

Supplemental Digital Content is available in the text.
Keywords: elastin, genes, mutation, stem cells, vascular diseases
Abstract
Objective:
Elastin gene deletion or mutation leads to arterial stenoses due to vascular smooth muscle cell (SMC) proliferation. Human induced pluripotent stem cells–derived SMCs can model the elastin insufficiency phenotype in vitro but show only partial rescue with rapamycin. Our objective was to identify drug candidates with superior efficacy in rescuing the SMC phenotype in elastin insufficiency patients.
Approach and Results:
SMCs generated from induced pluripotent stem cells from 5 elastin insufficiency patients with severe recurrent vascular stenoses (3 Williams syndrome and 2 elastin mutations) were phenotypically immature, hyperproliferative, poorly responsive to endothelin, and exerted reduced tension in 3-dimensional smooth muscle biowires. Elastin mRNA and protein were reduced in SMCs from patients compared to healthy control SMCs. Fourteen drug candidates were tested on patient SMCs. Of the mammalian target of rapamycin inhibitors studied, everolimus restored differentiation, rescued proliferation, and improved endothelin-induced calcium flux in all patient SMCs except one Williams syndrome. Of the calcium channel blockers, verapamil increased SMC differentiation and reduced proliferation in Williams syndrome patient cells but not in elastin mutation patients and had no effect on endothelin response. Combination treatment with everolimus and verapamil was not superior to everolimus alone. Other drug candidates had limited efficacy.
Conclusions:
Everolimus caused the most consistent improvement in SMC differentiation, proliferation and in SMC function in patients with both syndromic and nonsyndromic elastin insufficiency, and offers the best candidate for drug repurposing for treatment of elastin insufficiency associated vasculopathy.
Highlights.
Elastin insufficiency caused by smooth muscle cell proliferation is a vascular disorder with no approved therapies. Elastin insufficiency caused by 7q11.23 deletion or by point mutations in the elastin gene can be modeled in the dish using patient induced pluripotent stem cell-derived smooth muscle cells.
Compared with healthy cells, cells from 5 patients with elastin insufficiency were immature, hyperproliferative, did not respond to endothelin and 3-dimensional biowires generated from patient smooth muscle cells failed to compact.
Candidate drugs tested on induced pluripotent stem cell-derived smooth muscle cells showed that mTOR (mammalian target of rapamycin) inhibitor, everolimus, was the best at rescuing the abnormal phenotype by enhancing smooth muscle cell differentiation and functional maturation.
As a drug with a known safety profile, these findings provide supporting evidence for consideration of everolimus for treatment of elastin insufficiency syndromes, and ultimately, may also benefit patients with acquired vascular disorders.
Elastin is the dominant extracellular matrix protein in the arterial wall, allowing vascular elasticity, and facilitating the recoil essential to blood vessel physiology. It is mainly produced by smooth muscle cells (SMCs) during late fetal and early postnatal life, with minimal generation of elastin throughout adulthood.1 Chromosomal deletion of 7q11.23 that includes deletion of the elastin (ELN) gene causes Williams syndrome (WS) with supravalvar aortic stenosis (SVAS) and other large artery stenosis or generalized arteriopathy. This is related to increased proliferation of SMCs2 that are functionally immature. Heterozygous mutations of the ELN gene cause nonsyndromic SVAS, that is, SVAS without other systemic manifestations. The arterial narrowing often recurs despite surgery,3,4 and there are no drugs clinically approved to treat this condition. Novel therapies are being tested in animal models and human cells as was recently reviewed.5 A recent small clinical trial evaluating minoxidil treatment on patients with WS reported no positive improvement in vascular phenotype.6 Our goal was to find targeted therapies that can rescue the abnormal vascular phenotype in patients with elastin insufficiency (EI) using drugs approved by the Food and Drug Administration for other indications as a potential drug repurposing strategy.
Although mouse models of EI have greatly improved our overall understanding of elastin signaling, there are limitations in their use in drug screens. Eln−/− mice die shortly after birth due to severe aortic obstruction,7 and heterozygous mice are viable but do not exhibit SVAS.8 Interestingly, a transgenic mouse carrying a human version of ELN on a bacterial artificial chromosome recapitulates aortic thickening with heterozygosity suggesting that the human and mouse elastin gene, and elastin synthesis, are not regulated equivalently in the developing aorta, and highlights the need for human-relevant models.9–11 Patient induced pluripotent stem cells (iPSCs) provide human-relevant models while retaining the genetic background of the patient and provide a noninvasive and renewable cell source for study of phenotype and drug responses. Importantly, for the study of EI, the use of patient cells that still carry a functioning copy of the ELN gene facilitates the testing of drugs that promote elastin transcription. Human iPSCs have been widely used to study the function of susceptible genes in a variety of diseases, including cardiovascular diseases.12–15 The use of iPSCs also offers a highly useful platform for drug screening because of their potential for replicating in vivo drug safety and efficacy.16–19
Human iPSCs can successfully be differentiated into vascular SMCs with efficiencies exceeding 80%,20 and their functional properties can be studied as they respond to vasoactive agonists.21 SMCs derived from patient iPSCs have been used to model vascular disease, such as WS, SVAS, hypertension, Marfan and Hutchinson-Gilford Progeria syndromes.22–26 These iPSC-SMCs recapitulated the pathological phenotype of each disease and identified novel targets for treatment.22,23,25 In our previous report, we recapitulated the disease phenotype of EI using patient iPSC-derived SMCs from a single patient with WS. The SMCs were hyperproliferative, poorly differentiated, and poorly contractile compared with healthy control cells. The antiproliferative mTOR (mammalian target of rapamycin) inhibitor rapamycin rescued the differentiation and proliferation defects but did not improve contractile properties.22
The goal of the current study was to identify one or more drug classes that would rescue not just the phenotypic abnormalities but also functional abnormalities in the SMCs of patients with WS as well as those with ELN mutations. We generated iPSCs from 2 additional patients with WS and 2 patients with heterozygous ELN mutations, all of whom had infantile-onset severe disease. We studied the effect of 14 candidate drugs on SMC differentiation, proliferation, and calcium flux. Our results showed that drugs belonging to the class of mTOR inhibitors showed the greatest efficacy in rescuing not just phenotypic but also contractile abnormalities in EI patient SMCs.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Cell Source
De-identified patient with WS (WS2, WS3) and elastin mutation patient (ELN1, ELN2) skin fibroblasts were obtained from patients recruited through the SickKids Heart Centre Biobank Registry (Toronto, ON, Canada). WS1-iPSC line C and wild-type control 1 BJ iPSC were previously reported by us22; control 2 21P and control 3 19-2 iPSCs were previously reported as controls in autism studies.27,28 H9 human embryonic stem cells were obtained from the National Stem Cell Bank (WiCell Research Institute, Madison, WI). All investigations were conducted according to the Declaration of Helsinki principles, studies were approved by the Hospital for Sick Children institutional review board, and written informed consent was obtained from the patient/parent/legal guardians. Human embryonic stem cell and iPSC studies were approved by the Canadian Institute for Health Research Stem Cell Oversight Committee of Canada.
Generation of iPSCs and Pluripotency Characterization
All EI patient skin fibroblasts were reprogrammed with the OCT4, SOX2, KLF4, and C-MYC retrovirus vectors using the Early Transposon promoter and Oct (octamer-binding transcription factor)-4 and Sox2 enhancers (EOS) lentivirus protocol as previously published.29 In brief, the EOS system allows expression of reporter genes in pluripotent stem cells but not in primary fibroblasts, which aids isolation of reprogrammed iPSC lines. Emerging human iPSC colonies marked with EGFP and puromycin-resistance were isolated, expanded, and characterized. Controls 1 and 3 iPSCs were also reprogrammed with retrovirus and the EOS lentivirus. Control 2 was reprogrammed using a nonintegrative Sendai virus approach.27 In vitro differentiation into embryoid bodies was performed and analyzed for pluripotency as described.30
Deletion Confirmation Using Multiplex Ligation Probe Amplification and DNA Fluorescence In Situ Hybridization
We confirmed the genetic diagnosis of 7q11.23 deletion in DNA from WS patients using the SALSA multiplex ligation probe amplification Kit P029-A1 which contains 32 multiplex ligation probe amplification probes for 8 genes located in the WS critical region (MRC Holland), and the ELN gene mutation in nonsyndromic SVAS patients using Sanger sequencing. To verify the deletion of chromosome region 7q11.23 in the iPSCs, we performed karyotyping by G banding chromosome analysis with a 400 to 500 band resolution through The Centre For Applied Genomics, Hospital for Sick Children, Toronto, and fluorescence in situ hybridization. A deletion at chromosome 7q11.23 involving 50 oligonucleotide probes from position 72 404 049 to 73 771 409 bp, including ELN, was identified using the oligonucleotide-based array Comparative Genomic Hybridization (Molecular Cytogenetics Laboratory, Hospital for Sick Children, Toronto, Canada).
Variant Confirmation Using Sanger Sequencing
To confirm ELN mutation in iPSCs, polymerase chain reaction (PCR) products from patient and control iPSCs were sent to The Centre For Applied Genomics for Sanger sequencing. Primers for ELN variants were designed using National Center for Biotechnology Information. ELN1: 5′–GGGAAGGAGCAGGTAGATCAG–3′ and 5′–TCTATTGTGACCACCCCAGTC–3′; ELN2: 5′–ACAAGTCCCTTAATGAGTGTGTTG–3′ and 5′–GGAACAAAGGCCAAGTCCATC–3′. Primer-BLAST and primer specificity were confirmed using the Ensembl Project BLAST/BLAT. Chromatographs were analyzed using FinchTV.
Differentiation of iPSCs Into SMCs
Control and patient iPSCs were differentiated into SMCs using previously published protocols.22,31 iPSC colonies were digested using 1 mg/mL collagenase type IV and transferred to an ultralow attachment 6-well plate (Corning Incorp, Corning, NY) to generate embryoid bodies. Knockout DMEM was used for 2 days and replaced by DMEM/F12 supplemented with 10% fetal bovine serum and 1% glutamax. Six-day-old embryoid bodies were transferred to 6-well plates coated with 0.1% gelatin for an additional 6 days in DMEM +10% fetal bovine serum. The embryoid bodies were dissociated with 0.05% trypsin and cultured on Matrigel-coated 6-well plates in smooth muscle growth medium (medium 231 + smooth muscle growth supplement; Life Technologies, Carlsbad, CA). Cells were passaged when they reached 80% to 90% confluence. To induce differentiation to SMCs, cells were re-plated in smooth muscle differentiation medium (medium 231 + smooth muscle differentiation supplement; Life Technologies) on gelatin-coated plates for 6 days.
Immunocytochemistry and High Content Imaging Assays
Five thousand cells were plated per well in 96 well plates, and after differentiation was treated with dimethyl sulfoxide (DMSO) or candidate drugs (dissolved in DMSO) for 6 days and stained using high content cell imaging (Cellomics ArrayScan VTI, ThermoFisher Scientific, Ottawa, ON, Canada). Three independent experiments were performed using 3 technical replicates for each experiment. Cells were fixed with 4% paraformaldehyde, permeabilized, blocked, and incubated overnight with primary antibody, rabbit anti-smooth muscle 22 alpha (SM22α; Abcam, Cambridge, MA). Secondary goat- anti-rabbit IgG antibody conjugated with fluorescein isothiocyanate (Sigma-Aldrich, St Louis, MO) was added to the samples and incubated for an hour. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (Life Technologies). Fluorescence was analyzed using an ArrayScan VTI HCS Reader (ThermoFisher Scientific). Images were captured using the Volocity or the vHCS View Software.
xCELLigence Real-Time Cell Monitoring
The xCELLigence system (Roche/ACEA Biosciences, San Diego, CA) was used to continuously monitor cell proliferation. Cells were cultivated in culture plates equipped with microelectrodes, which allow the computer to measure mechanical impedance.32 The xCELLigence software (version 1.2.1.1.002) converts collected data to a cell index, as a measure of cell proliferation. Five thousand cells were seeded per well of xCELLigence E-96 plates in growth medium in 3 independent experiments conducted on 3 different days (and with 3 technical replicates per experiment). Plates were placed in the system, incubated at 37°C under 5% CO2 and monitored at 15-minute time intervals. After 24 hours, medium was changed to differentiation medium, which was replaced after 2 days. Cell impedance was recorded at 15-minute intervals for up to 145 hours. Candidate drugs were added after 24 hours in differentiation conditions and change in cell index as an indicator of cell number was compared between cases and control iPSC-SMCs and between treated and untreated SMCs.
Calcium Measurement
SMCs were loaded with 1 µmol/L fluo5F AM (Life Technologies) at 37°C for 30 minutes, then washed with Hank’s Balanced Salt Solution. Fluorescence was visualized using a Nikon TE-2000 epifluorescence microscope (Nikon Corporation, Tokyo, Japan). Images were collected and analyzed using Volocity image acquisition and analysis software (PerkinElmer Inc, Waltham, MA). Fluorescence intensity was measured after subtracting nonspecific fluorescence background in 50 individual cells per well in 3 wells. The experiments were repeated 3 times. Values were plotted against time using graphing software (Excel, Microsoft, Redmond, WA). After 10 seconds baseline, cells were exposed to 100 nmol/L of the calcium agonist, endothelin-1 (Sigma-Aldrich) to stimulate calcium influx which was continuously monitored for 3 minutes. The change in fluorescence intensity (ie, F–F0) was used to estimate intracellular calcium flux in response to endothelin in control and patient iPSC-SMCs and in DMSO versus drug-treated SMCs.
Smooth Muscle Biowire Assay
To generate 3-dimensional-bioengineered tissues from SMCs, that is, biowires, polydimethylsiloxane posts were made using the Sylgard 184 Elastomer Kit by Dow Corning Corporation. Forty-five grams of elastomer base was mixed with 3 g of curing agent to make the posts. This mixture was added on a metal template (Department of Mechanical and Industrial Engineering, University of Toronto), which was placed in a vacuum machine at −760 mm Hg for 1 hour and then heated at 80°C for another 1 hour. A collagen gel was prepared with 2.2 g/L sodium bicarbonate, 1 mol/L sodium hydroxide, 3 mg/mL collagen (Corning), and Matrigel (Corning). The collagen gel was added to a suspension of 500 000 SMCs. This mixture was added to a well containing the polydimethylsiloxane posts and maintained in the incubator. After 24 hours, differentiation medium was added into the well with 0.01% DMSO or 100 nmol/L rapamycin. Tissue remodeling resulting from tractional forces during gel compaction was assessed by measuring tissue width every day for 7 days. Both passive and active tension were assessed by measuring the silicone post displacement from the microscopically acquired video images over multiple time points. Multiple measurements were averaged to arrive at an average post displacement for each condition. Elastic modulus and post dimensions were described in the previous work.33 The post displacement was related to the bending force exerted by the tissue on the post using cantilever beam-partial uniform load equation as previously described. For accurate application of beam bending equations, the vertical position of the tissue on the post was determined by cutting through the tissue microwell and imaging at the end of the experiment. To determine tension, the force per width of the tissue that results from beam bending equations was converted into a point load and divided by the cross-sectional area of the tissue calculated from the experimentally measured average tissue diameter. Passive tension, that is, the tension exerted by tissues on the posts in a resting state, was calculated from the post displacement for resting tissues at day 7, whereas active tension was assessed from the maximum post displacement after endothelin stimulation (n=3 biological replicates for DMSO and rapamycin-treated control 1, WBS2, and ELN1).
Reverse Transcription-Quantitative PCR for ELN
Total RNA was extracted from control and patient cultured SMCs using the RNeasy Mini kit (QIAGEN, Toronto, Canada) from 3 independent experiments (and 3 technical replicates per experiment). cDNA was synthesized from 1 µg RNA with Superscript III first-strand synthesis (Life Technologies) in a 20-µL reaction volume. Primer sequences for ELN were 5′-CAAGGCTGCCAAGTACGG-3′ and 5′-CCAGGAACTAACCCAAACTGG-3′ for elastin; and 5′-GCTGAGAACGGGAAGCTTGT-3′ and 5′-TCTCCATGGTGGTGAAGACG-3′ for GAPDH. Real-time PCR was performed on a StepOnePlus Real-Time System (Life Technologies) using SYBR GreenER qPCR SuperMix (Life Technologies). Quantification of gene expression was assessed with the comparative cycle threshold (ΔΔCT) method. mRNA expression was compared between control and patient SMCs. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) for pluripotency genes was performed as described.30
Mass Spectrometry for ELN
Mass spectrometry on protein lysates from control and patient SMCs was performed in the SickKids Proteomics, Analytics Robotics & Chemical Biology Centre. Parallel reaction monitoring analysis was performed on a Q-Exactive HF-X hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific) outfitted with a nanospray source and EASY-nLC split-free nano-LC system (ThermoFisher Scientific). Lyophilized peptide mixtures were dissolved in 0.1% formic acid and loaded onto a 75 µm × 50 cm PepMax RSLC EASY-Spray column filled with 2 µm C18 beads (ThermoFisher Scientific) at a pressure of 900 bars. Peptides were eluted over 60 minutes at a rate of 250 nL/min using a 0% to 35% acetonitrile gradient in 0.1% formic acid. Peptides were introduced by nanoelectrospray into the spectrometer. The instrument consisted of a tandem mass spectrometry scan detecting precursor ions. Automatic gain control targets were 1×105 with a maximum ion injection time of 50 ms. The quadrupole isolation window was set to 0.5 m/z, and the normalized collision energy was set at 30. Heavy synthetic elastin peptides were spiked-in to ensure the right light elastin peptides were monitored and precisely quantified in the cell samples. Amounts were normalized to endogenous tubulin beta chain 5 and myosin heavy chain 9. The following elastin peptide sequences were measured and relative amounts were compared between control and patient SMCs—LPGGYGLPYTTGK [212, 224]; LPYGYGPGGVAGAAGK [225, 240]; AGYPTGTGVGPQAAAAAAAK [241, 260]; and VPGALAAAK [644, 652]. Mass spectrometry was repeated 3 times in all controls and patient iPSC-SMCs as well as in DMSO and rapamycin-treated patient cells.
RNA Sequencing
RNA was extracted from DMSO-treated and rapamycin-treated iPSC-SMCs, and sequenced using the Illumina HiSeq 2500 platform through The Centre For Applied Genomics. Read data that passed quality control checks were aligned to the human genome browser, UCSC hg19, using Tophat v. 2.0.11. Alignments were processed to extract raw read counts for genes using htseq-count v.0.6.1p2. Differential expression analysis was performed between DMSO and rapamycin-treated cells using EdgeR. Reads per kilobase of transcript per million generated were normalized for the size of each library and normalized for the length of the transcripts. Using supervised hierarchical clustering, raw fold difference between DMSO-treated and rapamycin-treated SMCs was calculated for the following SMC associated gene sets using gene ontology terms for differentiation (GO:0051145, GO:0051152); proliferation (GO:0048661, GO:1904707); and contraction (GO:0006940, GO:0006939).
Statistical Analysis
Statistical analyses were performed using the Student t test on data from 3 independent experiments (n=3) with the mean of each independent experiment derived from 3 technical replicates where appropriate. Analyses were done to measure differences in the phenotype between (1) 5 cases and 3 control iPSC-SMCs and (2) candidate drug and DMSO-treated SMCs for the following variables: protein expression, mRNA expression by RT-qPCR, cell proliferation, and calcium flux. Three dimensional smooth muscle biowire assays were analyzed using cells derived from 2 cases and 1 control. The normality and variance were not tested to determine whether the applied parametric tests were appropriate. Two-way ANOVA with Bonferroni post hoc test was used for smooth muscle biowire compaction analyses. Differences were considered statistically significant at P<0.05.
Results
Reprogramming of Patient iPSCs
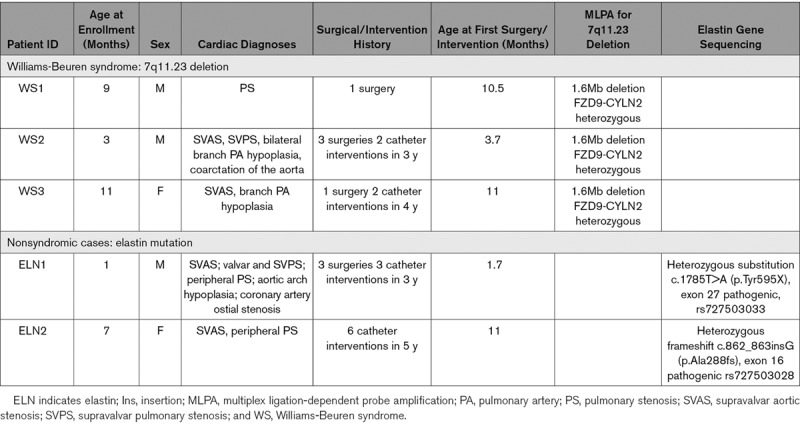
In addition to our previously reported WS (WS1) line,22 we reprogrammed skin fibroblasts from 4 patients with SVAS recruited through our Heart Centre Biobank. Two (WS2 and WS3) had 7q11.23 deletion confirmed by multiplex ligation probe amplification,22 while 2 had nonsyndromic SVAS due to heterozygous ELN mutations—one with a variant (substitution) in exon 27 (ELN1) and another with a frameshift variant (insertion) in exon 16 (ELN2), both predicted to induce a premature stop codon. Patient information, cardiac diagnoses, surgical history, and genetic test results are shown in the Table. In addition to the healthy BJ control line previously used by us (control 1),22 we also used lines from 2 additional unaffected individuals as controls—21P (control 2)27 and 19-2 (control 3).28 Fibroblasts do not express pluripotency genes,12–14,17–19 which we previously confirmed in WS1 lines.22 After reprogramming using the 4-factor retrovirus approach34 (Methods for details), all iPSC lines showed the hallmarks of pluripotent phenotype (Figure 1; Figure I in the Data Supplement). They expressed human pluripotency markers NANOG (homeobox protein NANOG), OCT-4, TRA-1-60 (T-cell receptor alpha locus), and SSEA-4 (stage specific embryo antigen 4) as shown by immunofluorescence and OCT4, NANOG, REX1 (reduced expression gene 1), and DNMT3B (DNA methyltransferase 3 beta) by RT-qPCR (Figure 1A and 1B; Figure IA and ID in the Data Supplement). The iPSCs gave rise to cells from all 3 germ layers upon in vitro differentiation into embryoid bodies and they all exhibited a normal karyotype (Figure 1C and 1D; Figure IB and IC in the Data Supplement). Short tandem repeat analyses were used to confirm that the iPSCs were from the correct patients (Table I in the Data Supplement). Assays were performed using one reprogrammed cell line derived from each of the 5 patients in accordance with Germain and Testa’s guidelines for optimal interpretation of iPSC-based studies.35
Figure 1.
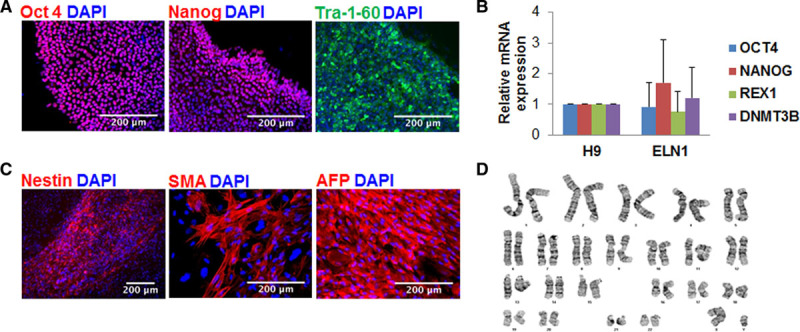
Pluripotency characterization of induced pluripotent stem cells (iPSCs) from the patient with an ELN variant (ELN1). A, iPSCs stained positive for pluripotency markers OCT (octamer-binding transcription factor)-4, NANOG, and TRA-1-60. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). B, Endogenous pluripotency genes were upregulated following successful reprogramming of ELN (elastin)-1 iPSCs as detected by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and comparable to pluripotency gene expression in human embryonic stem cells, H9. C, ELN1 iPSCs differentiated in vitro into all 3 germ layers. Immunocytochemistry showed nestin expression as an example of neuronal ectoderm, SMA (smooth muscle actin) expression for mesoderm, and AFP (α-fetoprotein) expression for endoderm. D, Normal G banding karyotype of ELN1 iPSCs. Results are shown as means and standard deviations from 3 independent replicates for each gene.
Table.
Patient Clinical and Genetic Data
Phenotype of iPSC-Derived SMCs
iPSCs were differentiated into SMCs using an established protocol.31 The morphological and functional phenotype were compared between patient iPSC-derived SMCs and healthy control iPSC-derived SMCs. Supporting data are provided in Table IIIA in the Data Supplement.
SMC Differentiation
We previously reported that control iPSC-SMCs showed downregulation of SM progenitor markers (CD34, Flk1 [fetal liver kinase 1], and Tie2 [tyrosine-protein kinase receptor]) and upregulation of SM differentiation markers (calponin, SM22a, smoothelin, myokardin, and telokin).22 We, therefore, selected SM22α staining to assess SMC differentiation using high content imaging. Control iPSCs generated 80% to 90% SM22α positive cells compared with patient iPSCs, which generated 45% to 68% SM22α-positive cells suggesting impaired differentiation in EI patients with the most severe differentiation abnormality seen in WS3 (Figure 2A and 2B).
Figure 2.
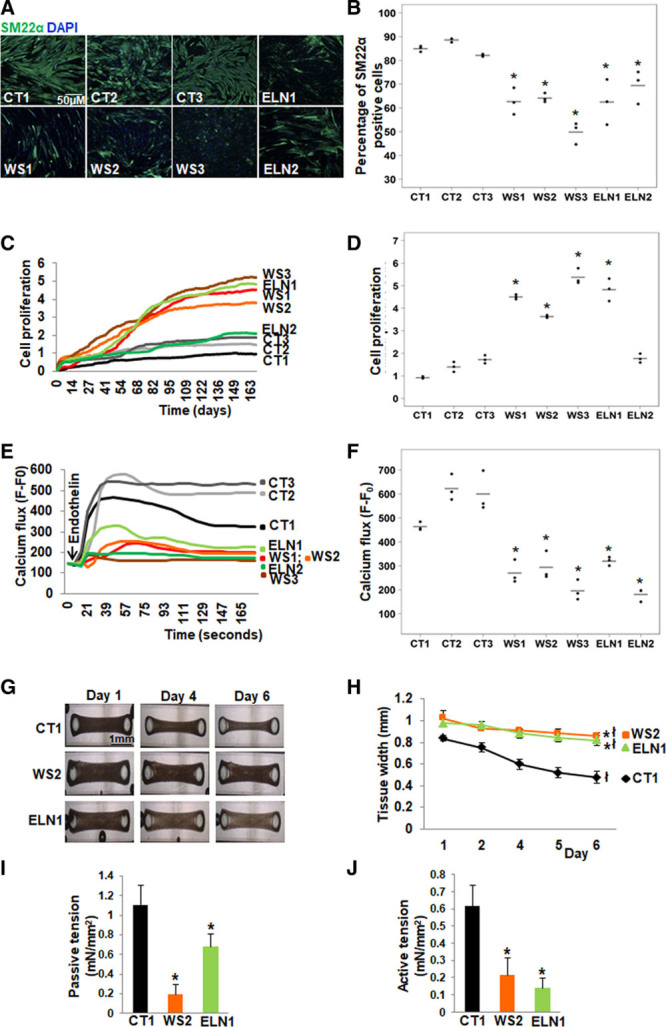
Abnormal smooth muscle cell (SMC) differentiation, proliferation and function in elastin insufficiency (EI) induced pluripotent stem cells (iPSC)-SMCs. A, SM22α (smooth muscle 22α) staining (representative images) and (B) quantification by high content imaging revealed the percentage of SM22α positive cells was lower in all patient with Williams syndrome (WS) and in ELN (elastin) patient SMCs compared with control SMCs. C, SMC proliferation measured by cell impedance (representative graph of cell index), and (D) quantification revealed increased proliferation in all 3 WS SMCs and in ELN1 patient SMCs compared to control SMCs. ELN2 patient SMC proliferation was not different from control SMCs. E, Calcium flux in response to endothelin (representative graph from 50 cells from an individual well) and (F) quantification from all replicates of maximum peak of mean fluorescence intensity after background correction (F–F0) showed lower calcium flux in response to endothelin in all patient SMCs compared with control SMCs (n=3 independent biological replicates and 3 technical replicates each for differentiation, proliferation, and calcium assays). G, Biowires generated from iPSC-SMCs from one control (CT1), one WS (WS2), and one ELN mutant patient (ELN1) showed failure of compaction of patient SMCs compared to control SMCs on day 6. H, Graph shows the change in the diameter of SMC-seeded biowires from day 1 to 6. All biowires showed some compaction by day 6, and the biowire diameter on day 6 remained significantly larger in WS2 and ELN1 patients compared to CT1 control. I, Passive tension at baseline was lower in patient SMC biowires compared with control. J, Active tension following treatment with endothelin was lower in patient SMC biowires compared to control (n=3 independent experiments). A–J, Supporting data are included in Table IIIA in the Data Supplement. *P<0.05, patient vs control, łP<0.05, day 6 vs 1.
SMC Proliferation
We used the xCELLigence system for real-time monitoring of cell proliferation. The system uses specially designed microtiter plates containing microelectrodes to noninvasively monitor dynamic changes in cell index of cultured cells that reflects changes in cell size and number. Cell index was higher suggesting higher proliferation in all patient SMCs except for ELN2-SMCs, which did not demonstrate hyperproliferation. WS3 was the most proliferative compared to controls (Figure 2C and 2D).
Endothelin-Induced Calcium Flux
SMC active function was evaluated by the ability of SMCs to increase calcium flux in response to a vasoactive agonist, endothelin. Cells on day 6 of differentiation were loaded with the Fluo5F calcium dye, and calcium transients were measured by the intensity of fluorescence before and after treatment with endothelin. All 3 controls responded to endothelin as seen by the rise in calcium flux within 30 seconds of treatment. While ELN1-SMCs showed a small increase in calcium flux following treatment, WS1 and WS2-SMCs indicated a smaller and delayed response. WS3 and ELN2-SMCs showed the lowest response to endothelin (Figure 2E and 2F).
Smooth Muscle Biowire Maturation and Contractility
Contractile response and SMC maturation were further assessed by generating 3-dimensional smooth muscle biowires from one control (control 1), one WS (WS2), and one ELN mutation (ELN1) patient SMCs. A mixture of SMC suspension and a collagen gel was added into a well containing a pair of polydimethylsiloxane posts to serve as fixation points and for measurements of contraction force. While control cells compacted after 6 days, both WS2 and ELN1 patient SMCs did not fully compact (Figure 2G and 2H). Passive tension was reduced in patient SMCs compared to control cells (Figure 2I). Biowires were then treated with endothelin and the change in active tension, that is, force was measured. Compared with control SMCs, tension generation in response to endothelin was lower in both patient SMCs (Figure 2J).
Elastin Gene and Protein Expression
ELN variants were confirmed in the differentiated iPSC-SMCs using Sanger sequencing (Figure II in the Data Supplement). We measured ELN mRNA expression in all iPSC-SMCs using RT-qPCR. Compared with control SMCs, elastin mRNA levels were significantly lower in all patient SMCs (Figure 3A). Elastin protein expression was then quantified by parallel reaction monitoring mass spectrometry using the sum of 4 different peptides. Elastin was significantly lower in all EI patient SMCs compared to controls except for ELN2-SMCs which showed a nonsignificantly lower ELN expression. WS3 had the lowest levels compared to controls (Figure 3B). To determine if the difference in ELN mRNA expression between ELN1 and ELN2-SMCs could be related to failure of nonsense-mediated decay of truncated elastin in ELN2, we quantified the amount of elastin peptides separately. Three peptides upstream of ELN1 (p.Tyr595X) and ELN2 (p.Ala288fs) variants and one peptide downstream of the variants, that is, VPGALAAAK (644, 652) were quantified. Because the downstream peptide was present in both ELN mutation patients, it indicates that mutated mRNA was being eliminated and that the baseline phenotype was likely not related to translation of structurally abnormal protein in ELN mutant SMCs (Figure 3C).
Figure 3.
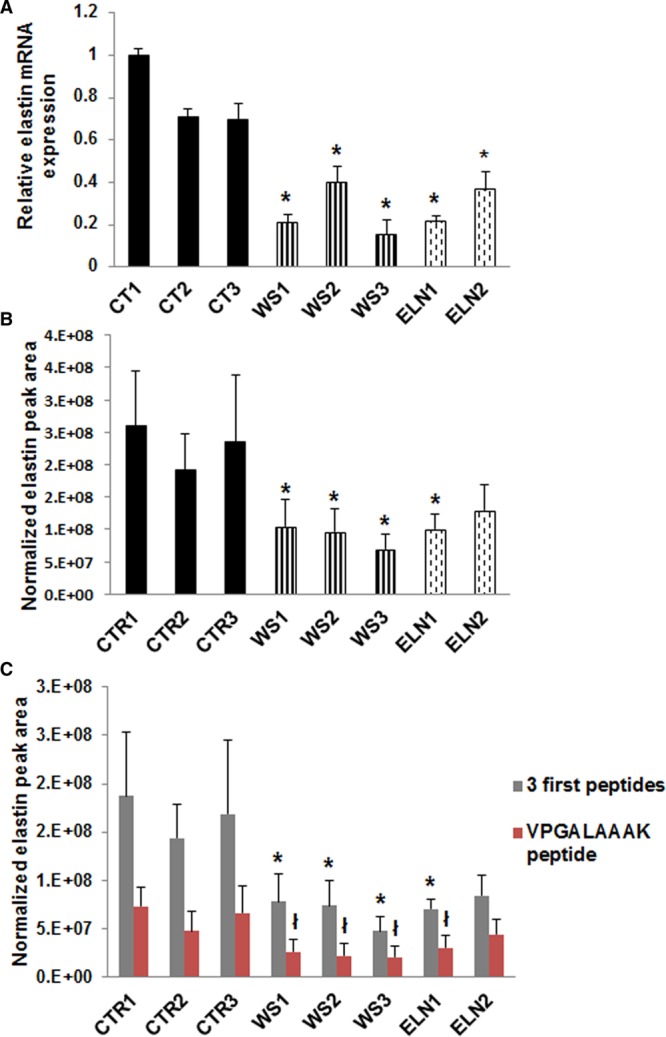
ELN expression was decreased in elastin insufficiency (EI) induced pluripotent stem cells (iPSC)-smooth muscle cells (SMCs). A, ELN mRNA expression by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was 20%–46% lower in patient iPSC-SMCs compared with all control SMCs. B, Parallel reaction monitoring mass spectrometry of the sum of peak area of 4 normalized elastin peptides showed lower elastin formation in all patient iPS-SMCs compared to controls (not statistically significant for ELN2). C, Quantification of 3 elastin peptides upstream of the elastin variants and one elastin peptide downstream of the elastin variants showed lower abundance of both upstream and downstream peptides in all patient cells (n=3 independent experiments). *P<0.05, patient vs control SMCs; †P<0.05, patient vs control SMCs for fourth peptide only. WS indicates Williams syndrome.
In summary, we successfully differentiated patient iPSCs into SMCs and recapitulated the abnormal EI phenotype. The iPSC-SMCs from the ELN2 patient with the frameshift variant had higher elastin levels than other patients and showed only mild abnormalities in differentiation and proliferation compared with other patients. The SMCs from the WS3 patient had lower elastin levels than other patients and showed the most severe abnormalities in SMC differentiation, proliferation, and calcium flux.
Response of Patient iPSC-SMCs to Rapamycin
We previously reported the ability of the mTOR inhibitor rapamycin to rescue abnormal SMC differentiation and hyperproliferation.22 To confirm that the SMCs being studied were functionally active and drug responsive, we performed a pilot experiment. We studied the effect of rapamycin on gene expression by RNAseq of rapamycin-treated and DMSO-treated SMCs from all patients and compared gene sets that were differentially expressed using gene ontology (P<0.05). Figure 4A shows the fold-change in gene expression between rapamycin and DMSO-treated SMCs. Compared with DMSO-treated SMCs, rapamycin-treated SMCs showed reduced expression of SMC proliferation genes and a variable increase in expression of SMC differentiation and contraction genes. Rapamycin also increased elastin protein levels as seen by mass spectrometry likely reflecting improved SMC maturation (Figure 4B). Transmission electron microscopy performed on WS2 and ELN1 smooth muscle biowires showed that rapamycin induced SMC elongation and the formation of myofilaments, the main feature of a differentiated SMC (Figure 4C and 4D) compared with DMSO-treated biowires. Although antibody staining was not performed, the presence of myofilaments was confirmed by an independent pathology service at our institution. Rapamycin also induced structural maturation in the form of biowire compaction in both WS2 and ELN1 patient biowires (Figure 4E and 4F). These findings confirmed that SMCs from EI patients were functionally active and drug responsive.
Figure 4.
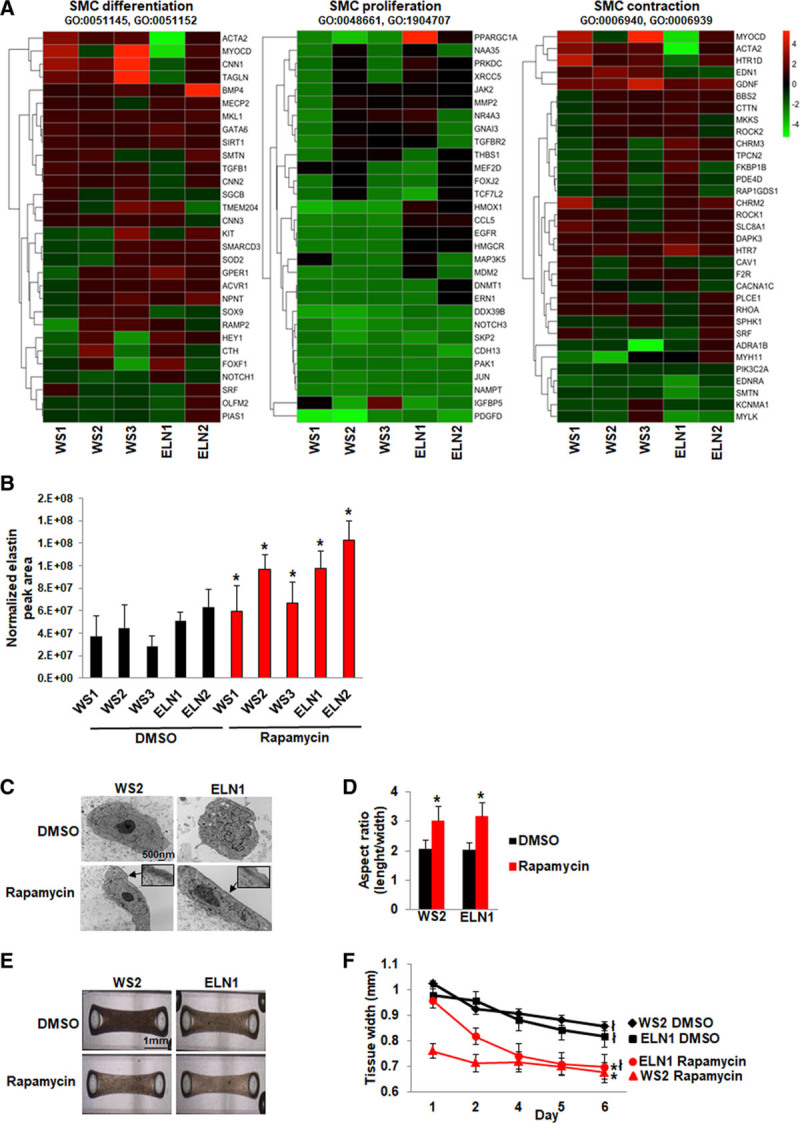
Phenotypic rescue by rapamycin. A, Heat map of RNA sequencing data from 5 patient induced pluripotent stem cells (iPSC)-smooth muscle cells (SMCs). Supervised hierarchical clustering showing raw fold difference in gene expression between dimethyl sulfoxide (DMSO) and corresponding rapamycin-treated SMCs for genes associated with SMC differentiation, SMC proliferation, and SMC contraction (P<0.05 between rapamycin vs DMSO-treated SMCs). Positive values indicate upregulation and negative values indicate downregulation compared to untreated SMCs. B, Elastin expression measured by mass spectrometry in 3 independent experiments increased in all patient SMCs after treatment with rapamycin. C, Transmission electron microscopy of patient smooth muscle biowires treated with DMSO or rapamycin. SMC maturation was observed after rapamycin treatment with the appearance of myofilaments (arrows and insets) and an elongated cell shape. D, The length to width ratio of the SMCs was higher in rapamycin compared to DMSO-treated biowires. E, Biowires treated with rapamycin showed greater compaction compared to DMSO-treated biowires by day 6. F, Comparison of tissue width from day 1 to 6 showed that both patient biowires showed compaction by day 6, but the compaction was greater in the rapamycin-treated compared to DMSO-treated biowires (*P<0.05, DMSO vs rapamycin-treated biowires, łP<0.05, day 6 vs 1; (n=3 independent experiments). WS indicates Williams syndrome.
Response of Patient iPSC-SMCs in Candidate Drug Screens
We explored additional drugs in the class of mTOR inhibitors as well as drugs from other classes that have the potential to target SMC differentiation, proliferation, and function and elastin synthesis, including calcium channel blockers (CCBs), other antiproliferative drugs, and proelastogenic drugs. Cells were treated with drug for 6 days throughout the differentiation process. Dose selection (Table II in the Data Supplement) was based on the dose that induced the optimum response without toxicity, as shown in Figure III in the Data Supplement. Significance tests were performed in comparison to DMSO-treated cells, which were not different from untreated cells (Table IIIB through IIID in the Data Supplement), and control cell lines were included to compare with the phenotype of healthy cells.
mTOR Inhibitors
Similar to rapamycin, the mTORC1 (mTOR complex 1) inhibitors, everolimus and temsirolimus, increased differentiation of all patient SMCs as depicted by significantly higher percentage of SM22α positive cells compared to DMSO-treated cells and to levels equivalent to the healthy control lines (Figure 5A). AZD0857, a dual inhibitor of mTORC1 and mTORC2, increased SM22α percentage in all WS patient cells but not in the ELN mutant patient cells. All mTOR inhibitors decreased cell impedance, a measure of SMC proliferation, in all WS and ELN1 cells but did not have an effect on ELN2 cells that were not hyperproliferative (Figure 5B). Compared with DMSO-treated cells, most of the mTOR inhibitors increased endothelin-induced calcium flux in one or more patient SMCs except WS3, which had lowest elastin levels and the most severe phenotype and did not respond to any mTOR inhibitor. Also, similar to our previous results, rapamycin did not improve calcium flux in WS1. Everolimus rescued calcium flux but not to control levels in a higher proportion of patient SMCs (4 of 5) but WS3 was a nonresponder (Figure 5C). Overall, mTOR inhibitors restored differentiation in EI iPSC-SMCs, rescued SMC proliferation and rescued contractile abnormalities but not to control levels, with everolimus showing the most consistent effects.
Figure 5.
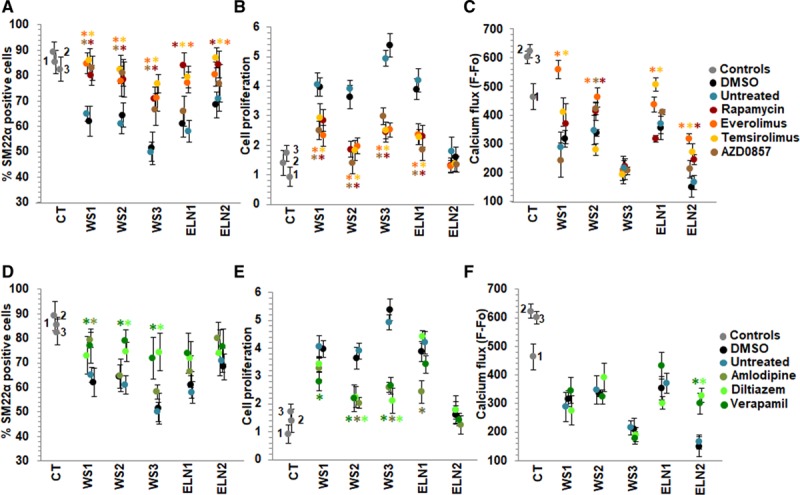
Effect of candidate drugs on smooth muscle cell (SMC) differentiation, proliferation, and calcium flux. A–C, mTOR (mammalian target of rapamycin) inhibitors. A, The percentage of SM22α (smooth muscle 22α) positive cells measured by high content imaging showed that dimethyl sulfoxide (DMSO)-treated elastin insufficiency (EI) patient SMCs express 50%–70% SM22α (black dots) similar to untreated patient cells (blue) in contrast to 80%–90% expressed in control SMCs (gray). Rapamycin (dark red), everolimus (orange), and temsirolimus (yellow) increased % of SM22α positive cells in all patients when compared to DMSO treatment (black). AZD0857 (brown) only increased SMC differentiation in Williams syndrome (WS) patient SMCs. B, All 4 mTOR inhibitors decreased SMC proliferation in all WS and in ELN (elastin)-1 cells. ELN2 cells were not hyperproliferative and did not show any further change in proliferation with mTOR inhibitors. C, Endothelin-induced calcium flux was increased by everolimus in WS1, WS2, ELN1, and ELN2-SMCs compared with DMSO-treated cells. Rapamycin only improved calcium flux in two patients (WS2, ELN2), temsirolimus in 3 patients (WS1, ELN1, ELN2), and AZD0857 in 1 patient (WS2). WS3 did not respond to any mTOR inhibitor. D–F, Calcium channel blockers. D, Verapamil (dark green) and diltiazem (bright green) increased % of SM22α positive cells only in 3 and 2 WS patients, respectively, but not in elastin mutation patients. E, Verapamil and diltiazem decreased SMC proliferation only in 3 and 2 WS patients, respectively, not in elastin mutation patients. Amlodipine (military green) treatment was associated with cell death (data not shown). F, Verapamil and diltiazem improved endothelin-induced calcium flux only in ELN2 patient SMCs (n=3 independent experiments, using 3 technical replicates for each experiment). A–F, Supporting data are shown in Table IIIB in the Data Supplement. *P<0.05, drug treatment vs DMSO.
Calcium Channel Blocker
We then investigated the effect of CCBs that are often used to treat hypertension in EI patients36,37 and have also been reported to reduce vascular SMC proliferation in a calcium channel-independent manner.38 Diltiazem and verapamil improved SMC differentiation and decreased cell proliferation in WS2 and WS3 patients, respectively, but not in ELN mutants. They improved endothelin response only in ELN2 cells (Figure 5D through 5F). Amlodipine induced cytotoxicity, that is, cell death in all patient SMCs even at low doses (Figure 5E).
Other Antiproliferative Drugs
We screened other antiproliferative drugs, including paclitaxel, fluvastatin, apocynin, and losartan. Although all 4 antiproliferative drugs decreased cell proliferation in all WS and in ELN1 SMCs, the effect on SMC differentiation was not significant for all drugs and all patients (except for the effect of paclitaxel on WS2 cells; Figure IVA and IVB in the Data Supplement). Fluvastatin induced cell toxicity at 3 different doses. These drugs were, therefore, not studied further for effect on endothelin response.
Proelastogenic Drugs
Dexamethasone, aldosterone, and retinyl acetate have previously been shown to increase elastin production39–41 but have not been tested in conditions of primary EI. The effect of these drugs on SMC proliferation differed. Aldosterone significantly decreased proliferation in all patients with WS; retinyl acetate was only effective on ELN1 and dexamethasone had no effect. The drugs did not improve SMC differentiation and were therefore not studied further for effect on endothelin response (Figure IVC and IVD and Table IIID in the Data Supplement).
Combination Therapy
As shown in Figure 5, everolimus restored SMC differentiation and rescued cell proliferation and calcium flux but not to the level of controls. To determine if the effects of everolimus could be further improved, we tested the effect of combining everolimus with a drug from a different class. Verapamil was selected as best in class agent based on its observed SMC responses. Combined treatment with the two drugs was not significantly better (Table IIIC in the Data Supplement) than everolimus alone in restoring SMC differentiation (Figure 6A), in rescuing proliferation (Figure 6B), and in improving calcium flux (Figure 6C). Moreover, combined treatment did not rescue endothelin-induced calcium flux in WS3, which had the most severe and unresponsive phenotype (Figure 6C).
Figure 6.
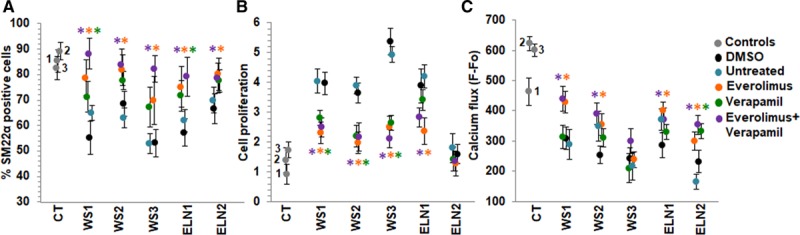
Combination treatment with a mTOR (mammalian target of rapamycin) inhibitor and a calcium channel blocker. A, Everolimus restored differentiation of SM22α (smooth muscle 22α) positive cells to the control cell level. Combination treatment with everolimus and verapamil (purple) was not superior to treatment with everolimus alone (orange) in any patient. Although there is a strong trend towards significance, we note that verapamil treatment (green) was less effective on Williams syndrome (WS)2 (P=0.068) and WS3 (P=0.059) than in Figure 5D. Verapamil was also more effective on ELN (elastin)-1 (P=0.037). This has no effect on the conclusion that everolimus alone rescued all patients. B, Everolimus rescued proliferation in all 4 patients exhibiting the hyperproliferation phenotype. Combination treatment was not significantly different to treatment with everolimus. C, Everolimus rescued endothelin-induced calcium flux in 4 patients but not to control levels, and WS3 did not respond. Combination treatment was not significantly different to treatment with everolimus. n=3 independent experiments, using 3 technical replicates for each experiment. A–C, Supporting data are provided in Table IIIC in the Data Supplement. *P<0.05, drug treatment vs DMSO.
In summary, mTOR inhibitors were the most effective drug class in rescuing the abnormal EI phenotype in patient SMCs with everolimus treatment associated with the highest and most consistent response rate across patient SMCs compared with any other drug.
Discussion
We generated iPSCs from a group of patients with EI, all of whom had a severe phenotype requiring multiple surgical and catheter re-interventions for progressive and recurrent vascular stenoses starting in infancy. iPSCs from the patients recapitulated the disease phenotype in vitro as demonstrated by abnormal SMC differentiation, increased proliferation, reduced compaction on 3-dimensional smooth muscle biowires, and reduced contractile response to endothelin. Candidate drug screens identified that mTOR inhibitors were the most effective drug class in patients with WS as well as ELN mutations, with everolimus showing the most favorable response. The severity of the SMC phenotype and the interindividual variability in drug response was associated with variable elastin levels between patients. This is the first systematic evaluation of clinically available drugs in correcting vascular SMC abnormalities in multiple patients with syndromic and nonsyndromic EI. Overall, our study showed how drug testing of SMCs derived from patient iPSCs can identify a potentially efficacious drug for use in these patients. The availability of effective drug therapy has the potential to reduce vascular stenosis or restenosis and reduce the need for invasive re-interventions and mortality in this severe disease.
Elastin Protein Levels and SMC Phenotype
As expected for haploinsufficient WS cells and for ELN nonsense/frameshift mutations that are subject to nonsense-mediated decay,3 ELN mRNA was significantly reduced in SMCs from all 5 patients. Elastin protein was also significantly reduced in these samples with lowest levels seen in WS3 SMCs and higher levels in ELN2-SMCs, which showed only a nonsignificant reduction in elastin. The lower amounts of elastin in WS3 may account for the more severe phenotype as well as the failure of both classes of drugs to rescue the response to endothelin. ELN2-SMCs were not hyperproliferative suggesting that their elastin levels were likely already sufficient to prevent a proliferative phenotype. We measured ELN peptides upstream and downstream of the variant but did not find evidence of incomplete nonsense-mediated decay. This suggests that the baseline phenotype was likely related to EI in all patients rather than to translation of structurally abnormal protein in ELN mutant SMCs. Although the presence of some compensatory translation pathway in ELN2 cannot be ruled out, detailed evaluation of post-transcriptional regulation of elastin was beyond the scope of this study.42,43
Everolimus Emerges as Best of Class mTOR Inhibitor for Rescue of SMC Differentiation, Proliferation, and Contractility in EI Patient SMCs
mTOR is a serine/threonine-protein kinase that forms 2 distinct complexes—mTORC1 that stimulates protein synthesis and cell cycle progression, while mTORC2 regulates the cytoskeleton and cell survival. Our findings that mTORC1 inhibitors, everolimus and temsirolimus, were more effective than a dual inhibitor of mTORC1 and mTORC2, AZD0857, supports a stronger role for pathways involved in cell proliferation as opposed to cell survival in mediating the EI phenotype. This is consistent with previous findings in a mouse model of EI where mTORC1 inhibition decreased SMC proliferation more effectively than mTORC2 inhibition.44 However, unlike the mouse study, our study also evaluated the effect of these analogs on SMC function through assessment of calcium flux in response to endothelin. The mechanism is hypothesized to be an insulin receptor substrate-1 feedback signal linked to mTOR inhibition that activates the AKT pathway and promotes contractile protein expression and inhibits SMC proliferation.45,46 Also, mTOR inhibition displaces FK506-binding protein 12 from ryanodine receptor 2 calcium release channel which can result in increased intracellular release of free calcium from the endoplasmic reticulum and may explain the increased endothelin-induced calcium flux in patient SMCs treated with mTOR inhibitors.47 Comparison of drug responses revealed that everolimus was the most effective of all analogs in improving SMC contractility in response to endothelin.
Other Drug Classes Are Less Effective Than mTOR Inhibitors in EI SMCs
CCBs are widely used to treat hypertension in patients with EI but their effect on SMC phenotype and function has not been systematically evaluated in patients with EI. Of the CCBs tested, verapamil and diltiazem rescued the differentiation and proliferation defects but only in patients with WS, not in elastin mutation patients. Also, CCBs did not rescue the poor response to endothelin. Additionally, amlodipine was associated with increased cell death. Although the mechanism of cell toxicity requires further study, this finding raises concerns about the appropriateness of clinical use of amlodipine as an antihypertensive agent in patients with EI. All the other drug classes tested only had antiproliferative effects and did not improve SMC differentiation and were, therefore, not considered candidates for further study. In addition, combination treatment with everolimus and verapamil was not superior than treatment with everolimus alone even in the everolimus-unresponsive WS3 cells suggesting an absence of synergy between these 2 drug classes.
Although treatment with everolimus significantly rescued the in vitro SMC phenotypes, evidence for efficacy will require a clinical trial in patients with EI with vascular stenoses, administered systemically or as drug-eluting stents in areas of discrete stenoses. Everolimus has a well-established safety profile with therapeutic blood levels maintained using therapeutic drug monitoring.48 Side-effects can be further minimized with targeted delivery using drug-eluting stents. As other novel treatment options emerge,5 their ability to rescue SMC phenotypes can be evaluated in our iPSC-derived patient SMCs, especially for efficacy in patient SMCs that fail to respond to everolimus.
Overall, these data confirm the effectiveness of mTOR inhibitors in general, and of everolimus in particular, as a best in class drug with potential for rescuing the vascular SMC phenotype in patients with EI. Although mTOR inhibitors are used clinically in drug-eluting stents to prevent and treat restenosis in adult coronary artery disease and systemically in children and adults to treat transplant coronary artery disease,49–51 our findings suggest that vascular stenoses caused by EI may respond better to everolimus. Since elastin variants may influence the vascular phenotype in patients with acquired vascular disorders, our study highlights the importance of assessing the underlying molecular basis of vascular disease to select the most effective mTOR inhibitor and identifies everolimus as an attractive candidate for repurposing to treat vascular stenosis in EI.
Acknowledgments
We acknowledge the Labatt Family Heart Centre Biobank for access to patient DNA and cells for this study, and The Centre for Applied Genomics, the SickKids imaging facility, and SickKids Proteomics, the Analytics Robotics & Chemical Biology Centre at the Hospital for Sick Children for their assistance with experimental assays. Yew Heng from the Division of Pathology, Department of Pediatric Laboratory Medicine, the Hospital for Sick Children acquired and interpreted electron microscopy images.
Sources of Funding
This work was supported by funding from the Canadian Institutes of Health Research (grant no. 126146 to Drs Mital and Ellis), SickKids Foundation Stem Cell Innovation Grant (to Drs Mital and Ellis), Ted Rogers Centre for Heart Research (to Dr Mital), and the Heart and Stroke Foundation of Ontario Chair (to Dr Mital).
Disclosures
M. Radisic is co-founder of TARA Biosystems Inc and holds equity in this company. The other authors report no conflicts.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- CCB
- calcium channel blocker
- DMSO
- dimethyl sulfoxide
- EI
- elastin insufficiency
- ELN
- elastin
- iPSC
- induced pluripotent stem cell
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex
- Oct
- octamer-binding transcription factor
- RT-qPCR
- reverse transcription-quantitative polymerase chain reaction
- SM22α
- smooth muscle 22 α
- SMC
- smooth muscle cell
- SVAS
- supravalvar aortic stenosis
- WS
- Williams syndrome
For Sources of Funding and Disclosures, see page 1338.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.119.313936.
References
- 1.Akhtar K, Broekelmann TJ, Miao M, Keeley FW, Starcher BC, Pierce RA, Mecham RP, Adair-Kirk TL. Oxidative and nitrosative modifications of tropoelastin prevent elastic fiber assembly in vitro. J Biol Chem. 2010;285:37396–37404. doi: 10.1074/jbc.M110.126789. doi: 10.1074/jbc.M110.126789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urbán Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urbán Z, Michels VV, Thibodeau SN, Davis EC, Bonnefont JP, Munnich A, Eyskens B, Gewillig M, Devriendt K, Boyd CD. Isolated supravalvular aortic stenosis: functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum Genet. 2000;106:577–588. doi: 10.1007/s004390000285. doi: 10.1007/s004390000285. [DOI] [PubMed] [Google Scholar]
- 4.Kececioglu D, Kotthoff S, Vogt J. Williams-Beuren syndrome: a 30-year follow-up of natural and postoperative course. Eur Heart J. 1993;14:1458–1464. doi: 10.1093/eurheartj/14.11.1458. doi: 10.1093/eurheartj/14.11.1458. [DOI] [PubMed] [Google Scholar]
- 5.Fhayli W, Boëté Q, Harki O, Briançon-Marjollet A, Jacob MP, Faury G. Rise and fall of elastic fibers from development to aging. Consequences on arterial structure-function and therapeutical perspectives. Matrix Biol. 2019;84:41–56. doi: 10.1016/j.matbio.2019.08.005. doi: 10.1016/j.matbio.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Kassai B, Bouyé P, Gilbert-Dussardier B, Godart F, Thambo JB, Rossi M, Cochat P, Chirossel P, Luong S, Serusclat A, et al. Minoxidil versus placebo in the treatment of arterial wall hypertrophy in children with Williams Beuren Syndrome: a randomized controlled trial. BMC Pediatr. 2019;19:170. doi: 10.1186/s12887-019-1544-1. doi: 10.1186/s12887-019-1544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 8.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res. 2007;101:523–531. doi: 10.1161/CIRCRESAHA.107.153510. doi: 10.1161/CIRCRESAHA.107.153510. [DOI] [PubMed] [Google Scholar]
- 10.Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer LA, et al. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med. 2009;1:50–65. doi: 10.1002/emmm.200900003. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osborne LR. Animal models of Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:209–219. doi: 10.1002/ajmg.c.30257. doi: 10.1002/ajmg.c.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 14.Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birket MJ, Ribeiro MC, Kosmidis G, Ward D, Leitoguinho AR, van de Pol V, Dambrot C, Devalla HD, Davis RP, Mastroberardino PG, et al. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell Rep. 2015;13:733–745. doi: 10.1016/j.celrep.2015.09.025. doi: 10.1016/j.celrep.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heilker R, Traub S, Reinhardt P, Schöler HR, Sterneckert J. iPS cell derived neuronal cells for drug discovery. Trends Pharmacol Sci. 2014;35:510–519. doi: 10.1016/j.tips.2014.07.003. doi: 10.1016/j.tips.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakai T, Naito AT, Kuramoto Y, Ito M, Okada K, Higo T, Nakagawa A, Shibamoto M, Yamaguchi T, Sumida T, et al. Phenotypic screening using patient-derived induced pluripotent stem cells identified Pyr3 as a candidate compound for the treatment of infantile hypertrophic cardiomyopathy. Int Heart J. 2018;59:1096–1105. doi: 10.1536/ihj.17-730. doi: 10.1536/ihj.17-730. [DOI] [PubMed] [Google Scholar]
- 19.Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–191. doi: 10.1002/emmm.201100194. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patsch C, Challet-Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17:994–1003. doi: 10.1038/ncb3205. doi: 10.1038/ncb3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halaidych OV, Cochrane A, van den Hil FE, Mummery CL, Orlova VV. Quantitative analysis of intracellular Ca2+ release and contraction in hipsc-derived vascular smooth muscle cells. Stem Cell Reports. 2019;12:647–656. doi: 10.1016/j.stemcr.2019.02.003. doi: 10.1016/j.stemcr.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinnear C, Chang WY, Khattak S, Hinek A, Thompson T, de Carvalho Rodrigues D, Kennedy K, Mahmut N, Pasceri P, Stanford WL, et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med. 2013;2:2–15. doi: 10.5966/sctm.2012-0054. doi: 10.5966/sctm.2012-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge X, Ren Y, Bartulos O, Lee MY, Yue Z, Kim KY, Li W, Amos PJ, Bozkulak EC, Iyer A, et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation. 2012;126:1695–1704. doi: 10.1161/CIRCULATIONAHA.112.116996. doi: 10.1161/CIRCULATIONAHA.112.116996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biel NM, Santostefano KE, DiVita BB, El Rouby N, Carrasquilla SD, Simmons C, Nakanishi M, Cooper-DeHoff RM, Johnson JA, Terada N. Vascular smooth muscle cells from hypertensive patient-derived induced pluripotent stem cells to advance hypertension pharmacogenomics. Stem Cells Transl Med. 2015;4:1380–1390. doi: 10.5966/sctm.2015-0126. doi: 10.5966/sctm.2015-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49:97–109. doi: 10.1038/ng.3723. doi: 10.1038/ng.3723. [DOI] [PubMed] [Google Scholar]
- 26.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deneault E, Faheem M, White SH, Rodrigues DC, Sun S, Wei W, Piekna A, Thompson T, Howe JL, Chalil L. Cntn5(-)(/+)or ehmt2(-)(/+)human ipsc-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife. 2019;8:e40092. doi: 10.7554/eLife.40092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, Wang Z, Alexandrova R, Pellecchia G, Wei W, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Reports. 2018;11:1211–1225. doi: 10.1016/j.stemcr.2018.10.003. doi: 10.1016/j.stemcr.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hotta A, Cheung AY, Farra N, Garcha K, Chang WY, Pasceri P, Stanford WL, Ellis J. EOS lentiviral vector selection system for human induced pluripotent stem cells. Nat Protoc. 2009;4:1828–1844. doi: 10.1038/nprot.2009.201. doi: 10.1038/nprot.2009.201. [DOI] [PubMed] [Google Scholar]
- 30.Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie CQ, Zhang J, Villacorta L, Cui T, Huang H, Chen YE. A highly efficient method to differentiate smooth muscle cells from human embryonic stem cells. Arterioscler Thromb Vasc Biol. 2007;27:e311–e312. doi: 10.1161/ATVBAHA.107.154260. doi: 10.1161/ATVBAHA.107.154260. [DOI] [PubMed] [Google Scholar]
- 32.Ke N, Wang X, Xu X, Abassi YA. The xCELLigence system for real-time and label-free monitoring of cell viability. Methods Mol Biol. 2011;740:33–43. doi: 10.1007/978-1-61779-108-6_6. doi: 10.1007/978-1-61779-108-6_6. [DOI] [PubMed] [Google Scholar]
- 33.Miklas JW, Nunes SS, Sofla A, Reis LA, Pahnke A, Xiao Y, Laschinger C, Radisic M. Bioreactor for modulation of cardiac microtissue phenotype by combined static stretch and electrical stimulation. Biofabrication. 2014;6:024113. doi: 10.1088/1758-5082/6/2/024113. doi: 10.1088/1758-5082/6/2/024113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hotta A, Cheung AY, Farra N, Vijayaragavan K, Séguin CA, Draper JS, Pasceri P, Maksakova IA, Mager DL, Rossant J, et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6:370–376. doi: 10.1038/nmeth.1325. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- 35.Germain PL, Testa G. Taming human genetic variability: transcriptomic meta-analysis guides the experimental design and interpretation of ipsc-based disease modeling. Stem Cell Reports. 2017;8:1784–1796. doi: 10.1016/j.stemcr.2017.05.012. doi: 10.1016/j.stemcr.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez Padial L, Barón-Esquivias G, Hernández Madrid A, Marzal Martín D, Pallarés-Carratalá V, de la Sierra A. Clinical experience with diltiazem in the treatment of cardiovascular diseases. Cardiol Ther. 2016;5:75–82. doi: 10.1007/s40119-016-0059-1. doi: 10.1007/s40119-016-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Black HR. Calcium channel blockers in the treatment of hypertension and prevention of cardiovascular disease: results from major clinical trials. Clin Cornerstone. 2004;6:53–66. doi: 10.1016/s1098-3597(04)80078-4. doi: 10.1016/s1098-3597(04)80078-4. [DOI] [PubMed] [Google Scholar]
- 38.Salabei JK, Balakumaran A, Frey JC, Boor PJ, Treinen-Moslen M, Conklin DJ. Verapamil stereoisomers induce antiproliferative effects in vascular smooth muscle cells via autophagy. Toxicol Appl Pharmacol. 2012;262:265–272. doi: 10.1016/j.taap.2012.04.036. doi: 10.1016/j.taap.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnett CP, Chitayat D, Bradley TJ, Wang Y, Hinek A. Dexamethasone normalizes aberrant elastic fiber production and collagen 1 secretion by Loeys-Dietz syndrome fibroblasts: a possible treatment? Eur J Hum Genet. 2011;19:624–633. doi: 10.1038/ejhg.2010.259. doi: 10.1038/ejhg.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bunda S, Liu P, Wang Y, Liu K, Hinek A. Aldosterone induces elastin production in cardiac fibroblasts through activation of insulin-like growth factor-I receptors in a mineralocorticoid receptor-independent manner. Am J Pathol. 2007;171:809–819. doi: 10.2353/ajpath.2007.070101. doi: 10.2353/ajpath.2007.070101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chailley-Heu B, Boucherat O, Barlier-Mur AM, Bourbon JR. FGF-18 is upregulated in the postnatal rat lung and enhances elastogenesis in myofibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;288:L43–L51. doi: 10.1152/ajplung.00096.2004. doi: 10.1152/ajplung.00096.2004. [DOI] [PubMed] [Google Scholar]
- 42.Qi YF, Shu C, Xiao ZX, Luo MY, Fang K, Guo YY, Zhang WB, Yue J. Post-Transcriptional control of tropoelastin in aortic smooth muscle cells affects aortic dissection onset. Mol Cells. 2018;41:198–206. doi: 10.14348/molcells.2018.2193. doi: 10.14348/molcells.2018.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ott CE, Grünhagen J, Jäger M, Horbelt D, Schwill S, Kallenbach K, Guo G, Manke T, Knaus P, Mundlos S, et al. MicroRNAs differentially expressed in postnatal aortic development downregulate elastin via 3’ UTR and coding-sequence binding sites. PLoS One. 2011;6:e16250. doi: 10.1371/journal.pone.0016250. doi: 10.1371/journal.pone.0016250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiao Y, Li G, Li Q, Ali R, Qin L, Li W, Qyang Y, Greif DM, Geirsson A, Humphrey JD, et al. mTOR (Mechanistic Target of Rapamycin) inhibition decreases mechanosignaling, collagen accumulation, and stiffening of the thoracic aorta in elastin-deficient mice. Arterioscler Thromb Vasc Biol. 2017;37:1657–1666. doi: 10.1161/ATVBAHA.117.309653. doi: 10.1161/ATVBAHA.117.309653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282:36112–36120. doi: 10.1074/jbc.M703914200. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 46.Jin Y, Xie Y, Ostriker AC, Zhang X, Liu R, Lee MY, Leslie KL, Tang W, Du J, Lee SH, et al. Opposing Actions of AKT (Protein Kinase B) isoforms in vascular smooth muscle injury and therapeutic response. Arterioscler Thromb Vasc Biol. 2017;37:2311–2321. doi: 10.1161/ATVBAHA.117.310053. doi: 10.1161/ATVBAHA.117.310053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Habib A, Karmali V, Polavarapu R, Akahori H, Cheng Q, Pachura K, Kolodgie FD, Finn AV. Sirolimus-FKBP12.6 impairs endothelial barrier function through protein kinase C-α activation and disruption of the p120-vascular endothelial cadherin interaction. Arterioscler Thromb Vasc Biol. 2013;33:2425–2431. doi: 10.1161/ATVBAHA.113.301659. doi: 10.1161/ATVBAHA.113.301659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arena C, Troiano G, Zhurakivska K, Nocini R, Lo Muzio L. Stomatitis and everolimus: a review of current literature on 8,201 patients. Onco Targets Ther. 2019;12:9669–9683. doi: 10.2147/OTT.S195121. doi: 10.2147/OTT.S195121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Windecker S, Remondino A, Eberli FR, Jüni P, Räber L, Wenaweser P, Togni M, Billinger M, Tüller D, Seiler C, et al. Sirolimus-eluting and paclitaxel-eluting stents for coronary revascularization. N Engl J Med. 2005;353:653–662. doi: 10.1056/NEJMoa051175. doi: 10.1056/NEJMoa051175. [DOI] [PubMed] [Google Scholar]
- 50.Poon M, Badimon JJ, Fuster V. Overcoming restenosis with sirolimus: from alphabet soup to clinical reality. Lancet. 2002;359:619–622. doi: 10.1016/s0140-6736(02)07751-6. doi: 10.1016/s0140-6736(02)07751-6. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki T, Kopia G, Hayashi S, Bailey LR, Llanos G, Wilensky R, Klugherz BD, Papandreou G, Narayan P, Leon MB, et al. Stent-based delivery of sirolimus reduces neointimal formation in a porcine coronary model. Circulation. 2001;104:1188–1193. doi: 10.1161/hc3601.093987. doi: 10.1161/hc3601.093987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.