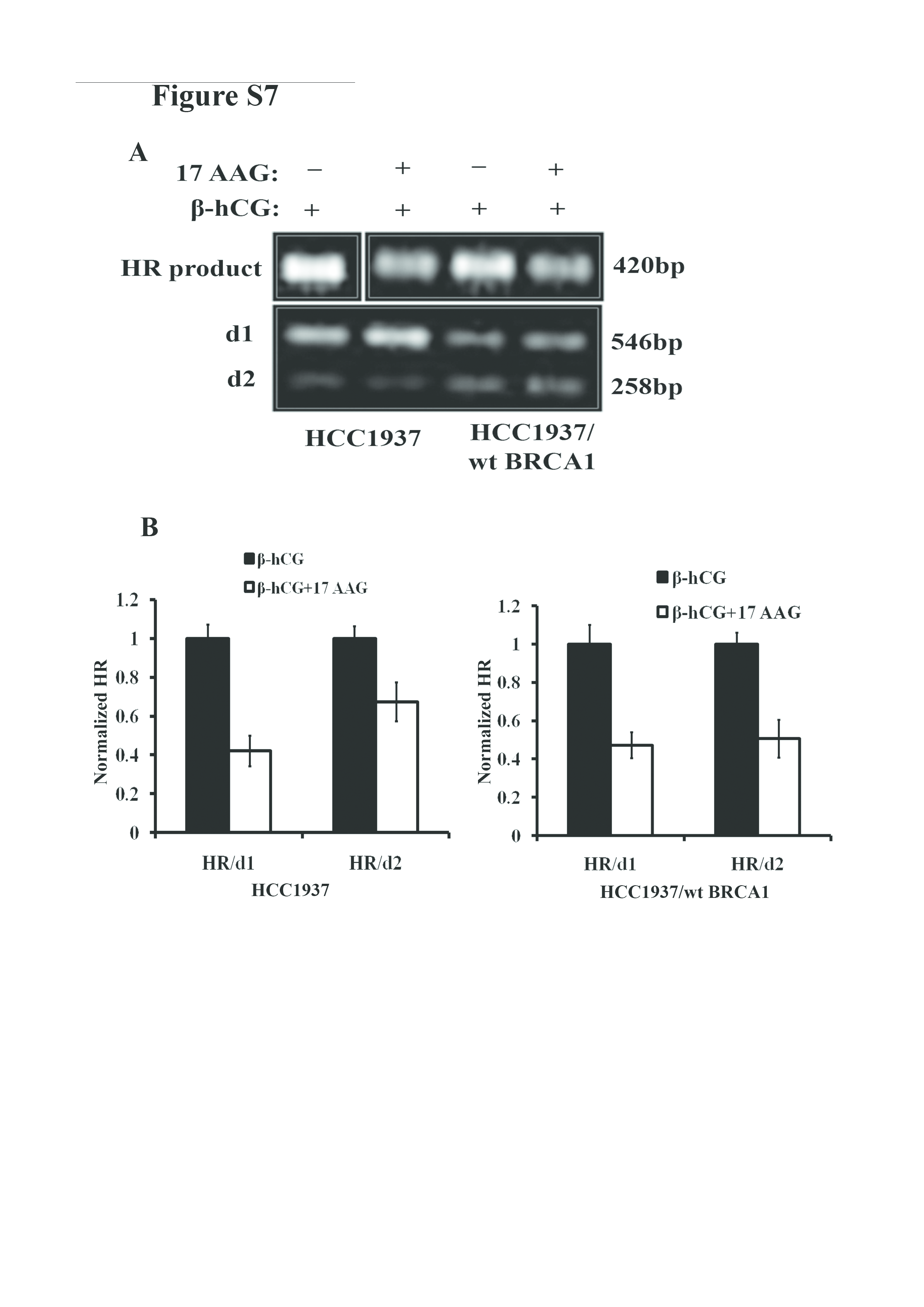
Recently, the concept of hypomorphic BRCA1 has evolved in which the mutant BRCA1 retains partial tumor suppressive activity (18,19). As β-hCG induces mutant but not wild-type BRCA1, we analyzed the level of HR in both HCC1937 and HCC1937/wt BRCA1 cell lines. In the presence of β-hCG, the level of HR has been increased in mutant cells, whereas it was reduced in wild-type cells (Figure 5A and B). Further, effect of 17AAG on HR was assessed in both the cell lines as it affects the stability of mutant and wild-type BRCA1. As expected, addition of 17AAG along with β-hCG rescues HR only in HCC1937 whereas HR level was reduced in HCC1937/wt BRCA1 (Figure 5C, Supplementary Figure 7A and B, available at Carcinogenesis Online). To further confirm that mutant BRCA1 still possess HR function, mutant BRCA1 has been stably silenced using BRCA1 shRNA and HR level was analyzed. A significant reduction in the level of HR was observed clearly indicating the role of mutant BRCA1 in HR (Figure 5D). Addition of 17AAG along with β-hCG did not show any difference in the ability of HR, when mutant BRCA1 is stably silenced in HCC1937 (Figure 5E). The influence on HR in BRCA1 mutant and wild-type cells with respect to β-hCG and 17AAG is depicted in Figure 5F. All these observations clearly demonstrate that mutant BRCA1 could be hypomorphic and induce HR in the presence of β-hCG.
Figure 5.

β-hCG induces HR in BRCA1 mutant but not in wild-type cells. (A) HR assay showing the level of recombination in HCC1937 and HCC1937/wt BRCA1 upon supplementation of β-hCG. d1 and d2 plasmid were taken as internal control. The d1 and d2 were obtained as 546bp and 258bp products whereas recombination product (HR) was obtained at 420bp. (B) HR levels were normalized to HCC1937 and quantitation of HR was given on right panel. (C) Quantitative Real Time PCR (qRT-PCR) analysis showing the level of HR in HCC1937 and HCC1937/wt BRCA1 cells treated with 17AAG in the presence or absence of β-hCG. HR levels were normalized to HCC1937 + 17AAG. (D) qRT-PCR analysis showing the level of HR in HCC1937 and mutant BRCA1 stably silenced HCC1937 cells (HCC1937 sh BRCA1). (E) qRT-PCR analysis showing the level of HR in HCC1937 and mutant BRCA1 stably silenced HCC1937 cells (HCC1937 sh BRCA1) treated with 17AAG in the presence or absence of β-hCG. (F) Pictorial depiction of HR in BRCA1 mutant and wild-type cells with respect to β-hCG. Full blots of cropped images are included in supplementary information. **P < 0.01.
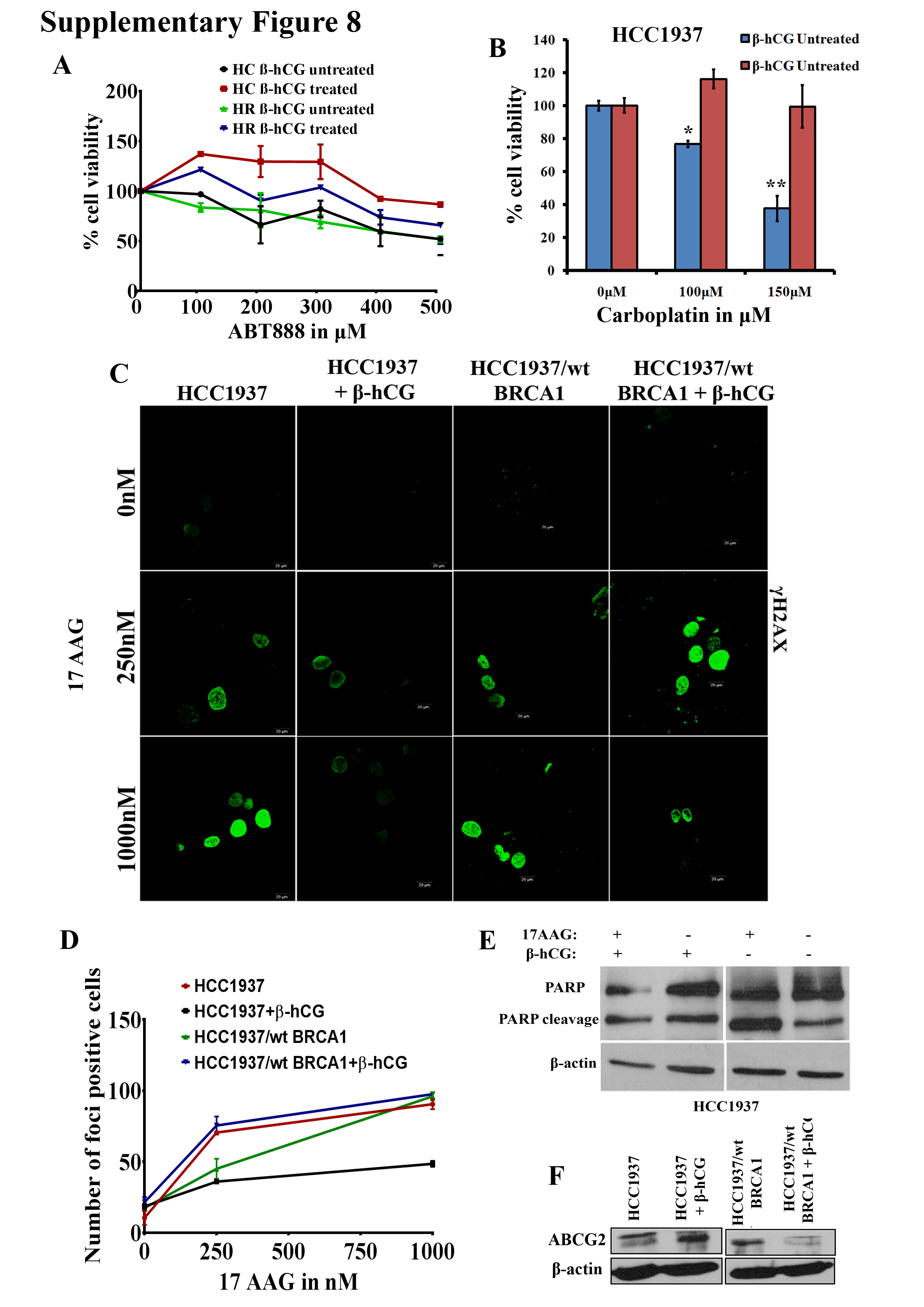
Overexpression of mutant BRCA1 has been reported in chemoresistant clones of breast cancer cell lines (17). Conversely, we analyzed whether the rescue of HR repair through β-hCG-mediated stabilization of mutant BRCA1 induces chemoresistance. The colony-forming ability was increased in the presence of β-hCG when treated with increasing concentrations of 17AAG only in HCC1937 whereas it was reduced in HCC1937/wt BRCA1 (Figure 6A). Further, by cell proliferation assay, it has been observed that β-hCG supplementation makes the mutant BRCA1 cells, resistant and wild-type BRCA1 cells sensitive to 17AAG and PARP inhibitor, ABT888 (Figure 6B and Supplementary Figure 8A, available at Carcinogenesis Online). Also, mutant BRCA1 cells are resistant to carboplatin upon β-hCG supplementation in HCC1937 (Supplementary Figure 8B, available at Carcinogenesis Online). Alternatively, silencing of endogenous β-hCG makes the BRCA1 mutant cells sensitive to 17AAG (Figure 6C). In addition, stable knockdown of mutant BRCA1 makes the BRCA1-mutant cells sensitive to 17AAG (Figure 6D). γH2A.X foci formation is an indicator of DNA damage response. β-hCG could reduce the γH2A.X foci formation induced by 17AAG in HCC1937 but not in HCC1937/wt BRCA1 (Supplementary Figure 8C and D and Supplementary Figure 9, available at Carcinogenesis Online). Also, PARP cleavage was found to be high in β-hCG unsupplemented HCC1937 cells treated with 17AAG in comparison to β-hCG supplemented HCC1937 cells treated with 17AAG (Supplementary Figure 8E, available at Carcinogenesis Online). In addition, the multidrug resistance protein ABCG2 was increased in β-hCG overexpressing HCC1937 (HCC1937 β) but decreased in β-hCG overexpressing HCC1937/wt BRCA1 (HCC1937/wt BRCA1 β), which clearly confirms that in the presence of β-hCG, BRCA1 mutant cells are highly resistant whereas BRCA1 wild-type cells are highly sensitive to HSP90 and PARP inhibitors (Supplementary Figure 8F, available at Carcinogenesis Online). All these observations clearly demonstrate β-hCG–HSP90–mutant BRCA1 cascades in inducing drug resistance in BRCA1-defective breast cancers.
Figure 6.

β-hCG induces drug resistance in BRCA1 mutant cells. (A) Colony-forming ability (adherent colonies) of HCC1937 and HCC1937/wt BRCA1 in the presence or in the absence of 5 IU of β-hCG after treating with indicated doses of 17AAG for 72 h. (B) Cell proliferation of HCC1937 and HCC1937/wt BRCA1 cells treated with increasing concentrations of 17AAG for 96 h in the presence or in the absence of β-hCG as assessed by MTS assay. (C) Cell proliferation of β-hCG-silenced HCC1937 cells treated with increasing concentrations of 17AAG for 72 h as assessed by MTS assay. (D) Cell proliferation of BRCA1 stably silenced HCC1937 cells treated with increasing concentrations of 17AAG for 72 h as assessed by MTS assay. (E) The level of phosphorylation of TGFBRII in HCC1937 and HCC1937/wt BRCA1cells treated with and/or without 17AAG in the presence or absence of β-hCG. (F) Western blot analysis of P-CHK2 and γ-H2A.X in HCC1937 and MCF7 treated with 17AAG or SB-431542 or combination of both. (G) Cell proliferation of HCC1937 and MCF7 treated with combination of 17AAG and SB-431542 as assessed by MTS assay. Full blots of cropped images are included in supplementary information.
β-hCG–HSP90–TGFBRII axis in inducing tumorigenesis of BRCA1 mutant cancers
β-hCG has been documented to induce tumorigenesis via TGFBRII in BRCA1-defective cancer cells (12). HSP0 has been already reported to stabilize TGFBRII by direct interaction (20). As β-HSP90 is induced in β-hCG, we intended to unveil the role of β-hCG-induced HSP90 in TGFBRII signaling. Hence, cells were treated with 17AAG along with β-hCG. Severe inhibition of P-TGFBRII was observed in both HCC1937 and HCC1937/wt BRCA1 upon inhibiting HSP90 with 17AAG. The inhibition of P-TGFBRII was rescued when β-hCG was supplemented along with 17AAG in HCC1937 but not in HCC1937/wt BRCA1 (Figure 6E). This shows that HSP90-mediated stabilization of TGFBRII is essential for β-hCG-induced TGFBRII signaling in BRCA1-defective condition. Further, combination of 17AAG and TGFBRII inhibitor, SB-431542, has induced comparatively higher levels of DNA damage than 17AAG or SB-431542 alone as marked by γH2A.X accumulation (Figure 6F). Also, cell viability was severely impaired in BRCA1-defective HCC1937 than BRCA1 wild-type MCF-7 upon treating with a combination of 17AAG and SB-431542 (Figure 6G). Targeting β-hCG-mediated HSP90–TGFBRII axis by a combination of 17AAG and SB-431542 could prove effective treatment strategy for β-hCG expressing BRCA1-defective cancers (Supplementary Figure 10, available at Carcinogenesis Online).
Discussion
We have already demonstrated that β-hCG overexpression is observed in BRCA1-defective cancer cells. Further, it was shown that wild-type BRCA1 could bind to the promoter and inhibit the expression of β-hCG and also that the overexpression of β-hCG in BRCA1 mutant cells could phosphorylate TGFBRII and induce proliferation and invasion (12). hCG and breast cancers are well debated terms with respect to its tumorigenic and antitumorigenic effects. It has been reviewed that although the full-length hCG exhibits protective effect against breast cancers, β-hCG expression proves to be tumorigenic and hence an ideal target for breast cancer therapy (21,22). In this study, β-hCG induces proliferation in BRCA1 mutant cells but not in cells that have wild-type BRCA1. Exposure to β-hCG or to physiological conditions that induce β-hCG, such as pregnancy, could induce tumorigenesis in BRCA1-mutated individuals. However, pregnancy could be protective in BRCA1 wild-type individuals. Previous studies supporting the induction of apoptosis by β-hCG have essentially used the BRCA1 wild-type cells (7). The controversial ‘dual role’ of β-hCG in contextually promoting or inhibiting tumorigenesis can be explained by this study (23). Here, we have observed that β-hCG could induce the overexpression of mutant BRCA1 protein and inhibition of wild-type BRCA1 protein by two separate mechanisms. There was a reduction in mRNA expression of BRCA1 in both the BRCA1 mutant (HCC1937 and SUM149) as well as the BRCA1 wild-type (HCC1937/wt BRCA1 and MCF10A) cell lines upon β-hCG supplementation. We analyzed whether the repression of wild-type BRCA1 by β-hCG was through Wnt/β-catenin signaling pathway as the activation of canonical Wnt signaling is known to be capable of repressing the expression of BRCA1 mRNA (16). Transcriptional inhibition of BRCA1 mRNA can occur either by β-catenin-Slug or LSD1-mediated pathway. As Slug is overexpressed in BRCA1 wild-type cells, Slug-mediated repression could be the reason for inhibition of BRCA1 in these cells. Presence of β-hCG induces recruitment of LSD1 to the promoter of mutant BRCA1, which could be the reason for inhibition of BRCA1 mRNA in HCC1937 cells. Correspondingly a high expression of LSD1 protein was observed in BRCA1 conditional knockout mouse tissues. A higher expression of LSD1 was observed in WAP Cre conditional knockout mouse tissues as β-hCG was found to be higher in WAP Cre than in MMTV Cre mouse models (12). We have also seen that decreased ubiquitination might be the reason for increased expression of mutant BRCA1 protein upon β-hCG supplementation. Here, the interesting point is that the mutant but not wild-type BRCA1 protein is stabilized by HSP90 in the presence of β-hCG. HSP90 is already reported to stabilize the cancer-related mutant proteins including mutant BRCA1 and has been implicated in the process of tumorigenesis (17). Here, we observed that supplementation of β-hCG can induce direct interaction between mutant BRCA1 and HSP90 proteins. Our observations have clearly demonstrated that the presence of β-hCG induces HSP90-mediated stabilization of mutant BRCA1.
There are multiple pathways by which HR could be restored by BRCA1 upon replication arrest. As we have already seen that β-hCG induces mutant but not wild-type BRCA1, the functional consequence of the change in BRCA1 protein expression was analyzed in terms of HR. In contrast to what is expected, mutant BRCA1 still retains residual levels of HR that was induced upon supplementation of β-hCG. On the contrary, this treatment reduced HR repair efficiency in wild-type BRCA1 condition. Although 17AAG reduces HR in both the cell lines, supplementation of β-hCG restores HR only in HCC1937 as mutant BRCA1 is stabilized even in the presence of 17 AAG.
Even though full-length BRCA1 is absent in HCC1937, the splice variants may be partially functional resulting in partial restoration of HR (24). Also it has been reported that 53BP1 loss can restore HR in the absence of wild-type BRCA1 (25,26). Either of this could be the reason for partial HR restoration in BRCA1-defective cells upon β-hCG supplementation. Restoration of wild-type BRCA1 in HCC1937 by secondary mutation has been excluded, as HR is reduced in wild-type BRCA1 condition (HCC1937/wt BRCA1) upon β-hCG supplementation. In support of this, we observed an increase in cell proliferation and colony-forming ability upon supplementing β-hCG in 17AAG treated HCC1937 over HCC1937/wt BRCA1. Increase in the HR in the presence β-hCG might have resulted in drug resistance in HCC1937. It has been reported that 17AAG in combination with olaparib downregulated BRCA1 and/or RAD51 protein levels and induced significantly more γH2AX activation (27). In this study, we have shown that 17AAG-induced γH2AX was reduced upon supplementation of β-hCG, which could be the reason for increased HR in BRCA1-defective cells. Correspondingly, the BRCA1-defective but not wild-type cells become resistant to 17AAG in the presence of β-hCG.
β-hCG has been documented to induce tumorigenesis via TGFBRII in BRCA1-defective cancer cells (12). As HSP90 is known to induce Smad2/3 signaling by stabilizing TGFBRII (20), the induction of HSP90 by β-hCG could be the reason for TGFBRII signaling in addition to direct phosphorylation of TGFBRII by β-hCG in BRCA1-defective condition. Even in the presence of HSP90 inhibitor, β-hCG mediated phosphorylation of TGFBRII only in BRCA1-mutated cells, which may be the root cause for tumor proliferation in such cells. Mechanistically, stabilization of mutant BRCA1 by β-hCG could be an important event in the tumorigenesis of BRCA1-mutated triple negative breast tumors.
More research needs to be conducted to substantiate the effect of β-hCG with respect to BRCA1 in breast cancer. As it has been reported that 30% of breast cancer cases show BRCA1 promoter hyper methylation, the effect of β-hCG in breast cancer would possibly address larger population of patients with breast cancer. Also, preclinical trials with mouse models to understand the effect of pregnancy-induced breast tumorigenesis in BRCA1 mutation carriers need to be evaluated.
Conclusion
In breast cancers, β-hCG expression is highly controversial with literatures supporting both its protective and tumorigenic effects. It has also been reported that if a woman is a BRCA1 mutation carrier, then the risk of breast cancer at an early age is increased with full-term pregnancies. In this study, a combination of HSP90 inhibitor and TGFBRII inhibitor has shown to sensitize β-hCG expressing BRCA1-defective breast cancers to cell death. Targeting the β-hCG–HSP90–TGFBRII axis could prove an effective treatment strategy for BRCA1-mutated breast tumors.
Funding
Intramural grant from Rajiv Gandhi Centre for Biotechnology, Kerala State Council for Science Technology and Environment (016/SRSHS/2011/CSTE); grant-in-aid from the Board of Research in Nuclear Sciences (No. 2009/37/5/BRNS/1620 and No. 37(1)/14/16/2014); Indian Council for Medical Research (No. 53/20/2012-BMS); and Science and Engineering Research Board, Department of Science and Technology, Government of India (EMR/2017/002222 to P.S.)
Conflict of Interest Statement: None declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We acknowledge Professor Chu Xia Deng, Faculty of Health Sciences, University of Macau, China, for providing transgenic mice. The Senior Research Fellowships awarded by University Grants Commission to S.K.S., Indian Council for Medical Research to S.K.H., Council for Scientific and Industrial Research to R.N., Government of India, are duly acknowledged. The support from Kerala University is also acknowledged.
Glossary
Abbreviations
- ChIP
Chromatin Immunoprecipitation
- hCG
human Chorionic Gonadotropin
- HR
homologous recombination
- mRNA
messenger RNA
References
- 1. Russo J., et al. (2005) The protective role of pregnancy in breast cancer. Breast Cancer Res., 7, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meier-Abt F., et al. (2014) How pregnancy at early age protects against breast cancer. Trends Mol. Med., 20, 143–153. [DOI] [PubMed] [Google Scholar]
- 3. Meier-Abt F., et al. (2013) Parity induces differentiation and reduces Wnt/Notch signaling ratio and proliferation potential of basal stem/progenitor cells isolated from mouse mammary epithelium. Breast Cancer Res., 15, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jernström H., et al. (1999) Pregnancy and risk of early breast cancer in carriers of BRCA1 and BRCA2. Lancet, 354, 1846–1850. [DOI] [PubMed] [Google Scholar]
- 5. Andrieu N., et al. ; EMBRACE; GENEPSO; GEO-HEBON; IBCCS Collaborators Group (2006) Pregnancies, breast-feeding, and breast cancer risk in the International BRCA1/2 Carrier Cohort Study (IBCCS). J. Natl. Cancer Inst., 98, 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartge P., et al. (2002) Breast cancer risk in Ashkenazi BRCA1/2 mutation carriers: effects of reproductive history. Epidemiology, 13, 255–261. [DOI] [PubMed] [Google Scholar]
- 7. Lopez D., et al. (2008) Purified human chorionic gonadotropin induces apoptosis in breast cancer. Mol. Cancer Ther., 7, 2837–2844. [DOI] [PubMed] [Google Scholar]
- 8. He L.Z., et al. (2004) A novel human cancer vaccine elicits cellular responses to the tumor-associated antigen, human chorionic gonadotropin beta. Clin. Cancer Res., 10, 1920–1927. [DOI] [PubMed] [Google Scholar]
- 9. Delves P.J., et al. (2007) Designing a new generation of anti-hCG vaccines for cancer therapy. Mol. Cell. Endocrinol., 260-262, 276–281. [DOI] [PubMed] [Google Scholar]
- 10. Li Z., et al. (2013) Human chorionic gonadotropin β induces migration and invasion via activating ERK1/2 and MMP-2 in human prostate cancer DU145 cells. PLoS One, 8, e54592. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Yuri T., et al. (2014) Human chorionic gonadotropin suppresses human breast cancer cell growth directly via p53-mediated mitochondrial apoptotic pathway and indirectly via ovarian steroid secretion. Anticancer Res., 34, 1347–1354. [PubMed] [Google Scholar]
- 12. Sengodan S.K., et al. (2017) BRCA1 regulation on β-hCG: a mechanism for tumorigenicity in BRCA1 defective breast cancer. Oncogenesis, 6, e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nair R.S., et al. (2016) Increased sensitivity of BRCA defective triple negative breast tumors to plumbagin through induction of DNA Double Strand Breaks (DSB). Sci. Rep., 6, 26631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mullan P.B., et al. (2006) The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene, 25, 5854–5863. [DOI] [PubMed] [Google Scholar]
- 15. Deng C.X. (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res., 34, 1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu Z.Q., et al. (2012) Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA., 109, 16654–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson N., et al. (2013) Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA., 110, 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sonnenblick A., et al. (2015) An update on PARP inhibitors—moving to the adjuvant setting. Nat. Rev. Clin. Oncol., 12, 27–41. [DOI] [PubMed] [Google Scholar]
- 19. Aly A., et al. (2011) BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J. Mol. Cell Biol., 3, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wrighton K.H., et al. (2008) Critical regulation of TGFbeta signaling by Hsp90. Proc. Natl. Acad. Sci. USA., 105, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuler-Toprak S., et al. (2017) Human chorionic gonadotropin and breast cancer. Int. J. Mol. Sci., 18:1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gehring C., et al. (2016) The controversial role of human chorionic gonadotropin in the development of breast cancer and other types of tumors. Breast, 26, 135–140. [DOI] [PubMed] [Google Scholar]
- 23. Toniolo P., et al. (2010) Human chorionic gonadotropin in pregnancy and maternal risk of breast cancer. Cancer Res., 70, 6779–6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu Y., et al. (2016) BRCA1-deficient breast cancer cell lines are resistant to MEK inhibitors and show distinct sensitivities to 6-thioguanine. Sci. Rep., 6, 28217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bouwman P., et al. (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol., 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bunting S.F., et al. (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi Y.E., et al. (2014) Sublethal concentrations of 17-AAG suppress homologous recombination DNA repair and enhance sensitivity to carboplatin and olaparib in HR proficient ovarian cancer cells. Oncotarget, 5, 2678–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.