

Abstract
Objective
A cross-sectional study was performed to evaluate whether quantitative oculomotor measures correlate with disease severity in late-onset GM2 gangliosidosis (LOGG) and assess cognition and sleep as potential early nonmotor features.
Methods
Ten patients with LOGG underwent quantitative oculomotor recordings, including measurements of the angular vestibulo-ocular reflex (VOR), with results compared to age- and sex-matched controls. Disease severity was assessed by ataxia rating scales. Cognitive/neuropsychiatric features were assessed by the cerebellar cognitive affective syndrome (CCAS) scale, Cerebellar Neuropsychiatric Rating Scale, and sleep quality evaluated using subjective sleep scales.
Results
Oculomotor abnormalities were found in all participants, including 3/10 with clinically normal eye movements. Abnormalities involved impaired saccadic accuracy (5/10), abnormal vertical (8/10) and horizontal (4/10) pursuit, reduced optokinetic nystagmus (OKN) responses (7/10), low VOR gain (10/10), and impaired VOR cancellation (2/10). Compared to controls, the LOGG group showed significant differences in saccade, VOR, OKN, and visually enhanced VOR gains. Severity of saccadic dysmetria, OKN, and VOR fixation-suppression impairments correlated with ataxia scales (p < 0.05). Nine out of ten patients with LOGG had evidence of the CCAS (5/10 definite, 2/10 probable, 2/10 possible). Excessive daytime sleepiness was present in 4/10 and 8/10 had poor subjective sleep quality.
Conclusions
Cerebellar oculomotor abnormalities were present in all patients with LOGG, including those with normal clinical oculomotor examinations. Saccade accuracy (dorsal cerebellar vermis localization), fixation suppression, and OKN gain (cerebellar flocculus/paraflocculus localization) correlated with disease severity, suggesting that quantitative oculomotor measurements could be used to track disease progression. We found evidence of the CCAS, suggesting that cerebellar dysfunction may explain the cognitive disorder in LOGG. Sleep impairments were prevalent and require further study.
Late-onset GM2 gangliosidoses (LOGG), comprising late-onset Tay-Sachs (LOTS) and Sandhoff disease, are rare (<50 known US patients) inherited neurodegenerative lysosomal storage disorders due to deficiency in β-hexosaminidase A (LOTS) and A and B (Sandhoff disease), and are believed to be clinically indistinguishable.1 Symptoms include progressive ataxia, anterior horn cell disease, and psychiatric symptoms with cognitive dysfunction, hypothesized to represent, at least in part, the cerebellar cognitive affective syndrome (CCAS).2 Brain MRI reveals cerebellar atrophy, even in patients without cerebellar motor signs. Pathology reveals cerebellar vermian and hemispheric atrophy, without brainstem involvement.1 New outcome measures and scales have been developed to assess cerebellar pathology.
We assessed 3 areas poorly delineated in LOGG, which may represent potential early motor and nonmotor clinical biomarkers: oculomotor pathology, cognitive performance/neuropsychiatric symptoms, and sleep. Oculomotor abnormalities are found in LOTS,3,4 are early signs of motor dysfunction in cerebellar diseases,5–7 predating more obvious clinical ataxia,6,7 and quantitative oculomotor analysis has been suggested as a potential biomarker in LOGG.3 Cognitive impairment may precede motor signs in metabolic ataxias8 and other neurodegenerative diseases9 and while common in LOTS, there has been little formal study.4,8,10–12 Sleep disorders are common in neurodegenerative disorders13–15 and may predate motor symptom onset.16 There is cerebellar involvement in normal sleep, sleep disorders are common in related ataxias,17,18 and functional and structural cerebellar changes are found in common sleep disorders.18 Our hypothesis is that these 3 features may be present in patients with LOGG, even in the absence of recognized clinical symptoms.
Methods
Standard protocol approvals, registrations, and patient consents
This study was approved by the Partners Human Studies Committee and all patients provided written informed consent prior to participation.
Participants
Patients were identified and recruited through the National Tay-Sachs and Allied Diseases Association. We performed a cross-sectional study at the Massachusetts General Hospital comparing a cohort of patients ≥18 years old with genetically confirmed LOGG with age- and sex-matched controls. We enrolled 10 patients with LOGG (7 LOTS, 3 Sandhoff disease) between April and September 2017. All patients were genetically confirmed. Each patient with LOGG received a detailed clinical examination performed by a movement disorders specialist with expertise in the assessment of ataxia (C.D.S.). Each patient underwent eye movement recordings at the Jenks Vestibular Laboratory at the Massachusetts Eye and Ear Infirmary using standard clinical paradigms,19 and these included measurements of saccades (latency, amplitude, and peak velocity); horizontal and vertical pursuit (sinusoidal visual stimuli at 0.2 Hz and 0.4 Hz); optokinetic nystagmus (OKN) tested with sinusoidal (0.05 Hz) visual stimulation; vestibulo-ocular reflex (VOR) testing in the dark using sinusoidal yaw-axis rotations over a 0.01–1.0 Hz frequency range20; and fixation-suppression (focus on a visual target rotating with the head) and visual-augmentation (focus on the stationary surround) of the VOR during sinusoidal 0.05 Hz yaw rotation. We also assessed 10 age- and sex-matched healthy control participants. We assessed disease severity with candidate clinical ataxia rating scales21 including the Brief Ataxia Rating Scale (BARS),22 Scale for the Assessment and Rating of Ataxia (SARA),23 Friedreich's Ataxia Rating Scale (FARS),24 and the Elstein LOTS severity scale (LOTS-SS).11 We also used a measure of functional status, the Functional Staging for Ataxia, as assessment of ataxia disease stage, as well as the patient-reported FARS Part 2 Activities of Daily Living (ADL) score, providing a multidomain assessment of disability.24 Cognitive/neuropsychiatric features were assessed by the novel CCAS Scale (CCAS-S)25 and Cerebellar Neuropsychiatric Rating Scale (CNRS).26 Subjective sleep quality was evaluated with the Pittsburgh Sleep Quality Index (PSQI)27 and the Epworth Sleepiness Scale (ESS).28 All participants underwent 3T MRI as part of an ongoing natural history study in LOGG. There were no missing data. Statistical analysis was carried out using SAS 9.4 (SAS Institute, Cary, NC). We performed descriptive statistics, t tests for comparison of continuous variables, Pearson correlation coefficients, and linear regression modeling to control for potential confounders, including patient age.
Data availability
We will share deidentified data upon request to the corresponding author. Data will be shared in Excel format and include scores and quantitative measures across the various neurologic domains assessed.
Results
Clinical and imaging features
The demographics of the LOGG cohort with markers of disease severity and the controls are detailed in table 1. The clinical characteristics, including disease stage and functional status, clinical oculomotor findings, and imaging findings, of the LOGG cohort are presented in table 2. Some of the patients with LOGG were related, with a group of 3/7 LOTS and 2/3 Sandhoff disease being siblings. Given the rare nature of this disease, we do not mention their subject numbers to avoid identifying these individuals. Clinically, patients with LOTS and patients with late-onset Sandhoff disease had frequent weakness, with a predilection for involvement of the triceps and quadriceps, resulting in gait impairment, sometimes requiring a walking aid. In comparison, cerebellar motor features were mainly present in LOTS and were generally mild. This involved a generally mild, nasal, and slightly stuttering dysarthria (only seen in LOTS, with normal speech in 3/3 patients with Sandhoff disease), mild appendicular dysmetria, and common but generally mild cerebellar eye signs, almost exclusively present in LOTS. In addition, 5/7 patients with LOTS had a slightly jerky, high-frequency postural and action tremor, which was not seen in the patients with Sandhoff disease. Mood or anxiety symptoms were present in 3/7 patients with LOTS and 2/3 patients with Sandhoff disease. It is notable that all patients with LOTS had considerable global cerebellar atrophy on imaging, in comparison to visually normal cerebellar volume in the patients with Sandhoff disease. MRI of the LOGG cohort is included in figure 1.
Table 1.
Demographic details and disease severity in late-onset GM2 gangliosidosis (LOGG) and control groups

Table 2.
Clinical and imaging characteristics of the late-onset GM2 gangliosidosis cohort


Figure 1. MRI of the late-onset GM2 gangliosidosis (LOGG) cohort.

Each row includes axial, coronal, and sagittal MRI sequences. LOTS = late-onset Tay-Sachs.
Clinical ataxia scales in LOGG
The late-onset Sandhoff disease cohort were older and tended to have less severe ataxia as measured by the disease rating scales than their LOTS counterparts, although the sample size was too small to make any assertions. Using the Pearson correlation coefficient, there was strong correlation among the 3 ataxia rating scales (BARS, SARA, FARS), r > 0.90, p ≤ 0.0005. However, these scales correlated considerably less well with the LOTS-SS (r = 0.72–0.80, p < 0.05), likely due to its reduced sensitivity for assessing clinical ataxia and inclusion of psychiatric symptoms and weakness.
Oculomotor abnormalities in LOGG
Eye movement abnormalities were evident on clinical examination in 7 of 10 patients with LOGG and typically included impaired pursuit and dysmetric saccades (table 2). There was a greater clinical burden of oculomotor abnormalities in LOTS (6/7) than in Sandhoff disease (1/3). The most severely affected patient (who had LOTS) had extensive clinical oculomotor abnormalities, including saccadic slowing and square wave jerks. Another patient with LOTS had difficulty with the voluntary initiation of pursuit eye movements on examination. No patient described clinical visual symptoms, despite abnormal oculomotor examinations, which is typical for such dysfunction in other ataxias.
Quantitative oculomotor abnormalities were found in all participants, including the 3 participants with normal oculomotor clinical examination results. The incidence of eye movement abnormalities appeared to be quantitatively similar in the LOTS and Sandhoff disease groups, although this was limited by the small sample size. Compared to age-matched normative values, deficits involved both voluntary and reflexive eye movements. Voluntary eye movement deficits included abnormal saccadic gain (amplitude of eye movement/amplitude of target movement) (5/10), abnormal vertical (8/10) and horizontal (4/10) pursuit gain (eye velocity/target velocity), and impaired fixation-suppression of the VOR (2/10). Reflexive eye movement deficits included reduced gain (eye velocity/head velocity) of the VOR (10/10) but normal VOR phase and time constant and reduced OKN gain (eye velocity/OKN stimulus velocity) (7/10). Compared to the normal control group, the LOGG group had significantly abnormal saccade (t test, p = 0.036), VOR (t test, p < 0.001), visually augmented VOR (t test, p < 0.001), and OKN (t test, p = 0.001) gains. Saccade latencies were longer in the LOGG group but this difference was not significant (t test, p = 0.08). All other eye movement measurements did not differ significantly between the LOGG and control groups (p > 0.05 for all comparisons).
Correlations between oculomotor abnormalities and markers of disease severity
Ataxia scales were adjusted for clinical weakness, to appropriately assess degree of ataxia as opposed to inappropriately high scoring in the setting of weakness, precluding assessment of lower extremity dysmetria. Significant correlations were found only for adjusted ataxia scores, which provide a more accurate reflection of the severity of the clinical cerebellar motor syndrome. Saccade gain was negatively correlated with all 3 disease scales as smaller saccade gains were associated with higher scores (BARS [r = −0.76], SARA [r = −0.67], and FARS [r = −0.65], all with p < 0.05) (figure 2, A–C). In contrast, saccade latency and peak velocity did not correlate with any of the disease severity scales. OKN gain was also negatively correlated with the 3 scales (BARS, SARA, and FARS [r = −0.89, r = −0.90, and r = −0.87, respectively], all p < 0.002) (figure 2, D–F). The gain of the VOR during the fixation-suppression task (viewing a head-stationary target) was positively correlated with the SARA score (r = 0.67, p = 0.03) and the FARS showed a similar pattern but did not reach significance (r = 0.59, p = 0.07) and the BARS did not reach significance (p = 0.101) (figure 2, G–I). All of these correlations remained significant after controlling for age. The other eye movement measurements were not correlated with the disease severity scales.
Figure 2. Oculomotor measures vs ataxia rating scale scores.

To demonstrate differences in the cohort, late-onset Tay-Sachs (LOTS) displayed in blue and late-onset Sandhoff disease (LOSD) in red. Saccade gain vs Brief Ataxia Rating Scale (BARS) (A), Scale for the Assessment and Rating of Ataxia (SARA) (B), Friedreich's Ataxia Rating Scale (FARS) (C); optokinetic nystagmus (OKN) gain vs BARS (D), SARA (E), FARS (F); vestibulo-ocular reflex (VOR) gain during fixation-suppression task vs BARS (G), SARA (H), FARS (I).
To assess whether these 3 oculomotor measures were correlated with clinical features beyond ataxia severity in what is a multidomain process, we compared the eye movements to a measure of functional status using the patient-reported FARS ADL score, which provides a global assessment of functioning across several domains. We found that the FARS ADL score correlated with OKN gain (r = −0.66, p = 0.0361) and with the gain of the VOR during the fixation-suppression task (r = 0.69, p = 0.0282) but not with saccade gain (p = 0.5111), as shown in figure 3, and these correlations remained significant (p < 0.05) after controlling for patient age.
Figure 3. Saccade, optokinetic nystagmus (OKN), and vestibulo-ocular reflex (VOR) fixation-suppression gains vs patient-reported functional assessment.

Functional assessment proxy, Friedreich's Ataxia Rating Scale Part 2 Activities of Daily Living (FARS ADL) score against oculomotor parameters: saccade gain vs FARS ADL (A), OKN gain vs FARS ADL (B), and VOR gain during fixation-suppression task vs FARS ADL (C). LOSD = late-onset Sandhoff disease; LOTS = late-onset Tay-Sachs.
Cognitive/neuropsychiatric features in LOGG
There was evidence of frequently asymptomatic cognitive deficits with abnormal CCAS-S scores in all but one participant (9/10): 5 fulfilled criteria for definite CCAS (3 or more failed tests), 2 for probable CCAS (2 failed tests), and 2 for possible CCAS (1 failed test) by the assessment criteria.25 The individual scores and tasks failed are shown in table 2 and examples of the cube copying abnormalities in the LOGG cohort are shown in figure 4. Only one patient described memory or cognitive difficulties, despite the presence of clearly abnormal scale scores. The CCAS-S scores did not correlate with clinical ataxia scores. The pattern of deficits included abnormal executive function in 6/10 (Go/No-Go, working memory tested by reverse digit span, and mental flexibility tested by category switching), visuospatial deficits tested by the cube drawing, and linguistic impairment evident in phonemic and semantic fluency tasks. Verbal recall was deficient in 7/10 patients, 4 of whom were not able to provide the word even with multiple choice cues. Whereas mean CCAS-S total sores were lower in those with LOTS (93 ± 15.8) compared to patients with Sandhoff disease (101 ± 9.5), the difference in this small cohort was not significant (p = 0.45). CCAS-S scores did not correlate with the sleep scales (ESS r = 0.27, p = 0.44, PSQI r = 0.37, p = 0.29); patients with worse CCAS-S scores tended to have better sleep scores.
Figure 4. Cube drawing examples as part of the cerebellar cognitive affective syndrome scale in the late-onset GM2 gangliosidosis cohort.
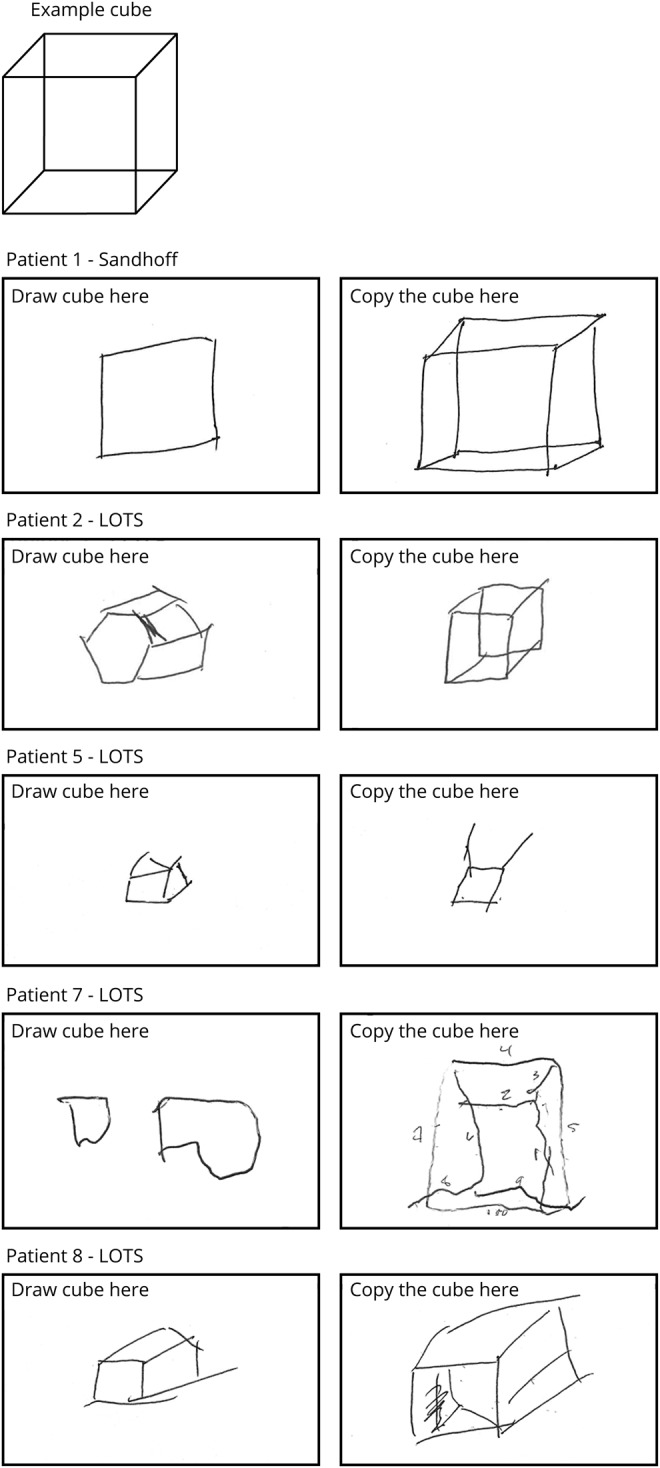
Spontaneous uncued cube drawing (left) and copying the example cube (right). Numbers drawn in patient 7 are to count number of lines and not produced by the patient. LOTS = late-onset Tay-Sachs.
There were high levels of psychiatric symptoms using the novel CNRS26 across all affective domains of the CCAS. All participants were reported by caregivers to have some psychiatric symptomatology, and this was pronounced in 3 participants. There was a higher burden of symptoms related to emotional control and social skill set, with less in the autism and psychosis spectrum, and little in attention control. The patients with Sandhoff disease had more psychiatric symptoms (mean total CNRS score 33.7 ± 8.7) than those with LOTS (mean total CNRS score 17.3 ± 15.3), but this difference was not significant (p = 0.13). The total CNRS score did not correlate with worsening CCAS-S, disease stage, or clinical ataxia severity.
Sleep quality and excessive daytime sleepiness in LOGG
There was poor subjective sleep quality (PSQI score ≥5) in 8/10 participants, which did not correlate with ataxia severity or disease stage. There were also significantly delayed subjective times of sleep onset and sleep offset in the sleepiest patients, indicating a delayed sleep phase syndrome. There were high levels of excessive daytime sleepiness, with 4/10 scoring ≥10 on the ESS. In one patient with late-onset Sandhoff disease, who had the highest sleep scores, this was likely related to concurrent, considerable obstructive sleep apnea. The sleep data in this small sample were skewed by this patient, although there were no significant differences between the LOTS/Sandhoff disease groups even when this patient was excluded.
Discussion
LOTS and late-onset Sandhoff disease are very rare genetic disorders that have important, distinct, and pathognomonic clinical features, although data come mainly from LOTS and there are few case reports/case series of patients with late-onset Sandhoff disease.29–35 In keeping with our hypotheses, we demonstrated abnormalities in oculomotor function, cognitive/neuropsychiatric symptoms, and sleep problems throughout the clinical spectrum of the patients with LOGG assessed, even in the absence of clinical symptoms.
Overview of clinical features of LOGG
These disorders are difficult to diagnose, given their clinical heterogeneity and significantly older age at onset in comparison to the typical infantile and juvenile forms, and comprise progressive mixed motor and movement disorder, as well as nonmotor symptoms. Psychiatric symptoms can be prominent, sometimes resulting in institutionalization,4,12 although these features were mildly represented in our cohort. Clinical clues include a combination of weakness, with a somewhat peculiar predilection for the triceps and quadriceps, leading to difficulties locking knees and causing the frequently early need for walking aids. Patients tend to have a slightly nasal dysarthria, sometimes with a stuttering quality, and stuttering can be an initial manifestation.36,37 There is also a frequent, jerky, high-frequency postural tremor. It was notable that in our cohort, all patients with Sandhoff disease had normal speech and did not have postural tremor, in comparison to their LOTS counterparts. Patients with LOGG may be misdiagnosed with motor neuron disease or purely psychiatric disease in the early stages; however, the pattern of weakness and the presence of cerebellar atrophy on imaging (mainly in LOTS) can help point to the diagnosis.38 Our results demonstrate that patients with LOGG displayed disturbances across several motor and nonmotor domains, including oculomotor dysfunction, as well as cognitive/neuropsychiatric and sleep dysfunction.
Oculomotor abnormalities in LOGG
In prior studies of the oculomotor features in LOGG, eye movement abnormalities were noted in 27% of one cohort of LOTS4 and included supranuclear gaze palsy, poor convergence, impaired OKN responses and VOR, internuclear ophthalmoplegia, and saccadic abnormalities.3 Rucker et al.3 conducted the only large-scale study addressing quantitative oculomotor analysis in LOTS and found occasional square wave jerks and saccades that could be hypometric, hypermetric, or multistep. Discriminating features in this population included transient saccadic decelerations and premature termination of the saccadic pulse,3 which was not apparent on bedside examination. In other ataxias, eye movement abnormalities are common,7,39 may be the sole manifestation of early5 and premanifest disease,6 and particular patterns may help to distinguish between different ataxias,39 or differentiate cerebellar disorders from other neurodegenerative disorders.40 Furthermore, in a recent study assessing the burden of clinical oculomotor features on examination in a population of patients with spinocerebellar ataxia at their first clinical visit, the BARS oculomotor score (number of eye movement abnormalities including at rest, pursuit, saccades, nystagmus, and saccadic slowing) correlated with ataxia scales as well as measures of functional impairment.7
In our study, oculomotor abnormalities were found across the spectrum of LOGG. Quantitative oculomotor abnormalities were present in all patients with LOGG, even when eye movements appeared normal on clinical examination. Specifically, eye movement abnormalities in LOGG that localize to the cerebellar flocculus and paraflocculus5 included pursuit, OKN, VOR gain, and visual suppression and enhancement of the VOR. Conversely, there was no evidence of cerebellar nodulus/uvula dysfunction given the normal VOR dynamics (phase, time constant), although more specific tests of this brain region such as tilt suppression of the VOR were not utilized.41,42 Saccade dysmetria was a prominent finding in LOGG and indicates damage in the dorsal vermis of the cerebellum, where saccade amplitude is controlled.43 Further quantitative MRI analysis of our cohort may help delineate the precise pattern of cerebellar involvement.
The severity of several oculomotor abnormalities correlated with clinical ataxia severity. Quantitative assessment of saccadic, OKN, and VOR fixation-suppression gains correlated with worsening disease severity, as measured by ataxia rating scales. The gait and stance abnormalities in LOGG are frequently related more to weakness, as opposed to ataxia, given the predilection for involvement of the quadriceps in the lower extremities. As gait and stance assessments form a disproportionate part of overall severity in ataxia rating scale scores, this suggests that overall disease severity from both weakness and ataxia is encapsulated in these measurements. We also performed assessments that were not purely cerebellar, to form a broader assessment of disease severity. Measures assessing functional status (FARS ADL score) correlated with OKN and VOR measures, suggesting that these quantitative oculomotor measures may also have relevance to patient disability and noncerebellar features of disease. Quantitative oculography may therefore represent a useful biomarker of disease severity, suggesting its potential utility to quantify response to therapy for future clinical trials. A limitation of the current study is the low sample size in this exceptionally rare disorder, which may have obscured other potentially significant associations. Further work is ongoing to assess the change in these eye movement measures over time, as it will be important to determine if they represent a useful marker of disease progression. Oculomotor measures may improve power for assessment of disease progression, given their objective, quantifiable nature, and could thus reduce sample size requirements, and have been suggested as potential outcome measures for future clinical trials in other ataxias.44
Cognitive/psychiatric features of LOGG
Nonmotor features frequently predate motor dysfunction in neurodegenerative movement disorders and we focused on 2 particular nonmotor symptom domains—cognitive function/neuropsychiatric symptom burden—as well as sleep quality as a proxy for sleep dysfunction. The CCAS describes a constellation of cognitive and psychiatric symptoms involving impairment of executive functions, visual-spatial organization, and memory, as well as affective domains of attention control, emotional control, autism spectrum, psychosis spectrum, and social skill set.2 Previous studies suggest that these features are frequently abnormal in patients with LOTS.4,12 Studies of cognition in LOTS have revealed impairments in executive functioning, involving processing speed, visual sequencing and set shifting, memory, and verbal impairments.10,11 Deficits range from mild memory deficits to significant global cognitive decline and dementia and have been found in up to 47% of patients.4,11,12 In a prospective study by Zaroff et al.10 on the cognitive/neuropsychiatric features in LOTS, no relationship was found between the cognitive deficits and neuromuscular/cerebellar dysfunction. In this study, it was suggested that these deficits may represent the CCAS, given that all of those studied had evidence of cerebellar dysfunction.10 Psychiatric symptoms are prominent in LOGG and can be debilitating. They include frank psychosis (sometimes misdiagnosed as schizophrenia), bipolar affective disorder, other mood disorders, as well as autistic traits including childlike behavior and poor social interaction.12 In its most extreme form, there have been rare cases of schizophreniform-appearing catatonia.45
In this study, there was evidence of the cognitive and neuropsychiatric aspects of the CCAS in LOGG throughout the spectrum of disease (table 3). These impairments were identified using scales designed for this purpose (the CCAS-S25 and the CNRS26) and showed significant abnormalities, often despite lack of patient or relative/caregiver cognitive concerns. This may therefore suggest that using sensitive scales to detect the CCAS may reveal the early onset of cognitive changes, which may even predate motor manifestation, as can be seen in other neurodegenerative movement disorders.9 Notably, 4 patients were unable to recall words even from multiple choice lists, which is atypical for cerebellar cognition, raising the possibility that these more severe amnestic deficits arose from cerebral hemisphere dysfunction rather than the cerebellum itself.2,25
Table 3.
Cerebellar cognitive affective syndrome scale (CCAS-S) scores and individual cognitive tasks in the late-onset GM2 gangliosidosis cohort

Evidence for sleep disorders in LOGG
We performed a first study assessing sleep in LOGG. In the related ataxias, a variety of sleep disorders has been found, including REM sleep behavior disorder, restless legs syndrome, periodic limb movements of sleep, excessive daytime sleepiness, and obstructive and central sleep apnea.17,18 In this study, patients with LOGG reported widespread poor subjective sleep quality and excessive daytime sleepiness, suggesting possible circadian rhythm dysfunction. Formal sleep studies in LOGG are warranted, although we acknowledge that there may be a mismatch of reported sleep quality with results of formal sleep studies.46 This may also represent a further potential early disease marker.
Clinical differences between LOTS and late-onset Sandhoff disease
The LOTS population demonstrated more cerebellar motor features including more prominent quantitative oculomotor findings (with minimal clinical eye movement abnormalities in patients with Sandhoff disease), more cognitive deficits, and a higher burden of psychiatric symptoms. In addition, although not quantitatively assessed, the dysarthria and tremor seen frequently in our patients with LOTS were not present in patients with late-onset Sandhoff disease. Furthermore, the patients with LOTS had marked cerebellar volume loss on MRI, whereas on MRI the cerebellum of the patients with Sandhoff disease appeared morphologically grossly normal (figure 1). There are case reports of patients with late-onset Sandhoff disease with a cerebellar phenotype,29,31 however, suggesting that our patients may have been of the lower motor neuron neuromuscular phenotype. These findings suggest clear differences between these 2 diseases, which involve different genes, and although 2 of 3 patients with Sandhoff disease and 3 of 7 patients with LOTS were related in our cohort, this requires further study.
Future directions
Given the correlation of oculomotor abnormalities and disease severity, our data suggest that eye movement recordings may represent a marker of disease severity in LOGG and a potential early sign of motor disease manifestation, in keeping with other metabolic and cerebellar disorders. It is possible that such a measure may allow the diagnosis of clinically manifest disease in gene-positive individuals at an earlier stage and make it possible to track the slow clinical progression and response to interventions in a sensitive and quantitative manner. We also found widespread evidence of 2 common nonmotor symptoms (cognitive dysfunction/burden of psychiatric symptoms and sleep quality), both of which were abnormal on testing despite the absence of clinical symptoms. No participant described visual symptoms of oculomotor abnormalities, and patients rarely described cognitive or sleep symptoms. This supports our hypothesis that these subtle motor (oculomotor abnormalities) and nonmotor (cognitive/neuropsychiatric and sleep) features are not only highly prevalent in LOGG but are either asymptomatic (oculomotor abnormalities) or deficits are generally unrecognized by patients. Therefore, these features may serve as markers of early disease and may respond to potential future therapeutic agents, suggesting that assessment for these features in clinical trials will be important.
Further research in patients with LOGG is required to help answer these important questions, particularly for those who are still early in their clinical course. Current conceptions of LOTS and late-onset Sandhoff disease are that they are clinically indistinguishable. In contrast, our studies suggest that there may be clinical and radiologic distinctions between the 2 forms of LOGG. Assessment of larger populations of patients with LOGG will be necessary to determine whether LOTS can be differentiated from late-onset Sandhoff disease as we have observed, as well as to better delineate the other important features we have highlighted. Our approach could also be applied to other rare metabolic ataxias and degenerative cerebellar disorders.
Study funding
The study was not industry-sponsored. Supported by U Penn Orphan Disease Center Million Dollar Bike Ride grant MDBR-17-131-NTSAD. This work was conducted with support from MGH Division of Clinical Research (DCR) Biostatistics Program and Harvard Catalyst, The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH award UL 1TR002541), and financial contributions from Harvard University and its affiliated academic health care centers.
Disclosure
C. Stephen has received financial support from Pfizer, Sanofi-Genzyme, Bristol-Myers Squibb, Biogen, and Biohaven for the conduct of clinical trials. D. Balkwill, P. James, E. Haxton, and K. Sassower report no disclosures relevant to the manuscript. J. Schmahmann receives consulting fees from Cadent Pharmaceuticals and grant support from the National Ataxia Foundation and the Ataxia Telangiectasia Children's Project and received financial support from Biohaven for conduct of a clinical trial in the ataxias. F. Eichler receives consulting fees from Ionis Pharmaceuticals and SwanBio Therapeutics and grant support from National Tay-Sachs and Allied Diseases and Cystinosis Research Foundation and has received financial support from bluebird bio and Minoryx Therapeutics for the conduct of clinical trials. R. Lewis receives fees as a consultant for Decibel Therapeutics and has received research support from Med El. Go to https://n.neurology.org/lookup/doi/10.1212/WNL.0000000000008959 for full disclosures.
Acknowledgment
The authors thank the NTSAD for facilitating recruitment of patients; the patients and their relatives/caregivers for their participation; and Jason Macmore, laboratory manager from the Schmahmann Laboratory for Neuroanatomy and Cerebellar Neurobiology, for help in the production of figures.
Glossary
- BARS
Brief Ataxia Rating Scale
- CCAS
cerebellar cognitive affective syndrome
- CCAS-S
cerebellar cognitive affective syndrome scale
- CNRS
Cerebellar Neuropsychiatric Rating Scale
- ESS
Epworth Sleepiness Scale
- FARS
Friedreich's Ataxia Rating Scale
- FARS ADL
Friedreich's Ataxia Rating Scale Part 2 Activities of Daily Living
- LOGG
late-onset GM2 gangliosidosis
- LOTS
late-onset Tay-Sachs
- LOTS-SS
LOTS severity scale
- OKN
optokinetic nystagmus
- PSQI
Pittsburgh Sleep Quality Index
- SARA
Scale for the Assessment and Rating of Ataxia
- VOR
vestibulo-ocular reflex
Appendix. Authors
References
- 1.Kaback MM, Desnick RJ. Hexosaminidase A deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews [Internet]. Seattle: University of Washington; 2011:1993–2016. [Google Scholar]
- 2.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain 1998;121:561–579. [DOI] [PubMed] [Google Scholar]
- 3.Rucker J, Shapiro B, Han Y, et al. Neuro-ophthalmology of late-onset Tay–Sachs disease (LOTS). Neurology 2004;63:1918–1926. [DOI] [PubMed] [Google Scholar]
- 4.MacQueen GM, Rosebush PI, Mazurek MF. Neuropsychiatric aspects of the adult variant of Tay-Sachs disease. J Neuropsychiatr Clin Neurosci 1998;10:10–19. [DOI] [PubMed] [Google Scholar]
- 5.Lewis RF, Zee DS. Ocular motor disorders associated with cerebellar lesions: pathophysiology and topical localization. Rev Neurol 1993;149:665–677. [PubMed] [Google Scholar]
- 6.Velázquez-Pérez L, Seifried C, Abele M, et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol 2009;120:632–635. [DOI] [PubMed] [Google Scholar]
- 7.Stephen CD, Schmahmann JD. Eye movement abnormalities are ubiquitous in the spinocerebellar ataxias. Cerebellum 2019;18:1130–1136. [DOI] [PubMed] [Google Scholar]
- 8.Degos B, Nadjar Y, Amador Mdel M, et al. Natural history of cerebrotendinous xanthomatosis: a paediatric disease diagnosed in adulthood. Orphanet J Rare Dis 2016;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulsen JS. Cognitive impairment in Huntington disease: diagnosis and treatment. Curr Neurol Neurosci Rep 2011;11:474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaroff C, Neudorfer O, Morrison C, Pastores G, Rubin H, Kolodny E. Neuropsychological assessment of patients with late onset GM2 gangliosidosis. Neurology 2004;62:2283–2286. [DOI] [PubMed] [Google Scholar]
- 11.Elstein D, Doniger G, Simon E, Korn-Lubetzki I, Navon R, Zimran A. Neurocognitive testing in late-onset Tay–Sachs disease: a pilot study. J Inherit Metab Dis 2008;31:518–523. [DOI] [PubMed] [Google Scholar]
- 12.Frey LC, Ringel SP, Filley CM. The natural history of cognitive dysfunction in late-onset GM2 gangliosidosis. Arch Neurol 2005;62:989–994. [DOI] [PubMed] [Google Scholar]
- 13.Abbott SA, Videnovic A. Chronic sleep disturbance and neural injury: links to neurodegenerative disease. Nat Sci Sleep 2016;8:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iranzo A. Sleep in neurodegenerative diseases. Sleep Med Clin 2016;11:1–18. [DOI] [PubMed] [Google Scholar]
- 15.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016;354:1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazar AS, Panin F, Goodman AO, et al. Sleep deficits but no metabolic deficits in premanifest Huntington's disease. Ann Neurol 2015;78:630–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedroso JL, Braga-Neto P, Felício AC, et al. Sleep disorders in cerebellar ataxias. Arq Neuropsiquiatr 2011;69:253–257. [DOI] [PubMed] [Google Scholar]
- 18.DelRosso LM, Hoque R. The cerebellum and sleep. Neurol Clin 2014;32:893–900. [DOI] [PubMed] [Google Scholar]
- 19.Markely BA. Introduction to electronystagmography for END technologists. Am J Electroneurodiagnostic Technol 2007;47:178–189. [PubMed] [Google Scholar]
- 20.Wall C III. The sinusoidal harmonic acceleration rotary chair test: theoretical and clinical basis. Neurol Clin 1990;8:269–285. [PubMed] [Google Scholar]
- 21.Puga AC, Hamed A, Kissell J, et al. Functional Performance in Late-Onset GM2 Gangliosidosis (Tay-Sachs and Sandhoff Diseases), Longitudinal Data over 3 Consecutive Years. WORLD Symposium, February 5–9, 2018, San Diego, CA.
- 22.Schmahmann JD, Gardner R, MacMore J, Vangel MG. Development of a brief ataxia rating scale (BARS) based on a modified form of the ICARS. Mov Disord 2009;24:1820–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitz-Hübsch T, Du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 24.Lynch DR, Farmer JM, Tsou AY, et al. Measuring Friedreich ataxia: complementary features of examination and performance measures. Neurology 2006;66:1711–1716. [DOI] [PubMed] [Google Scholar]
- 25.Hoche F, Guell X, Vangel MG, Sherman JC, Schmahmann JD. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain 2018;141:248–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoche F, Guell X, Sherman JC, Vangel MG, Schmahmann JD. Cerebellar contribution to social cognition. Cerebellum 2016;15:732–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buysse DJ, Reynolds CF, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatr Res 1989;28:193–213. [DOI] [PubMed] [Google Scholar]
- 28.Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 1991;14:540–545. [DOI] [PubMed] [Google Scholar]
- 29.Delnooz CC, Lefeber DJ, Langemeijer SM, et al. New cases of adult-onset Sandhoff disease with a cerebellar or lower motor neuron phenotype. J Neurol Neurosurg Psychiatry 2010;81:968–972. [DOI] [PubMed] [Google Scholar]
- 30.Schnorf H, Gitzelmann R, Bosshard NU, Spycher M, Waespe W. Early and severe sensory loss in three adult siblings with hexosaminidase A and B deficiency (Sandhoff disease). J Neurol Neurosurg Psychiatry 1995;59:520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tazen S, Goldman J, Fahn S. Cerebellar ataxia in adult-onset Sandhoff disease: a case report. Mov Disord 2012;27(suppl 1):S195. [Google Scholar]
- 32.Chardon JW, Bourque PR, Geraghty MT, Boycott KM. Very late-onset Sandhoff disease presenting as Kennedy disease. Muscle Nerve 2015;52:1135–1136. [DOI] [PubMed] [Google Scholar]
- 33.Thomas PK, Young E, King RH. Sandhoff disease mimicking adult-onset bulbospinal neuronopathy. J Neurol Neurosurg Psychiatry 1989;52:1103–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshizawa T, Kohno Y, Nissato S, Shoji S. Compound heterozygosity with two novel mutations in the HEXB gene produces adult Sandhoff disease presenting as a motor neuron disease phenotype. J Neurol Sci 2002;195:129–138. [DOI] [PubMed] [Google Scholar]
- 35.Kang SY, Song SK, Lee JS, Choi JC, Kang JH. Adult Sandhoff disease with 2 mutations in the HEXB gene presenting as brachial amyotrophic diplegia. J Clin Neuromuscul Dis 2013;15:47–51. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro BE, Natowicz MR. Late-onset Tay-Sachs disease presenting as a childhood stutter. J Neurol Neurosurg Psychiatry 2009;80:94–95. [DOI] [PubMed] [Google Scholar]
- 37.Grim KK, Phillips GD, Renner DR. Dysarthria and stutter as presenting symptoms of late-onset Tay-Sachs disease in three siblings. Mov Disord Clin Pract 2015;2:289–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barritt AW, Anderson SJ, Leigh PN, Ridha BH. Late-onset Tay-Sachs disease. Pract Neurol 2017;17:396–399. [DOI] [PubMed] [Google Scholar]
- 39.Moscovich M, Okun MS, Favilla C, et al. Clinical evaluation of eye movements in spinocerebellar ataxias: a prospective multicenter study. J Neuroophthalmol 2015;35:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willard A, Lueck CJ. Ocular motor disorders. Curr Opin Neurol 2014;27:75–82. [DOI] [PubMed] [Google Scholar]
- 41.Kheradmand A, Zee DS. Cerebellum and ocular motor control. Front Neurol 2011;2:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hain TC, Zee DS, Maria BL. Tilt suppression of vestibulo-ocular reflex in patients with cerebellar lesions. Acta Otolaryngol 1988;105:13–20. [DOI] [PubMed] [Google Scholar]
- 43.Barash S, Melikyan A, Sivakov A, Zhang M, Glickstein M, Thier P. Saccadic dysmetria and adaptation after lesions of the cerebellar cortex. J Neurosci 1999;19:10931–10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodríguez-Labrada R, Velázquez-Pérez L, Auburger G, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord 2016;31:570–578. [DOI] [PubMed] [Google Scholar]
- 45.Rosebush PI, MacQueen GM, Clarke JT, Callahan JW, Strasberg PM, Mazurek MF. Late-onset Tay-Sachs disease presenting as catatonic schizophrenia: diagnostic and treatment issues. J Clin Psychiatry 1995;56:347–353. [PubMed] [Google Scholar]
- 46.Bianchi MT, Williams KL, McKinney S, Ellenbogen JM. The subjective–objective mismatch in sleep perception among those with insomnia and sleep apnea. J Sleep Res 2013;22:557–568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We will share deidentified data upon request to the corresponding author. Data will be shared in Excel format and include scores and quantitative measures across the various neurologic domains assessed.