

Abstract
Objective
Precise genetic analyses were conducted with ring finger protein 213 (RNF213) in relation to a particular clinical phenotype in Chinese patients with moyamoya disease (MMD) to determine whether heterozygosity is responsible for the early-onset and severe form of this disease.
Methods
A case–control study for RNF213 p.R4810K involving 1,385 Chinese patients with MMD and 2,903 normal control participants was performed. Correlation analyses between genotype and phenotype or different clinical features were also statistically explored.
Results
An obvious trend was observed: the carrying rate of RNF213 p.R4810K gradually decreased when moving from coastal cities in northeast, north, and east China to southern cities or inland areas. Higher frequencies of p.R4810K were observed in patients with MMD compared with control participants (odds ratio, 48.1; 95% confidence interval, 29.1–79.6; p = 1.6 × 10−141). In addition, the onset age of all patients with the GA and AA genotypes were lower than with the GG genotype, and the median onset age was 40.0, 36.0, and 11.5 years with GG, GA, and AA, respectively, thereby confirming that those with GA or AA could acquire MMD during early life stages. Patients with MMD with the GA genotype were more susceptible to posterior cerebral artery (PCA) involvement compared to those with the GG genotype (38.4% vs 23.3%, p = 8.3 × 10−7).
Conclusions
Strong evidence suggests that the carrying rate of RNF213 p.R4810K is closely related MMD risk in China and has given rise to an earlier onset age and more severe PCA involvement.
Moyamoya disease (MMD) is a nonatherosclerotic cerebrovascular structural abnormality that extends to all age groups and occurs worldwide, and a high prevalence has been observed in East Asian descendants. The etiology and pathogenesis of MMD remain unknown. The locus p.R4810K of finger protein 213 (RNF213) was first identified as a pathogenic gene in Japanese familial MMD by studies conducted by Liu et al.,1 and case–control correlation analyses in an East Asian population showed that p.R4810K was also a pathogenic gene of MMD in China, Japan, and Korea.1 Furthermore, some genetic studies have shown that the variant exists in 95% of Japanese familial cases, 80% of Japanese and Korean sporadic cases, and 20% of Chinese sporadic cases.2–9 In addition, results regarding the clinical relevance of p.R4810K indicated that the AA genotype corresponded to a significantly earlier onset age and a more severe form of MMD,5,6,10,11 and Miyatake et al.6 reported that the GA genotype predicts a relatively late onset age and mild clinical course in Japanese patients. It is well-known that MMD is identified as a typical unbalanced disease,12–14 and prevalence of clinical characteristics, long-term outcomes, and other specific demographic features of Chinese MMD also differ from those of other ethnicities.15 However, potential effects related to RNF213 p.R4810K on various clinical phenotypes of MMD in China have yet to be systematically studied. This study aimed to clarify whether RNF213 p.R4810K was potentially associated with clinical phenotypes based on a large-sample clinical phenotype analysis of Chinese MMD.
Methods
Sample size and statistical power
The minor allele frequency of RNF213 p.R4810K was 0.003, with an odds ratio (OR) of 33.65 in Chinese cases16; type I error and statistical power was 0.05 and 0.8, respectively. The calculation indicated that 57 cases and 114 control participants can assure the statistical power of 0.8. The statistical power was 1.0 according to the sample size of 1,385 cases and 2,903 control participants included in the present study.
Study population
A total of 1,400 Chinese patients who had been newly or previously diagnosed with MMD were recruited in this study, from 27 provinces and cities throughout the country. The patients were consecutively enrolled at the Department of Neurosurgery, Fifth Medical Centre, Chinese PLA General Hospital (former 307th Hospital of the PLA), Beijing, and the Department of Neurology, the First Affiliated Hospital of Anhui Medical University, China, from January 2000 to July 2018. MMD diagnosis was determined using digital subtraction angiography (DSA) or magnetic resonance angiography (MRA) or MRI, which were combined with common clinical interviews of relevant and available relatives of patients. Diagnostic criteria for definite MMD diagnosis were based on the criteria of the Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis, Health Labor Sciences Research Grant for Research on Measures for Intractable Diseases, Japan.17 Patients with other basic diseases, such as arteriosclerosis, autoimmune disease, meningitis, or Down syndrome, were excluded from the study. A total of 2,903 unrelated control participants were recruited (1,238 men and 1,665 women, with a mean age of 62.18 ± 14.24 years). Control participants had no typical MMD symptoms, and were not screened by conventional DSA, MRA, CT angiography, or other tests. The low prevalence of MMD in the Chinese population was assumed not to affect the results of the association study.
To characterize the clinical features of patients with MMD, the clinical records of all previously reviewed cases were collected and examined scrupulously, including hospital charts, clinic notes, and radiologic studies. We ascertained the medical history, onset age, onset symptoms, native location, nationality, diagnosis method, and posterior cerebral artery (PCA) involvement combined with angiographic stage. Angiographic stage was evaluated according to the Suzuki classification.
DNA extraction and single nucleotide polymorphism (SNP) genotyping
After informed consent was obtained, 10 mL peripheral vein blood was extracted from available patients with MMD and normal control participants, placed in EDTA Na4 anticoagulant tubes, and stored in a freezer at −80°C until analysis. Genomic DNA was extracted from blood samples with a Blood Genetic DNA Mini Kit (CWBIO, Beijing, China). The concentration of the 4303 DNA was tested by NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA) and then diluted to working concentrations of 5 ng/μL for genotyping and validation.
Genotyping of p.R4810K in all participants was conducted using TaqMan Probe (TaqMan SNP Genotyping Assays; Applied Biosystems, Foster City, CA) and a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). The total system contained 5 μL: 2.0 μL purified genomic DNA, 2.5 μL TaqPath ProAmp Master Mixes (Applied Biosystems), 0.1 μL 40 × SNP genotyping assay, and 0.4 μL deoxyribonuclease-free water. The appropriate PCR thermal cycling conditions were as follows: maintained for 5 minutes for initial denature/enzyme activation, 40 cycles of 5 seconds at 95°C for denaturation, and 1 minute at 60°C for annealing and extension. After each PCR amplification, an endpoint plate read was conducted using the QuantStudio 6 Flex Real-Time PCR System. The genotype of each sample was confirmed based on the fluorescence signals.
Statistical analysis
PS: Power and Sample Size software was used to calculate the required size. Statistical analyses were performed using SPSS 20.0 (SPSS Inc., Chicago, IL). Patients lacking information for each clinical feature were excluded from the final statistical analyses. In order to compare and analyze differences in onset age in relation to various phenotype variables, we categorized patients into 2 groups: childhood onset (0–17 years old) and adult onset (≥18 years old). Difference analyses between the RNF213 genotype and each categorical phenotype variable, such as clinical symptoms or age at onset, unilateral/bilateral distribution of vasculopathy, and Suzuki classification, were performed by χ2 tests (χ2 with Fisher exact test was used when warranted). Non-normal distributions or those with p > 0.05 based on the test of homogeneity of variances with continuous variables were compared using the Mann-Whitney U test and Kruskal-Wallis test. In all analyses, p values were calculated using 2-tailed statistical tests.
Standard protocol approvals, registrations, and patient consents
The ethical issues involved in this study were examined and approved by the Ethics Committee of China Medical University, the Fifth Medical Centre, Chinese PLA General Hospital, and the First Affiliated Hospital of Anhui Medical University, and all participants and immediate family members signed written informed consent forms.
Data availability
With the permission of the corresponding authors and their institutions, combined with the relevant documents, all data used for analysis will be shared after ethics approval if requested by other investigators for reasonable purposes of replicating procedures and results.
Results
Demographics and clinical presentation
The clinical and demographic characteristics of 1,400 patients with MMD and 2,903 normal control participants are summarized in table 1. The median age at symptom onset was 35.2 years (range 1–71 years). The female-to-male ratio was 1.2:1. Two age peaks at 5–9 and 45–49 years, with a higher frequency observed in the adult peak, are presented in figure 1. In the present cohort, 97.1% of patients had bilateral disease, 2.9% had unilateral MMD, and 1.6% of patients had a family history of MMD (table 1). TIA was the predominant clinical manifestation, followed by cerebral infarction and cerebral hemorrhage. The majority of patients presented with Suzuki angiographic stage 4 or 5 (61.3%). In addition, the median initial ages at definite MMD and unilateral MMD onset were 38.0 and 40.5 years, respectively; this trend is presented in figure 1B.
Table 1.
Sample demographics of patients with moyamoya disease (MMD) and normal control participants
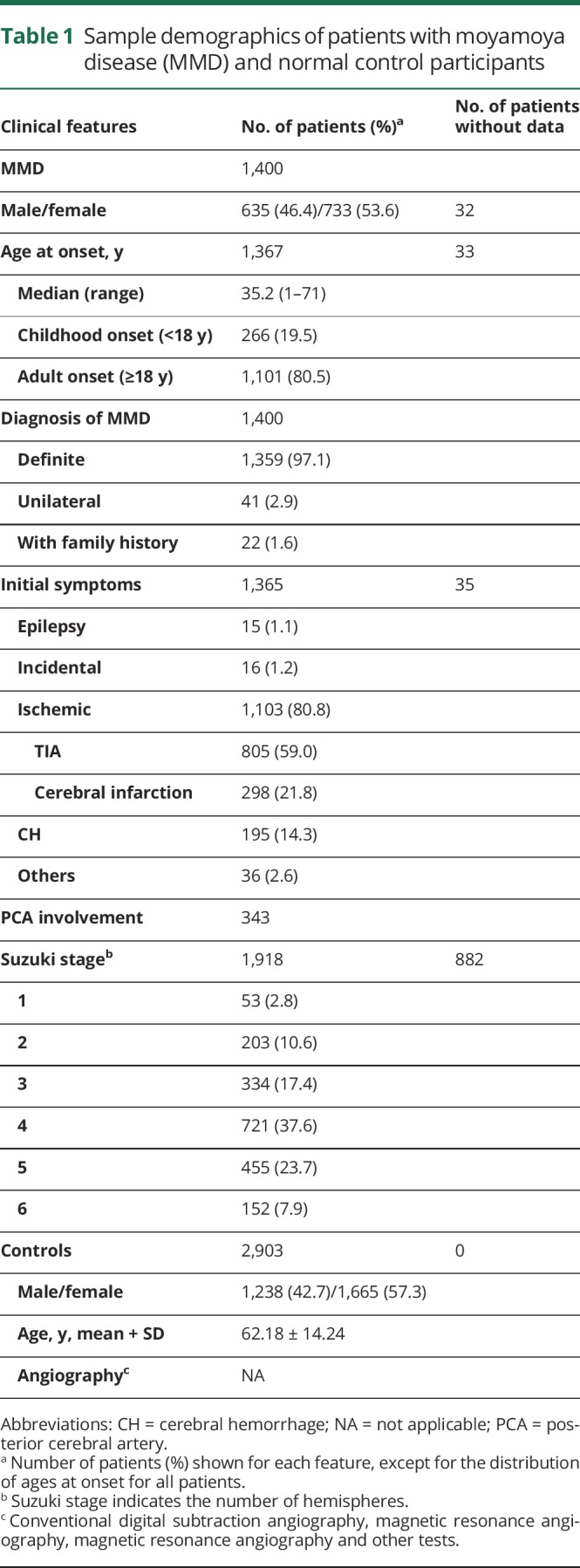
Figure 1. Age distribution patterns in Chinese moyamoya disease.

(A) Dual peak distribution of moyamoya disease in all, male, and female groups. (B) The initial age distribution of moyamoya disease in all, bilateral, and unilateral cases.
Higher carrying rate of RNF213 p.R4810K in Eastern and Northeast China
The birthplace distribution of the 1,352 patients with MMD carrying RNF213 p.R4810K is presented in figure 2. More than half of the cases (56.0%) originated from the Central Plains and surrounding regions in China, such as the Henan, Shandong, and Hebei provinces (26.2%, 15.5%, and 14.3%, respectively). The mutation rate in cases carrying RNF213 p.R4810K in our study suggested that higher rates were distributed in coastal provinces in Eastern and Northeast China, such as the Shandong, Jiangsu, and Liaoning provinces (37.2%, 37.0%, and 33.3%, respectively).
Figure 2. Distribution of 1,352 cases with moyamoya disease in China.

Values given inside and outside parentheses indicate the number of moyamoya patients and the carry rate (%) of RNF213 p.R4810K in each region, respectively.
RNF213 p.R4810K increased the risks of MMD
We next examined association between RNF213 p.R4810K and MMD. As shown in table 2, both the variant and minor alleles substantially increased the risk for MMD (dominant model, G/A + A/A vs G/G: OR, 54.0; 95% confidence interval [CI], 32.5–90.0; p = 1.5 × 10−144; allele model, A vs G: OR, 48.1; 95% CI, 29.1–79.6; p = 1.6 × 10−141). Although no homozygous mutations were detected in the control samples, the results further confirmed that homozygosity conferred strong effects. Both the variant and minor alleles occurred significantly more frequently in childhood-onset patients than in the adult-onset group (34.1% vs 20.7%, p = 9.3 × 10−6; 17.8% vs 10.4%, p = 7.6 × 10−6). Regarding the initial manifestation, except for A allele increased the risk of TIA (p = 3.8 × 10−2); among incidental, hemorrhagic, and ischemic cases, no associations have been found between the p.R4810K genotype or allele and these symptoms. Moreover, compared with patients with MMD with no family history, the frequency of variant p.R4810K genotypes and the minor allele occurred more frequently in patients with family history (dominant model, p = 1.9 × 10−2; allele model, p = 3.2 × 10−2; table 2). However, no observed tendencies were found to correlate with bilateral and unilateral MMD (table 2).
Table 2.
Genotype and allele distribution of p.R4810K of RNF213 in patients with moyamoya disease (MMD) and normal control participants

Correlations between RNF213 p.R4810K and clinical phenotype
The age at onset of all patients with GA and AA was significantly lower than in those with GG (p < 0.05, figure 3A). The median age at onset was 11.5, 36.0, and 40.0 in AA, GA, and GG, respectively, and the cumulative incidence of MMD was higher with AA and GA than with GG in almost all age distributions (figure 3B). Either GA or AA was observed at higher frequencies than GG in patients with childhood onset (27.5% vs 16.6%, p = 3.4 × 10−5; 66.7% vs 16.6%, p = 9.0 × 10−3, respectively; figure 4). In addition, AA was more common in patients with childhood onset than GA (66.7% vs 27.5%, p = 5.6 × 10−2; figure 4), and this further confirmed that patients carrying the A allele could acquire MMD during early life stages. Patients with MMD with a family history were more frequent with GA than in the GG group (p = 1.8 × 10−2). Furthermore, a significant difference suggested that the AA genotype was more common than GA in those with epilepsy (16.7% vs 0.7%, p = 5.8 × 10−2), and it was observed that the AA genotype was slightly more common than GG (16.7% vs 1.2%, p = 0.07); except for epilepsy, other clinical presentations, including incidental, ischemic, and hemorrhagic, have not been found to be significantly different among AA, GA, or GG genotypes. In addition, some patients with MMD with GA were more susceptible to PCA involvement compared to those in the group with GG (38.4% vs 23.3%, p = 8.3 × 10−7).
Figure 3. Correlation between p.R4810K genotype and age at onset of moyamoya disease.

(A) A box plot of the age at onset among 3 patient groups with mutant homozygote (AA), heterozygote (GA), or wild-type (GG) genotypes of the p.R4810K variant. (B) Cumulative incidence curve of the 3 patient groups with homozygote (AA), heterozygote (GA), or wild-type (GG) genotypes of the p.R4810K variant.
Figure 4. Correlations between p.R4810K genotype and clinical characteristics.

The clinical characteristics of moyamoya disease for the 3 patient groups with homozygote (AA), heterozygote (GA), or wild-type (GG) genotypes of the p.R4810K variant (1,385 patients). The numbers of patients in each group are indicated above the respective bars. Bil = bilateral vasculopathy; CH = cerebral hemorrhage; CI = cerebral infarction; Epi = epilepsy; FH = family history; PCA = posterior cerebral artery.
Discussion
We confirmed that clear risk of MMD was attributed to RNF213 p.R4810K based on the largest sample of Chinese patients studied to date. More importantly, this is the first study to report the distributions of birthplaces and carrying rates in Chinese patients with MMD and significant differences in genotype–phenotype correlations between people of China and other countries. Both GA and AA could predict an early age at onset and PCA involvement. This is based on the speculation that the effect of founder mutation of RNF213 p.R4810K and both heterozygous and homozygous changes in Chinese MMD onset are uncommon, as the feature differed from that of Japanese patients.
The trend observed in the geographical distribution map of all patients with MMD from 27 Chinese provinces showed a gradual decrease from areas with high carrying rates, such as coastal cities in northeast, north, and east China, to areas with low rates, such as southern cities or inland areas. This corresponds with the assumption that the cities with the highest carrying rates are near Japan or Korea, which may be related to human migration routes,18 and further demonstrates effects exhibited by ancestral mutations in the RNF213 p.R4810K gene of MMD. Therefore, the racial significance and genetic background of this disease warrant further clarification of the focus of this study through nationwide or global research to increase the reliability of these findings.
A case-control study was also performed and revealed that the carrying rate of RNF213 p.R4810K of MMD was 22.7%, lower than that in the Japanese (90%) and the Korean (79%) populations, but these results were similar to that of the Chinese population (23%) indicated in the reports by Liu et al.1 Furthermore, we identified the allele frequency of p.R4810K to be 0.5% in Chinese people, which was much lower than that of the Japanese (1.36%) and the Korean (1.36%) populations and in accordance with the findings of Liu et al.1 on distribution of p.R4810K in Eastern and Southeastern Asian populations.19 The genetic background of this disease clearly differs among various countries despite each belonging to an Asian population. The lower incidence of MMD in the Chinese population may be due to a lower frequency of the founder mutation. The results of our study also verified the results of the previous study by Liu et al.1 and our recent meta-analysis.16
It was worth noting that the childhood carrying rate was clearly higher than that of adult patients, and AA groups also displayed the youngest median age at onset. Therefore, we speculated that the homozygous p.R4810K variant of RNF213 may also predict early onset of MMD in Chinese patients, which is similar to the results of studies in some various countries,5,6,10 but the largest divergence was found when both GA and AA patients showed significantly younger ages in China than that of GG patients, which suggested that the mutation affected more patients with MMD. Further, Miyatake et al.6 reported that GA predicted a relatively late onset age and clinical course in Japanese patients. Most Chinese patients with MMD with AA (66.7%) had TIA at initial presentation; this was unlike Japanese patients, who displayed infarction as the most common presentation in the AA group.5 No significant difference was found in the severity of initial symptoms between patients carrying the RNF213 p.R4810K mutation and noncarriers. We originally analyzed the relationship between RNF213 p.R4810K and initial symptoms and found that the A allele increased the risk of TIA; however, a significant result was not obtained. Nevertheless, a low percentage of incidental cases were found in this study, perhaps due to different factors. Regardless, these patients should remain the focus of further study, and angiography or MRA, combined with routine DSA, are necessary to identify additional patients.
Moreover, patients with a family history of MMD were more likely to carry the p.R4810K variant in the current study, and this result supported the results of some previous studies.1,2,20 Interestingly, the ages of patients with family history were largely concentrated in the 5–9 age range, and these results coincided with that of Zhang et al.,21 who found that Chinese familial MMD can occur in patients and their third-degree relatives, of which first-degree relatives are the most affected. Doctors should pay more attention to the family histories of patients with MMD during the process of diagnosis and expand the scope of screening in high-risk groups, especially younger patients. Nevertheless, posterior circulation is a significant collateral arterial route in the earlier stages of MMD22 and is treated as an important predictor for outcomes of patients with MMD.23,24 Several studies have demonstrated that the prevalence of PCA involvement in MMD was 18.1%–36.2% in pediatric patients and 21.2%–43.4% in adult patients at initial diagnosis.24–26 Our study showed similar results. In addition, Kim and Kim11 found that PCA territorial involvement was more frequently observed in RNF213-positive patients than in RNF213-negative patients. Our results further showed that GA patients were more susceptible to PCA involvement than were wild-type patients. This result differed from that of Miyatake et al.5; however, we considered that this may be attributed to differences in genotype distribution between Chinese and Japanese patients or the limited number of AA patients in China, so we concluded that the p.R4810K mutation may cause more severe and wider vasculopathy in the brain. Meanwhile, other poor prognostic factors, such as cerebral infarction, were more commonly identified in Japanese and Korean patients with AA,5,10 but we detected epilepsy more frequently in Chinese patients. Therefore, future research should further expand the sample size to verify the reliability of these results, especially in patients with the AA genotype.
This study had certain limitations that should be noted. First, we only analyzed relationships between RNF213 p.R4810K and clinical features. However, MMD is a complex disease with genetic heterogeneity and a classic pattern of inheritance, and single gene-focused study cannot sufficiently elucidate its genetic susceptibility; meanwhile, the function of single genes or the clinical features of MMD may be dependent on the synergy of other genetic variations. Second, this is a study based on a single-center hospital, so selection bias is unavoidable. Third, we have chosen the Suzuki staging system to classify the severity of MMD,27,28 but it has been reported that this system was based mainly on intensified serial angiographic changes or decreases in moyamoya vessels, and clear disadvantages remain in the accuracy of classification based on angiographic findings.29,30 Therefore, the staging system defined by Mugikura et al.30 should be used for precise staging in the future.
To our knowledge, this study concerning the relationship between the RNF213 p.R4810K genotype and clinical phenotype was based on the largest case sample globally and the largest range in China to date. From the perspective of our research results, either homozygous or heterozygous RNF213 p.R4810K can predict the early age at onset of MMD and PCA involvement in Chinese patients with MMD. Continuous and timely follow-up observation may be helpful for clinicians and families to implement effective measures for slowing disease progression and further prevent the occurrence of adverse outcomes to the greatest extent prior to the diagnosis of new definite clinical cases.
Study funding
Supported by the National Natural Science Foundation of China (grant no. 81573240) and the Liaoning Provincial Undergraduate Training Program for Innovation and Entrepreneurship of China (grant no. 201810159184).
Disclosure
Y. Wang, Z. Zhang, L. Wei, Q. Zhang, Z. Zou, L. Yang, D. Li, M. Shang, C. Han, M. Mambiya, X. Bao, L. Qian, F. Hao, K. Zhang, H. Wang, S. Liu, M. Liu, F. Zeng, F. Nie, and K. Wang report no disclosures relevant to the manuscript. W. Liu is funded by the National Natural Science Foundation of China (grant no. 81573240) and the Liaoning Provincial Undergraduate Training Program for Innovation and Entrepreneurship of China (grant no. 201810159184). L. Duan reports no disclosures relevant to the manuscript. Go to https://n.neurology.org/lookup/doi/10.1212/WNL.0000000000008901 for full disclosures.
Acknowledgment
The authors thank the participants for their cooperation in this study.
Glossary
- CI
confidence interval
- DSA
digital subtraction angiography
- MMD
moyamoya disease
- MRA
magnetic resonance angiography
- OR
odds ratio
- PCA
posterior cerebral artery
- SNP
single nucleotide polymorphism
Appendix. Authors
References
- 1.Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One 2011;6:e22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 2011;56:34–40. [DOI] [PubMed] [Google Scholar]
- 3.Wu Z, Jiang H, Zhang L, et al. Molecular analysis of RNF213 gene for moyamoya disease in the Chinese Han population. PLoS One 2012;7:e48179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Zhang Z, Liu W, et al. Impacts and interactions of PDGFRB, MMP-3, TIMP-2, and RNF213 polymorphisms on the risk of Moyamoya disease in Han Chinese human subjects. Gene 2013;526:437–442. [DOI] [PubMed] [Google Scholar]
- 5.Miyatake S, Miyake N, Touho H, et al. Homozygous c.14576G>A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology 2012;78:803–810. [DOI] [PubMed] [Google Scholar]
- 6.Miyatake S, Touho H, Miyake N, et al. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G>A variant in RNF213 showed varying clinical course and severity. J Hum Genet 2012;57:804–806. [DOI] [PubMed] [Google Scholar]
- 7.Miyawaki S, Imai H, Takayanagi S, Mukasa A, Nakatomi H, Saito N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke 2012;43:3371–3374. [DOI] [PubMed] [Google Scholar]
- 8.Miyawaki S, Imai H, Shimizu M, et al. Genetic variant RNF213 c.14576G>A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke 2013;44:2894–2897. [DOI] [PubMed] [Google Scholar]
- 9.Cecchi AC, Guo D, Ren Z, et al. RNF213 rare variants in an ethnically diverse population with Moyamoya disease. Stroke 2014;45:3200–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim EH, Yum MS, Ra YS, et al. Importance of RNF213 polymorphism on clinical features and long-term outcome in moyamoya disease. J Neurosurg 2016;124:1221–1227. [DOI] [PubMed] [Google Scholar]
- 11.Kim WH, Kim SD. Posterior circulation involvement and collateral flow pattern in moyamoya disease with the RNF213 polymorphism. Childs Nerv Syst 2019;35:309–314. [DOI] [PubMed] [Google Scholar]
- 12.Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol 2008;7:1056–1066. [DOI] [PubMed] [Google Scholar]
- 13.Miao W, Zhao PL, Zhang YS, et al. Epidemiological and clinical features of Moyamoya disease in Nanjing, China. Clin Neurol Neurosurg 2010;112:199–203. [DOI] [PubMed] [Google Scholar]
- 14.Fujimura M, Sonobe S, Nishijima Y, et al. Genetics and biomarkers of moyamoya disease: significance of RNF213 as a susceptibility gene. J Stroke 2014;16:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan L, Bao XY, Yang WZ, et al. Moyamoya disease in China: its clinical features and outcomes. Stroke 2012;43:56–60. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Mambiya M, Li Q, et al. RNF213 p.R4810K polymorphism and the risk of moyamoya disease, intracranial major artery stenosis/occlusion, and quasi-moyamoya disease: a meta-analysis. J Stroke Cerebrovasc Dis 2018;27:2259–2270. [DOI] [PubMed] [Google Scholar]
- 17.Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir 2012;52:245–266. [DOI] [PubMed] [Google Scholar]
- 18.Abdulla MA, Ahmed I, Assawamakin A, et al. Mapping human genetic diversity in Asia. Science 2009;326:1541–1545. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Hitomi T, Kobayashi H, Harada KH, Koizumi A. Distribution of moyamoya disease susceptibility polymorphism p.R4810K in RNF213 in East and Southeast Asian populations. Neurol Med Chir 2012;52:299–303. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Liu Y, Zhang D, et al. RNF213 as the major susceptibility gene for Chinese patients with moyamoya disease and its clinical relevance. J Neurosurg 2017;126:1106–1113. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Zhang R, Duan L. Clinical features of familial moyamoya disease in China. Int J Cerebrovasc Dis 2016:114–116. [Google Scholar]
- 22.Miyamoto S, Kikuchi H, Karasawa J, Nagata I, Ikota T, Takeuchi S. Study of the posterior circulation in moyamoya disease: clinical and neuroradiological evaluation. J Neurosurg 1984;61:1032–1037. [DOI] [PubMed] [Google Scholar]
- 23.Mugikura S, Takahashi S, Higano S, et al. The relationship between cerebral infarction and angiographic characteristics in childhood moyamoya disease. AJNR Am J Neuroradiol 1999;20:336–343. [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada I, Himeno Y, Suzuki S, Matsushima Y. Posterior circulation in moyamoya disease: angiographic study. Radiology 1995;197:239–246. [DOI] [PubMed] [Google Scholar]
- 25.Mugikura S, Higano S, Shirane R, Fujimura M, Shimanuki Y, Takahashi S. Posterior circulation and high prevalence of ischemic stroke among young pediatric patients with moyamoya disease: evidence of angiography-based differences by age at diagnosis. AJNR Am J Neuroradiol 2011;32:192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funaki T, Takahashi JC, Takagi Y, et al. Impact of posterior cerebral artery involvement on long-term clinical and social outcome of pediatric moyamoya disease. J Neurosurg Pediatr 2013;12:626–632. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki J, Kodama N. Moyamoya disease: a review. Stroke 1983;14:104–109. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease: disease showing abnormal net-like vessels in base of brain. Arch Neurol 1969;20:288–299. [DOI] [PubMed] [Google Scholar]
- 29.Hishikawa T, Tokunaga K, Sugiu K, Date I. Assessment of the difference in posterior circulation involvement between pediatric and adult patients with moyamoya disease. J Neurosurg 2013;119:961–965. [DOI] [PubMed] [Google Scholar]
- 30.Mugikura S, Takahashi S, Higano S, Shirane R, Sakurai Y, Yamada S. Predominant involvement of ipsilateral anterior and posterior circulations in moyamoya disease. Stroke 2002;33:1497–1500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
With the permission of the corresponding authors and their institutions, combined with the relevant documents, all data used for analysis will be shared after ethics approval if requested by other investigators for reasonable purposes of replicating procedures and results.