

Abstract
Primary hyperoxaluria is a rare monogenic disorder characterized by excessive hepatic production of oxalate leading to recurrent nephrolithiasis, nephrocalcinosis and progressive kidney damage. Most patients with primary hyperoxaluria are diagnosed after clinical suspicion based on symptoms. Since some patients are detected by family screening following detection of an affected family member, we compared the clinical phenotype of these two groups. Patients with primary hyperoxaluria types 1, 2, and 3 enrolled in the Rare Kidney Stone Consortium Primary Hyperoxaluria Registry were retrospectively analyzed following capture of clinical and laboratory results in the Registry. Among 495 patients with primary hyperoxaluria, 47 were detected by family screening. After excluding 150 patients with end stage kidney disease at diagnosis, 300 clinical suspicion and 45 family screening individuals remained. Compared to patients with clinical suspicion, those identified by family screening had significantly fewer stones at diagnosis (mean 1.2 vs. 3.6), although initial symptoms occurred at a similar age (median age 6.1 vs. 7.6 years). Urinary oxalate did not differ between these groups. The estimated glomerular filtration rate at diagnosis and its decline over time were similar for the two groups. Altogether, five of 45 in family screening and 67 of 300 of clinical suspicion individuals developed end stage kidney disease at last follow-up. Thus, patients with primary hyperoxaluria identified through family screening have significant disease despite no outward clinical suspicion at diagnosis. Since promising novel treatments are emerging, genetic screening of family members is warranted because they are at significant risk for disease progression.
Keywords: primary hyperoxaluria, genetic testing, kidney stones
Graphical Abstract
INTRODUCTION:
Primary hyperoxaluria (PH) is a rare autosomal recessive monogenic disorder characterized by excessive hepatic production of oxalate leading to recurrent nephrolithiasis, nephrocalcinosis, and progressive kidney damage, often resulting in end-stage kidney disease (ESKD). Mutations in three genes have been identified that cause the three known types of PH: AGXT leading to PH type 1 (PH1), GRHPR leading to PH type 2 (PH2), and HOGA1 leading to PH type 3 (PH3).1 Other patients have clinical features consistent with a genetic cause of hyperoxaluria but lack mutations in any of these 3 genes.
Most patients are diagnosed with PH after initially presenting with clinical symptoms (kidney stones, nephrocalcinosis and/or chronic kidney disease (CKD)) and found to have markedly elevated urine and/or plasma oxalate, and lack known gastrointestinal causes of enteric hyperoxaluria. Morbidity and mortality of this autosomal recessive disorder is quite high, with PH1 generally being most severe, followed by PH2, then PH3. Therefore, once an affected proband is identified, screening of all siblings is generally advised either via staged biochemical measurement of urinary oxalate and genetic screening if hyperoxaluria is present, or immediate genetic screening for the mutation(s) identified in the proband. If an affected sibling is identified and hyperoxaluria confirmed, treatments including hyper-hydration and use of crystallization inhibitors (citrate or neutral phosphorus) could be initiated earlier, even prior to any disease manifestations, though the value of a primary prevention strategy has never been assessed in PH.
In In the current study we hypothesized that PH patients identified by familial screening (FS) might have a milder phenotype than those diagnosed due to clinical suspicion (CS). We used data in the Rare Kidney Stone Consortium PH Registry to test this hypothesis.
RESULTS:
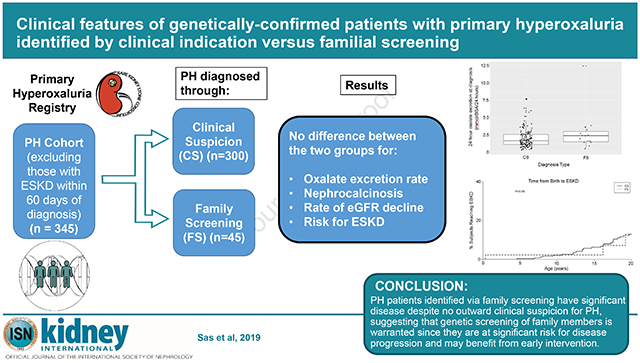
Among the 495 genetically confirmed PH patients in the RKSC registry at the time of this analysis, 345 remained after 150 were excluded due to development of ESKD within 60 days of diagnosis. The entire cohort including those with ESKD was analyzed separately. Among those who did not develop ESKD within 60 days of diagnosis, 45 (13%) were identified by FS, and the remaining 300 diagnosed due to CS (Figure 1). Sensitivity analysis that compared censoring deaths vs. treating death as an event yielded similar results. Table 1 displays clinical and imaging data for the two groups. Follow-up was 10.1 and 12.0 years in the CS and FS groups, respectively (p=0.35). As expected, those identified by FS were younger at diagnosis (median age 3.9 vs. 7.5 yrs for FS and CS cases, respectively, p<0.001). However, initial symptoms occurred at a similar age for both groups (median age 3.2 vs. 4.2 yrs for FS and CS cases, p=0.18).
Figure 1:
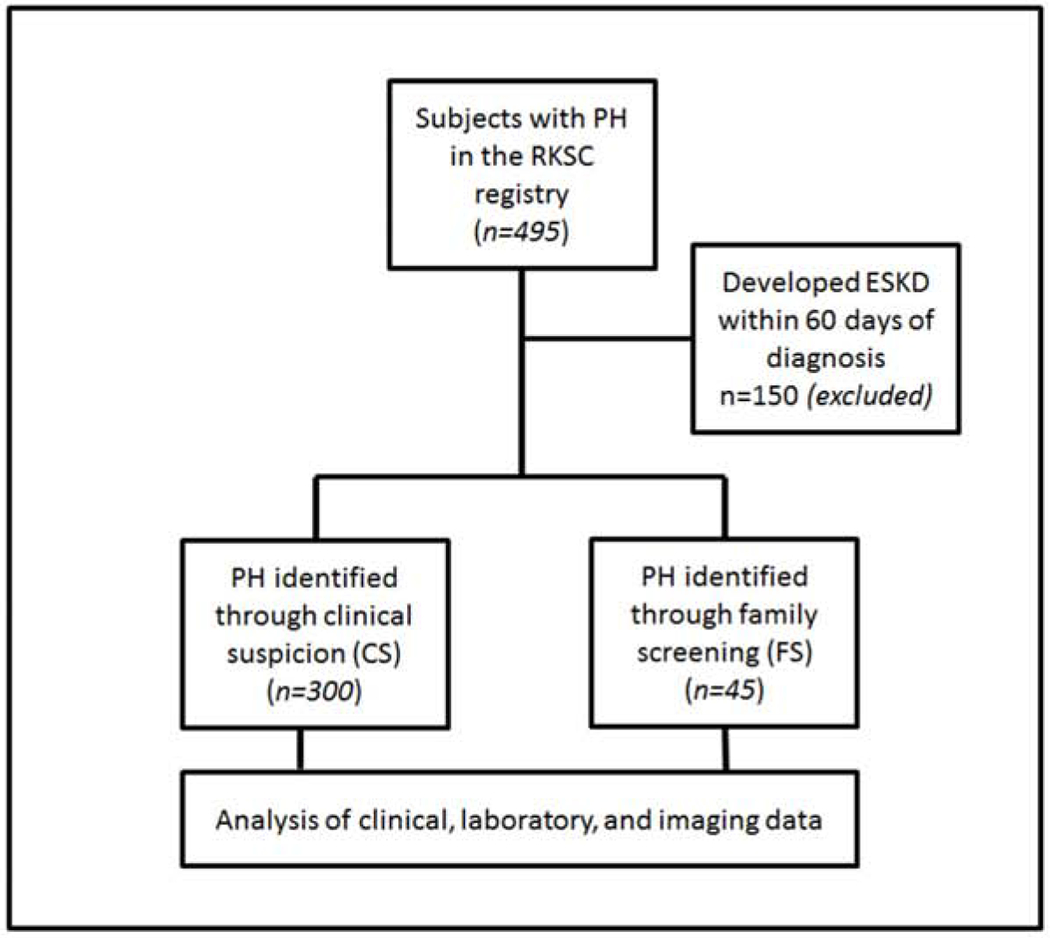
Study flowchart of subjects with primary hyperoxaluria (PH) identified either through clinical suspicion (CS) or family screening (FS).
Table 1:
Clinical, imaging, and laboratory data from all primary hyperoxaluria patients who did not develop ESKD within 60 days of diagnosis.
| Diagnosis Type | ||||
|---|---|---|---|---|
| Clinical Suspicion (N=300) | Family Screening (N=45) | Total (N=345) | P-value | |
| PH type, n (%) | 0.271 | |||
| PH 1 | 210 (70.0%) | 32 (71.1%) | 242 (70.1%) | |
| PH 2 | 34 (11.3%) | 8 (17.8%) | 42 (12.2%) | |
| PH 3 | 56 (18.7%) | 5 (11.1%) | 61 (17.7%) | |
| Gender, n (%) | 0.201 | |||
| Male | 177 (59.0%) | 22 (48.9%) | 199 (57.7%) | |
| Female | 123 (41.0%) | 23 (51.1%) | 146 (42.3%) | |
| Follow-up duration (years) | 0.351 | |||
| N | 300 | 45 | 345 | |
| Mean (SD) | 10.1 (12.01) | 12.0 (12.94) | 10.4 (12.13) | |
| Median | 5.5 | 6.6 | 5.7 | |
| Range | 0.0, 64.1 | 0.0, 46.7 | 0.0, 64.1 | |
| Age at first symptoms (years) | 0.182 | |||
| N | 263 | 26 | 289 | |
| Mean (SD) | 7.6 (8.72) | 6.1 (8.59) | 7.5 (8.71) | |
| Median | 4.2 | 3.2 | 4.1 | |
| Range | 0.1, 41.5 | 0.1, 39.9 | 0.1, 41.5 | |
| Age at diagnosis (years) | <0.0012 | |||
| N | 300 | 45 | 345 | |
| Mean (SD) | 14.8 (16.39) | 8.0 (9.70) | 13.9 (15.84) | |
| Median | 7.5 | 3.9 | 6.9 | |
| Range | 0.0, 70.7 | 0.0, 39.4 | 0.0, 70.7 | |
| Nephrocalcinosis present at diagnosis | 0.981 | |||
| No | 253 (84.3%) | 38 (84.4%) | 291 (84.3%) | |
| Yes | 47 (15.7%) | 7 (15.6%) | 54 (15.7%) | |
| Nephrocalcinosis prior to ESKD, n (%) | 0.561 | |||
| No | 219 (73.0%) | 31 (68.9%) | 250 (72.5%) | |
| Yes | 81 (27.0%) | 14 (31.1%) | 95 (27.5%) | |
| Stones passed prior to diagnosis, n (%) | <.0011 | |||
| No | 162 (54.0%) | 39 (86.7%) | 201 (58.3%) | |
| Yes | 138 (46.0%) | 6 (13.3%) | 144 (41.7%) | |
| Number of stone passage episodes | 0.792 | |||
| N | 95 | 10 | 105 | |
| Mean (SD) | 2.4 (2.09) | 1.7 (0.82) | 2.3 (2.01) | |
| Median | 1 | 1.5 | 1 | |
| Range | 1.0, 11.0 | 1.0, 3.0 | 1.0, 11.0 | |
| Total # stones imaged at diagnosis | 0.0052 | |||
| N | 142 | 13 | 155 | |
| Mean (SD) | 3.6 (4.01) | 1.2 (1.64) | 3.9 (8.58) | |
| Median | 2.5 | 0 | 2 | |
| Range | 0.0, 21.0 | 0.0, 4.0 | 0.0, 100.0 | |
| Number of urological procedures for stones | 0.422 | |||
| N | 83 | 7 | 90 | |
| Mean (SD) | 3.9 (4.50) | 2.3 (1.80) | 3.8 (4.37) | |
| Median | 2 | 2 | 2 | |
| Range | 1.0, 29.0 | 1.0, 6.0 | 1.0, 33.0 | |
| eGFR at diagnosis (mL/min/1.73m2) | 0.662 | |||
| N | 181 | 22 | 203 | |
| Mean (SD) | 75.6 (31.16) | 83.1 (47.00) | 85.2 (124.93) | |
| Median | 69.6 | 73.6 | 70.1 | |
| Range | 17.0, 260.6 | 28.9, 251.1 | 17.0, 1790.3 | |
| eGFR slope (mL/min/year) | 0.862 | |||
| N | 187 | 31 | 218 | |
| Mean (SD) | −1.2 (11.67) | −5.8 (22.81) | −1.9 (13.83) | |
| Median | −0.3 | −0.4 | −0.4 | |
| Range | −84.8, 46.7 | −94.4, 30.8 | −94.4, 46.7 | |
| End-stage kidney disease, n (%) | 0.171 | |||
| No | 233 (77.7%) | 39 (86.7%) | 272 (78.8%) | |
| Yes | 67 (22.3%) | 6 (13.3%) | 73 (21.2%) | |
| Plasma oxalate at diagnosis (mcmol/L) | 0.012 | |||
| N | 39 | 4 | 43 | |
| Mean (SD) | 9.0 (11.67) | 27.3 (17.50) | 10.7 (13.19) | |
| Median | 5.8 | 25.4 | 8.1 | |
| Range | 0.0, 68.0 | 10.3, 48.0 | 0.0, 68.0 | |
| 24-hour oxalate excretion (mmol/BSA/24 hours) at diagnosis | 0.122 | |||
| N | 162 | 15 | 177 | |
| Mean (SD) | 2.0 (1.30) | 2.8 (2.82) | 2.0 (1.50) | |
| Median | 1.7 | 2.4 | 1.7 | |
| Range | 0.3, 7.7 | 0.5, 12.5 | 0.3, 12.5 | |
| Average 24-hour oxalate excretion (mmol/BSA/24 hours) prior to ESKD | 0.712 | |||
| N | 233 | 34 | 267 | |
| Mean (SD) | 1.6 (1.01) | 1.8 (1.13) | 1.7 (1.03) | |
| Median | 1.4 | 1.4 | 1.4 | |
| Range | 0.2, 7.6 | 0.3, 5.7 | 0.2, 7.6 | |
| 24-hour calcium excretion (mg/kg/24 hours) at diagnosis | 0.892 | |||
| N | 140 | 6 | 146 | |
| Mean (SD) | 1.7 (1.49) | 1.7 (1.26) | 1.7 (1.48) | |
| Median | 1.2 | 1.8 | 1.2 | |
| Range | 0.1, 9.5 | 0.1, 3.2 | 0.1, 9.5 | |
| 24-hour citrate excretion (mg/BSA/24 hours) at diagnosis | 0.582 | |||
| N | 118 | 5 | 123 | |
| Mean (SD) | 523.1 (423.79) | 590.4 (814.87) | 525.8 (440.67) | |
| Median | 432.4 | 281.9 | 423 | |
| Range | 10.5, 1912.8 | 51.6, 2035.8 | 10.5, 2035.8 | |
| 24-hour urine volume (mL/BSA/24 hours) at diagnosis | 0.522 | |||
| N | 145 | 12 | 157 | |
| Mean (SD) | 2477.6 (1209.36) | 2201.2 (913.26) | 2456.5 (1189.24) | |
| Median | 2240.8 | 1821.5 | 2213.9 | |
| Range | 645.4, 7007.0 | 1193.2, 3576.6 | 645.4, 7007.0 | |
Chi-Square
Kruskal Wallis
Urinary biochemical parameters did not differ between the 2 groups (Table 1). Mean 24-hour oxalate excretion at diagnosis was similar (2.0 for CS vs. 2.8 mmol/1.73m2/24 hours for FS, p=0.12; Figure 2). Although plasma oxalate concentration was higher in the FS group, only 4 subjects had a plasma oxalate result at the time of diagnosis. Those identified by FS had fewer stones by imaging at diagnosis (mean 1.2 vs. 3.6, p=0.005). Similarly, the FS group had a lower percentage of subjects who had passed stones prior to diagnosis (13.3% vs. 46.0%, p<0.001). The percentage of subjects with nephrocalcinosis did not differ between the two groups (27.0% in CS vs. 31.1% in FS, p=0.56).
Figure 2: 24-hour urinary oxalate excretion between CS and FS.
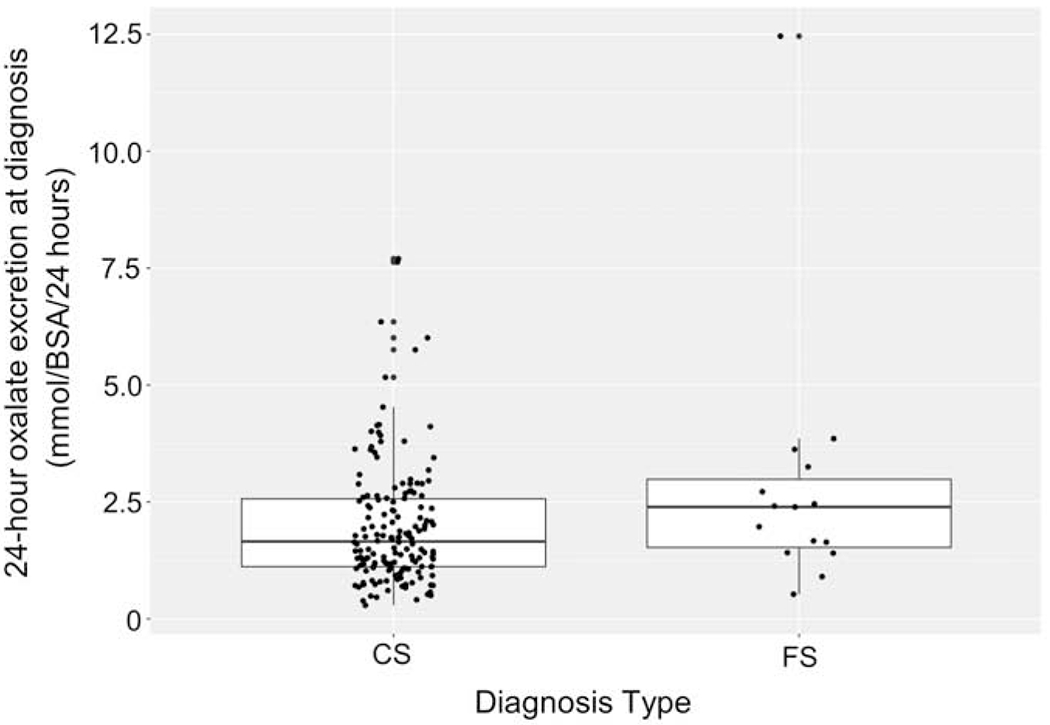
Median daily oxalate excretion was similar between the two groups (p=0.12).
eGFR at the time of diagnosis and loss of kidney function over time (eGFR slope) were similar between the two groups. Altogether, 13.3% (6/45) of FS patients and 22.3% (67/300) of CS developed ESKD at last follow-up (p=0.17) (Figure 3).
Figure 3: Kaplan-Meier curve for risk of ESKD between CS and FS.
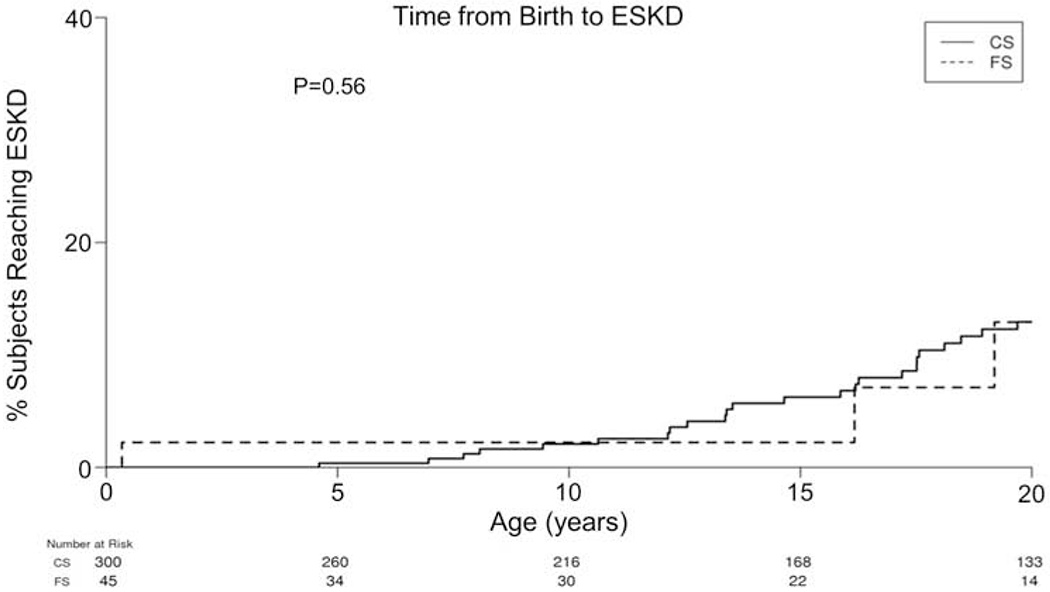
Risk of progression to ESKD was similar between the two (p=0.56)
The entire cohort (n=495) included 150 subjects who developed ESKD within 60 days of diagnosis was also subjected to a sensitivity analysis (Table 2). In this larger group, 47 (10%) were identified by FS. As expected, the mean eGFR at the time of diagnosis was significantly lower in the CS subgroup (55.0 vs. 76.9 mL/min/1.73m2, p=0.02). Otherwise, differences in clinical values between the CS and FS groups paralleled those present in the smaller cohort without ESKD within 60 days of diagnosis.
Table 2:
Clinical, imaging, and laboratory data from all primary hyperoxaluria patients.
| Diagnosis Type | ||||
|---|---|---|---|---|
| Clinical Suspicion (N=448) | Family Screening (N=47) | Total (N=495) | P-value | |
| PH type, n (%) | 0.281 | |||
| PH 1 | 349 (77.9%) | 34 (72.3%) | 383 (77.4%) | |
| PH 2 | 43 (9.6%) | 8 (17.0%) | 51 (10.3%) | |
| PH 3 | 56 (12.5%) | 5 (10.6%) | 61 (12.3%) | |
| Gender, n (%) | 0.241 | |||
| Male | 250 (55.8%) | 22 (46.8%) | 272 (54.9%) | |
| Female | 198 (44.2%) | 25 (53.2%) | 223 (45.1%) | |
| Follow-up duration (years) | 0.031 | |||
| N | 448 | 47 | 495 | |
| Mean (SD) | 8.4 (10.80) | 11.9 (12.69) | 8.7 (11.02) | |
| Median | 4.6 | 6.6 | 4.7 | |
| Range | 0.0, 64.1 | 0.0, 46.7 | 0.0, 64.1 | |
| Age at first symptoms (years) | 0.122 | |||
| N | 394 | 27 | 421 | |
| Mean (SD) | 9.4 (10.92) | 6.5 (8.63) | 9.2 (10.80) | |
| Median | 4.9 | 3.7 | 4.8 | |
| Range | 0.1, 53.0 | 0.1, 39.9 | 0.1, 53.0 | |
| Age at diagnosis (years) | <.0012 | |||
| N | 448 | 47 | 495 | |
| Mean (SD) | 18.0 (18.08) | 8.6 (10.03) | 17.1 (17.69) | |
| Median | 10.6 | 4 | 9.6 | |
| Range | 0.0, 74.0 | 0.0, 39.4 | 0.0, 74.0 | |
| Nephrocalcinosis present at diagnosis | 0.492 | |||
| Yes | 363 (81.0%) | 40 (85.1%) | 403 (81.4%) | |
| No | 85 (19.0%) | 7 (14.9%) | 92 (18.6%) | |
| Stones passed prior to diagnosis, n (%) | <0.0011 | |||
| No | 264 (58.9%) | 40 (85.1%) | 304 (61.4%) | |
| Yes | 184 (41.1%) | 7 (14.9%) | 191 (38.6%) | |
| Number of stone passage episodes | 0.892 | |||
| N | 106 | 10 | 116 | |
| Mean (SD) | 2.3 (2.09) | 1.7 (0.82) | 2.3 (2.07) | |
| Median | 1 | 1.5 | 1 | |
| Range | 1.0, 11.0 | 1.0, 3.0 | 1.0, 11.0 | |
| Total # stones imaged at diagnosis | 0.021 | |||
| N | 185 | 14 | 199 | |
| Mean (SD) | 3.8 (7.90) | 1.4 (1.74) | 3.6 (7.65) | |
| Median | 2 | 0.5 | 2 | |
| Range | 0.0, 100.0 | 0.0, 4.0 | 0.0, 100.0 | |
| Number of urological procedures for stones | 0.562 | |||
| N | 89 | 7 | 96 | |
| Mean (SD) | 3.7 (4.40) | 2.3 (1.80) | 3.6 (4.28) | |
| Median | 2 | 2 | 2 | |
| Range | 1.0, 29.0 | 1.0, 6.0 | 1.0, 33.0 | |
| eGFR at diagnosis (mL/min/1.73m2) | 0.022 | |||
| N | 282 | 24 | 307 | |
| Mean (SD) | 55.0 (43.27) | 76.9 (49.49) | 56.7 (44.10) | |
| Median | 53.9 | 71.2 | 55.8 | |
| Range | 1.9, 324.7 | 5.8, 251.1 | 1.9, 324.7 | |
| eGFR slope (mL/minute/year) | 0.921 | |||
| N | 195 | 31 | 226 | |
| Mean (SD) | −1.3 (11.49) | −5.8 (22.81) | −1.9 (13.62) | |
| Median | −0.3 | −0.4 | −0.4 | |
| Range | −84.8, 46.7 | −94.4, 30.8 | −94.4, 46.7 | |
| End-stage renal failure, n (%) | 0.0011 | |||
| No | 241 (53.8%) | 39 (83.0%) | 280 (56.6%) | |
| Yes | 207 (46.2%) | 8 (17.0%) | 215 (43.4%) | |
| End-stage renal failure 60 Days after dx, n (%) | <.0011 | |||
| No | 300 (67.0%) | 45 (95.7%) | 345 (69.7%) | |
| Yes | 148 (33.0%) | 2 (4.3%) | 150 (30.3%) | |
| Plasma oxalate at diagnosis (mcmol/L) | 0.921 | |||
| N | 88 | 6 | 94 | |
| Mean (SD) | 53.7 (54.63) | 36.1 (21.75) | 52.6 (53.25) | |
| Median | 30 | 36.6 | 33.1 | |
| Range | 0.0, 200.0 | 10.3, 69.7 | 0.0, 200.0 | |
| Plasma oxalate average prior to ESKD (mcmol/L) | 0.051 | |||
| N | 146 | 26 | 172 | |
| Mean (SD) | 18.1 (30.55) | 8.5 (10.65) | 16.6 (28.63) | |
| Median | 7.1 | 3.1 | 6.3 | |
| Range | 0.0, 160.5 | 0.9, 48.0 | 0.0, 160.5 | |
| 24-hour oxalate excretion (mmol/BSA/24 hours) at diagnosis | 0.342 | |||
| N | 187 | 17 | 204 | |
| Mean (SD) | 1.9 (1.30) | 2.6 (2.76) | 2.0 (1.48) | |
| Median | 1.6 | 2 | 1.6 | |
| Range | 0.1, 7.7 | 0.1, 12.5 | 0.1, 12.5 | |
| 24-hour calcium excretion (mg/kg/24 hours) at diagnosis | 0.752 | |||
| N | 158 | 6 | 164 | |
| Mean (SD) | 1.6 (1.45) | 1.7 (1.26) | 1.6 (1.44) | |
| Median | 1 | 1.8 | 1.1 | |
| Range | 0.1, 9.5 | 0.1, 3.2 | 0.1, 9.5 | |
| 24-hour citrate excretion (mg/BSA/24 hours) at diagnosis | 0.692 | |||
| N | 132 | 5 | 137 | |
| Mean (SD) | 494.2 (417.27) | 590.4 (814.87) | 497.7 (433.09) | |
| Median | 407.7 | 281.9 | 397 | |
| Range | 10.5, 1912.8 | 51.6, 2035.8 | 10.5, 2035.8 | |
| 24-hour urine volume (mL/BSA/24 hours) at diagnosis | 0.322 | |||
| N | 169 | 13 | 182 | |
| Mean (SD) | 2447.9 (1240.17) | 2054.0 (1022.72) | 2419.8 (1227.70) | |
| Median | 2260.2 | 1670.2 | 2241.3 | |
| Range | 74.3, 7007.0 | 288.4, 3576.6 | 74.3, 7007.0 | |
Chi-Square
Kruskal Wallis
The sample size was insufficient to allow subgroup analysis of the different forms of PH detected by CS vs FS.
DISCUSSION:
The current study compared the clinical course of PH patients genetically diagnosed because of CS versus those detected by FS after a proband had been diagnosed. The results indicate that those identified via FS were younger with fewer stones at the time of diagnosis compared to those with clinical indications for testing. Nevertheless, the presence of NC at initial evaluation and ultimate clinical course after diagnosis of both groups appeared similar, including the rate of GFR loss over time. Thus, severity of urinary stone disease (USD) and symptoms at the time of diagnosis does not imply a more benign long-term prognosis.
It was interesting to note that some of the FS group were entirely asymptomatic when diagnosed, while others had experienced symptomatic USD prior to their diagnosis, and PH had not previously been considered. Potential reasons for this include an incomplete work-up for USD, lower index of suspicion for the possibility of PH until the proband was diagnosed, and initial unwillingness of the patient to have genetic testing.
PH is a rare autosomal recessive monogenic disorder characterized by marked hyperoxaluria, often with a severe phenotype. Patients frequently experience kidney stones, nephrocalcinosis, and ESKD. While it is extraordinarily rare for patients with PH3 to reach ESKD, 57% of PH1 patients reach ESKD by age 40 and 14-25% of PH2 patients have reached ESKD.2,3 Current evidence suggests that the degree of hyperoxaluria at diagnosis as well as presence of nephrocalcinosis increase ESKD risk, while kidney stone risk may be less dependent on degree of hyperoxaluria in this patient population.4,5 Current treatments include maintaining a dilute urine by increased fluid intake, as well as liberal use of crystallization inhibitors, including potassium citrate or neutral phosphorus.6,7 Other treatment strategies require an individualized approach based on patient-specific risk factors. Pyridoxine is a cofactor for alanine:glyoxylate aminotransferase, the aberrant enzyme in PH1, and pharmacological doses of pyridoxine can significantly reduce urinary oxalate excretion in a subset of PH1 patients with specific mutations.8 Despite these measures, the risk of ESKD is high, especially in PH type 1.
Because of the severe phenotype associated with PH, once a proband has been identified it is recommended that all siblings should be screened for the disorder. It has previously been assumed that probands are identified first because they have a more severe phenotype than unaffected siblings, despite a shared genotype. Causes of an earlier presentation and more severe phenotype could include environmental cofactors or genetic modifiers. In the current study, patients who underwent genetic testing because of CS were older and had more stones at the time of diagnosis than the FS group, as might be expected. However, biochemical characteristics of those patients identified by FS were very similar to those identified because of CS, suggesting that the underlying mutation is the most important determinant of these features. In addition, subsequent loss of GFR was similar between the 2 groups.
The current study strongly supports family screening in PH once a proband is diagnosed, since even though these individuals are often asymptomatic at diagnosis, their ultimate clinical course is similar to those that present with symptoms. Initiation of currently-available treatments at an earlier age may have benefits. In addition, promising treatments for PH, including siRNA-based therapeutics, are now entering phase III clinical trials, while work continues to identify other innovative agents for treatment, like stiripentol.2,9 The benefit of early genetic diagnosis and treatment has been established for other pediatric disease processes,10–12 and these novel treatment possibilities may offer significantly improved clinical outcomes for PH patients preemptively identified through genetic screening.
Initial screening can be conducted by measurement of urine oxalate excretion or random oxalate/creatinine and renal ultrasound with confirmation by genetic testing if hyperoxaluria, stones, or nephrocalcinosis are found. An algorithm to guide steps in diagnosis has been published.1 It is important to note that access to health care and diagnostic testing is vital for diseases like PH and unfortunately is not equally available throughout the world. It is also important to consider that genetic testing can have significant negative impacts and should be done thoughtfully. While the Genetic Information Act of 2008 prevents discrimination based on genetic testing results for employment and health insurance, it does not offer protection for other types of insurance including life, disability, or long-term care.13 Thus potential benefits must be weighed against the potential risks and fully discussed with the patient/parents to allow informed consent before performing genetic testing.
Our study has several strengths and weaknesses. This was a retrospective analysis of clinical registry data, a voluntary data set that is inherently incomplete. Screening was not performed in all siblings and it is possible that there are siblings with genetic disease who, indeed, manifest a milder phenotype for reasons yet to be explained. Nevertheless, data from a large registry of affected PH patients with genetically confirmed diagnoses was available for this study that supports the importance of familial screening. While we recognize that primary hyperoxaluria is a rare disease, the primary finding that patients with genetic disease discovered through family screening had similar disease severity as the proband may be applicable to many rare diseases.
CONCLUSION:
Patients identified through family PH screening have a similar disease severity as those identified due to CS. Although those detected by FS have fewer stones at the time of diagnosis, their biochemical and clinical characteristics including loss of kidney function over 10 years following diagnosis, appear very similar to those whose diagnosis was made due to clinical suspicion. Thus, familial screening is important to identify PH patients who are likely to benefit from early initiation of treatment.
METHODS:
Study population
This was a retrospective observational study. Clinical and laboratory information were collected from PH patients in the RKSC PH registry enrolled between 2003 and 2018.14 Demographics, clinical, and laboratory data were abstracted from the Registry. In the current study PH1 was confirmed by mutations of the AGXT gene, PH2 by mutations of GRHPR, and PH3 by mutations of HOGA1. Probands diagnosed after the onset of end stage kidney disease (ESKD) were excluded from this analysis.
All 24-hour urine values were from baseline (0-60 days after diagnosis) or during follow-up (> 60 days until last available prior to ESKD). Renal function was assessed using the Full Age Spectrum (FAS) GFR equation since the cohort spans age ranges from infancy to late adulthood.15 ESKD or renal failure was defined as an eGFR <15 ml/min/1.73m2 or start of dialysis or kidney transplantation. Episodes of stone passage and urological procedures for stones were self-reported.
Statistical Methods
Results were expressed as median (25th, 75th) for continuous variables and as percentages for categorical variables. Comparisons between groups for the categorical variables were performed with the Chi-square test for categorical variables and the Kruskal-Wallis test for continuous variables. To maximize data for analysis, the value closest to diagnosis between 3 years prior and 60 days after diagnosis was used.
The percentage of subjects free of ESKD following PH diagnosis was estimated using the Kaplan-Meier method. Factors associated with renal survival were estimated by univariate analyses using Cox’s proportional hazard model with log-rank tests and trend tests used for comparison between subgroups. ESKD was considered the event and censored on death or loss to follow-up. We further assessed the same model treating deaths as events, rather than censoring them, as a sensitivity analysis. P values < 0.05 were considered to be statistically significant. All calculations were performed using SAS (version 9.4).
Acknowledgements:
This work was funded by the Rare Kidney Stone Consortium (U54DK83908), which is part of Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). This consortium was funded through collaboration between NCATS, and the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: All of the authors declare no competing interests.
REFERENCES:
- 1.Edvardsson VO, Goldfarb DS, Lieske JC, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28(10):1923–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sas DJ, Harris PC, Milliner DS. Recent advances in the identification and management of inherited hyperoxalurias. Urolithiasis. 2019;47(1):79–89. [DOI] [PubMed] [Google Scholar]
- 3.Garrelfs SF, Rumsby G, Peters-Sengers H, et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow-up. Kidney Int. 2019. [DOI] [PubMed] [Google Scholar]
- 4.Tang X, Bergstralh EJ, Mehta RA, Vrtiska TJ, Milliner DS, Lieske JC. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int. 2015;87(3):623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao F, Bergstralh EJ, Mehta RA, et al. Predictors of Incident ESRD among Patients with Primary Hyperoxaluria Presenting Prior to Kidney Failure. Clin J Am Soc Nephrol. 2016;11(1):119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallson PC, Rose GA, Sulaiman S. Raising urinary citrate lowers calcium oxalate and calcium phosphate crystal formation in whole urine. Urol Int. 1983;38(3):179–181. [DOI] [PubMed] [Google Scholar]
- 7.Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331(23):1553–1558. [DOI] [PubMed] [Google Scholar]
- 8.Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005;67(5):1704–1709. [DOI] [PubMed] [Google Scholar]
- 9.Le Dudal M, Huguet L, Perez J, et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. J Clin Invest. 2019;129(6):2571–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bockenhauer D, Bichet DG. Nephrogenic diabetes insipidus. Curr Opin Pediatr. 2017;29(2):199–205. [DOI] [PubMed] [Google Scholar]
- 11.Mak DY, Sykes J, Stephenson AL, Lands LC. The benefits of newborn screening for cystic fibrosis: The Canadian experience. J Cyst Fibros. 2016;15(3):302–308. [DOI] [PubMed] [Google Scholar]
- 12.Mei D, Parrini E, Marini C, Guerrini R. The Impact of Next-Generation Sequencing on the Diagnosis and Treatment of Epilepsy in Paediatric Patients. Mol Diagn Ther. 2017;21(4):357–373. [DOI] [PubMed] [Google Scholar]
- 13.Adams DR, Eng CM. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N Engl J Med. 2018;379(14):1353–1362. [DOI] [PubMed] [Google Scholar]
- 14.Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290–296. [DOI] [PubMed] [Google Scholar]
- 15.Pottel H, Hoste L, Dubourg L, et al. An estimated glomerular filtration rate equation for the full age spectrum. Nephrol Dial Transplant. 2016;31(5):798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]