

Abstract
The HIV-1 nucleocapsid (NC) protein represents an excellent molecular target for the development of antiretrovirals by virtue of its well-characterized chaperone activities, which play pivotal roles in essential steps of the viral life cycle. Our ongoing search for candidates able to impair NC binding/annealing activities led to the identification of peptidyl-anthraquinones as a promising class of nucleic acid ligands. Seeking to elucidate the inhibition determinants and increase the potency of this class of compounds, we have now explored the effects of chirality in the linker connecting the planar nucleus to the basic side chains. We show here that the non-natural linker configuration imparted unexpected TAR RNA targeting properties to the 2,6-peptidyl-anthraquinones and significantly enhanced their potency. Even if the new compounds were able to interact directly with the NC protein, they manifested a consistently higher affinity for the TAR RNA substrate and their TAR-binding properties mirrored their ability to interfere with NC-TAR interactions. Based on these findings, we propose that the viral Tat protein, sharing the same RNA substrate but acting in distinct phases of the viral life cycle, constitutes an additional druggable target for this class of peptidyl-anthraquinones. The inhibition of Tat-TAR interaction for the test compounds correlated again with their TAR-binding properties, while simultaneously failing to demonstrate any direct Tat-binding capabilities. These considerations highlighted the importance of TAR RNA in the elucidation of their inhibition mechanism, rather than direct protein inhibition. We have therefore identified anti-TAR compounds with dual in vitro inhibitory activity on different viral proteins, demonstrating that it is possible to develop multitarget compounds capable of interfering with processes mediated by the interactions of this essential RNA domain of HIV-1 genome with NC and Tat proteins.
Graphical Abstract

INTRODUCTION
Current anti-HIV therapeutics tend to exhibit desirable potency and selectivity, as well as undesirable clinical limitations, which stem from putative toxic effects associated with long-term treatments and the relentless emergence of resistant strains. As a consequence, there is an urgent need to develop new therapeutic agents against essential viral determinants that are not currently targeted by available drugs.1–3 The HIV-1 nucleocapsid (NC) protein represents an attractive and promising target by virtue of its highly conserved nature among viral clades and its vital roles in a range of processes of the viral lifecycle.4 This relatively small, basic protein is characterized by two CCHC-type zinc-finger domains that are highly conserved and confer the ability to directly interact with viral nucleic acids and to catalyze essential structural rearrangements.4 In particular, its interaction with susceptible substrates can induce transient melting of base-pairing, which makes previously paired strands available for reannealing in different patterns conducive to more thermodynamically stable conformations.5–10 An example of this activity is provided by the rearrangement of the transactivation responsive element (TAR), an essential domain of the viral genome, which is characterized by a relatively stable bulge–loop structure (Figure 1). Annealing of TAR RNA to a reverse-transcribed complementary DNA (cTAR, Figure 1) is an obligatory step of the reverse transcription process of viral genome. This operation is mediated by NC, which is responsible for melting the secondary structures of TAR and cTAR and promoting the formation of the more stable heteroduplex TAR/cTAR (Figure 1), thus allowing for the minus strand-transfer and reverse transcription processes to proceed.11
Figure 1.
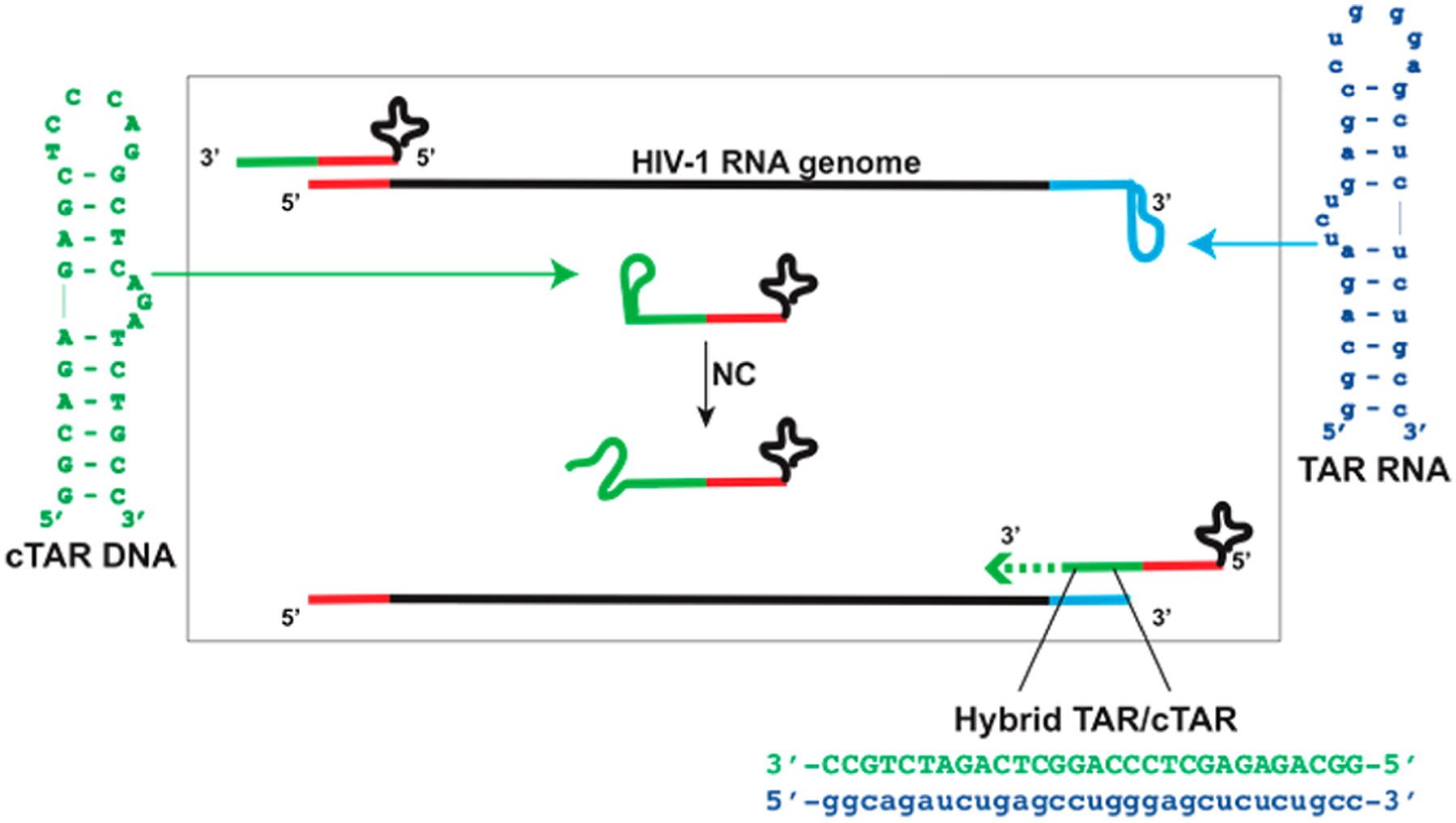
Constructs replicating TAR RNA (blue) and cTAR DNA (green) employed in the study. The scheme describes a key step of the reverse transcription of viral RNA, in which NC destabilizes these stem-loop structures to promote their annealing into a stable TAR/cTAR heteroduplex.
These essential chaperone properties make NC a potential target for ligands capable of either preventing its interaction with the target substrate or stabilizing the substrate structure to forestall the expected rearrangements. Based on this hypothesis, we identified 2,6-dipeptidyl anthraquinones (2,6-AQ) as a class of potent anti-NC agents in vitro.12–14 The chemical structure of these compounds is characterized by an anthraquinone nucleus bearing aminoacyl linkers at positions 2 and 6, which connect to terminal anchor groups containing a set of different positively charged moieties (e.g., linear or cyclic alkyl chains, as well as heterocyclic or aminophenyl chains, Figure 2). Acting as threading intercalators, we proved that these compounds interfere with NC activity by stabilizing the rather dynamic structures of TAR and cTAR.12,13
Figure 2.
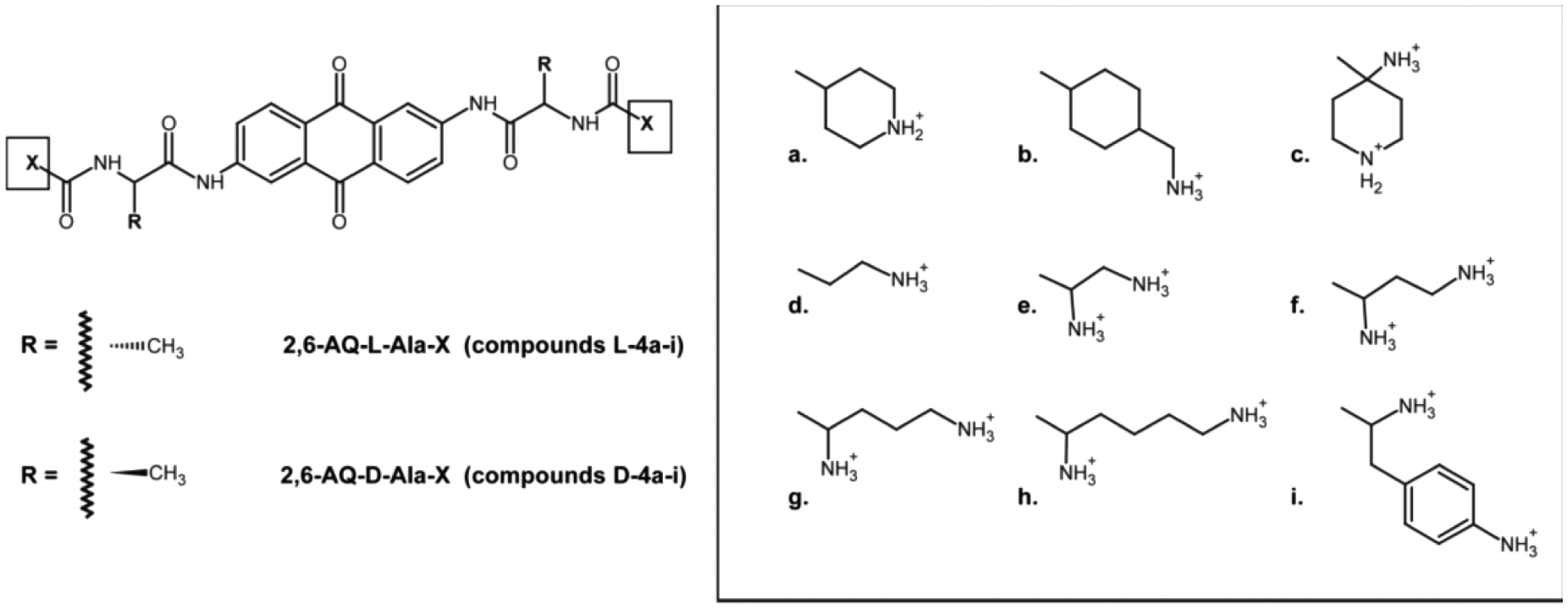
Newly synthesized 2,6-dipeptidyl anthraquinones considered in this study (i.e., 2,6-AQ-d-Ala-X, D-4a-i). For reference purposes, the chemical structures of compounds investigated earlier are also provided (i.e., 2,6-AQ-l-Ala-X, L-4a-i).12
A thorough structure–activity relationship (SAR) analysis of related series of 2,6-dipeptidyl anthraquinones, which bore a wide variety of aminoacyl linkers, identified key structural requirements necessary for the development of putative NC inhibitors12,13 The studies underscored the importance of the length of the highly basic terminal residues and the flexibility of the linear linkers connecting them to the aromatic nucleus to achieve the desired binding and stabilization of the cognate nucleic acid substrates. However, when we compared Gly and l-Ala linkers in side chains of fixed optimal length, we observed a sizable loss of inhibitory activity in spite of their similar nucleic acid binding and stabilizing properties. This observation suggested that either the linker configuration or the presence of a branching methyl group in Ala could represent important factors in determining inhibition.
In this report, we tested the significance of linker chirality by synthesizing a new complete series of 2,6-disubstituted anthraquinones, which contained Ala linker residues in the non-natural d configuration (i.e., 2,6-AQ-d-Ala-X, D-4a-i, Figure 2). We then evaluated their activity in vitro by employing the same assays developed earlier to test the analogous series with opposite linker configuration (i.e., 2,6-AQ-l-Ala-X, L-4a-i, Figure 2).12 This modus operandi enabled direct comparisons with previously reported results, which allowed us to detect the sometimes subtle effects of chirality. The experiments revealed unexpected properties that suggested additional mechanisms of action for this class of compounds and, based on these findings, pointed toward alternative potential targets. The results were discussed in the context of our SAR analysis of peptidyl-anthraquinones and used to glean a possible outlook for the development of actual therapeutics from these types of structures.
CHEMISTRY
Synthesis of 2,6-AQ-d-Ala-X Anthraquinone Derivatives.
Compounds D-4a-i (Figure 2) were obtained according to the synthetic strategy summarized in Scheme 1, which included appropriate modifications of a previous procedure reported for 2,6-disubstituted anthraquinones.12 Briefly, the general strategy is based on the condensation of the 2,6-diaminoanthraquinone nucleus 1 with Fmoc-protected acyl chloride 2 in the presence of pyridine in DMF. Each reaction mixture was submitted to overnight stirring at room temperature. The resulting material was then dried and treated with 33% diethylamine in THF to remove the Fmoc protecting group. The material was successively reacted with trifluoroacetic acid in H2O (9:1; v/v) to obtain intermediate 3, which was in turn coupled with the protected amino acid of choice in the presence of HBTU and DMAP in DMF. The Fmoc and Boc protecting groups were removed by using 33% diethylamine in THF and trifluoroacetic acid in H2O (9:1; v/v), respectively, to produce the desired compounds 4a–i.
Scheme 1.
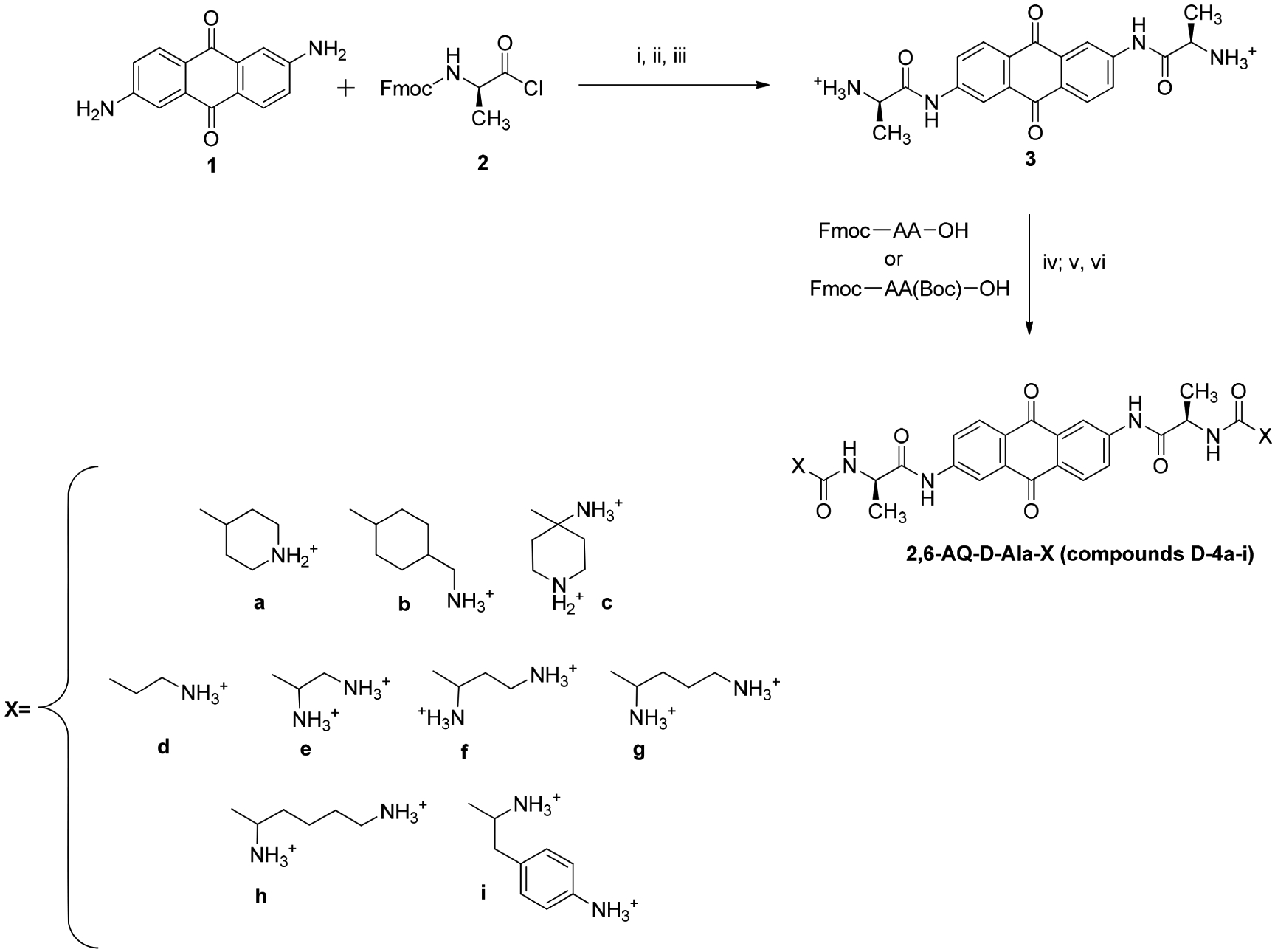
General Synthetic Pathway for the Preparation of the Anthraquinone Derivatives Included in the Studya
aReagents and conditions: (i) Pyridine, DMF; (ii) 33% Diethylamine, THF; (iii) TFA/H2O; (iv) HBTU, DMAP, DMF; (v) 33% Diethylamine, THF; (vi) TFA/H2O.
Fmoc-d-Ala-Cl (2) was prepared from commercially available Fmoc-d-Ala-OH, which was treated with an excess of thionyl chloride in CH2Cl2, and then heated by microwave irradiation in a sealed vessel for 8 min (Scheme 2). We found that increasing temperature, reaction time, or microwave power did not increase the product yield, but rather induced reagent decomposition.
Scheme 2.
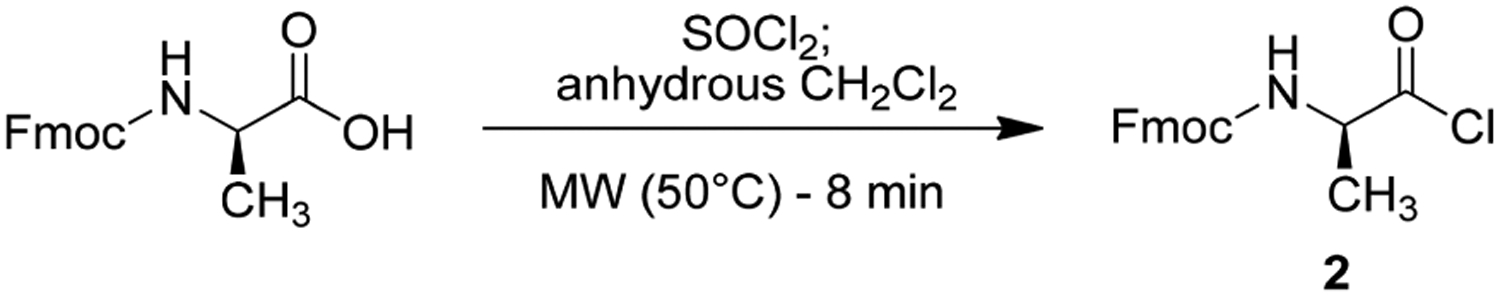
Preparation of Intermediate 2
The final compounds were purified by preparative RP-HPLC and fully characterized by mass spectrometry, 1H NMR, and 13C NMR. The data recorded for all final compounds were consistent with their proposed structures.
RESULTS AND DISCUSSION
Effects of 2,6-Dipeptidyl Anthraquinones on NC-Mediated Melting of Individual TAR and cTAR Structures.
The effects of the new series of 2,6-AQ-d-Ala-X derivatives (D-4a-i in Figure 2) on the NC-induced destabilization of individual TAR and cTAR structures were evaluated by using an adaptation of a high throughput screening (HTS) approach described earlier.12–15 Briefly, the assay employed nucleic acid substrates consisting of 29-mer constructs that replicated the apical portion of the TAR domain of viral RNA and its complementary DNA sequence (i.e., TAR and cTAR, Figure 1). The 5′ and 3′-ends of each construct were appropriately labeled with either a fluorophore or a quencher, which enabled the determination of the stem-melting activity of NC from the observed fluorescence emission. In particular, the emission recorded in the absence of ligand provided a measure of the baseline activity of NC, which minimized quenching by dissociating the double-stranded stem and increasing the distance between fluorophore and quencher. In the presence of ligand, instead, decreasing fluorescent emission was caused by inhibition of the NC’s melting activity, which left a higher proportion of substrate in the stem-loop form conducive to quenching.12,13,16 In addition to initial baseline determinations, control experiments were also carried out to ensure that the compounds of interest were incapable of inducing direct quenching in the absence of NC (see Experimental Section). A series of determinations were completed with increasing amounts of ligand to determine the concentration that induced 50% reduction of the baseline activity (expressed as IC50).12–14,16
The data obtained from the D-4a-i anthraquinones revealed that derivatives bearing doubly charged linear-alkyl side chains possessed potent inhibitory properties (e.g., D-4e-h in Table 1). In particular, the lowest IC50 values were exhibited in both TAR and cTAR determinations by the D-4g and D-4h compounds, with the latter providing 3.35 and 2.16 μM on the respective assay. Compound D-4b with a monocharged cyclic alkyl side chain showed somewhat intermediate potency with preferential activity on TAR, whereas all other compounds bearing cyclic aliphatic or aromatic moieties displayed lower potency. A close examination of these results suggested that increasing the length of the linear side-chain may increase the potency according to a D-4h > D-4g > D-4f > D-4e relative scale, which agreed with the results of our previous SAR study on compounds with either Gly or l-Ala linkers.12,13
Table 1.
Inhibition of NC-Mediated Melting of Individual TAR and cTAR Structures
| Compound | IC50 TARa (μM) | IC50 cTARa (μM) |
|---|---|---|
| D-4a | 39.7 ± 3.99 | 24.9 ± 2.20 |
| D-4b | 8.79 ± 0.11 | 12.8 ± 1.09 |
| D-4c | 51.3 ± 5.01 | 32.1 ± 2.76 |
| D-4d | 33.7 ± 2.05 | 19.9 ± 1.58 |
| D-4e | 10.5 ± 1.50 | 13.8 ± 1.50 |
| D-4f | 9.24 ± 0.49 | 6.52 ± 0.47 |
| D-4g | 3.27 ± 0.44 | 4.76 ± 0.68 |
| D-4h | 3.35 ± 0.51 | 2.16 ± 0.37 |
| D-4i | 47.4 ± 1.34 | 20.2 ± 0.78 |
Values are the mean of data obtained from three experiments, each performed in triplicate
A direct comparison between corresponding IC50 values observed for the analogous 2,6-AQ-d-Ala-X and 2,6-AQ-l-Ala-X12 series (Figure 3) revealed that compounds bearing linear terminal alkyl residues (i.e., 4d–h) were generally more potent when the linker was in the non-natural d-configuration rather than their l-configuration counterparts. This was remarkable in particular when TAR was assayed rather than cTAR (Figure S1 in Supporting Information). 2,6-AQ-d-Ala-X compounds bearing cyclic or heterocyclic side chains (4a, 4c, and 4i) displayed in all cases weaker activity than the corresponding l-Ala-X counterparts, with the exception of the aminomethyl-cyclohexyl D-4b, whose behavior on TAR in this assay clusters within the flexible linear-alkyl category. These differences between the activity of the l-Ala and the d-Ala series of compounds highlight the significant impact of the stereochemistry of the chiral amino acidic linker on the 2,6-dipeptidyl anthraquinones structure–activity relationship, therefore answering our initial question on the importance of the configuration of the connecting Ala moiety in NC inhibition.
Figure 3.
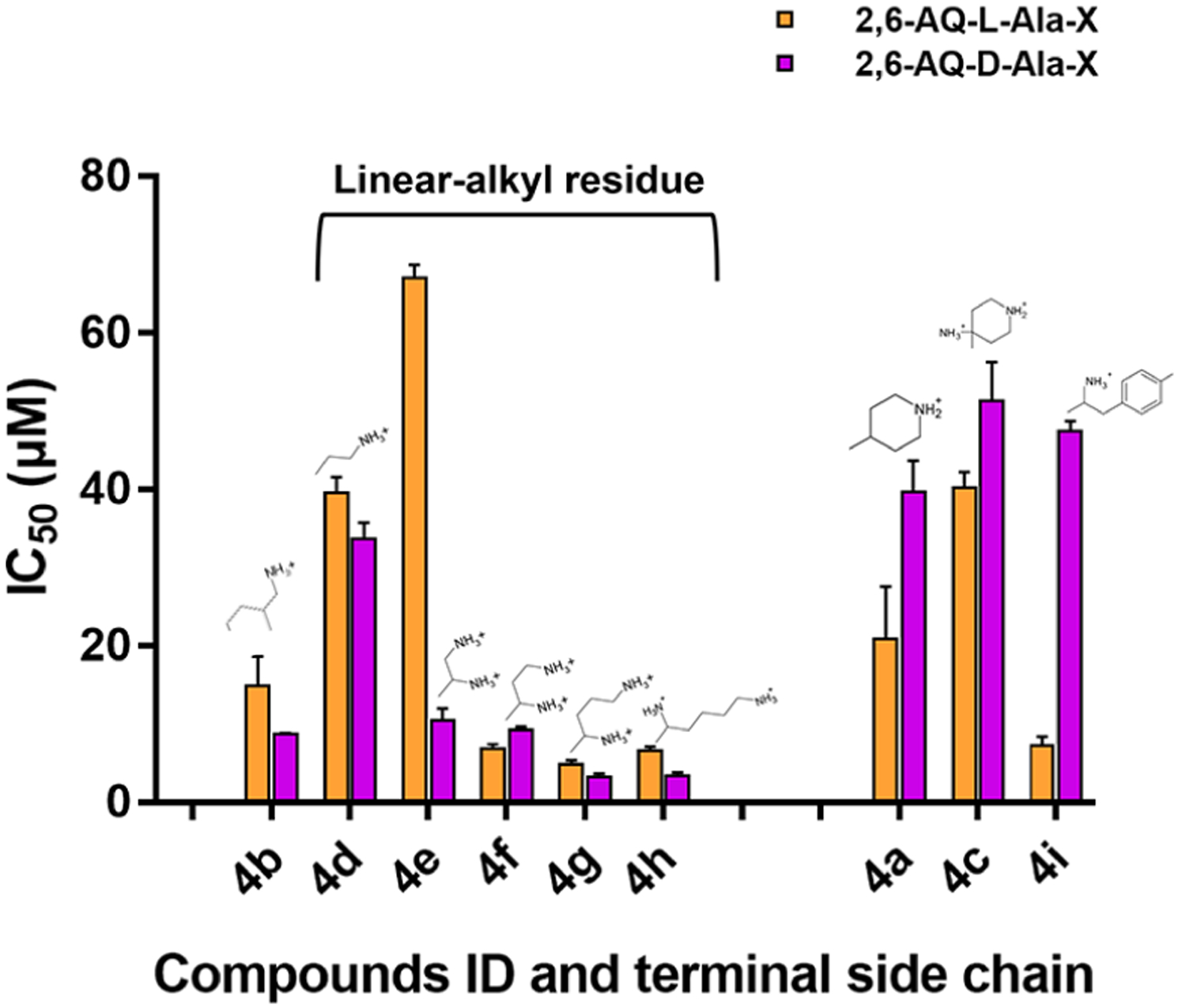
IC50 values observed for the effects of 2,6-AQ-d-Ala-X (purple, this study) and 2,6-AQ-l-Ala-X (yellow, ref 12) compounds on the NC-induced helix destabilization of TAR structure (see Experimental Section).
Effects of 2,6-Dipeptidyl Anthraquinones on NC-Mediated Annealing of TAR and cTAR Constructs.
The nucleic acid chaperone activities of NC are substantiated by its ability to destabilize double-stranded regions of RNA structure and enable their reannealing into more stable pairing arrangements. In the TAR/cTAR system, initial melting of the intrastrand pairs that define their individual stem-loop structures can lead to interstrand annealing with formation of a stable TAR/cTAR heteroduplex.11,17 The ability to monitor this process provided us with the opportunity to evaluate the effects of the 2,6-AQ-d-Ala-X anthraquinones on the annealing activity of NC in vitro. We employed the nucleocapsid annealing mediated electrophoresis (NAME) assay described earlier,12,18 which was carried out according to alternative protocols involving preincubation of ligand with either the NC protein or the nucleic acid substrates (see Experimental Section). Our tests revealed that only compounds D-4e-h possessed detectable inhibitory activity in these experiments (see Figure S2 of Supporting Information). Furthermore, d-Ala compounds were more active upon nucleic acid preincubation, in analogy with previous observations obtained from other series of peptidyl anthraquinones.12,18 Tests were repeated using a wider range of ligand concentrations to obtain IC50 values as a measure of their ability to interfere with annealing activity under the selected experimental conditions (see representative gel for D-4h in Figure S3 of Supporting Information and Table 2).
Table 2.
Inhibition of NC-Mediated Annealing of TAR/cTAR Assessed by NAME Assay
| compound | IC50 NAMEa (μM) |
|---|---|
| D-4a | >100 |
| D-4b | >100 |
| D-4c | >100 |
| D-4d | >100 |
| D-4e | 76.7 ± 8.61 |
| D-4f | 78.8 ± 10.8 |
| D-4g | 37.5 ± 13.6 |
| D-4h | 28.9 ± 4.59 |
| D-4i | >100 |
Values represent the mean and standard error of the mean (SEM) obtained from experiments performed in triplicate
The results showed that linear anthraquinones D-4e-h possessed inhibitory activities consistent with a D-4h > D-4g > D-4f ≈ D-4e relative scale of potency. Not surprisingly, this scale matched that observed for the potency of inhibition of NC-induced melting of TAR and cTAR, which was also correlated with the length of the doubly charged side chain. Compound D-4h was the most potent inhibitor of both melting and annealing activities with respective IC50s of 3.35 and 28.9 μM (Tables 1 and 2). This was not the case for its 2,6-AQ-l-Ala-X analog L-4h, which was significantly less active both in the melting and in the annealing assay (i.e., IC50 of 6.61 and 80.4 μM).12 The outcome of these experiments further indicates that the non-natural d configuration of Ala-linkers is a strong inhibition determinant of 2,6-dipeptidyl-anthraquinone compounds.
Binding Modes of 2,6-Dipeptidyl Anthraquinones to Individual TAR and cTAR Structures.
In the formerly analyzed l-Ala linker series, the NC inhibitory properties mirrored their nucleic acid binding capabilities, providing strong evidence that the mechanism of action involve stabilization of TAR and cTAR secondary structure through intercalation of their anthraquinone systems.12,13 With the new d-Ala series, we therefore evaluated the effects of varying the linker configuration on the binding features on TAR and cTAR, employing electrospray ionization mass spectrometry (ESI-MS) analysis under nondenaturing conditions as performed for the l-Ala series (see Experimental Section).12,13,19–21
Initial determinations were carried out in the absence of ligand to verify the experimental masses of the TAR and cTAR constructs, which matched very closely those calculated from the respective sequences (i.e., 9286.2 u experimental versus 9286.3 Da monoisotopic mass calculated for TAR; 8884.5 u versus 8884.5 Da for cTAR). Subsequent analyses were conducted on sample solutions that contained, respectively, 1 and 10 μM concentrations of nucleic acid substrate and 2,6-dipeptidyl anthraquinone ligand (i.e., a 1:10 molar ratio). Representative ESI-MS spectra obtained from mixtures of D-4h with either TAR or cTAR are provided in Figure 4, which displays only the regions containing the 6- or 5-charge state for the sake of clarity. Experimental and calculated monoisotopic masses for all the detected species are provided in Tables S1 and S2 of Supporting Information.
Figure 4.
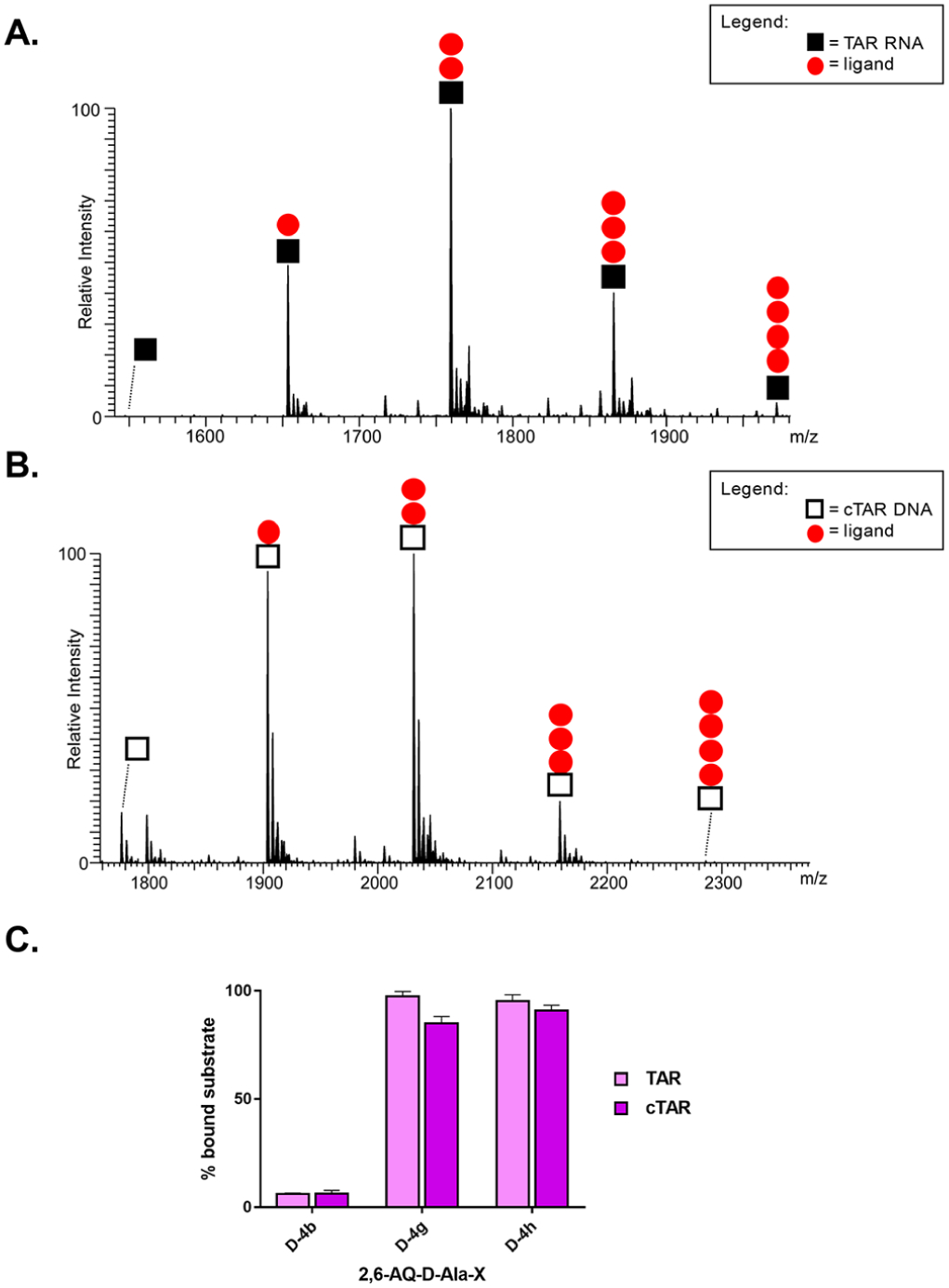
Representative ESI-MS spectra of samples containing a 1:10 molar ratio of either TAR (A) or cTAR (B) and D-4h (red ●) compound in 150 mM ammonium acetate (see Experimental Section). Symbol ◼ corresponds to TAR RNA substrate and ◻ to cTAR DNA substrate. The stoichiometries of the observed complexes are indicated in each spectrum. Signals of lower intensity detected near free/bound species consist of typical sodium and ammonium adducts. (C) Histograms displaying the percentages of bound substrate observed in A and B spectra and from Figure S4 for D-4b and D-4h. The indicated percentages of bound substrate provide a measure of the relative affinities of d-analogues for either nucleic acid substrate.
Consistent with the ability of ESI-MS to observe intact noncovalent complexes between nucleic acids and peptidyl-anthraquinones,12,14 all spectra contained signals corresponding to free unbound substrates, as well as stable complexes with different stoichiometries. The observed binding patterns did not reveal significant cooperativity for either substrate (Figures 4 and S4). In all cases, the coexistence of free TAR or cTAR with complexes of different stoichiometries indicated that binding to the second site was initiated before saturation of the first was complete, consistent with the presence of independent binding sites with equivalent affinities on either construct. The results revealed a clear correlation between binding stoichiometry and the inhibition of annealing activity revealed by the NAME assays. In particular, the D-4g and D-4h compounds, which possessed the most pronounced inhibitory properties, produced TAR complexes containing up to 3 and 4 equiv of ligand, respectively. By contrast, the D-4b compound with the least pronounced inhibitory properties produced only 1:1 complexes with TAR under the same experimental conditions. In the case of the cTAR substrate, D-4g and D-4h produced complexes with up to 4:1 stoichiometries, whereas D-4b produced again only 1:1 complexes.
These results are quite consistent with what was previously reported for the l-Ala series. We then proceeded to calculate the relative binding affinities of the various compounds for the different substrates, employing the percentage of bound substrate observed in each spectrum (see Experimental Section).12 The representative histograms in Figure 4C revealed that D-4g and D-4h possessed comparable affinities that were much greater than those afforded by D-4b under the same experimental conditions (i.e., D-4g ≈ D-4h ≫ D-4b relative scale). Surprisingly, D-4g and D-4h displayed a slightly greater affinity for the RNA than for the DNA substrate, which contrasted with the behavior manifested by the corresponding l-series analogues. In fact, the histograms obtained from the data generated in our previous studies12,13 clearly demonstrated that analogous compounds with l-Ala linker (named L-4g, L-4h, and L-4a) possessed greater affinities for cTAR than for TAR (Figure S5 of Supporting Information).
The unexpected but detectable selectivity for the RNA rather than for the DNA construct manifested by the d-analogues had never been observed before and suggested further investigation. We evaluated under the same experimental conditions the binding properties of the strong TAR binder D-4h and its l-counterpart L-4h to different RNA substrates. We selected the stem-loop SL3 sequence, a RNA domain of the HIV-1 genome packaging signal, owing to the presence of an apical loop,19,20,22 and a random 16-mer double-stranded RNA (dsRNA) sequence. As performed in the presence of TAR, ESI-MS analyses were conducted on sample solutions that contained, respectively, 1 and 10 μM concentrations of nucleic acid substrate and 2,6-dipeptidyl anthraquinone ligand (not shown). We calculated the relative binding affinities of the compounds for the different substrates, employing the percentage of bound substrate observed in each spectrum (see Experimental Section). The representative histograms in Figure S6 (see Supporting Information) compare the binding affinities of selected compounds for TAR (Figure 4C) with those observed for SL3 and for the dsRNA. Once again the d-analogue displayed a slightly greater affinity for all the three RNA substrates compared to its l-counterpart, corroborating our hypothesis that the non-natural conformation of the linker in the side chains is important to achieve RNA binding. Noteworthy, the data clearly revealed that the test compounds possess a remarkable greater binding affinities for the bulge-loop TAR structure than for both the stem-loop SL3 sequence and the double-stranded RNA, highlighting their preferential binding to dynamic bulged regions of RNA structures.
The increased selectivity toward TAR induced by the d-Ala linker series suggests a possible strategy for designing RNA-specific ligands, which deserves further investigation. In addition, it entails the potential inhibition of other factors that interact with this region of viral genome during the HIV-1 lifecycle, as we will discuss in the final paragraph.
Multifaceted Aspects of 2,6-Dipeptidyl Anthraquinone Inhibition.
Although the results obtained to this point revealed excellent correlation between efficient inhibition of NC-mediated reactions and nucleic acid binding, they do not satisfactorily explain the discrepancy between the different potencies in NC inhibition by the d- and l-Ala series. For this reason, we tested whether direct binding to NC protein by 2,6-AQ-d-Ala-X ligands could account for additional inhibitory effects. The experiments were carried out by comparing samples that contained full-length NC and selected analogues of the d- or l-series (see Experimental Section). Also in this case, ESI-MS analyses were carried out under nondenaturing conditions to enable the detection of intact complexes with fully metalated NC (see Experimental Section).23 Control experiments in the absence of ligand afforded a mass of 6489.1 u, which matched very closely the monoisotopic value of 6488.9 Da calculated from the sequence and including two Zn(II) ions. Representative spectra obtained in the presence of selected 2,6-AQ-d-Ala-X anthraquinones are shown in Figure 5, while those obtained in the presence of their 2,6-AQ-l-Ala-X counterparts are reported in Figure S7 (Supporting Information). For the sake of clarity, only the regions corresponding to the 4+ charge states were plotted on the same intensity scale to enable a direct comparison of the abundances of the respective complexes. Observed and expected masses for all the detected species are reported in Table S3 (Supporting Information).
Figure 5.
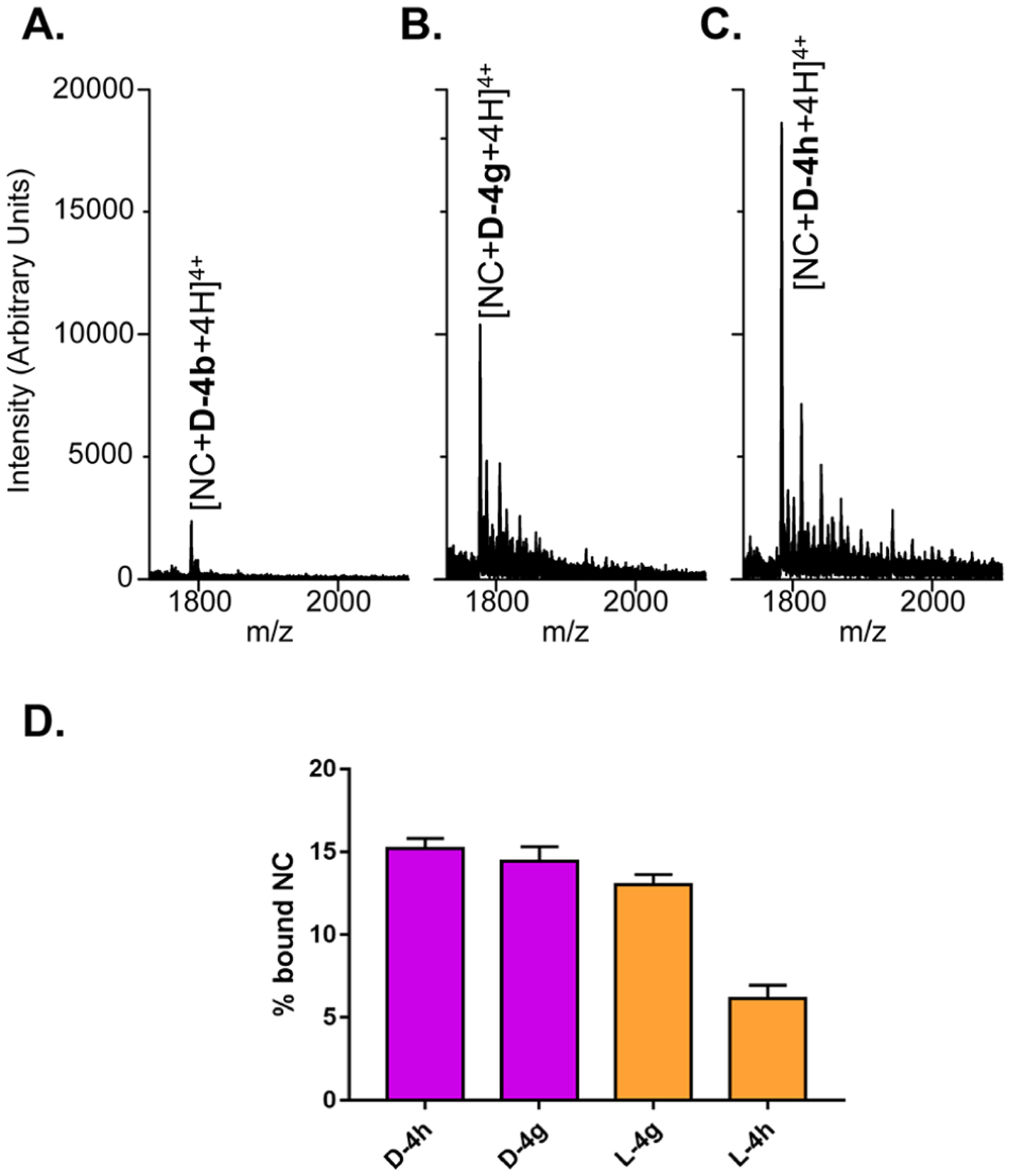
Representative ESI-MS spectra from samples containing full-length NC and ligand D-4b (A), D-4g (B), and D-4h (C), each in 1:10 substrate to ligand ratio (see Experimental Section). Weaker signals near the main peak are typical sodium and ammonium adducts. Only the region containing the 4+ charge state of complexes is reported for clarity. (D) Histograms comparing the fractional occupancy of d-analogues obtained from the above spectra with that of their l-counterparts obtained from the data in Figure S5 (Supporting Information).
The results clearly showed that the ligands were capable of binding to full-length NC to form stable 1:1 complexes, with those containing compound D-4g and D-4h displaying greater abundances than those containing D-4b.19,24 The masses observed for these complexes were consistent with the presence of two Zn(II) ions, which demonstrated that ligand binding did not interfere at all with metal coordination (Table S3). The fact that these species were detected with the same charge state allowed us to estimate the partitioning between unbound and bound species in the sample from their respective signal intensities (see Experimental Section).19,24 This treatment revealed that D-4g and D-4h produced fractional occupancies of 14.4 ± 0.93 and 15.2 ± 0.60, respectively, whereas D-4b provided only 3.57 ± 0.08. Under the same conditions, the corresponding L-4g and L-4h (l-Ala linker series) analogues afforded occupancy values of 13.3 ± 0.60 and 6.14 ± 0.79, which were decidedly lower (Figure 5C). The relative abundances observed for the complexes of NC with d-Ala and l-Ala compounds (Figures 5 and S5, respectively) provided a putative D-4h > D-4g > L-4g > L-4h relative scale of binding affinities for NC protein, which accurately matched the ranking of the IC50s for annealing inhibition provided by the NAME assays (see Table 2 and ref 12). The fact that corresponding d-Ala and l-Ala analogues were consistently at the opposite ends of these putative scales suggested direct correlations between linker stereochemistry and NC binding, leading to enhanced inhibitory activities.
Inhibition of the NC Binding to TAR by 2,6-Dipeptidyl Anthraquinones.
NC is a TAR binding protein: the ability of selected 2,6-dipeptidyl anthraquinones to bind directly to NC or to TAR raises several possibilities on the mechanism of NC inhibition by these ligands. To gain a greater understanding of the mechanism of action of d-Ala derivatives, we evaluated these scenarios in systematic fashion by analyzing samples in which preformed (i) NC-TAR, (ii) ligand-NC, or (iii) ligand-TAR complexes were challenged by addition of the remaining component (Scheme 3). This set of binding experiments was initially performed in the presence of the strongest NC and TAR binder, D-4h. The outcome of the experiments was determined by performing ESI-MS under the same conditions employed previously to evaluate ligand binding, which enabled the unambiguous identification of all species present at equilibrium in solution.
Scheme 3.
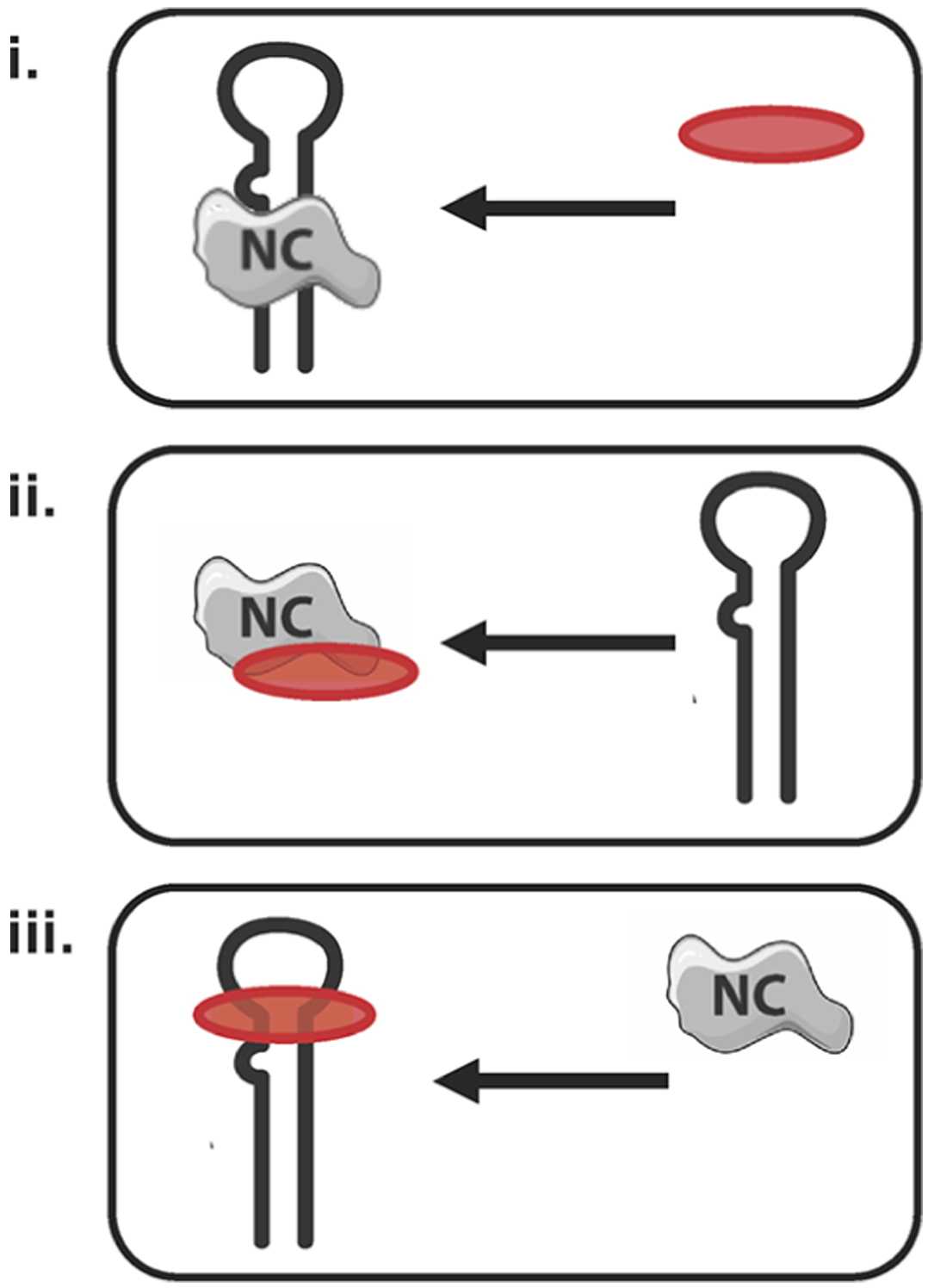
Schematic Representation of the Experimental Procedures Employed to Evaluate the Inhibition of NC Binding to TAR by Selected Ligandsa
aThe red oval represents the ligand. (i) Displacement of NC●TAR complex was analyzed by adding ligand to the preformed protein-RNA complex; (ii) interaction of NC with TAR substrate was assayed after preincubation of the protein with ligand; (iii) NC protein was added to preformed TAR-ligand complexes.
Data in Figure 6 were obtained from a sample corresponding to case i in Scheme 3, which was prepared by incubating equimolar amounts of NC and TAR (i.e., 6 μM concentration of each) for 15 min, and then added of a 1:10 molar ratio of D-4h ligand (i.e., final 60 μM concentration). The signal observed for the NC•TAR complex upon ligand addition in Figure 6 was significantly weaker than that obtained for the same species in a control experiment carried out in the absence of ligand (data not shown). At the same time, strong signals were detected for the D-4h•TAR species formed by displacement of the initial NC-TAR complex. It is clear that upon NC displacement the ligand D-4h forms several D-4h•TAR complexes, with the same stoichiometries found in Figure 4A. Interestingly, we observe for the first time the formation of ternary complexes between NC protein, TAR RNA, and D-4h ligand at different stoichiometries, with the notable exception of the ternary complex containing 4 ligand molecules. We can therefore assume that at least one of the four ligand-TAR binding sites competes with NC binding site to TAR.
Figure 6.

ESI-MS spectrum of samples obtained by adding ligand D-4h to the preformed TAR●NC complex (case i in Scheme 3). Lower intensity signals near free/bound species consist of typical sodium and ammonium adducts. Gray ▲ corresponds to NC protein; ◼ to the TAR RNA substrate; and red ● to the D-4h ligand. For the sake of clarity only the 6- and 7-charge states of the binary (RNA-ligand) and ternary (RNA-ligand-protein) complexes are labeled in the spectrum.
In analogous fashion, we analyzed the outcome of experiments obtained by treating preformed D-4h•NC complex with TAR (case ii in Scheme 3); again, this led to the detection of abundant D-4h•TAR formed by complex displacement. Finally, treating preformed D-4h•TAR with NC (case iii in Scheme 3) provided signals corresponding to the initial species, with intensity matching that observed in control experiments in the absence of NC. Taken together, these results revealed that D-4h•TAR was the most favorable complex under each scenario, consistent with the relatively higher affinity of this ligand for the TAR substrate.
This is true also for the other strong TAR-binder D-4g. Figure 7 reports a representative histogram comparing the results from samples of D-4h, D-4g and D-4b, a poor TAR binder. The relative abundance of the NC•TAR complexes in the various samples corresponding to case i in Scheme 3 was utilized to compare the inhibition effects manifested by the ligands (see Experimental Section).
Figure 7.
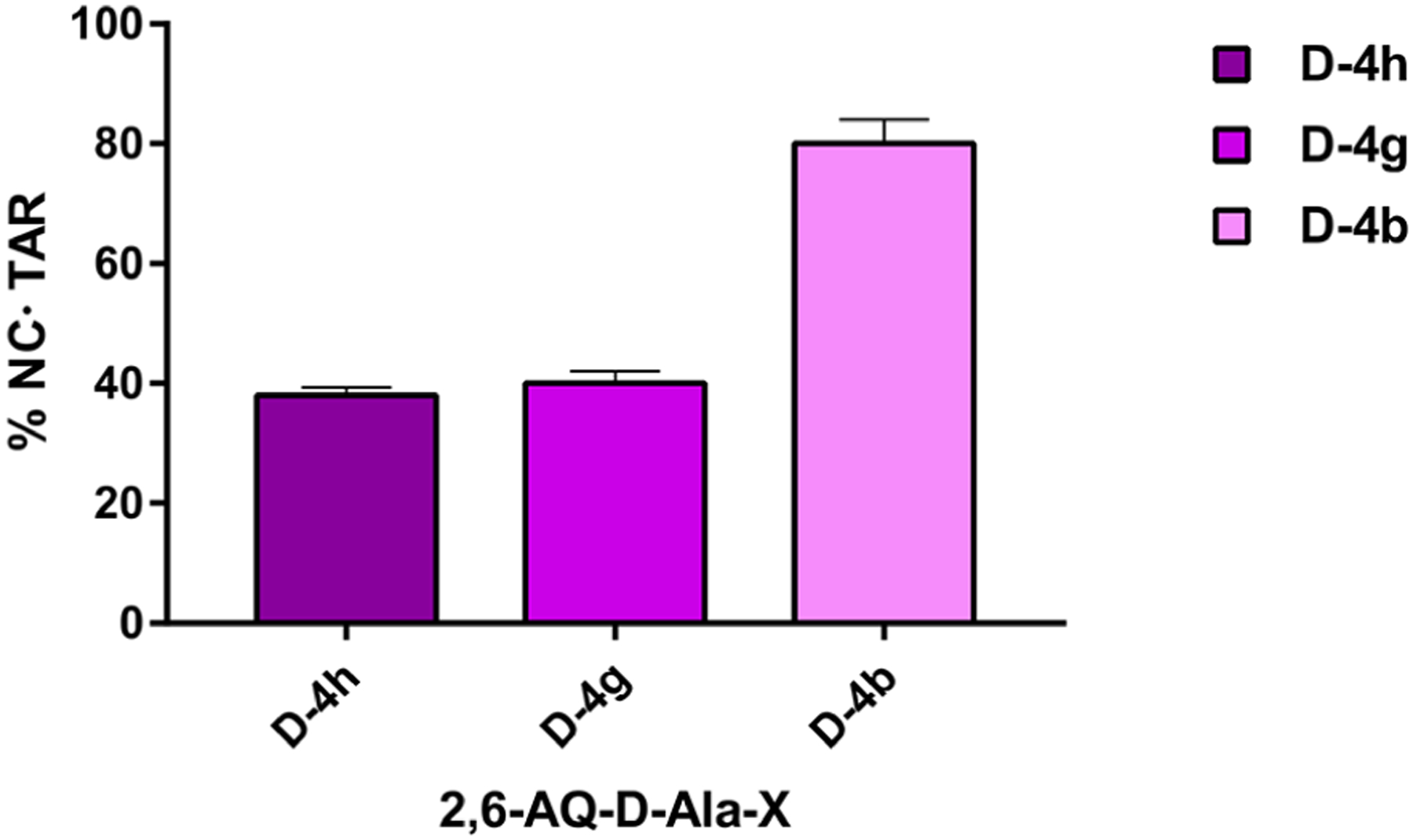
Representative histogram comparing the percentage of NC protein bound to TAR RNA (NC●TAR complexes) in the presence of ligand D-4h with those of D-4g and D-4b. Data were obtained from ESI-MS analyses of samples prepared following the procedure i, shown in Scheme 3 (see Experimental Section).
The relative scale of competition for NC displacement from TAR observed for the test compounds was D-4h > D-4g ≫ D-4b, which precisely mirrored the ranking for annealing inhibition provided by the NAME assays (Table 2 and ref 12), once more revealing the correlation between TAR binding and NC-mediated annealing inhibition.
2,6-Dipeptidyl Anthraquinone Effects on Tat-TAR Interactions.
The observed ability of 2,6-AQ-d-Ala-X compounds to establish specific interactions with the TAR structure suggested that these ligands might be capable of interfering with other functions enacted by this domain in the viral lifecycle. We tested this hypothesis by examining their putative effects on the specific binding interactions between TAR RNA and a peptide mimicking the HIV-1 trans-activator of transcription (Tat) factor. This protein can promote the efficient transcription of integrated proviral genome,25–28 as well as support the annealing activities of NC during reverse transcription, thus earning itself the definition of nucleic acids annealer.29,30 For this reason, we evaluated the effects of selected d-Ala analogues on the formation of the Tat-TAR complex by using a fluorescence-quenching approach. The assay relied on a TAR construct labeled with an appropriate quencher and a short stretch (aa 48–57) of the Tat protein corresponding to the RNA binding region,31,32 which was instead labeled with a fluorescent dye (see Experimental Section).14,33,34 In this way, the formation of the binding complex produced effective quenching, whereas its ligand-induced inhibition enabled the detection of fluorescence emission. As reported in Table 3, anthraquinones D-4e-h bearing linear alkyl side-chains strongly inhibited the formation of Tat-TAR complex with a potency ranking that matched that of NC annealing inhibition, i.e., D-4h > D-4g > D-4f > D-4e. In particular, the strongest Tat-TAR inhibitors were again the D-4g and D-4h compounds, which were also the most active TAR binders and NC inhibitors.
Table 3.
Inhibition of Tat-TAR Complex Formation
| compound | Kia (μM) |
|---|---|
| D-4b | 1.31 ± 0.18 |
| D-4e | 0.79 ± 0.18 |
| D-4f | 0.54 ± 0.08 |
| D-4g | 0.14 ± 0.01 |
| D-4h | 0.11 ± 0.01 |
Values are the mean ± SEM of data obtained by three experiments performed in triplicate.
Inspired by the results obtained from the investigation of NC, we tested the possibility that the ligands may be capable of binding the Tat peptide, as observed for NC. Following the same strategy, the experiments were carried out mixing Tat with selected analogues of the D-series, and then analyzing the samples by ESI-MS under nondenaturing conditions. The results showed unambiguously that the test ligands were not capable of binding directly to the Tat peptide (data not shown), thus indicating that the ability of d-analogues to interfere with the Tat-TAR complex was based exclusively on their TAR-binding properties.
CONCLUSIONS
This study was prompted by an earlier observation that the activity of 2,6-disubstituted anthraquinones significantly benefited from replacing l-Ala with Gly residues in the linkers between anthraquinone nucleus and charged side-chain groups.12 We hypothesized that the methyl group of l-Ala or its configuration was responsible for decreasing the ability to inhibit NC. To test this hypothesis we synthesized and analyzed a series of 2,6-AQ-d-Ala-X compounds containing non-natural d-Ala residues. The results demonstrated that this modification restored the inhibitory properties, with d-Ala compounds displaying the same type of correlation with side-chain length observed for the potent Gly analogues. Indeed, as the length of the linear alkyl side-chain increased in the 2,6-AQ-d-Ala-X series, inhibition increased according to a D-4h > D-4g > D-4f > D-4e relative scale of potency. In all assays, D-4h proved to be the most potent inhibitor of the series by achieving inhibition of NC activities in vitro at concentrations comparable to those observed in earlier studies for the most promising hits of the 2,6-AQ-ß-Ala-Orn and 2,6-AQ-Gly-Lys series.12
In addition to providing new insights into the structure–activity relationships of 2,6-dipeptidyl anthraquinones, this study gave us the opportunity to further investigate the putative mechanism of action of this class of compounds. At first sight, the inhibitory properties of ligands acting as threading intercalators could be ascribed to their marked binding to both TAR and cTAR secondary structures. In contrast, the detectable selectivity for the TAR substrate manifested by the d-analogues was never observed before and highlighted the importance of this RNA structure to elucidate the NC inhibition mechanism. RNA-binding small molecules are an important and highly challenging area of therapeutic research. Several classes of RNA binders have been reported,35–48 but many of them display nonspecificity in RNA binding.49 The presence of d-Ala linker in 2,6-dipeptidyl anthraquinones leading to RNA recognition and TAR selectivity therefore constitutes a new possible strategy for the development of RNA-specific ligands, which deserves further investigation. Pursuing by NMR study and docking experiments the high-resolution structural determination of ligand-RNA complexes will be necessary to understand at the molecular level the steric effects introduced by the non-natural stereochemical configuration.
Although the compounds tested in the study demonstrated the ability to interact directly with the NC protein, their binding affinities for the protein were consistently inferior to those manifested by the same compounds for the TAR construct. Indeed, the detection of ternary complexes containing protein, RNA, and ligands was consistent with the possible presence of distinct binding sites onto the TAR structure. The exception is represented by D-4h, which might share at least a putative binding site with the NC protein (Figure 6). Considering that this compound was also the most potent inhibitor in the series (Tables 1 and 2), it would be tempting to attribute its enhanced activity to the contribution of a putative competition mechanism.
Additionally, the TAR-binding agents displayed the same potency scale observed for NC when tested with the Tat-TAR system, while simultaneously failing to demonstrate any direct Tat-binding capabilities. These considerations confirmed that the mechanism of 2,6-dipeptidyl anthraquinones is based on their nucleic acid tropism, rather than protein inhibition. For this reason, they should be more appropriately defined as TAR-targeted candidates capable of interfering with processes mediated by the interactions of this essential domain of HIV-1 genome with NC and Tat proteins. Since Tat and NC are multifunctional auxiliary proteins of HIV-1, both acting through interaction with the TAR sequence, we believe that agents capable of interfering with those critical steps are expected to block HIV-1 replication at multiple levels. Dual targeting through the same RNA substrate opens the possibility to consider d-Ala-peptidyl-anthraquinones as pleiotropic inhibitors, able to interfere not only with NC-mediated reactions during reverse transcription, but also with the Tat mediated processes. The concomitant inhibition of different targets within the same pathway would possibly increase the genetic barrier required to gain resistance toward these putative drugs.
In the end, the absence of binding with the Tat protein represented a glaring but important contrast with the observed NC-binding properties. Although our results are a very small sampling, the selectivity toward the NC protein could constitute the basis for further developing the d-series as “true” anti-NC compounds. In this direction, additional studies are currently underway to evaluate the effects of new terminal residues to improve the direct interaction of 2,6-dipeptidyl anthraquinones with the hydrophobic plateau of NC protein, thus combining in the same molecules different mechanisms contributing to the overall NC inhibition.
EXPERIMENTAL SECTION
Materials and Methods.
For compound synthesis, purification (preparative HPLC) and chemical characterization (analytical HPLC, ESI-MS, and NMR) we used the same procedures, conditions, reactants and reagents reported in our previous paper.12 Microwave reactions were performed using a microwave oven (ETHOS 1600, Milestone) especially designed for organic synthesis.
TAR is the 29-mer RNA sequence 5′-GGCAGAUCUGAG-CCUGGGAGCUCUCUGCC-3′ and cTAR is its DNA complementary sequence 5′-GGCAGAGAGCTCCCAGGC-TCAGATCTGCC-3′. SL3 is the 20-mer sequence 5′-GGACUAGCGGAGGCUAGUCC-3′.22 Duplex construct dsRNA was obtained by annealing the 16-mer complementary sequences 5′-UAGGGGGAAGCUUGG-3′ and 5′-CCAAAGCUUCCCCCUA-3′. All RNA and DNA oligonucleotides were purchased from Metabion International AG (Martinsried, Germany). The full-length recombinant NC protein was obtained in house as previously reported.20
(R)-(9H-Fluoren-9-yl)methyl 1-chloro-1-oxopropan-2-yl-carbamate (2; Fmoc-d-Ala-Cl).
Commercially available Fmoc-d-Ala-OH (Aldrich, 2.5 g, 8.03 mmol) was dissolved in anhydrous dichloromethane (40 mL) in a two-neck flask. Excess SOCl2 (5 mL; 68.54 mmol) was added dropwise. The mixture was transferred into a sealed vessel and heated by microwave irradiation at 50 °C (300W power) for 8 min (30 s ramping step; 7 min and 30 s holding step). Solvent was evaporated under reduced pressure and the residue was purified by crystallization from n-hexane affording 2.60 g of pure 2a as a white solid. Yield: 98%.
N,N′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)bis(2-aminoacetamide)-bistrifluoroacetate (3).
Fmoc-d-Ala-Cl (2a, 6,39 g, 19,4 mmol) and pyridine (1.27 mL, 15.8 mmol) were slowly added to a solution of 2,6-diaminoanthraquinone (1, 350 mg, 1.94 mmol) in DMF (80 mL). The reaction mixture was stirred at room temperature for 24 h. Solvent was removed by reduced pressure distillation. The residue was purified by crystallization from ethyl acetate, thus obtaining bis((9H-fluoren-9-yl)methyl) ((2R,2′R)-((9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl))bis(1-oxopropane-2,1-diyl))-dicarbamate (2,6-Fmoc-d-Ala-anthraquinone) as an orange powder (1.47 g, 1.78 mmol, yield 92%), which was used in the next step without any further purification. This compound was reacted with a 33% diethylamine solution in THF (60 mL) for 2 h. Solvent was again distilled off under reduced pressure and the residue was finally reacted with a solution of trifluoroacetic acid in water (9:1, v/v, 20 mL) for 1 h. Reaction mixture was then added with diethyl ether (60 mL). The precipitate was collected by centrifugation and dried to obtain 862 mg of intermediate 3a as an intense orange solid. Yield 73% (overall). 1H NMR (DMSO-d6): δ 11.28 (bs, 2H), 8.49 (d, 2H), 8.30–8.20 (m, 8H), 8.06 (dd, 2H), 2.69 (d, 6H), 2.50 (m, 2h). 13C NMR (DMSO-d6): δ 181.2, 165.6, 143.6, 134.2, 128.4, 128.2, 123.4, 115.5, 49.4, 16.6. ESI-MS: 380.15 [M + H]+; 191.18 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl)-bis(1-oxopropane-2,1-diyl)-dipiperidine-4-carboxamide-bis-trifluoroacetate (D-4a).
Intermediate 3a (250 mg, 0.42 mmol) was dissolved in dry DMF (18.6 mL), and DMAP (545.56 mg, 3.24 mmol), 1-Fmocpiperidine-4-carboxylic acid (Fmoc-Inp-OH, 1.00 g, 3.20 mmol), and HBTU (1.36 g, 3.6 mmol) were added. The resulting solution was stirred at room temperature for 24 h, then poured into Et2O and centrifuged. The solid obtained was washed with Et2O, and the final product was dried in vacuo and used in the next step without any further purification. In order to remove the Fmoc-protecting group, the solid obtained was reacted with a 33% diethylamine solution in THF (30 mL) for 2 h, poured into Et2O, and centrifuged. The solid obtained was washed with Et2O and water and then dried in vacuo. Subsequently, the obtained solid was reacted with a solution of trifluoroacetic acid in water (9:1, v/v, 20 mL) for 1 h and then poured into Et2O. The resulting suspension was centrifuged. The solid obtained was washed with Et2O and dried in vacuo. Purification by preparative RP-HPLC afforded the pure compound D-4a as an intense orange solid. Yield: 76%. Orange solid. K′ (HPLC): 5,5. 1H NMR (DMSO-d6, 400 MHz): δ 10.71 (s, 2H), 8.71 (t, 2H), 8.44 (bs, 4H), 8.33 (d, 2H), 8.14 (bs, 4H), 8.03 (dd, 2H), 4.37 (m, 2H), 3.24 (m, 4H), 2.84 (m, 4H), 1.83 (m, 4H), 1.68 (m, 4H), 1.31 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 181.79, 172.07, 168.73, 144.89, 134.82, 129.04, 128.68, 124.11, 116.44, 51.90, 50.02, 40.59, 38.72, 28.68, 23.02, 18.31. ESI-MS: 603.3 [M + H]+; 302.1 [M + 2H]++.
(1R,1′ R,4S,4′S)-N,N′-((2S,2′S)-1,1′-(9,10-dioxo-9,10-dihy-droanthracene-2,6-diyl)-bis(azanediyl)bis-(1oxopropane2,1diyl))bis(4(aminomethyl)cyclohexane-carboxamide)-bis-trifluoroacetate (D-4b).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and 4-(Fmoc-aminomethyl)-cyclohexanecarboxylic acid (N-Fmoc-tranexamic acid). Yield: 65%. Orange solid. K′ (HPLC): 6.4. 1H NMR (DMSO-d6, 400 MHz): δ 10.69 (s, 2H), 8.47 (t, 2H), 8.18 (d, 2H), 8.16 (d, 2H), 8.05 (dd, 2H), 7.77 (bs, 6h), 4.36 (m, 2H), 2.85 (m, 4H), 2.64 (m, 2H), 1.76 (m, 2H), 1.67 (m, 8H), 1.31 (d, 6H), 0.88 (m, 8H). 13C NMR (DMSO-d6, 400 MHz): δ 182.02, 172.43, 171.83, 144.69, 135.03, 129.18, 128.52, 124.11, 116.41, 51.82, 43.97, 38.02, 35.67 29.66, 29.13, 17.95. ESI-MS: 659.1 [M + H]+; 330.2 [M + 2H]++.
N,N′-(1,1′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)-bis(azanediyl)bis(1-oxo-propane-2,1-diyl))bis(4-aminopiperidine-4-carboxamide)-tetra-trifluoroacetate (D-4c).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and 1-Fmoc-4-(Fmoc-amino)-piperidine-4-carboxylic acid (Fmoc-Pip(Fmoc)-OH). Yield: 63%. Orange solid. K′ (HPLC): 4.7. 1H NMR (DMSO-d6, 400 MHz): δ 10.69 (s, 2H), 8.46 (t, 2H), 8.31 (d, 2H), 8.18 (bs, 4H), 8.15 (d, 2H), 8.03 (dd, 2H), 7.90 (bs, 6H), 4.34 (m, 2H), 3.58 (m, 4H), 3.14 (m, 4H), 2.85 (m, 4h), 2.71 (m, 4h), 1.31 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 182.04, 171.37, 169.08, 145.22, 135.00, 129.21, 128.59, 124.19, 116.45, 56.87, 51.23, 35.72, 28.96, 17.94. ESI-MS: 633.4 [M + H]+; 317.2 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl)bis(1-oxopropane-2,1-diyl)bis(2,4-diaminobutanamide)-tetra-trifluoroacetate (D-4d).
We followed the synthetic procedures reported for D-4a, starting from the intermediate 3c and Fmoc-β-Alanine-OH (Fmoc-β-Ala-OH). Yield: 81%. Yellow solid. K′ (HPLC): 4.1. 1H NMR (DMSO-d6, 400 MHz): δ 10.85 (s, 2h), 8.91 (t, 2H), 8.48 (d, 2H), 8.18 (d, 2H), 8.07 (dd, 2H), 7.78 (bs, 6H), 4.54 (m, 2H), 2.82 (m, 4H), 1.75 (m, 4H), 1.38 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 181.99, 171.16, 169.46, 144.96, 134.87, 129.25, 128.61, 124.07, 116.55, 51.33, 36.02, 33.03, 18.13. ESI-MS: 523.5 [m + H]+; 261.2 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl)-bis(1-oxopropane-2,1-diyl)bis(2,3-diaminopropanamide)-tetra-trifluoroacetate (D-4e).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and Nα-Fmoc-Nβ-Boc-L-diaminopropionic acid (Fmoc-Dap(Boc)-OH). Yield: 86%. Orange solid. K′ (HPLC): 4.8. 1H NMR (DMSO-d6, 400 MHz): δ 10.92 (s, 2H), 9.00 (t, 2H), 8.47 (bs, 6H), 8.19 (bs, 6H), 8.18 (d, 2H), 8.06 (d, 2H), 8.04 (dd, 2H), 4.55 (m, 2H), 4.21 (m, 2H), 3.15 (m, 4H), 1.43 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 181.81, 172.169, 158.77, 144.65, 134.81, 129.07, 128.85, 124.26, 116.61, 50.82, 50.36, 40.15, 18.30. ESI-MS: 553.5 [M + H]+; 277.2 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl)-bis(1-oxopropane-2,1-diyl)bis(2,4-diaminobutanamide)-tetra-trifluoroacetate (D-4f).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and Nα-Fmoc-Nγ-Boc-L-diaminobutyric acid (Fmoc-Dab(Boc)-OH). Yield: 79%. Orange solid. K′ (hPLC): 4.8. 1H NMR (DMSO-d6, 400 MHz): δ 10.81 (s, 2H), 8.87 (t, 2H), 8.45 (bs, 6H), 8.17 (d, 2H), 8.16 (d, 2H), 8.04 (dd, 2H), 7.78 (bs, 6H), 4.53 (m, 2H), 3.82 (m, 2H), 2.84 (m, 4H), 1.75 (m, 4h), 1.41 (d, 6h). 13C NMR (DMSO-d6, 400 MHz): δ 181.88, 172.10, 168.69, 145.33, 134.87, 129.11, 128.73, 124.24, 116.55, 50.86, 50.31, 35.41, 29.48, 18.13. ESI-MS: 581.4 [M + H]+; 291.3 [M + 2H]++.
N,N ′-(1,1′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)-bis(azanediyl)bis(1-oxo-propane-2,1-diyl))bis(2,5-diaminopentanamide)-tetra-trifluoroacetate (D-4g).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and Nα-Fmoc-Nδ-Boc-l-ornithine (Fmoc-Orn(Boc)-OH). Yield: 81%. Orange solid. K′ (HPLC): 4.8. 1H NMR (DMSO-d6, 400 MHz): δ 10.88 (s, 2H), 8.86 (t, 2H), 8.46 (bs, 6H), 8.18 (d, 2H), 8.16 (d, 2H), 8.06 (dd, 2H), 7.85 (bs, 6H), 4.51 (m, 2H), 3.85 (m, 2H), 3.16 (m, 4H), 2.83 (m, 4H), 1.77 (m, 4H), 1.41 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 181.99, 169.77, 168.13, 144.74, 134.24, 129.09, 128.69, 124.24, 116.49, 52.13, 50.24, 38.84, 28.44, 22.79, 18.18. ESI-MS: 609.4 [M + H]+; 305.4 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-Dioxo-9,10-dihydro-anthracene-2,6-diyl)bis(azanediyl)-bis(1-oxopropane-2,1-diyl)bis(2,6-diaminohexanamide)-tetra-trifluoroacetate (D-4h).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and Nα-Fmoc-Nε-Boc-l-Lysine (Fmoc-Lys(Boc)-OH. Yield: 83%. Orange solid. K′ (HPLC): 4.9. 1H NMR (DMSO-d6, 400 MHz): δ 10.86 (s, 2H), 8.86 (t, 2H), 8.44 (bs, 6H), 8.18 (d, 2H), 8.16 (d, 2H), 8.06 (dd, 2h), 7.78 (bs, 6H), 4.50 (q, 2h), 3.85 (m, 2H), 2.75 (m, 4H), 1.72 (m, 4H), 1.54 (m, 4H), 1.40 (d, 6H), 1.33 (m, 4H). 13C NMR (DMSO-d6, 400 MHz): δ 181.87, 172.19, 168.90, 158.75, 144.93, 134.84, 129.09, 128.67, 124.08, 116.44, 52.22, 50.04, 38.97, 30.88, 26.93, 21.48, 18.12. ESI-MS: 636.9 [M + H]+; 319.1 [M + 2H]++.
N,N′-(2S,2′S)-1,1′-(9,10-Dioxo-9,10-dihydroanthracene-2,6-diyl)bis(azanediyl)bis(1-oxopropane-2,1-diyl)bis(2-amino-3-(4-aminophenyl)propanamide)-tetra-trifluoroacetate(D-4i).
We followed the synthetic procedures reported for D-4a, starting from intermediate 3a and Fmoc-4-(Boc-amino)-l-phenylalanine (Fmoc-4-(NH-Boc)Phe-OH). Yield: 69%. Orange solid. K′ (HPLC): 5.1. 1H NMR (DMSO-d6, 400 MHz): δ 10.74 (s, 2H), 8.74 (t, 2H), 8.44 (bs, 6H), 8.18 (d, 2H), 8.15 (d, 2H), 8.02 (dd, 2H), 7.01 (d, 4H), 6.94 (d, 4H), 6.72 (bs, 6h), 4.44 (m, 2H), 4.00 (m, 2H), 2.88 (m, 4h), 1.22 (d, 6H). 13C NMR (DMSO-d6, 400 MHz): δ 182.05, 169.72, 168.36, 158.32, 144.45, 134.95, 130.94, 129.03, 128.84, 128.58, 123.93, 118.84, 116.52, 53.93, 50.31, 36.87, 18.03. ESI-MS: 705.2 [M + H]+; 353.1 [m + 2H]++.
Inhibition of NC-Mediated Destabilization of TAR and cTAR Stem.
The identification of inhibitors able to impair the NC chaperone activity on TAR and cTAR was performed by means of the high throughput screening (HTS) previously described.13,14,16
Inhibition of NC-Mediated TAR/cTAR Annealing.
We investigated the ability of the newly synthesized 2,6-AQ-d-Ala-X anthraquinones to impair the annealing activity of full-length NC protein activity monitoring the annealing of TAR with cTAR through the nucleocapsid annealing mediated electrophoresis (NAME) assay described earlier,12,18 which was carried out according to alternative protocols involving preincubation of ligand with either the NC protein or the nucleic acid substrates.12 Selected compounds were further analyzed using different sets of concentrations in the oligopreincubation mode to accurately determine their potency in inhibiting NC-mediated annealing activity.13
Direct Binding of Ligands to Individual TAR and cTAR.
We evaluated the binding properties of selected 2,6-AQ-d-Ala-X anthraquinones to either TAR or cTAR employing electrospray ionization mass spectrometry (ESI-MS) analysis under nondenaturing conditions as previously reported for the l-Ala series.12,13,19–21 The determination of free and bound RNA or DNA, necessary to evaluate the binding affinity of compounds to TAR and cTAR, was assessed from the relative abundances, expressed as percentage and compared.24 The binding properties of selected compounds were evaluated also toward the stem-loop SL3 and the double-stranded RNA. SL3 construct was heated to 95 °C for 5 min and then ice-cooled in order to assume the proper hairpin structure. The dsRNA was also heated and then slowly cool to room temperature in order to form the RNA duplex. ESI-MS experiments were performed under the same experimental conditions used in the presence of TAR.
Direct Binding of Ligands to NC.
The direct binding of selected 2,6-AQ-d-Ala-X anthraquinones to the full-length NC protein was assessed by ESI-MS in positive ion mode via direct infusion nanospray ionization on a Synapt G2 HDMS traveling-wave ion mobility spectrometry (IMS) mass spectrometer (Waters, Manchester, UK).50 Typical final mixtures contained up to 10:1 2,6-dipeptidyl-anthraquinone:NC molar ratio in 150 mM ammonium acetate (pH 7.5). Mass Lynx (v 4.1, SCN781) software was used to process data. To evaluate the binding affinity of compounds to the full-length NC, free and complexed protein abundances in each experiment were calculated as reported earlier for TAR and cTAR, and were finally expressed as percentage and compared.
Inhibition of NC Binding to TAR.
Possible effects induced by 2,6-dipeptidyl anthraquinones on the specific binding of NC protein to TAR substrate were evaluated by analyzing samples in which preformed i, NC-TAR; ii, ligand-NC; or iii, ligand-TAR complexes were challenged by addition of the remaining component as exemplified in Scheme 3. With the concentration of each component kept constant in the three different procedures, only the order of incubation changed to evaluate all the possible scenarios. Samples were prepared by incubating equimolar amounts of NC and TAR (i.e., 6 μM concentration of each) and a 1:10 molar ratio of each ligand (i.e., final 60 μM concentration) in 150 mM ammonium acetate (pH 7.5). ESI-MS performed under nondenaturing conditions was applied to unambiguously identify all species present at equilibrium in solution. In order to characterize all the reaction products, all samples were analyzed in both positive and negative ion modes via direct infusion nanospray ionization on a Synapt G2 HDMS traveling-wave ion mobility spectrometry (IMS) mass spectrometer (Waters, Manchester, UK), using the same conditions used for the binding analysis of ligands to NC. Data were processed by using Mass Lynx (v 4.1, SCN781) software. To evaluate the binding of the full-length NC to TAR construct, free and complexed protein abundances in each experiment were calculated, expressed as percentage and compared.
Inhibition of Tat/TAR Complex Formation.
The effect of anthraquinone derivatives on the Tat/TAR complex was evaluated using a FRET-based competition assay, as previously described.14,33,51
Supplementary Material
ACKNOWLEDGMENTS
A.S. has received funding from the European Union’s Horizon 2020 Research and Innovation programme under the Marie Skłodowska-Curie grant agreement No. 751931.
ABBREVIATIONS USED
- HIV
Human Immunodeficiency Virus
- NC
nucleocapsid protein
- TAR
transactivation response element
- cTAR
DNA sequence complementary to TAR
- RT
reverse transcriptase
- SAR
structure–activity relationship
- FQA
fluorescence quenching assay
- SI
selectivity index
- HTS
high throughput screening
- IC50
50% inhibitory concentration
- NAME
nucleocapsid annealing-mediated electrophoresis
- SDS
sodium dodecyl sulfate
- EDTA
ethylenediaminetetraacetic acid
- PAGE
polyacrylamide gel electrophoresis
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00104.
IC50 values; Inhibition of the TAR/cTAR annealing reaction; ESI-MS spectra; Monoisotopic mass (Da) of complexes detected by ESI-MS analysis; Comparison of the percentages of complexes calculated from the ESI-MS spectra (PDF)
The authors declare no competing financial interest
REFERENCES
- (1).Mori M, Kovalenko L, Lyonnais S, Antaki D, Torbett BE, Botta M, Mirambeau G, and Mely Y (2015) Nucleocapsid Protein: A Desirable Target for Future Therapies Against HIV-1. Curr. Top. Microbiol. Immunol 389, 53–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Richman DD (2014) Editorial Commentary: Extending HIV drug resistance testing to low levels of plasma viremia. Clin. Infect. Dis 58 (8), 1174–5. [DOI] [PubMed] [Google Scholar]
- (3).Dolgin E (2014) Long-acting HIV drugs advanced to overcome adherence challenge. Nat. Med 20 (4), 323–4. [DOI] [PubMed] [Google Scholar]
- (4).Darlix JL, Garrido JL, Morellet N, Mely Y, and de Rocquigny H (2007) Properties, functions, and drug targeting of the multifunctional nucleocapsid protein of the human immunodeficiency virus. Adv. Pharmacol 55, 299–346. [DOI] [PubMed] [Google Scholar]
- (5).Guo J, Henderson LE, Bess J, Kane B, and Levin JG (1997) Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J. Virol 71 (7), 5178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Johnson PE, Turner RB, Wu ZR, Hairston L, Guo J, Levin JG, and Summers MF (2000) A mechanism for plus-strand transfer enhancement by the HIV-1 nucleocapsid protein during reverse transcription. Biochemistry 39 (31), 9084–91. [DOI] [PubMed] [Google Scholar]
- (7).Levin JG, Guo J, Rouzina I, and Musier-Forsyth K (2005) Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol 80, 217–86. [DOI] [PubMed] [Google Scholar]
- (8).Mougel M, Houzet L, and Darlix JL (2009) When is it time for reverse transcription to start and go? Retrovirology 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rajendran A, Endo M, Hidaka K, Tran PL, Mergny JL, Gorelick RJ, and Sugiyama H (2013) HIV-1 nucleocapsid proteins as molecular chaperones for tetramolecular antiparallel G-quadruplex formation. J. Am. Chem. Soc 135 (49), 18575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thomas JA, and Gorelick RJ (2008) Nucleocapsid protein function in early infection processes. Virus Res. 134 (1—2), 39–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Darlix JL, Godet J, Ivanyi-Nagy R, Fosse P, Mauffret O, and Mely Y (2011) Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol 410 (4), 565–81. [DOI] [PubMed] [Google Scholar]
- (12).Frecentese F, Sosic A, Saccone I, Gamba E, Link K, Miola A, Cappellini M, Cattelan MG, Severino B, Fiorino F, et al. (2016) Synthesis and in Vitro Screening of New Series of 2,6-Dipeptidyl-anthraquinones: Influence of Side Chain Length on HIV-1 Nucleocapsid Inhibitors. J. Med. Chem 59 (5), 1914–24. [DOI] [PubMed] [Google Scholar]
- (13).Sosic A, Frecentese F, Perissutti E, Sinigaglia L, Santagada V, Caliendo G, Magli E, Ciano A, Zagotto G, Parolin C, et al. (2013) Design, synthesis and biological evaluation of TAR and cTAR binders as HIV-1 nucleocapsid inhibitors. MedChemComm 4 (10), 1388–1393. [Google Scholar]
- (14).Sosic A, Sinigaglia L, Cappellini M, Carli I, Parolin C, Zagotto G, Sabatino G, Rovero P, Fabris D, and Gatto B (2016) Mechanisms of HIV-1 Nucleocapsid Protein Inhibition by Lysyl-Peptidyl-Anthraquinone Conjugates. Bioconjugate Chem. 27 (1), 247–56. [DOI] [PubMed] [Google Scholar]
- (15).Shvadchak V, Sanglier S, Rocle S, Villa P, Haiech J, Hibert M, Van Dorsselaer A, Mely Y, and de Rocquigny H (2009) Identification by high throughput screening of small compounds inhibiting the nucleic acid destabilization activity of the HIV-1 nucleocapsid protein. Biochimie 91 (7), 916–923. [DOI] [PubMed] [Google Scholar]
- (16).Sosic A, Cappellini M, Sinigaglia L, Jacquet R, Deffieux D, Fabris D, Quideau S, and Gatto B (2015) Polyphenolic C-glucosidic ellagitannins present in oak-aged wine inhibit HIV-1 nucleocapsid protein. Tetrahedron 71 (20), 3020–3026. [Google Scholar]
- (17).Kanevsky I, Chaminade F, Chen Y, Godet J, Rene B, Darlix JL, Mely Y, Mauffret O, and Fosse P (2011) Structural determinants of TAR RNA-DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. Nucleic Acids Res. 39 (18), 8148–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sosic A, Cappellini M, Scalabrin M, and Gatto B (2015) Nucleocapsid Annealing-Mediated Electrophoresis (NAME) Assay Allows the Rapid Identification of HIV-1 Nucleocapsid Inhibitors. J. Visualized Exp No. 95, 1 DOI: 10.3791/52474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Turner KB, Hagan NA, and Fabris D (2006) Inhibitory effects of archetypical nucleic acid ligands on the interactions of HIV-1 nucleocapsid protein with elements of Psi-RNA. Nucleic Acids Res. 34 (5), 1305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Turner KB, Hagan NA, Kohlway AS, and Fabris D (2006) Mapping noncovalent ligand binding to stemloop domains of the HIV-1 packaging signal by tandem mass spectrometry. J. Am. Soc. Mass Spectrom 17 (10), 1401–11. [DOI] [PubMed] [Google Scholar]
- (21).Turner KB, Kohlway AS, Hagan NA, and Fabris D (2009) Noncovalent probes for the investigation of structure and dynamics of protein-nucleic acid assemblies: the case of NC-mediated dimerization of genomic RNA in HIV-1. Biopolymers 91 (4), 283–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pappalardo L, Kerwood DJ, Pelczer I, and Borer PN (1998) Three-dimensional folding of an RNA hairpin required for packaging HIV-1. J. Mol. Biol 282 (4), 801–18. [DOI] [PubMed] [Google Scholar]
- (23).Fabris D, Zaia J, Hathout Y, and Fenselau C (1996) Retention of Thiol Protons in Two Classes of Protein Zinc Ion Coordination Centers. J. Am. Chem. Soc 118, 12242–12243. [Google Scholar]
- (24).Hagan N, and Fabris D (2003) Direct mass spectrometric determination of the stoichiometry and binding affinity of the complexes between nucleocapsid protein and RNA stem-loop hairpins of the HIV-1 Psi-recognition element. Biochemistry 42 (36), 10736–45. [DOI] [PubMed] [Google Scholar]
- (25).Nabel G, and Baltimore D (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326 (6114), 711–3. [DOI] [PubMed] [Google Scholar]
- (26).Berkhout B, Silverman RH, and Jeang KT (1989) Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 59 (2), 273–82. [DOI] [PubMed] [Google Scholar]
- (27).Weeks KM, Ampe C, Schultz SC, Steitz TA, and Crothers DM (1990) Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science 249, 1281–1285. [DOI] [PubMed] [Google Scholar]
- (28).Rana TM, and Jeang KT (1999) Biochemical and functional interactions between HIV-1 Tat protein and TAR RNA. Arch. Biochem. Biophys 365 (2), 175–85. [DOI] [PubMed] [Google Scholar]
- (29).Boudier C, Humbert N, Chaminade F, Chen Y, de Rocquigny H, Godet J, Mauffret O, Fosse P, and Mely Y (2014) Dynamic interactions of the HIV-1 Tat with nucleic acids are critical for Tat activity in reverse transcription. Nucleic Acids Res. 42 (2), 1065–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Boudier C, Storchak R, Sharma KK, Didier P, Follenius-Wund A, Muller S, Darlix JL, and Mely Y (2010) The mechanism of HIV-1 Tat-directed nucleic acid annealing supports its role in reverse transcription. J. Mol. Biol 400 (3), 487–501. [DOI] [PubMed] [Google Scholar]
- (31).Churcher M, Lamont C, Hamy F, Dingwall C, Green SM, Lowe AD, Butler P, Gait MJ, and Karn J (1993) High Affinity Binding of TAR RNA by the Human Immunodeficiency Virus Type-1 Tat Protein Requires Base-pairs in the RNA Stem and Amino Acid Residues Flanking the Basic Region. J. Mol. Biol 230, 90. [DOI] [PubMed] [Google Scholar]
- (32).Cao H, Tamilarasu N, and Rana TM (2006) Orientation and affinity of HIV-1 Tat fragments in Tat-TAR complex determined by fluorescence resonance energy transfer. Bioconjugate Chem. 17 (2), 352–8. [DOI] [PubMed] [Google Scholar]
- (33).Manfroni G, Gatto B, Tabarrini O, Sabatini S, Cecchetti V, Giaretta G, Parolin C, Del Vecchio C, Calistri A, Palumbo M, et al. (2009) Synthesis and Biological Evaluation of 2-Phenyl-quinolones Targeted at Tat/TAR Recognition. Bioorg. Med. Chem. Lett 19, 714–717. [DOI] [PubMed] [Google Scholar]
- (34).Tabarrini O, Massari S, Daelemans D, Stevens M, Manfroni G, Sabatini S, Balzarini J, Cecchetti V, Pannecouque C, and Fravolini A (2008) Structure-activity relationship study on anti-HIV 6-desfluoroquinolones. J. Med. Chem 51 (17), 5454–8. [DOI] [PubMed] [Google Scholar]
- (35).Arya SK, Guo C, Josephs SF, and Wong-Staal F (1985) Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 229 (4708), 69–73. [DOI] [PubMed] [Google Scholar]
- (36).Jin Y, Watkins D, Degtyareva NN, Green KD, Spano MN, Garneau-Tsodikova S, and Arya DP (2016) Arginine-linked neomycin B dimers: synthesis, rRNA binding, and resistance enzyme activity. MedChemComm 7 (1), 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kamphan A, Gong C, Maiti K, Sur S, Traiphol R, and Arya DP (2017) Utilization of chromic polydiacetylene assemblies as a platform to probe specific binding between drug and RNA. RSC Adv 7 (66), 41435–41443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kumar S, Kellish P, Robinson WE Jr., Wang D, Appella DH, and Arya DP (2012) Click dimers to target HIV TAR RNA conformation. Biochemistry 51 (11), 2331–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kumar S, Ranjan N, Kellish P, Gong C, Watkins D, and Arya DP (2016) Multivalency in the recognition and antagonism of a HIV TAR RNA-TAT assembly using an aminoglycoside benzimidazole scaffold. Org. Biomol. Chem 14 (6), 2052–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ranjan N, and Arya DP (2016) Linker dependent intercalation of bisbenzimidazole-aminosugars in an RNA duplex; selectivity in RNA vs. DNA binding. Bioorg. Med. Chem. Lett 26 (24), 5989–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ranjan N, Kellish P, King A, and Arya DP (2017) Impact of Linker Length and Composition on Fragment Binding and Cell Permeation: Story of a Bisbenzimidazole Dye Fragment. Biochemistry 56 (49), 6434–6447. [DOI] [PubMed] [Google Scholar]
- (42).Shaw NN, and Arya DP (2008) Recognition of the unique structure of DNA:RNA hybrids. Biochimie 90 (7), 1026–39. [DOI] [PubMed] [Google Scholar]
- (43).Watkins D, Kumar S, Green KD, Arya DP, and Garneau-Tsodikova S (2015) Influence of linker length and composition on enzymatic activity and ribosomal binding of neomycin dimers. Antimicrob. Agents Chemother 59 (7), 3899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Aboul-ela F, Karn J, and Varani G (1995) The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol 253, 313–32. [DOI] [PubMed] [Google Scholar]
- (45).Aboul-ela F, Karn J, and Varani G (1996) Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 24 (20), 3974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, and Varani G (2009) Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U. S. A 106 (29), 11931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Davis B, Afshar M, Varani G, Murchie AIH, Karn J, Lentzen G, Drysdale M, Bower J, Potter AJ, Starkey ID, et al. (2004) Rational design of inhibitors of HIV-1 TAR RNA through the stabilization of electrostatic *hot spots*. J. Mol. Biol 336, 343–356. [DOI] [PubMed] [Google Scholar]
- (48).Gallego J, and Varani G (2001) Targeting RNA with small-molecule drugs: therapeutic promise and chemical challenges. Acc. Chem. Res 34, 836–843. [DOI] [PubMed] [Google Scholar]
- (49).Chow CS, and Bogdan FM (1997) A Structural Basis for RNAminus signLigand Interactions. Chem. Rev 97 (5), 1489–1514. [DOI] [PubMed] [Google Scholar]
- (50).Lippens JL, Mangrum JB, McIntyre W, Redick B, and Fabris D (2016) A simple heated-capillary modification improves the analysis of non-covalent complexes by Z-spray electrospray ionization. Rapid Commun. Mass Spectrom 30 (6), 773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tabarrini O, Massari S, Daelemans D, Meschini F, Manfroni G, Bottega L, Gatto B, Palumbo M, Pannecouque C, and Cecchetti V (2010) Studies of anti-HIV transcription inhibitor quinolones: identification of potent N1-vinyl derivatives. ChemMed-Chem 5 (11), 1880–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.