

Rigidity-dependent force generation involves no mechanosensing and is enslaved to a rigidity-independent contractile displacement.
Abstract
Cells’ ability to apply contractile forces to their environment and to sense its mechanical properties (e.g., rigidity) are among their most fundamental features. Yet, the interrelations between contractility and mechanosensing, in particular, whether contractile force generation depends on mechanosensing, are not understood. We use theory and extensive experiments to study the time evolution of cellular contractile forces and show that they are generated by time-dependent actomyosin contractile displacements that are independent of the environment’s rigidity. Consequently, contractile forces are nonmechanosensitive. We further show that the force-generating displacements are directly related to the evolution of the actomyosin network, most notably to the time-dependent concentration of F-actin. The emerging picture of force generation and mechanosensitivity offers a unified framework for understanding contractility.
INTRODUCTION
The mechanical properties of the extracellular matrix (ECM) play critical roles in the most fundamental cellular processes (1). Matrix rigidity, for instance, affects cell polarization, migration, growth, death, and differentiation (2–6). Probing ECM mechanical properties (“mechanosensing”) and feeding the outcome back into cells (“mechanotransduction”) are processes that occur via integrin-based cell-matrix adhesions. These multimolecular complexes bind the ECM at the extracellular side and attach to the actomyosin cytoskeleton at the intracellular side (see Fig. 1A). The cytoskeleton is composed of a network of actin filaments (F-actin) and myosin motors, where the latter actively generate contractile forces. It is well established that with increased ECM stiffness, larger contractile forces are transmitted to the matrix (7–10). The common view is that, at any point in time, this transmission is regulated through the strength of the adhesions (8). The formation of large adhesions (classically referred to “focal adhesions”) on stiff matrices is typically accompanied by the formation and growth of thick actin stress fibers that are attached to them (3). Collectively, these processes have marked effects on cellular properties and functions. Still, although actomyosin-based cellular contractility is regarded as a fundamental mechanosensing mechanism, a comprehensive understanding of both the contractile force generation process and mechanosensitivity and their interrelations is currently missing. In particular, whether the contractile force generation process is mechanosensitive, i.e., regulated through feedback from the cell’s probing/sensing of the rigidity, is not known.
Fig. 1. A simple model of cellular contractility.
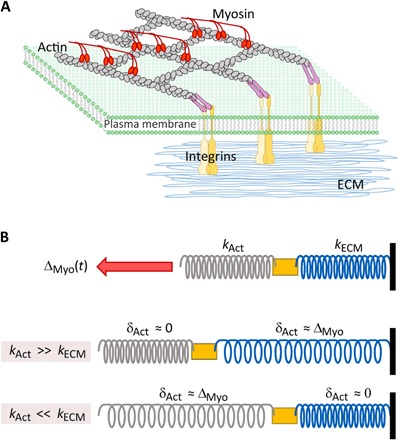
(A) A schematic of the cellular force generation machinery. Shown are the integrin molecules, which are attached to the ECM at their extracellular side and to actin structures (indirectly, through adapter proteins—purple rods) at the cellular side. Myosin motors that attach to the actin filaments generate a time-dependent displacement, ∆(t) (red arrow). (B) Top: An abstraction of (A) in which a spring of effective rigidity kECM, representing the ECM, is connected in series to another spring of effective rigidity kAct, representing the actin structures. The contractile displacement ∆(t), which is applied to the actin structures, is also shown. Color code as in (A). Middle: The degree to which the ECM or the actin structures are deformed depends on their relative rigidity. For kAct ≫ kECM, the myosin-generated contractile displacement ∆(t) is accommodated by ECM deformation, δECM ≈ ∆, while the actin filaments composing the actin structures slide past each other as rigid objects with no internal deformation, δAct ≈ 0. Bottom: In the opposite limit (relevant for experiments performed on rigid ECM, e.g., plastic/glass plates), kAct ≪ kECM, the myosin-generated contractile displacement ∆(t) is accommodated by stretching the actin filaments composing the actin structures, δAct ≈ ∆, while the rigid ECM is not deformed, δECM ≈ 0.
Here, we address the basic nature and time evolution of cellular contractile forces through a simple mathematical model combined with extensive experiments using cells adhering to micropillar arrays (Fig. 2A) (7). We show that, for a broad range of physiologically relevant ECM rigidities, the contractile force is proportional to the ECM rigidity as a result of simple mechanics. The proportionality factor is an intrinsic, cell-specific time-dependent contractile displacement that is rigidity independent. Namely, we demonstrate that cellular contractile forces are themselves generically nonmechanosensitive. This fundamental observation implies that information about ECM rigidity is internally and simply encoded at each point in time in the magnitude of the contractile force. We further show that the time-dependent contractile displacement is determined by the time evolution of the F-actin concentration, indicating that the dynamics of myosin motors are not the rate-limiting factor in the cellular force generation process. The emerging picture of cellular contractility is shown to explain and unify various existing observations.
Fig. 2. Pillar array experiments reveal cell-specific, time-dependent, rigidity-independent contractile displacements.
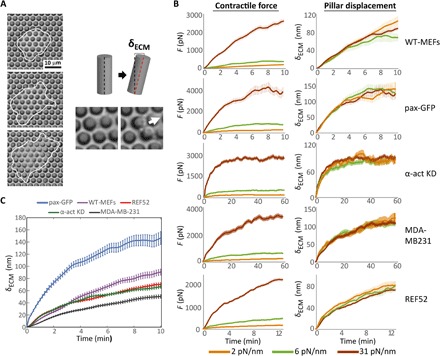
We used low-illumination bright-field time-lapse imaging to track live cells for several hours as they spread on the pillars to measure δECM(t). (A) Left: Frames from a time-lapse movie of a mouse fibroblast spreading on an array of 2-μm-diameter pillars coated with fibronectin (cell borders marked by the white line). Right: When the cell forms adhesions on the pillars, it pulls on them; pillar displacement (δECM) can be observed by tracking their tops over time. (B) Means ± SEM of the time-dependent contractile forces [F(t, kECM); left] and pillar displacement [δECM(t) = F(t, kECM)/kECM; right] on pillars of three widely different rigidities (corresponding to different pillar heights, the rigidity values appear on the legend). For all cell lines tested, δECM(t) = F(t, kECM)/kECM collapsed onto a single master curve (right). n > 50 pillars from >8 cells in each case. (C) Averages of the pillar displacement data from all three rigidities [(B), right] reveal cell-specific contractile displacements.
RESULTS
A simple model of cellular contractility
The major component of the cellular contractile force machinery is actomyosin networks, which are made up of force transmitting actin structures and force generating myosin motors (Fig. 1A). For adherent cells, the generated contractile force is transferred across the plasma membrane to the ECM through integrins (Fig. 1A). To model how contractile forces are generated, we first assume that the myosin motors intrinsically generate a time-dependent contractile displacement, ∆(t, kECM), which they impose at time t on the actin structures to which they attach and which may depend on the effective rigidity of the ECM, kECM. Whether the contractile displacement depends on kECM is a major question to be addressed below. Note that the existence of a contractile displacement is in line with the fundamental property of myosin motors, which display a typical working stroke size (namely, distance) (11). ∆(t, kECM) is accommodated by the displacement δAct(t) of the actin structure and by the deformation δECM(t) of the ECM, i.e., ∆(t) = δAct(t) + δECM(t). At any time t, the integrin adhesions are strong enough to withstand the forces applied to them.
The contractile force F(t, kECM) satisfies the coarse-grained force balance equation F(t, kECM) = kAct δAct(t) = kECM δECM(t), where kAct is the effective rigidity of the actin structures [Fig. 1B, top, note S1, and Schwarz et al. (12)]. This, in turn, implies that (for simplicity reasons, we treat kAct as a constant, although it is time dependent; see also note S2). To proceed, we further assume that for a broad range of physiologically relevant conditions (namely, relatively soft environments), the effective rigidity of actin structures is significantly larger than the rigidity of the ECM, i.e., kAct ≫ kECM. With this assumption, to be extensively tested below, we obtain
| (1) |
This equation shows that the dependence of the contractile force on the external rigidity may have two qualitatively different origins. First, the possible mechanosensitive contribution—if it exists—is contained within the contractile displacement ∆(t, kECM). Second, and independently of the possible kECM dependence of ∆(t, kECM), there exists also a nonmechanosensitive contribution—the overall prefactor constant kECM, which emerges from a linear force-displacement relation of the ECM. Consequently, Eq. 1 demonstrates that the mere existence of a dependence of F(t, kECM) on the external rigidity kECM does not immediately indicate that the contractile force is mechanosensitive. The mean and maximal force levels were reported earlier to be proportional to kECM for epithelial cells (13), but the question of time-dependent mechanosensitivity remained unanswered. Equation 1 offers a clean procedure to address this question and extract the possible mechanosensitive part of the contractile force by plotting the force F(t, kECM) normalized by kECM, F(t, kECM)/kECM—a procedure that is followed in our experiments described below.
Note that F(t, kECM)/kECM simply equals δECM(t), which implies that in the regime kAct ≫ kECM, the model predicts also δECM(t) ≈ ∆(t, kECM) and δAct(t) ≈ 0 (see Fig. 1B, middle). That is, in this case, the actin filaments/structures are predicted to experience no internal deformation but rather to slide one relative to another as rigid objects (as they polymerize/depolymerize at their edges), and the ECM deformation δECM(t) allows direct access to the myosin-generated collective contractile displacement ∆(t, kECM). Last, note that in the opposite regime, kAct ≪ kECM, relevant to cells adhering to glass/plastic plates, the model predicts δAct(t) ≈ ∆(t, kECM) and δECM(t) ≈ 0 (see Fig. 1B, bottom). Notably, in this regime, our model predicts the saturation force F(t → ∞, kECM) to be independent of kECM (see note S2), in line with previous results (14).
Direct experimental tests demonstrate that cellular contractile forces are nonmechanosensitive
To test whether ∆(t, kECM) in Eq. 1 depends on kECM, i.e., whether it is mechanosensitive, we performed extensive experiments on cells adhering to arrays of fibronectin-coated flexible polydimethylsiloxane (PDMS) micropillars of 2 μm in diameter (see Materials and Methods and Fig. 2A). These arrays allow measuring the time-dependent contractile force as a function of rigidity that is varied only by the pillars’ height while maintaining the material from which they are formed (hence, the surface chemistry) and their cross-sectional area fixed. We monitored the cells from the very initial stages of attachment to the pillars, and as they were spreading, we tracked the cell-driven pillar deflections, which is simply δECM(t) (Fig. 2A). We used pillars with rigidities that vary by more than 15-fold, kECM = 2,6,31 pN/nm, equivalent to effective elastic moduli in the range Eeff ≈ 1.5 to 22 kPa (see Materials and Methods), thus allowing us to test the relevance of the relation kAct ≫ kECM to a broad range of physiological conditions (15). The validity of this relation will not be assessed directly, as there are significant uncertainties about the typical value of kAct; instead, we will indirectly assess it through the relation δECM(t) ≈ F(t, kECM)/kECM.
To test the generality of our model, we used five cell lines that displayed distinct phenotypic behaviors when adhering to soft and rigid matrices (fig. S1): wild-type mouse embryonic fibroblasts (WT-MEFs), MEFs stably expressing paxillin–green fluorescent protein (pax-GFP), MEFs with stable knockdown of α-actinin (α-act KD), rat embryonic fibroblasts (REF52), and the human breast adenocarcinoma cell line MDA-MB231. For all cell lines, we measured δECM(t) for multiple pillars from the initial rise of pillar displacement until the release (fig. S2), averaged them, and superimposed the results for the widely different rigidities. The outcome, shown in Fig. 2B, is remarkable: For each cell line, the displacement function collapses on a master curve that is independent of the rigidity, despite the more than 15-fold variation in the latter. Thus, δECM is time dependent but kECM independent, and F(t, kECM) ≈ kECM δECM(t) ≈ kECM ∆(t). This result, which indicates that the contractile force F trivially depends on kECM through a linear spring relation and hence is nonmechanosensitive, is one of the major results of this paper.
The contractile displacement, ∆(t), which is independent of kECM, features not only generic properties that are cell line independent but also quantitative differences between cell lines. In all cell lines, ∆(t) exhibits a gradual increase with time, until a plateau is reached on a time scale of 5 to 10 min. The exact time to reach the plateau and the plateau level itself does depend on the cell line (Fig. 2C). Moreover, while WT-MEFs and pax-GFP cells often released the pillars (i.e., decrease in the displacement/force) shortly after reaching the plateau, α-act KD and MDA-MB231 cells maintained the pillar displacements for much longer periods (tens of minutes on average) (Fig. 2B), and REF52 often showed a second increase in ∆(t) several minutes after reaching the plateau (fig. S3).
Nonmechanosensitive contractile displacements are directly related to the time-dependent concentration of F-actin
The most outstanding questions emerging from the findings presented above are related to the origin of the nonmechanosensitive, intrinsic time-dependent contractile displacement ∆(t), in particular, its typical time scale and plateau level. The generated stress, i.e., F divided by the adhesion area, for different rigidities is presented in fig. S4, where it is shown to vary from ~0.5 to ~5 kPa, while the mean adhesion area featured significantly less variation (the measured range is 0.25 to 0.75 μm2). Consequently, adhesions of similar area can sustain a relatively broad range of forces [as was previously observed (10)], suggesting that adhesions are not the limiting factor for force generation. Thus, we turn our focus to the actomyosin cytoskeleton. Since myosin motors drive contractile force generation, it is logical to assume that the displacement time scales and plateau levels are controlled by the gradual increase in myosin activity. However, myosin is very rapidly activated and recruited to actin filaments at the cell edge (16), and, moreover, myosin motors generate a quantum of contractile displacement (the working stroke size) on a time scale of milliseconds (11), orders of magnitude faster than the time scale characterizing ∆(t). This may suggest that it is not the myosin motors per se, but rather the structural evolution of the actin network, to which the myosin motors attach, which might underlie ∆(t). Thus, we turned to characterize the evolution of the actin networks during force generation using the simplest quantity, i.e., the local (near pillar) concentration (number density) of F-actin, CF-actin(t).
To that end, we expressed the F-actin reporter tdTomato-tractin (17) in WT-MEFs, α-act KD cells, and MDA-MB-231 cells, and performed pillar displacement assays in parallel to tracking the fluorescence level of tractin around each pillar over time, which is a measure of CF-actin(t) (Fig. 3A). As the contractile displacement features a characteristic time of ~10 min (cf. Fig. 2B), short time variations in δECM(t) and CF-actin(t) were smoothed out using a moving average window (see Materials and Methods); as shown in Fig. 3B, both curves appear highly correlated at all times. Such high correlation between δECM(t) and CF-actin(t) of individual pillars is observed across the different cell lines and rigidities, as shown in Fig. 3C. To test whether this relation persists to shorter time scales, we analyzed the CF-actin(t) on δECM(t) data without any smoothing (Fig. 3D). The results reveal that both CF-actin(t) and δECM(t) synchronously oscillate approximately every 4 min, regardless of cell type (Fig. 3E). In >80% of the cases when a rise in displacement was observed, it was accompanied by a simultaneous rise in CF-actin. As shown in fig. S5, the slightest increase in F-actin density was paralleled by an increase in the contractile displacement, and moreover, short time scale disassembly of F-actin (decrease in tractin intensity) was paralleled by a drop in displacement. These results were further substantiated by experiments in which CF-actin(t) has been significantly and promptly reduced using the actin polymerization inhibitor latrunculin A, leading to the disappearance of contractile displacements, ∆(t) → 0 (fig. S6).
Fig. 3. Correlation between F-actin density and pillar displacement.
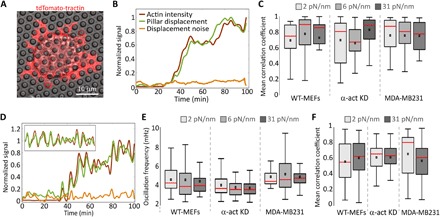
(A) Micrograph of a mouse fibroblast (WT-MEF) expressing tdTomato-tractin and spreading on 2-μm-diameter pillars. (B) Example of low-pass–filtered curves of pillar displacement and of tractin intensity around the same pillar over time. (C) Mean correlation coefficients of pillar displacement and tractin intensity such as in (B). n > 30 from >5 cells in each case. The amplitude of the displacement noise, obtained by measuring the magnitude of the displacement (irrespective of its direction) of a pillar that was not in contact with the cell throughout the experiment, is added for reference. (D) Non–low-pass–filtered pillar displacement and tractin intensity over time curves reveal simultaneous oscillations in both. Inset shows the same data (starting from the initial rise of both signals) after subtraction of the low-pass filter curves in each case (i.e., minus the so-called direct current component). Colors are as in (B) (see legend there). (E) Mean frequency of pillar displacement oscillations. The frequency was calculated using Fourier transform. Tractin oscillated at a similar frequency in all cases (not shown). (F) Mean correlation coefficients of actin and myosin density between the pillars.
Together, these observations support the simple yet quite remarkable relation
| (2) |
which suggests that the density of actin filaments essentially determines the contractile displacement generated by the actomyosin network. This implies, as hypothesized above, that myosin motors are not the rate-limiting factor in contractile force generation, i.e., that there are enough of them available at any point in time and that once attachment sites on the actin filaments are available, the myosin motors attach to them and generate displacement rapidly. This picture predicts that the densities of F-actin and myosin motors are highly correlated at any point in time, independently of ECM rigidity.
To test this prediction, we seeded cells on the pillar arrays with the two extreme rigidities (2 and 31 pN/nm) and fixed them after 30 min, during the contractile phase of spreading (18). We then stained the cells for F-actin and for phosphorylated (activated) myosin light chain (p-myosin). Using super-resolution microscopy (19), we observed large (micron-scale) actin structures between the pillars, which we classified as network-associated filaments, and small (nanometer-scale) unstructured filaments (fig. S7, A and B). p-myosin was arranged in clusters (fig. S7C), consistent with the appearance of myosin minifilaments (9, 20). We then tested the correlation between the density of the large F-actin structures and p-myosin between the pillars and found high positive correlation regardless of rigidity and cell type (correlation coefficients of 0.6 to 0.7 in all cases; Fig. 3F), as predicted. Since each pillar in the collected images represents a different point in time during the displacement process, this indicates that whenever actin filaments are available, myosin motors operate on them.
Cell-type dependence arises from structural differences in the actin cytoskeleton
The dependence of ∆(t) on CF-actin(t) implies that any possible cell-type dependence is encapsulated in the proportionality factor in Eq. 2. To test this, we measured the rate of change of CF-actin(t) and of δECM(t) [which measures ∆(t)] during the simultaneous short-term oscillations and plotted the average dδECM/dt and dCF−actin/dt against each other to extract the proportionality factor. Note that by measuring the relative changes in F-actin concentration, we could disregard any differences in tractin transfection efficiency and in F-actin levels between cells. The resulting graphs exhibit cell-type dependence; in particular, the proportionality factor of MDA-MB231 is significantly higher than that of the other two cell lines (Fig. 4A). This finding indicates that the degree to which the displacements follow changes in F-actin density varies between cell types.
Fig. 4. Structural differences in F-actin organization correlate with displacement response to CF−actin(t).
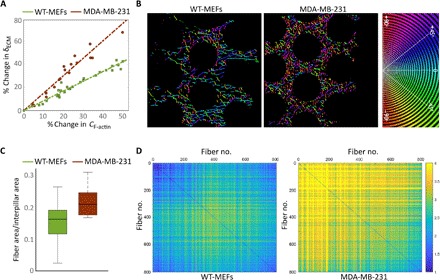
(A) The rate of change (in percentage) of δECM, which quantifies d∆/dt, as a function of rate of change (in percentage) of CF-actin, which quantifies dCF−actin/dt. Measurements were taken during each rise in the simultaneous oscillations (Fig. 3D), averaged for each rigidity (n > 60 data points from >15 pillars from >4 cells in each case), and all data points from all three rigidities are plotted here for WT-MEFs and MDA-MB-231 cells. For visual clarity, the α-act KD data (which are closer to those of WT-MEFs than to these of MDA-MB-231 cells) are not shown. (B) Processed super-resolution images of large actin filaments at the cell edge color-coded for angles (see Materials and Methods for details). Only part of the cell edge is shown in each case; the right side of each image is outside of the cell. α-act KD cells displayed similar fiber distribution to that of WT-MEFs (not shown). (C) Ratio between the area occupied by the large actin fibers and the interpillar area at the cell edge. MDA-MB-231 networks were ~50% denser compared to WT-MEFs (P < 0.001). (D) WT-MEFs display highly parallel fibers compared to MDA-MB-231. Images such as those shown in (B) were analyzed as follows: The largest 800 fibers in each image were arranged according to size in ascending order, and the interfiber slope differences were calculated for each fiber against all other fibers in each image. These differences are represented here by color-coded plots. The images shown are the average of 40 images in each case. Lower values (blue hues) represent parallel fibers. α-act KD showed a similar distribution to that of WT-MEFs (not shown).
To shed light on this cell-type dependence, we used the super-resolution images of F-actin and p-myosin to characterize the assembled networks in WT-MEF, α-act KD, and MDA-MB-231 cells (fig. S7). We found that across the three cell lines, actin at the cell edges between pillars assembled into networks that occupied similar areas (0.2 to 0.35 μm2 per network) and similar complexity of branching (fig. S7D). The number of p-myosin clusters per network in MDA-MB-231 cells was 30 to 40% lower than the other two cells lines (fig. S7E), possibly related to the relatively low displacement rate (on time scales of tens of minutes) of MDA-MB-231 cells (Fig. 2C). This, however, cannot explain the higher proportionality factor in Eq. 2 (which relates to time scales of seconds) observed for MDA-MB-231 cells compared to the other cell lines, cf. Fig. 4A. The cell-type dependence of the proportionality factor instead implies that the degree to which changes in CF-actin are translated into contractile displacement depends on the geometrical properties of the assembled actin network. Analysis of the spatial organization of F-actin in the cells (Fig. 4B) revealed that whereas MDA-MB-231 cells display highly dense networks, WT-MEFs typically form long, parallel, and more highly spaced filaments (Fig. 4, C and D) (α-act KD cells were similar to WT-MEFs, fig. S7F). Thus, although additional work is required to quantitatively relate the geometry of assembled networks to the proportionality factor, the observed structural differences are likely to be related to the different proportionality factor in Eq. 2 of WT-MEFs and MDA-MB-231 cells (i.e., different effective force transmission in response to changes in CF-actin).
Predictions for well-spread cells in steady state and consistency with existing literature
The experiments described above were performed on adherent cells that had not yet reached their well-spread steady state with mature actin stress fibers that span a sizable area of the cell. On the other hand, our coarse-grained model makes no explicit reference to the specific actin structures that mediate the contractile displacement. Consequently, we expect the model to remain valid also for well-spread cells in steady state. To test this prediction, we used REF52 cells, which were previously shown to generate large forces at steady state (10) and allowed them to attach to the pillars for 4 hours before imaging. These measurements showed that as in the case of early spreading, δECM(t) generated by well-spread REF52 cells collapses on a master curve that is rigidity independent (Fig. 5A), strongly supporting the generality of our results. The time-dependent displacement featured a similar time scale (~10 min) in both the steady state (Fig. 5A) and early spreading regime (Fig. 2B), possibly suggesting that the contractile displacement ∆(t) in the two regimes is intrinsically related. To test this, we plot one against the other in Fig. 5B. The result reveals that the two are (predominantly) linearly related to one another, demonstrating that the generated time-dependent displacement in the two regimes is in fact the very same function multiplied by a constant (the slope of the linear relation). The slope indicates that the early spreading displacements are six- to sevenfold smaller than δECM(t) of the cells at steady state. This may imply that, at steady state, when the cell edge extends and forms a new adhesion on a previously nondisplaced pillar, the actin filaments that emanate from the new adhesion connect to preassembled stress fibers. Consistent with this prediction, the deflected pillars in cells at steady state were connected to thick actin stress fibers (Fig. 5C and fig. S8) that were not observed in the early stages of spreading (Fig. 5D and note S2).
Fig. 5. Cells at steady-state generate rigidity-independent contractile displacements that are six- to sevenfold larger than early spreading displacements.
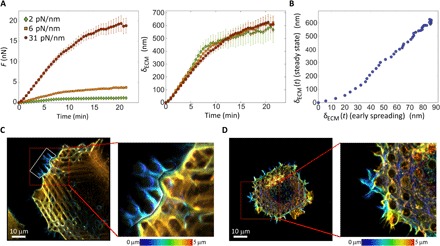
(A) F(t, kECM) and δECM(t) = F(t, kECM)/kECM curves (left and right, respectively) of REF52 cells under well-spread steady-state conditions. (B) δECM(t) of early spreading REF52 cells (cf. Fig. 2C) versus δECM(t) of REF52 under well-spread steady-state conditions (cf. panel A). (C and D) Z-stack projections of REF52 cells on pillars (31 pN/nm) stained for F-actin color-coded for depth; (C) cell after 5 hours of spreading. The brightness of the region marked by white borders was enhanced for purpose of clarity. Thick actin stress fibers (yellow-orange hues) are observed 2 to 4 μm above the fibers directly surrounding the pillar (blue hues). (D) Cell after 30 min of spreading. No stress fibers are observed, and the actin structures are much less organized compared to steady-state condition (C).
This remarkable generality is also consistent with the previous results of Trichet et al. (10), who reported F(t, kECM) for cells under well-spread steady-state conditions over a broad range of pillar rigidities kECM. To show this, we first plot of Trichet et al. (10) in fig. S9 (A and B), observing a collapse on a single master curve, as we predict and in agreement with our data in Fig. 5A. Moreover, in fig. S9C, we plot our δECM(t) (Fig. 5A) against that of Trichet et al. (10) (fig. S9B), which once more reveals a linear relation, indicating that it is again the very same time-dependent contractile displacement (here, as both measurements were conducted under steady-state conditions, the slope is small, slightly below 2). Consequently, we conclude that cellular contractility under well-spread steady-state conditions is controlled by the very same intrinsic (nonmechanosensitive) displacement ∆(t), lending strong support to the generality of the emerging picture.
These results in the steady-state regime may have another intriguing implication. The independence of the intrinsic time-dependent contractile displacement ∆(t) on the rigidity kECM implies that ∆(t) is in fact also independent of the length of the stress fibers that are attached to the pillars through focal adhesions. This is immediately inferred from the clear dependence of the cell size and consequently of the length of stress fibers, on the rigidity kECM as observed in figure 4 of Trichet et al. (10). On the other hand, one may assume that like in muscle cells (21, 22), myosin II motors apply their contractile displacement everywhere along the stress fiber, implying that ∆(t) increases with the length of the stress fiber. Our results [as well as those of Trichet et al. (10)] indicate that this is not the case but rather that stress fibers of different lengths generate the very same contractile displacement ∆(t). This leads to the activity localization hypothesis, which suggests that myosin II motors apply their contractile displacement to a rather localized region of the stress fiber and that the size of this region does not depend on the overall length of the stress fiber.
DISCUSSION
Our results reveal basic aspects of cellular force generation and mechanosensitivity, providing a novel framework to address various questions related to these important cellular processes. The starting point for our investigation is the widespread view that cellular contractile forces are mechanosensitive, i.e., that cells regulate these forces through sensing the rigidity of the ECM. This view is based on previous extensive observations that show that contractile forces depend on ECM rigidity: The larger the rigidity is, the larger cellular contractile forces are (10, 23, 24). We show that cellular forces are generated through intrinsic nonmechanosensitive contractile displacements. That is, independently of the ECM rigidity, a particular cell type applies a well-defined time-dependent displacement to its environment (Fig. 2). This implies that the contractile force that is required to reach a certain displacement is proportional to the ECM rigidity but that this dependence is purely mechanistic, i.e., resulting from a simple linear (Hookean) force-rigidity relation. Consequently, cellular contractile forces are themselves nonmechanosensitive.
These findings have major implications for our understanding of cellular force generation and mechanosensitivity. They show that while contractile cellular forces are not mechanosensitive, information about ECM rigidity is simply encoded in them. Hence, any mechanosensitive cellular process should be sensitive to the contractile force (e.g., its instantaneous magnitude or time rate of change), feeding back the rigidity information into other cellular processes. Moreover, as our findings show that the nonmechanosensitive force generation process emerges from intrinsic time-dependent displacements, any attempt to understand cellular force generation should focus on the intrinsic time-dependent displacements. As a first step toward achieving this important goal, we showed that the time-dependent contractile displacement is directly and universally related to the time evolution of actomyosin networks. In particular, it is shown to be causally related to the density of the actin filaments near the adhesion sites, and the degree to which changes in this density are translated into displacement is shown to depend on the cell type–specific spatial organization and structure of the networks formed by these filaments (Figs. 3 and 4).
Our results reveal a large degree of universality in the time-dependent contractile displacement. In particular, we show that very different cellular states—those pertaining to the early spreading process and those pertaining to steady-state spreading conditions—give rise to essentially the very same time-dependent contractile displacement, differing only by an overall amplitude (Fig. 5). These results are fully consistent with the experimental results of Trichet et al. (10), whose reanalysis also demonstrated nonmechanosensitive contractile displacements that differ by only a multiplicative constant from our own measurements in the well-spread steady state (for the same cell type, here, REF52). Our results also strongly support the relation kAct ≫ kECM (used to derive Eq. 1), which indicates that over a broad range of conditions, the actin structures involved in the force generation process are much stiffer than the ECM and do not deform at all during the process. This is consistent with previous in vitro measurements that showed that actin networks can be up to 40-fold stiffer than the loads that they bear (25).
The biophysical picture emerging from our findings is consistent with other available observations in the literature and hence provides a unifying picture of cellular contractility. For example, while the inferred relation kAct ≫ kECM is relevant to a broad range of physiological conditions, experiments performed on cells adhering to ultrastiff substrates (e.g., plastic/glass) are in the opposite regime of kAct(t, kECM) ≪ kECM. Under these conditions, where the ECM cannot be deformed, we predict that the displacement ∆(t) is accommodated by stretching the actin structures, δAct(t) ≈ ∆(t) > 0. Consistent with this prediction, in laser ablation experiments applied to cells adhering to glass plates (26), stress fibers are observed to instantaneously retract by an amount comparable to the saturation level of δECM(t → ∞) ≈ ∆(t → ∞) observed in Fig. 5A and fig. S9B (see also note S2). This observation further supports the major result that the contractile displacement ∆(t) is an intrinsic, nonmechanosensitive property of cells (independently of ECM rigidity and independently of whether the displacement is accommodated by the ECM or by F-actin structures). It is important to stress that our use of ECM pillars whose effective rigidity varies with their height alone, keeping their material and surface properties entirely fixed, enables us to cleanly isolate the effect of the external rigidity, ruling out any possible intervening surface chemistry effects.
Previous studies of force regulation as a function of ECM rigidity have given rise to the “integrin clutch model,” which predicts that contractile forces depend on the number of attached clutches (integrins) to the moving, polymerization-driven, actin fibers (27–29). Our results show that the nonmechanosensitive contractility critically depends on the density of F-actin, as well as its spatial organization, making no explicit reference to the polymerization-driven flow of actin. Because we have not measured the number of engaged clutches at each time point during the contractile displacement process (nor is it possible to the best of our knowledge to accurately perform these measurements), the relation between the density of F-actin and adhesion dynamics should be further explored (e.g., in light of the common view that F-actin is recruited at the adhesion sites).
Our findings also give rise to various important questions. First, there is a need to go beyond the found d∆(t)/dt ~ dCF−actin(t)/dt relation (which in itself should be further studied) to understand the origin of some of the properties of ∆(t). In particular, the activity localization hypothesis associated with ∆(t) should be further tested in future work, together with the origin of the characteristic time scale and plateau levels of ∆(t), including their cell-type dependence. In addition, the mechanism behind the short-term oscillatory behavior of F-actin (characterized by a time of ~4 min) should be clarified, potentially in relation to the cyclic activation/deactivation of actin polymerization factors, as was recently observed in contracting secretory vesicles (30). The relation between this oscillatory time scale and the lifetime of adhesions during force loading, including the turnover time of proteins within them (31, 32), should be further explored. Last, our main result, F(t, kECM) ≈ kECM ∆(t), shows that mechanosensing is driven by cells through ∆(t) and that the sensing itself is performed through the contractile force F(t, kECM). How the latter feeds back into various internal cellular processes should be further explored with particular focus on the time-dependent accumulation of rigidity signals, rather than exclusively on their magnitude. This may shift our view of mechanosensitivity toward the dynamic nature of this process.
MATERIALS AND METHODS
Cells culture and reagents
WT-MEFs (RPTPα+/+ cells) (33) and α-act KD cells (34) were a gift from M. Sheetz [Mechanobiology Institute (MBI) Singapore and the University of Texas Medical Branch]; REF52 cells (35) and pax-GFP cells (Ilkf/f fibroblasts stably expressing paxillin–enhanced GFP) (36) were a gift from B. Geiger (Weizmann Institute of Science); MDA-MB-231 cells (37) were a gift from Y. Shaked (Technion). All cells were cultured at 37°C in a 5% CO2 incubator in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and penicillin-streptomycin (100 IU/ml; all reagents were from Biological Industries).
Plasmids and transfections
The YFP-Tpm2.1 plasmid was a gift from P. Gunning (the University of New South Wales). The tdTomato-tractin plasmid was a gift from M. Sheetz (MBI Singapore and the University of Texas Medical Branch). Transfections were carried out 1 day before measurements using the NEPA21 Electroporator (Nepa Gene) according to the manufacturer’s instructions, with ~106 cells per reaction and 10 μg of DNA.
Pillar and soft gel fabrication
In the pillar experiments, the external rigidity kECM is controlled by varying the height of the pillars, while keeping their cross-sectional area and intrinsic chemical properties fixed. All pillars had a diameter of 2 μm and heights of 5.3, 9.4, or 13.2 μm. We used 2-μm-diameter pillars, as these can be used to measure the long-term time-dependent forces that are generated after initial formation and reinforcement of the adhesions (7, 9). The center-to-center spacing between pillars was 4 μm. Pillar bending stiffness, kECM, was calculated by Euler-Bernoulli beam theory
where D and L are the diameter and length of the pillar, respectively, and E is the Young’s modulus of the material (2 MPa for the PDMS used here).
Using a common relation to estimate an effective elastic modulus that corresponds to a given rigidity , where a is the radius of the pillars (14), we obtain effective elastic moduli in the range Eeff ≈ 1.5 to 22 kPa. The latter is well within the relevant physiological regime, ranging from endothelial tissues to cartilage (15), thus allowing us to test the relevance of the relation kAct(t, kECM) ≫ kECM to a broad range of physiological conditions.
Pillar fabrication was performed by pouring PDMS (mixed at 10:1 with its curing agent, Sylgard 184; Dow Corning) into silicon molds [fabricated as previously described (7)] with holes at fixed depths and distances. The molds were then placed, face down, onto glass-bottom 35-mm dishes (#0 coverslip, Cellvis), which were incubated at 65°C for 12 hours to cure the PDMS. The molds were next peeled off while immersing in ethanol to prevent pillar collapse. The ethanol was replaced with phosphate-buffered saline, and human plasma full-length fibronectin (Merck) was added to the dish at a final concentration of 10 μg/μl for a 1-hour incubation at 37°C. Next, residual fibronectin was washed away by replacing the buffer to Hanks’ balanced salt solution (HBSS) buffer (Biological Industries) supplemented with 20 mM Hepes (pH 7.2). One- and 40-kPa substrates (fig. S1) were fabricated using Sylgard 52-276, at a ratio of 1:1.1 and 1:3.2, respectively, according to the measurements performed by Ou et al. (38).
Pillar displacement measurements
One day before the pillar experiments, cells were sparsely plated to minimize cell-cell interactions before replating. The following day, cells were trypsinized, centrifuged with growth medium, and then resuspended and preincubated in HBSS/Hepes at 37°C for 30 min before the experiment. Cells were then spread on the fibronectin-coated pillars. In all cases, we made sure that the cells were isolated when plated on the substrates.
Time-lapse imaging of cells spreading on the pillars was performed using an inverted microscope (Leica DMIRE2) at 37°C using a 63× 1.4 numerical aperture (NA) oil immersion objective. Bright-field images were recorded every 10 s with a Retiga EXi Fast 1394 charge-coupled device camera (QImaging). The microscope and camera were controlled by Micro-Manager software (39). For each cell, a movie of 1 to 3 hours was recorded. To minimize photodamage to the cells, a 600-nm long-pass filter was inserted into the illumination path.
Tracking of pillar movements over time was performed with ImageJ (National Institutes of Health) using the Nanotracking plugin, as described previously (9). Briefly, the cross-correlation between the pillar image in every frame of the movie and an image of the same pillar from the first frame of the movie was calculated, and the relative x and y position of the pillar in every frame of the movie was obtained. To consider only movements of pillars from their zero position, we only analyzed pillars that were not in contact with the cell at the start of the movie, and that during the movie, the cell edge reached to them. Drift correction was performed using data from pillars far from any cell in each movie. For each pillar, the displacement curve was generated by MATLAB (MathWorks).
Parallel measurements of tdTomato-tractin intensity and pillar displacements were performed on a Zeiss LSM800 confocal microscope using a 63× 1.4 NA objective at 37°C. Images were taken every 30 s, and measurements of pillar movements were performed as described above. Measurements of tdTomato-tractin intensity were taken around each pillar of interest in a circular area with a radius of 2 μm. Smoothing of the data in Fig. 3B was performed using a moving average with a window size of 30 frames (5 min). Analyses of correlation between changes in F-actin density and in pillar displacement during the short-term oscillations (Fig. 3D) were performed only on displacements that began below the noise level (~20 nm).
Fluorescence microscopy
For immunofluorescence microscopy, cells were plated on fibronectin-coated pillars, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Immunolabeling was performed with primary antibodies against p-myosin and paxillin (Abcam) and with Alexa Fluor 488– or Alexa Fluor 555–conjugated secondary antibodies. Images were taken with a Zeiss LSM800 confocal microscope using a 63× 1.4 NA objective.
For super-resolution analyses [super-resolution radial fluctuations (SRRF)] (19), a region of interest of 12 μm by 12 μm was imaged using the Airyscan function on the LSM800 confocal microscope. A series of 25 images was taken at 5 frames/s; these were processed using the Airyscan algorithm using the ZEN Blue software (Zeiss), followed by further processing using the SRRF algorithm (19). The trainable Weka segmentation plugin for Fiji (40) was then used to define the actin fibers between the pillars (Fig. 4), and thresholding was used to define the paxillin (fig. S4) or p-myosin (fig. S7) clusters above background fluorescence levels. Fiber orientation analysis was performed using the Ridge Detection and OrientationJ plugins for Fiji.
Statistical analysis
MATLAB (MathWorks) was used for data analysis and graph plotting. All ensemble average pillar displacement curves are shown with error bars representing the SEM. In the boxplots shown in the figures, the central mark indicates the median, the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively, and the whiskers extend to the most extreme data points not considered outliers; outliers are not shown.
Supplementary Material
Acknowledgments
We thank Y. Lubomirsky (Weizmann Institute of Science) for discussions related to Eq. 1, N. Kaplan (Technion–Israel Institute of Technology) for discussions on data analysis, and S. Bershadsky (MBI, Singapore) for discussions related to theoretical aspects of this work. Funding: H.W. acknowledges support from the Israel Science Foundation (1738/17) and from the Rappaport Institute. H.W. is an incumbent of the David and Inez Myers Career Advancement Chair in Life Sciences. E.B. acknowledges support from the William Z. and Eda Bess Novick Young Scientist Fund and from the Harold Perlman Family. Author contributions: L.F. performed the experiments and analyzed the data. L.K. and A.M. assisted in data analysis and pillar experiments, respectively. A.L. and E.B. conceived and developed the theory. E.B and H.W. conceived and planned the experiments. E.B. and H.W. wrote the manuscript. All authors discussed the results and contributed to the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz6997/DC1
REFERENCES AND NOTES
- 1.Iskratsch T., Wolfenson H., Sheetz M. P., Appreciating force and shape—The rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 15, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo C. M., Wang H. B., Dembo M., Wang Y. L., Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144–152 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prager-Khoutorsky M., Lichtenstein A., Krishnan R., Rajendran K., Mayo A., Kam Z., Geiger B., Bershadsky A. D., Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol. 13, 1457–1465 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Wang H.-B., Dembo M., Wang Y.-L., Substrate flexibility regulates growth and apoptosis of normal but not transformed cells. Am. J. Physiol. Cell Physiol. 279, C1345–C1350 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri O., Koshy S. T., Branco da Cunha C., Shin J. W., Verbeke C. S., Allison K. H., Mooney D. J., Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 13, 970–978 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Doyle A. D., Kutys M. L., Conti M. A., Matsumoto K., Adelstein R. S., Yamada K. M., Micro-environmental control of cell migration–Myosin IIA is required for efficient migration in fibrillar environments through control of cell adhesion dynamics. J. Cell Sci. 125, 2244–2256 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghassemi S., Meacci G., Liu S., Gondarenko A. A., Mathur A., Roca-Cusachs P., Sheetz M. P., Hone J., Cells test substrate rigidity by local contractions on submicrometer pillars. Proc. Natl. Acad. Sci. U.S.A. 109, 5328–5333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elosegui-Artola A., Oria R., Chen Y., Kosmalska A., Pérez-González C., Castro N., Zhu C., Trepat X., Roca-Cusachs P., Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 18, 540–548 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Wolfenson H., Meacci G., Liu S., Stachowiak M. R., Iskratsch T., Ghassemi S., Roca-Cusachs P., O’Shaughnessy B., Hone J., Sheetz M. P., Tropomyosin controls sarcomere-like contractions for rigidity sensing and suppressing growth on soft matrices. Nat. Cell Biol. 18, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trichet L., Le Digabel J., Hawkins R. J., Vedula S. R., Gupta M., Ribrault C., Hersen P., Voituriez R., Ladoux B., Evidence of a large-scale mechanosensing mechanism for cellular adaptation to substrate stiffness. Proc. Natl. Acad. Sci. U.S.A. 109, 6933–6938 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaya M., Higuchi H., Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science 329, 686–689 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Schwarz U. S., Erdmann T., Bischofs I. B., Focal adhesions as mechanosensors: The two-spring model. Biosystems 83, 225–232 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Saez A., Buguin A., Silberzan P., Ladoux B., Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J. 89, L52–L54 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghibaudo M., Saez A., Trichet L., Xayaphoummine A., Browaeys J., Silberzan P., Buguin A., Ladoux B., Traction forces and rigidity sensing regulate cell functions. Soft Matter 4, 1836–1836 (2008). [Google Scholar]
- 15.Cox T. R., Erler J. T., Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 4, 165–178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shutova M., Yang C., Vasiliev J. M., Svitkina T., Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLOS ONE 7, e40814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belin B. J., Goins L. M., Mullins R. D., Comparative analysis of tools for live cell imaging of actin network architecture. Bioarchitecture. 4, 189–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfenson H., Yang B., Sheetz M. P., Steps in mechanotransduction pathways that control cell morphology. 81, 585–605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gustafsson N., Culley S., Ashdown G., Owen D. M., Pereira P. M., Henriques R., Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 7, 12471 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohner J., Rupprecht J.-F., Hu J., Mandriota N., Saxena M., de Araujo D. P., Hone J., Sahin O., Prost J., Sheetz M. P., Large and reversible myosin-dependent forces in rigidity sensing. Nat. Phys. , 689–695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ter Keurs H. E. D., The interaction of Ca2+ with sarcomeric proteins: Role in function and dysfunction of the heart. Am J Physiol Hear. Circ Physiol. 302, H38–H50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livne A., Geiger B., The inner workings of stress fibers - from contractile machinery to focal adhesions and back. J. Cell Sci. 129, 1293–1304 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Oakes P. W., Bidone T. C., Beckham Y., Skeeters A. V., Ramirez-San Juan G. R., Winter S. P., Voth G. A., Gardel M. L., Lamellipodium is a myosin-independent mechanosensor. Proc. Natl. Acad. Sci. U.S.A. 115, 2646–2651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarasewicz E., Oakes R. S., Aviles M. O., Straehla J., Chilton K. M., Decker J. T., Wu J., Shea L. D., Jeruss J. S., Embryonic stem cell secreted factors decrease invasiveness of triple-negative breast cancer cells through regulome modulation. Cancer Biol. Ther. 19, 271–281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bieling P., Li T. D., Weichsel J., McGorty R., Jreij P., Huang B., Fletcher D. A., Mullins R. D., Force feedback controls motor activity and mechanical properties of self-assembling branched actin networks. Cell 164, 115–127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell R. J., Xia S. L., Dickinson R. B., Lele T. P., Sarcomere mechanics in capillary endothelial cells. Biophys. J. 97, 1578–1585 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan C. E., Odde D. J., Traction dynamics of filopodia on compliant substrates. Science 322, 1687–1691 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Elosegui-Artola A., Trepat X., Roca-Cusachs P., Control of mechanotransduction by molecular clutch dynamics. Trends Cell Biol. 28, 356–367 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Case L. B., Waterman C. M., Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 17, 955–963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segal D., Zaritsky A., Schejter E. D., Shilo B.-Z., Feedback inhibition of actin on Rho mediates content release from large secretory vesicles. J. Cell Biol. 217, 1815–1826 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfenson H., Bershadsky A., Henis Y. I., Geiger B., Actomyosin-generated tension controls the molecular kinetics of focal adhesions. J. Cell Sci. 124, 1425–1432 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavelin I., Wolfenson H., Patla I., Henis Y. I., Medalia O., Volberg T., Livne A., Kam Z., Geiger B., Differential effect of actomyosin relaxation on the dynamic properties of focal adhesion proteins. PLOS ONE 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang G., Huang A. H., Cai Y., Tanase M., Sheetz M. P., Rigidity sensing at the leading edge through αvβ3 integrins and RPTPα. Biophys. J. 90, 1804–1809 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roca-Cusachs P., del Rio A., Puklin-Faucher E., Gauthier N. C., Biais N., Sheetz M. P., Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. U.S.A. 110, E1361–E1370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavalcanti-Adam E. A., Volberg T., Micoulet A., Kessler H., Geiger B., Spatz J. P., Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 92, 2964–2974 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elad N., Volberg T., Patla I., Hirschfeld-Warneken V., Grashoff C., Spatz J. P., Fässler R., Geiger B., Medalia O., The role of integrin-linked kinase in the molecular architecture of focal adhesions. J. Cell Sci. 126, 4099–4107 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Gingis-Velitski S., Loven D., Benayoun L., Munster M., Bril R., Voloshin T., Alishekevitz D., Bertolini F., Shaked Y., Host response to short-term, single-agent chemotherapy induces matrix metalloproteinase-9 expression and accelerates metastasis in mice. Cancer Res. 71, 6986–6996 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Ou G., Thakar D., Tung J. C., Miroshnikova Y. A., Dufort C. C., Gutierrez E., Groisman A., Weaver V. M., Visualizing mechanical modulation of nanoscale organization of cell-matrix adhesions. Integr. Biol. 8, 795–804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N., Computer control of microscopes using μmanager. Curr. Protoc. Mol. Biol. 92, 14.20.1–14.20.17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arganda-Carreras I., Kaynig V., Rueden C., Eliceiri K. W., Schindelin J., Cardona A., Sebastian Seung H., Trainable weka segmentation: A machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Yeh Y.-C., Ling J.-Y., Chen W.-C., Lin H.-H., Tang M.-J., Mechanotransduction of matrix stiffness in regulation of focal adhesion size and number: Reciprocal regulation of caveolin-1 and β1 integrin. Sci. Rep. 7, 15008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz6997/DC1