

Summary
The Gram-positive bacterium Staphylococcus aureus is a versatile pathogen that can sense and adapt to a wide variety of environments within the human host, in part through its 16 two-component regulatory systems. The ArlRS two-component system has been shown to affect many cellular processes in S. aureus, including autolysis, biofilm formation, capsule synthesis, and virulence. Yet the molecular details of this regulation remained largely unknown. We used RNA-sequencing to identify the ArlRS regulon, and found 70% overlap with that of the global regulator MgrA. These genes included cell wall-anchored adhesins (ebh, sdrD), polysaccharide and capsule synthesis genes, cell wall remodeling genes (lytN, ddh), the urease operon, genes involved in metal transport (feoA, mntH, sirA), anaerobic metabolism genes (adhE, pflA, nrdDG), and a large number of virulence factors (lukSF, lukAB, nuc, gehB, norB, chs, scn, and esxA). We show that ArlR directly activates expression of mgrA and identify a probable ArlR binding site (TTTTCTCAT-N4-TTTTAATAA). A highly similar sequence is also found in the spx P2 promoter, which was recently shown to be regulated by ArlRS. We also demonstrate that ArlS has kinase activity toward ArlR in vitro, although it has slower kinetics than other similar histidine kinases.
Keywords: Staphylococcus aureus, virulence factors, histidine kinase, RNA sequencing, binding site, regulon
Abbreviated Summary
Methicillin Resistant Staphylococcus aureus (MRSA) is a leading cause of bloodstream infections, pneumonia, and surgical site infections. One of the hallmarks of S. aureus is its diverse array of virulence factors, such as secreted toxins, cell wall-anchored adhesins, and immune evasion factors. We demonstrate that the ArlRS two-component system regulates more than 200 genes, including many virulence factors, primarily through controlling expression of the global regulators MgrA and Spx.
Introduction
The Gram-positive pathogen Staphylococcus aureus is a commensal that colonizes the nostrils, throat, and skin of ~30% of the population (Mertz et al., 2009, Gorwitz et al., 2008, Miller & Diep, 2008). It causes a broad spectrum of invasive infections, including osteomyelitis, bacteremia, endocarditis, and pneumonia (Lowy, 1998), and it is one of the leading causes of healthcare-associated infections such as surgical site infections and central line-associated bloodstream infections (Weiner et al., 2016). S. aureus is such a versatile pathogen in part because of its large arsenal of virulence factors that help it to establish a foothold in diverse body sites and ultimately spread to other sites. These virulence factors include secreted toxins and enzymes (Tam & Torres, 2019), cell wall-anchored adhesins (Foster et al., 2014), and a variety of proteins involved in immune evasion (Thammavongsa et al., 2015). Expression of many of these virulence genes is controlled by two-component systems (TCS) (Haag & Bagnoli, 2017) such as the agr quorum-sensing system (Thoendel et al., 2011), as well as SaeRS (Liu et al., 2016), SrrAB (Yarwood et al., 2001), and ArlRS (Liang et al., 2005). These TCSs allow S. aureus to sense extracellular conditions and change gene expression accordingly. A typical TCS consists of a membrane bound histidine kinase that responds to a particular stimulus, causing it to autophosphorylate. It then transfers that phosphate to a cytoplasmic response regulator protein, which can then bind DNA and affect transcription of a subset of genes (Stock et al., 2000).
The ArlRS TCS was originally identified as a regulator of autolysis (autolysis-related locus) and biofilm formation (Fournier & Hooper, 2000). Mutations in arlRS lead to increased biofilm formation (Toledo-Arana et al., 2005), but also decreased clumping in the presence of human plasma (Walker et al., 2013) and decreased adhesion to host extracellular matrix proteins (Kwiecinski et al., 2019). ArlRS is also important for virulence in a number of animal models of infection, including systemic infection in mice (Benton et al., 2004, Radin et al., 2016), infectious endocarditis in rabbits (Walker et al., 2013), and a mouse catheter-associated biofilm infection (Burgui et al., 2018). Despite its importance in infection, the molecular details of the ArlRS TCS and specifically what it regulates are largely unknown. An early microarray study suggested that ArlRS directly or indirectly regulated over 100 genes, including other regulators like the agr quorum-sensing system and the LytSR TCS (Liang et al., 2005). Since then, ArlRS has been shown to activate expression of the global regulator MgrA (Luong & Lee, 2006, Crosby et al., 2016). MgrA in turn regulates a variety genes, including those for surface proteins and secreted virulence factors (Crosby et al., 2016). ArlRS was shown to affect amino acid metabolism and sensitivity to the Zn-, Mn-, and Fe-binding protein calprotectin (Radin et al., 2016). It was also reported that ArlRS is required for leukocidin expression, perhaps through regulation of agr (Harper et al., 2018). Recent work showed that ArlRS also activates expression of Spx, a transcriptional regulator involved in the response to oxidative stress and antibiotic resistance (Bai et al., 2019). ArlR was shown to directly bind to the spx promoter region, although the exact binding site was not determined (Bai et al., 2019).
ArlS is a membrane-bound histidine kinase with an extracellular domain that senses an unknown signal. Upon activation, ArlS autophosphorylates a conserved histidine residue (H242), and then transfers this phosphate to an aspartate residue within ArlR (D52) (Villanueva et al., 2018, Delgado et al., 1993, Stock et al., 2000). ArlR is an OmpR/PhoB-type response regulator protein that binds DNA upon phosphorylation by ArlS. Many histidine kinases have both kinase and phosphatase activity toward their cognate response regulator, and the dominant activity depends on the presence of their specific signal and the balance between kinase/phosphatase activities (Casino et al., 2010). The phosphatase activity serves as a way to reset signaling activity after the stimulus is gone, and also suppresses non-specific signaling from other histidine kinases and inorganic phosphate donors like acetyl-phosphate (Huynh & Stewart, 2011, Siryaporn & Goulian, 2008, Hentschel et al., 2014, Wayne et al., 2012). Distinct residues within the catalytic domain of histidine kinases are required for kinase and phosphatase activity (Willett & Kirby, 2012, Dutta et al., 2000, Wayne et al., 2012).
In this work we used RNA-seq to investigate the ArlRS regulon and compare it to the group of genes regulated by MgrA (Crosby et al., 2016). The two regulons largely overlap, indicating that MgrA is one of the major targets of ArlR. We also confirm that the disulfide stress regulator Spx is regulated by ArlRS, but not by MgrA. Both mgrA and spx promoters share a potential ArlR binding site that is located 11 bp upstream of the −10 sequence, suggesting that they are directly regulated by ArlR. Lastly, we investigated the kinase and phosphatase activity of ArlS using targeted point mutations.
Results
Analysis of the ArlRS regulon
We previously determined the MgrA regulon using RNA-sequencing (Crosby et al., 2016), and in this work we sought to compare this to the ArlRS regulon under similar growth conditions. S. aureus USA300 strain LAC and an isogenic arlRS deletion mutant were grown in tryptic soy broth to mid-log phase (OD600 of 1.5), and changes in gene expression were determined using RNA-sequencing. Using a ≥2-fold cutoff limit and false discovery rate (FDR)-corrected p-value of <0.05, 126 genes were up-regulated and 124 genes were down-regulated in the arlRS mutant compared to LAC (Table S1). As expected, mgrA expression was down ~5-fold in the arlRS mutant, confirming that ArlRS activates mgrA expression. Expression of spx was down 2.7-fold in the arlRS mutant, and no change in expression was seen for the agrBDCA operon or the lytSR TCS (Table S1).
There was substantial overlap between the ArlRS regulon and that of MgrA. Of the 250 genes regulated by ArlRS, 70% were also differentially expressed in the mgrA mutant, and in all cases the direction of regulation was the same (Table S2). Focusing on the genes regulated ≥4-fold by ArlRS, 55 out of 57 (96%) were also regulated ≥ 2-fold by MgrA (Fig. 1). Up-regulated genes included the cell surface adhesins ebh, sdrD, fmtB, and sasC, cell wall hydrolase genes such as lytN, the urease operon genes ureABC, putative polysaccharide synthesis loci cap1ABC and SAUSA300_0130–0133, and the Mn transporter mntH. Down-regulated genes in common between ArlRS and MgrA included the capsule biosynthetic genes cap5A and cap5D, and a variety of secreted virulence factors, such as lukSF (Panton Valentine Leukocidin), lukAB, chemotaxis inhibitor chs (CHIPS), complement inhibitor scn (SCIN), and nuclease (nuc). Both the capsule operon (Luong & Lee, 2006) and the leukocidins (Harper et al., 2018) have been previously reported to be regulated by ArlRS. The first ten genes in the type VII secretion system gene cluster, starting with esxA, were down 3–6 fold in the arlRS mutant, consistent with a previous report that esxA is regulated by ArlRS (Schulthess et al., 2012). Three transcriptional regulators, sarV, atlR, and SAUSA300_1202, were also repressed by both ArlRS and MgrA, suggesting that there are likely several levels to the ArlRS regulatory cascade, and that some genes may be indirectly regulated by MgrA. ArlRS-regulated genes that appeared to be independent of MgrA included the putative peptidoglycan hydrolase SAUSA300_0739, and the catechol siderophore transport genes sstB and sstC (Morrissey et al., 2000, Beasley et al., 2011).
Fig. 1. RNA-seq analysis of the ArlRS regulon and comparison to that of MgrA.

All genes with a fold change ≥ 4 in the arlRS mutant are shown, grouped by predicted function. For comparison, the change in expression of these genes in the mgrA mutant grown under the same conditions is also shown (Crosby et al., 2016).
To confirm a subset of the RNA-seq results, we constructed GFP transcriptional fusions for 12 of these genes, 6 predicted to be up-regulated and 6 down-regulated (Fig. 2). Construction of fusion plasmids was based on predicted promoter sequences from Prados et al. (Prados et al., 2016) or other published promoter sequences where possible. Reporter expression at 24 h is shown in Fig. 2, and representative growth curves for a subset of these reporters are shown in Supplemental Fig. 1. As expected based on previous results (Crosby et al., 2016), expression from the upstream mgrA promoter (mgrA P2) was strongly dependent on ArlRS, but was not autoregulated (Fig. 2A). Expression of nuclease (nuc), the leukocidins lukA and lukS, the type VII secretion gene esxA, and the secreted lipase gehB were all significantly down in the arlRS mutant and could be complemented (Fig. 2B–F), verifying the RNA-seq results. All five of these genes were also significantly affected by the mgrA deletion (Fig. 2B–F), suggesting that ArlRS may indirectly regulate their expression through MgrA. Likewise, the up-regulated genes that we investigated, ebh, mntH, sdrD, lytN, SAUSA300_0130, and ureA all showed significantly higher expression in both the arlRS and mgrA mutants, and the arlRS effect could be complemented (Fig. 2G–L). We have previously shown that under these growth conditions ~90% of MgrA expression is driven by the ArlRS-regulated upstream promoter mgrA P2 (Crosby et al., 2016). In the arlRS mutant there is residual low-level expression of MgrA from the downstream mgrA P1 promoter, which likely explains the difference in target gene expression in the arlRS and mgrA mutants.
Fig. 2. Verification of selected RNA-seq results using transcriptional reporters.

Plasmids containing the promoter regions fused to GFP were introduced into the wild-type LAC strain (black), arlRS mutant (blue), chromosomally complemented arlRS mutant (gray), and mgrA mutant (red). Gene name or locus tag (strain USA300_FPR3757) is indicated above each graph. Cultures were grown for 24 h and the average fluorescence signal and standard deviation was calculated from at least three independent experiments. Statistical significance relative to LAC was calculated with Prism using a one-way ANOVA with Dunnett’s test for multiple comparisons. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001
Identifying the ArlRS binding site
The RNA-seq data suggest that ArlR likely directly regulates expression of mgrA, although the exact binding site remains unknown. The mgrA gene has two promoters (Fig. 3A), and we have previously shown that expression from only the upstream promoter, mgrA P2, is activated by ArlR (Crosby et al., 2016). Two potential transcription start sites (TSSs) for mgrA P2 have been identified (Ingavale et al., 2003, Prados et al., 2016), located 300 and 302 bp upstream of the mgrA start codon. Prados et al. (Prados et al., 2016) selected the adenine 300 bp upstream as the “representative TSS” based on its lower p-value in their work, so we used that as the mgrA P2 TSS going forward. There is a consensus −10 sequence (TATAAT) centered 11 bp upstream of this TSS, but this promoter appears to lack an identifiable −35 sequence. Expression from this promoter requires arlRS (Fig. 3B), suggesting that ArlR may compensate for the poor −35 site.
Fig. 3. Determining minimum mgrAP2 promoter length required for activation by ArlRS.

Sequentially smaller fragments of the mgrA P2 promoter region were cloned into pCM11 in front of GFP (A), and expression was measured at 24 h in wild-type strain LAC (B). Numbers correspond to the plasmid numbers in (A). The average fluorescence signal and standard deviation was calculated from at least three independent experiments. Statistical analysis is as described for Fig. 2. (C) Sequences of the four shortest reporters, showing only the region upstream of the transcription start site (TSS). The predicted −10 site is indicated above the sequences.
Our first step in locating the ArlR binding site was to clone a series of truncated mgrA P2 promoter fragments in front of GFP (Fig. 3A–C). The shortest fragment with full activity in the LAC wild-type strain, pHC137, included 72 bp upstream of the TSS. Shorter promoter fragments showed decreasing activity, and the shortest reporter, pHC146 (32 bp upstream of the TSS) had virtually no activity. This information suggested that ArlR likely binds somewhere between 30 and 72 bp upstream of the TSS. Next, we took the shortest fully active reporter, pHC137, and constructed staggered 6-bp mutations across this entire region (Fig. 4A–B). When these mutated reporters were introduced into LAC, six reporters showed minor decreases in expression, and seven consecutive reporters had almost no activity (Fig. 4A). These reporters with minimal activity had mutations spanning a region located ~45–25 bp upstream of the TSS, overlapping the area where a −35 sequence would normally be (Fig. 4B). We determined that this region was likely to contain the ArlR binding site. We then made a series of 2 bp mutations spanning this region to better define the ArlR binding site (Fig. 5A–B). One of these reporters, pHC198, containing a 2 bp change 46 and 45 bp upstream of the TSS, actually had higher activity, while the remaining reporters had much lower activity than the wild-type sequence (pHC137). The reason for the higher activity of pHC198 is unclear, but perhaps introducing these mutations helps ArlR to bind productively to the mgrA P2 promoter by disrupting another potential binding position. Mutations in the middle of the binding site, pHC194, pHC195, and pHC196, were less detrimental than mutations on either side, suggesting a symmetrical binding site separated by a short gap. We surmised that the likely ArlR binding site is TTTTCTCAT-N4-TTTTAATAA, located 44 to 23 bp upstream of the TSS. This near direct repeat is consistent with other OmpR-family response regulators, which often bind direct repeats separated by a 4–5 bp spacer (de Been et al., 2008).
Fig. 4. Mutating potential ArlR binding site.
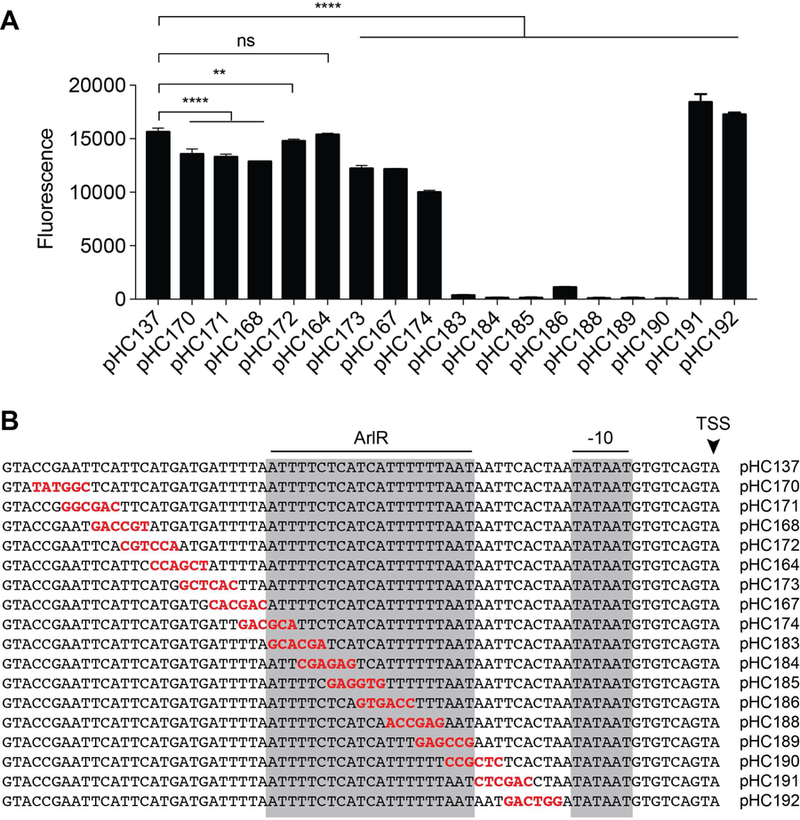
6 bp mutations were introduced into pHC137, an mgrA P2 reporter with full activity, and expression was tested at 24 h in wild-type strain LAC (A). (B) Sequences of each promoter fragment, showing the 6 bp mutation in red. The inferred ArlR binding site is indicated by the gray box, along with the predicted −10 site and transcription start site. The average fluorescence signal and standard deviation was calculated from at least three independent experiments.
Fig. 5. Refining the ArlR binding site.

2 bp mutations were introduced into the mgrA P2 reporter pHC137, based on the results of Fig. 4. Expression was measured at 24 h in strain LAC as before. The refined ArlR binding site and −10 sequence are indicated by the gray boxes. The average fluorescence signal and standard deviation was calculated from at least three independent experiments.
To show direct binding of ArlR to this potential binding site we used electrophoretic mobility shift assays (EMSAs). Increasing concentrations of phosphorylated ArlR (0 to 2 μg, which corresponds to 0 to 7.2 μM) were incubated with an IRDye labeled 50 nt DNA probe overlapping the proposed binding site, or a probe directly upstream of the first probe (Fig. 6). ArlR was able to bind to the probe containing the proposed binding site and showed minimal binding to the upstream non-specific probe. This indicates that ArlR likely directly regulates mgrA expression and that we have identified the correct location of the binding site.
Fig. 6. EMSA showing binding of ArlR to the mgrA P2 promoter.

(A) Purified, His-tagged ArlR was incubated with 10 nM IRDye700-labeled probes, and then separated by PAGE. (B) Location of probes 2 and 3 within the mgrA P2 promoter, as well as the predicted ArlR binding site. The images are representative of two replicates.
Spx regulation
It was recently shown that ArlRS activates expression of the transcriptional regulator spx (Bai et al., 2019). Spx interacts directly with RNA polymerase and is involved in the disulfide stress response in Firmicutes (Hillion & Antelmann, 2015). Expression of spx was down 2.7-fold in the arlRS mutant in our RNA-seq experiment (Table S1. A spx reporter (pHC169) was constructed containing the majority of the intergenic region upstream of spx fused to GFP. Expression of this reporter was significantly lower in the arlRS mutant and could be complemented (Fig. 7A). However, expression in the mgrA mutant was similar to wild-type (Fig. 7A), suggesting that ArlRS may directly regulate spx. The spx gene has two promoters within this upstream region, a distal promoter P1 and a proximal promoter P2 (Fig. 7B) (Pamp et al., 2006, Jousselin et al., 2013, Donegan et al., 2014, Prados et al., 2016). To investigate spx regulation by ArlRS further, we constructed separate reporters for each promoter. Time courses using each of these three reporters in the wild-type LAC background and the arlRS mutant demonstrated that only spx P2 is regulated by ArlRS (Fig. 7C–E). In the absence of arlRS there was essentially no expression from this P2 reporter, similar to the mgrA P2 promoter, whose expression requires ArlRS. Alignment of mgrA P2 and spx P2 demonstrated that both promoters have very similar sequences in the ArlR binding region (Fig. 7F). The first repeat is identical, and in the second repeat seven out of nine nucleotides are identical. In addition, the spacing between the proposed ArlR binding site and canonical −10 sequence is identical in the two promoters (Fig. 7F), lending strength to our proposed ArlR binding site.
Fig. 7. ArlRS regulate expression of spx P2.

(A) Expression of spx-GFP reporter pHC169 in the indicated S. aureus strains after 24 h of growth. The average fluorescence signal and standard deviation was calculated from three independent experiments. (B) Schematic of the major spx promoters determined by Prados et al. (Prados et al., 2016), as well as the transcriptional reporters used below. (C-E) Time course of each spx reporter in wild-type LAC (black) or the arlRS mutant (blue). (F) Alignment of mgrA P2 and spx P2, showing the potential ArlR binding site. The growth curve is representative of two replicates.
Search for other promoters containing a potential ArlR binding site
Alignment of the mgrA P2 and spx P2 promoters suggests a potential ArlR consensus binding site TTTTCTCAT-N4-TTTTAATNN. A search for ≥90% matches to this sequence in the S. aureus USA300_FPR3757 genome yielded 38 hits (Table S3). Only 13 of these hits were located upstream of a nearby gene and in the same orientation. This set of 13 included mgrA and spx, as expected. The only other overlap with our RNA-seq experiment was SAUSA300_0130, a putative UDP-glucose 4-epimerase and the first gene in a possible five-gene glycan synthesis locus. Two of the 13 potential ArlR binding sites were located just upstream of SAUSA300_0130 (Supplemental Fig. 2). Although the location of the SAUSA300_0130 promoter is unknown, these binding sites are likely too close to the start codon to substitute for a −35 promoter element. In addition, expression of SAUSA300_0130 was up ~23-fold in the arlRS mutant, suggesting that ArlR might repress expression of SAUSA300_0130. Our RNA-seq results and SAUSA300_0130-GFP fusion (Fig. 2K) indicate that SAUSA300_0130 is repressed by MgrA. It seems unusual, but possible, that this potential glycan synthesis operon would be repressed both directly and indirectly (through MgrA) by ArlRS.
ArlS kinase/phosphatase activity
In this study, we also characterized ArlS histidine kinase enzyme kinetics that dictate its ability to regulate the phosphorylation state of its cognate response regulator ArlR. Although ArlS has already been identified as a HisKA (or Subtype 1A) family member (e.g. ScanProsite (de Castro et al., 2006)), the N-terminal region has not been annotated. Homology modeling using the Phyre2 (Kelley et al., 2015) server predicts that ArlS is a multi-domain protein (Fig. 8A) containing two transmembrane spanning regions (from residue 1 to 33 and 154 to 186) and a putative extracellular sensor or Cache domain (Anantharaman & Aravind, 2000) (residue 34 to 153). The second transmembrane region extends into the cytoplasmic HAMP (named for histidine kinases, adenylyl cyclases, methyl-accepting chemotaxis proteins and Phosphatases) domain (Parkinson, 2010). The catalytic core of ArlS consists of dimerization and histidine phosphotransfer (DHp) and catalytic ATP binding (CA) domains. It has been reported that 75–85% of histidine kinases from 1500 sequenced bacterial genomes belong to the HisKA family (Trajtenberg et al., 2010, Willett & Kirby, 2012). The HisKA family of histidine kinases is characterized by the presence of an H-box consensus motif (Stock et al., 1989) within the DHp domain that contains the phosphorylatable histidine residue: H E X R T/N P, where X is a hydrophobic residue. Within this same motif, the conserved threonine/asparagine residue has been shown to be important for phosphatase activity (Willett & Kirby, 2012, Huynh & Stewart, 2011).
Fig. 8. Characterization and activity of ArlS and ArlR proteins.

(A) Domain architecture of ArlS and ArlR proteins. ArlS is a membrane bound histidine kinase with extracellular sensing domain, transmembrane HAMP domain and cystoplasmic DHpCA domain. ArlR is predicted to be an OmpR-like response regulator with receiver domain and winged helix-turn-helix DNA binding domain. (B) Coomassie staining showing purified ArlS (DHpCA, ~26 kDa; HAMP-DHpCA, ~31 kDa) and ArlR (~26 kDa) proteins. (C) Autophosphorylation of ArlS HAMP-DHpCA (left panel) and DHpCA (right panel). (D) Phosphotransfer of ArlS HAMP-DHpCA (left panel) and DHpCA (right panel) to full-length ArlR. All assays were performed at least three times and the images are representative data.
Our data show that, like other HisKA proteins, ArlS is a dual function enzyme with kinase and phosphatase activity that regulates the phosphorylation state of ArlR. We purified full-length ArlR (~26 kDa), as well as soluble ArlS constructs corresponding to just the DHp and CA domains (DHpCA, ~26 kDa), or the HAMP, DHp and CA domains (HAMP-DHpCA, ~31 kDa) (Fig. 8B). Fig. 8C shows the autophosphorylation kinetics of ArlS DHpCA and HAMP-DHpCA constructs. Although most studies performed on histidine kinases have focused on the autophosphorylation activity of the DHpCA domain, recent data indicate that the HAMP domain can influence kinase and phosphatase activity (Wang et al., 2013). Fig. 8C (left and right panels) indicates that ArlS HAMP-DHpCA and ArlS DHpCA have similar autophosphorylation rates. Moreover, the HAMP-DHpCA and DHpCA domains alone have slow autophosphorylation kinetics (t1/2 = 15–20 minutes) compared to t1/2 ~30–60 seconds for other histidine kinases, such as WalK (Gutu et al., 2010), CrdS (Willett & Kirby, 2011), and AgrC (Srivastava et al., 2014) (Fig. S4A). ArlS also undergoes phosphotransfer to ArlR as shown in Fig. 8D (left and right panels). 50% of the phosphoryl group transfers to ArlR within 1 minute of the reaction when ArlR is incubated with ArlS HAMP-DHpCA. The phosphotransfer kinetics is further enhanced when ArlR is phosphorylated in the absence of HAMP. 50% phosphoryl group is transferred within 15 seconds of the reaction compared to 1 minute when the HAMP domain is present (Fig. S4B–C). In contrast to the autophosphorylation kinetics, the phosphatase kinetics is very similar to that observed in other histidine kinases (Willett & Kirby, 2012, Gutu et al., 2010, Willett & Kirby, 2011, Srivastava et al., 2014). Also, of note is the phosphatase activity of ArlS, which is evident by the disappearance of phosphorylated ArlR over time (shown by arrow in Fig. 8D). Similar to the phosphotransfer activity, the phosphatase activity was enhanced without the HAMP domain.
In order to further characterize the ArlRS two-component system, we investigated the importance of the key residues in ArlS involved in its kinase and phosphatase function. The signature phosphorylated histidine is clearly important for kinase activity, while the conserved threonine/asparagine residue is important for phosphatase activity (Willett & Kirby, 2012, Huynh & Stewart, 2011). Based on the amino acid sequence alignment with other histidine kinases (Fig. 9A), we hypothesized that His242 and Thr246 are important for autophosphorylation and phosphatase activity of ArlS, respectively. Additionally, we examined the role of the conserved phosphorylation site in ArlR, Asp52 (Fig. 9B). We used our chromosomally complemented LAC ΔarlRS strain to construct alanine substitutions in place of His242 and Thr246 in ArlS, providing single-copy versions of these variants in a ΔarlRS background. As expected, the mgrA P2-GFP reporter pHC68 had no activity in the arlRS mutant and was fully complemented with a wild-type copy of arlRS on the chromosome (Fig. 9C). Mutating His242 to an alanine in ArlS decreased expression of the mgrA reporter by ~80%, and was comparable to complementing the arlRS mutation with arlR alone (Fig. 9C), indicating that His242 is required for ArlS kinase activity. Conversely, mutating Thr246 to an alanine increased mgrA expression two-fold compared to wild-type ArlRS, demonstrating that Thr246 is crucial for ArlS phosphatase function (Fig. 9C). A His242/Thr246 double mutant had similar activity to the His242 single mutant (Fig. 9C). We also showed that the conserved phosphorylation site in ArlR, Asp52, is important for ArlR function. Mutation of Asp52 to either an alanine or a glutamate decreased mgrA expression by 92–96% (Fig. 9C). For some OmpR-family response regulators, replacement of this conserved aspartate with glutamate mimics phosphorylation and results in a constitutively active state (Delaune et al., 2012, Lan & Igo, 1998), but this does not appear to be true for ArlR.
Fig. 9. Identification of residues important for ArlS kinase and phosphatase function.
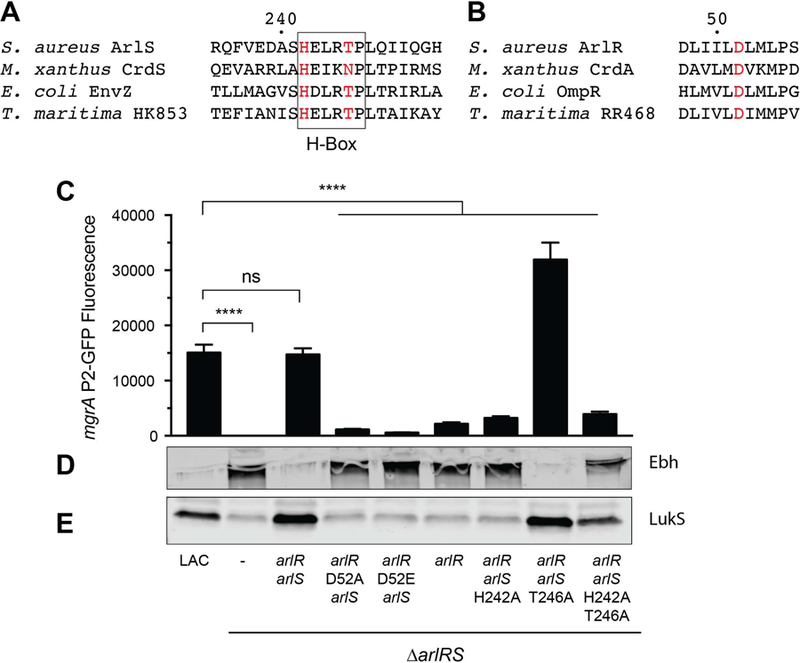
(A) Alignment of the Dimerization and Histidine Phosphotransfer (DHp) domains of hisitine kinases from various two-component systems. Numbering corresponds to the ArlS sequence. (B) Alignment of a portion of the receiver domains of response regulators centered on the phosphorylated aspartic acid residue, D52 in ArlR. Numbering corresponds to ArlR sequence. (C) S. aureus LAC strains with an arlRS deletion and chromosomal complementation with various versions of arlR and arlS as indicated. ArlS H242 lacks kinase activity in vitro, and ArlS T246A is deficient in phosphatase activity in vitro. Bar graph shows expression of mgrA from the mgrA P2-GFP promoter fusion pHC68 after 24 h of growth in TSB. Values represent averages and standard deviations of three independent experiments. Lower panels show Western blots of Ebh (D) and LukS (E) from culture supernatants, and are representative of two replicates.
We investigated protein expression levels of two genes under the control of the ArlRS/MgrA cascade, the large surface protein Ebh (Fig. 9D) and the PVL leukocidin component LukS (Fig. 9E). LukS is down-regulated in the arlRS mutant (Fig. 1, 2D), and its expression in the various arlR and arlS point mutations showed a similar pattern to that of the mgrA reporter (Fig. 9E). Ebh, on the other hand, is directly repressed by MgrA (Fig. 2G) (Crosby et al., 2016), and its expression showed the opposite pattern as mgrA and LukS (Fig. 9D).
In vitro kinase assays showed that the autophosphorylation activity of the ArlS H242A mutant is completely abolished, while the WT control had activity (Fig. S3). Hence, H242 is critical for the ArlRS two-component system to be activated in vitro and in vivo. In vitro kinase and phosphotransfer experiments were also performed with the ArlS T246A mutant. The kinase activity of the mutant (Fig. 10A) was similar to ArlS wild type, but the phosphotransfer data (shown by arrow in Fig. 10B) demonstrated that the T246A mutation prevents dephosphorylation of ArlR as shown by the presence of phosphorylated ArlR up to 60 minutes in the reaction. This is in contrast to ArlS wild type that showed loss of phosphorylated ArlR at the same time point (Fig. 8D, left panel). Both the ArlS H242A and T246A proteins contain the expected secondary structure as assessed by circular dichroism spectra, suggesting that they are properly folded (Fig. S5). Altogether, the ArlS H242 residue is essential for kinase activity, while the T246 residue controls the phosphatase activity of this histidine kinase.
Fig.10. ArlS has both kinase and phosphatase activity.
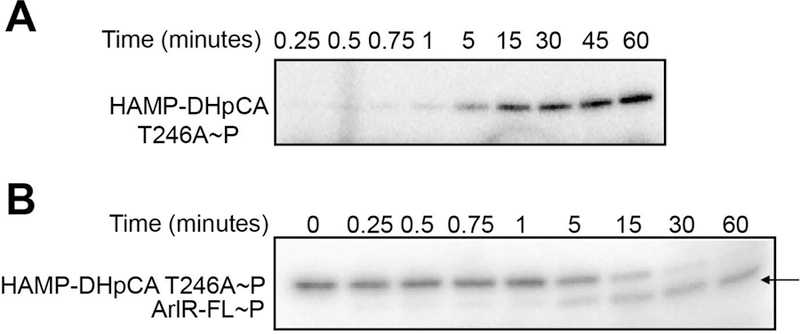
(A) Auto-phosphorylation of the ArlS HAMP-DhpCA T246A mutant. (B) Phosphotransfer from the ArlS HAMP-DhpCA T246A mutant to full-length ArlR. Arrow indicates the phosphorylated ArlR band at 60 min, demonstrating the loss of ArlS phosphatase activity in the T246A mutant. The assays were performed at least three times and the images are representative data.
Alternative activation of ArlR
Although the ArlRS TCS was required for expression of mgrA P2, we observed that ArlR alone, in the absence of ArlS, was still able to drive expression of MgrA at ~15–20% of wild type levels (Fig. 9C, Fig. 11). Mutating the phosphorylation site in arlR to an alanine decreased mgrA expression to levels similar to the arlRS mutant (Fig. 11). This has been observed previously with other response regulators and suggests non-specific phosphorylation from other histidine kinases or inorganic phosphate sources such as acetyl phosphate (Gao & Stock, 2013, Klein et al., 2007). It has recently been shown that the histidine kinase GraS may be able to substitute for ArlS in vivo, although direct phosphorylation of ArlR by GraS was not examined (Villanueva et al., 2018). We investigated if GraS could act as a non-cognate histidine kinase for ArlR using our mgrA P2-GFP reporter plasmid. Deletion of graRS in the arlR+ strain did not significantly decrease expression of mgrA (Fig. 11), suggesting that there is not significant cross talk between ArlRS and GraRS.
Fig. 11. Alternative activation of ArlR.

S. aureus LAC strains with deletions in arlRS and graRS, and chromosomal complementation with various versions of arlR and arlS as indicated. Expression of mgrA from the mgrA P2-GFP promoter fusion pHC68 was measured after 24 h of growth in TSB. Values represent averages and standard deviations of three independent experiments. Significance was determined by ANOVA using Tukey’s test for multiple comparisons.
Discussion
The ArlRS TCS was connected with autolysis and biofilm phenotypes nearly 20 years ago (Fournier & Hooper, 2000, Toledo-Arana et al., 2005), and its importance in virulence has been observed in several animal models of infection (Benton et al., 2004, Walker et al., 2013, Radin et al., 2016, Burgui et al., 2018, Kwiecinski et al., 2019). Yet the molecular details of gene regulation by ArlRS have remained largely unknown. In this study we clarified the ArlRS regulon, defined a likely ArlR binding site, and investigated the kinase and phosphatase kinetics of ArlS. It has been shown previously that ArlRS activates expression of the global regulator MgrA (Luong & Lee, 2006, Crosby et al., 2016), and in this work RNA-sequencing analysis of the ArlRS regulon demonstrated that there is considerable overlap with the MgrA regulon (Crosby et al., 2016). In total, 70% of the genes regulated by ArlRS were also regulated in the same direction by MgrA (Table S2), and 96% of genes with ≥4-fold change in expression showed overlap with the MgrA regulon (Fig. 1). We demonstrate for the first time that ArlR directly activates mgrA expression, and identify a potential ArlR binding site (Fig. 6). The ArlRS TCS was also recently shown to directly regulate spx expression (Bai et al., 2019). We show that this regulation occurs at the spx P2 promoter, which contains a similar ArlR binding site (Fig. 7).
These results indicate that ArlRS functions at the top of a regulatory cascade (Fig. 12) in which ArlR directly activates expression of the important global transcriptional regulators MgrA and Spx. Both MgrA and Spx are redox sensitive (Hillion & Antelmann, 2015), suggesting that ArlRS may be involved in the oxidative stress response. MgrA is in the MarR/OhrR family and contains a conserved redox-sensing cysteine residue. Oxidation of this cysteine residue by reactive oxygen species results in MgrA dissociation from DNA (Chen et al., 2006). Spx is a thiol stress sensor, and it is primarily regulated at the posttranslational level via degradation by the ClpXP proteasome (Pamp et al., 2006, Donegan et al., 2014). The Spx protein is only stabilized when a CXXC motif near its N-terminus is oxidized, and in this oxidized state it binds to RNA polymerase to affect gene expression (Hillion & Antelmann, 2015). Spx is a key stress-response regulator that controls expression of thioredoxin reductase (trxB), the ica locus for poly-N-acetylglucosamine synthesis, and TrfA, a Clp proteasome adaptor involved in resistance to cell wall-active antibiotics (Pamp et al., 2006, Jousselin et al., 2013, Bai et al., 2019). Under the conditions of our RNA-seq experiment we did not see regulation of trxB, icaADBC, or trfA. This could be due to the relatively low fold-change in spx expression in the arlRS mutant, combined with the strong posttranslational regulation of Spx protein levels (Pamp et al., 2006, Donegan et al., 2014). It is possible that the effect of an arlRS mutant on Spx function would be more apparent under redox stress conditions.
Fig. 12. Scheme for the ArlRS regulon.
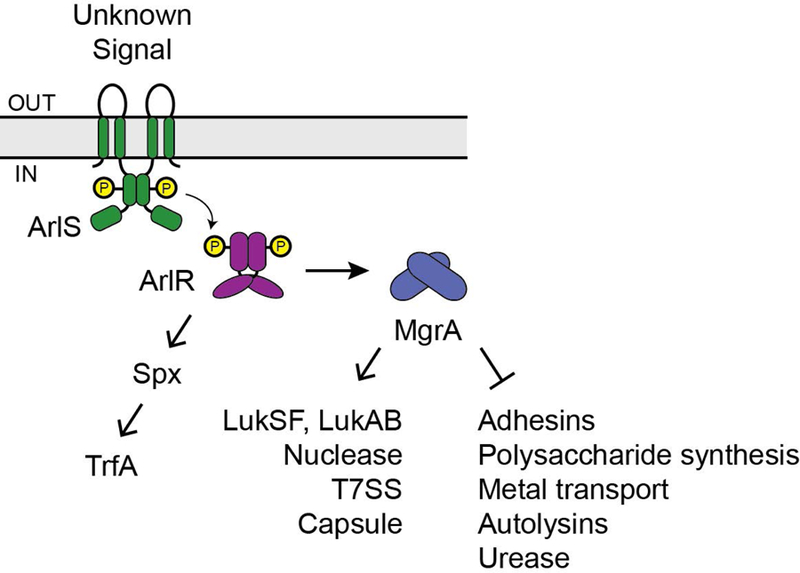
ArlR appears to directly activate expression of the transcriptional regulators mgrA and spx. MgrA then either directly or indirectly regulates over 100 genes, some of which are indicated.
In contrast to previous studies (Liang et al., 2005, Harper et al., 2018, Ingavale et al., 2005, Yan et al., 2019), we did not see evidence that ArlRS controls expression of the agr quorum-sensing system. We speculate that some of the apparent down-regulation of agr seen in other studies could result from strain- and media-dependent growth delays in the arlRS mutant (Liang et al., 2005, Radin et al., 2016), since agr expression is highly dependent on growth phase. There is partial overlap in the ArlRS and agr regulons, such as lukSF, lukAB, and the urease and capsule operons (Thoendel et al., 2011). However, there are many genes in the agr regulon that are not regulated by ArlRS, suggesting that the overlap may be indicative of complex regulation of virulence factors in S. aureus, rather than direct regulation of agrBDCA expression by ArlRS.
Genes repressed by ArlRS-MgrA
A variety of genes and operons are strongly repressed by the ArlRS-MgrA cascade (Fig. 1 and 12). These include two direct targets of MgrA repression, the transcriptional regulator sarV (Manna et al., 2004) and the giant surface protein ebh (Crosby et al., 2016). Other cell surface adhesins regulated by this cascade include sdrD, fmtB, and sasC (Fig. 1). We have previously shown that regulation of surface proteins by the ArlRS-MgrA cascade has a large effect on multicellular behavior, including clumping, adhesion to host proteins, and biofilm formation (Walker et al., 2013, Crosby et al., 2016, Kwiecinski et al., 2019). ArlRS-MgrA also affects extracellular polysaccharide production. In agreement with previous work (Luong & Lee, 2006), we see activation of several genes in the capsule biosynthesis operon (Fig. 1). ArlRS-MgrA appears to simultaneously repress expression of two uncharacterized putative polysaccharide biosynthesis gene clusters, cap1ABC and SAUSA300_0130–0133. Whether these hypothetical clusters are actually involved in capsule or polysaccharide biosynthesis remains to be determined.
The ArlRS TCS was originally identified as a regulator of autolysis (Fournier & Hooper, 2000), although the exact mechanism remains unknown. Consistent with previous results, we observe ArlRS-MgrA mediated repression of the autolysin lytN, but not lytM or atl (Luong et al., 2006, Memmi et al., 2012). Similarly to Luong et al. (Luong et al., 2006), we saw that AtlR is overexpressed in the arlRS mutant. AtlR is a repressor of Atl expression (Houston et al., 2011), however this should result in decreased autolytic activity in the arlRS mutant. In addition, we see modest regulation of the LysM/CHAP domain containing enzyme SAUSA300_0739 (Osipovitch et al., 2015) by ArlRS but not MgrA.
Our results show that the ArlRS-MgrA cascade represses expression of the urease gene cluster, consistent with previous observations (Liang et al., 2005, Luong et al., 2006). S. aureus relies on urease for maintaining pH homeostasis in acidic conditions (Zhou et al., 2019). ArlRS and MgrA also appear to be important in controlling divalent cation acquisition. Genes encoding the ferrous iron transporter FeoA, the catechol siderophore transporter SstABCD (Morrissey et al., 2000, Beasley et al., 2011), and the staphyloferrin B receptor SirA (Grigg et al., 2010) were all repressed by ArlRS. In addition, the manganese transporter MntH (Horsburgh et al., 2002) was up ~8-fold in the arlRS mutant. Divalent cation acquisition is critical inside the host, where metals are often limiting (Cassat & Skaar, 2012), but the contribution of ArlRS-dependent regulation of these processes during S. aureus disease is not yet known.
Genes activated by ArlRS-MgrA
The genes that are activated by ArlRS are predominantly enzymes involved in anaerobic metabolism or secreted virulence factors. In all cases these genes were also down-regulated in the mgrA mutant, suggesting that regulation occurs via MgrA (Fig. 1 and 12). Regulation by both ArlRS and MgrA was confirmed using transcriptional reporters for a subset of these genes: nuc, lukA, lukS, esxA, and gehB (Fig. 2). Previous work has shown that MgrA can act as a repressor, and the MgrA binding site has been identified (Manna et al., 2004, Crosby et al., 2016). It is not clear if MgrA can also function as an activator, but apparent activation by MgrA could be the result of indirect regulation. MgrA is known to repress expression of the transcriptional regulator SarV (Manna et al., 2004), and our results suggest that the DNA-binding proteins AtlR and SAUSA300_1202 are also repressed by ArlRS-MgrA (Fig 1). It is also possible that stable regulatory RNAs play a role in mediating expression of secreted virulence factors (Morrison et al., 2012, Das et al., 2016).
Not all secreted virulence factors are regulated by ArlRS-MgrA, though. Notably, alpha hemolysin (hla) showed no change in gene expression in the arlRS or mgrA mutants. This is in contrast to reports that MgrA regulates Hla expression (Ingavale et al., 2005, Sun et al., 2011), but consistent with an earlier microarray analysis of the MgrA regulon in strain Newman (Luong et al., 2006). We also observed a 2.6-fold increase in protein A (spa) gene expression in the arlRS mutant, consistent with the 54-fold increase in spa expression in the mgrA mutant and previous reports that spa is repressed by ArlRS (Merino et al., 2009) or MgrA (Ingavale et al., 2005, Luong et al., 2006). However, this is in contrast to the large decrease in protein A expression recently reported for an arlRS mutant grown in chemically defined medium (Villanueva et al., 2018), suggesting that regulation of protein A expression is complex and highly dependent on growth conditions.
Identification of the ArlR binding site
By using a series of site directed mutants in the mgrA P2 reporter we have identified what is likely to be the ArlR binding site (Fig. 6). ArlR is a member of the OmpR family of response regulators, which typically bind to direct repeats separated by a 4–5 bp spacer (de Been et al., 2008). This spacing allows the DNA binding domains of each monomer to bind in a head-to-tail orientation along one face of the DNA helix (Blanco et al., 2002). Consistent with this, the potential ArlR binding site appears to be almost a direct repeat separated by a 4 bp gap (TTTTCTCAT-N4-TTTTAATAA in the mgrA P2 promoter). This sequence is nearly identical in the spx P2 promoter, and in both promoters, there is an 11 bp gap between the −10 sequence and the potential ArlR binding site (Fig. 7F). This binding site appears to be in place of a −35 sequence, as neither mgrA P2 or spx P2 has a recognizable −35 sequence. This spacing and localization is similar to that of the PhoB response regulator in E. coli (Makino et al., 1993), another member of the OmpR family. PhoB-dependent promoters also lack a consensus −35 sequence and require PhoB for expression (Makino et al., 1988). PhoB appears to interact directly with the housekeeping sigma factor to facilitate gene expression, rather than binding farther upstream in the promoter region and interacting with the alpha subunit of RNA polymerase (Makino et al., 1993).
A search of the S. aureus USA300 genome for ≥90% matches to the consensus binding sequence TTTTCTCAT-N4-TTTTAATNN resulted in only four hits that occurred in the same orientation within likely promoter regions. Besides mgrA and spx, the other two hits were upstream of the putative UDP-glucose 4-epimerase SAUSA300_0130. While expression from mgrA P2 and spx P2 is activated by ArlR binding, it appears that expression of SAUSA300_0130 may be directly repressed by both ArlR and MgrA.
Kinase and phosphatase activity of ArlS
Our in vivo and in vitro results demonstrate that ArlS has both kinase and phosphatase activity (Fig. 8, 9, and 10), which is typical of HisKA family histidine kinases (Willett & Kirby, 2012). ArlS appears to have slow autophosphorylation kinetics. Interestingly, the HAMP domain inhibited the kinetics of phosphotransfer and phosphatase activity. Although the biochemical and structural bases for these observations are not entirely clear, it may be that ArlS requires a lipid membrane environment for full activity. In addition, the in vitro experiments did not include the extracellular Cache domain and the signal(s) needed to activate ArlS. Thus, the presence of the lipid membrane, the extracellular domain and bound ligand likely affect the kinetics of ArlS activity in a physiologically relevant manner.
ArlS may function predominantly as a kinase during normal in vitro growth. Elimination of ArlS kinase activity by mutating H242 decreased mgrA expression ~5-fold, whereas mutation of T246, which is critical for phosphatase activity, increased mgrA expression ~2-fold (Fig. 9C). Consistent with this, Ebh and LukS protein levels in the ArlS T246A phosphatase mutant appear to be similar to wild-type levels (Fig. 9D, 9E), indicating that phosphatase activity is not playing a large role in gene expression under these conditions. For any TCS the phosphatase activity is finely tuned to suppress non-specific phosphorylation by non-cognate histidine kinases and inorganic phosphate donors (Gao & Stock, 2017). Indeed, in the absence of ArlS we do see some non-specific activation of ArlR (Fig. 9C, Fig. 11). A previous report suggested that there may be cross talk between ArlRS and the GraRS TCS (Villanueva et al., 2018), however our in vivo reporters did not indicate that GraS is involved in activation of ArlR (Fig. 11). Future work could investigate if this low-level background activation of ArlR is due to chemical phosphorylation of ArlR or cross talk with a different TCS or kinase.
In this work we clarified how ArlRS regulates gene expression and ultimately virulence. ArlRS appears to be at the top of a regulatory cascade, and it affects gene expression through activation of the transcriptional regulators MgrA and Spx. In our RNA-seq experiment MgrA regulated >100 genes (Crosby et al., 2016), although it is not yet clear how many of these genes are directly regulated by MgrA. There are most likely several levels of regulation in this cascade, potentially including both transcriptional regulators like SarV and small RNAs. In contrast, we did not observe changes in expression of genes in the Spx regulon under the growth conditions used here. Examining the effect of ArlRS under redox stress conditions may aid in identifying new roles for this TCS.
Experimental Procedures
Reagents and growth conditions
S. aureus strains and plasmids used in this work are listed in Table 1. S. aureus was cultured in tryptic soy broth (TSB) and E. coli was cultured in lysogeny broth (LB) at 37 °C with shaking at 200 rpm. Antibiotics were added to the media at the following concentrations: chloramphenicol (Cam), 10 μg/mL; erythromycin (Erm), 5 μg/mL; and tetracycline (Tet), 1 μg/mL. E. coli strains with plasmids were maintained on media supplemented with ampicillin at 100 μg/mL; kanamycin, 50 μg/mL; or spectinomycin at 50 μg/mL.
Table 1.
Bacterial strains and plasmids
| Strain/plasmid | Genotype/properties | Reference |
|---|---|---|
| E. coli | ||
| DH5α | Cloning strain | Protein Express |
| DC10B | Cloning strain (dcm−) | (Monk et al., 2012) |
| ER2566 | Protein expression strain | NEB |
| BL21 (DE3) | Protein expression strain | Novagen |
| S. aureus | ||
| RN4220 | Restriction deficient cloning host | (Nair et al., 2011) |
| AH1263 | USA300 CA-MRSA ErmS (LAC*) | (Boles et al., 2010) |
| AH1975 | LAC* ΔarlRS | (Walker et al., 2013) |
| AH3052 | LAC* Δspa | (Ibberson et al., 2014) |
| AH3056 | LAC* ΔarlRS Δspa | (Crosby et al., 2016) |
| AH3244 | LAC* ΔarlRS Φ11::LL29 arlRS | (Kwiecinski et al., 2019) |
| AH3275 | LAC* ΔarlRS Φ11::LL29 arlR D52A, arlS | This work |
| AH3277 | LAC* ΔarlRS Φ11::LL29 arlR D52E, arlS | This work |
| AH3278 | LAC* ΔarlRS Δspa Φ11::LL29 arlRS | This work |
| AH3280 | LAC* ΔarlRS Δspa Φ11::LL29 arlR D52A, arlS | This work |
| AH3281 | LAC* ΔarlRS Δspa Φ11::LL29 arlR D52E, arlS | This work |
| AH3455 | LAC* ΔmgrA::tetM | (Crosby et al., 2016) |
| AH5242 | LAC* ΔarlRS Φ11::LL29 arlR | This work |
| AH5243 | LAC* ΔarlRS Φ11::LL29 arlR, arlS H242A | This work |
| AH5244 | LAC* ΔarlRS Φ11::LL29 arlR, arlS T246A | This work |
| AH5271 | LAC* ΔarlRS Φ11::LL29 arlR, arlS H242A T246A | This work |
| AH5360 | LAC* ΔarlRS Δspa Φ11::LL29 arlR | This work |
| AH5361 | LAC* ΔarlRS Δspa Φ11::LL29 arlR, arlS H242A | This work |
| AH5362 | LAC* ΔarlRS Δspa Φ11::LL29 arlR, arlS T246A | This work |
| AH5363 | LAC* ΔarlRS Δspa Φ11::LL29 arlR, arlS H242A T246A | This work |
| AH5407 | LAC* ΔarlRS ΔgraRS Φ11::LL29 arlR | This work |
| AH5434 | LAC* ΔarlRS Φ11::LL29 arlR D52A | This work |
| Plasmids | ||
| pCEMCS | Empty vector control for pCM11, ErmR | (Malone et al., 2009) |
| pCM11 | sGFP expression vector, ErmR | (Lauderdale et al., 2010) |
| pCM28 | Empty vector control for pCM29, CamR | (Boles et al., 2010) |
| pCM29 | sGFP expression vector, CamR | (Pang et al., 2010) |
| pCM38 | Pnuc-GFP reporter, CamR | (Yan et al., 2019) |
| pLL29 | S. aureus integration vector, TetR | (Luong & Lee, 2007) |
| pET21a | Overexpression vector, AmpR | Novagen |
| pET28a | Overexpression vector, KanR | Novagen |
| pTEV5 | Overexpression vector, N-terminal Tev-cleavable His6 tag, AmpR | (Rocco et al., 2008) |
| pHC07 | pET28a-arlR, KanR | This work |
| pHC24 | pLL29-arlRS, SpecR | (Kwiecinski et al., 2019) |
| pHC28 | pLL29-arlR D52A, arlS, SpecR | This work |
| pHC29 | pLL29-arlR D52E, arlS, SpecR | This work |
| pHC35 | pTEV5-arlR, AmpR | This work |
| pHC68 | mgrA P2-sGFP fusion, 251 bp insert, ErmR | (Crosby et al., 2016) |
| pHC136 | mgrA P2-sGFP fusion, 156 bp insert, ErmR | This work |
| pHC137 | mgrA P2-sGFP fusion, 123 bp insert, ErmR | This work |
| pHC138 | mgrA P2-sGFP fusion, 97 bp insert, ErmR | This work |
| pHC139 | PureA-sGFP fusion, CamR | This work |
| pHC140 | P1566-sGFP fusion, CamR | This work |
| pHC141 | P0130-sGFP fusion, CamR | This work |
| pHC144 | PesxA-sGFP fusion, CamR | This work |
| pHC145 | mgrA P2-sGFP fusion, 110 bp insert, ErmR | This work |
| pHC146 | mgrA P2-sGFP fusion, 83 bp insert, ErmR | This work |
| pHC147 | PlukA-sGFP fusion, CamR | This work |
| pHC148 | PlukS-sGFP fusion, CamR | This work |
| pHC149 | Pebh-sGFP fusion, CamR | This work |
| pHC150 | PmntH-sGFP fusion, CamR | This work |
| pHC151 | PsdrD-sGFP fusion, CamR | This work |
| pHC152 | PlytN-sGFP fusion, CamR | This work |
| pHC159 | pLL29-arlR, SpecR | This work |
| pHC161 | pLL29-arlR, arlS H242A, SpecR | This work |
| pHC162 | pLL29-arlR, arlS T246A, SpecR | This work |
| pHC164 | mgrA P2-sGFP scramble 2, ErmR | This work |
| pHC166 | pLL29-arlR, arlS H242A T246A, SpecR | This work |
| pHC167 | mgrA P2-sGFP scramble 3, ErmR | This work |
| pHC168 | mgrA P2-sGFP scramble 4, ErmR | This work |
| pHC169 | Pspx-sGFP fusion, CamR | This work |
| pHC170 | mgrA P2-sGFP scramble 5, ErmR | This work |
| pHC171 | mgrA P2-sGFP scramble 6, ErmR | This work |
| pHC172 | mgrA P2-sGFP scramble 7, ErmR | This work |
| pHC173 | mgrA P2-sGFP scramble 8, ErmR | This work |
| pHC174 | mgrA P2-sGFP scramble 9, ErmR | This work |
| pHC176 | spx P1-sGFP fusion, CamR | This work |
| pHC177 | spx P2-sGFP fusion, CamR | This work |
| pHC178 | PgehB-sGFP fusion, CamR | This work |
| pHC183 | mgrA P2-sGFP scramble 10, ErmR | This work |
| pHC184 | mgrA P2-sGFP scramble 11, ErmR | This work |
| pHC185 | mgrA P2-sGFP scramble 12, ErmR | This work |
| pHC186 | mgrA P2-sGFP scramble 13, ErmR | This work |
| pHC188 | mgrA P2-sGFP scramble 14, ErmR | This work |
| pHC189 | mgrA P2-sGFP scramble 15, ErmR | This work |
| pHC190 | mgrA P2-sGFP scramble 16, ErmR | This work |
| pHC191 | mgrA P2-sGFP scramble 17, ErmR | This work |
| pHC192 | mgrA P2-sGFP scramble 18, ErmR | This work |
| pHC194 | mgrA P2-sGFP scramble 19, ErmR | This work |
| pHC195 | mgrA P2-sGFP scramble 20, ErmR | This work |
| pHC196 | mgrA P2-sGFP scramble 21, ErmR | This work |
| pHC198 | mgrA P2-sGFP scramble 22, ErmR | This work |
| pHC199 | mgrA P2-sGFP scramble 23, ErmR | This work |
| pHC200 | mgrA P2-sGFP scramble 24, ErmR | This work |
| pHC201 | mgrA P2-sGFP scramble 25, ErmR | This work |
| pHC202 | mgrA P2-sGFP scramble 26, ErmR | This work |
| pHC203 | mgrA P2-sGFP scramble 27, ErmR | This work |
| pHC204 | mgrA P2-sGFP scramble 28, ErmR | This work |
| pHC205 | mgrA P2-sGFP scramble 29, ErmR | This work |
| pHC206 | mgrA P2-sGFP scramble 30, ErmR | This work |
| pET21-ArlS1 | ArlS HAMP-DHpCA (amino acids 187–451) in pET21a-His-Tev, AmpR | This work |
| pET21-ArlS2 | ArlS DHpCA (amino acids 231–451) in pET21a-His-Tev, AmpR | This work |
| pET21-ArlS3 | ArlS HAMP-DHpCA H242A, AmpR | This work |
| pET21-ArlS4 | ArlS HAMP-DHpCA T246A, AmpR | This work |
| pET21-ArlS5 | ArlS HAMP-DHpCA H242A T246A, AmpR | This work |
Recombinant DNA and genetic techniques
E. coli DH5α and DC10B were used as a cloning hosts for plasmid constructions. Restriction enzymes, DNA ligase, and Phusion DNA polymerase were purchased from New England Biolabs. The plasmid mini-prep and gel extraction kits were purchased from Invitrogen or Qiagen. Lysostaphin, used for S. aureus DNA extractions, was purchased from Sigma. Plasmids were purified from S. aureus RN4220 or E. coli DC10B and electroporated into S. aureus LAC strains as described previously (Monk et al., 2012, Lofblom et al., 2007). Bacteriophage transductions between S. aureus strains were performed with phage 11 as described previously (Novick, 1991). All oligonucleotides were ordered from IDT (Coralville, IA) and are listed in Supplementary Table 4. DNA sequencing was performed at the University of Iowa DNA Core Facility or the Molecular Biology Service Center at the University of Colorado Anschutz Medical Campus.
RNA-sequencing
Triplicate cultures of LAC and LAC ΔarlRS were grown to an OD600 of 1.5 in TSB, harvested by centrifugation, and washed briefly with RNAprotect Bacterial Reagent (Qiagen) to stabilize the RNA. Cell pellets were then resuspended in PBS and lysed with lysostaphin for 1 h at room temperature prior to RNA purification using the Qiagen RNeasy Mini kit. RNA was then treated with Turbo DNA-free (Invitrogen), and assessed for quality using a Bioanalyzer (Agilent). rRNA was depleted using the Ribo-Zero rRNA Removal Kit for Gram-positive bacteria (Illumina). cDNA libraries were generated at the University of Iowa Genomics Division using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Samples were barcoded, pooled, and sequenced in 100×100 paired end reads using a HiSeq 2000 sequencer (Illumina). The resulting sequences were aligned to the USA300_FPR3757 genome sequence and differential expression was quantified using CLC Genomics Workbench ver. 12 (Qiagen). Genes were considered differentially expressed if they showed a ≥2-fold change and a false discovery rate (FDR)-corrected P value ≤ 0.05. Raw data from an identical comparison of gene expression in LAC ΔmgrA::tet and LAC (Crosby et al., 2016) were reanalyzed using CLC Genomics Workbench. Sequence files and analysis have been deposited in the NCBI GEO database under accession numbers GSE130777 (ΔmgrA vs LAC) and GSE130887 (ΔarlRS vs LAC).
Promoter fusion plasmids
All mgrA promoter-sGFP transcriptional reporters were generated in the shuttle vector pCM11 (Lauderdale et al., 2010). Fragments of the mgrA promoter were amplified from S. aureus strain LAC genomic DNA using the primers listed in Table S4, and digested with HindIII and KpnI before ligating into pCM11. Promoter fusions for all other genes were generated in plasmid pCM29 (Pang et al., 2010). Promoter fragments were amplified using the primers listed in Table S4 and digested with XbaI and KpnI before ligating into pCM29. In all cases promoter fragments were cloned upstream of an optimized ribosome binding site and codon optimized gene for superfolder GFP. Site directed mutagenesis was performed using modified versions of the QuikChange (Stratagene) method (Liu & Naismith, 2008, Zheng et al., 2004) and the primers listed in Table S4.
To assess expression, overnight cultures were diluted 1:100 in TSB containing the appropriate antibiotic in a black 96-well plate, and plates were incubated at 37 °C with shaking in a humidified microtiter plate shaker (Stuart). A Tecan Infinite M200 plate reader was used to periodically measure OD600 and fluorescence intensity with excitation at 495 nm and emission at 515 nm. For each experiment values from triplicate wells were averaged after subtracting background fluorescence from strains carrying an empty vector control (pCEMCS for erythromycin resistant reporters and pCM28 for chloramphenicol resistant reporters). For graphs of 24 h time points, averages and standard deviations were calculated from at least three independent experiments. Statistical significance was determined using a one-way ANOVA with a Dunnett test for correction for multiple comparisons (GraphPad Prism).
arlRS chromosomal complementation
The arlRS chromosomal complementation strain AH3244 has been described previously (Kwiecinski et al., 2019). Point mutations were introduced into arlR or arlS using the QuikChange mutagenesis method (Stratagene), using the primers listed in Table S4 and pHC24 (pLL29-arlRS) as a template. A construct expressing arlR alone was also generated by amplifying arlR with primers HC84 and HC708. The product was cloned into pLL29 using BamHI and HindIII. In all cases the plasmids were electroporated into S. aureus RN4220 containing pLL2787, resulting in integration of pLL29 into the phage 11 attachment site. Integrated arlRS constructs were then transduced into S. aureus LAC, and integration was confirmed by PCR as described previously (Luong & Lee, 2007).
Protein Expression and Purification
The DNA fragments of HAMP-DHpCA region (amino acids 187–451) and DHpCA domain (amino acids 231–451) of ArlS from S. aureus LAC Strain were amplified by PCR and sub-cloned into a modified pET21a plasmid (Novagen) containing an N-terminal 6× His-tag followed by a recombinant tobacco etch virus (rTEV) protease cleavage site (ENLYFQG). The DNA fragment of full-length ArlR (amino acids 1–219) was amplified by PCR and cloned into a modified pET31b plasmid (Novagen) (pTEV5), which has an N-terminal 6× His-tag followed by a rTEV protease cleavage site. The ArlS HAMP-DHpCA H242A and ArlS HAMP-DHpCA T246A mutants were created by oligonucleotide-directed mutagenesis. The mutants were confirmed by Sanger sequencing (University of Iowa, Iowa Institute of Human Genetics Genomic Division). E. coli strain BL21(DE3) (Novagen) cells containing ArlS or ArlR plasmids were grown in Luria Broth with 100ug/mL ampicillin and the protein expression was induced with 0.5 mM isopropyl-1-thio-D-glactopyranoside at 18 °C overnight. The cells were harvested by centrifugation and re-suspended in buffer containing 50 mM Tris-HCl pH 7.5, 250 mM NaCl and 10 mM imidazole. All proteins were purified by nickel-affinity chromatography (Nickel Sepharose 6 fast flow media, GE Healthcare). The N-terminal 6x His-tag of all proteins was removed by incubation with rTEV protease at 4 °C overnight. Size-exclusion chromatography (Superdex 75, GE Healthcare) was performed as a final purification step and eluted in a buffer containing 25 mM Tris pH 7.5, 150 mM NaCl, 5 % glycerol, 0.5 mM EDTA and 1 mM dithiothreitol (DTT). Proteins were ~95% pure as judged by SDS-PAGE as shown in Fig. 8B.
Kinase and phosphotransfer assays
Enzyme assays were followed by radioactivity (32P) using previously described protocols (Willett & Kirby, 2011). Briefly, auto-phosphorylation reactions used 5 µM ArlS HAMP-DHpCA or ArlS DhpCA incubated with ATP mix [0.25 μM [γ−32P]-ATP and 250 μM cold ATP (Sigma) in 1X kinase buffer (25 mM Tris pH 7.6, 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, 50 mM KCl, 1 mM DTT)] for 1 hour. The reactions were quenched at desired time points by addition of SDS-PAGE loading buffer. The samples were then resolved by SDS-PAGE and the radioactive bands visualized using a phosphor screen (GE healthcare Typhoon FLA 9500). The bands were quantified using ImageJ software.
Phosphotransfer reactions used 5 µM of auto-phosphorylated ArlS HAMP-DhpCA and ArlS DhpCA prepared as described above. Auto-phosphorylated ArlS HAMP-DhpCA or DhpCA was then incubated with 5 µM full-length ArlR. The phospho-transfer reaction was allowed to proceed for 1 hour and individual samples quenched at desired time points by addition of SDS-PAGE loading buffer. The samples were resolved by SDS-PAGE and the radioactive bands visualized on a phosphor screen (GE healthcare Typhoon FLA 9500). The autophosphorylation and phosphotransfer assays were repeated at least three times and representative images are shown.
Electrophoretic mobility shift assay
Full-length arlR was cloned into the overexpression vector pET28a using primers HC41 and HC42 (Table S4) and transformed into the overexpression strain ER2566. This strain was grown in LB supplemented with kanamycin and expression was induced using 0.5 mM IPTG at 25 °C overnight. Cells were harvested and re-suspended in buffer containing 50 mM Tris-HCl pH 7.5, 250 mM NaCl, 10 mM imidazole, and 5% glycerol. Cells were lysed using BugBuster (Millipore) and sonication, before purification of His6-ArlR by nickel-affinity chromatography (HIS-Select Nickel Affinity Gel, Sigma). Purified ArlR was concentrated and dialyzed against storage buffer containing 25 mM Tris pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM dithiothreitol, and 0.5 mM EDTA.
EMSA experiments were modified from the protocol described by Bai et al. (Bai et al., 2019). DNA probes 5’ end-labeled with IRDye700 were generated by annealing 50 nt oligos HC656 and HC657 (probe 2), and HC789 and HC790 (probe 3). Prior to DNA binding, ArlR was phosphorylated for 20 min using 50 mM acetyl phosphate. Increasing concentrations of ArlR (0 to 2 μg) were incubated with 10 nM DNA probe for 20 min at 25 °C in 10 μL reactions containing 20 mM HEPES pH 7.6, 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM dithiothreitol, 0.2 % Tween 20, and 30 mM KCl. Reactions were then separated on a 0.5X TBE/8% polyacrylamide gel at 4 °C, using 0.75X TBE running buffer, before imaging on a Licor Odyssey CLx Imaging System. EMSA experiments were repeated twice and a representative image is shown.
Western blotting
S. aureus LAC strains lacking protein A (Δspa) were grown overnight in TSB, and then subcultured 1:100 into RPMI medium (Gibco) supplemented with 1% casamino acids. After growing at 37 °C for 8 h with shaking, 1.6 ml of culture were removed, the cells were pelleted, and the supernatant was transferred to a new tube. Secreted proteins were precipitated by adding 178 μL of trichloroacetic acid, vortexing briefly, and leaving at 4 °C overnight. Precipitated proteins were then centrifuged for 10 m at 17,000g, washed twice with 100% ethanol, and allowed to air dry. Pellets were resuspended in SDS-PAGE loading buffer and run on 4–20% gradient SDS-PAGE gels (Bio-Rad). Proteins were then transferred to nitrocellulose membranes using the high molecular weight setting on a Trans-blot Turbo transfer system (Bio-Rad). Membranes were blocked with TBST plus 5% milk, and then probed using a 1:10,000 dilution of either anti-Ebh antibodies (Walker et al., 2013) or anti-LukS antibodies (IBT Bioservices). Blots were then washed and probed with a 1:10,000 dilution of IRDye 680 goat anti-rabbit secondary antibody (LiCor) before imaging on a LiCor Odyssey CLx Imaging System.
Supplementary Material
Table S1. Genes regulated by ArlRS
Table S2. RNA-seq comparison
Table S3. Binding site search
Table S4. Primers used in this work
Acknowledgments
We thank J. Bair and E. Snir at the University of Iowa Genomics Division of the Institute of Human Genetics for the library preparation and sequencing. J. Kavanaugh and other members of the Horswill lab provided helpful discussion and technical advice. H. Crosby was supported by an NIH Training Grant T32 AI007511 and American Heart Association postdoctoral fellowship 15POST25720016. J. Kwiecinski was supported by a Swedish Society for Medical Research postdoctoral fellowship and an American Heart Association postdoctoral fellowship 17POST33670580. Research was supported by NIH NIAID grants AI083211 (ARH), AI135305 (EJF), and AI141490 (ARH and EJF).
References
- Anantharaman V, and Aravind L. (2000) Cache - a signaling domain common to animal Ca(2+)-channel subunits and a class of prokaryotic chemotaxis receptors. Trends in biochemical sciences 25: 535–537. [DOI] [PubMed] [Google Scholar]
- Bai J, Zhu X, Zhao K, Yan Y, Xu T, Wang J, Zheng J, Huang W, Shi L, Shang Y, Lv Z, Wang X, Wu Y, and Qu D. (2019) The role of ArlRS in regulating oxacillin susceptibility in methicillin-resistant Staphylococcus aureus indicates it is a potential target for antimicrobial resistance breakers. Emerging microbes & infections 8: 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley FC, Marolda CL, Cheung J, Buac S, and Heinrichs DE. (2011) Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by Staphyloferrin A, Staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infection and immunity 79: 2345–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, and Buysse JM. (2004) Large-scale identification of genes required for full virulence of Staphylococcus aureus. Journal of bacteriology 186: 8478–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AG, Sola M, Gomis-Ruth FX, and Coll M. (2002) Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10: 701–713. [DOI] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Roth AJ, and Horswill AR. (2010) Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PloS one 5: e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgui S, Gil C, Solano C, Lasa I, and Valle J. (2018) A Systematic Evaluation of the Two-Component Systems Network Reveals That ArlRS Is a Key Regulator of Catheter Colonization by Staphylococcus aureus. Frontiers in microbiology 9: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casino P, Rubio V, and Marina A. (2010) The mechanism of signal transduction by two-component systems. Current opinion in structural biology 20: 763–771. [DOI] [PubMed] [Google Scholar]
- Cassat JE, and Skaar EP. (2012) Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Seminars in immunopathology 34: 215–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PR, Bae T, Williams WA, Duguid EM, Rice PA, Schneewind O, and He C. (2006) An oxidation-sensing mechanism is used by the global regulator MgrA in Staphylococcus aureus. Nature chemical biology 2: 591–595. [DOI] [PubMed] [Google Scholar]
- Crosby HA, Schlievert PM, Merriman JA, King JM, Salgado-Pabon W, and Horswill AR. (2016) The Staphylococcus aureus Global Regulator MgrA Modulates Clumping and Virulence by Controlling Surface Protein Expression. PLoS pathogens 12: e1005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Lindemann C, Young BC, Muller J, Osterreich B, Ternette N, Winkler AC, Paprotka K, Reinhardt R, Forstner KU, Allen E, Flaxman A, Yamaguchi Y, Rollier CS, van Diemen P, Blattner S, Remmele CW, Selle M, Dittrich M, Muller T, Vogel J, Ohlsen K, Crook DW, Massey R, Wilson DJ, Rudel T, Wyllie DH, and Fraunholz MJ. (2016) Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proceedings of the National Academy of Sciences of the United States of America 113: E3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Been M, Bart MJ, Abee T, Siezen RJ, and Francke C. (2008) The identification of response regulator-specific binding sites reveals new roles of two-component systems in Bacillus cereus and closely related low-GC Gram-positives. Environmental microbiology 10: 2796–2809. [DOI] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, and Hulo N. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic acids research 34: W362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaune A, Dubrac S, Blanchet C, Poupel O, Mader U, Hiron A, Leduc A, Fitting C, Nicolas P, Cavaillon JM, Adib-Conquy M, and Msadek T. (2012) The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infection and immunity 80: 3438–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado J, Forst S, Harlocker S, and Inouye M. (1993) Identification of a phosphorylation site and functional analysis of conserved aspartic acid residues of OmpR, a transcriptional activator for ompF and ompC in Escherichia coli. Molecular microbiology 10: 1037–1047. [DOI] [PubMed] [Google Scholar]
- Donegan NP, Marvin JS, and Cheung AL. (2014) Role of adaptor TrfA and ClpPC in controlling levels of SsrA-tagged proteins and antitoxins in Staphylococcus aureus. Journal of bacteriology 196: 4140–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Yoshida T, and Inouye M. (2000) The critical role of the conserved Thr247 residue in the functioning of the osmosensor EnvZ, a histidine Kinase/Phosphatase, in Escherichia coli. The Journal of biological chemistry 275: 38645–38653. [DOI] [PubMed] [Google Scholar]
- Foster TJ, Geoghegan JA, Ganesh VK, and Hook M. (2014) Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nature reviews. Microbiology 12: 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier B, and Hooper DC. (2000) A new two-component regulatory system involved in adhesion, autolysis, and extracellular proteolytic activity of Staphylococcus aureus. Journal of bacteriology 182: 3955–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, and Stock AM. (2013) Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proceedings of the National Academy of Sciences of the United States of America 110: 672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, and Stock AM. (2017) Quantitative Kinetic Analyses of Shutting Off a Two-Component System. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, Jensen BJ, Killgore G, Tenover FC, and Kuehnert MJ. (2008) Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. The Journal of infectious diseases 197: 1226–1234. [DOI] [PubMed] [Google Scholar]
- Grigg JC, Cheung J, Heinrichs DE, and Murphy ME. (2010) Specificity of Staphyloferrin B recognition by the SirA receptor from Staphylococcus aureus. The Journal of biological chemistry 285: 34579–34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutu AD, Wayne KJ, Sham LT, and Winkler ME. (2010) Kinetic characterization of the WalRKSpn (VicRK) two-component system of Streptococcus pneumoniae: dependence of WalKSpn (VicK) phosphatase activity on its PAS domain. Journal of bacteriology 192: 2346–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag AF, and Bagnoli F. (2017) The Role of Two-Component Signal Transduction Systems in Staphylococcus aureus Virulence Regulation. Current topics in microbiology and immunology 409: 145–198. [DOI] [PubMed] [Google Scholar]
- Harper L, Balasubramanian D, Ohneck EA, Sause WE, Chapman J, Mejia-Sosa B, Lhakhang T, Heguy A, Tsirigos A, Ueberheide B, Boyd JM, Lun DS, and Torres VJ. (2018) Staphylococcus aureus Responds to the Central Metabolite Pyruvate To Regulate Virulence. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentschel E, Mack C, Gatgens C, Bott M, Brocker M, and Frunzke J. (2014) Phosphatase activity of the histidine kinases ensures pathway specificity of the ChrSA and HrrSA two-component systems in Corynebacterium glutamicum. Molecular microbiology 92: 1326–1342. [DOI] [PubMed] [Google Scholar]
- Hillion M, and Antelmann H. (2015) Thiol-based redox switches in prokaryotes. Biological chemistry 396: 415–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh MJ, Wharton SJ, Cox AG, Ingham E, Peacock S, and Foster SJ. (2002) MntR modulates expression of the PerR regulon and superoxide resistance in Staphylococcus aureus through control of manganese uptake. Molecular microbiology 44: 1269–1286. [DOI] [PubMed] [Google Scholar]
- Houston P, Rowe SE, Pozzi C, Waters EM, and O’Gara JP. (2011) Essential role for the major autolysin in the fibronectin-binding protein-mediated Staphylococcus aureus biofilm phenotype. Infection and immunity 79: 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TN, and Stewart V. (2011) Negative control in two-component signal transduction by transmitter phosphatase activity. Molecular microbiology 82: 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibberson CB, Jones CL, Singh S, Wise MC, Hart ME, Zurawski DV, and Horswill AR. (2014) Staphylococcus aureus hyaluronidase is a CodY-regulated virulence factor. Infection and immunity 82: 4253–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingavale S, van Wamel W, Luong TT, Lee CY, and Cheung AL. (2005) Rat/MgrA, a regulator of autolysis, is a regulator of virulence genes in Staphylococcus aureus. Infection and immunity 73: 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingavale SS, Van Wamel W, and Cheung AL. (2003) Characterization of RAT, an autolysis regulator in Staphylococcus aureus. Molecular microbiology 48: 1451–1466. [DOI] [PubMed] [Google Scholar]
- Jousselin A, Kelley WL, Barras C, Lew DP, and Renzoni A. (2013) The Staphylococcus aureus thiol/oxidative stress global regulator Spx controls trfA, a gene implicated in cell wall antibiotic resistance. Antimicrobial agents and chemotherapy 57: 3283–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols 10: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AH, Shulla A, Reimann SA, Keating DH, and Wolfe AJ. (2007) The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. Journal of bacteriology 189: 5574–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiecinski JM, Crosby HA, Valotteau C, Hippensteel JA, Nayak MK, Chauhan AK, Schmidt EP, Dufrene YF, and Horswill AR. (2019) Staphylococcus aureus adhesion in endovascular infections is controlled by the ArlRS-MgrA signaling cascade. PLoS pathogens 15: e1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan CY, and Igo MM. (1998) Differential expression of the OmpF and OmpC porin proteins in Escherichia coli K-12 depends upon the level of active OmpR. Journal of bacteriology 180: 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderdale KJ, Malone CL, Boles BR, Morcuende J, and Horswill AR. (2010) Biofilm dispersal of community-associated methicillin-resistant Staphylococcus aureus on orthopedic implant material. Journal of orthopaedic research : official publication of the Orthopaedic Research Society 28: 55–61. [DOI] [PubMed] [Google Scholar]
- Liang X, Zheng L, Landwehr C, Lunsford D, Holmes D, and Ji Y. (2005) Global regulation of gene expression by ArlRS, a two-component signal transduction regulatory system of Staphylococcus aureus. Journal of bacteriology 187: 5486–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, and Naismith JH. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC biotechnology 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Yeo WS, and Bae T. (2016) The SaeRS Two-Component System of Staphylococcus aureus. Genes 7. [Google Scholar]
- Lofblom J, Kronqvist N, Uhlen M, Stahl S, and Wernerus H. (2007) Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. Journal of applied microbiology 102: 736–747. [DOI] [PubMed] [Google Scholar]
- Lowy FD. (1998) Staphylococcus aureus infections. The New England journal of medicine 339: 520–532. [DOI] [PubMed] [Google Scholar]
- Luong TT, Dunman PM, Murphy E, Projan SJ, and Lee CY. (2006) Transcription Profiling of the mgrA Regulon in Staphylococcus aureus. Journal of bacteriology 188: 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TT, and Lee CY. (2006) The arl locus positively regulates Staphylococcus aureus type 5 capsule via an mgrA-dependent pathway. Microbiology 152: 3123–3131. [DOI] [PubMed] [Google Scholar]
- Luong TT, and Lee CY. (2007) Improved single-copy integration vectors for Staphylococcus aureus. Journal of microbiological methods 70: 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino K, Amemura M, Kim SK, Nakata A, and Shinagawa H. (1993) Role of the sigma 70 subunit of RNA polymerase in transcriptional activation by activator protein PhoB in Escherichia coli. Genes & development 7: 149–160. [DOI] [PubMed] [Google Scholar]
- Makino K, Shinagawa H, Amemura M, Kimura S, Nakata A, and Ishihama A. (1988) Regulation of the phosphate regulon of Escherichia coli. Activation of pstS transcription by PhoB protein in vitro. Journal of molecular biology 203: 85–95. [DOI] [PubMed] [Google Scholar]
- Malone CL, Boles BR, Lauderdale KJ, Thoendel M, Kavanaugh JS, and Horswill AR. (2009) Fluorescent reporters for Staphylococcus aureus. Journal of microbiological methods 77: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna AC, Ingavale SS, Maloney M, van Wamel W, and Cheung AL. (2004) Identification of sarV (SA2062), a new transcriptional regulator, is repressed by SarA and MgrA (SA0641) and involved in the regulation of autolysis in Staphylococcus aureus. Journal of bacteriology 186: 5267–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmi G, Nair DR, and Cheung A. (2012) Role of ArlRS in autolysis in methicillin-sensitive and methicillin-resistant Staphylococcus aureus strains. Journal of bacteriology 194: 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, Lopez JA, Foster TJ, Penades JR, and Lasa I. (2009) Protein A-mediated multicellular behavior in Staphylococcus aureus. Journal of bacteriology 191: 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz D, Frei R, Periat N, Zimmerli M, Battegay M, Fluckiger U, and Widmer AF. (2009) Exclusive Staphylococcus aureus throat carriage: at-risk populations. Archives of internal medicine 169: 172–178. [DOI] [PubMed] [Google Scholar]
- Miller LG, and Diep BA. (2008) Clinical practice: colonization, fomites, and virulence: rethinking the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 46: 752–760. [DOI] [PubMed] [Google Scholar]
- Monk IR, Shah IM, Xu M, Tan MW, and Foster TJ. (2012) Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JM, Miller EW, Benson MA, Alonzo F 3rd, Yoong P, Torres VJ, Hinrichs SH, and Dunman PM. (2012) Characterization of SSR42, a novel virulence factor regulatory RNA that contributes to the pathogenesis of a Staphylococcus aureus USA300 representative. Journal of bacteriology 194: 2924–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey JA, Cockayne A, Hill PJ, and Williams P. (2000) Molecular cloning and analysis of a putative siderophore ABC transporter from Staphylococcus aureus. Infection and immunity 68: 6281–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, and Cheung AL. (2011) Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. Journal of bacteriology 193: 2332–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP. (1991) Genetic systems in staphylococci. Methods in enzymology 204: 587–636. [DOI] [PubMed] [Google Scholar]
- Osipovitch DC, Therrien S, and Griswold KE. (2015) Discovery of novel S. aureus autolysins and molecular engineering to enhance bacteriolytic activity. Applied microbiology and biotechnology 99: 6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamp SJ, Frees D, Engelmann S, Hecker M, and Ingmer H. (2006) Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. Journal of bacteriology 188: 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, and Nauseef WM. (2010) agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. Journal of innate immunity 2: 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JS. (2010) Signaling mechanisms of HAMP domains in chemoreceptors and sensor kinases. Annual review of microbiology 64: 101–122. [DOI] [PubMed] [Google Scholar]
- Prados J, Linder P, and Redder P. (2016) TSS-EMOTE, a refined protocol for a more complete and less biased global mapping of transcription start sites in bacterial pathogens. BMC genomics 17: 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radin JN, Kelliher JL, Parraga Solorzano PK, and Kehl-Fie TE. (2016) The Two-Component System ArlRS and Alterations in Metabolism Enable Staphylococcus aureus to Resist Calprotectin-Induced Manganese Starvation. PLoS pathogens 12: e1006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco CJ, Dennison KL, Klenchin VA, Rayment I, and Escalante-Semerena JC. (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess B, Bloes DA, and Berger-Bachi B. (2012) Opposing roles of sigmaB and sigmaB-controlled SpoVG in the global regulation of esxA in Staphylococcus aureus. BMC microbiology 12: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siryaporn A, and Goulian M. (2008) Cross-talk suppression between the CpxA-CpxR and EnvZ-OmpR two-component systems in E. coli. Molecular microbiology 70: 494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava SK, Rajasree K, Fasim A, Arakere G, and Gopal B. (2014) Influence of the AgrC-AgrA complex on the response time of Staphylococcus aureus quorum sensing. Journal of bacteriology 196: 2876–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AM, Robinson VL, and Goudreau PN. (2000) Two-component signal transduction. Annual review of biochemistry 69: 183–215. [DOI] [PubMed] [Google Scholar]
- Stock JB, Ninfa AJ, and Stock AM. (1989) Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiological reviews 53: 450–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zhou L, Zhao BC, Deng X, Cho H, Yi C, Jian X, Song CX, Luan CH, Bae T, Li Z, and He C. (2011) Targeting MgrA-mediated virulence regulation in Staphylococcus aureus. Chemistry & biology 18: 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam K, and Torres VJ. (2019) Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiology spectrum 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thammavongsa V, Kim HK, Missiakas D, and Schneewind O. (2015) Staphylococcal manipulation of host immune responses. Nature reviews. Microbiology 13: 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoendel M, Kavanaugh JS, Flack CE, and Horswill AR. (2011) Peptide signaling in the staphylococci. Chemical reviews 111: 117–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Merino N, Vergara-Irigaray M, Debarbouille M, Penades JR, and Lasa I. (2005) Staphylococcus aureus develops an alternative, ica-independent biofilm in the absence of the arlRS two-component system. Journal of bacteriology 187: 5318–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajtenberg F, Grana M, Ruetalo N, Botti H, and Buschiazzo A. (2010) Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. The Journal of biological chemistry 285: 24892–24903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva M, Garcia B, Valle J, Rapun B, Ruiz de Los Mozos I, Solano C, Marti M, Penades JR, Toledo-Arana A, and Lasa I. (2018) Sensory deprivation in Staphylococcus aureus. Nature communications 9: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JN, Crosby HA, Spaulding AR, Salgado-Pabon W, Malone CL, Rosenthal CB, Schlievert PM, Boyd JM, and Horswill AR. (2013) The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS pathogens 9: e1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sang J, Wang J, Su M, Downey JS, Wu Q, Wang S, Cai Y, Xu X, Wu J, Senadheera DB, Cvitkovitch DG, Chen L, Goodman SD, and Han A. (2013) Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLoS biology 11: e1001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne KJ, Li S, Kazmierczak KM, Tsui HC, and Winkler ME. (2012) Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Molecular microbiology 86: 645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, and Sievert DM. (2016) Antimicrobial-Resistant Pathogens Associated With Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infection control and hospital epidemiology 37: 1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett JW, and Kirby JR. (2011) CrdS and CrdA comprise a two-component system that is cooperatively regulated by the Che3 chemosensory system in Myxococcus xanthus. mBio 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett JW, and Kirby JR. (2012) Genetic and biochemical dissection of a HisKA domain identifies residues required exclusively for kinase and phosphatase activities. PLoS genetics 8: e1003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wang Q, Teng M, and Li X. (2019) The DNA-binding mechanism of the TCS response regulator ArlR from Staphylococcus aureus. Journal of structural biology. [DOI] [PubMed]
- Yarwood JM, McCormick JK, and Schlievert PM. (2001) Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. Journal of bacteriology 183: 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Baumann U, and Reymond JL. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic acids research 32: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Bhinderwala F, Lehman MK, Thomas VC, Chaudhari SS, Yamada KJ, Foster KW, Powers R, Kielian T, and Fey PD. (2019) Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS pathogens 15: e1007538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes regulated by ArlRS
Table S2. RNA-seq comparison
Table S3. Binding site search
Table S4. Primers used in this work