

Abstract
Circulatory protein glycosylation is a biomarker of multiple disease and inflammatory states and has been applied in the clinic for liver dysfunction, heart disease and diabetes. With the notable exception of antibodies, the liver produces most of the circulatory glycoproteins, including the acute phase proteins released as a function of the inflammatory response. Among these proteins is β-galactoside α2,6-sialyltransferase (ST6Gal1), an enzyme required for α2,6-linked sialylation of glycoproteins. Here, we describe a hepatocyte-specific conditional knockout of ST6Gal1 (H-cKO) using albumin promoter-driven Cre-lox recombination. We confirm the loss of circulatory glycoprotein α2,6 sialylation and note no obvious dysfunction or pathology in young H-cKO mice, yet these mice show robust changes in plasma glycoprotein fucosylation, branching and the abundance of bisecting GlcNAc and marked changes in a number of metabolic pathways. As H-cKO mice aged, they spontaneously developed fatty liver disease characterized by the buildup of fat droplets in the liver, inflammatory cytokine production and a shift in liver leukocyte phenotype away from anti-inflammatory Kupffer cells and towards proinflammatory M1 macrophages. These findings connect hepatocyte and circulatory glycoprotein sialylation to the regulation of metabolism and inflammation, potentially identifying the glycome as a new target for liver-driven disease.
Keywords: hepatocyte, inflammation, liver metabolism, macrophage, ST6Gal1
Introduction
Protein glycosylation is a fundamental pathway in all cells and occurs predominantly within the endoplasmic reticulum and Golgi apparatus, collectively known as the secretory pathway. With the notable exception of albumin, essentially all secreted and membrane proteins are glycosylated, including the majority of liver-produced plasma proteins. As a result, the liver is a central factor in the regulation of circulatory homeostasis, and the plasma protein glycome is increasingly used as a target for biomarker research (Adamczyk et al. 2012; Kaul et al. 2012; Ruhaak et al. 2013; Maverakis et al. 2015), especially in cancer (An et al. 2009). For example, we recently demonstrated that changes in the plasma glycome were predictive of cardiovascular disease among HIV+ individuals (Oswald et al. 2019), while many others have reported differences ranging from the fucosylation of haptoglobin in pancreatic cancer (Miyoshi and Nakano 2008) to alterations in prostate-specific antigen glycosylation in prostate cancer patient sera (Tabarés et al. 2006).
The liver is central to the inflammatory cascade, in part through its role in the acute phase response (APR). The APR is a system-wide response to infection, tissue injury and inflammation characterized by the release of acute phase proteins (APPs) into the circulation and can be triggered by inflammatory cytokines such as IL-6 and leukemia inhibitory factor (LIF) (Mayer et al. 1993; Schmidt-Arras and Rose-John 2016). In addition to the widely recognized APPs such as C-reactive protein and serum amyloid A, β-galactoside-α2,6-sialylatransferase 1 (ST6Gal1) is also upregulated and released into circulation as an APP (Weinstein et al. 1987; Appenheimer et al. 2003). ST6Gal1 is expressed ubiquitously and is the only member of its family responsible for the α2,6 sialylation of proteins outside of the central nervous system (Harduin-Lepers et al. 2005; Lehoux et al. 2010). This enzyme has been extensively studied and recently gained attention due to its roles in IgG sialylation (Jones et al. 2012), myelopoiesis (Jones et al. 2010) and cancer (Schultz et al. 2016) and is known to be active within the plasma (Jones et al. 2016). Indeed, mice with targeted manipulation of the P1 region of its promoter have modestly reduced liver ST6Gal1 expression (Appenheimer et al. 2003) and alterations in myelopoiesis (Jones et al. 2010), while germline deletion results in known defects in B cell signaling (Hennet et al. 1998; Crocker et al. 2007; Meyer et al. 2018).
Glycoprotein sialylation influences APP behavior through indirect properties, such as folding, stability and solubility (Varki 2008; Varki and Gagneux 2012), as well as specific and direct interactions, including sialylated glycoproteins acting as decoys for pathogens such as influenza and malaria (Adams et al. 1992; Gagneux and Varki 1999). Sialylation is also an intrinsic marker of protein and cellular origin and age, and turnover is promoted by the hepatic asialoglycoprotein receptor (ASGPR). Thus, the ASGPR functions to recognize both potentially foreign glycans and the glycans of proteins that have spontaneously hydrolyzed due to age or other conditions (Yuan et al. 2015). Finally, sialylated glycans are the ligands for the Siglec family of proteins, which are broadly involved in dampening immune responses through their ability to recruit phosphatases to sites of receptor signaling in plasma membranes (Zhou et al. 2018), including the B cell receptor (Crocker et al. 2007), and are the primary target for selectin interactions during leukocyte homing (Johnson et al. 2013).
In order to gain a better understanding of the relationship between α2,6 sialylation, circulatory glycoproteins and inflammation, we generated a hepatocyte-specific ST6Gal1 conditional knockout mouse (H-cKO) using the lox-Cre recombinase system powered by the albumin promoter. We found an absence of gross pathology in young and resting mice, but observed large, unexpected changes in the plasma glycome. In addition to the expected loss of α2,6 sialylation, plasma glycoproteins also showed significant changes in fucosylation and bisecting GlcNAc residues. However, at approximately 6 months of age, resting mice began to show an inflammatory signature characterized by changes in liver macrophages and the production of IL-6. By 52 weeks, H-cKO mice developed a clear and spontaneous nonalcoholic fatty liver disease with steatohepatitis (NAFLD/NASH), suggesting a long-term dysregulation of liver metabolism and chronic inflammation. Consistent with this interpretation, we found that the H-cKO mouse had more liver inflammation, as measured by TNFα and MCP-1 production, when driven to obesity using a high fat diet. Further, RNA-seq analysis of liver from young mice prior to the development of pathology or inflammation revealed that the glycosylation changes induced by the removal of hepatocyte ST6Gal1 resulted in a dysregulation of liver metabolic pathways, ultimately driving inflammation and disease. Our findings demonstrate the importance of the liver and circulatory glycoprotein α2,6 sialylation via ST6Gal1 catalysis in the maintenance of metabolic and inflammatory homeostasis.
Results
In order to investigate the role of ST6Gal1 in the liver, we crossed mice expressing the Cre recombinase under the control of the hepatocyte-specific albumin promoter (Postic et al. 1999) with mice carrying the ST6Gal1 locus with flanking loxP sites (ST6Gal1f/f) (Hennet et al. 1998), thereby creating a hepatocyte-specific conditional knockout of ST6Gal1 (H-cKO) (Figure 1A). Mice were bred to homozygosity on both loci (Figure 1B). Using quantitative RT-PCR on mRNA isolated from multiple tissues, we confirmed the selective and near complete loss of ST6Gal1 in the liver. Low levels of ST6Gal1 transcript remained in the liver (Figure 1C), consistent with expression among nonhepatocyte lineages, including endothelial and Kupffer cells (Crispe 2011).
Fig. 1.
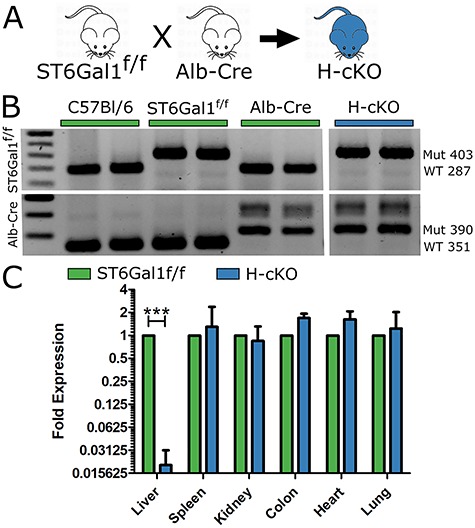
Generation of the hepatocyte conditional knockout (H-cKO) mouse. (A) Scheme for breeding of the H-cKO mouse through the crossing of the ST6Gal1f/f and Alb-Cre strains. (B) PCR confirmation of the genotype of the H-cKO mouse. (C) Analysis of ST6Gal1 RNA in 8–12-week-old mouse livers via quantitative PCR (qPCR) of ST6Gal1f/f and H-cKO strains demonstrated a substantial ablation of liver transcript without effects elsewhere in the mouse. Student’s T test, ***P < 0.001.
Functional validation of the ST6Gal1 ablation was performed by comparing glycoprotein sialylation from H-cKO and ST6Gal1f/f control mice. Lectin-based western analysis using the α2,6-sialic acid-specific lectin SNA shows a significant reduction in α2,6-sialylation among liver proteins (Figure 2A) and an even more dramatic loss of α2,6-sialylation among plasma glycoproteins (Figure 2B). Confocal microscopy with SNA further revealed the loss of hepatocyte α2,6-sialylation in H-cKO livers compared to controls (Figure 2C–D). Consistent with low but not absent mRNA levels of ST6Gal1 (Figure 1C), the vascular endothelium and the liver sinusoidal epithelial cells (LSECs) remained α2,6-sialylated in H-cKO mice (Figure 2C–D). Importantly, no other tissue in these mice, including brain, heart, lung, kidney and spleen, were distinguishable by SNA staining (Figures 2 and S1). Together, these data demonstrate the selectivity and penetrance of the knockout.
Fig. 2.
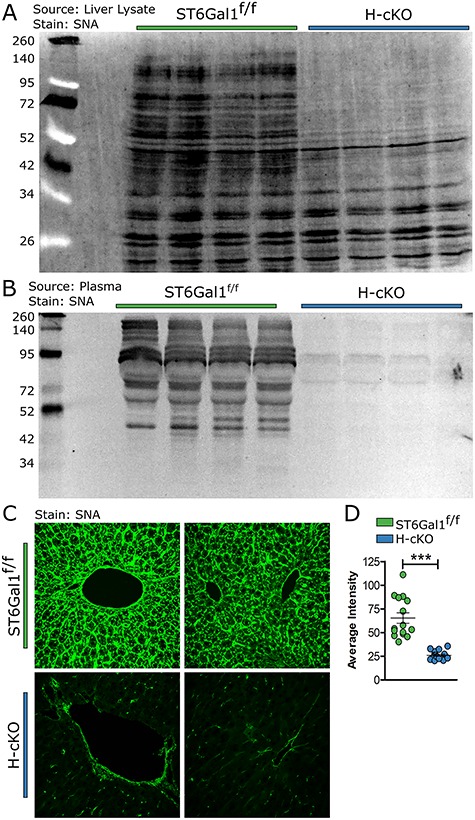
Removal of ST6Gal1 from hepatocytes resulted in the loss of α2,6 sialic acid linkages in liver protein and plasma protein and in the vast majority of liver cells. (A) A blot of liver lysate protein from 8–12-week-old ST6Gal1f/f and H-cKO mice stained with SNA demonstrated loss of α2,6 sialylation in H-cKO mice. (B) Blotting plasma protein with SNA demonstrated the loss of α2,6 sialylation in circulatory protein. (C) Liver section stained with SNA-FITC and imaged via confocal analysis showed loss of α2,6 sialylation among hepatocytes, but not other liver cells. (D) Quantification of average pixel intensity for SNA staining of liver. Student’s T test, ***P < 0.001.
Next, we performed a complete necropsy of young adult H-cKO and control ST6Gal1f/f mice between 8 and 12 weeks of age. We found no difference in blood chemistry or IgG titer, including markers of liver function in these mice (Figure 3A and B). Likewise, we found no differences between blood cellularity, with the vast majority of both H-cKO and ST6Gal1f/f samples falling within defined normal bounds (Figure 3C). Histological evaluation of all major tissues (Figures 3 and S2), including the liver (Figure 3D), also revealed no phenotypic differences in young mice.
Fig. 3.
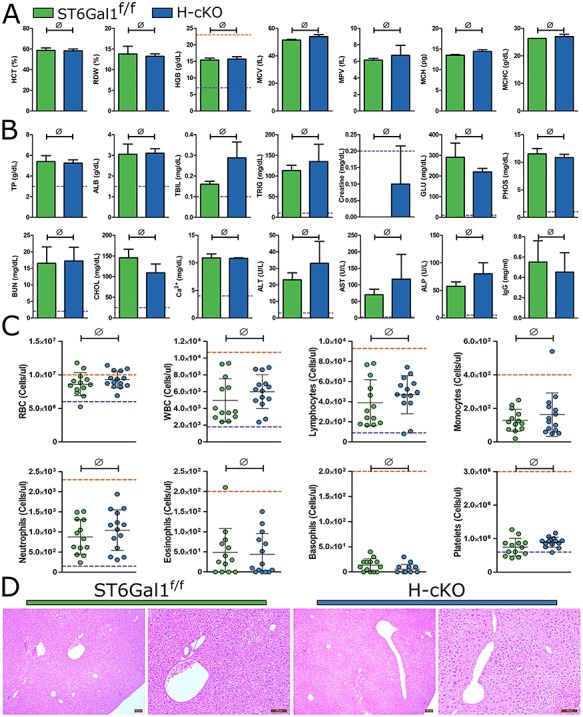
Necropsy of H-cKO mouse reveals no baseline pathology or abnormalities. (A) Analysis of blood composition measuring RDW, MCV, hemoglobin, MCH, MCHC, MPV and MCT in 8–12-week-old wild-type and H-cKO mice. (B) Liver and kidney function analysis measuring plasma concentrations of total bilirubin, aspartate aminotransferase, urea nitrogen, alanine aminotransferase, glucose, creatine, alkaline phosphatase, albumin, cholesterol, phosphate, total protein, Ca2+, triglycerides and IgG. (C) Complete blood count (CBC) analysis of blood cell distributions in wild-type and mutant mice. (D) Histological observation via H&E staining of mouse liver, no abnormalities identified. Student’s T test, ∅: P ≥ 0.05. On each graph, each dot represents one mouse. Purple and orange lines indicate upper and lower edges of normal range, respectively.
In order to profile the glycans on glycoproteins in the circulation, a lectin-based ELISA we previously developed for high-throughput glycan analyses (Oswald et al. 2019) was performed on isolated plasma from young adult mice. A total of 11 lectins covering a broad range of glycan determinants were used. As expected, α2,6-linked sialic acid (SNA) was sharply reduced in H-cKO plasma, while total N- and O-linked glycans (ConA, Jacalin and WGA) were indistinguishable; however, a number of unexpected differences were noted (Figure 4A). A substantial increase in bisecting GlcNAc (PHA-E) was seen along with decreased core and branched fucosylation (LCA, PSA and AAL). Increased α2,3-sialylation (MAL-II) and N-glycan branching (PHA-L and DSL) were also apparent in H-cKO samples.
Fig. 4.
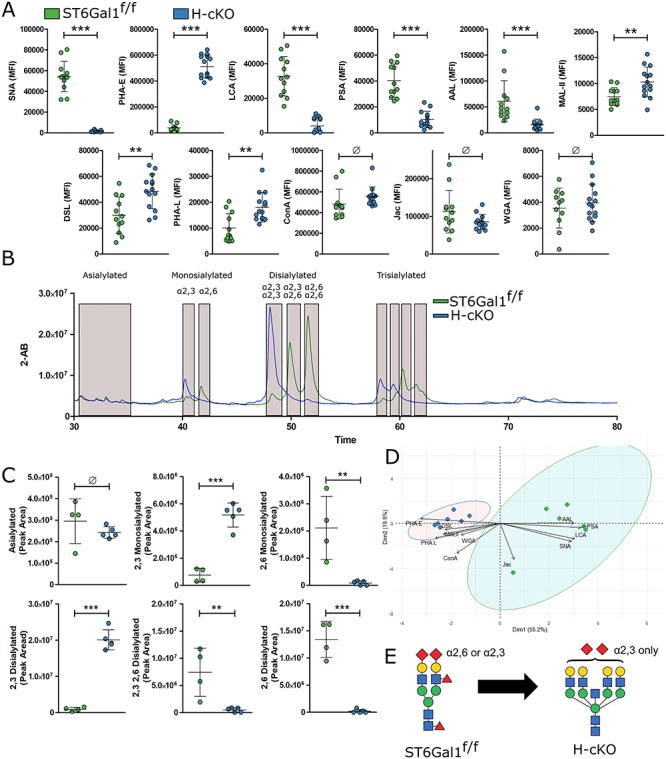
Hepatocyte conditional knockout of ST6Gal1 leads to many changes in circulatory glycoprotein carbohydrate structures. (A) Results of a multiplexed panel of lectins applied in a multiplexed ELISA format, covering a broad specificity of glycoforms. Stains included SNA, MAL-II, ConA, Jac, WGA, DSL, PHA-E, PHA-L, AAL, PSA and LCA. (B) N-linked glycans from total plasma protein were released, labeled, separated and quantified by HILIC with 2-AB labeling. (C) Quantification of the above labeled peaks demonstrated the clear shift from α2,6 sialylated species in the ST6Gal1f/f mouse to α2,3 sialylated species in the H-cKO mouse. (D) PCA analysis demonstrates that H-cKO plasma glycosylation differed dramatically from ST6Gal1f/f glycosylation in a distinct, consistent pattern. (E) Generalized characteristics of complex N-linked glycan changes included a loss of α2,6 sialic acid, but a maintenance of total sialic acid, as well as loss of core and/or branched fucose, increased branching and increased bisection. All mice were 8–12 weeks of age. Student’s T test, **P < 0.01, ***P < 0.001, Ø: P ≥ 0.05.
Since α2,6- and α2,3-linked sialylation often compete for the same underlying glycan substrates, increases in MAL-II staining associated with decreases in SNA could be expected. To determine the extent to which α2,3-linked sialylation compensated for the loss of α2,6-linked sialylation, we performed quantitative hydrophilic liquid chromatography (HILIC) on isolated N-glycans from total plasma glycoprotein samples. We found a strong shift away from α2,6 sialylation and towards α2,3 sialylation in the H-cKO plasma, although the total amount of sialic acid was similar (Figure 4B and C). Multi-dimensional analysis further demonstrated a clear and strong separation between the H-cKO and ST6Gal1f/f control mice (Figure 4D) dominated by a single axis (principle component 1). These data illustrate a global shift from smaller, α2,6-sialylated and fucosylated glycans to larger (more branching), bisected and α2,3-sialylated and glycans lacking fucose (Figure 4E), which is reminiscent of glycans associated with an assortment of pathologies, inflammation and diseases (Blomme et al. 2009; Blomme et al. 2011; Novokmet et al. 2014; Clerc et al. 2016).
With unexpected changes to plasma glycoprotein glycans in the young adult H-cKO mice, we performed transcriptomic analysis of liver samples to better understand the origin of those differences. We found more than 200 differentially expressed genes (DEGs) between H-cKO and ST6Gal1f/f control mice (Figures 5A and B and S1). Surprisingly, none of these genes are related to protein glycosylation (Figure 5C and D). Not a single glycosyltransferase, glycosidase or other gene implicated in glycan synthesis, including nucleotide-sugar transporters or enzymes responsible for their synthesis, was significantly altered. These data suggested that the changes in glycosylation were more likely associated with underlying and indirect metabolic differences not reflected in the transcriptome. Consistent with this interpretation, gene ontology (GO) enrichment analysis of the top 50 differentially regulated genes revealed a number of patterns, including pathways involved in cellular redox, lipid synthesis and storage, molecules associated with iron binding, regulation of gluconeogenesis and the acute phase response (Figures 5 and S2–S4). Gene set enrichment analysis (GSEA) further highlighted a number of other metabolic and catabolic pathways, including MTOR signaling (Figure 5E), glycolysis (Figure 5F), fatty acid metabolism (Figure 5G), xenobiotic compound processing (Figure 5H) and peroxisome (Figure 5I). Finally, both TGFβ (Figure 5J) and complement (Figure 5K) pathways were altered, but there was no detected immune phenotype identified that might suggest an inherent proinflammatory bias in these mice.
Fig. 5.
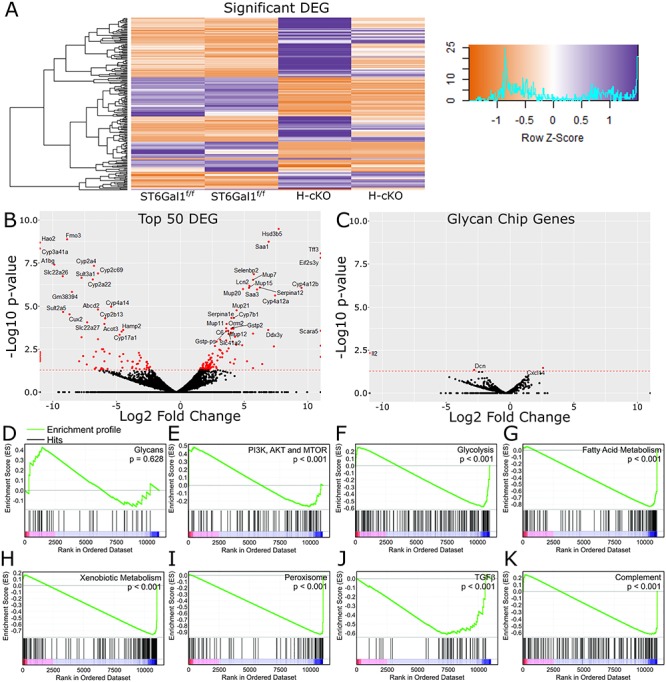
Changes in ST6Gal1 expression leads to broad liver transcriptional change. (A) RNA-seq analysis from young 8–12-week-old ST6Gal1f/f or H-cKO mouse liver samples, showing all 214 differentially expressed genes identified. (B) Volcano plot labeled with the top 50 differentially expressed genes. (C) Volcano plot of genes implicated in the synthesis of N-glycans, showing a distinct lack of change. (D–K) Gene set enrichment analysis (GSEA) confirmed a lack of change in glycosylation (D), but a stronger than expected association and enrichment with several metabolic pathways, including mTOR (E), glycolysis (F), fatty acid metabolism (G), processing of xenobiotic compounds (H) and peroxisome associated genes (I). Two immune pathways, TGFβ (J) and complement protein expression (K) are likewise associated.
With the variety of redox and metabolic changes present in young H-cKO mice revealed by transcriptomics, we hypothesized that H-cKO mice may show an abnormal phenotype if given a high fat diet (HFD). ST6Gal1f/f and H-cKO mice were placed on a 60% fat diet for 15 weeks starting at approximately 8 weeks of age and compared to animals on control chow. A clear buildup of fat in the liver of HFD mice was visible by H&E and Oil Red O staining, although no clear differences between control ST6Gal1f/f and H-cKO mice were observed (Figure 6A). The HFD mice gained considerable weight, with males at 200% their starting weight and females reaching 150% by the end of the experiment (Figure 6B and C). Accounting for grams of food eaten, there were no differences between the groups, but when the food was scaled for caloric content, it is clear that HFD mice consumed about 50% more calories per day than the controls (Figure 6D). Nonfasting blood glucose levels were also monitored throughout the time course of the study. We found no statistically significant change in male or female HFD mice compared to normal diet (Figure 6E and F); however, glucose challenge of fasting mice demonstrated that obese HFD male (Figure 6G), but not female (Figure 6H), mice were unable to control blood glucose normally. Mirroring the pathology (Figure 6A), no difference was seen between ST6Gal1f/f and H-cKO mice in these measurements (Figure 6B–H).
Fig. 6.
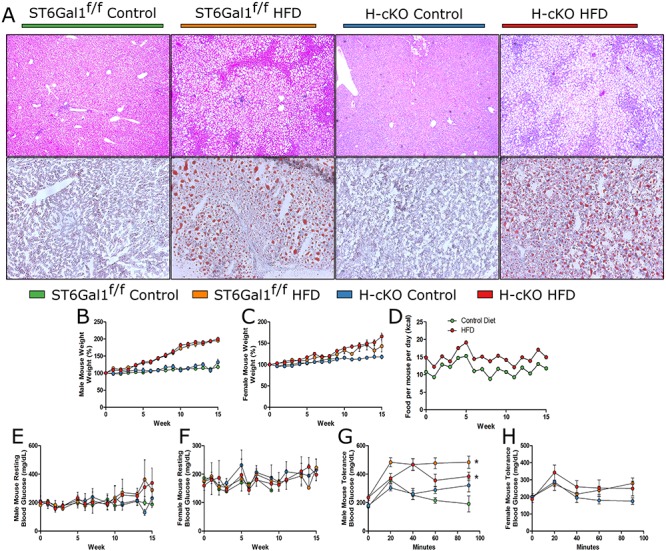
High fat diet leads to fatty liver in both ST6Gal1f/f and H-cKO mice. (A) Histological analysis via H&E and Oil Red O demonstrates severe fat accumulation in both the ST6Gal1f/f and H-cKO mice on HFD. (B–C) Male mice on HFD gained a considerable (~80%) more weight than their control diet counterparts, to about 200% their initial weight. Female mice on HFD gained about 150% of their initial weight. Weight gain for either group was not dependent on genotype. (D) HFD mice ate approximately 50% more calories than control diet mice. (E–F) Resting blood glucose measurements for male and female mice were not significant impacted by genotype or HFD. Male (G) but not female (H) HFD mice showed significant glucose intolerance in fasting mice challenged with glucose. Mice were approximately 23–26 weeks of age at harvest. AUC, Student’s T test, *P < 0.05.
Analyses of blood chemistry showed that all of the mice in the study maintained analyte levels within normal bounds (Figure 7A), although bilirubin (TBIL) was slightly elevated in H-cKO mice, regardless of diet, compared to ST6Galf/f controls. Glucose (GLU) and aspartate aminotransferase (AST), a marker of liver damage and stress, also trended up in both HFD groups and the control H-cKO. These data potentially suggest the development of subclinical liver stress in the control H-cKO mice between 12 weeks (Figure 3B) and approximately 26 weeks (Figure 7A) of age. None of these differences reached statistical significance.
Fig. 7.
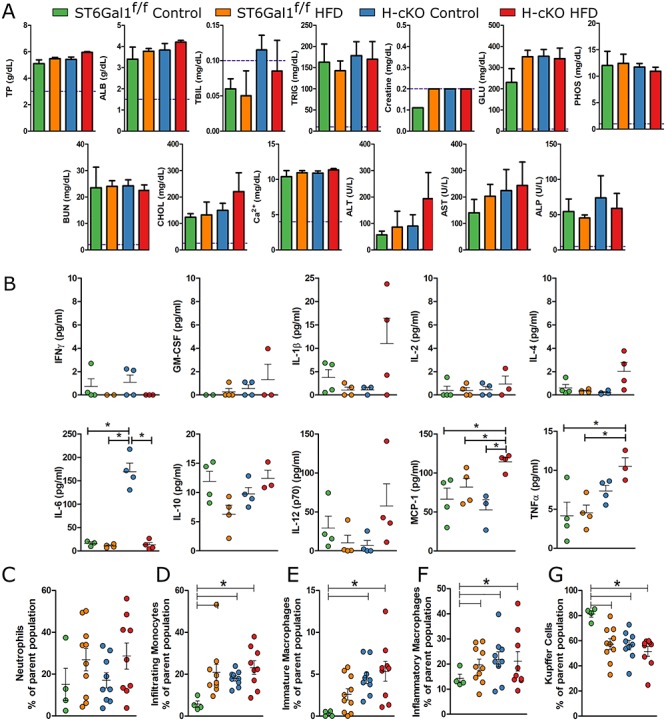
High fat diet study reveals inflammatory profile in H-cKO and HFD mice. (A) Blood chemistry analysis showed no clinical level blood chemistry perturbation in HFD mice, although trends are that TBIL, ALT and AST may be increased in H-cKO mice. (B) Cytokine analyses on the plasma of the ST6Gal1f/f and H-cKO mice on control or HFD revealed several cytokines associated with NAFLD or liver inflammation (IL-6, MCP-1, TNFα) were increased in H-cKO mice. (C) Neutrophil infiltration of livers was not different. (D–G) Flow cytometric profiling of CD45+CD11b+Ly6C− cells from the liver reveals a clear trend away from anti-inflammatory Kupffer cells and a trend towards infiltrating and proinflammatory macrophages in HFD mice but also in control H-cKO mice. Mice were approximately 23–26 weeks of age at harvest. Student’s T test, *P < 0.05, Ø: P ≥ 0.05. On each graph, each dot represents one mouse.
In measuring circulating cytokine concentrations, plasma-localized IFNγ, GM-CSF, IL-2, IL-10 and IL-12p70 were not different between all groups (Figure 7B). In HFD H-cKO mice, IL-1β and IL-4 trended up but did not reach significance. In contrast, TNFα and MCP-1 were both increased in HFD H-cKO mice compared to ST6Gal1f/f mice on either diet. Perhaps most interestingly, we discovered that IL-6 was significantly elevated in control diet H-cKO mice, but none of the other groups (Figure 7B). Similar to the AST levels, IL-6 may reflect an underlying and age-dependent liver stress in H-cKO mice, as it has long been associated with liver inflammation and the APR.
Flow cytometry was used to assess liver leukocyte infiltration and phenotype in order to determine whether nonalcoholic steatohepatitis (NASH) had developed in these mice. We found no differences in CD45+CD11b+Ly6G+ neutrophil recruitment (Figure 7C). In addition, CD45+CD11b+Ly6G− immune cells were sorted into Ly6C+F4/80−-infiltrating monocytes, Ly6C+F4/80+ proinflammatory macrophages, Ly6C−F4/80+ Kupffer cells or Ly6C−F4/80− patrolling monocytes (Figures 7 and S1) (Stijlemans et al. 2015). In ST6Gal1f/f animals, HFD induced increases in infiltrating monocytes (Figure 7D), patrolling monocytes (Figure 7E) and proinflammatory/M1 macrophages (Figure 7F) with a concomitant decrease in anti-inflammatory/M2 Kupffer cells (Figure 7G). This is consistent with the development of NASH (Morinaga et al. 2015). Interestingly, we found that H-cKO mice showed the same NASH phenotype regardless of diet. Together with the blood chemistry trends (Figure 7A) and cytokine production (Figure 7B), these data strongly suggested that by 26 weeks of age H-cKO mice develop subclinical but detectable liver stress and inflammation and that HFD did not exacerbate this effect.
Our data has shown that young, 12-week-old adult H-cKO mice have significant changes in plasma glycoprotein glycosylation (Figure 4) and transcriptional reprogramming suggestive of metabolic differences (Figure 5); however, these mice had normal blood chemistry (Figure 3A and B) and cellularity (Figure 3C), and no observable liver pathology (Figure 3D). By the age of 26 weeks, however, blood chemistry begins to change (Figure 7A), high concentrations of IL-6 are released into the circulation (Figure 7B) and significant changes in monocyte and macrophage populations (Figure 7D–G) are seen, collectively suggesting that glycosylation changes lead to abnormal metabolism and low-grade but progressive inflammation. In order to better characterize this trend, H-cKO and ST6Gal1f/f control mice were allowed to age to 1 year under normal housing and feeding conditions.
We discovered that old H-cKO mice developed severe nonalcoholic fatty liver disease (NAFLD) with 100% penetrance across both sexes, although no difference in overall weight was seen. Liver histology by H&E (Figure 8A) and Oil Red O (Figure 8B) revealed a clear and dramatic buildup of fat droplets in the H-cKO livers, although blood chemistry remained within normal bounds (Figure 8C). Twelve-week- and 52-week-old ST6Gal1f/f and H-cKO mice cytokine concentrations in the plasma were also measured. As expected, 12-week-old mice showed no IL-6 above baseline, but remarkably, the 52-week-old H-cKO mice also did not have elevated IL-6 (Figure 8D). In fact, none of the cytokines tested were statistically difference between the groups. Finally, flow cytometric analysis of the monocyte and macrophage population showed a significant increase in infiltrating monocytes (Figure 8E), some increase in patrolling monocytes (Figure 8F), no change in inflammatory macrophages (Figure 8G) and a concomitant decrease in Kupffer cells (Figure 8H) when comparing 12-week- vs. 52-week-old H-cKO mice and H-cKO vs. ST6Gal1f/f controls, all of which suggests a dysregulation of liver tolerance and a skew towards inflammation. As with younger mice, no difference was apparent in the neutrophil population (Figure 8I). These data demonstrate that loss of ST6Gal1 expression in hepatocytes spontaneously leads to progressive liver dysfunction characteristic of NAFLD with NASH.
Fig. 8.
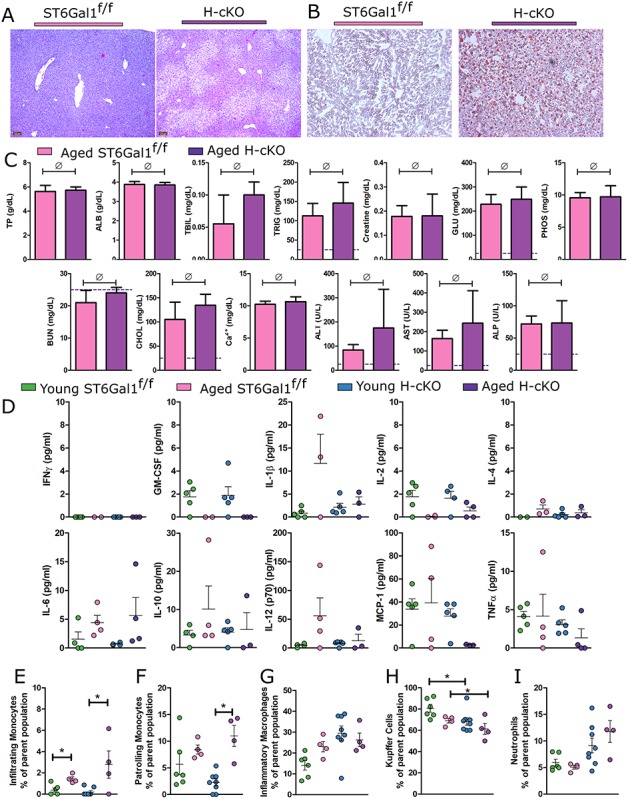
Aged H-cKO mice develop spontaneous fatty liver disease. (A) H&E imaging and (B) Oil Red O staining of liver tissues processed from aged mice (approximately 52 weeks of age) demonstrate NAFLD pathology. (C) Blood chemistry analysis revealed no elevation of parameters beyond those expected in a healthy mouse. (D) A 10-plex panel of proinflammatory cytokines was probed for signs of inflammation in the plasma of the ST6Gal1f/f and H-cKO mice at young or old age. (E) Analysis of PMN infiltration into liver revealed no differences. (F–I) Flow cytometric profiling of CD45+CD11b+Ly6C− cells from liver reveals a decrease in Kupffer cells and an increase in proinflammatory associated macrophages in H-cKO mice compared to either their young (8–12 weeks of age) or ST6Gal1f/f controls. Student’s T test, *P < 0.05, ∅: P ≥ 0.05. On each graph, each dot represents one mouse.
Discussion
In this study, we report the creation of a mouse carrying a hepatocyte-specific conditional knockout of the α2,6-sialyltransferase ST6Gal1 (H-cKO). In-depth analysis verified that this mouse completely lacked ST6Gal1 expression in hepatocytes, but not in other cells or tissues in the liver or elsewhere. In young adult mice, we found a lack of detectable pathology in any tissue, including the liver, and normal leukocyte distribution and phenotype in the liver and blood. However, there was a profound change not only in the sialylation pattern of the liver tissue and secreted circulatory glycoproteins but also in the fucosylation, bisecting GlcNAc and N-glycan branching by lectin ELISA. Remarkably, these changes are not associated with detectable alterations in the transcriptional regulation of any glycosyltransferase, glycosidase, nucleotide-sugar transporter or other enzymatic pathways directly involved with protein glycosylation. This raises the possibility that a complex network of glycosyltransferases exists in the Golgi, such that with the absence of a single glycosyltransferase, significantly different glycoforms can be generated without altering other glycosyltransferase levels. Perhaps more importantly, our findings demonstrate that transcriptional changes, or lack thereof, cannot be reliably used as a surrogate for regulated changes in protein glycosylation within a cell.
There are a number of potential explanations for how the loss of α2,6-linked sialylation in hepatocytes and their secreted glycoproteins alters glycan composition beyond sialylation. It is possible that the Golgi apparatus is disrupted or reorganized in ways that promote differential glycan maturation or that the behavior of Golgi enzymes, themselves glycoproteins, is influenced by their own sialylation pattern (Freeze and Ng 2011). It is also possible that sialylation of substrate glycans increases or decreases Golgi enzyme function through altering substrate binding and specificity, as shown in structural findings demonstrating the potent influence of adjacent substrate sugars on enzymatic activity (Kadirvelraj et al. 2018). For example, MGAT3, the enzyme responsible for adding bisecting GlcNAc, may be inhibited by α2,6-linked sialylation, and thus loss of ST6Gal1 may promote greater MGAT3 activity, and subsequently more bisected glycans, within the cell. Likewise, FUT8 may prefer α2,6-sialylated species in vivo, leading to a reduction in α1,6 core fucosylation in ST6Gal1-null mice. The more likely explanation, however, is that changes in the underlying metabolism of the cell are driving the differences (Blomme et al. 2009). Consistent with this notion, transcriptomic analysis revealed significant changes in a number of metabolic pathways, including fatty acid metabolism, glycolysis and gluconeogenesis. As an example, if more glucose is shunted into the nucleotide-sugar donor pathway, the increased levels of UDP-GlcNAc may cause increased enzymatic activity of MGAT enzymes, resulting in increased N-glycan branching.
By virtue of the anti-inflammatory environment established by Kupffer cells (Bilzer et al. 2006; Movita et al. 2012; Crispe 2014), the liver is intimately involved in the regulation of systemic immune tone (Robinson et al. 2016), the prevention of sepsis (Yan et al. 2014) and the promotion of tolerance (Thomson and Knolle 2010). In fact, Kupffer cells have a unique, strongly immune nonreactive phenotype (Movita et al. 2012) and have been implicated in the promotion of dietary tolerance (Crispe 2014), avoidance of obesity associated inflammation (Neyrinck et al. 2009) and generation of regulatory T cells (You et al. 2008). The emergence of high concentrations of IL-6 in the plasma, which is associated with liver inflammation (Schmidt-Arras and Rose-John 2016), and the shift toward proinflammatory M1 macrophages and away from Kupffer cells in 26-week-old H-cKO mice, is therefore a key sign that these mice are becoming inflamed with age. Indeed, we found similar changes in the macrophage population in wild-type and H-cKO mice fed a high fat diet, further supporting a model in which H-cKO mice have underlying metabolic dysfunction influencing the immune tone of the mice.
Nonalcoholic fatty liver disease (NAFLD) is the first stage of liver disease that is strongly associated with metabolic syndrome and diabetes. The low-grade inflammatory responses that are linked to obesity and over nutrition are termed metabolic inflammation, or simply “metaflammation,” and represent an important but poorly understood mechanism leading to many deleterious health outcomes (Caputo et al. 2017). The liver is key to metaflammation, as it is involved in the regulation of glucose and insulin tolerance (Han et al. 2016) and the production of systemic inflammatory mediators (Robinson et al. 2016), but the buildup of fat, loss of immune tolerance and eventual loss of normal liver function are common. Given the metabolic changes evident in the H-cKO mouse, it is clear that this mouse is susceptible to liver metaflammation, even at a relatively young age. In the H-cKO metaflammation phenotype, there is a clear disruption in CYP family enzymes involved in both fatty acid oxidation and xenobiotic processing, with 19 CYP family member enzymes differentially regulated in the livers of our mice. This indicated that there is some disrupted signaling in the regulation of these enzymes—one possibility is that the change in cell surface glycosylation of cell surface and plasma proteins has increased the uptake of protein by the ASGPR, leading to increased protein turnover via synthesis and breakdown in hepatocytes, resulting in cellular stress. Although incompletely understood, the H-cKO phenotype therefore demonstrates a previously unknown connection between liver glycans, plasma protein carbohydrates and the metabolic state of the liver, collectively leading to hepatic dysregulation and NAFLD.
Untreated, NAFLD can develop into nonalcoholic steatohepatitis (NASH), liver cirrhosis or hepatocellular carcinoma (HCC). With an estimated global prevalence of over 25%, NAFLD represents a massive health burden (Younossi et al. 2016). The progression from NAFLD to NASH in human patients can take many years. In the H-cKO mice, the glycan-initiated metabolic dysfunction in the liver took approximately half of the normal 2-year mouse lifespan to develop fully into NAFLD with NASH when provided a normal healthy diet. IL-6 has a complicated relationship with liver regulation and homeostasis in that it is well established as a key element in the APR, can be produced by hepatocytes (Norris et al. 2014) and has been associated with both protection (Cressman et al. 1996) and progression (Yamaguchi et al. 2010) in NAFLD (Braunersreuther et al. 2012). Although mice were not allowed to age beyond 52 weeks, the loss of IL-6 production at 1 year is consistent with chronic IL-6 signaling, which has been associated with the dysregulation of hepatic metabolism, the progression of NAFLD and the production of other inflammatory cytokines including TNFα (Gavito et al. 2016). It could also signal the destruction and dysfunction of hepatocytes due to lipotoxicity concomitant with NAFLD and NASH and suggests continued progression towards cirrhosis and potentially HCC is likely in H-cKO mice.
Our study places the regulation of liver and circulatory protein glycosylation near the heart of NAFLD and NASH through its effects upon normal metabolic pathways. Indeed, loss of ST6Gal1 in hepatocytes had a profound impact upon liver physiology, although the H-cKO mouse did not phenocopy another ST6Gal1 mutant mouse model. Mice with a disrupted P1 promoter region upstream of ST6Gal1 (ΔP1) has been described as an ablation of liver-released circulatory ST6Gal1 (Dalziel et al. 1999; Appenheimer et al. 2003; Lau et al. 2006; Jones et al. 2010; Nasirikenari et al. 2010; Jones et al. 2012; Nasirikenari et al. 2014), yet the originating publication describing this mouse showed only a reduction of some, but not all, ST6Gal1 transcripts in the liver and a reduction but not ablation of circulatory activity. Changes in ST6Gal1 expression were also not confined to the liver (Appenheimer et al. 2003). The discrepancies between H-cKO and ΔP1 mouse phenotypes are compounded by the ΔP1 mouse hepatocyte and circulatory glycoprotein glycans, insofar as they maintain plasma protein SNA staining and have no other major changes (Appenheimer et al. 2003). Combining observations of the altered myelopoiesis reported for the ΔP1 ST6Gal1 mouse (Lau et al. 2006; Nasirikenari et al. 2010) juxtaposed with the change in hematopoietic cell surface glycosylation (Nasirikenari et al. 2014), lack of plasma glycome changes and no observation of liver disease, we hypothesize that the phenotype reported for the ΔP1 ST6Gal1 mouse may be the result of the reduction in ST6Gal1 in other tissues or cells, as opposed to the complete hepatocyte-specific ablation in the H-cKO mice.
There are 24 major plasma glycoproteins of hepatic origin, comprising at least 20 mg/mL of the total in human plasma (Clerc et al. 2016). Given the links between the glycome and disease (Zhou et al. 2018; Reily et al. 2019), plasma glycosylation has become a target for biomarker research. Our work recently revealed that changes in glycosylation can predict the onset of cardiovascular disease among HIV-infected individuals (Oswald et al. 2019), whereas work dating back to 1993 has shown that glycoform-specific variations of haptoglobin are associated with rheumatoid arthritis (Thompson et al. 1993) and work as recent as 2017 has identified novel glycan biomarkers of pancreatic cancer (Barnett et al. 2017). If loss of α2,6-sialylation among hepatocytes and hepatic plasma glycoproteins preceded inflammation, changes in metabolism, liver dysfunction and ultimately the development of NAFLD with NASH, then glycomic changes characterizing multiple disease states may be playing active roles in disease progression rather than simply acting as disease markers. Here, our direct evidence of glycosylation influencing the development of metaflammation and metabolic dysfunction paves the way for future studies that dissect the relationship between diet and immunity that underlies much of the disease burden seen in the modern world.
Experimental procedures
Animal care and use
ST6Gal1f/f (stock 006901) and Alb-Cre (stock 003574) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in the Case Western Reserve University Animal Resource Center in pathogen free conditions in accordance with Institutional Animal Care and Use Committee mandates. The H-cKO (ST6Gal1f/f × Alb-Cre+/+) was generated by crossing ST6Gal1f/f mice with Alb-Cre mice, crossing progeny and maintaining a homozygous strain. All mice were on the C57Bl/6 background. All mouse experiments were designed to include mice of male and female sex-aged between 8 and 12 weeks, except where noted. Mice were considered to be at old age between 44 and 52 weeks. Mice were labeled via tattoo on their ears shortly after weaning.
Sample collection
Mice were sacrificed using carbon dioxide per institutional animal care guidelines. Blood was drawn from mice via terminal cardiac puncture immediately following euthanasia. For serum, blood was allowed to clot at room temperature for 1 h; for plasma, blood was drawn into 0.1 volumes of 3.2% sodium citrate to prevent coagulation and allowed to rest at room temperature for 15 min. Blood was centrifuged at 2000 × g for 15 min, and the plasma or serum portion was aspirated from the tube. For cellular analyses from blood, blood was collected in 0.1 volumes of 3.2% sodium citrate, allowed to rest and room temperature for 15 min and then processed for flow cytometry or analyzed on the Hemavet system (Drew Scientific). Organ biopsies were taken from the mice and immediately frozen in liquid nitrogen for either RNA or protein preparation, fixed in 10% formalin for FFPE analysis or embedded in Tissue-Tek Optimal Cutting Temperature (OCT, Sakura) compound and frozen.
PCR
Tail DNA was purified using the DNeasy Blood and Tissue kit (Qiagen) according to manufacturer instructions. Genotypes of mice were tested using protocols published on The Jackson Laboratory website.
qPCR
RNA for qPCR was extracted from the indicated organ using the RNeasy Blood and Tissue Kit (Qiagen) according to manufacturer instructions. mRNA reverse transcription to cDNA was performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). qPCR was performed using SYBR Green Supermix (Bio-Rad) in a CFX96 (Bio-Rad) Thermal-Cycler. Primers were as follows:
ST6Gal1 forward GTCCTGGTCTTTCTCCTGTTT
ST6Gal1 reverse: GCTCTTCGGCATCTGGAATA
GAPDH forward ACAGTCCATGCCATCACTGCC
GAPDH reverse GCCTGCTTCACCACCTTCTTG
Necropsy services
The mouse necropsy was performed at the Charles River Laboratories. Mice were shipped to the facility and sacrificed on-site. Analyses performed were gross observation of tissues, removal of tissue biopsies for FFPE, complete blood counts and a panel of blood chemistry. Additional plasma samples were generated in lab and sent to Charles River Laboratories for blood chemistry analysis.
Bright-field microscopy
FFPE embedding, sectioning and H&E staining were performed at the CWRU Cancer Center Histology Services Core via standard procedure. Frozen sectioning and Oil Red O staining were performed at AML Laboratories. Light micrographs were acquired on a Leica DM IL LED Microscope (090–135.002) using a 10× eyepiece and either a 4× or 10× objective. All images displayed are representative of at tissues stained from at least three distinct biological replicates.
Confocal microscopy
Sections of tissue were deparaffinized and rehydrated as described previously (Jones et al. 2016). Slides were blocked in Carbohydrate-Free Blocking Solution (Vector) and stained with SNA-FITC (Vector) at 0.5 μg/mL for 1 h. The background signal was reduced via TrueVIEW Autofluorescence Quenching Kit (Vector). Slides were fixed using VECTASHIELD Hardset Antifade Mounting Medium (Vector). Confocal imaging was performed on a SP5 Laser Scanning Confocal Microscope (Leica). Quantification of mean pixel intensity was performed using ImageJ.
Western blotting
Tissue biopsies (as collected above) were weighed to extract protein from 5–10 mg of tissue at a time using 0.5–1 mL of RIPA Buffer with protease inhibitor cocktail (Roche). Frozen tissue was added to a Dounce homogenizer with buffer, plunged with a grinding motion 40–50 times, drawn through a 27-gauge needle five times and spun at 15,000 × g for 5 min at 4°C, and the supernatant collected. Western blotting performed as previously described (Jones et al. 2016). Briefly, 1 μL of plasma or 20 μg of tissue homogenate was separated using SDS-PAGE, transferred to Immobilon-FL PVDF membranes (Millipore, IPFL0010), blocked with Carbo-Free Blocking Solution (Vector, SP-5040) and stained with SNA-FITC (0.5 μg/mL; Vector, FL-1301). Blots were imaged using ImageQuant LAS 4000 mini (GE).
Flow cytometry
Flow cytometry was performed as described previously (Jones et al. 2016). Briefly, tissue biopsies of liver were mashed through a 100-micron filter, and cells were resuspended. Liver samples were treated with red blood cell lysis buffer and separated using a 33% Percoll solution as has been described (Stijlemans et al. 2015). Cells were stained with anti-CD45-FITC (BioLegend, 147710), anti-CD11b-Brilliant Violet 605 (BD Biosciences, 562015), anti-Ly6G-Alex Fluor 647 (BioLegend, 127610), anti-F4/80-Brilliant Violet 711 (BD Biosciences, 565612) or anti-Ly6C-biotin (BioLegend, 128003) and Streptavidin-APC-eFluor 780 (eBioscience, 47-4317-82). Cells were washed, then resuspended in MACS buffer (2 mM EDTA, 0.5% FBS in PBS). Cells were analyzed using the Attune NxT in the Cytometry and Imaging Microscopy Shared Resource of the Case Comprehensive Cancer Center.
Multiplex lectin ELISA
Multiplex lectin ELISA was performed as previously described (Oswald et al. 2019). Briefly, samples were coated to a 384-well high-binding ELISA plate (Greiner Bio-One, 784077) at 0.5 μg/mL in carbonate coating buffer (0.1 M NaHCO3, 0.03 M NaCO3, pH 9.5) overnight at 4°C. The plates were blocked with Carbohydrate-Free Blocking Solution (Vector, SP-5040), then incubated with divalent ion solution (1 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2) for 10 min. Lectins were diluted in Carbohydrate-Free Blocking Solution and incubated for 1 h. Europium-conjugated streptavidin (Perkin Elmer 1244-360) was diluted in europium dilution buffer (50 mM Tris, 150 mM NaCl, 20 μM EDTA) to 100 ng/mL and incubated for 1 h. Enhancement solution (Perkin Elmer, 4001-0010) and analyzed using time-resolved fluorescence on a Victor 3V 1420 multilabel counter (Perkin Elmer).
Glycan 2-AB labeling and HILIC chromatography
Glycan 2-AB labeling was performed as described elsewhere (Xia et al. 2005). Briefly, glycoproteins were denatured by heating to 95°C for 5 min, then digested with trypsin (Thermo Fisher) overnight at 37°C. Trypsin was inactivated by heating to 95°C for 5 min. N-Glycans were enzymatically removed from the sample by treatment with PNGase F (NEB) overnight at 37°C. Glycans were separated from peptides by reversed-phase chromatography through a C18 cartridge (Thermo Fisher, 60108-390) according to manufacturer instructions, then dried in a Vertis lyophilizer overnight. Purified glycan was labeled using 50 μg/μL 2-AB and 60 μg/μL sodium cyanoborohydride in a 3:7 solution of acetic acid and DMSO at 65°C for 3 h.
Hydrophilic interaction liquid chromatography (HILIC) was performed using a Dionex ICS-5000+ system and a Zorbax NH2 column (Agilent 880952-708) and 10% to 80% gradient of acetonitrile and 100 mM ammonium formate pH 4.4 over 80 min. 2-AB fluorescence was detected with an excitation of 330 nm and emission of 420 nm.
High fat diet study
Mice were fed on either a 60% fat diet (Research Diets, D12492) or a 10% control diet (Research Diets, D12450B) from an age of 8 weeks. At the beginning of each week, the mice were moved to a clean cage and their food replaced. At this time, the mice were weighed and their resting blood glucose was measured.
Glucose testing
Mouse blood glucose was tested by creating a small cut in the tip of a mouse’s tail using a razor blade, then measuring the glucose in the drop of blood on a CareTouch Glucometer (Future Diagnostics LLC). For standard measurements, mice were not fasted prior to procedure. For glucose tolerance measurements, mice were fasted for 8 h prior to measuring blood glucose. Glucose challenge was administered at 1 mg/kg via i.p. injection, and blood glucose measured at 15, 30, 60 and 90 min.
Statistical methods
Statistical analyses were performed in excel or R. Data was considered statistically significant at *P < 0.05, **P < 0.01, ***P < 0.001 for Student’s T tests. R plots were generated using the ggplot package. Heatmaps were created using the heatmap.2 package. RNA-seq analysis was performed by LC Sciences via their PolyA RNA sequencing service. GSEA was performed using the 2.2.4 version of GSEA from the broad institute, as has been described (Daly et al. 2003; Subramanian et al. 2005) using the Diff_of_Classes metric against the Hallmark, KEGG or GO databases. Additional plots were created using version 5 of GraphPad Prism.
Supplementary Material
Acknowledgements
We thank Carlos A. Alvarez and Julie Y. Zhou for endless valuable scientific feedback and input and their assistance in editing and proofreading of the manuscript. We thank Jennifer Mikulan for the histological technical support. This research was supported by the Tissue Resources Shared Resource of the Case Comprehensive Cancer center as well as the Cytometry and Imaging Microscopy Core Facility of the Case Comprehensive Cancer center (P30CA043703). We also thank Alex Huang, MD, PhD for support in H&E and Oil Red O histological imaging.
Abbreviations
ST6Gal1, β-galactoside α2,6-sialyltransferase 1; H-cKO, hepatocyte-specific conditional knockout of ST6Gal1; LPS, lipopolysaccharide; ASGPR, asialoglycoprotein receptor; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; 2-AB, 2-aminobenzamide; HILIC, hydrophilic liquid chromatography; PCA, principal component analysis; SNA, Sambucus nigra agglutinin; MAL-II, Maackia amurensis lectin II; RCA120, Ricinus communis agglutinin I; WGA, wheat germ agglutinin; Jac, jacalin; Con A, concanavalin A; PHA-E, Phaseolus vulgaris erythroagglutinin; PHA-L, Phaseolus vulgaris leucoagglutinin; DSL, Datura stramonium lectin; LCA, Lens culinaris agglutinin; AAL, Aleuria aurantia lectin; HCT, hematocrit; RDW, red cell distribution width; HGB, hemoglobin; MCV, mean corpuscular volume; MPV, mean platelet volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; TP, total protein; ALB, albumin; TBIL, total bilirubin; TRIG, triglycerides; GLU, glucose; PHOS, phosphate; BUN, blood urea nitrogen; CHOL, cholesterol; ALT, alanine transaminase; AST, alanine aminotransferase; ALP, alkaline phosphatase; RBC, red blood cells; WBC, white blood cells; GI, gastrointestinal tract; HFD, high fat diet.
Funding
National Institute of General Medical Sciences (GM115234 to B.A.C.); National Institute of Allergy and Infectious Diseases (AI089474 to D.M.O.)
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
D.M.O., experimental design, data collection and analysis, manuscript writing; M.B.J., experimental design, manuscript editing; B.A.C., experimental design, data analysis, manuscript writing, funding.
References
- Adamczyk B, Tharmalingam T, Rudd PM. 2012. Glycans as cancer biomarkers. Biochim Biophys Acta - Gen Subj. 1820(9):1347–1353. [DOI] [PubMed] [Google Scholar]
- Adams JH, Sim BK, Dolan SA, Fang X, Kaslow DC, Miller LH. 1992. A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci. 89(15):7085–7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An HJ, Kronewitter SR, de Leoz MLA, Lebrilla CB. 2009. Glycomics and disease markers. Curr Opin Chem Biol. 13(5–6):601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenheimer MM, Huang RY, Chandrasekaran EV, Dalziel M, Hu YP, Soloway PD, Wuensch SA, Matta KL, Lau JTY. 2003. Biologic contribution of P1 promoter-mediated expression of ST6Gal I sialyltransferase. Glycobiology. 13(8):591–600. [DOI] [PubMed] [Google Scholar]
- Barnett D, Liu Y, Partyka K, Huang Y, Tang H, Hostetter G, Brand RE, Singhi AD, Drake RR, Haab BB. 2017. The CA19-9 and Sialyl-TRA antigens define separate subpopulations of pancreatic Cancer cells. Sci Rep. 7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilzer M, Roggel F, Gerbes AL. 2006. Role of Kupffer cells in host defense and liver disease. Liver Int. 26(10):1175–1186. [DOI] [PubMed] [Google Scholar]
- Blomme B, Van Steenkiste C, Callewaert N, Van Vlierberghe H. 2009. Alteration of protein glycosylation in liver diseases. J Hepatol. 50(3):592–603. [DOI] [PubMed] [Google Scholar]
- Blomme B, van Steenkiste C, Grassi P, Haslam SM, Dell A, Callewaert N, van Vlierberghe H. 2011. Alterations of serum protein N-glycosylation in two mouse models of chronic liver disease are hepatocyte and not B cell driven. Am J Physiol - Gastrointest Liver Physiol. 300(5):G833–G842. [DOI] [PubMed] [Google Scholar]
- Braunersreuther V, Viviani GL, Mach F, Montecucco F. 2012. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 18(8):727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo T, Gilardi F, Desvergne B. 2017. From chronic overnutrition to metaflammation and insulin resistance: Adipose tissue and liver contributions. FEBS Lett. 591(19):3061–3088. [DOI] [PubMed] [Google Scholar]
- Clerc F, Reiding KR, Jansen BC, Kammeijer GSM, Bondt A, Wuhrer M. 2016. Human plasma protein N-glycosylation. Glycoconj J. 33(3):309–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. 1996. Liver failure and defective hepatocyte regeneration in interleukin-6- deficient mice. Science (80- ). 274(5291):1379–1383. [DOI] [PubMed] [Google Scholar]
- Crispe IN. 2011. Liver antigen-presenting cells. J Hepatol. 54(2):357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispe IN. 2014. Kupffer cells in immune tolerance. Encycl Med Immunol. 623–628. [Google Scholar]
- Crocker PR, Paulson JC, Varki A. 2007. Siglecs and their roles in the immune system. Nat Rev Immunol. 7(4):255–266. [DOI] [PubMed] [Google Scholar]
- Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 34(3):267–273. [DOI] [PubMed] [Google Scholar]
- Dalziel M, Lemaire S, Ewing J, Kobayashi L, Lau JTY. 1999. Hepatic acute phase induction of murine β-galactoside α2,6 sialyltransferase (ST6Gal I) is IL-6 dependent and mediated by elevation of exon H-containing class of transcripts. Glycobiology. 9(10):1003–1008. [DOI] [PubMed] [Google Scholar]
- Freeze HH, Ng BG. 2011. Golgi Glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol. 3(9):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux P, Varki A. 1999. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology. 9(8):747–755. [DOI] [PubMed] [Google Scholar]
- Gavito AL, Bautista D, Suarez J, Badran S, Arco R, Pavón FJ, Serrano A, Rivera P, Decara J, Cuesta AL et al. 2016. Chronic IL-6 administration desensitizes IL-6 response in liver, causes hyperleptinemia and aggravates steatosis in diet-induced-obese mice. PLoS One. 11(6):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HS, Kang G, Kim JS, Choi BH, Koo SH. 2016. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med. 48(3):e218–e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harduin-Lepers A, Mollicone R, Delannoy P, Oriol R. 2005. The animal sialyltransferases and sialyltransferase-related genes: A phylogenetic approach. Glycobiology. 15(8):805–817. [DOI] [PubMed] [Google Scholar]
- Hennet T, Chui D, Paulson JC, Marth JD. 1998. Immune regulation by the ST6Gal sialyltransferase. Proc Natl Acad Sci U S A. 95(8):4504–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Jones MB, Ryan SO, Cobb BA. 2013. The regulatory power of glycans and their binding partners in immunity. Trends Immunol. 34(6):290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Nasirikenari M, Feng L, Migliore MT, Choi KS, Kazim L, Lau JTY. 2010. Role for hepatic and circulatory ST6Gal-1 sialyltransferase in regulating myelopoiesis. J Biol Chem. 285(32):25009–25017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Nasirikenari M, Lugade AA, Thanavala Y, Lau JTY. 2012. Anti-inflammatory IgG production requires functional P1 promoter in β-galactoside α2,6-sialyltransferase 1 (ST6Gal-1) gene. J Biol Chem. 287(19):15365–15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Oswald DM, Joshi S, Whiteheart SW, Orlando R, Cobb BA. 2016. B-cell-independent sialylation of IgG. Proc Natl Acad Sci U S A. 113(26):7207–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadirvelraj R, Yang JY, Sanders JH, Liu L, Ramiah A, Prabhakar PK, Boons GJ, Wood ZA, Moremen KW. 2018. Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc Natl Acad Sci U S A. 115(18):4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Hutfless S, Liu L, Bayless TM, Marohn MR, Li X. 2012. Serum anti-glycan antibody biomarkers for inflammatory bowel disease diagnosis and progression: A systematic review and meta-analysis. Inflamm Bowel Dis. 18(10):1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JTYY, Nasirikenari M, Segal BH, Ostberg JR, Urbasic A, Lau JTYY. 2006. Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal I. Blood. 108(10):3397–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux S, Groux-Degroote S, Cazet A, Dhaenens CM, Maurage CA, Caillet-Boudin ML, Delannoy P, Krzewinski-Recchi MA. 2010. Transcriptional regulation of the human ST6GAL2 gene in cerebral cortex and neuronal cells. Glycoconj J. 27(1):99–114. [DOI] [PubMed] [Google Scholar]
- Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB. 2015. Glycans in the immune system and the altered Glycan theory of autoimmunity: A critical review. J Autoimmun. 57:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer P, Geissler K, Ward M, Metcalf D. 1993. Recombinant human leukemia inhibitory factor induces acute phase proteins and raises the blood platelet counts in nonhuman primates. Blood. 81(12):3226–3233. [PubMed] [Google Scholar]
- Meyer SJ, Linder AT, Brandl C, Nitschke L. 2018. B cell Siglecs-news on signaling and its interplay with Ligand binding. Front Immunol. 9(December):2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi E, Nakano M. 2008. Fucosylated haptoglobin is a novel marker for pancreatic cancer: Detailed analyses of oligosaccharide structures. Proteomics. 8(16):3257–3262. [DOI] [PubMed] [Google Scholar]
- Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ et al. 2015. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes. 64(4):1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJM, Janssen HLA, Boonstra A. 2012. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J Leukoc Biol. 92(4):723–733. [DOI] [PubMed] [Google Scholar]
- Nasirikenari M, Chandrasekaran EV, Matta KL, Segal BH, Bogner PN, Lugade AA, Thanavala Y, Lee JJ, Lau JTY. 2010. Altered eosinophil profile in mice with ST6Gal-1 deficiency: An additional role for ST6Gal-1 generated by the P1 promoter in regulating allergic inflammation. J Leukoc Biol. 87(3):457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasirikenari M, Veillon L, Collins CC, Azadi P, Lau JTY. 2014. Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 sialyltransferase. J Biol Chem. 289(10):7178–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. 2009. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem Biophys Res Commun. 385(3):351–356. [DOI] [PubMed] [Google Scholar]
- Norris CA, He M, Kang LI, Ding MQ, Radder JE, Haynes MM, Yang Y, Paranjpe S, Bowen WC, Orr A et al. 2014. Synthesis of IL-6 by hepatocytes is a normal response to common hepatic stimuli. PLoS One. 9(4):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novokmet M, Lukić E, Vučković F, Durić Ž, Keser T, Rajšl K, Remondini D, Castellani G, Gašparović H, Gornik O et al. 2014. Changes in IgG and total plasma protein glycomes in acute systemic inflammation. Sci Rep. 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald DM, Sim ES, Baker C, Farhan O, Debanne SM, Morris NJ, Rodriguez BG, Jones MB, Cobb BA. 2019. Plasma glycomics predict cardiovascular disease in patients with ART-controlled HIV infections. FASEB J. 33(2):1852–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 274(1):305–315. [DOI] [PubMed] [Google Scholar]
- Reily C, Stewart TJ, Renfrow MB, Novak J. 2019. Glycosylation in health and disease. Nat Rev Nephrol. 15(6):346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MW, Harmon C, O’Farrelly C. 2016. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 13(3):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak LR, Miyamoto S, Lebrilla CB. 2013. Developments in the identification of glycan biomarkers for the detection of cancer. Mol Cell Proteomics. 12(4):846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Arras D, Rose-John S. 2016. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol. 64(6):1403–1415. [DOI] [PubMed] [Google Scholar]
- Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, Conner MG, Arend RC, Yoon KJ, Klug CA et al. 2016. The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 76(13):3978–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stijlemans B, Sparkes A, Abels C, Keirsse J, Brys L, Elkrim Y, Baetselier P, Beschin A, Ginderachter J. 2015. Murine liver myeloid cell isolation protocol. Bio-Protocol. 5(10):1–10. [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarés G, Radcliffe CM, Barrabés S, Ramírez M, Aleixandre N, Hoesel W, Dwek RA, Rudd PM, Peracaula R, de Llorens R. 2006. Different glycan structures in prostate-specific antigen from prostate cancer sera in relation to seminal plasma PSA. Glycobiology. 16(2):132–145. [DOI] [PubMed] [Google Scholar]
- Thompson S, Dargan E, Griffiths ID, Kelly CA, Turner GA. 1993. The glycosylation of haptoglobin in rheumatoid arthritis. Clin Chim Acta. 220(1):107–114. [DOI] [PubMed] [Google Scholar]
- Thomson AW, Knolle PA. 2010. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 10(11):753–766. [DOI] [PubMed] [Google Scholar]
- Varki A. 2008. Sialic acids in human health and disease. Trends Mol Med. 14(8):351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Gagneux P. 2012. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 1253(1):16–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J, Lee EU, McEntee K, Lai PH, Paulson JC. 1987. Primary structure of beta-galactoside alpha 2,6-sialyltransferase. Conversion of membrane-bound enzyme to soluble forms by cleavage of the NH2-terminal signal anchor. J Biol Chem. 262(36):17735–17743. [PubMed] [Google Scholar]
- Xia B, Kawar ZS, Ju T, Alvarez RA, Sachdev GP, Cummings RD. 2005. Versatile fluorescent derivatization of glycans for glycomic analysis. Nat Methods. 2(11):845–850. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, Okanoue T, Yoshikawa T. 2010. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Investig. 90(8):1169–1178. [DOI] [PubMed] [Google Scholar]
- Yan J, Song L, Shulin L. 2014. The role of the liver in sepsis. Int Rev Immunol. 33(6):498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Q, Cheng L, Kedl RM, Ju C. 2008. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 48(3):978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. 2016. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 64(1):73–84. [DOI] [PubMed] [Google Scholar]
- Yuan S, Li Q, Zhang Y, Huang C, Wu H, Li Y, Liu Y, Yu N, Zhang H, Lu G et al. 2015. Changes in anti-thyroglobulin IgG glycosylation patterns in Hashimoto’s thyroiditis patients. J Clin Endocrinol Metab. 100(2):717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JY, Oswald DM, Oliva KD, Kreisman LSC, Cobb BA. 2018. The Glycoscience of immunity. Trends Immunol. 39(7):523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.