

Abstract
Autophagy, an adaptive catabolic process, plays a cytoprotective role in enabling cellular homeostasis in the innate and adaptive immune systems. Neutrophils, the most abundant immune cells in circulation, are professional killers that orchestrate a series of events during acute inflammation. The recent literature indicates that autophagy has important roles in regulating neutrophil functions, including differentiation, degranulation, metabolism and neutrophil extracellular trap formation, that dictate neutrophil fate. It is also becoming increasingly clear that autophagy regulation is critical for neutrophils to exert their immunological activity. However, evidence regarding the systematic communication between neutrophils and autophagy is insufficient. Here, we provide an updated overview of the function of autophagy as a regulator of neutrophils and discuss its clinical relevance to provide novel insight into potentially relevant treatment strategies.
Keywords: Autophagy, Neutrophil, Innate immunity, NETs, Clinical application, Neutrophil extracellular trap
Background
Neutrophils, the most abundant category of polymorphonuclear leukocytes in circulation, are professional killers during acute inflammation [1]. Neutrophils patrol the blood continuously and recruit cells to sites of inflammation at the onset of injury [2]. Activated neutrophils use multiple strategies, including phagocytosis, autophagy, degranulation, reactive oxygen species (ROS) release and neutrophil extracellular trap (NET) formation, to destroy infectious threats [3]. They modulate the innate immune response and have prolonged activity in ongoing inflammation in numerous diseases. Lately, neutrophils have come under increased scrutiny due to surprising findings regarding their heterogeneity and plasticity [4] and their abilities to be long-lived and regulate adaptive immune responses [5].
Autophagy, interpreted as self-eating from the Greek words auto, meaning “self”, and phagein, meaning “to eat”, is an evolutionarily conserved degradative process [6] that follows four major routes: macroautophagy, microautophagy, chaperone-mediated autophagy [7], and non-canonical autophagy (8). Macroautophagy (hereafter referred to as autophagy) utilizes autophagosomes, which are double-membrane vesicles, to isolate portions of the cytoplasm, such as those containing harmful and unwanted material, and deliver them to lysosomes for degradation [9]. Autophagy is defined as non-selective or selective; selective autophagy targets various cargoes for degradation and includes organelle-specific autophagy (such as mitophagy, pexophagy, and reticulophagy) and xenophagy (the degradation of microorganisms) [10]. Macroautophagy, the best-characterized form of autophagy, is the major route of autophagy in neutrophils and will hereafter be referred to simply as autophagy. Autophagy is essential to maintain host health and helps the capture and clearance of invading pathogens by the immune system.
The cognition of the immunological functions of neutrophil autophagy has increased remarkably in recent years, and it is now appreciated that the primordial functions of autophagy have evolved and been incorporated into multiple innate and adaptive immune pathways [11]. Accumulating evidence indicates that autophagy has important roles in regulating neutrophil functions, including degranulation, metabolism, and NET formation. However, knowledge of the systematic relationship between neutrophils and autophagy is insufficient. In this review, we summarize the updated function of autophagy as a regulator of neutrophils and then focus on its clinical relevance to provide a novel therapeutic strategy.
Review
Mechanisms of autophagy
Autophagy, an adaptive catabolic process, plays a cytoprotective role to enable cellular homeostasis [12]. Neutrophil autophagy occurs at a basal level in nutrient-rich conditions, but can be markedly induced in response to many cellular stresses, including starvation, endoplasmic reticulum (ER) stress, oxidative stress, and exposure to certain chemicals, radiation, and hypoxia [10, 13, 14]. In the innate immune response, neutrophils utilize autophagy as an antimicrobial effector function, which can be regulated in the process of a memory response [15].
Steps in the autophagic pathway and autophagy-related proteins
Neutrophil autophagy involves the following pivotal steps: signal induction, membrane nucleation, cargo targeting, vesicle expansion, autophagosome formation, fusion with the lysosome, cargo degradation, and nutrient recycling [16] (Fig. 1).
Figure 1.
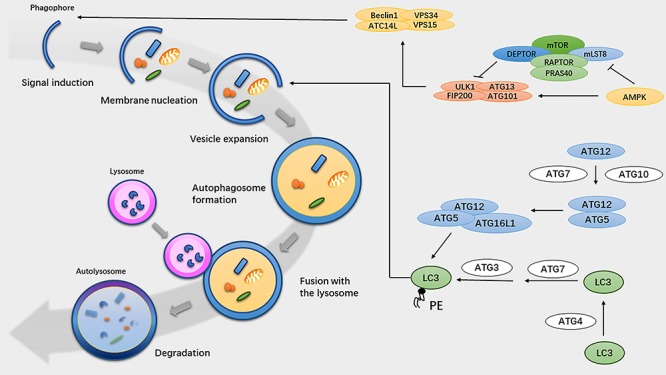
Overview of the autophagy pathway. Autophagy proceeds through several steps: signal induction, membrane nucleation, cargo targeting, vesicle expansion, autophagosome formation, fusion with the lysosome, cargo degradation, and nutrient recycling. Key proteins that regulate autophagy in mammals are shown on the right. Autophagosome activation is mediated by three initial signaling complexes: the ULK1, PI3KC3, and ATG16L1 complexes. Autophagosome elongation is regulated by two ubiquitin-like conjugate systems: the ATG12–ATG5–ATG16L1 and LC3–PE complexes. AMPK AMP-activated protein kinase, mTOR mammalian target of rapamycin, LC3 microtubule-associated protein 1 light chain 3, PRAS40, 40 kDa Pro-rich AKT substrate, RAPTOR regulatory-associated protein of mTOR, AMP adenosine-triphosphate, VPS vacuolar protein sorting, ULK1 unc-51 like autophagy activating kinase 1, ATG autophagy-related protein, FIP200 family kinase-interacting protein of 200 kDa, PE phosphatidylethanolamine
Autophagosome formation is the key event in the autophagy pathway. The autophagosome is activated by three initial signaling complexes: the serine/threonine protein kinase unc-51-like autophagy-activating kinase 1 (ULK1) complex (comprising ULK1, a focal adhesion kinase (FAK) family kinase-interacting protein of 200 kDa (FIP200, also known as RB1CC1), autophagy-related protein (ATG) 13 and ATG101, [17–19]); the phosphoinositide 3-kinase catalytic subunit type III (PI3KC3) complex (comprising Beclin-1, vacuolar protein sorting (VPS) 34, VPS15 and ATG14L [20]); and the ATG16L1 complex (comprising ATG16L1, ATG5, and ATG12, [21]). The serine/threonine kinase mammalian target of rapamycin complex 1 (mTORC1) is phosphorylated under conditions of nutrient excess, whereas nutrient deprivation and immune signaling activation lead to the inhibition of mTORC1 phosphorylation and the activation of the Beclin-1–VPS34 complex by ULK1 complex formation [22, 23].
During the phagophore membrane nucleation step, phosphatidylinositol-3-phosphate (PtdIns3P)-rich regions are produced by the lipid kinase VPS34 and recruited to the surface of donor membranes, including the ER, Golgi apparatus, ER–mitochondria contact sites, endosomes, and plasma membrane [24–28]. The ULK1 complex recruits Beclin-1, ATG14L, and phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4), along with the phagophore, to the phagophore assembly site (PAS). PtdIns3P is recognized by PtdIns3P-binding factor WD repeat (WDR) domain phosphoinositide-interacting protein 1 (WIPI1)–WIPI4 [29]. ATG9 plays a role in PAS formation and expansion by momentarily interacting with omegasomes [30, 31].
Autophagosome elongation is regulated by two ubiquitin-like conjugate systems: the ATG12–ATG5–ATG16L1 and microtubule-associated protein 1 light chain 3-phosphatidylethanolamine (LC3-PE) complexes. ATG12–ATG5 conjugation is activated by ATG7 and ATG10 [32]. The ATG16L complex is generated subsequently and non-covalently binds to the ATG5–ATG12 conjugate [21]. In parallel, LC3 is cleaved at its C-terminal arginine by the ATG4 protease to expose a glycine residue [33]. There are seven mammalian orthologs of ATG8 (LC3A, microtubule-associated protein 1 light chain 3 beta (LC3B), LC3C, GABA Type A Receptor Associated Protein (GABARAP), GABA Type A Receptor Associated Protein Like 1 (GABARAPL1), GABARAPL2, and GABARAPL3; these are referred to collectively as LC3 in this review) [34]. The ATG16L1 complex conjugates LC3 to phosphatidylethanolamine (PE) to form the second ubiquitin-like conjugate system [35], which involves ATG7 and ATG3 [36]. LC3 regulates membrane tethering and fusion and recruits membranes to extend the isolation membrane and form autophagosomes; thus, LC3 is widely used as a microscopic detection marker [34, 37].
During the final maturation step, autophagosomes become degradative autolysosomes. The small guanosine triphosphatase (GTPase) Ras-related protein 7 (RAB7, [38]), the autophagosomal SNARE protein syntaxin 17 [39] and the lysosomal SNARE vesicle-associated membrane protein 8 (VAMP8), as well as lysosomal membrane proteins such as lysosomal-associated membrane glycoprotein 2 (LAMP2) [40, 41], are required for autophagosome and lysosome fusion. Lysosomes break the inner autophagosomal membrane, and lysosomal hydrolases regulate the degradation of autophagosome cargo.
Regulation of the autophagy pathway
Autophagy is an intricate physiological mechanism that can be regulated by numerous molecules and pathways. Nutrient signaling mediates autophagy progression by targeting mTOR [42]. The Class I phosphatidylinositol-3-kinase (PI3K)–also known as Protein Kinase B (AKT) pathway negatively regulates autophagy by activating mTOR [43]. Adenosine-triphosphate (AMP)-activated protein kinase (AMPK) inhibits mTOR and phosphorylates ULK1 to promote autophagy [6]. mTOR regulates the activation of autophagy. The Beclin-1-interacting complex also mediates autophagy progression, and upregulating PI3P promotes autophagosomal membrane nucleation [44]. Other interacting factors, such as ATG9, may also regulate this process [34]. Autophagosome elongation is mainly mediated by the ATG12–ATG5–ATG16L1 and LC3–PE complexes [45].
Therefore, neutrophil autophagy is an intricate catabolic process that involves the following pivotal steps: signal induction, membrane nucleation, cargo targeting, vesicle expansion, autophagosome formation, fusion with the lysosome, cargo degradation, and nutrient recycling. Several molecules and pathways are involved in regulating autophagy progression.
Autophagy-mediated regulation of neutrophil differentiation
Neutrophil differentiation is dependent on extensive cytoplasmic and nuclear remodeling. Every second, approximately 1×106 neutrophils are generated in the bone marrow of humans. Exquisite regulation is required for granulopoiesis. Multipotent progenitors (MPPs) give rise to granulocyte–monocyte progenitors (GMPs) and myeloblasts (MBs), which are considered the initial granulocyte precursors [46]. Through the promyelocyte (MC), metamyelocyte (MM), and band cell (BC) stages, MBs differentiate into mature polymorphonuclear neutrophils (PMNs) [4, 47]. Autophagy was shown to be involved in neutrophil differentiation and proliferation in bone marrow and lymphoid organs in a mouse model and to have different effects on various stages of differentiation [48, 49].
During early granulopoiesis, autophagy is essential for normal and induced neutrophil differentiation by governing these energy-consuming changes. Neutrophil differentiation depends on free fatty acids generated by autophagy, and these free fatty acids support mitochondrial respiration. Autophagy-deficient (ATG7 knockout at the GMP–MB stage) neutrophil precursors show excessive glycolysis, lipid droplet accumulation, and adenosine-triphosphate (ATP) depletion but no change in glycolytic activity [47]. However, autophagy controls the earliest precursor stage, and defects at the hematopoietic stem cell (HSC) level (Lyz2-Cre-mediated ATG5 deletion) cause the rapid production of granulocyte precursors, which do not show morphological changes or functional deficiencies [49].
Autophagy is important for differentiation in response to specific stimulants, including granulocyte colony-stimulating factor (G-CSF) and all-trans retinoic acid (ATRA). G-CSF induces autophagy in both neutrophils and HSCs in human and mouse models. Autophagy protects neutrophils from cytokine-induced stress to prolong their life span and counteract neutropenia after cytotoxic chemotherapy. Autophagy ensures HSC survival for transplantation [50]. Beclin-1-independent autophagy is positively correlated with ATRA-induced neutrophil differentiation [51]. The expression of ATG3, ATG4D, ATG5, and WIPI1 is increased by the Ets family hematopoietic transcription factor PU.1, which is important for neutrophil differentiation [52, 53]. PU.1 regulates autophagy via microtubule-associated protein 1S (MAP1S, also known as C19ORF5), which plays a role in microtubule dynamics [54]. Damage-regulated autophagy modulator-1 (DRAM-1) is involved in ATRA-induced neutrophil differentiation [55].
In general, autophagy has different effects on the different stages of normal neutrophil differentiation. During early granulopoiesis, autophagy is essential for neutrophil differentiation because it governs energy-consuming changes. At the HSC level, autophagy controls the earliest precursor stage. Autophagy is important for differentiation in response to specific stimulants, including G-CSF and ATRA.
Autophagy-mediated regulation of neutrophil lifespan
Neutrophils, which are terminally differentiated cells in the innate immune system, have a short life span. In the absence of activation, neutrophils are constitutively committed to apoptosis. Autophagy is considered a “double-edged sword” that controls the fate of neutrophils. Autophagy can help cells survive by detecting oxidative stress and eliminating damaged cellular components [56]; however, it also jeopardizes cell survival and accelerates cell death. Here, we elaborate on the effect of autophagy on apoptotic and neutrophil-derived giant phagocytes (Gϕ) to explore the mystery between autophagy and neutrophil lifespan.
Autophagy and apoptosis
Autophagy and apoptosis, two classical pathways of programmed cell death, exhibit complex interactions. Apoptosis can suppress autophagy for digesting crucial autophagy-related proteins [57, 58], and autophagy can both promote and suppress apoptosis. On the one hand, autophagy can prevent neutrophils from undergoing intrinsic apoptotic by multiple mechanisms, such as mitophagy and the degradation of pro-apoptotic proteins. Autophagy can suppress ER stress and help neutrophils survive apoptotic stimuli [59]. A reduced autophagy capacity results in a compensatory increase in apoptosis [60]. 3-Methyladenine (MA) and chloroquine (CQ), two autophagy inhibitors, markedly promote spontaneous neutrophil apoptosis by downregulating Mcl-1 without affecting B-cell lymphoma-XL (BCL-XL) [61]. On the other hand, autophagy can facilitate apoptosis. Caspase 8 can activate autophagosomes by serving as a platform [60]. Caspase 3 and poly ADP-ribose polymerase (PARP) can be activated by autophagy via a Fas-associated exogenous pathway. Autophagy also degrades endogenous inhibitors of apoptosis. Although autophagy has pro-apoptotic activity, this activity is relatively weaker than its anti-apoptotic activity. Ultimately, autophagy has an inhibitory effect on apoptosis.
Many molecules, such as ATG5, BCL-2, and p53, simultaneously modulate autophagy and apoptosis [62]. ATG5 and BCL-2 might be the key regulators. ATG5, an important regulator of autophagy, can translocate from the cytoplasm to mitochondria and activate caspases. The proteolytic cleavage of ATG5 weakens autophagy and induces apoptosis. In mice, BCL-2, an anti-apoptotic protein, inhibits pro-apoptotic members of the BCL-2 family and inhibits Beclin-1 to interfere with autophagy. High BCL-2 levels can abolish the pro-apoptotic function of truncated ATG5 [63].
Autophagy and neutrophil-derived Gϕ
Autophagy is involved in the development of the long-living subset of neutrophils, neutrophil-derived Gϕ, which are positive for CD15, CD66b, CD63, CD11b, myeloperoxidase (MPO), and neutrophil elastase (NE) [64]. Autophagy is a basic characteristic of these cells and largely controls their formation. Gϕ act as cellular “cannibals” to eliminate dead neutrophils and debris. The phagocytic centers of Gϕ include LC3B-coated vacuoles and microtubule-associated protein 1 light chain beta (LCB) aggregates. LC3B accumulation increases during oxidized LDL (oxLDL) internalization. Treatment with 3-methyladenine (3MA) and bafilomycin A1 (BafA1), two specific autophagy inhibitors, arrests the development of Gϕ [65].
In short, autophagy should be considered an important mechanism that prolongs the lifespan of cells. Autophagy exhibits bidirectional regulation of apoptosis, but in general, it inhibits apoptosis. Autophagy facilitates the development of long-living neutrophil subsets, such as neutrophil-derived Gϕ.
Autophagy-mediated regulation of neutrophil degranulation
Autophagy regulates neutrophil-mediated inflammation and affects neutrophil degranulation and ROS production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase nitrogen oxide (NOX) [66]. There are more than four types of granules in neutrophils [67]. Primary and tertiary granules originate from the ER–Golgi network. Primary, or azurophilic, granules store MPO, β-glucuronidase, elastase, and other antimicrobial factors. Tertiary (gelatinase) granules contain matrix metalloprotease-9 (MMP-9). Secondary (specific) granules originate from both the ER–Golgi and endocytosis and contain lactoferrin and MMP-9. These granules express significant molecules, including CD11b/CD18 and complement receptors, that are key for neutrophil migration and activation on the membrane; another such important protein in secondary granules is flavocytochrome b558, a component of the NOX machinery [68]. The fusion of secondary granules can produce ROS by activating the NOX complex. Autophagy deficiency disrupts neutrophil degranulation, especially that of secondary and tertiary granules. Knockout of ATG5 or ATG7 inhibits degranulation, indicating that this event likely occurs through autophagy, not an ATG-independent mechanism. Granule fusion helps induce NOX-mediated ROS generation, and ROS are vital for neutrophil apoptosis. Under inflammatory conditions, ER stress can induce neutrophil autophagy, and autophagy can inhibit ER stress [59]. Autophagy downregulates ROS and increases neutrophil circulation to control inflammation.
Autophagy-mediated regulation of NETs
NETs are an important neutrophil antimicrobial effector in response to various stimuli [69]. Classical NET release occurs in a complex series of orchestrated events regulated in five stages, including ROS generation, peptidyl arginine deiminase-4 activation, distinct vesicle formation, chromatin decondensation and active extrusion of the DNA/histone/cathelicidin antimicrobial peptide cocktail into the extracellular space [70]. Classical autophagy plays a multilayer regulatory role in NET formation. Autophagy participates in pathogen elimination as an alternative degradative platform to phagosomes with lysosomes [71]. Autophagy is involved in the externalization of membrane-bound and cytosolic proteins, thereby influencing stage 3 of NET vacuolization [60, 72]. Autophagy may also play a role in inhibiting the respiratory burst, blocking cytoskeletal dynamics and chromatin decondensation and inducing histone citrullination in stages 1, 4 and 5 of NET formation [73–75]. However, chaperone-mediated autophagy (CMA) has no effect on NET formation [76].
Is autophagy the answer?
According to the classical theory, autophagy in indisputably involved in neutrophil NET formation. However, a controversial point of view was recently proposed: a key molecule in autophagy, rather than autophagy itself, is involved in the regulation of neutrophil NET formation. Is autophagy the answer? To answer this question, we analyzed all autophagy-related molecules involved in NET generation and outlined the potential mechanism in Table 1. We found no evidence contradicting the notion that PI3K–AKT–mTOR signaling affects NET formation in the early stage of autophagy. The focus of the debate is whether ATG5 is essential for NET formation and whether the later stage of autophagy has the same effect as the early stage. ATG5 levels increase when autophagy induces NET formation [43, 77]. ATG5-dependent autophagy is required for NET formation [60], which is inhibited by 3MA and wortmannin. However, neutrophils from ATG5-knockout mice show normal NET release. The late autophagy inhibitors BafA1 and CQ had no effect on normal NET formation [78]. From the perspective of signaling pathways, NET formation was not regulated by autophagy but rather by the PI3K–AKT–mTOR pathway, which interacts with signaling cascades initiated by triggers of NET formation. However, considering the metabolism of neutrophils, autophagy has a positive effect on NET formation. Thus, we deduced that autophagy affects NET formation, but this hypothesis has not been proven.
Table 1.
Autophagy-related molecules involved in NET formation
| Key molecule | Species | Model | Observed effect of autophagy on NET | Associated mechanism | Reference |
|---|---|---|---|---|---|
| mTOR | Human | Patients with ST segment elevation acute myocardial infarction (STEMI) and controls | NET formation was induced by the inhibition of mTOR and the induction of autophagy | The effect of polyP on autophagy and NET formation is dose-dependent | [42] |
| mTOR | Human | Neutrophils stimulated with ANCA-positive IgG | Autophagy promoted NET formation | ANCA-positive IgG-induced NET formation is enhanced by rapamycin and suppressed by 3MA | [79] |
| mTOR | Human | Patients with lupus nephritis | Autophagy affected NET formation and release | The mTOR inhibitors rapamycin and WYE-354 (which can induce autophagy) increase the number of SYTOX-positive neutrophils and the expression of NE | [80] |
| mTOR, Beclin-1 | Human | Patients with systemic lupus erythaematosus | NET formation activated neutrophils through autophagy | Pharmacologic inhibition of autophagy prevents intracellular chromatin decondensation, which is necessary for NETosis and NET formation | [81] |
| mTOR | Human | Neutrophils stimulated with fMLP | The mTOR pathway plays a pivotal role in NET formation by regulating autophagy downstream of FPR signaling | NET inducer PMA activates autophagy | [74] |
| ATG5, mTOR | Mouse | Aged mice, TLR2 KO mice and MyD88 KO mice | TLR2 ligand-induced NETosis in aged mice was compromised by an ATG5 defect, leading to a subsequent impairment of autophagy | Pharmacologic inhibition of mTOR accelerates NET release | [60] |
| mTOR | Mouse | Mincle−/− mice | Mincle regulated autophagy to control NET formation | Rapamycin initiates autophagy by inhibiting mTOR and increases PMA-induced NET formation | [82] |
| PI3K, ATG5 | Human | Human neutrophils with the D39 strain of Pneumococcus | Extensive NET formation in neutrophils induced by pneumococci was critically dependent on autophagy | Pneumococci induce autophagy in neutrophils in a PI3K-dependent manner requiring ATG5 | [43] |
| ATG5, Beclin-1 | Human | Septic patients | Autophagy primed neutrophils for increased NET formation | In neutrophils, the expression of autophagy-related proteins, such as ATG5 and Beclin-1, increases during sepsis | [77] |
| ATG5 | Mouse | ATG5-deficient mice | ATG5-dependent autophagy was not required for extracellular DNA trap formation. The late autophagy inhibitors BafA1 and CQ had no effect on NETs | 3MA and wortmannin inhibit ET formation by blocking class I PI3Ks | [78] |
| ATG5, Beclin-1 | Mouse | Mouse model of sepsis | Neutrophil autophagy enhanced survival in a mouse model of sepsis via increased NET formation | Neutrophil autophagy is not normally induced in septic mice | [77] |
| PI3K | Human | Neutrophils stimulated with CAH, CALY, or CAIY | Autophagy was involved in NET release induced by C. albicans morphotypes | Pre-treatment with WT, which suppresses autophagy through the persistent inhibition of class III PI3Ks, significantly reduces NET formation | [83] |
| PI3K | Human | Patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) | Anti-LAMP-2 antibody-induced NET formation involved autophagy | LAMP-2 is a major constituent of the lysosomal membrane and plays a role in autophagy | [84] |
| PI3K | Human | Patients with gout | NET formation was associated with autophagy-related signaling in gout | PI3K inhibition prevents NET formation | [85] |
| PI3K | Human | Patients with CDG | NET-mediated cell death required both autophagy and superoxide generation | Autophagy prevents intracellular chromatin decondensation, which is essential for NETosis and NET formation | [86] |
| PI3K | Caprine | Healthy caprine | The inhibition of PMN autophagy by blocking the PI3K-mediated signaling pathway failed to influence tachyzoite-induced NETosis | Parasite-triggered NETosis is independent of NOX, SOCE, ERK1/2 and p38 MAPK activity | [87] |
| DDIT4/REDD1 | Human | Active ulcerative colitis patient and control | NETs were positively regulated by autophagy, and the autophagy inhibitors BafA1 and HCQ abolished NET release | REDD1 is a key inducer of autophagy-mediated NETosis | [88] |
| V-ATPase | Human | Patients with H. pylori-positive gastritis | Clarithromycin upregulated autophagy to promote NET formation | PMNs pretreated with BafA1 prior to stimulation with clarithromycin show reduced NET formation | [89] |
| HMGB1/RAGE | Human | Patient with acute myocardial infarction | High-mobility group protein B1 (HMGB1) promoted autophagy and primed neutrophils for NET generation | Wortmannin and 3MA abrogate NET formation elicited by activated platelets or HMGB1 | [90] |
| G6PD | Human | Taiwanese Hakka | Autophagy was involved in PMA-stimulated NET formation | ROS can elicit NETosis independent of autophagy | [91] |
| HMGB1 | Human | Neutrophils in thrombosis | Tissue factor (TF) localized in autophagosomes prior to extracellular delivery via NETs | Autophagy functions as a secretory mechanism for the externalization of membrane-bound or cytosolic proteins in NETs | [72] |
| HMGB1 | Human | Patients with Gram-negative sepsis | Autophagy mediated the delivery of TF to NETs in sepsis patients | Autophagy regulates the translocation of certain neutrophil proteins, including TF and HMGB1, to NETs | [92] |
| HMGB1, RAGE | Mouse | RAGE−/− mouse model | NETs were upregulated in pancreatic cancer through RAGE-dependent autophagy pathways | Circulating HMGB1 induces autophagy intrinsically and extrinsically by binding to RAGE | [93] |
| P140 | Mouse | MRL/lpr mouse model | Neutrophil NET formation was not directly influenced by the CMA-targeting peptide P140 | P140 does not influence NET formation, cytokine/chemokine production, or CMA in neutrophils | [76] |
| NFκB | Mouse | Gulo−/− mice | Autophagy was necessary for the induction of intracellular chromatin decondensation during PMA-induced NETosis | ATG3, ATG5, ATG6, ATG7, and ATG8 mRNA levels were significantly upregulated in PMNs from VitC-deficient mice | [73] |
AAV associated vasculitis, ANCA antineutrophil cytoplasmic antibodies, CMA chaperone-mediated autophagy, HMGB1 high-mobility group protein B1, LAMP2 lysosomal-associated membrane glycoprotein 2, mTOR mammalian target of rapamycin, NETs, neutrophil extracellular traps, NFkB nuclear factor kappa-light-chain-enhancer of activated B cells, PMN polymorphonuclear neutrophil, REDD1 regulated in development and DNA damage response-1, STEMI ST-Elevation myocardial infarction, TF tissue factor, ATG autophagy-related protein, PI3K phosphoinositide 3-kinase, RAGE receptor for advanced glycation end products, FPR formyl peptide receptor, CQ chloroquine, 3MA 3-methyladenine, NOX nitrogen oxide, ERK1/2, extracellular regulated protein kinases 1/2, ROS reactive oxygen species, PMN polymorphonuclear neutrophil. FADD Fas-associated death domain
mTOR, a bridge between autophagy and NETs
The PI3K–AKT–mTOR axis connects autophagy and NET induction and has a considerably influence on both processes. mTOR is a serine/threonine kinase that regulates cellular stress, growth, and autophagy. Inactivation of mTOR by dephosphorylation of Ser-2448 downregulates autophagy [42]. Inorganic polyphosphate induces NET formation, and interferon lambda-1(IFN-λ1)/interleukin-29 (IL-29) treatment exerts the opposite effect by targeting mTOR. Rapamycin and WYE-354, autophagy inducers that inhibit mTOR, promote NET formation by autophagy downstream of formyl peptide receptor (FPR) signaling in human neutrophils [79, 80]. The increase in NET release is sensitive to the inhibition of respiratory burst and the blockade of cytoskeletal dynamics [74].
Autophagy and ROS, dependent or independent factors?
Autophagy is dependent on ROS production by NOX. NOX2-generated ROS promote antibacterial autophagy, which recruits microtubule-associated proteins 1A/1B light chain 3 (LC3) to the phagosome via inhibiting the PI3K-I–Akt–mTOR signaling pathway [94]. ROS, and more specifically O22−, stimulate cellular autophagy and accelerate intracellular pathogen clearance [81]. NETosis, a distinct form of cell death, requires autophagy and superoxide generation [95, 96]. Interestingly, both ROS and autophagy have independent effects on NET formation, and a lack of either axis results in apoptosis [74]. On the one hand, NET release depends on NOX2 activation by the Raf–MEK–ERK pathway [97]. 3MA cannot inhibit ROS-induced NET formation. On the other hand, as reported, Mincle regulates NET formation by mediating autophagy without affecting ROS [82]. Candida albicans hyphal (CAH) cells upregulate NET formation through autophagy in a ROS-independent manner in the early phase [83]. Inhibiting either autophagy or ROS results in impaired NET formation due to impeded intracellular chromatin decondensation [81, 86]; thus, we can target autophagy to regulate NET formation but preserve ROS production to maintain innate immune defense.
In summary, classical autophagy has a multilayer regulatory function in NET formation and a positive effect on NET formation, considering the metabolism of neutrophils and the kinetics of NET formation. The PI3K–AKT–mTOR axis connects autophagy and NET induction, and mTOR inhibition promotes NET formation via autophagy. Autophagy is dependent on ROS production by NOX but has an independent effect on NET formation.
Autophagy-mediated regulation of neutrophils in bacterial infection
Neutrophils are considered the most effective and abundant front-line defenders, especially at the early phase of infection. Neutrophils play a significant role in pathogen clearance through multiple antimicrobial strategies, including the exocytosis-mediated release of antimicrobial molecules, phagocytosis, neutrophil NET formation, and autophagy. Autophagy is a significant pathway for the degradation of content trapped by phagocytosis. When pathogens escape phagosomes, autophagy becomes the only weapon available to neutrophils to capture the intracellular “escaped prisoner.” Autophagy plays a role in the detection and elimination of invasive pathogens such as intracellular bacteria, viruses, and parasites [98]. During autophagy, pathogens are engulfed by the autophagosome membrane and then degraded by lysosomes. Autophagic death can reduce intracellular pathogen survival and cytokine production. Autophagy of infected neutrophils regulates cytokine production to control inflammation [99]. Autophagy and NET formation have decisive effects on the fate of neutrophils that are invaded by pathogens. Neutrophils can selectively release NETs by discriminating different bacteria, which is dependent on autophagy [100].
Xenophagy
Xenophagy is a specialized form of autophagy in which invading bacteria are recognized, captured, and killed, and it plays a significant role in the bacteriostatic activity of neutrophils. Downregulating xenophagy increases the susceptibility to bacterial infection and decreases the resistance to pathogens. As reported previously, xenophagy is vital for the α-defensin-mediated killing of Bacillus anthracis bacilli by neutrophils [101]. ATG5−/− mice are more susceptible than wild-type mice to Listeria monocytogenes and Toxoplasma gondii and show increased susceptibility to viral encephalitis [102]. Autophagy-deficient myeloid cells are more susceptible toC. albicans infection [103]. However, bacteria subvert neutrophils by manipulating xenophagy to help bacterial spread via the bloodstream [104, 105]. Pathogens counter host attacks by sabotaging xenophagy at the degradation step to prolong their lifespan. Pathogens also utilize autophagy as a mechanism to acquire nutrients [43]. In adherent-invasive Escherichia coli (AIEC)-infected neutrophils, autophagy is downregulated, which limits the antibacterial inflammatory response [106]. Burkholderia pseudomallei induces xenophagy in a T3SS-dependent manner and show significantly increased survival in neutrophils in which autophagy is inhibited [107]. Both live and dead bacteria can induce autophagy [83]. Interestingly, specific differences between live and dead bacteria are key to determining whether bacteria escape autophagy. As reported previously, autophagosomes are considered a niche for intracellular S. aureus replication, which results in pathogen dissemination. S. aureus can survive autophagy and be transported by neutrophils in the bloodstream [108]. In the study of the scavenger receptor-induced autophagy elimination of Listeria and Yersinia, autophagy often failed to eliminate live bacteria in vivo [109] (Fig. 2).
Figure 2.
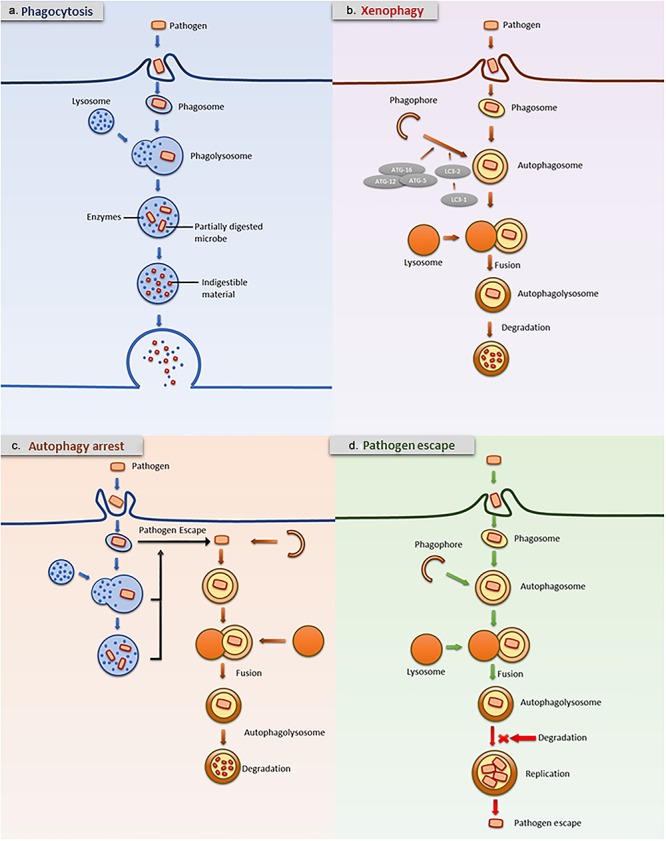
Autophagy arrest and pathogen escape. Neutrophils utilize multiple antimicrobial strategies to eliminate bacteria, including phagocytosis (a) and autophagy (b). Autophagy plays a role in detecting and eliminating invasive pathogens. In neutrophil autophagy, pathogens are engulfed by autophagosome membranes and degraded by lysosomes. Autophagy is an important pathway for the degradation of phagocytosed content. When pathogens escape phagosomes, they can be captured by autophagy (c). However, autophagy often fails to kill live bacteria. Autophagosomes are considered a niche for pathogen replication, resulting in dissemination (d). LC3 microtubule-associated protein 1 light chain 3, ATG autophagy-related protein
Macroautophagy-independent effect of ATG5
There is increasing focus on the macroautophagy-independent effect of ATG5 in M. tuberculosis (Mtb) infection. ATG5 plays a special role in Mtb infection and limits intracellular bacterial growth [110]. However, the conventional autophagy pathway does not have the same effect, suggesting that autophagy is not the essential reason for restricting Mtb. Our analysis indicated two possible explanations for this observation. One explanation is that ATG5 regulates this autophagy process in a manner independent of other ATG genes. Another possible explanation is that ATG5 limits Mtb infection in an autophagy-independent manner. ATG5 is involved in other vesicle-trafficking processes, including endocytosis, protein secretion, and LC3-associated phagocytosis [111, 112]. Proteolytic cleavage of ATG5 induces apoptosis [108]. ATG5 associates with BCL-XL and Fas-associated death domain (FADD) to regulate cell death, which is linked to inflammation and infection [113, 114]. ATG5 also interacts with many proteins involved in pathogenesis [115]. Further research is needed to reach the final conclusion.
Autophagy plays a role in the detection and elimination of invasive pathogens. Intracellular bacteria, viruses, and parasites are involved. Autophagy induces autophagic death to reduce intracellular pathogen survival and cytokine production. Bacteria subvert neutrophils by manipulating xenophagy to increase their spread via the bloodstream. Autophagy often fails to eliminate live bacteria in vivo. ATG5 plays a unique role in Mtb infection and limits intracellular bacterial growth. Neutrophil autophagy is now considered a novel therapeutic target in infection.
Neutrophil autophagy and aseptic inflammation
Neutrophils are the first line of innate immunity and migrate to sites of inflammation upon activation. Neutrophil autophagy is involved in this biological process. As reported, autophagy can be activated under aseptic inflammatory conditions, as evidenced by strong LC3 immunoreactivity [99, 116]. The relationship between neutrophil autophagy and inflammation is intricate [117]. Autophagy is a protective mechanism in neutrophil-mediated inflammation, and blocking autophagy may cause uncontrolled inflammation [118]. Autophagy decreases cytokine production and downregulates neutrophil influx to control inflammation [119, 120]. Autophagy limits degranulation and affects ROS generation, resulting in the downregulation of apoptosis and influencing neutrophil tissue infiltration [109]. Many signaling molecules are involved in both autophagy and apoptosis and regulate the balance between these two processes, as described previously in detail [119].
Neutrophil autophagy and cancer
Neutrophils are significantly involved in the pathophysiology of cancer and differentiate into anti-tumor neutrophils (N1) and pro-tumor neutrophils (N2) neutrophils, two subsets with pro-tumorigenic and tumor suppressor effects during oncogenesis [121]. Similar to neutrophil subsets, neutrophil autophagy also has pro-tumorigenic and anti-tumorigenic functions depending on the tumor environment. Neutrophil autophagy influences the progression of solid tumors. For example, in human hepatocellular carcinoma (HCC), neutrophil autophagy is activated by extracellular regulated protein kinases 1/2 (ERK1/2), p38, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. The upregulation of autophagy maintains mitochondrial stabilization and sustains neutrophil survival in the tumor microenvironment. Neutrophil autophagy promotes tumor growth by increasing the levels of the pro-metastatic proteins oncostatin M (OSM) and MMP-9 and promoting cancer cell migration [122]. However, in hematological malignancies, neutrophil autophagy is essential for blocking disease progression. In acute myelocytic leukemia (AML), autophagy is essential for neutrophil differentiation. Impairing autophagy limits neutrophil degranulation and decreases the release of inflammatory molecules [52, 54]. Neutrophil autophagy may be a novel target for oncotherapy, but the goal of upregulating or downregulating neutrophil autophagy as a therapeutic depends on the type of tumor.
Clinical applications
Neutrophil autophagy has clinical relevance in many pathologies, including inflammation, cancer and, infectious diseases. So neutrophil autophagy is placed under the spotlight for pharmacologists and clinicians [11]. The pharmacological regulation of neutrophil autophagy may represent a novel strategy for the treatment of certain diseases [6] (Table 2).
Table 2.
Pharmacologic regulation of neutrophil autophagy and potential applications
| Pharmaceutical | Associated mechanism | Potential application | Reference |
|---|---|---|---|
| IVIG | Upregulates autophagy | Infectious diseases | [123] |
| Vitamin D | Upregulates autophagy | Tuberculosis | [124] |
| Carbamazepine | Upregulates autophagy | Infectious diseases | [124] |
| Valproic acid | Upregulates autophagy | Infectious diseases | [125] |
| Celastrol | Upregulates autophagy | Colitis | [126] |
| Rapamycin | Inhibits mTOR and upregulates autophagy | Cigarette smoke exposure | [119] |
| PolyP | Inhibits mTOR and upregulates autophagy | Arterial STEMI thrombi | [42] |
| SLC37A4 | Inhibits mTOR and upregulates autophagy | Glycogen storage disease | [127] |
| Clarithromycin | Upregulates autophagy and increases NET formation | Infectious diseases | [89] |
| NLRP32/2 and NLRP3 inhibitor | Downregulates autophagy and upregulates phagocytosis | Sepsis | [128] |
| Ethyl pyruvate | Downregulates autophagy and granule release | Lipopolysaccharide-induced acute lung injury | [129] |
| Low-molecular-weight heparin (LMWH) | Inhibits autophagy and NET formation | Inflammation | [130] |
| Bafilomycin A1 and HCQ | Inhibits autophagy and NET formation | Active ulcerative colitis | [88] |
| Chloroquine (CQ) | Inhibits autophagy and NET formation | Pancreatic cancer | [93] |
| 3-Methyladenine (3MA) | Downregulates autophagy and apoptosis | ANCA-associated vasculitis | [84] |
| 2-Morpholin-4-yl-8-phenyl-chromen-4-one | Downregulates autophagy and apoptosis | ANCA-associated vasculitis | [84] |
3MA 3-methyladenine, CQ chloroquine, IVIG intravenous immunoglobulin, LMWH low-molecular-weight heparin, mTOR mammalian target of rapamycin, NLRP32/2 NLR family pyrin domain containing 32/2, NLRP3 NLR family pyrin domain containing 3, SLC37A4 solute carrier family 37 member 4, NET neutrophil extracellular trap, STEMI ST-Elevation myocardial infarction, ANCA antineutrophil cytoplasmic antibodies
Infectious diseases
Neutrophil autophagy is now considered a novel therapeutic target in infection. The majority of pharmacological agents stimulate autophagy, including intravenous immunoglobulin (IVIG), clarithromycin, vitamin D, carbamazepine, and valproic acid. IVIG boosts the ability of neutrophils to kill multidrug-resistant bacteria and eliminate drug-sensitive strains by stimulating autophagy. Inhibiting autophagy weakens the antibacterial activity of neutrophils [123, 131]. Clarithromycin plays an immunomodulatory role as an inducer of neutrophil autophagy to amplify NET formation [89]. Vitamin D can prevent exacerbated inflammation by upregulating autophagy to suppress the release of pro-inflammatory cytokines during Mtb infection [132]. Carbamazepine and valproic acid are autophagy inducers that show similar effects [133]. NLR family pyrin domain containing 32/2 (NLRP32/2) and NLR family pyrin domain containing 3 (NLRP3) inhibitors downregulate autophagy and upregulate phagocytosis to improve bacterial clearance in polymicrobial sepsis [128].
Respiratory diseases
In respiratory diseases, neutrophil autophagy plays a crucial role in smoking, acute lung injury, emphysematous, asthma, and fibrosis. Increased neutrophil autophagy is a marker of smoke exposure and a protective mechanism of the lung. The mTOR inhibitor rapamycin increases autophagy in neutrophils and reduces alveolar inflammation during cigarette smoke exposure [119]. Ethyl pyruvate is a novel potential treatment for lipopolysaccharide-induced acute lung injury that inhibits neutrophil autophagy and downregulates granule release to alleviate disease progression [129]. IFN regulatory factor 3 (IRF3) activates autophagy, phagocytosis and apoptosis in neutrophils, and the IRF3 pathway is considered to ameliorate the lipopolysaccharide-triggered exacerbation of emphysematous [48]. Autophagy prevents NF-κB activation, and the inhibition of autophagy reduces lung injury after prolonged low-pressure mechanical ventilation [134]. In asthma patients, autophagy augments the effects of immune cells, including neutrophils, and increases asthma severity [135]. Inhibiting autophagy can reduce neutrophil migration, and autophagy is becoming a new therapeutic target in corticosteroid-resistant asthma patients [136]. In pulmonary fibrosis patients, neutrophil autophagy is a target for controlling fibrosis by regulating NET formation [137].
Cardiovascular diseases
Neutrophil autophagy is a novel therapeutic target in cardiovascular disease, including arterial ST-Elevation myocardial infarction (STEMI) thrombi and antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis. In arterial STEMI thrombi, autophagy regulates NETs, which contribute to disease development. mTOR inhibition by polyP counteracts the effects of IFN-λ1/IL-29 treatment, resulting in the inhibition of NET formation. IL-29 and polyP are important regulators of thromboinflammation that control neutrophil autophagy [42]. Low-molecular-weight heparin (LMWH) inhibits autophagy and NET formation in activated neutrophils to mobilize granule content. LMWH is the preferred treatment for inflammation, and autophagy provides the theoretical basis for the use of this agent [130]. In ANCA-associated vasculitis, the autophagy inhibitors 3MA and 2-morpholin-4-yl-8-phenyl-chromen-4-one can attenuate the decreased apoptosis rate. NET formation and autophagy are associated during autoimmune attacks [84]. In thrombosis, autophagosomes engulf tissue factor (TF) before it is delivered extracellularly in NETs. HMGB1, a component of the autophagy pathway, is involved in the externalization of membrane-bound and cytosolic proteins in NETs [72].
Endocrine diseases
The regulation of neutrophil autophagy has shown a considerable curative effect in endocrine diseases. In diabetes, the increases in apoptosis and ROS production in neutrophils may be related to the suppression of autophagy by mTOR. In a diabetic rat model, neutrophils expressed low levels of autophagy-related molecules such as LC3B and had few punctate structures labelled as autophagosomal membranes. Autophagy is considered to regulate neutrophil-induced inflammation by reducing MPO activity and ROS production [138]. Glycogen storage disease type Ib (GSD-Ib), an autosomal recessive glycogen storage disorder with a phenotype of metabolic and myeloid defects, is caused by glucose-6-phosphate translocase (G6PT) deficiency and results in severe congenital neutropenia. Solute carrier family 37 member 4(SLC37A4) and G6PT are key activators of neutrophil autophagy that negatively regulate mTORC1. SLC37A4 increases the interaction between N-terminal Venus-tagged ULK1 (ULK1-VN) and C-terminal Venus-tagged ATG9 (ATG9-VC) to affect mTORC1 function through calcium mobilization. Neutrophil autophagy may be a potential target for the treatment of neutropenia in GSD-Ib [127]. In fasted volunteers, neutrophils activated autophagy to counteract the nutrient deprivation, whereas leukocyte subpopulations showed only a significant increase in the number of LC3B+ puncta. Therefore, neutrophil autophagy is an indicator of autophagy at the whole-body level under starved pathological conditions [139].
Digestive diseases
Neutrophil autophagy is an effective therapeutic target in digestive inflammation and cancer. In active ulcerative colitis, autophagy is thought to control neutrophil-driven inflammation. Regulated in development and DNA damage response-1 (REDD1), a component of the autophagy pathway, was reported to be a vital inducer of autophagy-mediated NETosis and NET-associated IL-1β release. The autophagy inhibitors BafA1 and HCQ are used to negatively regulate NET release [88]. In pancreatic cancer, inhibiting autophagy with CQ or genetic ablation of RAGE decreased the propensity for NET formation, serum DNA levels, and citrullinated histone H3 expression. Patients treated with an autophagy inhibitor show decreased NET formation in the pancreatic tumor microenvironment and in peripheral blood, leading to lesion control [93]. Autophagy defects are an important factor in the pathogenesis of Crohn’s disease (CD), and autophagy plays a crucial role in host defense against infection. As reported previously, the ATG16L1 variant T300 AD decreases phagocytic and bactericidal activities, resulting in CD [140]. Celastrol ameliorates inflammation via the PI3K–Akt–mTOR pathway, which is implicated in autophagy [126].
Conclusion
Autophagy has received considerable attention in the field of cellular immunology over the past five years. Autophagy evolved as a metabolic and quality control system in neutrophils; it is a first line of defense against microbial infection and affects many aspects of neutrophil biology, including differentiation, lifespan, degranulation, and NET formation. Neutrophil autophagy is an attractive target for the development of new treatments of multiple diseases, including inflammation, cancer and infectious diseases. However, the existing understanding of neutrophil autophagy is still in its infancy, and the explicit functions of ATG proteins and the particular regulatory mechanisms underlying autophagy on neutrophils are still being explored. Whether every component of autophagy is involved in NET formation and whether all the components of canonical autophagy participate in xenophagy remain unknown. It is difficult to determine whether the induction or suppression of autophagy is better for pathogen clearance from neutrophils. The mechanism by which bacteria inhibit autophagosome–lysosome fusion and the involved bacterial effectors still need to be fully elucidated. Further research is needed to clarify the hypotheses regarding the detailed function of autophagy in neutrophils.
Acknowledgments
Not applicable.
Declarations
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81772135 and No. 81471903) and the Jiangsu Natural Science Foundation (No. BE2017695).
Availability of data and material
Not applicable.
Authors’ contributions
All authors contributed to the literature search, drafted the article, and contributed important intellectual content. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest
The authors declare that they have no competing financial interests.
References
- 1. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011;11:519–31. [DOI] [PubMed] [Google Scholar]
- 2. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 2011;12:1035–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 2016;16:378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol 2014;15:602–11. [DOI] [PubMed] [Google Scholar]
- 5. Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity 2017;46:15–28. [DOI] [PubMed] [Google Scholar]
- 6. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:1845–6. [DOI] [PubMed] [Google Scholar]
- 7. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011;27:107–32. [DOI] [PubMed] [Google Scholar]
- 8. Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 2011;13:7–12. [DOI] [PubMed] [Google Scholar]
- 9. Mizushima N. Autophagy: process and function. Genes Dev 2007;21:2861–73. [DOI] [PubMed] [Google Scholar]
- 10. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 2014;12:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013;13:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gong L, Devenish RJ, Prescott M. Autophagy as a macrophage response to bacterial infection. IUBMB Life 2012;64:740–7. [DOI] [PubMed] [Google Scholar]
- 13. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41. [DOI] [PubMed] [Google Scholar]
- 14. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 2018;19:349–64. [DOI] [PubMed] [Google Scholar]
- 15. Narni-Mancinelli E, Soudja SM, Crozat K, Dalod M, Gounon P, Geissmann F, et al. Inflammatory monocytes and neutrophils are licensed to kill during memory responses in vivo. PLoS Pathog 2011;7:e1002457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008;181:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009;5:973–9. [DOI] [PubMed] [Google Scholar]
- 19. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008;19:5360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009;11:385–96. [DOI] [PubMed] [Google Scholar]
- 21. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008;19:2092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013;15:741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 2015;59:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2013;2:e00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010;141:656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013;495:389–93. [DOI] [PubMed] [Google Scholar]
- 27. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009;11:1433–7. [DOI] [PubMed] [Google Scholar]
- 28. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010;12:747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: Essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci 2015;128:207–17. [DOI] [PubMed] [Google Scholar]
- 30. Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012;23:1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006;119:3888–900. [DOI] [PubMed] [Google Scholar]
- 32. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000;408:488–92. [DOI] [PubMed] [Google Scholar]
- 33. Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 2000;151:263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol 2015;16:1014–24. [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 1998;273:33889–92. [DOI] [PubMed] [Google Scholar]
- 36. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004;36:2503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007;130:165–78. [DOI] [PubMed] [Google Scholar]
- 38. Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004;117:4837–48. [DOI] [PubMed] [Google Scholar]
- 39. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012;151:1256–69. [DOI] [PubMed] [Google Scholar]
- 40. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000;406:902–6. [DOI] [PubMed] [Google Scholar]
- 41. Eskelinen EL, Illert AL, Tanaka Y, Schwarzmann G, Blanz J, Von Figura K, et al. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell 2002;13:3355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chrysanthopoulou A, Kambas K, Stakos D, Mitroulis I, Mitsios A, Vidali V, et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J Pathol 2017;243:111–22. [DOI] [PubMed] [Google Scholar]
- 43. Ullah I, Ritchie ND, Evans TJ. The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun 2017;23:413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014;15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ferguson TA, Green DR. Autophagy and phagocytosis converge for better vision. Autophagy 2014;10:165–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Borregaard N. Neutrophils, from marrow to microbes. Immunity 2010;33:657–70. [DOI] [PubMed] [Google Scholar]
- 47. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 2017;47:466–80 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ishii T, Hosoki K, Nikura Y, Yamashita N, Nagase T, Yamashita N. IFN regulatory factor 3 potentiates emphysematous aggravation by lipopolysaccharide. J Immunol 2017;198:3637–49. [DOI] [PubMed] [Google Scholar]
- 49. Rozman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ 2015;22:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leveque-El Mouttie L, Vu T, Lineburg KE, Kuns RD, Bagger FO, Teal BE, et al. Autophagy is required for stem cell mobilization by G-CSF. Blood 2015; 125: 2933–6. [DOI] [PubMed] [Google Scholar]
- 51. Trocoli A, Mathieu J, Priault M, Reiffers J, Souquere S, Pierron G, et al. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011;7:1108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin J, Britschgi A, Schlafli AM, Humbert M, Shan-Krauer D, Batliner J, et al. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid Med Cell Longev 2018;2018:1482795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brigger D, Proikas-Cezanne T, Tschan MP. WIPI-dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis 2014;5:e1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haimovici A, Brigger D, Torbett BE, Fey MF, Tschan MP. Induction of the autophagy-associated gene MAP1S via PU.1 supports APL differentiation. Leuk Res 2014;38:1041–7. [DOI] [PubMed] [Google Scholar]
- 55. Humbert M, Mueller C, Fey MF, Tschan MP. Inhibition of damage-regulated autophagy modulator-1 (DRAM-1) impairs neutrophil differentiation of NB4 APL cells. Leuk Res 2012;36:1552–6. [DOI] [PubMed] [Google Scholar]
- 56. Dyugovskaya L, Berger S, Polyakov A, Lavie P, Lavie L. Intermittent hypoxia affects the spontaneous differentiation in vitro of human neutrophils into long-lived Giant phagocytes. Oxid Med Cell Longev 2016;2016:9636937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S, et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012;17:810–20. [DOI] [PubMed] [Google Scholar]
- 58. Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 2010;17:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu H, et al. Endoplasmic reticulum stress of neutrophils is required for ischemia/reperfusion-induced acute lung injury. J Immunol 2015;195:4802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y, et al. Aging-related Atg5 defect impairs neutrophil extracellular traps formation. Immunology 2017;151:417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pliyev BK, Menshikov M. Differential effects of the autophagy inhibitors 3-methyladenine and chloroquine on spontaneous and TNF-alpha-induced neutrophil apoptosis. Apoptosis 2012;17:1050–65. [DOI] [PubMed] [Google Scholar]
- 62. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside job: Staphylococcus aureus host–pathogen interactions. Int J Med Microbiol 2018;308:307–24. [DOI] [PubMed] [Google Scholar]
- 64. Lavie L, Dyugovskaya L, Polyakov A, Rogovoy O, Leder E. Development and identification of a novel subpopulation of human neutrophil-derived Giant phagocytes in vitro. J Vis Exp 2017;119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dyugovskaya L, Berger S, Polyakov A, Lavie L. The development of giant phagocytes in long-term neutrophil cultures. J Leukoc Biol 2014;96:511–21. [DOI] [PubMed] [Google Scholar]
- 66. Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep 2015;12:1731–9. [DOI] [PubMed] [Google Scholar]
- 67. Borregaard N. Development of neutrophil granule diversity. Ann N Y Acad Sci 1997;832:62–8. [DOI] [PubMed] [Google Scholar]
- 68. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 2012;30:459–89. [DOI] [PubMed] [Google Scholar]
- 69. Cools-Lartigue J, Spicer J, Najmeh S, Ferri L. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci 2014;71:4179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cooper PR, Palmer LJ, Chapple IL. Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol 2000 2013;63:165–97. [DOI] [PubMed] [Google Scholar]
- 71. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 2011;18:581–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis—the journey of TF through NETs. Front Immunol 2012;3:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA 3rd, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients 2013;5:3131–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Itakura A, McCarty OJ. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol 2013;305:C348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Murai M. Neutrophil cell death in response to infection and its relation to coagulation. J Intensive Care 2013;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maueroder C, Schall N, Meyer F, Mahajan A, Garnier B, Hahn J, et al. Capability of neutrophils to form NETs is not directly influenced by a CMA-targeting peptide. Front Immunol 2017;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med 2017;196:577–89. [DOI] [PubMed] [Google Scholar]
- 78. Germic N, Stojkov D, Oberson K, Yousefi S, Simon HU. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology 2017;152:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sha LL, Wang H, Wang C, Peng HY, Chen M, Zhao MH. Autophagy is induced by anti-neutrophil cytoplasmic abs and promotes neutrophil extracellular traps formation. Innate Immun 2016;22:658–65. [DOI] [PubMed] [Google Scholar]
- 80. Wang L, Law HK. The role of autophagy in lupus nephritis. Int J Mol Sci 2015;16:25154–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pan Q, Gao C, Chen Y, Feng Y, Liu WJ, Liu HF. Update on the role of autophagy in systemic lupus erythematosus: s novel therapeutic target. Biomed Pharmacother 2015;71:190–3. [DOI] [PubMed] [Google Scholar]
- 82. Sharma A, Simonson TJ, Jondle CN, Mishra BB, Sharma J. Mincle-mediated neutrophil extracellular trap formation by regulation of autophagy. J Infect Dis 2017;215:1040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol 2016;7:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tang S, Zhang Y, Yin SW, Gao XJ, Shi WW, Wang Y, et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin Exp Immunol 2015;180:408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS One 2011;6:e29318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2011;21:290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Villagra-Blanco R, Silva LMR, Gartner U, Wagner H, Failing K, Wehrend A, et al. Molecular analyses on Neospora caninum-triggered NETosis in the caprine system. Dev Comp Immunol 2017;72:119–27. [DOI] [PubMed] [Google Scholar]
- 88. Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, et al. REDD1/autophagy pathway is associated with neutrophil-driven IL-1beta inflammatory response in active ulcerative colitis. J Immunol 2018;200:3950–61. [DOI] [PubMed] [Google Scholar]
- 89. Konstantinidis T, Kambas K, Mitsios A, Panopoulou M, Tsironidou V, Dellaporta E, et al. Immunomodulatory role of clarithromycin in Acinetobacter baumannii infection via formation of neutrophil extracellular traps. Antimicrob Agents Chemother 2016;60:1040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 2014;12:2074–88. [DOI] [PubMed] [Google Scholar]
- 91. Cheng ML, Ho HY, Lin HY, Lai YC, Chiu DT. Effective NET formation in neutrophils from individuals with G6PD Taiwan-Hakka is associated with enhanced NADP(+) biosynthesis. Free Radic Res 2013;47:699–709. [DOI] [PubMed] [Google Scholar]
- 92. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One 2012;7:e45427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther 2015;22:326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: s nexus of cellular homeostasis. Redox Biol 2015;6:472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Manda-Handzlik A, Bystrzycka W, Wachowska M, Sieczkowska S, Stelmaszczyk-Emmel A, Demkow U, et al. The influence of agents differentiating HL-60 cells toward granulocyte-like cells on their ability to release neutrophil extracellular traps. Immunol Cell Biol 2018;96:413–25. [DOI] [PubMed] [Google Scholar]
- 96. Suzuki E, Maverakis E, Sarin R, Bouchareychas L, Kuchroo VK, Nestle FO, et al. T cell-independent mechanisms associated with neutrophil extracellular trap formation and selective autophagy in IL-17A-mediated epidermal hyperplasia. J Immunol 2016;197:4403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Paiva CN, Bozza MT. Are reactive oxygen species always detrimental to pathogens? Antioxid Redox Signal 2014;20:1000–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chargui A, El May MV. Autophagy mediates neutrophil responses to bacterial infection. APMIS 2014;122:1047–58. [DOI] [PubMed] [Google Scholar]
- 99. Avagliano L, Massa V, Zullino S, Doi P, Marconi AM, Ferrazzi E, et al. Inflammation modulates LC3 expression in human preterm delivery. J Matern Fetal Neonatal Med 2017;30:698–704. [DOI] [PubMed] [Google Scholar]
- 100. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol 2016;7:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ramachandran G, Gade P, Tsai P, Lu W, Kalvakolanu DV, Rosen GM, et al. Potential role of autophagy in the bactericidal activity of human PMNs for Bacillus anthracis. Pathog Dis 2015;73:ftv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wang C, Mendonsa GR, Symington JW, Zhang Q, Cadwell K, Virgin HW, et al. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci U S A 2012;109:11008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun 2015;6:5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Torraca V, Mostowy S. Zebrafish infection: from pathogenesis to cell biology. Trends Cell Biol 2018;28:143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. O'Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of autophagy in phagocytes facilitates Staphylococcus aureus bloodstream infection. Infect Immun 2015;83:3445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One 2012;7:e51727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rinchai D, Riyapa D, Buddhisa S, Utispan K, Titball RW, Stevens MP, et al. Macroautophagy is essential for killing of intracellular Burkholderia pseudomallei in human neutrophils. Autophagy 2015;11:748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Horn J, Stelzner K, Rudel T, Fraunholz M. Inside job: Staphylococcus aureus host–pathogen interactions. Int J Med Microbiol 2018;308:607–24. [DOI] [PubMed] [Google Scholar]
- 109. Pfeiler S, Khandagale AB, Magenau A, Nichols M, Heijnen HF, Rinninger F, et al. Distinct surveillance pathway for immunopathology during acute infection via autophagy and SR-BI. Sci Rep 2016;6:34440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015;528:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol 2012;22:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007;450:1253–7. [DOI] [PubMed] [Google Scholar]
- 113. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006;8:1124–32. [DOI] [PubMed] [Google Scholar]
- 114. Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012;287:12455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010;466:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhu L, Yang J, Zhang J, Peng B. The presence of autophagy in human periapical lesions. J Endod 2013;39:1379–84. [DOI] [PubMed] [Google Scholar]
- 117. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med 2017;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol 2017;14:963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Xu XC, Wu YF, Zhou JS, Chen HP, Wang Y, Li ZY, et al. Autophagy inhibitors suppress environmental particulate matter-induced airway inflammation. Toxicol Lett 2017;280:206–12. [DOI] [PubMed] [Google Scholar]
- 120. Gabrion A, Hmitou I, Moshous D, Neven B, Lefevre-Utile A, Diana JS, et al. Mammalian target of rapamycin inhibition counterbalances the inflammatory status of immune cells in patients with chronic granulomatous disease. J Allergy Clin Immunol 2017;139:1641–9 e6. [DOI] [PubMed] [Google Scholar]
- 121. Coffelt SB, Wellenstein MD, Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer 2016;16:431–46. [DOI] [PubMed] [Google Scholar]
- 122. Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, et al. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol 2015;62:131–9. [DOI] [PubMed] [Google Scholar]
- 123. Matsuo H, Itoh H, Kitamura N, Kamikubo Y, Higuchi T, Shiga S, et al. Intravenous immunoglobulin enhances the killing activity and autophagy of neutrophils isolated from immunocompromised patients against multidrug-resistant bacteria. Biochem Biophys Res Commun 2015;464:94–9. [DOI] [PubMed] [Google Scholar]
- 124. Vyas SP, Goswami R. Striking the right immunological balance prevents progression of tuberculosis. Inflamm Res 2017;66:1031–56. [DOI] [PubMed] [Google Scholar]
- 125. Vural A, Kehrl JH. Autophagy in macrophages: Impacting inflammation and bacterial infection. Scientifica (Cairo) 2014;2014:825463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhao J, Sun Y, Shi P, Dong JN, Zuo LG, Wang HG, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol 2015;26:221–8. [DOI] [PubMed] [Google Scholar]
- 127. Cappello AR, Curcio R, Lappano R, Maggiolini M, Dolce V. The physiopathological role of the exchangers belonging to the SLC37 family. Front Chem 2018;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol 2017;198:1253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhu Q, Wang H, Wang H, Luo Y, Yu Y, Du Q, et al. Protective effects of ethyl pyruvate on lipopolysaccharide induced acute lung injury through inhibition of autophagy in neutrophils. Mol Med Rep 2017;15:1272–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Manfredi AA, Rovere-Querini P, D'Angelo A, Maugeri N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res 2017;123:146–56. [DOI] [PubMed] [Google Scholar]
- 131. Itoh H, Matsuo H, Kitamura N, Yamamoto S, Higuchi T, Takematsu H, et al. Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. J Leukoc Biol 2015;98:107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yu X, Li C, Hong W, Pan W, Xie J. Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications. Cell Signal 2013;25:1272–8. [DOI] [PubMed] [Google Scholar]
- 133. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med 2015;7:127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lopez-Alonso I, Aguirre A, Gonzalez-Lopez A, Fernandez AF, Amado-Rodriguez L, Astudillo A, et al. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol 2013;304:L844–52. [DOI] [PubMed] [Google Scholar]
- 135. Pham DL, Kim SH, Losol P, Yang EM, Shin YS, Ye YM, et al. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J Intern Med 2016;31:375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, et al. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy 2017;47:57–70. [DOI] [PubMed] [Google Scholar]
- 137. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018;14:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol 2015;194:5407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pietrocola F, Demont Y, Castoldi F, Enot D, Durand S, Semeraro M, et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy 2017;13:567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gutierrez A, Scharl M, Sempere L, Holler E, Zapater P, Almenta I, et al. Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn's disease. Gut 2014;63:272–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.