

Supplemental Digital Content is available in the text.
Keywords: animals, atherosclerosis, autophagy, glycolysis, mice
Abstract
Objective:
Intraplaque neovascularization is an important feature of unstable human atherosclerotic plaques. However, its impact on plaque formation and stability is poorly studied. Because proliferating endothelial cells generate up to 85% of their ATP from glycolysis, we investigated whether pharmacological inhibition of glycolytic flux by the small-molecule 3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) could have beneficial effects on plaque formation and composition.
Approach and Results:
ApoE−/− (apolipoprotein E deficient) mice treated with 3PO (50 µg/g, ip; 4×/wk, 4 weeks) showed a metabolic switch toward ketone body formation. Treatment of ApoE−/−Fbn1C1039G+/− mice with 3PO (50 µg/g, ip) either after 4 (preventive, twice/wk, 10 weeks) or 16 weeks of Western diet (curative, 4×/wk, 4 weeks) inhibited intraplaque neovascularization by 50% and 38%, respectively. Plaque formation was significantly reduced in all 3PO-treated animals. This effect was independent of intraplaque neovascularization. In vitro experiments showed that 3PO favors an anti-inflammatory M2 macrophage subtype and suppresses an M1 proinflammatory phenotype. Moreover, 3PO induced autophagy, which in turn impaired NF-κB (nuclear factor-kappa B) signaling and inhibited TNF-α (tumor necrosis factor-alpha)–mediated VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1) upregulation. Consistently, a preventive 3PO regimen reduced endothelial VCAM-1 expression in vivo. Furthermore, 3PO improved cardiac function in ApoE−/−Fbn1C1039G+/− mice after 10 weeks of treatment.
Conclusions:
Partial inhibition of glycolysis restrained intraplaque angiogenesis without affecting plaque composition. However, less plaques were formed, which was accompanied by downregulation of endothelial adhesion molecules—an event that depends on autophagy induction. Inhibition of coronary plaque formation by 3PO resulted in an overall improved cardiac function.
Highlights.
By using a mouse model of advanced atherosclerosis that develops intraplaque neovascularization (ApoE−/−Fbn1C1039G+/− mice), we found that partial inhibition of glycolysis impairs neovascularization in plaques.
Partial inhibition of glycolysis restrains atherosclerotic plaque formation independent of intraplaque neovascularization and without affecting plaque composition.
Partial inhibition of glycolysis has a positive impact on cardiac function when administered in a preventive manner.
Partial inhibition of glycolysis increases the number of autophagosomes in endothelial cells.
Partial inhibition of glycolysis reduces endothelial VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1) expression in an autophagy-dependent way.
Partial inhibition of glycolysis favors an anti-inflammatory M2 macrophage subtype and suppresses an M1 proinflammatory phenotype in vitro.
Blood vessels constitute the largest network in our body to ensure a continuous blood flow toward different organs and tissues. However, despite their role in supplying nutrients and oxygenated blood, new vessels that branch off from existing vessels (a process known as neovascularization) could also play a detrimental role in various ischemic and inflammatory diseases including atherosclerosis.1 Because intraplaque microvessels are immature and leaky,2 they facilitate infiltration of lipids, inflammatory mediators, and erythrocytes. Consequently, intraplaque neovascularization has been linked to plaque progression.3,4 Intraplaque neovascularization is initiated in advanced plaques by hypoxia and emerges when the size or inflammatory burden of the plaque exceeds a critical threshold. Clinical trials targeting neovascularization in pathological settings mainly focus on blocking VEGF (vascular endothelial growth factor) signaling.5 However, these studies have reported moderate success due to drug resistance or adverse effects.6 Recent evidence indicates that proliferating endothelial cells (ECs) generate up to 85% of their ATP from glycolysis,7 suggesting that EC metabolism is an attractive alternative target to reduce neovascularization.8 Pharmacological inhibition of glycolytic flux by intraperitoneal injection of the small-molecule 3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) reduces vessel sprouting in EC spheroids, zebra fish embryos, mouse retina, and models of inflammation.9 3PO dose dependently reduces glycolysis in ECs but by no more than 35% to 40%, thus less than the nonmetabolizable glucose analog 2-deoxy-D-glucose, which reduces glycolysis by ≈80%.9 Because 3PO reduces cellular fructose-2,6-bisphosphate levels, it has been proposed that 3PO targets PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3).10 However, more recent findings indicate that 3PO does not bind PFKFB3 and may act through mechanisms that are unrelated to PFKFB3 inhibition.11 Importantly, suppression of glycolysis by 3PO occurs without lowering the energy charge or increasing oxygen consumption and without abrogating side metabolic pathways such as the pentose phosphate pathway necessary for NADPH (nicotinamide adenine dinucleotide phosphate) production. Although inhibition of glycolysis by 3PO is partial and transient, it is sufficient to reduce neovascularization.9
Intraplaque neovascularization is a typical feature of advanced human atherosclerotic plaques but is rarely observed in animal models.12 Nonetheless, our group has recently reported that ApoE−/− (apolipoprotein E deficient) mice, containing a heterozygous mutation (C1039G+/−) in the Fbn1 (fibrillin-1) gene, display substantial intraplaque neovascularization in the brachiocephalic artery and common carotid arteries.13 Because Fbn1 is the major structural component of extracellular microfibrils in the vessel wall, neovascularization in ApoE−/−Fbn1C1039G+/− mice probably occurs because elastin fragmentation allows microvessel sprouting from the adventitial vasa vasorum through the media into the intimal lesion.14 Moreover, the high degree of stenosis and the presence of activated macrophages in plaques of ApoE−/−Fbn1C1039G+/− mice likely results in intraplaque hypoxia and triggers the growth of new vessels from the adventitia.14,15 In the present study, we explored the possibility of inhibiting intraplaque angiogenesis with 3PO in ApoE−/−Fbn1C1039G+/− mice and studied its potential plaque stabilizing effects.
Materials and Methods
The authors declare that all supporting data are available within the article (and its Data Supplement).
Mice
Female ApoE−/− mice were fed a Western diet (WD; Altromin; C1000 diet supplemented with 20% milk fat and 0.15% cholesterol, No. 100171), starting at the age of 8 weeks, and treated with glycolysis inhibitor 3PO (50 mg/kg, ip, 4×/wk) or vehicle (dimethyl sulfoxide) for 4 weeks. The animals were housed in a temperature-controlled room with a 12-hour light/dark cycle and had free access to water and food (either normal laboratory diet [ssniff, R/M-H] or WD). To perform a glucose tolerance test, mice were fasted for 16 hours and injected with glucose (1 g/kg, ip). Blood glucose levels were determined and plotted in function of time. Insulin action was evaluated in vivo by injection of insulin (Novorapid, 1 U/kg, ip). Food and water intake of mice that were individually housed in metabolic cages (Tecniplast; floor area, 200 cm2) were monitored for 24 hours. Mice in metabolic cages were weighted at the start of the experiment and after 24 hours in the cage. Fasting and nonfasting blood glucose levels were analyzed with a hand-held glucometer (OneTouch Ultra, range, 20–600 mg/dL; Lifescan) by taking a droplet of blood from the tip of the mouse’s tail. At the end of the experiment, blood samples were obtained from the retro-orbital plexus of anesthetized mice (sodium pentobarbital, 75 mg/kg; ip). Subsequently, mice were sacrificed with sodium pentobarbital (250 mg/kg, ip). Plasma samples were analyzed with an automated Vista 1500 System (Siemens Healthcare Diagnostics) for liver enzymes, total cholesterol, and triglycerides. Insulin and β-hydroxybutyrate were determined with a mouse insulin ELISA kit (80-INSMS-E01; ALPCO) and β-hydroxybutyrate assay kit (ab83390; Abcam), respectively. To assess autophagy induction by 3PO in vivo, GFP-LC3 (green fluorescent protein-microtubule-associated protein light chain 3) #53 transgenic mice16 were treated with 3PO (50 mg/kg, ip, 4×/wk) or vehicle (dimethyl sulfoxide). After 2 weeks, the autophagic flux inhibitor chloroquine was administered (50 mg/kg, ip), and mice were sacrificed 3 hours later using an overdose of sodium pentobarbital (250 mg/kg, ip). Liver and heart samples were isolated for immunohistochemical detection of LC3.
For atherosclerosis studies, 3PO (50 mg/kg, ip) or solvent (dimethyl sulfoxide) was administered to female WD-fed ApoE−/−Fbn1C1039G+/− mice, starting either after 4 (2×/wk, 10 weeks, preventive regimen) or after 16 weeks of WD (4×/wk, 4 weeks, curative regimen; Figure I in the Data Supplement). Female mice were chosen because the Fbn1 mutation frequently leads to aortic dissection in male ApoE−/−Fbn1C1039G+/− mice but not in female ApoE−/−Fbn1C1039G+/− mice.13 In addition, ApoE−/− mice on WD were treated with 3PO as described above (preventive regimen) to test the effects of 3PO on plaque size and composition in the absence of intraplaque neovascularization. All animal procedures were conducted according to the guidelines for experimental atherosclerosis studies described in a scientific statement of the American Heart Association and the ATVB Council.17,18 Experiments were approved by the ethics committee of the University of Antwerp.
Histology
After euthanasia, the proximal aorta, aortic arch, carotid artery, and heart were collected. Tissues were fixed in 4% formalin for 24 hours, dehydrated overnight in 60% isopropanol, and subsequently embedded in paraffin. The plaque formation index in ApoE−/−Fbn1C1039G+/− mice was calculated on longitudinal sections of the carotid artery by using the following formula: (∑ total plaque length/∑ total vessel length)×100. Because plaques of ApoE−/− mice are less advanced as compared with ApoE−/−Fbn1C1039G+/− mice, the plaque formation index in ApoE−/− mice was measured on longitudinal sections of the aortic arch (instead of the carotid artery). Hematoxylin-eosin staining was performed on cross sections of the carotid artery of ApoE−/−Fbn1C1039G+/− mice to analyze plaque thickness and percentage necrosis. The plaque thickness was assessed by taking the mean value of 10 random measurements in the respective area. Necrosis was defined as acellular areas filled with necrotic clefts and necrotic debris. Immunohistochemical staining for anti-VWF (von Willebrand factor; PC054; Binding Site) and Ter-119 (anti–Ter-119, 550565; BD Biosciences) were performed to detect plaque ECs and erythrocytes, respectively. Autophagosome formation in liver and heart of 3PO-treated GFP-LC3 mice was measured via LC3 immunostaining using rabbit anti-LC3 antibody (3868S; Cell Signaling). Plaque composition was analyzed with Sirius red and anti–α-SMC actin (A2547; Sigma-Aldrich) staining to detect collagen and vascular smooth muscle cells, respectively. Macrophages and macrophage polarization were examined by immunohistochemistry using anti-MAC3 (01781D; Pharmingen) and antibodies against M1/M2 markers (anti-Egr2 [early growth response 2], PA5-27814 [ThermoFisher Scientific]; anti-GPR18 [G-protein−coupled receptor 18], PA5-23218 [ThermoFisher Scientific]; anti-Arg1 [arginase 1], PA5-29645 [ThermoFisher Scientific]; anti-CD38 [cluster of differentiation 38], MBS129421 [MyBioSource]). Quantification of immunostains was done from 10 random images per section using ImageJ software. The occurrence of myocardial infarctions (defined as large fibrotic areas) and coronary plaques was analyzed on Masson trichrome staining (transversal sections). Expression of adhesion molecule VCAM-1 (vascular cell adhesion molecule-1) was analyzed using anti–VCAM-1 (ab134047; Abcam). En face Oil Red O staining was performed on the carotid artery and aortic arch of ApoE−/−Fbn1C1039G+/− mice.
Echocardiography
Transthoracic echocardiograms were performed on anesthetized mice (isoflurane, 4% for induction and 2.5% for maintenance) at the end of the experiment using a VEVO2100 (VisualSonics), equipped with a 25-MHz transducer. Left ventricular internal diameter during diastole (LVIDd) and left ventricular internal diameter during systole (LVIDs) were measured, and fractional shortening ([LVIDd–LVIDs]/LVIDd×100) was calculated.
Cell Culture
Human aortic ECs (HAOECs; Sigma-Aldrich) were cultured in Endothelial Cell Growth Medium (PromoCell) supplemented with 2% fetal bovine serum, 0.4% EC growth supplement, 0.1 ng/mL epidermal growth factor, 1 ng/mL basic fibroblast growth factor, 90 µg/mL heparin, and 1 µg/mL hydrocortisone. To investigate the expression of VCAM-1 and ICAM-1 (intercellular adhesion molecule-1), HAOECs were stimulated with 20 ng/mL human TNF-α (tumor necrosis factor-alpha). A transcription factor assay kit was used to detect the NF-κB (nuclear factor-kappa B) transcription factor DNA binding activity in nuclear extracts according to the manufacturer’s instructions (ab133112; Abcam). For siRNA-mediated silencing of the autophagy pathway, HAOECs were seeded into 6-well plates and transfected at 75% confluency with 2.5 mL Opti-MEM Reduced Serum Medium (Thermo Fisher) containing 40 nmol/L ATG7 (autophagy-related 7) siRNA (Dharmacon) and 2.5 µL Lipofectamine RNAiMAX (Thermo Fisher) for 6 hours.
Real-Time RT-PCR
Total RNA was isolated using an Isolation II RNA mini kit (Bioline) according to the manufacturer’s instructions. Reverse transcription was performed with a Sensifast cDNA Synthesis Kit (Bioline). Thereafter, Taqman gene expression assay (Applied Biosystems) for CD38 (assay ID: Mm01220906_m1), Gpr18 (assay ID: Mm01224541_m1), Egr2 (assay ID: Mm00456650_m1), and Arg1 (assay ID: Mm00475988_m1) was performed in duplicate on an ABI prism 7300 sequence detector system (Applied Biosystems). The parameters for polymerase chain reaction amplification were 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Relative expression of mRNA was calculated using the comparative threshold cycle method. All data were normalized for quantity of cDNA input by performing measurements on the endogenous reference gene β-actin.
Western Blot Analyses
Cells were lysed in an appropriate volume of Laemmli sample buffer (Bio-Rad) containing β-mercaptoethanol (Sigma-Aldrich) and boiled for 5 minutes. Protein samples were then loaded onto precasted Bolt 4% to 12% Tris-Bis gels (Invitrogen) and after electrophoresis transferred to Immobilon-FL PVDF membranes (Millipore) according to standard procedures. Membranes were blocked for 1 hour with Odyssey blocking buffer (LI-COR Biosciences) diluted 1:5 with PBS. After blocking, membranes were probed overnight at 4°C with primary antibodies diluted in Odyssey blocking buffer, followed by 1-hour incubation with IRDye-labeled secondary antibodies at room temperature. Antibody detection was achieved using an Odyssey SA infrared imaging system (LI-COR Biosciences). The intensity of the protein bands was quantified using Image Studio software. The following primary antibodies were used: anti–β-actin (ab8226; Cell Signaling), anti-mTOR (2972; Cell Signaling), anti–phospho-mTOR (S2448; 2971; Cell Signaling), anti-p70 S6 kinase (9202; Cell Signaling), anti–phospho-p70 S6 kinase (Thr389; 9205; Cell Signaling), anti-LC3 (clone 5F10, 0231-100/LC3-5F10; Nanotools), anti–VCAM-1 (ab134047; Abcam), anti-ATG7 (8558S; Cell Signaling), anti–NF-κB (8242; Cell Signaling), anti-phospho–NF-κB (3039; Cell Signaling) and anti–ICAM-1 (ab179707; Abcam). IRDye-labeled secondary antibodies (goat anti-mouse IgG, 926-68070, and goat anti-rabbit IgG, 926-32211) were purchased from LI-COR Biosciences.
Transmission Electron Microscopy
Tissue samples were fixed in 2.5% glutaraldehyde, 0.1 M sodium cacodylate, and 0.05% CaCl2 (pH 7.4) and further processed for transmission electron microscopy as described previously,19 with a minor modification (extra staining with 1% tannic acid in veronal acetate for 1 hour after OsO4 postfixation). A FEI Tecnai microscope was used to examine ultrathin sections at 80 to 120 kV. Autophagic vacuoles were quantified on 5 different images taken at random per section.
Statistics
All data are expressed as mean±SEM. Statistical analyses were performed using SPSS software (version 25; SPSS, Inc). Statistical tests are specified in the figure and table legends. When parametric statistics (ANOVA, Student t test) were used, a test of normality (Shapiro-Wilk test) and a test for equal variances (Levene test) was performed. In every case, the data passed normality and equal variance tests. If not, nonparametric statistics (Mann-Whitney U test, Kruskal-Wallis test) were used. A χ2 test was used to determine whether there was an association between categorical variables (ie, whether the variables are independent or related). A Fisher exact test was used when sample sizes were small (ie, when 1 of the 4 cells of a 2×2 table had <5 observations) to test whether 2 categorical variables were associated with each other or not. A Mann-Whitney U test was used to compare differences between 2 independent groups when the dependent variable was either ordinal or continuous but not normally distributed. A Kruskal-Wallis test was used as an alternative for a 1-way ANOVA if the assumptions of the latter were violated (ie, data not normally distributed or unequal variances). Differences were considered significant at P<0.05.
Results
3PO Is Not Toxic but Causes a Metabolic Switch in Mice
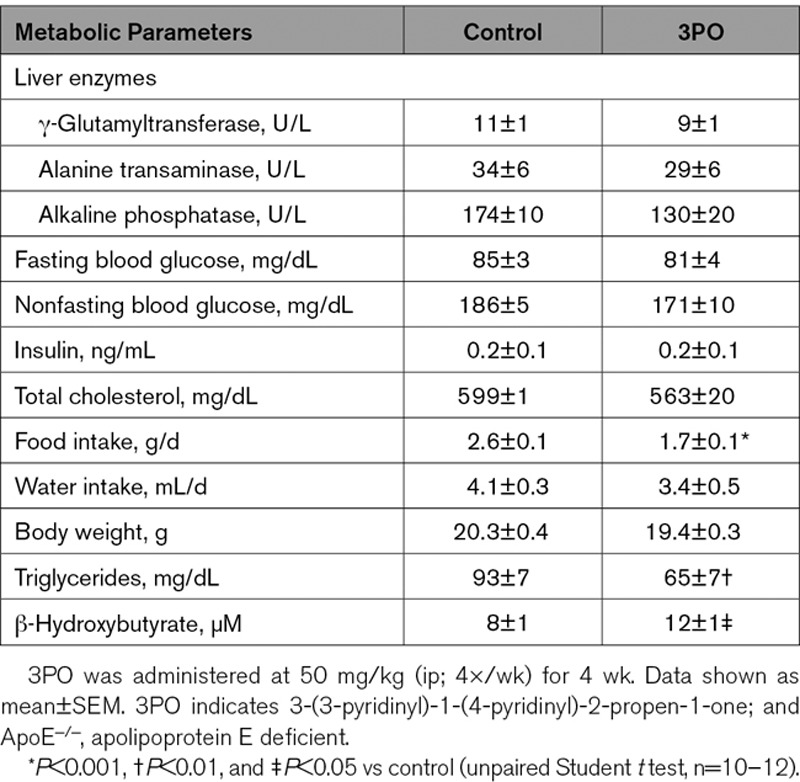
To evaluate whether glycolysis inhibitor 3PO affects general metabolism, female ApoE−/− mice were fed a WD and treated with 3PO (50 mg/kg, ip, 4×/wk) or vehicle for 4 weeks. Plasma analysis did not reveal changes in the level of liver enzymes (GTT, ALT, and ALP), fasting and nonfasting blood glucose, insulin or total cholesterol (Table 1). Moreover, according to a glucose and insulin tolerance test, glucose absorption and insulin receptor sensitivity were not different in 3PO-treated mice as compared with vehicle-treated controls (Figure II in the Data Supplement). Experiments in metabolic cages showed that water intake was not altered, although food consumption significantly decreased in 3PO-treated mice (P=0.0002; Table 1). Also body weight tended to decrease, although this effect was not statistically significant (P=0.1241). Levels of plasma triglycerides clearly decreased (P=0.0086), whereas those of ketone body β-hydroxybutyrate increased (P=0.0443; Table 1). Because starvation is a well-known trigger of autophagy induction, we evaluated whether reduced food intake after 3PO treatment stimulates autophagy in liver and heart, which are one of the most sensitive organs to starvation-induced autophagy.16 Unlike complete food withdrawal that strongly evoked autophagy, autophagosome staining was negative in liver and heart of 3PO-treated GFP-LC3 mice (Figure III in the Data Supplement).
Table 1.
Metabolic Parameters of ApoE−/− Mice Treated for 4 wk With 3PO or Vehicle (Dimethyl Sulfoxide)
3PO Inhibits Neovascularization in Plaques of ApoE−/−Fbn1C1039G+/− Mice
3PO (50 mg/kg, ip) or vehicle was administered to ApoE−/− Fbn1C1039G+/− mice starting either after 4 WD (2×/wk, 10 weeks, preventive regimen) or 16 weeks of WD (4×/wk, 4 weeks, curative regimen). As shown by anti-VWF staining of plaques in the carotid arteries of ApoE−/−Fbn1C1039G+/− mice, 3PO reduced the number of mice with intraplaque neovascularization with almost 40% (curative regimen) and 50% (preventive regimen; Figure 1A). Moreover, the number of microvessels per plaque significantly decreased (Figure 1A). Intraplaque microvessels were not observed in the proximal aorta. An anti–Ter-119 staining showed that microvessels were leaky and released erythrocytes into the plaque (Figure 1B). The number of intraplaque hemorrhages was reduced after treatment with 3PO in the preventive setting (control, 2.25 [0.0–7.0]; 3PO, 0.00 [0.0–3.0]/mm2; P=0.03). A decrease in the number of intraplaque hemorrhages was also observed in a curative 3PO regimen, although this effect was not significant (control, 9.57 [0.0–25.53]; 3PO, 0.0 [0.0–23.09]/mm2; P=0.46).
Figure 1.
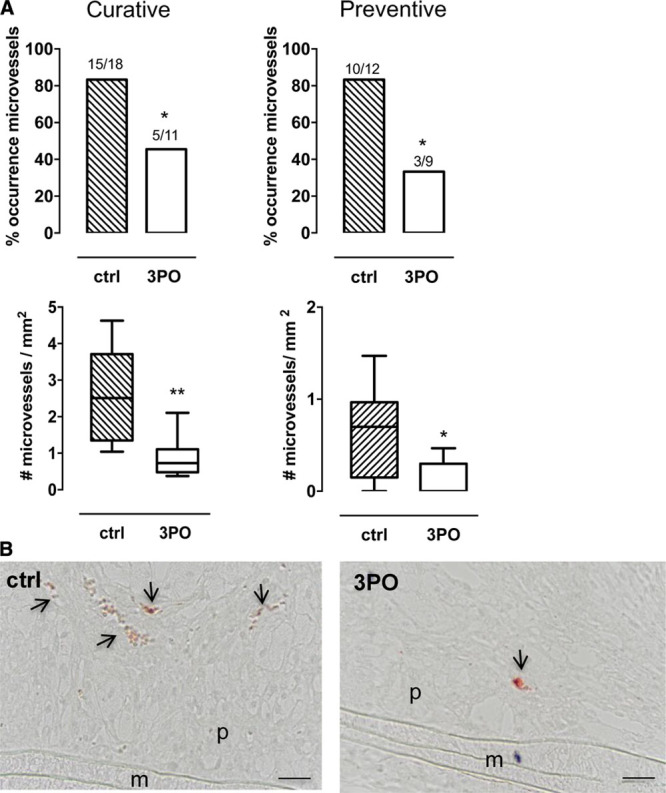
3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) inhibits intraplaque neovascularization in the carotid artery of ApoE−/−Fbn1C1039G± mice. A, Differences in the occurrence (number of mice) and the number of intraplaque microvessels of control (ctrl) and 3PO-treated mice that underwent a curative or preventive 3PO regimen. *P<0.05, **P<0.01 vs ctrl (χ2 [top] or Mann-Whitney U test [bottom]; ctrl, n=8–10; 3PO, n=9–13). B, Anti–Ter-119 staining of atherosclerotic plaques in the carotid artery of ctrl and 3PO-treated mice (curative regimen). Microvessels are marked by arrows. Scale bar=50 µm. M indicates media; and P, plaque.
3PO Inhibits Atherosclerotic Plaque Formation Independent of Intraplaque Neovascularization and Without Affecting Plaque Composition
An analysis of total cholesterol did not reveal significant differences between 3PO-treated ApoE−/−Fbn1C1039G+/− mice and untreated controls (curative regimen, 507±58 versus 602±58 mg/dL; preventive regimen, 454±24 versus 527±33 mg/dL). 3PO did not change plaque thickness and plaque necrosis (Table 2; Figure IV in the Data Supplement). Moreover, the smooth muscle cell and macrophage content of plaques and the percentage of total plaque collagen was not different between control and 3PO-treated animals (Table 2; Figure IV in the Data Supplement). Nonetheless, plaque formation was reduced in both the curative regimen and preventive regimen as illustrated by the plaque formation index (Figure 2A) and en face Oil Red O stainings (Figure 2B). Importantly, ApoE−/−Fbn1C1039G+/− mice that contained plaques without obvious intraplaque neovascularization revealed similar inhibition of plaque formation (Figure 2A), suggesting that 3PO may control atherosclerotic plaque formation independent of intraplaque microvessel growth. In line with this finding, 3PO was able to inhibit plaque formation in the aortic arch of regular ApoE−/− mice that do not develop intraplaque neovascularization (control, 53.9±4.8%; 3PO, 38.6±5.2%; P=0.04).
Table 2.
Thickness and Composition of Atherosclerotic Plaques in the Carotid Artery of ApoE−/−Fbn1C1039G+/− Mice

Figure 2.
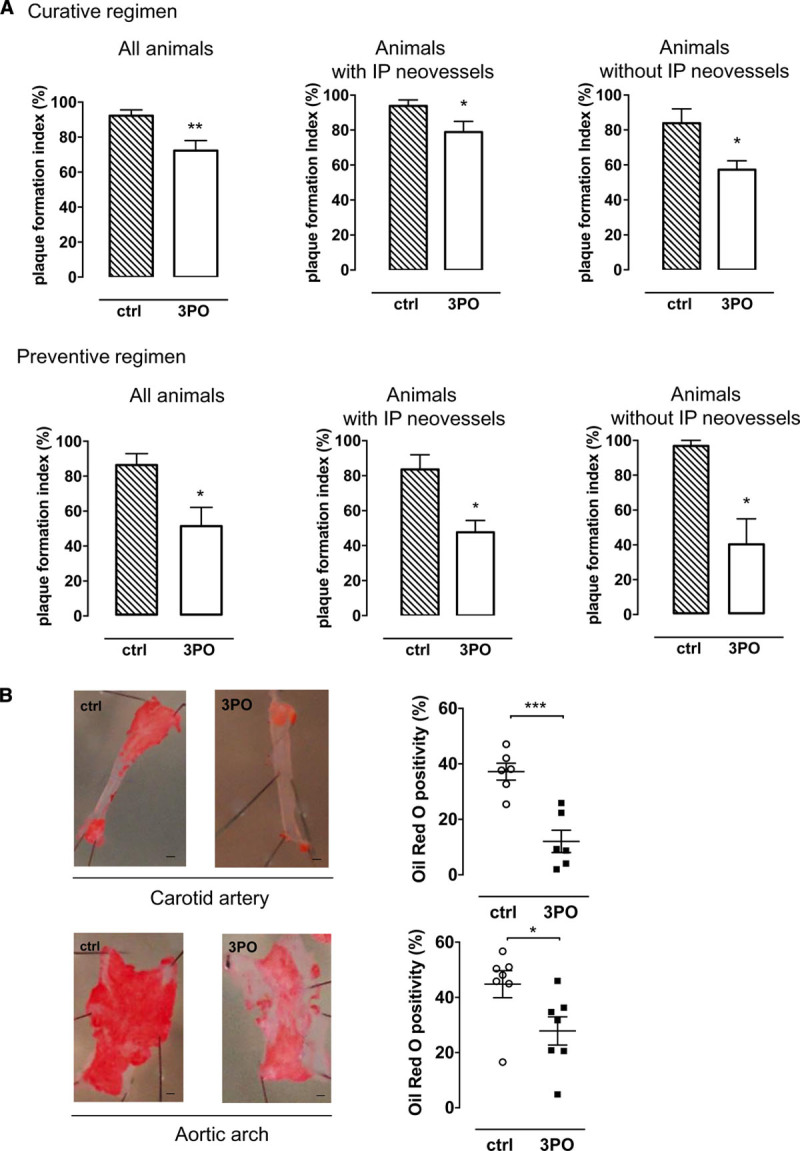
3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) inhibits atherosclerotic plaque formation in ApoE−/−Fbn1C1039G± mice. A, Plaque formation index of the right carotid artery as a measure of plaque occurrence in control (ctrl) and 3PO-treated mice that underwent a curative or preventive 3PO regimen. Plaque formation index is shown for all animals (left) of the ctrl group (n=12) and 3PO-treated group (n=9), for mice with intraplaque microvessels (middle; 10/12 ctrl mice and 3/9 treated mice) or for mice without intraplaque microvessels (right; 2/12 ctrl and 6/9 treated mice). *P<0.05, **P<0.01 vs ctrl (Mann-Whitney U test). B, En face Oil Red O staining of the carotid artery and aortic arch of ctrl (n=6–7) and 3PO-treated vessels (n=6–7). Scale bar=1.5 mm. *P<0.05, ***P<0.001 vs ctrl (independent samples t test). IP indicates intraplaque.
3PO Has a Positive Impact on Cardiac Function When Administered in a Preventive Manner
Unlike standard ApoE−/− mice or ApoE−/−Fbn1C1039G+/− mice on normal laboratory diet, ApoE−/−Fbn1C1039G+/− mice on WD develop (sometimes highly stenotic) coronary artery plaques, which negatively affect the heart.13 Therefore, cardiac function and structure were assessed in ApoE−/−Fbn1C1039G+/− mice after 3PO treatment via echocardiography and histology, respectively. In the curative setting, measurements of the LVIDd (control, 4.6±0.2 µm; 3PO, 4.5±0.2 µm) and the LVIDs (control, 3.5±0.2 µm; 3PO, 3.2±0.3 µm) did not show significant differences between control and treated animals. The fractional shortening was also similar between both groups (control, 23.1±3.0; 3PO, 29.3±3.7 µm). However, the heart/body weight ratio was significantly different between 3PO-treated animals and controls (control, 9.8±0.8%; 3PO, 6.9±0.3%; P=0.0081). The preventive regimen resulted in significantly smaller LVIDs and LVIDd with an increased fractional shortening (Figure VA through VD in the Data Supplement). Analysis of fibrotic areas on heart sections did not show a decrease in the occurrence of myocardial infraction (Fisher exact test: curative, ctrl [2/15] versus 3PO [2/12]; P=1.00; preventive, ctrl [2/10] versus 3PO [0/9]; P=0.47). The number of mice with coronary plaque formation tended to decrease in the curative setting (Fisher exact test: ctrl [12/14] versus 3PO [7/12] mice; P=0.19). It was not significant in the preventive setting, even though a 50% decrease in the occurrence of coronary plaque formation was observed in animals after 10 weeks of treatment (Fisher exact test: ctrl [6/10] versus 3PO [3/9] mice; P=0.36). Further analysis of coronary arteries (in preventive regimen) showed that perivascular fibrosis decreased after treatment with 3PO (Figure VE in the Data Supplement).
3PO Increases the Number of Autophagosomes in EC
3PO is a glycolysis inhibitor that leads to moderate ATP depletion.9 To investigate whether this metabolic stress condition triggers autophagy, HAOECs were stimulated in vitro with 3PO for 16 hours. Levels of the autophagosomal marker protein LC3-II increased in a concentration-dependent manner (Figure 3A) and stimulated formation of autophagic vesicles as shown by transmission electron microscopy (Figure 3B). In contrast to the well-known autophagy inducer everolimus that stimulates autophagy via mTOR inhibition, the phosphorylation status of mTOR or its downstream substrate p70S6K was not affected by 3PO (Figure 3C).
Figure 3.

3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) stimulates autophagosome formation in endothelial cells independent of mTOR. A, Human aortic endothelial cells (HAOECs) were stimulated with 3PO (5–20 µmol/L) for 16 h followed by Western blot analysis for the autophagosomal marker protein LC3. Protein levels of LC3-I and LC3-II were quantified relative to the reference protein β-actin. *P<0.05 vs untreated control (ctrl) cells (1-way ANOVA, followed by Dunnett test, n=3). B, Detection and quantification of autophagic vacuoles (arrows) in 3PO-treated HAOECs (20 µmol/L 3PO, 16 h) using transmission electron microscopy. Scale bar=500 nm. ***P<0.001 (Mann-Whitney U test; n=3). C, Evaluation of the phosphorylation status of mTOR and its downstream target p70S6K in 3PO-treated HAOECs via Western blotting. Everolimus (EV; 10 µmol/L) was used as a positive control. LC3 indicates microtubule-associated protein light chain 3; and mTOR, mammalian target of rapamycin.
3PO Reduces Endothelial VCAM-1 Expression in an Autophagy-Dependent Way
One of the early steps in plaque formation is the upregulation of adhesion molecules such as VCAM-1 followed by monocyte infiltration.20,21 Because 3PO inhibits plaque formation, we investigated whether VCAM-1 expression could be inhibited by 3PO. To this end, HAOECs were stimulated with TNF-α in the presence or absence of 3PO. TNF-α–treated cells clearly upregulated VCAM-1 and ICAM-1 protein expression, yet upregulation of both proteins was significantly impaired in the presence of 3PO (Figure 4). Moreover, NF-κB signaling was blunted by 3PO, given the significantly lower levels of phosphorylated NF-κB and a reduced DNA binding activity of NF-κB in 3PO-treated cells (Figure VI in the Data Supplement). Silencing of the essential autophagy gene ATG7 promoted VCAM-1 and ICAM-1 expression and could neither be enhanced by TNF-α nor inhibited by 3PO (Figure 4). ApoE−/−Fbn1C1039G+/− and ApoE−/− mice treated with 3PO (preventive regimen) revealed a decreased expression of VCAM-1 at the luminal EC surface of the plaque (Figure 4).
Figure 4.

3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) impairs TNF-α (tumor necrosis factor-alpha)–mediated upregulation of VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1) in endothelial cells via autophagy. A, Quantitative PCR and Western blot analysis of VCAM-1, ICAM-1, and ATG7 (autophagy-related 7) expression, either in wild-type (siCTRL-treated) human aortic endothelial cells (HAOECs; left) or siATG7-treated HAOECs (right) and exposed to hTNF-α (20 ng/mL) in the presence or absence of 3PO (10–20 µmol/L) for 24 h. β-actin was used as a reference gene. *P<0.05, **P<0.01 (1-way ANOVA followed by Dunnett test, n=3). B, Quantification of VCAM-1–positive endothelial cells in the carotid artery of ApoE−/−Fbn1C1039G+/− and the aortic arch of ApoE−/− (apolipoprotein E deficient) mice treated with 3PO or solvent (control [ctrl]) for 10 wk (preventive regimen). *P<0.05, ***P<0.001 vs ctrl (unpaired Student t test, n=7–12).
3PO Promotes an M2 Macrophage Subtype In Vitro
Because 3PO regulates inflammation and may affect macrophage polarization,22 gene expression of M1 and M2 exclusive genes23 was analyzed in vitro by real-time polymerase chain reaction in 3PO-treated mouse macrophages. 3PO did not change gene expression of M1 genes CD38 and Gpr18 but significantly upregulated expression of M2 genes Egr2 and Arg1 in a concentration-dependent manner (Figure 5). Furthermore, stimulation of an M1 phenotype by IFN-γ(interferon-gamma)/LPS (lipopolysaccharide) was inhibited by 3PO (Figure 5). Because of aspecific and unreliable antibody staining, the abovementioned M1/M2 markers failed to translate to macrophages in plaques of vehicle- or 3PO-treated carotid arteries and confirms recent statements in literature that valid in vivo M1/M2 surface markers remain to be discovered.24
Figure 5.

3PO (3-[3-pyridinyl]-1-[4-pyridinyl]-2-propen-1-one) promotes a macrophage M2 phenotype. Bone marrow–derived macrophages from ApoE−/− (apolipoprotein E deficient) mice were treated in vitro with 3PO (10–20 μM) or a mixture of IFN-γ (interferon-gamma; 20 ng/mL) and LPS (lipopolysaccharide; 100 ng/mL) for 24 h. Subsequently, mRNA expression of M1 exclusive genes CD38 (cluster of differentiation 38) and Gpr18 (G-protein−coupled receptor 18; A) or the M2-specific genes Erg2 (early growth response 2) and Arg1 (arginase 1; B) was analyzed by real-time polymerase chain reaction. *P<0.05 (Kruskal-Wallis followed by Mann-Whitney U test, n=4).
Discussion
There is an obvious association between intraplaque neovascularization and plaque vulnerability in advanced human plaques.25 However, the causality and impact of intraplaque neovascularization on plaque destabilization are poorly studied. In oncology, research is far more ahead on this topic, and several antiangiogenic strategies have been tested in an experimental setup.26 To date, blocking VEGF was the primary strategy for reducing neovascularization.27,28 Unfortunately, limited efficacy and adverse effects have been downsizing its success, even when multiple blockers were used simultaneously.5,29 Therefore, a fundamentally different approach is required to reboost antiangiogenic therapies. Given that ECs rely on glycolysis for up to 85% of their energy demand,7 targeting EC metabolism may represent an attractive new strategy to inhibit neovascularization.8,30 Transient and partial inhibition of glycolysis in proliferating ECs by the small-molecule 3PO inhibits pathological angiogenesis without interfering with the metabolism of healthy cells.9 In line with this statement, experimental evidence from the present study indicates that treatment of ApoE−/− mice with 3PO for 4 weeks caused neither substantial adverse effects nor changes in general metabolism. Reduced food intake was observed, yet circulating liver enzymes, blood glucose, insulin, and total cholesterol were not affected. Also a glucose and insulin tolerance test was perfectly normal. However, 3PO caused a decrease in the level of circulating triglycerides and a significant rise in β-hydroxybutyrate levels, indicating a metabolic switch from glucose to fatty acid–derived ketones to provide sufficient energy. Lack of major side effects was also reported recently by Beldman et al31 after treatment of atherosclerotic ApoE−/− mice for 6 weeks with 3PO (25 mg/kg, ip, 3×/wk).
Once safe administration of 3PO was evident; we next investigated the effect of 3PO on the formation of intraplaque microvessels in a mouse model of advanced atherosclerosis (ie, ApoE−/− mice with a heterozygous mutation in the fibrillin-1 gene [Fbn1C1039G+/−], yielding large plaques with a highly unstable phenotype and extensive intraplaque neovascularization).13 Analysis of intraplaque neovascularization in the carotid artery revealed a significant decrease in the occurrence and the amount of microvessels in ApoE−/−Fbn1C1039G+/− mice treated with 3PO. These results are in line with the effect of 3PO in cancer tissue, where 3PO inhibits vascular sprouting in pathological angiogenesis.7 Because inhibition of angiogenesis in malignant tissue can affect tumor growth,32 it was interesting to investigate the impact of 3PO on plaque size and vulnerability. After 4 weeks of treatment, we could not observe a difference in plaque composition between control and treated animals. These results fed the presumption that even though 4 weeks of treatment is enough to inhibit intraplaque neovascularization, it may have been too short to have any impact on plaque size or composition. Therefore, we decided to repeat the experiment in a preventive manner with 3PO being administered over a longer period of time. Given that ip injections are stressful for ApoE−/−Fbn1C1039G+/− mice, they were injected with 3PO for a maximum of 10 weeks.33 The preventive treatment regimen confirmed the results that we obtained after the curative treatment regimen with an even larger decrease in intraplaque neovascularization. Nonetheless, we could not observe a difference in parameters defining plaque vulnerability such as changes in macrophages, smooth muscle cells, and total collagen. Because intraplaque microvessels are considered a potential entry site for erythrocytes, lipids, and inflammatory mediators in the plaque,34,35 it was highly unexpected that, after 10 weeks of treatment, plaque composition was similar between control and treated animals.
Importantly, although the plaque size and composition remained unchanged, the occurrence of plaques as measured by the plaque formation index was significantly reduced in 3PO-treated animals. After 10 weeks of treatment, a 35% reduction in plaque formation was observed in the carotid artery. Given that neovascularization occurs after a certain degree of hypoxia, plaque progression is already in an advanced state. A decrease in the plaque formation index demonstrates that EC metabolism plays an important role in the early stages of plaque development, apart from angiogenesis. Indeed, a 3PO-mediated reduction in plaque development was also observed in regular ApoE−/− mice, which develop plaques without intraplaque neovascularization. When ECs undergo inflammatory activation, the upregulation of adhesion molecules such as VCAM-1 represents an important trigger in early lesion development, as they attract and encourage monocytes to enter the lesion.36 In the present study, we provide in vitro and in vivo evidence that 3PO interferes with the upregulation of VCAM-1 and ICAM-1. In vitro experiments with ECs, preactivated with TNF-α and treated with 3PO, revealed impaired NF-κB signaling and reduced VCAM-1 (and ICAM-1) expression levels. Moreover, a preventive 3PO regimen was able to reduce VCAM-1 expression in the endothelium in vivo. A study performed in cancer research consolidates these results as 3PO reduced the expression of adhesion molecules concomitant with tumor vessel normalization due to an altered EC proinflammatory signature.37 Nonetheless, the question remains why changes in VCAM-1 expression in 3PO-treated mice do not alter plaque composition. Our data seem to suggest that 3PO only impairs initiation of plaque development but not plaque progression. Indeed, one may speculate that once early plaques have formed, downregulation of VCAM-1 expression in ECs by 3PO does not significantly affect further leukocyte recruitment and plaque progression. Histological data from human plaques indicate that in early lesions, VCAM-1 and ICAM-1 are predominantly expressed by the endothelium, whereas in more advanced lesions, the majority of VCAM-1 expression is found in subsets of intimal VSMCs and macrophages.38 These findings may explain why downregulation of adhesion molecules by 3PO in ECs mainly affects the initial stage of atherosclerosis development but not further steps of plaque progression.
Interestingly, the metabolic stress caused by 3PO stimulated autophagosome formation in ECs and led to induction of autophagy. Similar observations were previously reported in cancer cells.39 Our results indicate that downregulation of endothelial VCAM-1 (and ICAM-1) expression by 3PO depends on autophagy induction. Expression of the adhesion molecules was not downregulated by 3PO in TNF-α–treated ECs in which expression of the essential autophagy gene ATG7 was silenced. On the contrary, VCAM-1 (and ICAM-1) expression was upregulated in ATG7-deficient ECs, suggesting that autophagy suppresses expression of these adhesion molecules. Previously, we reported similar data with the biguanide metformin. The latter compound attenuates expression of the EC adhesion molecules ICAM-1 and VCAM-1, as well as formation of atherosclerotic plaques via autophagy induction.40 Downregulation of ATG7 gene expression prevented TNF-α–induced upregulation of VCAM-1 expression and monocyte adhesion to ECs. Both metformin and 3PO govern the expression of cell adhesion molecules in ECs by inhibiting NF-κB activation. Our findings support previous data showing that endothelial autophagy is atheroprotective and limits atherosclerotic plaque formation by preventing endothelial apoptosis, senescence, and inflammation.41–43
Apart from the atheroprotective effects on ECs, several lines of evidence indicate that 3PO also negatively influences inflammation.22,31,44–46 In the present study, we could demonstrate that 3PO favors an anti-inflammatory M2 macrophage subtype and suppresses an M1 proinflammatory phenotype in vitro. This finding is consistent with previous reports showing a strong linear correlation between glycolytic flux and proinflammatory activation of macrophages (as measured by TNF-α production)22 and 3PO-mediated suppression of T-cell activation.44 Moreover, inhibition of glycolysis by 3PO has profound effects on cell viability of M1 macrophages.22
Given the importance of neovascularization in the ischemic myocardium after an acute myocardial infarction, we analyzed cardiac function and morphology.47 The inhibition of angiogenesis by targeting cell metabolism is still in a preclinical stage, thus literature regarding this topic is still lacking. After a curative treatment regimen, no significant differences were observed in cardiac function. However, after 10 weeks of treatment (preventive regimen), we observed an improved cardiac morphology and function (smaller LVIDs and LVIDd with increased fractional shortening). Moreover, even though only a small nonsignificant reduction was observed in the occurrence of myocardial infarctions, the occurrence of coronary plaques was decreased by 50% in treated animals. These findings are consistent with the overall effect of 3PO on plaque formation, in this case, coronary plaques. As such, it is conceivable that the overall improved cardiac function is a result of less coronary plaque formation. To further explain the improved cardiac function after 3PO treatment (preventive regimen), we also documented the level of perivascular fibrosis of coronary arteries. This parameter was significantly reduced by 3PO. Because coronary perivascular fibrosis can be caused by an impaired coronary blood flow,48 a lower degree of stenosis in the coronary arteries of 3PO-treated mice probably contributes to this finding. Similar observations were recently made in the same mouse model using lipid-lowering therapy.49 Apart from improved cardiac function through less coronary plaque formation and coronary perivascular fibrosis, it is important to note that autophagy induction has beneficial effects on the heart.50Because 3PO promotes autophagy, it is plausible to assume that autophagy induction by 3PO also contributes to improved cardiac function. However, given the lack of autophagosome formation in 3PO-treated GFP-LC3 mice, we consider this hypothesis unlikely, though cannot completely rule out this possibility.
In conclusion, we were able to inhibit intraplaque neovascularization with 3PO. However, the reduction in intraplaque microvessels did not exert a significant effect on plaque composition. Surprisingly, 3PO reduced the formation of plaques, not in size but in frequency. Less plaques were formed in the carotid artery indicating that 3PO already has an effect in the early onset of atherosclerosis by downregulating EC adhesion molecules. In addition, fewer coronary plaques were formed, which resulted in overall improved cardiac function. Interestingly, inhibition of key steps in glycolysis with small molecules has recently provided a novel area of cancer research and has been proven effective in slowing the proliferation of cancer cells, with PFK158 as a first-in-human and first-in-class PFKFB3 inhibitor in a phase I clinical trial (NCT02044861). Compared with cancer, the EC metabolism in atherosclerosis is largely unexplored, though is an emerging target that may offer attractive therapeutic avenues to counteract plaque progression.8,51,52
Acknowledgments
We would like to thank Dr Bart Peeters, Sofie De Moudt, Anne-Elise Van Hoydonck, Hermine Fret, and Rita Van den Bossche for technical help. We are grateful to Dr Bronwen Martin for critical reading of the manuscript.
Sources of Funding
This work was supported by the University of Antwerp (DOCPRO-BOF [Doctoraatsprojecten Bijzonder Onderzoeksfonds]), the Hercules Foundation (grant No. AUHA/13/03), and the Horizon 2020 program of the European Union–Marie Sklodowska Curie actions–ITN–MOGLYNET (Innovative Training Network–Modulation of Glycolytic Flux as a New Approach for Treatment of Atherosclerosis and Plaque Stabilization: a Multidisciplinary Study; grant No. 675527).
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- 3PO
- 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- ApoE−/−
- apolipoprotein E deficient
- EC
- endothelial cell
- Fbn1
- fibrillin-1
- HAOEC
- human aortic endothelial cell
- ICAM-1
- intercellular adhesion molecule-1
- LVIDd
- left ventricular internal diameter during diastole
- LVIDs
- left ventricular internal diameter during systole
- NF-κB
- nuclear factor-kappa B
- PFKFB3
- 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3
- TNF-α
- tumor necrosis factor-alpha
- VCAM-1
- vascular cell adhesion molecule-1
- VEGF
- vascular endothelial growth factor
- WD
- Western diet
For Sources of Funding and Disclosures, see page 1179.
These authors contributed equally to this article.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.119.313692.
References
- 1.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, Bauersachs J. Vasa vasorum angiogenesis: key player in the initiation and progression of atherosclerosis and potential target for the treatment of cardiovascular disease. Front Immunol. 2018;9:706. doi: 10.3389/fimmu.2018.00706. doi: 10.3389/fimmu.2018.00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VW, Virmani R, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–1527. doi: 10.1016/j.jacc.2008.12.056. doi: 10.1016/j.jacc.2008.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 5.Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17:471–494. doi: 10.1007/s10456-014-9420-y. doi: 10.1007/s10456-014-9420-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017;12:18–34. doi: 10.1016/j.redox.2017.01.007. doi: 10.1016/j.redox.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 8.Pircher A, Treps L, Bodrug N, Carmeliet P. Endothelial cell metabolism: a novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis. 2016;253:247–257. doi: 10.1016/j.atherosclerosis.2016.08.011. doi: 10.1016/j.atherosclerosis.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 9.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 11.Boyd S, Brookfield JL, Critchlow SE, Cumming IA, Curtis NJ, Debreczeni J, Degorce SL, Donald C, Evans NJ, Groombridge S, et al. Structure-based design of potent and selective inhibitors of the metabolic kinase PFKFB3. J Med Chem. 2015;58:3611–3625. doi: 10.1021/acs.jmedchem.5b00352. doi: 10.1021/acs.jmedchem.5b00352. [DOI] [PubMed] [Google Scholar]
- 12.Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, De Meyer GRY. Animal models of atherosclerosis. Eur J Pharmacol. 2017;816:3–13. doi: 10.1016/j.ejphar.2017.05.010. doi: 10.1016/j.ejphar.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Van der Donckt C, Van Herck JL, Schrijvers DM, Vanhoutte G, Verhoye M, Blockx I, Van Der Linden A, Bauters D, Lijnen HR, Sluimer JC, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. 2015;36:1049–1058. doi: 10.1093/eurheartj/ehu041. doi: 10.1093/eurheartj/ehu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Herck JL, De Meyer GRY, Martinet W, Van Hove CE, Foubert K, Theunis MH, Apers S, Bult H, Vrints CJ, Herman AG. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation. 2009;120:2478–2487. doi: 10.1161/CIRCULATIONAHA.109.872663. doi: 10.1161/CIRCULATIONAHA.109.872663. [DOI] [PubMed] [Google Scholar]
- 15.Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Role of lipids and intraplaque hypoxia in the formation of neovascularization in atherosclerosis. Ann Med. 2017;49:661–677. doi: 10.1080/07853890.2017.1366041. doi: 10.1080/07853890.2017.1366041. [DOI] [PubMed] [Google Scholar]
- 16.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. doi: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, 3rd, Rosenfeld ME, Virmani R American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; and Council on Basic Cardiovascular Sciences. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/ATV.0000000000000062. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 18.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement From ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. doi: 10.1161/ATVBAHA.117.309524. doi: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinet W, Timmermans JP, De Meyer GRY. Methods to assess autophagy in situ–transmission electron microscopy versus immunohistochemistry. Methods Enzymol. 2014;543:89–114. doi: 10.1016/B978-0-12-801329-8.00005-2. doi: 10.1016/B978-0-12-801329-8.00005-2. [DOI] [PubMed] [Google Scholar]
- 20.Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191–203. doi: 10.1016/s0021-9150(03)00097-2. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 21.Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, Rudd JH, Narula J, Enriquez JA, Través PG, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. 2015;35:1463–1471. doi: 10.1161/ATVBAHA.115.305551. doi: 10.1161/ATVBAHA.115.305551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front Immunol. 2019;10:1084. doi: 10.3389/fimmu.2019.01084. doi: 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Vries MR, Quax PH. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr Opin Lipidol. 2016;27:499–506. doi: 10.1097/MOL.0000000000000339. doi: 10.1097/MOL.0000000000000339. [DOI] [PubMed] [Google Scholar]
- 26.Rajabi M, Mousa SA. The role of angiogenesis in cancer treatment. Biomedicines. 2017;5:34. doi: 10.3390/biomedicines5020034. doi: 10.3390/biomedicines5020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Veken B, De Meyer GRY, Martinet W. Intraplaque neovascularization as a novel therapeutic target in advanced atherosclerosis. Expert Opin Ther Targets. 2016;20:1247–1257. doi: 10.1080/14728222.2016.1186650. doi: 10.1080/14728222.2016.1186650. [DOI] [PubMed] [Google Scholar]
- 28.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(suppl 3):4–10. doi: 10.1159/000088478. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 29.Moreo A, Vallerio P, Ricotta R, Stucchi M, Pozzi M, Musca F, Meani P, Maloberti A, Facchetti R, Di Bella S, et al. Effects of cancer therapy targeting vascular endothelial growth factor receptor on central blood pressure and cardiovascular system. Am J Hypertens. 2016;29:158–162. doi: 10.1093/ajh/hpv077. doi: 10.1093/ajh/hpv077. [DOI] [PubMed] [Google Scholar]
- 30.Ali L, Schnitzler JG, Kroon J. Metabolism: the road to inflammation and atherosclerosis. Curr Opin Lipidol. 2018;29:474–480. doi: 10.1097/MOL.0000000000000550. doi: 10.1097/MOL.0000000000000550. [DOI] [PubMed] [Google Scholar]
- 31.Beldman TJ, Malinova TS, Desclos E, Grootemaat AE, Misiak ALS, van der Velden S, van Roomen CPAA, Beckers L, van Veen HA, Krawczyk PM, et al. Nanoparticle-aided characterization of arterial endothelial architecture during atherosclerosis progression and metabolic therapy. ACS Nano. 2019;13:13759–13774. doi: 10.1021/acsnano.8b08875. doi: 10.1021/acsnano.8b08875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015;21:267–273. doi: 10.1097/PPO.0000000000000138. doi: 10.1097/PPO.0000000000000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roth L, Rombouts M, Schrijvers DM, Lemmens K, De Keulenaer GW, Martinet W, De Meyer GRY. Chronic intermittent mental stress promotes atherosclerotic plaque vulnerability, myocardial infarction and sudden death in mice. Atherosclerosis. 2015;242:288–294. doi: 10.1016/j.atherosclerosis.2015.07.025. doi: 10.1016/j.atherosclerosis.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 34.Michel JB, Virmani R, Arbustini E, Pasterkamp G. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J. 2011;32:1977–1985, 1985a, 1985b, 1985c. doi: 10.1093/eurheartj/ehr054. doi: 10.1093/eurheartj/ehr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 36.Davies MJ, Gordon JL, Gearing AJ, Pigott R, Woolf N, Katz D, Kyriakopoulos A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol. 1993;171:223–229. doi: 10.1002/path.1711710311. doi: 10.1002/path.1711710311. [DOI] [PubMed] [Google Scholar]
- 37.Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 2016;30:968–985. doi: 10.1016/j.ccell.2016.10.006. doi: 10.1016/j.ccell.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, McCarty J, Ferguson M, Hudkins K, Benjamin CD. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993;92:945–951. doi: 10.1172/JCI116670. doi: 10.1172/JCI116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klarer AC, O’Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, Clem B, Chesney J, Telang S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014;2:2. doi: 10.1186/2049-3002-2-2. doi: 10.1186/2049-3002-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michiels CF, Apers S, De Meyer GRY, Martinet W. Metformin attenuates expression of endothelial cell adhesion molecules and formation of atherosclerotic plaques via autophagy induction. Annals of Clinical & Experimental Metabolism. 2016;1:1001. [Google Scholar]
- 41.Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C, Pic I, Dupont N, Busse J, Stark K, et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A. 2017;114:E8675–E8684. doi: 10.1073/pnas.1702223114. doi: 10.1073/pnas.1702223114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salminen A, Kaarniranta K. Glycolysis links p53 function with NF-kappaB signaling: impact on cancer and aging process. J Cell Physiol. 2010;224:1–6. doi: 10.1002/jcp.22119. doi: 10.1002/jcp.22119. [DOI] [PubMed] [Google Scholar]
- 43.Zhang R, Li R, Liu Y, Li L, Tang Y. The glycolytic enzyme PFKFB3 controls TNF-α-induced endothelial proinflammatory responses. Inflammation. 2019;42:146–155. doi: 10.1007/s10753-018-0880-x. doi: 10.1007/s10753-018-0880-x. [DOI] [PubMed] [Google Scholar]
- 44.Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses T cell activation. J Transl Med. 2012;10:95. doi: 10.1186/1479-5876-10-95. doi: 10.1186/1479-5876-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boscá L, González-Ramos S, Prieto P, Fernández-Velasco M, Mojena M, Martín-Sanz P, Alemany S. Metabolic signatures linked to macrophage polarization: from glucose metabolism to oxidative phosphorylation. Biochem Soc Trans. 2015;43:740–744. doi: 10.1042/BST20150107. doi: 10.1042/BST20150107. [DOI] [PubMed] [Google Scholar]
- 46.McGarry T, Biniecka M, Gao W, Cluxton D, Canavan M, Wade S, Wade S, Gallagher L, Orr C, Veale DJ, et al. Resolution of TLR2-induced inflammation through manipulation of metabolic pathways in Rheumatoid Arthritis. Sci Rep. 2017;7:43165. doi: 10.1038/srep43165. doi: 10.1038/srep43165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khurana R, Simons M, Martin JF, Zachary IC. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005;112:1813–1824. doi: 10.1161/CIRCULATIONAHA.105.535294. doi: 10.1161/CIRCULATIONAHA.105.535294. [DOI] [PubMed] [Google Scholar]
- 48.Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. 2012;60:416–421. doi: 10.1016/j.jjcc.2012.06.009. doi: 10.1016/j.jjcc.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Roth L, Rombouts M, Schrijvers DM, Martinet W, De Meyer GRY. Cholesterol-independent effects of atorvastatin prevent cardiovascular morbidity and mortality in a mouse model of atherosclerotic plaque rupture. Vascul Pharmacol. 2016;80:50–58. doi: 10.1016/j.vph.2016.01.007. doi: 10.1016/j.vph.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res. 2018;123:803–824. doi: 10.1161/CIRCRESAHA.118.312208. doi: 10.1161/CIRCRESAHA.118.312208. [DOI] [PubMed] [Google Scholar]
- 51.Theodorou K, Boon RA. Endothelial cell metabolism in atherosclerosis. Front Cell Dev Biol. 2018;6:82. doi: 10.3389/fcell.2018.00082. doi: 10.3389/fcell.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perrotta P, Emini Veseli B, Van der Veken B, Roth L, Martinet W, De Meyer GRY. Pharmacological strategies to inhibit intra-plaque angiogenesis in atherosclerosis. Vascul Pharmacol. 2019;112:72–78. doi: 10.1016/j.vph.2018.06.014. doi: 10.1016/j.vph.2018.06.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.