Abstract
In diesem Kapitel werden zunächst die für die Neurointensivmedizin wesentlichen bakteriellen Infektionen (Meningitis, spinale und Hirnabszesse, Spondylodiszitis, septisch-embolische Herdenzephalitis) abgehandelt, die trotz gezielt eingesetzter Antibiotika und neurochirurgischer Therapieoptionen noch mit einer erheblichen Morbidität und Mortalität behaftet sind. Besonderheiten wie neurovaskuläre Komplikationen, die Tuberkulose des Nervensystems, Neuroborreliose, Neurosyphilis und opportunistische Infektionen bei Immunsuppressionszuständen finden hierbei besondere Berücksichtigung. Der zweite Teil dieses Kapitels behandelt akute und chronische Virusinfektionen des ZNS sowie in einem gesonderten Abschnitt die HIVInfektion und HIV-assoziierte Krankheitsbilder sowie Parasitosen und Pilzinfektionen, die in Industrieländern seit Einführung der HAART bei HIV zwar eher seltener, aber mit zunehmender Globalisierung auch in unseren Breiten immer noch anzutreffen sind.
Zum Einstieg
In diesem Kapitel werden zunächst die für die Neurointensivmedizin wesentlichen bakteriellen Infektionen (Meningitis, spinale und Hirnabszesse, Spondylodiszitis, septisch-embolische Herdenzephalitis) abgehandelt, die trotz gezielt eingesetzter Antibiotika und neurochirurgischer Therapieoptionen noch mit einer erheblichen Morbidität und Mortalität behaftet sind. Besonderheiten wie neurovaskuläre Komplikationen, die Tuberkulose des Nervensystems, Neuroborreliose, Neurosyphilis und opportunistische Infektionen bei Immunsuppressionszuständen finden hierbei besondere Berücksichtigung.
Der zweite Teil dieses Kapitels behandelt akute und chronische Virusinfektionen des ZNS sowie in einem gesonderten Abschnitt die HIV-Infektion und HIV-assoziierte Krankheitsbilder sowie Parasitosen und Pilzinfektionen, die in Industrieländern seit Einführung der HAART bei HIV zwar eher seltener, aber mit zunehmender Globalisierung auch in unseren Breiten immer noch anzutreffen sind.
Contributor Information
Stefan Schwab, Phone: +4949+49 9131 85 345 63, Email: stefan.schwab@uk-erlangen.de.
Peter Schellinger, Email: Peter.Schellinger@muehlenkreiskliniken.de.
Christian Werner, Email: wernerc@uni-mainz.de.
Andreas Unterberg, Email: Andreas.Unterberg@med.uni-heidelberg.de.
Werner Hacke, Phone: +4949+496221568211, Email: werner.hacke@med.uni-heidelberg.de.
Matthias Klein, Email: Matthias.Klein@med.uni-muenchen.de
Hans-Walter Pfister, Email: Hans-Walter.Pfister@med.uni-muenchen.de
Erich Schmutzhard, Email: Erich.Schmutzhard@i-med.ac.at
Uta Meyding-Lamadé, Email: meyding-lamade.uta@khnw.de
Corinna Schranz, Email: cschranz@gmx.de
Gabriele Arendt, Email: arendtg@uni-duesseldorf.de
Raimund Helbok, Email: raimund.helbok@uki.at
Bettina Pfausler, Email: b.pfausler@i-med.ac.at
André Grabowski, Email: grabowski@neuropraxis-ffm.de
Bodo Kress, Email: isemann.ingrid@khnw.de
Ronny Beer, Email: ronny.beer@i-med.ac.at
Literatur
Zu 32.1
- 1.Ajdukiewicz KM, Cartwright KE, Scarborough M, et al. Glycerol adjuvant therapy in adults with bacterial meningitis in a high HIV seroprevalence setting in Malawi: a double-blind, randomised controlled trial. Lancet Infect Dis. 2011;11:293–300. doi: 10.1016/S1473-3099(10)70317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auburtin M, Wolff M, Charpentier J, et al. Detrimental role of delayed antibiotic administration and penicillin-nonsusceptible strains in adult intensive care unit patients with pneumococcal meningitis: the PNEUMOREA prospective multicenter study. Crit Care Med. 2006;34:2758–2765. doi: 10.1097/01.CCM.0000239434.26669.65. [DOI] [PubMed] [Google Scholar]
- 3.Brouwer MC, Heckenberg SG, de Gans J, Spanjaard L, Reitsma JB, van de Beek D. Nationwide implementation of adjunctive dexamethasone therapy for pneumococcal meningitis. Neurology. 2010;75:1533–1539. doi: 10.1212/WNL.0b013e3181f96297. [DOI] [PubMed] [Google Scholar]
- 4.Brouwer MC, McIntyre P, de Gans J, Prasad K, van de BD. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2010;9:CD004405. doi: 10.1002/14651858.CD004405.pub3. [DOI] [PubMed] [Google Scholar]
- 5.Casado FJ, Blanco QA. ProcalcitoninA new marker for bacterial infection. An Esp Pediatr. 2001;54:69–73. doi: 10.1016/S1695-4033(01)78652-3. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri A, Martin PM, Kennedy PG, et al. EFNS guideline on the management of community-acquired bacterial meningitis: report of an EFNS Task Force on acute bacterial meningitis in older children and adults. Eur J Neurol. 2008;15:649–659. doi: 10.1111/j.1468-1331.2008.02193.x. [DOI] [PubMed] [Google Scholar]
- 7.de Gans J, van de Beek D. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347:1549–1556. doi: 10.1056/NEJMoa021334. [DOI] [PubMed] [Google Scholar]
- 8.Dubos F, Korczowski B, Aygun DA, et al. Serum procalcitonin level and other biological markers to distinguish between bacterial and aseptic meningitis in children: a European multicenter case cohort study. Arch Pediatr Adolesc Med. 2008;162:1157–1163. doi: 10.1001/archpedi.162.12.1157. [DOI] [PubMed] [Google Scholar]
- 9.Durand ML, Calderwood SB, Weber DJ, et al. Acute bacterial meningitis in adults. A review of 493 episodes. N Engl J Med. 1993;328:21–28. doi: 10.1056/NEJM199301073280104. [DOI] [PubMed] [Google Scholar]
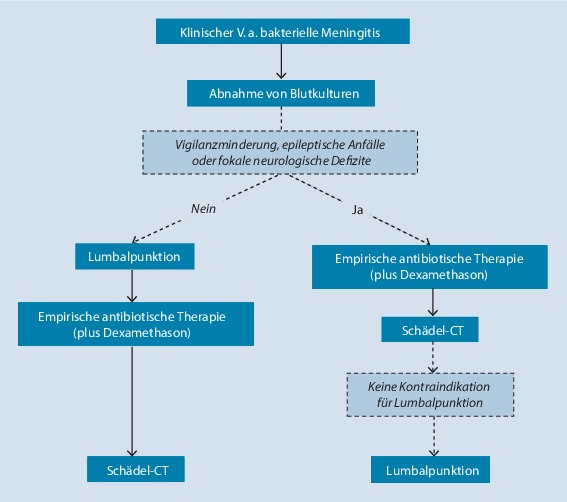
- 10.Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med. 2001;345:1727–1733. doi: 10.1056/NEJMoa010399. [DOI] [PubMed] [Google Scholar]
- 11.Heckenberg SG, de Gans J, Brouwer MC, et al. Clinical features, outcome, and meningococcal genotype in 258 adults with meningococcal meningitis: a prospective cohort study. Medicine (Baltimore) 2008;87:185–192. doi: 10.1097/MD.0b013e318180a6b4. [DOI] [PubMed] [Google Scholar]
- 12.Heckenberg SG, Brouwer MC, van der Ende A, van de Beek D. Adjunctive dexamethasone in adults with meningococcal meningitis. Neurology. 2012;79:1563–1569. doi: 10.1212/WNL.0b013e31826e2684. [DOI] [PubMed] [Google Scholar]
- 13.Imohl M, van der Linden M, Mutscher C, Reinert RR. Serotype distribution of invasive pneumococcal disease during the first 60 days of life. Vaccine. 2010;28:4758–4762. doi: 10.1016/j.vaccine.2010.04.097. [DOI] [PubMed] [Google Scholar]
- 14.Kastenbauer S, Pfister HW. Pneumococcal meningitis in adults: spectrum of complications and prognostic factors in a series of 87 cases. Brain. 2003;126(Pt 5):1015–1025. doi: 10.1093/brain/awg113. [DOI] [PubMed] [Google Scholar]
- 15.Klein M, Koedel U, Pfister HW. Oxidative stress in pneumococcal meningitis: a future target for adjunctive therapy? Prog Neurobiol. 2006;80:269–280. doi: 10.1016/j.pneurobio.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Klein M, Paul R, Angele B, Popp B, Pfister HW, Koedel U. Protein expression pattern in experimental pneumococcal meningitis. Microbes Infect. 2006;8:974–983. doi: 10.1016/j.micinf.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Klein M, Obermaier B, Angele B, et al. Innate immunity to pneumococcal infection of the central nervous system depends on toll-like receptor (TLR) 2 and TLR4. J Infect Dis. 2008;198:1028–1036. doi: 10.1086/591626. [DOI] [PubMed] [Google Scholar]
- 18.Klein M, Pfister HW, Leib SL, Koedel U. Therapy of community-acquired acute bacterial meningitis: the clock is running. Expert Opin Pharmacother. 2009;10:2609–2623. doi: 10.1517/14656560903277210. [DOI] [PubMed] [Google Scholar]
- 19.Klein M, Koedel U, Kastenbauer S, et al. Delayed cerebral thrombosis after initial good recovery from pneumococcal meningitis: past as prologue: delayed stroke as a parainfectious process of bacterial meningitis? Neurology. 2010;75:193–194. doi: 10.1212/WNL.0b013e3181e00e8e. [DOI] [PubMed] [Google Scholar]
- 20.Klein M, Koedel U, Pfefferkorn T, Zeller G, Woehrl B, Pfister HW. Arterial cerebrovascular complications in 94 adults with acute bacterial meningitis. Crit Care. 2011;15:R281. doi: 10.1186/cc10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koedel U, Klein M, Pfister HW. New understandings on the pathophysiology of bacterial meningitis. Curr Opin Infect Dis. 2010;23:217–223. doi: 10.1097/QCO.0b013e328337f49e. [DOI] [PubMed] [Google Scholar]
- 22.Kramer AH, Bleck TP. Neurocritical care of patients with central nervous system infections. Curr Treat Options Neurol. 2008;10:201–211. doi: 10.1007/s11940-008-0022-0. [DOI] [PubMed] [Google Scholar]
- 23.Krysan D. Serum procalcitonin levels aid in distinguishing bacterial from aseptic meningitis in children. J Pediatr. 2009;154:773. doi: 10.1016/j.jpeds.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 24.Molyneux EM, Walsh AL, Forsyth H, et al. Dexamethasone treatment in childhood bacterial meningitis in Malawi: a randomised controlled trial. Lancet. 2002;360:211–218. doi: 10.1016/S0140-6736(02)09458-8. [DOI] [PubMed] [Google Scholar]
- 25.Molyneux EM, Kawaza K, Phiri A, et al. Glycerol and Acetaminophen as Adjuvant Therapy Did Not Affect the Outcome of Bacterial Meningitis in Malawian Children. Pediatr Infect Dis J. 2014;33:214–216. doi: 10.1097/INF.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 26.Mourvillier B, Tubach F, van de Beek D, et al. Induced hypothermia in severe bacterial meningitis: a randomized clinical trial. JAMA. 2013;310:2174–2183. doi: 10.1001/jama.2013.280506. [DOI] [PubMed] [Google Scholar]
- 27.Nathan BR, Scheld WM. The potential roles of C-reactive protein and procalcitonin concentrations in the serum and cerebrospinal fluid in the diagnosis of bacterial meningitis. Curr Clin Top Infect Dis. 2002;22:155–165. [PubMed] [Google Scholar]
- 28.Nguyen TH, Tran TH, Thwaites G, et al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431–2440. doi: 10.1056/NEJMoa070852. [DOI] [PubMed] [Google Scholar]
- 29.Ntziora F, Falagas ME. Linezolid for the treatment of patients with central nervous system infection. Ann Pharmacother. 2007;41:296–308. doi: 10.1345/aph.1H307. [DOI] [PubMed] [Google Scholar]
- 30.Pfister HW, Feiden W, Einhaupl KM. Spectrum of complications during bacterial meningitis in adults. Results of a prospective clinical study. Arch Neurol. 1993;50:575–581. doi: 10.1001/archneur.1993.00540060015010. [DOI] [PubMed] [Google Scholar]
- 31.Pfister HW, Bühler R, Eiffert H, Nau R, Weber JR. Ambulant erworbene bakterielle (eitrige) Meningitis. Leitlinien für Diagnostik und Therapie in der Neurologie der DGN. Stuttgart: Thieme; 2012. pp. 494–504. [Google Scholar]
- 32.Proulx N, Frechette D, Toye B, Chan J, Kravcik S. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM. 2005;98:291–298. doi: 10.1093/qjmed/hci047. [DOI] [PubMed] [Google Scholar]
- 33.RKI (Robert Koch Institut) Empfehlung und Begründung einer postexpositionellen Meningokokken-Impfung. Epidemiol Bull. 2009;31:314–317. [Google Scholar]
- 34.RKI (Robert Koch Institut) (2010) Meningokokkenerkrankungen. RKI Ratgeber Infektionskrankheiten – Merkblätter für Ärzte. www.rki.de
- 35.RKI (Robert Koch Institut) Aktuelle Statistik meldepflichtiger Infektionskrankheiten, Deutschland. Epidemiol Bull. 2011;3:21. [Google Scholar]
- 36.RKI (Robert Koch Institut) Stellungnahme der Ständigen Impfkommission (STIKO) am Robert Koch-Institut (RKI) zum Stand der Bewertung des neuen Meningokokken-B-Impfstoffs Bexsero. Epidemiol Bull. 2013;49:495–498. [Google Scholar]
- 37.RKI (Robert Koch Institut) Jahresstatistik meldepflichtiger Infektionskrankheiten, Deutschland 2013. Robert Koch Institut. Epidemiol Bull. 2014;16:144. [Google Scholar]
- 38.Rupprecht TA, Pfister HW. Clinical experience with linezolid for the treatment of central nervous system infections. Eur J Neurol. 2005;12:536–542. doi: 10.1111/j.1468-1331.2005.01001.x. [DOI] [PubMed] [Google Scholar]
- 39.Sakushima K, Hayashino Y, Kawaguchi T, Jackson JL, Fukuhara S. Diagnostic accuracy of cerebrospinal fluid lactate for differentiating bacterial meningitis from aseptic meningitis: a meta-analysis. J Infect. 2011;62:255–262. doi: 10.1016/j.jinf.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Schut ES, Brouwer MC, de Gans J, Florquin S, Troost D, van de Beek D. Delayed cerebral thrombosis after initial good recovery from pneumococcal meningitis. Neurology. 2009;73:1988–1995. doi: 10.1212/WNL.0b013e3181c55d2e. [DOI] [PubMed] [Google Scholar]
- 41.Southwick FS. Septic thrombophlebitis of major dural venous sinuses. Curr Clin Top Infect Dis. 1995;15:179–203. [PubMed] [Google Scholar]
- 42.Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 2007;369:2196–2210. doi: 10.1016/S0140-6736(07)61016-2. [DOI] [PubMed] [Google Scholar]
- 43.van de Beek D, de Gans J, McIntyre P, Prasad K. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2007;1:CD004405. doi: 10.1002/14651858.CD004405.pub2. [DOI] [PubMed] [Google Scholar]
- 44.van de Beek D, Farrar JJ, de Gans J, et al. Adjunctive dexamethasone in bacterial meningitis: a meta-analysis of individual patient data. Lancet Neurol. 2010;9:254–263. doi: 10.1016/S1474-4422(10)70023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van de Beek D, Brouwer MC, Thwaites GE, Tunkel AR. Advances in treatment of bacterial meningitis. Lancet. 2012;380:1693–1702. doi: 10.1016/S0140-6736(12)61186-6. [DOI] [PubMed] [Google Scholar]
- 46.Weisfelt M, van de Beek D, Spanjaard L, Reitsma JB, de Gans J. Clinical features, complications, and outcome in adults with pneumococcal meningitis: a prospective case series. Lancet Neurol. 2006;5:123–129. doi: 10.1016/S1474-4422(05)70288-X. [DOI] [PubMed] [Google Scholar]
- 47.Winkler F, Kastenbauer S, Yousry TA, Maerz U, Pfister HW. Discrepancies between brain CT imaging and severely raised intracranial pressure proven by ventriculostomy in adults with pneumococcal meningitis. J Neurol. 2002;249:1292–1297. doi: 10.1007/s00415-002-0844-8. [DOI] [PubMed] [Google Scholar]
Zu 32.2
- 48.Baddley JW, Salzman D, Pappas PG. Fungal brain abscess in transplant recipients: epidemiologic microbiologic, and clinical features. Clin Transplant. 2000;16:419–424. doi: 10.1034/j.1399-0012.2002.02033.x. [DOI] [PubMed] [Google Scholar]
- 49.Bernal E, Munoz A, Nunez ML, Cano A (2010) Multidrug-resistant spinal tuberculosis and giant paraspinal abscess. Med Clin (Barc) Nov 10 (Epub ahead of print) [DOI] [PubMed]
- 50.Boviatsis EJ, Kouyialis AT, Stranjalis G, et al. CT-guided stereotactic biopsies of brain stem lesions: personal experience and literature review. Neurol Sci. 2003;24:97–102. doi: 10.1007/s10072-003-0093-3. [DOI] [PubMed] [Google Scholar]
- 51.Chang MC, Wu HT, Lee CH, et al. Tuberculous spondylitis and pyogenic spondylitis: comparative magnetic resonance imaging features. Spine. 2006;31:782–288. doi: 10.1097/01.brs.0000206385.11684.d5. [DOI] [PubMed] [Google Scholar]
- 52.Demir MK, Hakan T, Kilicoglu G, et al. Bacterial brain abscesses: prognostic value of an imaging severity index. Clin Radiol. 2007;62:564–572. doi: 10.1016/j.crad.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Greenberg BM. Central nervous system infections in the intensive care unit. Semin Neurol. 2008;28:682–689. doi: 10.1055/s-0028-1105976. [DOI] [PubMed] [Google Scholar]
- 54.Honda H, Warren DK. Central nervous system infections: meningitis and brain abscess. Infect Dis Clin North Am. 2009;23:609–623. doi: 10.1016/j.idc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 55.Jansson AK, Enblad P, Sjolin J. Efficacy and safety of cefotaxime in combination with metronidazole for empirical treatment of brain abscess in clinical practice: a retrospective study of 66 consecutive cases. Eur J Clin Microbiol Infect Dis. 2004;23:7–14. doi: 10.1007/s10096-003-1055-7. [DOI] [PubMed] [Google Scholar]
- 56.Kang K, Lim I, Roh JK. Positron emission tomographic findings in a tuberculous brain abscess. Ann Nucl Med. 2007;21:303–306. doi: 10.1007/s12149-007-0023-1. [DOI] [PubMed] [Google Scholar]
- 57.Kao PT, Tseng HK, Liu CP, et al. Brain abscess: clinical analysis of 53 cases. J Microbiol Immunol Infect. 2003;36:129–136. [PubMed] [Google Scholar]
- 58.Kupila L, Rantakokoo-Jalava K, Jalava J, et al. Aetiological diagnosis of brain abscesses and spinal infections: application of broad range bacterial polymerase chain reaction analysis. J Neurol Neurosurg Psychiatry. 2003;74:728–733. doi: 10.1136/jnnp.74.6.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kutlay M, Colak A, Yildiz S, et al. Stereotactic aspiration and antibiotic treatment combined with hyperbaric oxygen therapy in the management of bacterial brain abscess. Neurosurg. 2005;57:1140–1146. doi: 10.1227/01.NEU.0000186012.95462.E5. [DOI] [PubMed] [Google Scholar]
- 60.Le Moal G, Landron C, Grollier G, et al. Characteristics of brain abscess with isolation of anaerobic bacteria. Scand J Infect Dis. 2003;35:318–321. doi: 10.1080/00365540310000265. [DOI] [PubMed] [Google Scholar]
- 61.Leuthardt EC, Wippold FJ, Oswood MC, et al. Diffusion-weighted MR imaging in the preoperative assessment of brain abscesses. Surg Neurol. 2002;58:395–402. doi: 10.1016/S0090-3019(02)00929-1. [DOI] [PubMed] [Google Scholar]
- 62.Livraghi S, Melancia JP, Antunes jL The management of brain abscesses. Adv Tech Stand Neurosurg. 2003;28:285–313. doi: 10.1007/978-3-7091-0641-9_5. [DOI] [PubMed] [Google Scholar]
- 63.Lu CH, Chang WN, Lui CC. Strategies for the management of bacterial brain abscess. J Clin Neurosci. 2006;13:979–985. doi: 10.1016/j.jocn.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 64.Lumbiganon P, Chaikitpinyo A. Antibiotics for brain abscesses in people with cyanotic congenital heart disease. Cochrane Database Syst Rev. 2007;18:CD004469. doi: 10.1002/14651858.CD004469.pub2. [DOI] [PubMed] [Google Scholar]
- 65.McClelland S, Hall WA. Postoperative central nervous system infection: incidence and associated factors in 2111 nerosurgical procedures. Clin Infect Dis. 2007;45:55–59. doi: 10.1086/518580. [DOI] [PubMed] [Google Scholar]
- 66.Mueller-Mang C, Castillo M, Mang TG, et al. Fungal versus bacterial brain abscesses: is diffusion-weighted MR imaging a useful tool in the differential diagnosis? Neuroradiol. 2007;49:651–657. doi: 10.1007/s00234-007-0242-0. [DOI] [PubMed] [Google Scholar]
- 67.Pfister HW, Feiden W, Einhäupl KM. The spectrum of complications during bacterial meningitis in adults: Results of a prospective clinical study. Arch Neurol. 1993;50:575–580. doi: 10.1001/archneur.1993.00540060015010. [DOI] [PubMed] [Google Scholar]
- 68.Saez-Llorens X. Brain abscess in children. Semin Pediatr Infect Dis 2003. 2003;14:108–114. doi: 10.1053/spid.2003.127227. [DOI] [PubMed] [Google Scholar]
- 69.Skoutelis AT, Gogos CA, Maraziotis TE, et al. Managment of brain abscesses with sequential intravenous/oral antibiotic therapy. Eur J Clin Microbiol Infect Dis. 2000;19:332–335. doi: 10.1007/s100960050489. [DOI] [PubMed] [Google Scholar]
- 70.Su TM, Lan CM, Tsai YD, et al. Multilocated pyogenic brain abscess: experience in 25 patients. Neurosurgery. 2003;52:1075–1079. doi: 10.1227/01.NEU.0000057696.79800.1D. [DOI] [PubMed] [Google Scholar]
- 71.Tahir MZ, Hassan RU, Enam SA. Management of an extensive spinal epidural abscess from C-1 to the sacrum. J Neurosurg Spine. 2010;13:780–783. doi: 10.3171/2010.5.SPINE09545. [DOI] [PubMed] [Google Scholar]
- 72.Tattevin P, Bruneel F, Clair B, et al. Bacterial brain abscesses: a retrospective study of 94 patients admitted to an intensive care Unit (1980–1999) Am J Med. 2003;115:143–146. doi: 10.1016/S0002-9343(03)00292-4. [DOI] [PubMed] [Google Scholar]
- 73.The “Infection in Neurosurgery” Working Party of the British Society for Antimicrobial Therapy The rational use of antibiotics in the treatment of brain abscess. Br J Neurosurg. 2000;14:525–530. doi: 10.1080/02688690020005527. [DOI] [PubMed] [Google Scholar]
- 74.Tsai YD, Chang WN, Shen CC, et al. Intracranial suppuration: a clinical comparison of subdural empyemas and epidural abscesses. Surg Neurol. 2003;59:191–196. doi: 10.1016/S0090-3019(02)01054-6. [DOI] [PubMed] [Google Scholar]
- 75.Tseng JH, Tseng MY. Brain abscess in 142 patients: factors influencing outcome and mortality. Surg Neurol. 2006;65:557–562. doi: 10.1016/j.surneu.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 76.Tung GA, Rogg JM. Diffusion-Weighted Imaging of Cerebritis. Am J Neuroradiol. 2003;24:1110–1113. [PMC free article] [PubMed] [Google Scholar]
- 77.Visani P, Schmutzhard E, Trinka E, et al. Subcortical deficit pattern after brain abscess: a neuropsychological study. Eur J Neurol. 2006;13:599–603. doi: 10.1111/j.1468-1331.2006.01234.x. [DOI] [PubMed] [Google Scholar]
- 78.Wang VY, Chou D, Chin C. Spine and spinal cord emergencies: vascular and infectious causes. Neuroimaging Clin N Am. 2010;20:639–650. doi: 10.1016/j.nic.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 79.Yilmaz MH, Mete B, Kantarci F, et al. Tuberculous, brucellar and pyogenic spondylitis: comparison of magnetic resonance imaging findings and assessment of its value. South Med J. 2007;100:613–614. doi: 10.1097/SMJ.0b013e3180600eaa. [DOI] [PubMed] [Google Scholar]
- 80.Younis RT, Lazar RH, Anand VK. Intracranial complications of sinusitis: a 15-year review of 39 cases. Ear Nose Throat J. 2002;81:636–638. [PubMed] [Google Scholar]
Zu 32.3
- 81.Aksamit AJ. Whipple’s disease of the central nervous system. Handb Clin Neurol. 2010;96:231–237. doi: 10.1016/S0072-9752(09)96014-6. [DOI] [PubMed] [Google Scholar]
- 82.Anderson M. Neurology of Whipple’s disease. J Neurol Neurosurg Psychiatry. 2000;68:2–5. doi: 10.1136/jnnp.68.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andronikou S, Smith B, Hatherhill M, et al. Definitive neuroradiological diagnostic features of tuberculous meningitis in children. Pediatr Radiol. 2004;34:876–885. doi: 10.1007/s00247-004-1237-1. [DOI] [PubMed] [Google Scholar]
- 84.Asselman V, Thienemann F, Pepper DJ, et al. Central nervous system disorders after starting antiretroviral therapy in South Africa. AIDS. 2010;24:2871–2876. doi: 10.1097/QAD.0b013e328340fe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Banerjee B, Petersen K. Psychosis following mycoplasma pneumonia. Mil Med. 2009;174:1001–1004. doi: 10.7205/MILMED-D-00-8209. [DOI] [PubMed] [Google Scholar]
- 86.Bhigjee AI, Padayachee R, Paruk H, et al. Diagnosis of tuberculous meningitis: clinical and laboratory parameters. Int J Infect Dis. 2007;11:348–354. doi: 10.1016/j.ijid.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 87.Black DF, Aksamit AJ, Morris JM. MR imaging of central nervous system Whipple disease: a 15-year review. AJNR Am J Neuroradiol. 2010;31:1493–1497. doi: 10.3174/ajnr.A2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boillat N, Greub G. Rickettsiosis: a clinical approach. Rev Med Suisse. 2007;16:1222–1227. [PubMed] [Google Scholar]
- 89.Brinar VV, Habek M. Rare infections mimicking MS. Clin Neurol Neurosurg. 2010;112:625–628. doi: 10.1016/j.clineuro.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 90.Bross JE, Gordon G. Nocardial Meningitis: Case Reports and Review. Rev. Infect Dis. 1991;13:160–165. doi: 10.1093/clinids/12.5.160. [DOI] [PubMed] [Google Scholar]
- 91.Christmann D, Hansmann Y, Remy V, et al. Tick-borne neurological diseases. Rev Neurol. 2002;158:993–997. [PubMed] [Google Scholar]
- 92.Chotmongkol V, Teerajetgul Y, Yodwut C. Cerebrospinal fluid adenosine deaminase activity for the diagnosis of tuberculous meningitis in adults. SE Asian J Trop Med Public Health. 2006;37:948–952. [PubMed] [Google Scholar]
- 93.Cunha BA, Pherez FM. C. pneumoniae community-acquired pneumonia (CAP) in mimicking Mycoplasma pneumonia meningoencephalitis complicated by asthma. Heart Lung. 2009;38:530–533. doi: 10.1016/j.hrtlng.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 94.Dumler J, Madigan JE, Pusterlia N, et al. Ehrlichioses in Humans: Epidemiology, clinical Presentations, Diagnosis and Treatment. Clin Infect. 2007;45:45–51. doi: 10.1086/518146. [DOI] [PubMed] [Google Scholar]
- 95.Fenollar F, Puechal X, Raoult D. Whipple’s disease. N Engl Med. 2007;356:55–56. doi: 10.1056/NEJMra062477. [DOI] [PubMed] [Google Scholar]
- 96.Garcia Monco JC. Tuberculosis and other mycobacterial Infections. In: Noseworthy JH, editor. Neurological Therapeutics – Principles and Practice. 2. Oxon: Informa Health Care Abingdon; 2006. pp. 1011–1020. [Google Scholar]
- 97.Godia CE, Esteve CM, Campello RA, et al. Cerebral saltwasting syndrome due to infectious diseases of the central nervous system. Med Clin. 2007;128:278–279. doi: 10.1016/S0025-7753(07)72562-9. [DOI] [PubMed] [Google Scholar]
- 98.Gouriet F, Lepidi H, Habib G, et al. From Cat Scratch disease to endocarditis, the possible natural history of Bartonella henselae infection. BMC Inf Dis. 2007;7:30–35. doi: 10.1186/1471-2334-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hainard A, Tiberti N, Robin X, et al. Matrix metalloproteinase-9 and intercellular adhesion molecule 1 are powerful staging markers for human African trypanosomiasis. Trop Med Int Health. 2010;16:119–126. doi: 10.1111/j.1365-3156.2010.02642.x. [DOI] [PubMed] [Google Scholar]
- 100.Hongo I, Bloch KC. Ehrlichia infection of the central nervous system. Curr Treat Options Neurol. 2006;8:179–184. doi: 10.1007/s11940-006-0008-8. [DOI] [PubMed] [Google Scholar]
- 101.Jha DK, Mishra V, Choudhary A, et al. Factors affecting the outcome of neuroendoscopy in patients with tuberculous meningitis hydrocephalus: a preliminary study. Surg Neurol. 2007;68:35–41. doi: 10.1016/j.surneu.2006.10.055. [DOI] [PubMed] [Google Scholar]
- 102.Inan AS, Ceran N, Erdem I, et al. Neurobrucellosis with transient ischemic attack, vasculopathic changes, intracerebral granulomas and basal ganglia infarction: a case report. J Med Case Reports. 2010;4:340–342. doi: 10.1186/1752-1947-4-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kalita J, Misra UK, Ranjan P. Predictors of long-term neurological sequelae of tuberculous meningitis: a multivariate analysis. Eur J Neurol. 2007;14:33–37. doi: 10.1111/j.1468-1331.2006.01534.x. [DOI] [PubMed] [Google Scholar]
- 104.Kazar J. Coxiella burnetii infection. Ann NY Academy Sci. 2005;1063:105–114. doi: 10.1196/annals.1355.018. [DOI] [PubMed] [Google Scholar]
- 105.Koskiniemi M. CNS manifestations associated with Mycoplasma pneumoniae infections: summary of cases at the University of Helsinki and review. Clin Infect Dis. 1993;17:52–57. doi: 10.1093/clinids/17.Supplement_1.S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kuo VC, Sloan LM, Emmett M. Central nervous system tuberculosis. Proc Bayl Univ Med Cent. 2010;23:359–360. doi: 10.1080/08998280.2010.11928652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee CH, Lui CC, Liu JW. Immune reconstitution syndrome in a patient with AIDS with paradoxically deteriorating brain tuberculoma. AIDS Patient Care. 2007;21:234–239. doi: 10.1089/apc.2006.0085. [DOI] [PubMed] [Google Scholar]
- 108.Lin JJ, Harn JH, Hsu YD, et al. Rapid diagnosis of tuberculous meningitis by polymerase chain reaction assay of cerebrospinal fluid. J Neurol. 1995;242:147–152. doi: 10.1007/BF00936887. [DOI] [PubMed] [Google Scholar]
- 109.Mahajan SK, Rolain JM, Kanga A, Raoult D. Scrub typhus involving central nervous system, India, 2004–2006. Emerg Infect Dis. 2010;16:1641–1643. doi: 10.3201/eid1610.100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Masoodi I, Farooq O, Ahmad I, et al. Acute disseminated encephalomyelitis as the first presentation of CNS tuberculosis: report of a case with brief review. Ger Med Sci. 2010;8:1–4. doi: 10.3205/000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Noyola DE, Holder DL, Fishman MA, et al. Recurrent encephalopathy in cat-scratch disease. Pediatr Infect Dis. 1999;18:567–568. doi: 10.1097/00006454-199906000-00024. [DOI] [PubMed] [Google Scholar]
- 112.Pappas G, Papadimitriou P, Akriditis N, et al. The new global map of human brucellosis. Lancet Infect Dis. 2006;6:91–99. doi: 10.1016/S1473-3099(06)70382-6. [DOI] [PubMed] [Google Scholar]
- 113.Pfister HW. Meningits. Stuttgart: Kohlhammer; 2002. [Google Scholar]
- 114.Prasad K, Singh MB. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev. 2008;2008:CD002244. doi: 10.1002/14651858.CD002244.pub3. [DOI] [PubMed] [Google Scholar]
- 115.Przybojewski AS, Wilmshurst J. Objective CT criteria to determine the presence of abnormal basal enhancement in children with suspected tuberculous meningitis. Pediatr Radiol. 2006;36:687–696. doi: 10.1007/s00247-006-0160-z. [DOI] [PubMed] [Google Scholar]
- 116.Rafi W, Venkataswamy MM, Ravi V, et al. Rapid diagnosis of tuberculous meningitis: a comparative evaluation of in-house PCR assays involving three mycobacterial DNA sequences, IS6110-MPB-64 and 65 kDa antigen. J Neurol Sci. 2007;252:163–168. doi: 10.1016/j.jns.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 117.Raoult D, Birg MI, La Scola B, et al. Cultivation of the bacillus of Whipple’s disease. N Engl J Med. 2000;342:620–625. doi: 10.1056/NEJM200003023420903. [DOI] [PubMed] [Google Scholar]
- 118.Renard D, Morales R, Heroum C. Tuberculous meningovasculitis. Neurology. 2007;68:1745. doi: 10.1212/01.wnl.0000263653.20798.09. [DOI] [PubMed] [Google Scholar]
- 119.Roca B, Bahamonde D. Tuberulous Meningitis Presenting with Unusually Severe Hyponatremia. The Mount Sinai J Med. 2006;73:1029–1030. [PubMed] [Google Scholar]
- 120.Schiffer JT, Sterling TR. Timing of antiretroviral therapy initiation in tuberculosis patients with AIDS: a decision analysis. J Acquir Immune Defic Syndr. 2007;44:229–234. doi: 10.1097/QAI.0b013e31802e2975. [DOI] [PubMed] [Google Scholar]
- 121.Schmutzhard E, et al. Atypische erregerbedingte Meningoencephalitis. In: Diener HC, et al., editors. Leitlinien für Diagnostik und Therapie in der Neurologie. 4. Stuttgart: Thieme; 2008. [Google Scholar]
- 122.Schmutzhard E, Pfister HW. Seltene bakterielle Infektionen des Nervensystems. Akt Neurol. 2001;28:373–382. doi: 10.1055/s-2001-17539. [DOI] [Google Scholar]
- 123.Schoeman J, Mansvelt E, Springer P, et al. Coagulant and fibrinolytic status in tuberculous meningitis. Pediatr Infect Di J. 2007;26:428–431. doi: 10.1097/01.inf.0000261126.60283.cf. [DOI] [PubMed] [Google Scholar]
- 124.Skiba V, Barner KC. Central nervous system manifestations of Q fever responsive to steroids. Mil Med. 2009;174:857–859. doi: 10.7205/MILMED-D-03-7108. [DOI] [PubMed] [Google Scholar]
- 125.Socan M. Chlamydia pneumoniae and meningoencephalitis. N Engl J Med. 1994;11:331–406. doi: 10.1056/NEJM199408113310618. [DOI] [PubMed] [Google Scholar]
- 126.Srikanth SG, Taly AB, Nagarajan K, et al. Clinicoradiological features of tuberculous meningitis in patients over 50 years of age. J Neurol Neurosurg Psychiatr. 2007;78:536–538. doi: 10.1136/jnnp.2006.095620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tezer H, Kara A, Haliloglu G, et al. Mycoplasma pneumonia-associated transverse myelitis with unexpected rapid response to macrolide therapy: a case report. Turk J Pediatr. 2008;50:585–588. [PubMed] [Google Scholar]
- 128.Thurtell MJ, Keed AB, Yan M, et al. Tuberculous cranial pachymeningitis. Neurology. 2007;68:298–300. doi: 10.1212/01.wnl.0000252367.99393.34. [DOI] [PubMed] [Google Scholar]
- 129.Wang VY, Chou D, Chin C. Spine and spinal cord emergencies: vascular and infectious causes. Neuroimaging Clin N Am. 2010;20:639–650. doi: 10.1016/j.nic.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 130.Yis U, Kurul SH, Cakmakci H, Dirik E. Mycoplasma pneumonia: nervous system complications in childhood and review of the literature. Eur J Pediatr. 2008;167:973–978. doi: 10.1007/s00431-008-0714-1. [DOI] [PubMed] [Google Scholar]
Zu 32.4
- 131.Aguero-Rosenfeld ME, Wang G, Schwartz I, Wormser GP. Diagnosis of Lyme borreliosis. Clin Microbiol Rev. 2005;18:484–509. doi: 10.1128/CMR.18.3.484-509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Blank LJ, Rompalo AM, Erbelding EJ, Zenilman JM, Ghanem KG. Treatment of syphilis in HIV-infected subjects: a systematic review of the literature. Sex Transm Infect. 2011;87:9–16. doi: 10.1136/sti.2010.043893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ghanem KG. Review: Neurosyphilis: A historical perspective and review. CNS Neurosci Ther. 2010;16:157–168. doi: 10.1111/j.1755-5949.2010.00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hajjaj KN. Status epilepticus revealing syphilitic meningoencephalitis. Acta Neurol Belg. 2010;110:263–267. [PubMed] [Google Scholar]
- 135.Halperin JJ. Diagnosis and treatment of the neuromuscular manifestations of Lyme disease. Curr Treat Options Neuro. 2007;9:93–100. doi: 10.1007/s11940-007-0035-0. [DOI] [PubMed] [Google Scholar]
- 136.Halperin JJ. Is neuroborreliosis a medical emergency? Neurocrit Care. 2006;4:260–266. doi: 10.1385/NCC:4:3:260. [DOI] [PubMed] [Google Scholar]
- 137.Halperin JJ, Shapiro ED, Logigian E, et al. Practice parameter: treatment of nervous system Lyme disease (an evidence based review): report of the Quality of Standards Subcommittee of the American Academy of Neurology. Neurology. 2007;69:91–102. doi: 10.1212/01.wnl.0000265517.66976.28. [DOI] [PubMed] [Google Scholar]
- 138.Jenke AC, Stoek LM, Zilbauer M, Wirth S, Borusiak P. Facial palsy: Etiology, outcome and management in children. Eur J Paediatr Neurol. 2011;15:209–213. doi: 10.1016/j.ejpn.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 139.Kobayashi J, Nakagawa Y, Tobisawa S, Isozaki E, Koide R. Deterioration of MRI findings related to Jarisch-Herxheimer reaction in a patient with neurosyphilis. J Neurol. 2011;258:699–701. doi: 10.1007/s00415-010-5808-9. [DOI] [PubMed] [Google Scholar]
- 140.Makhani N, Morris SK, Page AV, et al. A Twist On Lyme: The challenge of Diagnosing European Lyme Neuroborreliosis. J Clin Microbiol. 2011;49:455–457. doi: 10.1128/JCM.01584-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Richter D, Postic D, Sertour N, et al. Delineation of Borrelia burgdorferi sensu lato species by multilocus sequence analysis and confirmation of the delineation of Borrelia spielmanii sp. nov. Int J Syst Evol Microbiol. 2006;56:873–881. doi: 10.1099/ijs.0.64050-0. [DOI] [PubMed] [Google Scholar]
- 142.Robertson J, Guy E, Andrews N, et al. European multicenter study of immunoblotting in serodiagnosis of lyme borreliosis. J Clin Microbiol. 2000;38:2097–2102. doi: 10.1128/jcm.38.6.2097-2102.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Roos K. Neurosyphilis. In: Noseworthy JH, editor. Neurological Therapeutics-Principles and Practice. 2. Abingdon, Oxon: Informa Health Care; 2006. pp. 1021–1024. [Google Scholar]
- 144.Schmutzhard E, Pfausler B, Gasse T, et al. Verlauf und Langzeitfolgen der chronischen Neuroborreliose. Wien Med Wochenschr. 1995;145:183–186. [PubMed] [Google Scholar]
- 145.van Burgel ND, Oosterloo M, Kroon FP, van Dam AP. Severe course of Lyme neuroborreliosis in an HIV-1 positive patient; case report and review of the literature. BMC Neurol. 2010;10:117–121. doi: 10.1186/1471-2377-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wilske B. Epidemiology and diagnosis of Lyme Borreliosis. Ann Med. 2005;37:568–579. doi: 10.1080/07853890500431934. [DOI] [PubMed] [Google Scholar]
- 147.Wilske B, Fingerle V, Schulte-Spechtel U. Microbiological and serological diagnosis of Lyme Borreliosis. FEMS Immunol Med Microbiol. 2007;49:13–21. doi: 10.1111/j.1574-695X.2006.00139.x. [DOI] [PubMed] [Google Scholar]
- 148.Workowski KA, Berman SM. Sexually transmitted diseases treatment. Guidelines. 2010;59(RR12):1–110. [PubMed] [Google Scholar]
- 149.Wormser GP, Nadelman RB, Dattwyler RJ, et al. Practice guidelines for the treatment of Lyme disease. The Infectious Diseases Society of America. Clin Infect Dis. 2000;31:1–14. doi: 10.1086/314053. [DOI] [PubMed] [Google Scholar]
Zu 32.5
- 150.Abzug MJ, Cloud G, Bradley J, Sanchez PJ, Romero J, Powel D, Lepow M, Mani C, Capparelli EV, Blount S, Lakeman F, Whitley RJ, Kimberlin DW. Double blind placebo-controlled trial of pleconaril in infants with enterovirus meningitis. Pediatr Infect Dis J. 2003;22:335–341. doi: 10.1097/01.inf.0000059765.92623.70. [DOI] [PubMed] [Google Scholar]
- 151.Adamo MA, Deshaies EM. Emergency decompressive craniectomy for fulminating infectious encephalitis. J Neurosurg. 2008;108:174–176. doi: 10.3171/JNS/2008/108/01/0174. [DOI] [PubMed] [Google Scholar]
- 152.Armangue T, Leypoldt F, Dalmau J. Autoimmune encephalitis as differential diagnosis of infectious encephalitis. Curr Opin Neurol. 2014;27:361–368. doi: 10.1097/WCO.0000000000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol. 2014;75:317–323. doi: 10.1002/ana.24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol. 1994;36:823–829. doi: 10.1002/ana.410360605. [DOI] [PubMed] [Google Scholar]
- 155.Bossart KN, Zhu Z, Middleton D, et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. Plos Pathog. 2009;5:e1000642. doi: 10.1371/journal.ppat.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Casrouge A, Zhang SY, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 157.Center for Disease Control, Prevention Recovery of a Patient from Clinical Rabies. MMW. 2004;53:1171–1173. [Google Scholar]
- 158.C D C Update: Outbreak of Nipah virus – Malaysia and Singapore. MMWR. 1999;48:335–337. [PubMed] [Google Scholar]
- 159.Chadha MS, Comer JA, Lowe L, et al. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg Infect Dis. 2006;12:235–240. doi: 10.3201/eid1202.051247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chaudhuri A, Kennedy PG. Diagnosis and treatment of viral encephalitis. Postgrad Med J. 2002;78:575–583. doi: 10.1136/pmj.78.924.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cinque P, Bossolasco S, Lundkvist A. Molecular analysis of cerebrospinal fluid in viral diseases of the central nervous system. J Clin Virol. 2003;26:1–28. doi: 10.1016/S1386-6532(02)00173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Collaborative Antiviral Study Group, http://clinicaltrials.gov/show/NCT00031486: A Phase III Double-Blind, Placebo-Controlled Trial of Long Term Therapy of Herpes Simplex Encephalitis (HSE): An Evaluation of Valacyclovir (CASG-204), lead sponsor National Institute of Allergy and Infectious Diseases (NIAID)
- 163.Corssmit EP, Leverstein-van Hall MA, Portegies P, Bakker P. Severe neurological complications in association with Epstein-Barr virus infection. J Neurovirol. 1997;3:460–464. doi: 10.3109/13550289709031193. [DOI] [PubMed] [Google Scholar]
- 164.Defang GN, Khetawat D, Broder CC, Quinnan GV., Jr. Induction of neutralizing antibodies to Hendra and Nipah glycoproteins using a Venezuelan equine encephalitis virus in vivo expression system. Vaccine. 2010;29:212–220. doi: 10.1016/j.vaccine.2010.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.DGN – Deutsche Gesellschaft für Neurologie (2012) Leitlinie „Virale Meningoenzephalitiden“
- 166.Falcone S, Post MJ. Encephalitis, cerebritis, and brain abscess: pathophysiology and imaging findings. Neuroimaging Clin N Am. 2000;10:333–353. [PubMed] [Google Scholar]
- 167.Fusco D, Krawitz P, LaRussa P, Steinberg S, Gershon A. Jacobs J VZV meningitis following varicella vaccine. J Clin Virol. 2010;48:275–277. doi: 10.1016/j.jcv.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Granerod J, Cunningham R, Zuckerman M, et al. Causality in acute encephalitis: defining aetiologies. Epidemiol Infect. 2010;138:783–800. doi: 10.1017/S0950268810000725. [DOI] [PubMed] [Google Scholar]
- 169.Granerod J, Tam CC, Crowcroft NS, Davies NWS, Borchert M, Thomas SL. Challenge of the unknown: A systematic review of acute encephalitis in non-outbreak situations. Neurology. 2010;75:924–932. doi: 10.1212/WNL.0b013e3181f11d65. [DOI] [PubMed] [Google Scholar]
- 170.Gurley ES, Montgomery JM, Hossain MJ, et al. Person-to-person transmission of Nipah virus in Bangladeshi community. Emerg Infect Dis. 2007;13(7):1031–1037. doi: 10.3201/eid1307.061128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hacohen Y, Deiva K, Pettingill P, Waters P, Siddiqui A, Chretien P, Menson E, Lin JP, Tardieu M, Vincent A, Lim MJ. N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord. 2014;29:90–96. doi: 10.1002/mds.25626. [DOI] [PubMed] [Google Scholar]
- 172.Haglund M, Forsgren M, Lindh G, Lindquist L. A 10-year follow-up study of tick-borne encephalitis in the Stockholm area and a review of the literature: need for a vaccination strategy. Scand J Infect Dis. 1996;28:217–224. doi: 10.3109/00365549609027160. [DOI] [PubMed] [Google Scholar]
- 173.Hauser SL. Harrison’s Neurology in Clinical Medicine. New York: McGraw-Hill; 2006. [Google Scholar]
- 174.Henquell C, Chambon M, Bailly JL, Alcaraz S, De Champs C, Archimbaud C, Labbe A, Charbonne F, Peigue-Lafeuille H. Prospective analysis of 61 cases of enteroviral meningitis: interest of systematic genome detection in cerebrospinal fluid irrespective of cytologic examination results. J Clin Virol. 2001;21:29–35. doi: 10.1016/S1386-6532(00)00176-1. [DOI] [PubMed] [Google Scholar]
- 175.Kaiser R. Tick-borne encephalitis (TBE) in Germany and clinical course of the disease. Int J Med Microbiol. 2002;291(33):58–61. doi: 10.1016/S1438-4221(02)80012-1. [DOI] [PubMed] [Google Scholar]
- 176.Kaiser R. Langzeitprognose bei primär myelitischer Manifestation der FSME. Eine Verlaufsanalyse über 10 Jahre. Nervenarzt. 2011;82:1020–1025. doi: 10.1007/s00115-011-3254-2. [DOI] [PubMed] [Google Scholar]
- 177.Kamei S. Trends in the management of herpes simplex encephalitis. Rinsho Shinkeigaku. 2006;46:950–953. [PubMed] [Google Scholar]
- 178.Kennedy PGE. Viral Encephalitis. J Neurol. 2005;252:268–272. doi: 10.1007/s00415-005-0770-7. [DOI] [PubMed] [Google Scholar]
- 179.Kutleša M, Baršić B. Therapeutic hypothermia for severe adult Herpes simplex virus encephalitis. Wien Klin Wochenschr. 2012;124:855–858. doi: 10.1007/s00508-012-0302-2. [DOI] [PubMed] [Google Scholar]
- 180.Lee KE, Umapathi T, Tan CB, et al. The neurological manifestations of Nipah virus encephalitis, a novel paramyxovirus. Ann Neurol. 1999;464:28–32. [PubMed] [Google Scholar]
- 181.Leitlinie der DGPI. Varizellen-Zoster. www.uni-duesseldorf.de/WWW/AWMF/II/048
- 182.Leypoldt F, Titulaer MJ, Aguilar E, Walther J, Bönstrup M, Havemeister S, Teegen B, Lütgehetmann M, Rosenkranz M, Magnus T, Dalmau J. Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology. 2013;81:1637–1639. doi: 10.1212/WNL.0b013e3182a9f531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Livorsi D, Anderson E, Qureshi S, Howard M, Wang YF, Paredes F-C. Brainstem encephalitis: an unusual presentation of herpes simplex virus infection. J Neurol. 2010;257:1432–1437. doi: 10.1007/s00415-010-5600-x. [DOI] [PubMed] [Google Scholar]
- 184.Martinez-Torres F, Menon S, Pritsch M, Victor N, Jenetzky E, Jensen K, Schielke E, Schmutzhard E, de Gans J, Chung CH, Luntz S, Hacke W, Meyding-Lamadé U. Protocol for German trial of Acyclovir and corticosteroids in Herpes-simplex-virus-encephalitis (GACHE): a multicenter, multinational, randomized, double-blind, placebo-controlled German, Austrian and Dutch trial. BMC Neurol. 2008;8:40. doi: 10.1186/1471-2377-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Meyding-Lamade UK, Lamade WR, Wildemann BT, Sarto K, Hacke W. Herpes simplex virus encephalitis: chronic progressive cerebral magnetic resonance imaging abnormalities in patients despite good clinical recovery. Clin Infect Dis. 1999;28:148–149. doi: 10.1086/517183. [DOI] [PubMed] [Google Scholar]
- 186.Meyding-Lamadé U, Strank C. Herpesvirus infections of the central nervous system in immunocompromised patients. Ther Adv Neurol Disord. 2012;5:279–296. doi: 10.1177/1756285612456234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9:1097–1105. doi: 10.1016/S1474-4422(10)70209-X. [DOI] [PubMed] [Google Scholar]
- 188.Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A. Moscona A Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entr. PLoS Pathog. 2010;6:e1001168. doi: 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Reiber H, Felgenhauer K. Protein transfer at the blood cerebrospinal fluid barrier and the quantitation of the humoral immune response within the central nervous system. Clin Chim Acta. 1987;163:319–328. doi: 10.1016/0009-8981(87)90250-6. [DOI] [PubMed] [Google Scholar]
- 190.RKI – Robert-Koch-Institut (2013) www.rki.de/DE/Content/Infekt/EpidBull/Merkblaetter/Ratgeber_Tollwut.html
- 191.Robert-Koch-Institut RKI–. FSME: Risikogebiete in Deutschland. Epidemiol Bull. 2014;15:121–136. [Google Scholar]
- 192.Romero JR, Newland JG. Viral meningitis and encephalitis: traditional and emerging viral agents. Semin Pediatr Infect Dis. 2003;14:72–82. doi: 10.1053/spid.2003.127223. [DOI] [PubMed] [Google Scholar]
- 193.Schmutzhard E. Viral infections of the CNS with special emphasis on herpes simplex infections. J Neurol. 2001;248:469–477. doi: 10.1007/s004150170155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schwab S, Junger E, Spranger M, Dorfler A, Albert F, Steiner HH, Hacke W. Craniectomy: an aggressive treatment approach in severe encephalitis. Neurology. 1997;48:412–417. doi: 10.1212/WNL.48.2.412. [DOI] [PubMed] [Google Scholar]
- 195.Sener RN. Diffusion MRI in Rasmussen’s encephalitis, herpes simplex encephalitis, and bacterial meningoencephalitis. Comput Med Imaging Graph. 2002;26:327–332. doi: 10.1016/S0895-6111(02)00028-9. [DOI] [PubMed] [Google Scholar]
- 196.Sivertsen B, Christensen PB. Acute encephalitis. Acta Neurol Scand. 1996;93:156–159. doi: 10.1111/j.1600-0404.1996.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 197.Sköldenberg B, Forsgren M, Alestig K, Bergström T, Burman L, Dahlqvist E, Forkman A, Frydén A, Lövgren K, Norlin K, et al. Acyclovir versus vidarabine in herpes simplex encephalitis. Randomised multicentre study in consecutive Swedish patients. Lancet. 1984;29:707–711. doi: 10.1016/S0140-6736(84)92623-0. [DOI] [PubMed] [Google Scholar]
- 198.Solomon T, Michael BD, Smith PE, Sanderson F, Davies NWS, Hart IJ, Holland M, Easton A, Buckley C, Kneen R, Beeching Management of suspected viral encephalitis in adults – Association of British Neurologists and British Infection Association National Guidelines. J Infection. 2012;64:347–373. doi: 10.1016/j.jinf.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 199.Sommer JB, Heckmann JG, Kraus J, Neundörfer B, Erbguth FJ. Generalized brain edema in non-purulent meningoencephalitis. The anti-edema effect of therapy with dexamethasone. Nervenarzt. 2000;71:112–115. doi: 10.1007/s001150050016. [DOI] [PubMed] [Google Scholar]
- 200.Steiner I, Budka H, Chaudhuri A, Koskiniemi M, Sainio K, Salonen O, Kennedy PG (2010) Viral meningoencephalitis: a review of diagnostic methods and guidelines for management. Eur J Neurol 17(999) [DOI] [PubMed]
- 201.Tan CT, Goh KJ, Wong KT, et al. Relapsed and late-onset Nipah encephalitis. Ann Neurol. 2002;51:703–708. doi: 10.1002/ana.10212. [DOI] [PubMed] [Google Scholar]
- 202.Taskin E, Turgut M, Kilic M, et al. Serum procalcitonin and cerebrospinal fluid cytokines level in children with meningitis. Mediators of Inflammation. 2004;13:269–273. doi: 10.1080/09629350400003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thakur KT, Motta M, Asemota AO, Kirsch HL, Benavides D, Schneider EB, McArthur JC, Geocadin RG, Venkatesan A. Predictors of outcome in acute encephalitis. Neurology. 2013;81:793–800. doi: 10.1212/WNL.0b013e3182a2cc6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Thomson RB, Jr., Bertram H. Laboratory diagnosis of central nervous system infections. Infect Dis Clin North Am. 2001;15:1047–1071. doi: 10.1016/S0891-5520(05)70186-0. [DOI] [PubMed] [Google Scholar]
- 205.Tyler KL. Update on herpes simplex encephalitis. Rev Neurol Dis. 2004;1:169–178. [PubMed] [Google Scholar]
- 206.Tyler KL. Emerging Viral Infections of the Central Nervous System, Part 2. Arch Neurol. 2009;66:1065–1074. doi: 10.1001/archneurol.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Utley TF, Ogden JA, Gibb A, McGrath N, Anderson NE. The long-term neuropsychological outcome of herpes simplex encephalitis in a series of unselected survivors. Neuropsychiatry Neuropsychol Behav Neurol. 1997;10:180–189. [PubMed] [Google Scholar]
- 208.Wagner I, Staykov D, Volbers B, Kloska S, Dörfler A, Schwab S, Bardutzky J. Mild hypothermia for space-occupying Herpes simplex virus encephalitis. Minerva Anestesiol. 2011;77:371–374. [PubMed] [Google Scholar]
- 209.Whitley RJ, Lakeman AD, Nahmias A, Roizman B. Dna restriction-enzyme analysis of herpes simplex virus isolates obtained from patients with encephalitis. N Engl J Med. 1982;307:1060–1062. doi: 10.1056/NEJM198210213071706. [DOI] [PubMed] [Google Scholar]
- 210.Whitley RJ, Alford CA, Hirsch MS, Schooley RT, Luby JP, Aoki FY, Hanley D, Nahmias AJ, Soong SJ. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314:144–149. doi: 10.1056/NEJM198601163140303. [DOI] [PubMed] [Google Scholar]
- 211.Whitley RJ, Gnann JW. Viral encephalitis: familiar infections and emerging pathogens. Lancet. 2002;359:507–513. doi: 10.1016/S0140-6736(02)07681-X. [DOI] [PubMed] [Google Scholar]
- 212.WHO (ed) Rabies Bulletin Europe, www.who-rabies-bulletin.org
- 213.Wildemann B, Ehrhart K, Storch-Hagenlocher B, Meyding-Lamade U, Steinvorth S, Hacke W, Haas J. Quantitation of herpes simplex virus type 1 DNA in cells of cerebrospinal fluid of patients with herpes simplex virus encephalitis. Neurology. 1997;48:1341–1346. doi: 10.1212/WNL.48.5.1341. [DOI] [PubMed] [Google Scholar]
- 214.Wong M, Connolly AM, Noetzel MJ. Poliomyelitis-like syndrome associated with Epstein-Barr virus infection. Pediatr Neurol. 1999;20:235–237. doi: 10.1016/S0887-8994(98)00142-8. [DOI] [PubMed] [Google Scholar]
- 215.Wong KT, Shieh WJ, Kumar S, et al. Nipah Virus Pathology Working Group. Nipah virus Infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol. 2002;161:2153–2167. doi: 10.1016/S0002-9440(10)64493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zu 32.6
- 216.Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN neuroinfectious disease section. Neurology. 2013;80:1430–1438. doi: 10.1212/WNL.0b013e31828c2fa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Biogen idec (ed) www.tysabri.de/index.php?inhalt=tysabri.sicherheitsmanagement
- 218.Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukencephalopathy and other forms of JC virus disease. Nat Rev Neurol. 2010;6:667–679. doi: 10.1038/nrneurol.2010.164. [DOI] [PubMed] [Google Scholar]
- 219.Campbell H, Andrews N, Brown KE, Miller E. Review of the effect of measles vaccination on the epidemiology of SSPE. Int J Epidemiol. 2007;36:1334–1348. doi: 10.1093/ije/dym207. [DOI] [PubMed] [Google Scholar]
- 220.Clifford DB, Yiannoutsos C, Glicksman M, Simpson DM, Singer EJ, Piliero PJ, Marra CM, Francis GS, McArthur JC, Tyler KL, Tselis AC, Hyslop NE. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623–625. doi: 10.1212/WNL.52.3.623. [DOI] [PubMed] [Google Scholar]
- 221.Clifford DB, Nath A, Cinque P, Brew BJ, Zivadinov R, Gorelik L, Zhao Z, Duda P. A study of mefloquine treatment for progressive multifocal leukoencephalopathy: results and exploration of predic- tors of PML outcomes. J Neurovirol. 2013;19:351–358. doi: 10.1007/s13365-013-0173-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dahlhaus S, Hoepner R, Chan A, Kleiter I, Adams O, Lukas C, Hellwig K, Gold R. Disease course and outcome of 15 monocentrically treated natalizumab-associated progressive multifocal leukencephalopathy patients. J Neurol Neurosurg Psychiatry. 2013;84:1068–1074. doi: 10.1136/jnnp-2013-304897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gasnault J, Costagliola D, Hendel-Chavez H, Dulioust A, Pakianather S, Mazet AA, de Goer de Herve MG, Lancar R, Lascaux AS, Porte L, Delfraissy JF, Taoufik Y, ANRS 125 Trial Team Improved survival of HIV-1-infected patients with progressive multifocal leukoencephalopathy receiving early 5-drug combination antiretroviral therapy. PLoS One. 2011;6(6):e20967–125. doi: 10.1371/journal.pone.0020967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gorelik L, Lerner M, Bixler S, et al. Anti-JCV Antibodies: Implications for PML Risk Stratification. Ann Neurol. 2010;68:295–303. doi: 10.1002/ana.22128. [DOI] [PubMed] [Google Scholar]
- 225.Hauser S, et al. Harrison’s Neurology in Clinical Medicine. New York: McGraw-Hill; 2006. [Google Scholar]
- 226.Helbok R, Broessner G, Pfauler B, Schmutzhard E. Chronic meningitis. J Neurol. 2009;256:168–175. doi: 10.1007/s00415-009-0122-0. [DOI] [PubMed] [Google Scholar]
- 227.Khanna N, Elzi L, Mueller NJ, Garzoni C, Cavassini M, Fux CA, Vernazza P, Bernasconi E, Battegay M, Hirsch HH, Swiss HIV Cohort Study (2009) Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the Swiss HIV Cohort Study. Clin Infect Dis (48):1459–1466 [DOI] [PubMed]
- 228.Manning L, Laman M, Edoni H, Mueller I, Karunajeewa HA, Smith D, Hwaiwhanje I, Siba PM, Davis TM. Subacute sclerosing panencephalitis in papua new guinean children: the cost of continuing inadequate measles vaccine coverage. PLoS Negl Trop Dis. 2011;5:e932. doi: 10.1371/journal.pntd.0000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mentzer D, Prestel J, Adams O, et al. Case definition for progressive multifocal leukoencephalopathy following treatment with monoclonal antibodies. J Neurol Neurosurg Psychiatry. 2012;83:927–933. doi: 10.1136/jnnp-2012-302478. [DOI] [PubMed] [Google Scholar]
- 230.Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy in patients with rheumatic diseases: are patients with systemic lupus erythematosus at particular risk? Autoimmun Rev. 2008;8:144–146. doi: 10.1016/j.autrev.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 231.Nagoshi K, Sadakane A, Nakamura Y, Yamada M, Mizusawa H (2011) Duration of Prion Disease is Longer in Japan Than in Other Countries. J Epidemiol May 28, Epub ahead of print [DOI] [PMC free article] [PubMed]
- 232.Owens GP, Ritchie AM, Gilden DH, Borgoon MP, Becker D, Bennet JL. Measles virus-specific plasma cells are prominent in subacute sclerosing panencephalitis CSF. Neurology. 2007;68:1815–1819. doi: 10.1212/01.wnl.0000262036.56594.7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Schönberger K, Ludwig MS, Wildner M, Weissbrich B (2013) Epidemiology of subacute sclerosing panencephalitis (SSPE) in Germany from 2003 to 2009: a risk estimation. PLoS One 2013 Jul 9;8(7) [DOI] [PMC free article] [PubMed]
- 234.Schwab N, Schneider-Hohendorf T, Posevitz V, et al. L-Selectin is a possible biomarker for individual PML risk in natalizumab-treated MS patients. Neurology. 2013;81:865–871. doi: 10.1212/WNL.0b013e3182a351fb. [DOI] [PubMed] [Google Scholar]
- 235.Sharma S, Kumar S. Case report: Cervical spinal cord signal changes in a case of adult-onset subacute sclerosing panencephalitis. Indian J Radiol Imaging. 2010;20:202–204. doi: 10.4103/0971-3026.69358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Simpson DM. HIV-associated PML: Changing epidemiology and clinical approach. Cleve Clin J Med. 2011;78:24–27. doi: 10.3949/ccjm.78.s2.06. [DOI] [PubMed] [Google Scholar]
- 237.Tatli B, Ekici B, Ozmen M. Flurpirtine may Stopp the progressive course of subacute sclerosing panencephalitis. Med Hypotheses. 2010;75:576–577. doi: 10.1016/j.mehy.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 238.Warnke C, Menge T, Hartung HP, Racke MK, Cravens PD, Bennett JL, Frohman EM, Greenberg BM, Zamvil SS, Gold R, Hemmer B, Kieseier BC, Stüve O. Natalizumab and progressive multifocal leukencephalopathy: what are the causal factors and can it be avoided? Arch Neurol. 2010;67:923–930. doi: 10.1001/archneurol.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Warnke C, von Geldern G, Markwerth P, et al. (2014) Cerebrospinal Fluid JC Virus Antibody Index for Diagnosis of Natalizumab-Associated Progressive Multifocal Leukoencephalopathy. Ann Neurol 2014, Apr 11 (epub ahead of print) [DOI] [PMC free article] [PubMed]
- 240.Wattjes MP, Vennegoor A, Moster J, van Oosten BW, Barkhof F, Killestein J. Diagnosis of asymptomatic natalizumab-associated PML: are we between a rock and a hard place? J Neurol. 2014;261:1139. doi: 10.1007/s00415-014-7336-5. [DOI] [PubMed] [Google Scholar]
- 241.White MK, Khalil K. Pathogenesis of progressive multifocal leukoencephalopathy-revisited. J Infect Dis. 2011;203:578–586. doi: 10.1093/infdis/jiq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yilmaz C, Caksen H, Yilmaz N, Güven HS, Bayram I. Two cases of Subacute Sclerosing Panencephalitis associated with Brainstem involvement. Trop Pediatr. 2007;53:280–283. doi: 10.1093/tropej/fmm020. [DOI] [PubMed] [Google Scholar]
- 243.Yousry TA, Pelletier D, Cadavid D, et al. MRI pattern in natalizumab-associated progressive multifocal leukencephalopathy. Ann Neurol. 2012;72:779–787. doi: 10.1002/ana.23676. [DOI] [PubMed] [Google Scholar]
Zu 32.7
- 244.Anduze-Faris BM, Fillet AM, Gozlan J, Lancar R, Boukli N, Gasnault J, Caumes E, Livartowsky J, Matheron S, Leport C, Salmon D, Costagliola D, Katlama C. Induction and maintenance therapy of cytomegalovirus central nervous system infection in HIV-infected patients. Aids. 2000;14:517–524. doi: 10.1097/00002030-200003310-00007. [DOI] [PubMed] [Google Scholar]
- 245.Arendt G. Neurologische Komplikationen der HIV-Infektion in der Ära antiretroviraler Kombinationstherapien (cART) Retroviren Bulletin. 2013;1:2013. [Google Scholar]
- 246.Babiker AG, Emery S, Fätkenheuer G, Gordin FM, Grund B, Lundgren JD, Neaton JD, Pett SL, Phillips A, Touloumi G, et al. Considerations in the rationale, design and methods of the Strategic Timing of AntiRetroviral Treatment (START) study. Clin Trials. 2013;10(1):5–S36. doi: 10.1177/1740774512440342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bucher HC, Griffith L, Guyatt GH, Opravil M. Meta-analysis of prophylactic treatments againts pneumocystis carinii and toxoplasma encephalitis. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15:104–114. doi: 10.1097/00042560-199706010-00002. [DOI] [PubMed] [Google Scholar]
- 248.Cardo DM, Culver DH, Ciesielsky CA, Srivastava PU, Marcus R, Abiteboul D, Heptonstall J, Ippolito G, Lot F, McKibben PS, Bell DM. A case-control study of HIVseroconversion in health care workers after percutaneous exposure. Centers for Disease Control and Prevention Needlestick Surveillance Group. N Engl J Med. 1997;337:1485–1490. doi: 10.1056/NEJM199711203372101. [DOI] [PubMed] [Google Scholar]
- 249.Centers for Disease Control (1992) 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. Morbidity and Mortality Weekly Report 41 (RR-17): 1–19 [PubMed]
- 250.Chaisson RE, Keruly JC, Moore RD. Association of initial CD4 cell count and viral load with response to highly active antiretroviral therapy. J Am Med Assoc. 2000;248:3128–3129. doi: 10.1001/jama.284.24.3123. [DOI] [PubMed] [Google Scholar]
- 251.Dworkin MS, Wan PC, Hanson DL, Jones JL. Progressive multifocal leucoencephalopathy: improved survival of human immunodeficiency virus-infected patients in the protease inhibitor era. J Infect Dis. 1999;180:621–625. doi: 10.1086/314937. [DOI] [PubMed] [Google Scholar]
- 252.Henderson DK. Postexposure chemoprophylaxis for occupational exposures to the human immunodeficiency virus. J Am Med Assoc. 1999;281:931–936. doi: 10.1001/jama.281.10.931. [DOI] [PubMed] [Google Scholar]
- 253.Hoffmann C, Rockstroh JK, Kamps BS, editors. HIV.Net 2007. www.hiv.net. Wuppertal-Beyenburg: Steinhäuser; 2007. [Google Scholar]
- 254.Joint United Nations Programme on HIV/AIDS (UNAIDS) and World Health Organization (2010) UNAIDS Report on the global AIDS Epidemic 2010
- 255.Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, Gelman BB, McArthur JC, McCutchan JA, Morgello S, et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65:65–70. doi: 10.1001/archneurol.2007.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Louie M, Markowitz M. Goals and milestones during treatment of HIV-1 infection with antiretroviral therapy: a pathogenesis-based perspective. Antiviral Res. 2002;55:15–25. doi: 10.1016/S0166-3542(02)00022-0. [DOI] [PubMed] [Google Scholar]
- 257.Mellors JW, Rinaldo CRJ, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 258.Panel on Antiretroviral Guidelines for Adult and Adolescents (2006) Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. Department of Health and Human Services. October 10, 2006: 1–113
- 259.Portegies P, Solod L, Cinque P, Chaudhuri A, Begovac J, Everall I, Weber T, Bojar M, Martinez-Martin P, Kennedy PG. Guidelines for the diagnosis and management of neurological complications of HIV infection. Eur J Neurol. 2004;11:297–304. doi: 10.1111/j.1468-1331.2004.00856.x. [DOI] [PubMed] [Google Scholar]
- 260.Saag MS, Graybill RJ, Larsen RA, Pappas PG, Perfect JR, Powderly WG, Sobel JD, Dismukes WE. Practice guidelines for the management of cryptococcal disease. Infectious diseases Society of America. Clin Infect Dis. 2000;30:710–718. doi: 10.1086/313757. [DOI] [PubMed] [Google Scholar]
- 261.Sackto N, McArthur JC. Prospects for therapy of HIV-associated neurologic diseases. J Neurovirol. 1997;3:89–101. doi: 10.3109/13550289709015799. [DOI] [PubMed] [Google Scholar]
- 262.Schabet M, Herrlinger U, Weller M, Bamberg M, Clemens R, Dichgans J. Neue Entwicklungen in Diagnostik und Therapie primärer Non-Hodgkin-Lymphome des zentralen Nervensystems. Nervenarzt. 1997;68:298–308. doi: 10.1007/s001150050128. [DOI] [PubMed] [Google Scholar]
- 263.Wildemann B, Haas J, Lynen N, Stingele K, Storch-Hagenlocher B. Diagnosis of cytomegalovirus encephalitis in patients with AIDS by quantitation of cytomegalovirus genomes in cells of cerebrospinal fluid. Neurology. 1998;50:693–697. doi: 10.1212/WNL.50.3.693. [DOI] [PubMed] [Google Scholar]
Zu 32.7: Empfohlene Websites
- 264.Centers for Disease Control, United States Health Department: http://www.cdc.gov/hiv/resources/guidelines
- 265.Joint United Nations Programme on HIV/AIDS (UNAIDS): http://www.unaids.org [PubMed]
- 266.Robert Koch Institut: http://www.rki.de
- 267.AIDSinfo: U.S. Department of Health and Human Services (DHHS): http://aidsinfo.nih.gov/ [DOI] [PubMed]
- 268.Deutsch-Österreichische Leitlinien zur antiretroviralen Therapie der HIV-Infektion. http://www.daignet.de
- 269.Leitlinien der Deutschen Gesellschaft für Neurologie zur Diagnostik und Therapie HIV-1-assoziierter Erkrankungen. www.dgn.org
Zu 32.8
- 270.Balasegaram M, Harris S, Checchi F, et al. Melarsoprol versus eflornithine for treating late-stage Gambian trypanosomiasis in the Republic of the Congo. Bulletin WHO. 2006;84:783–791. doi: 10.2471/blt.06.031955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Birbeck GL, Beare N, Lewallen S, et al. Identification of malaria retinopathy improves the specificity of the clinical diagnosis of cerebral malaria: findings from a prospective cohort study. Am J Trop Med Hyg. 2010;82:231–234. doi: 10.4269/ajtmh.2010.09-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Birbeck GL, Molyneux ME, Kaplan PW, et al. Blantyre Malaria Project Epilepsy Study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: a prospective cohort study. Lancet Neurol. 2010;9:1173–1181. doi: 10.1016/S1474-4422(10)70270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Braakman H, Fred MM, van de Molengraft JJ, et al. Lethal African trypanosomiasis in a traveler: MRI and neuropathology. Neurology. 2006;66:1094–1096. doi: 10.1212/01.wnl.0000209306.41647.13. [DOI] [PubMed] [Google Scholar]
- 274.Burgos JM, Begher S, Silva HM, et al. Molecular identification of Trypanosoma cruzi I tropism for central nervous system in Chagas reactivation due to AIDS. Am J Trop Med Hyg. 2008;78:294–297. [PubMed] [Google Scholar]
- 275.Carod-Artal FJ. Tropical causes of epilepsy. Rev Neurol. 2009;49:475–482. [PubMed] [Google Scholar]
- 276.Chacko G. Parasitic diseases of the central nervous system. Semin Diagn Pathol. 2010;27:167–185. doi: 10.1053/j.semdp.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 277.Córdova E, Maiolo E, Corti M, Orduna T. Neurological manifestations of Chagas’ disease. Neurol Res. 2010;32:238–244. doi: 10.1179/016164110X12644252260637. [DOI] [PubMed] [Google Scholar]
- 278.Corpelet C, Vacher P, Coudore F, et al. Role of quinine in life-threatening Babesia divergens infection successfully treated with clindamycin. Eur J Clin Microbiol Infect Dis. 2005;24:74–75. doi: 10.1007/s10096-004-1270-x. [DOI] [PubMed] [Google Scholar]
- 279.Dauriac-Le Masson V, Chochon F, Demeret S, et al. Toxocara canis meningomyelitis. J Neurol. 2005;252:1267–1268. doi: 10.1007/s00415-005-0688-0. [DOI] [PubMed] [Google Scholar]
- 280.Davis TME, White NJ, Looareesuwan S, Silamut K, Warrell DA. Quinine pharmacokinetics in cerebral malaria: predicted plasma concentrations after rapid intravenous loading using a two compartment mode. Trans R Soc Trop Med Hyg. 1988;82:542–547. doi: 10.1016/0035-9203(88)90498-1. [DOI] [PubMed] [Google Scholar]
- 281.Del Brutto OH, Roos KL, Coffey CS, et al. Meta-Analysis: Cysticidal Drugs for Neurocysticercosis: Albendazole and Praziquantel. Ann Intern Med. 2006;145:43–51. doi: 10.7326/0003-4819-145-1-200607040-00009. [DOI] [PubMed] [Google Scholar]
- 282.Deo I, Robledo L, Meza A, et al. Encephalitis due to a free-living amoeba (Balamuthia mandrillaris), case report with literature review. Surg Neurol. 2000;53:611–616. doi: 10.1016/S0090-3019(00)00232-9. [DOI] [PubMed] [Google Scholar]
- 283.Dondorp AM, Fanello CI, Hendriksen IC, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomized trial. Lancet. 2010;376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Dondorp A, Nosten F, Stepniewska K, et al. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- 285.Ellrodt A, Halfon P, Le Bras P, Halimi P, Bouree P, Desi M, Caquet R. Multifocal central nervous system lesions in three patients with trichinosis. Arch Neurol. 1987;44:432–434. doi: 10.1001/archneur.1987.00520160064016. [DOI] [PubMed] [Google Scholar]
- 286.Dulac O. Cerebral malaria and epilepsy. Lancet Neurol. 2010;9:1144–1145. doi: 10.1016/S1474-4422(10)70278-7. [DOI] [PubMed] [Google Scholar]
- 287.Elsheikha HM, Khan NA. Protozoa traversal of the blood-brain barrier to invade the central nervous system. FEMS Microbiol. 2010;34:532–553. doi: 10.1111/j.1574-6976.2010.00215.x. [DOI] [PubMed] [Google Scholar]
- 288.Fan KJ, Pezeshkpour GH. Cerebral sparganosis. Neurology. 1986;36:1249–1251. doi: 10.1212/WNL.36.9.1249. [DOI] [PubMed] [Google Scholar]
- 289.Feely NM, Waghorn DJ, Dexter T, Gallen I, Chioidini P. Strongyloides stercoralis hyperinfection: difficulties in diagnosis and treatment. Anaesthesia. 2010;65:298–301. doi: 10.1111/j.1365-2044.2009.06196.x. [DOI] [PubMed] [Google Scholar]
- 290.Garcia HH, Pretell EJ, Gilman RH, et al. A trial of antiparasitic treatment to reduce the rate of seizures due to cerebral cysticercosis. N Engl J Med. 2004;350:249–258. doi: 10.1056/NEJMoa031294. [DOI] [PubMed] [Google Scholar]
- 291.Golenser J, McQuillain J, Hee L, et al. Conventional and experimental treatment of cerebral malaria. Internat J Parasitol. 2006;36:583–593. doi: 10.1016/j.ijpara.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 292.Hainard A, Tiberti N, Robin X, et al. Matrix metalloproteinase-9 and intercellulara adhesion molecule 1 are powerful staging markers for human African Trypanosomiasis. Trop Med Int Health. 2011;16:119–126. doi: 10.1111/j.1365-3156.2010.02642.x. [DOI] [PubMed] [Google Scholar]
- 293.Helbok R, Brenneis C, Beer R, et al. A rare case of Toxocara canis cerebral vasculitis. Europ J Neurol. 2007;14:117–120. doi: 10.1111/j.1468-1331.2006.01564.x. [DOI] [PubMed] [Google Scholar]
- 294.Herman JS, Chiodini PL. Gnathostomiasis, another emerging imported disease. Clin Microbiol Rev. 2009;22:484–492. doi: 10.1128/CMR.00003-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Häselbarth K, Tenter AM, Brade V, et al. First case of human babesiosis in Germany – Clinical presentation and molecular characterisation of the pathogen. Internat J Med Microbiol. 2007;297:197–204. doi: 10.1016/j.ijmm.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 296.Intapan PM, Khotsri P, Kanpittaya J, et al. Immunoblot diagnostic test for neurognathostomiasis. Am J Trop Med Hyg. 2010;83:927–929. doi: 10.4269/ajtmh.2010.10-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Katchanov J, Nawa Y. Helminthic invasion of the central nervous system: many roads lead to Rome. Parasitol Int. 2010;59:491–496. doi: 10.1016/j.parint.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 298.Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113:496–504. doi: 10.1172/JCI200421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Krishnan A, Karnad DR. Severe falciparum malaria: an important cause of multiple organ failure in Indian intensive care unit patients. Crit Care Med. 2003;31:2278–2284. doi: 10.1097/01.CCM.0000079603.82822.69. [DOI] [PubMed] [Google Scholar]
- 300.Khan NA. Novel in vitro and in vivo models to study central nervous system infections due to Acanthamoeba spp. Exp Parasitol. 2010;126:69–72. doi: 10.1016/j.exppara.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 301.Kristensson K, Nygard M, Bertini G, Bentivoglio African trypanosome infections of the nervous system: parasite entry and effects on sleep and synaptic functions. Prog Neurobiol. 2010;91:152–171. doi: 10.1016/j.pneurobio.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 302.Lackner P, Beer R, Broessner G, et al. Acute granulomatous acanthamoeba encephalitis in an immunocompetent patient. Neurocrit Care. 2010;12:91–94. doi: 10.1007/s12028-009-9291-z. [DOI] [PubMed] [Google Scholar]
- 303.Lacout A, Guidoux C, Carlier RY. Posterior reversible encephalopathy syndrome in neuro-malaria. Indian J Radiol Imaging. 2010;20:198–201. doi: 10.4103/0971-3026.69357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lee MB. Everyday and exotic foodborne parasites. Can J Infect Dis. 2000;11:155–158. doi: 10.1155/2000/120498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Lv S, Zhang Y, Steinmann P, Utzinger J. Helminth infections of the central nervous system occurring in Southeast Asia and the Far East. Adv Parasitol. 2010;72:351–408. doi: 10.1016/S0065-308X(10)72012-1. [DOI] [PubMed] [Google Scholar]
- 306.Mace SE. Central nervous system infections as a cause of an altered mental status? What is the pathogen growing in your central nervous system? Emerg Med Clin North Am. 2010;28:535–570. doi: 10.1016/j.emc.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 307.Marr JJ, Docampo R. Chemotherapy for Chagas’ disease: a perspective of current therapy and considerations for future research. Rev Infect Dis. 1986;8:884–903. doi: 10.1093/clinids/8.6.884. [DOI] [PubMed] [Google Scholar]
- 308.Michal A, Regli F, Campiche R, Cavallo RJ, de Crousaz G, Oberson R, Rabinowicz T. Cerebral coenurosis. Report of a case with arteritis. J Neurol. 1977;216:265–272. doi: 10.1007/BF00314050. [DOI] [PubMed] [Google Scholar]
- 309.Michelet L, Fleury A, Sciutto E, et al. Human neurocysticercosis: Comparision of different diagnostic tests in cerebrospinal fluid. J Clin Microbiol. 2011;49:195–200. doi: 10.1128/JCM.01554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Molyneux ME, Taylor TE, Wirima JJ, Borgstein A. Clinical features and prognostic indicators in paediatric cerebral malaria: a study of 131 comatose Malawian children. O J Med. 1989;71:441–459. [PubMed] [Google Scholar]
- 311.Nanda NC, Rath P, Acharya J, Mishra P, Mishra SK. Falciparum Malaria in Children-A Brief Report of 305 Patients from Rourkela, Eastern India. Indian J Pediatr. 2011;78:475–477. doi: 10.1007/s12098-010-0287-7. [DOI] [PubMed] [Google Scholar]
- 312.Nkouawa A, Sako Y, Itoh S, et al. Serological studies of neurologic helminthic infections in rural areas of southwest Cameroon: toxocariasis, cysticercosis and paragonimiasis. PLoS Negl Trop Dis. 2010;4:e732. doi: 10.1371/journal.pntd.0000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Onyeyili PA, Onwualu JE. Efficacy of combination of DFMO and diminazene aceturate in the treatment of late-stage Trypanosoma brucei infection in rats. Trop Med Parasitol. 1991;42:143–145. [PubMed] [Google Scholar]
- 314.Pitella JEH. Ischemic cerebral changes in the chronic chagasic cardiopathy. Arq Neuropsiquiatr. 1984;42:105–115. doi: 10.1590/S0004-282X1984000200002. [DOI] [PubMed] [Google Scholar]
- 315.Poltera AA. Pathology of human African trypanosomiasis with reference to experimental African trypanosomiasis and infections of the central nervous system. Br Med Bull. 1985;41:169–174. doi: 10.1093/oxfordjournals.bmb.a072045. [DOI] [PubMed] [Google Scholar]
- 316.Potchen MJ, Birbeck GL, Demarco JK, et al. Neuroimaging findings in children with retinopathy-confirmed cerebral malaria. Eur J Radiol. 2010;74:262–268. doi: 10.1016/j.ejrad.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Russegger L, Schmutzhard E. Spinal toxocara abscess. Lancet. 1989;2:398. doi: 10.1016/S0140-6736(89)90583-7. [DOI] [PubMed] [Google Scholar]
- 318.Saddler M, Barry M, Ternouth I, Emmanuel J. Treatment of severe malaria by exchange transfusion. N Engl J Med. 1990;322:58. doi: 10.1056/NEJM199001043220113. [DOI] [PubMed] [Google Scholar]
- 319.Saleem T, Rabbani M, Jamil B. Primary amoebic meningoencephalitis: two new cases from Pakistan. Trop Doct. 2009;39:242–243. doi: 10.1258/td.2009.090032. [DOI] [PubMed] [Google Scholar]
- 320.Sattar MA, Hoque HW, Amin MR, Faiz MA, Rahman MR. Neurological findings and outcome in adult cerebral malaria. Bangladesh Med Res Counc Bull. 2009;35:15–17. doi: 10.3329/bmrcb.v35i1.2313. [DOI] [PubMed] [Google Scholar]
- 321.Schmutzhard E. Parasitic diseases of the central nervous system. Nervenarzt. 2010;81:162–171. doi: 10.1007/s00115-009-2853-7. [DOI] [PubMed] [Google Scholar]
- 322.Schmutzhard E. Protozoal Infections. In: Noseworthy JH, editor. Neurological Therapeutics – Principles and Practice. Abingdon, Oxon: Informa Health Care; 2006. pp. 1116–1132. [Google Scholar]
- 323.Schmutzhard E, Mayr U, Rumpl E, Prugger M, Pohl P. Secondary cerebral amebiasis due to infection with Entamoeba histolytica. Eur Neurol. 1986;25:161–165. doi: 10.1159/000116003. [DOI] [PubMed] [Google Scholar]
- 324.Schmutzhard E, Boongird P, Vejjajiva A. Eosinophilic meningitis and radiculomyelitis in Thailand, caused by CNS invasion of Gnathostoma spingerum and Angiostrongylus cantonensis. J Neurol Neurosurg Psychiatry. 1988;51:80–87. doi: 10.1136/jnnp.51.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore) 1978;57:527–544. doi: 10.1097/00005792-197811000-00004. [DOI] [PubMed] [Google Scholar]
- 326.Silva AA, Roffe E, Santiago H, et al. Trypanosoma cruzi-triggered meningoencephalitis is a CCR1/CCR5-independent inflammatory process. J Neuroimmunol. 2007;184:156–163. doi: 10.1016/j.jneuroim.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 327.Solaymani-Mohammadi S, Lam MM, Zunt JR, Petri WA, et al. Entamoeba histolytica encephalitis diagnosed by PCR of cerebral spinal fluid. Trans R Soc Trop Med Hyg. 2007;101:311–313. doi: 10.1016/j.trstmh.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 328.Sotelo-Avila C. Naegleria and Acanthamoeba. Free-living amebas pathogenic for man. Perspect Pediatr Pathol. 1987;10:51–58. [PubMed] [Google Scholar]
- 329.Taelman H, Schaechter PJ, Marcelis L. Difluoromethylornithine, an effective new treatment of Gambian trypanosomiasis. Am J Med. 1987;82:607–614. doi: 10.1016/0002-9343(87)90107-0. [DOI] [PubMed] [Google Scholar]
- 330.Tran TH, Dolecek C, Pham PM, et al. Dihydroartemisinin-piperaquine against multidrug-resistant Plasmodium falciparum malaria in Vietnam: randomised clinical trial. Lancet. 2004;363:18–22. doi: 10.1016/S0140-6736(03)15163-X. [DOI] [PubMed] [Google Scholar]
- 331.Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasiotol. 2003;11:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 332.Utzinger J, Bergquist R, Olveda R, Zhou XN. Important helminth infections in Southeast Asia diversity, potential for control and prospects for elimination. Adv Parasitol. 2010;72:1–30. doi: 10.1016/S0065-308X(10)72001-7. [DOI] [PubMed] [Google Scholar]
- 333.Visvesvara GS. Amebic meningoencephalitides and keratitis: challenges in diagnosis and treatment. Curr Opin Infect Dis. 2010;23:590–594. doi: 10.1097/QCO.0b013e32833ed78b. [DOI] [PubMed] [Google Scholar]
- 334.Wang J, Qi H, Diao Z, et al. An outbreak of angiostrongyliasis cantonensis in Beijing. J Parasitol. 2010;96:377–381. doi: 10.1645/GE-2214.1. [DOI] [PubMed] [Google Scholar]
- 335.Wang P, Wu MC, Chen SJ, et al. Research development of the pathogenesis pathways for neuroschistosomiasis. Neurosci Bull. 2010;26:168–174. doi: 10.1007/s12264-010-0920-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Warrell DA, Looareesuwan S, Warrell MJ, Kasemarn P, Intaraprasert R, Bunnag D, Harinasuta T. Dexamethasone proves deleterious in cerebral malaria. A double blind trial in 100 comatose patients. N Engl J Med. 1982;306:313–319. doi: 10.1056/NEJM198202113060601. [DOI] [PubMed] [Google Scholar]
- 337.Watt G, Long GW, Ranoa CP, Adapon B, Fernando MT, Cross JH. Praziquantel in treatment of cerebral schistosomiasis. Lancet. 1989;2:262–263. doi: 10.1016/s0140-6736(86)90110-8. [DOI] [PubMed] [Google Scholar]
- 338.Wenzler T, Boykin DW, Ismail MA, et al. New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289 Antimicrobiob Agents. Chemother. 2009;53:4185–4192. doi: 10.1128/AAC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Winkler AS, Willingham AL, 3rd, Sikasunge CS, et al. Epilepsy and neurocysticercosis in sub-Saharan Africa. Wien Klin Wochenschr. 2009;3:3–12. doi: 10.1007/s00508-009-1242-3. [DOI] [PubMed] [Google Scholar]
- 340.Wittner M, Rowin KS, Tanowitz HB, et al. Successful chemotherapy of transfusion babesiosis. An Intern Med. 1982;96:601–604. doi: 10.7326/0003-4819-96-5-601. [DOI] [PubMed] [Google Scholar]
- 341.World Health Organization . Managment of Servere Malaria: A practical Handbook. 2. Geneva: World Health Organization; 2000. [Google Scholar]
- 342.Zimmerman GA, Castro-Faria-Neto H. Persistent cognitive impairment after cerebral malaria: models, mechanisms and adjunctive therapies. Expert Rev Anti Infect Ther. 2010;8:1209–1212. doi: 10.1586/eri.10.117. [DOI] [PubMed] [Google Scholar]
- 343.Zlobl TL. Amebiasis. Prim Care Update. 2001;8:65–68. doi: 10.1016/S1068-607X(00)00076-7. [DOI] [PubMed] [Google Scholar]
Zu 32.9
- 344.Abbott SP, et al. Fatal cerebral mycoses caused by the ascomycete Chaetomium strumarium. J Clin Microbiol. 1995;33:2692–2698. doi: 10.1128/jcm.33.10.2692-2698.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Al-Jehani H, Guiot MC, Torres C, Marcoux J. Scedosporium cerebral abscesses after extra-corporeal membrane oxygenation. Can J Neurol Sci. 2010;37:671–678. doi: 10.1017/S0317167100010878. [DOI] [PubMed] [Google Scholar]
- 346.Andreoni SC. Medical Mycology Atlas. Milano: S-Systems; 2004. [Google Scholar]
- 347.Bennett JE. Rapid diagnosis of candidiasis and aspergillosis. Rev Infect Dis. 1987;9:398–402. doi: 10.1093/clinids/9.2.398. [DOI] [PubMed] [Google Scholar]
- 348.Berger JR, Greenberg RN. Isolated central nervous system histoplasmosis in an immunocompetent patient: 53-month hiatus to diagnosis and treatment. J Neurovirol. 2010;16:472–474. doi: 10.1007/BF03210853. [DOI] [PubMed] [Google Scholar]
- 349.Black KE, Baden LR. Fungal infections of the CNS: treatment strategies for the immunocompromised patient. CNS Drugs. 2007;21:293–318. doi: 10.2165/00023210-200721040-00004. [DOI] [PubMed] [Google Scholar]
- 350.Blyth CC, Hale K, Palasanthiran P, O’Brian T, Bennett HMl Antifungal therapy in infants and children with proven, probable or suspected invasive fungal infections. Cochrane Database Syst Rev Feb. 2010;17:CD006343. doi: 10.1002/14651858.CD006343.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Christophe C, Azzi N, Bouche B, et al. Magnetic resonance imaging and angiography in cerebral fungal vasculitis. Neuropediatrics. 1999;30:218–220. doi: 10.1055/s-2007-973494. [DOI] [PubMed] [Google Scholar]
- 352.Cuenca-Estrella M, Gomez-Lopez A, Garcia-Effron G, et al. Combined activity in vitro of Caspofungin, amphothericin B, and azole agents against itraconazloe-resistant clinical isolates of Aspergillus fumigatus. Antimicrob Agents Chemother. 2005;49:1232–1235. doi: 10.1128/AAC.49.3.1232-1235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Diekema DJ, Messer SA, Hollis RJ, et al. Activities of caspofungin, itraconazole, posaconazole, ravuconazole, voriconazole, and amphotericin B against 448 recent clinical isolates of filamentous fungi. J Clin Microbiol. 2003;41:3623–3626. doi: 10.1128/JCM.41.8.3623-3626.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Dupont B. Overview of the lipid formulations of amphotericin B. J Antimicrob Chemother. 2002;49:31–36. doi: 10.1093/jac/49.suppl_1.31. [DOI] [PubMed] [Google Scholar]
- 355.Feltracco P, Barbieri S, Furnari M, et al. Central nervous system infectious complications early after liver transplantation. Transplant Proc. 2010;42:1216–1222. doi: 10.1016/j.transproceed.2010.03.108. [DOI] [PubMed] [Google Scholar]
- 356.Fica A, Diaz MC, Luppi M. Unsuccessful treatment with voriconazole of a brain abscess due to Cladophialophora bantiana. Scand J Infect Dis. 2003;35:892–893. doi: 10.1080/00365540310016736. [DOI] [PubMed] [Google Scholar]
- 357.Friedman JA, Wijdicks EF, Fulgham JR. Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review. Mayo Clin Proc. 2000;75:403–408. doi: 10.4065/75.4.403. [DOI] [PubMed] [Google Scholar]
- 358.Garcia RJ, Troya P, Edwards C. Invasive aspergillosis with central nervous system dissemination in a presumably immunocompetent, non-neutropenic patient: case report and review. South Med J. 2006;99:607–610. doi: 10.1097/01.smj.0000217123.43673.90. [DOI] [PubMed] [Google Scholar]
- 359.Glass HC, Wirrell E, Sarnat HB, et al. MRI findings in an immunocompromised boy with CNS fungal infection. Can J Neurol Sci. 2007;34:88–91. doi: 10.1017/S0317167100005850. [DOI] [PubMed] [Google Scholar]
- 360.Gottfredsson M, Perfect JR. Fungal meningitis. Semin Neurol. 2000;20:307–322. doi: 10.1055/s-2000-9394. [DOI] [PubMed] [Google Scholar]
- 361.Girmenia C, Martino P. New antifungal drugs and new clinical trials: interpreting results may be difficult. Curr Opin Oncol. 2003;15:283–288. doi: 10.1097/00001622-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 362.Herbrecht R, Denning DW, Paterson TF. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347:408–415. doi: 10.1056/NEJMoa020191. [DOI] [PubMed] [Google Scholar]
- 363.Javadi A, Ataei B, Koleini N, Saboori M. Central nervous system aspergillosis in an immunocompetent patient. Neurosciences (Riyadh) 2010;15:193–195. [PubMed] [Google Scholar]
- 364.Kowacs PA, Soares Silvado CE, Monteiro De Almeida S, et al. Infection of the CNS by Scedosporium apiospermum after near drowning. Report of a fatal case and analysis of its confounding factors. J Clin Pathol. 2004;57:205–207. doi: 10.1136/jcp.2003.8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Labadie EL, Hamilton RH. Survival improvement in coccidioidal meningitis by high-dose intrathecal amphotericin B. Arch Intern Med. 1986;146:2013–2018. doi: 10.1001/archinte.1986.00360220179029. [DOI] [PubMed] [Google Scholar]
- 366.Lazo A, Wilner HJ, Metes JJ. Craniofacial mucormycosis: computed tomographic and angiographic findings in two cases. Radiology. 1981;139:623–626. doi: 10.1148/radiology.139.3.7232728. [DOI] [PubMed] [Google Scholar]
- 367.Lowe JT, Hudson WR. Rhinocerebral phycomycosis and internal carotid artery thrombosis. Arch Otolaryngol. 1975;101:100–103. doi: 10.1001/archotol.1975.00780310022006. [DOI] [PubMed] [Google Scholar]
- 368.Mathews M, Paré L, Hasso A. Intraventricular cryptococcal cysts masquerading as racemose neurocysticercosis. Surg Neurol. 2007;67:647–649. doi: 10.1016/j.surneu.2006.10.049. [DOI] [PubMed] [Google Scholar]
- 369.Mazumder SA, Cleveland KO. Cryptococcal meningitis after neurosurgery. Am J Med Sci. 2010;339:582–583. doi: 10.1097/MAJ.0b013e3181deb19b. [DOI] [PubMed] [Google Scholar]
- 370.Moen MD, Lyseng-Willliamson KA, Scott LJ. Liposomal amphotericin B: a review of its use as empirical therapy in febrile neutropenia and in the treatment of invasive fungal infections. Drugs. 2009;69:361–392. doi: 10.2165/00003495-200969030-00010. [DOI] [PubMed] [Google Scholar]
- 371.Munoz P, Valerio M, Palomo J, et al. Infectious and non-infectious neurologic complications in heart transplant recipients. Medicine (Baltimore) 2010;89:166–175. doi: 10.1097/MD.0b013e3181dfa59c. [DOI] [PubMed] [Google Scholar]
- 372.Mylonakis E, Paliou M, Sax PE, et al. Central nervous system aspergillosis in patients with human iummuno-defficiency virus infection. Medicine. 2000;79:269–280. doi: 10.1097/00005792-200007000-00008. [DOI] [PubMed] [Google Scholar]
- 373.Nevado J, De Alarcon A, Hernandez A. Caspofungin: a new therapeutic option for fungal endocarditis. Clin Microbiol Infect. 2005;11:248–252. doi: 10.1111/j.1469-0691.2005.01078.x. [DOI] [PubMed] [Google Scholar]
- 374.Perea JR, Diaz De Rada BS, Quetglas EG, et al. Oral versus intravneous therapy in the treatment of systemic mycosis. Clin Microbiol Infect. 2004;1:96–106. doi: 10.1111/j.1470-9465.2004.00846.x. [DOI] [PubMed] [Google Scholar]
- 375.Perlroth J, Choi B, Spellberg B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol. 2007;45:321–346. doi: 10.1080/13693780701218689. [DOI] [PubMed] [Google Scholar]
- 376.Pfausler, Kampfl A, Berek K, Maier H, Aichner F, Schmutzhard E. Syndrome of the Anterior Spinal Artery as the Primary Manifestation of Aspergillosis. Infection. 1995;23:240–242. doi: 10.1007/BF01781206. [DOI] [PubMed] [Google Scholar]
- 377.Purkins L, Wood N, Ghahramani P, et al. Coadministration of voriconazole and phenytoin: pharmacokinetic interaction, safety, and toleration. Br J Clin Pharmacol. 2003;1:37–44. doi: 10.1046/j.1365-2125.2003.01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Raman Sharma R. Fungal infections of the nervous system: current perspective and controversies in management. Int J Surg. 2010;8:591–601. doi: 10.1016/j.ijsu.2010.07.293. [DOI] [PubMed] [Google Scholar]
- 379.Ramesha KN, Kate MP, Kesavadas C, et al. Fungal infections of the central nervous system in HIV-negative patients: experience from a tertiary referral center of South India. Ann Indian Acad Neurol. 2010;13:112–116. doi: 10.4103/0972-2327.64635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Raparia K, Powell SZ, Cernoch P, Takei H. Cerebral mycosis: 7-year retrospective series in a tertiary center. Neuropathology. 2010;30:218–223. doi: 10.1111/j.1440-1789.2009.01067.x. [DOI] [PubMed] [Google Scholar]
- 381.Revankar SG, Sutton DA, Rinaldi MG (2004) Primary central nervous system phaeohyphomycocis: a review of 101 cases. Clin Infect Dis (38):206–216 [DOI] [PubMed]
- 382.Rubio MC, de Ocariz IR, Gil J, et al. Potential fungicidal effect of voriconazole against Candida spp. Int J Antimicrob Agents. 2005;25:264–267. doi: 10.1016/j.ijantimicag.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 383.Ruchel R, et al. Cerebral Pseudallescheria mycosis after near-drowning. Mycoses. 1995;38:473–475. doi: 10.1111/j.1439-0507.1995.tb00022.x. [DOI] [PubMed] [Google Scholar]
- 384.Sakhuja V, Sud K, Kalra OP, et al. Central nervous system complications in renal transplant recipients in a tropical environment. J Neurol Sci. 2001;183:89–93. doi: 10.1016/S0022-510X(00)00485-8. [DOI] [PubMed] [Google Scholar]
- 385.Sasaki T, Mineta M, Kobayashi K, Ando M, Obata M. Zygomycotic invasion of the central nervous system. Jpn J Radiol. 2010;28:376–80. doi: 10.1007/s11604-010-0435-z. [DOI] [PubMed] [Google Scholar]
- 386.Schmutzhard E. Fungal Infections. In: Noseworthy JH, editor. Neurological Therapeutics-Principles and Practice. Abingdon, Oxon: Informa Healthcare; 2006. pp. 1085–1115. [Google Scholar]
- 387.Scully EP, Baden LR, Katz JT. Fungal brain infections. Curr Opin Neurol. 2008;21:347–352. doi: 10.1097/WCO.0b013e3282fee95b. [DOI] [PubMed] [Google Scholar]
- 388.Senzolo M, Ferronato C, Burra P. Neurologic complications after solid organ transplantation. Transpl Int. 2009;22:269–278. doi: 10.1111/j.1432-2277.2008.00780.x. [DOI] [PubMed] [Google Scholar]
- 389.Shamin MS, Enam SA, Ali R, Anwar S. Craniocerebral aspergillosis: a review of advances in diagnosis and management. J Pak Med Assoc. 2010;60:573–579. [PubMed] [Google Scholar]
- 390.Siegal JA, Cacayorinb ED, Nassif AS, et al. Cerebral mucormycosis: proton MR spectroscopy and MR imaging. Magn Reson Imaging. 2000;18:915–920. doi: 10.1016/S0730-725X(00)00180-6. [DOI] [PubMed] [Google Scholar]
- 391.Sun HY, Forrest G, Gupta KL, et al. Rhino-orbital-cerebral zygomycosis in solid organ transplant recipients. Transplantation. 2010;15:85–92. doi: 10.1097/TP.0b013e3181dde8fc. [DOI] [PubMed] [Google Scholar]
- 392.Van Hal SJ, Clezy K. Emergence of invasive cerebral aspergillosis in an HIV-positive patient on voriconazole therapy. HIV Med. 2005;6:45–46. doi: 10.1017/S0025727300026843. [DOI] [PubMed] [Google Scholar]
- 393.Varanasi NL, Baskaran I, Alangaden GJ, et al. Novel effect of voriconazole on conidiation of Aspergillus species. Int J Antimicrob Agens. 2004;23:72–79. doi: 10.1016/j.ijantimicag.2003.05.011. [DOI] [PubMed] [Google Scholar]
- 394.Vos MJ, Debets-Ossenkopp YJ, Claessen FA, et al. Cerebellar and medullar histoplasmosis. Neurology. 2000;54:1441. doi: 10.1212/WNL.54.7.1441. [DOI] [PubMed] [Google Scholar]
- 395.Weng CT, Lee NY, Liu MF, et al. A retrospective study of catastrophic invasive fungal infections in patients with systemic lupus erythematosus from southern Taiwan. Lupus. 2010;19:1204–1209. doi: 10.1177/0961203310368969. [DOI] [PubMed] [Google Scholar]
- 396.Wingard JR, Leather H. A new era of antifungal therapy. Biol Blood Marrow Transplant. 2004;10:73–90. doi: 10.1016/j.bbmt.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 397.Zivkovic Neuroimaging and neurologic complications after organ transplantation. J Neuroimaging. 2007;17:110–23. doi: 10.1111/j.1552-6569.2007.00097.x. [DOI] [PubMed] [Google Scholar]
Zu 32.10
- 398.Alhelm F, et al. Spondylitis/Spondylodiszitis. Radiologe. 2006;46:480–485. doi: 10.1007/s00117-006-1368-5. [DOI] [PubMed] [Google Scholar]
- 399.Belzunegui J, et al. Vertebral osteomyelitis in northern Spain. Report of 62 cases. Clin Exp Rheumatol. 1999;177:447–452. [PubMed] [Google Scholar]
- 400.Beronius M, Bergman B, Andersson R. Vertebral osteomyelitis in Göteborg, Sweden: a retrospective study of patients during 1990–95. Scand J Infect Dis. 2001;33:527–532. doi: 10.1080/00365540110026566. [DOI] [PubMed] [Google Scholar]
- 401.Chelsom J, Solberg CO. Vertebral osteomyelitis at a Norwegian university hospital 1987–97: clinical features, laboratory findings and outcome. Scand J Infect Dis. 1998;30:147–151. doi: 10.1080/003655498750003537. [DOI] [PubMed] [Google Scholar]
- 402.Cordonnier C, et al. Prospective study of patients presenting with acute partial transverse myelopathy. J Neurol. 2003;250:1447–1452. doi: 10.1007/s00415-003-0242-x. [DOI] [PubMed] [Google Scholar]
- 403.Debette S, de Sèze J, Pruvo J-P, Zephir H, Pasquier F, Leys D, Vermersch P. Long-term outcome of acute and subacute myelopathies. J Neurol. 2009;256:980–988. doi: 10.1007/s00415-009-5058-x. [DOI] [PubMed] [Google Scholar]
- 404.De Seze J, et al. Acute Myelopathies. Clinical. Laboratory and outcome profiles in 79 patients. Brain. 2001;124:1509–1521. doi: 10.1093/brain/124.8.1509. [DOI] [PubMed] [Google Scholar]
- 405.Duncan S, Halfpenny C, Galea I. CNS inflammation other than multiple sclerosis: How likely is diagnosis? Neurology. 2014;82:1187–1189. doi: 10.1212/WNL.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Erickson BJ, Weinshenker BG. Compressive myelopathy mimicking transverse myelitis. Neurologist. 2010;16:120–122. doi: 10.1097/NRL.0b013e3181c29f12. [DOI] [PubMed] [Google Scholar]
- 407.Frohmann EM, Wingerchuk DM. Clinical Practice. Transverse Myelitis. N Engl J Med. 2010;363:564–572. doi: 10.1056/NEJMcp1001112. [DOI] [PubMed] [Google Scholar]
- 408.Grabowski A. Manual Neurologische Akut- und Intensivmedizin. Stuttgart: Schattauer; 2013. [Google Scholar]
- 409.Greenberg BM, et al. Idiopathic transverse myelitis. Corticosteroids, plasma exchange, or cyclophosphamid. Neurology. 2007;68:1614–1617. doi: 10.1212/01.wnl.0000260970.63493.c8. [DOI] [PubMed] [Google Scholar]
- 410.Ho EL. Infectious Etiologies of Myelopathy. Semin Neurol. 2012;32:154–160. doi: 10.1055/s-0032-1322588. [DOI] [PubMed] [Google Scholar]
- 411.Honnorat J, Antoine J-C. Paraneoplastic neurological syndromes. Orphanet J Rare Dis. 2007;2:22. doi: 10.1186/1750-1172-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Irani DN, Kerr DA. 14-3-3 protein in the cerebrospinal fluid of patients with acute transverse myelitis. Lancet. 2000;355(9207):901. doi: 10.1016/S0140-6736(99)04745-5. [DOI] [PubMed] [Google Scholar]
- 413.Jacob A, Weinshenker BG. An Approach to the Diagnosis of Acute Tranverse Myelitis. Semin Neurol. 2008;28:105–120. doi: 10.1055/s-2007-1019132. [DOI] [PubMed] [Google Scholar]
- 414.Kalita J, Misra UK, Mandal SK. Prognostic predictors of acute transverse myelitis. Acta Neurol Scand. 1998;98:60–63. doi: 10.1111/j.1600-0404.1998.tb07379.x. [DOI] [PubMed] [Google Scholar]
- 415.Kalita J, Misra UK. Neurophysiological studies in acute transverse myelitis. J Neurol. 2000;247:943–948. doi: 10.1007/s004150070051. [DOI] [PubMed] [Google Scholar]
- 416.Kalita J, Misra UK. Is methyl prednisolone useful in acute transverse myelitis? Spinal Cord. 2001;39:471–476. doi: 10.1038/sj.sc.3101190. [DOI] [PubMed] [Google Scholar]
- 417.Karussis D, Petrou P. The spectrum of post-vaccination inflammatory CNS demyelinating syndromes. Autoimm Rev. 2014;13:215–224. doi: 10.1016/j.autrev.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 418.Krishnan C, et al. Transverse Myelitis: pathogenesis, diagnosis and treatment. Front Biosci. 2004;9:1483–1499. doi: 10.2741/1351. [DOI] [PubMed] [Google Scholar]
- 419.Lewis M, Howdle The neurology of liver failure. Q J Med. 2003;96:623–633. doi: 10.1093/qjmed/hcg110. [DOI] [PubMed] [Google Scholar]
- 420.Mealy M, Wingerchuck D, Palace J, Greenberg B, Levy B. Comparison of Relapse and Treatment Failure Rates Among Patients With Neuromyelitis Optica. Multicenter Study of Treatment Efficacy. JAMA Neurology. 2014;71:324–330. doi: 10.1001/jamaneurol.2013.5699. [DOI] [PubMed] [Google Scholar]
- 421.Meyding-Lamadé U, et al. Akute spinale Entzündungen. Intensivmed. 2005;42:337–344. doi: 10.1007/s00390-005-0552-6. [DOI] [Google Scholar]
- 422.Rotbart HA. Viral meningitis. Semin Neurol. 2000;20:277–292. doi: 10.1055/s-2000-9427. [DOI] [PubMed] [Google Scholar]
- 423.Schellinger PD, Schmutzhard E, Fiebach JB, Pfausler B, Maier H, Schwab S. Poliomyelitic-like illness in central european encephalitis. Neurology. 2000;55:299–302. doi: 10.1212/WNL.55.2.299. [DOI] [PubMed] [Google Scholar]
- 424.Schlossbauer T, Panteleon A, Becker-Gaab C. Entzündliche Wirbelsäulenerkrankungen als Ursache für Rückenschmerzen. Radiologe. 2006;46:468–479. doi: 10.1007/s00117-006-1387-2. [DOI] [PubMed] [Google Scholar]
- 425.Schmalstieg W, Weinshenker BG. Approach to acute or subacute myelopathy. Neurology Clinical Practice. 2010;75(1):2–S8. doi: 10.1212/WNL.0b013e3181fb3638. [DOI] [PubMed] [Google Scholar]
- 426.Sellner J, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–1032. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- 427.Transverse Myelitis Consortium Working Group Neurology Proposed diagnostic citeria and nosology of acute tranverse myelitis. Neurology. 2002;59:499–505. doi: 10.1212/WNL.59.4.499. [DOI] [PubMed] [Google Scholar]
- 428.West T, Hess C, Cree B. Acute transverse Myelitis: Demyelinating, Inflammatory, and infectious Myelopathies. Semin Neurol. 2012;32:97–113. doi: 10.1055/s-0032-1322586. [DOI] [PubMed] [Google Scholar]
- 429.Wingerchuk DM, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 430.Wong SH, et al. Myelopathy but normal MRI: where next? Pract Neurol. 2008;8:90–102. doi: 10.1136/jnnp.2008.144121. [DOI] [PubMed] [Google Scholar]