

A new method to estimate growth rate of microbial strains from metagenomics provides insights into strain-level interactions.
Abstract
We developed a method for strain-level metagenomic estimation of growth rate (SMEG) for inferring growth rates of bacterial subspecies, or strains, from complex metagenomic samples. We applied our method, which is based on both reference strains and de novo approaches, to different gut metagenomic datasets, accurately identifying an outbreak-associated Escherichia coli strain and a previously unidentified association of an Akkermansia muciniphila strain in cancer immunotherapy responders. SMEG resolves strain-specific growth rates from mixtures of commensal or pathogenic strains to provide new insights into microbial interactions and disease associations at the strain level. SMEG is available for download at https://github.com/ohlab/SMEG.
INTRODUCTION
Growth rate inferences from shotgun metagenomic data are valuable for understanding microbial activity in situ, for example, new inferences in irritable bowel disease, type 2 diabetes, and microbial antagonism in the skin (1, 2). Because rapidly growing cells accumulate genome copies at the origin of replication (ori) compared to the terminus (ter) region (1), it is possible to use the ratio of sequence reads in these two regions as a proxy for growth rate. Several tools, such as PTR (1), GRiD (2), iRep (3), and DEMIC (4), have been developed to perform growth rate analysis based on the ori/ter ratio. While these approaches have been valuable for examining the population-level behavior of species, a substantial amount of the genetic diversity within and between ecosystems occurs at the subspecies, or strain, level. Moreover, multiple genetically heterogeneous strains can stably coexist in a community, with fluxes in strain relative abundance occurring during environmental transition states like infections (5). The ability to identify strains of a species that are metabolically active or actively replicating in a mixed population would propel new insights into strain-level interactions, how strains can dominate a population during an infection, identification of antimicrobial resistant strains, and how different individuals may harbor strains that affect their susceptibility to disease.
To date, no method that is capable of estimating strain-specific growth rates from metagenomic datasets exists. Here, we developed strain-level metagenomic estimation of growth rate (SMEG) for subspecies and strain-specific measurement of growth rate, allowing application to microbes of interest in a microbial community. After benchmarking SMEG’s accuracy, we applied our algorithm to different biological applications to demonstrate the versatility and importance of such a tool.
RESULTS AND DISCUSSION
Overview of SMEG
SMEG estimates of growth rate are conceptually based on ori/ter ratios, but ori and ter themselves are typically too highly conserved between strains to allow usage for differential coverage. Our algorithm takes a novel approach using differential coverage patterns of single-nucleotide polymorphisms (SNPs) in universally conserved or core genes to estimate strain-specific growth. The SMEG pipeline consists of two major steps, which first builds a profile of strain-specific differentiating SNPs for each species of interest (“coordinate set”) and then estimates growth rate from metagenomic data with complementary de novo and reference-based approaches (Fig. 1, A and B).
Fig. 1. The SMEG pipeline.
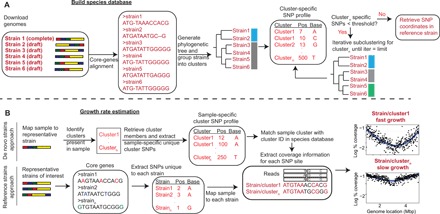
(A) Building the species database. Complete and draft-quality genomes of member strains available for a species are used to build the species database. Core genes are extracted, and multiple sequence alignment is conducted to generate a phylogenetic tree. Strains are grouped into clusters, and SMEG then generates cluster-specific unique SNPs, which are SNPs shared between a given proportion of cluster members but absent in strains from other clusters. Clusters having insufficient number of unique SNPs are iteratively subclustered to re-generate a unique SNPs profile, or “SNP coordinate set.” (B) Growth rate estimation. SMEG estimates growth rate using either a de novo or reference-based approach. In the de novo approach, SMEG maps reads to the representative strain to detect clusters in a sample. SMEG then re-generates a sample-specific, unique SNP profile using only the detected clusters, increasing the number of discriminatory SNPs usable to calculate growth rate. Then, nucleotide coverage is determined at each cluster’s unique SNP site across a variable sliding window to calculate the peak-to-trough ratio (PTR).
To build an SNP coordinate set for each species of interest (Fig. 1A), we created a genome database using available complete and draft genomes from the National Center for Biotechnology Information (NCBI) GenBank. We next conducted core genome alignment and subsequently clustered strains based on their phylogenetic distance. The rationale behind generating clusters is to reduce the SNP dimensionality of the genome database and increase robustness of strain assignment by identifying the most likely consensus SNPs unique to a cluster. The underlying biological assumption is that strains constituting a cluster have high phylogenetic relatedness and will have similar phenotypic properties like growth rate in a sample. For each cluster, we then generated a cluster-specific SNP profile, defined as SNPs shared between a given proportion of cluster members but absent in strains from other clusters (fig. S1A). Each of these discriminatory SNPs is assigned a consensus “quality” score based on the proportion of strains in that cluster harboring that SNP at that coordinate. Cutoffs based on these quality scores (“SNP assignment threshold”) can then be used to modulate the number of discriminatory SNPs per cluster, with lower cutoffs increasing the number of SNPs that can be used in a cluster’s analysis but decreasing the precision. Then, for clusters having an insufficient number of unique SNPs (user-defined threshold), we further subgroup those clusters and re-generate a unique SNP profile iteratively until the threshold is met, or for a maximum of three iterations to limit excessive subclustering (Fig. 1A). Last, SNP coordinates in a representative strain selected randomly based on genome completeness are retrieved for metagenomic read mapping. This coordinate map of SNPs allows rapid profiling of metagenomic reads.
After generating the SNP coordinate set, strain growth rate is calculated from metagenomic data using either a de novo or reference-based estimation approach (Fig. 1B). In the de novo approach, we seek to identify clusters in a sample without a priori knowledge of strain composition, which will allow detection of strains belonging to a cluster before growth estimation, even if genome sequences of such strains are absent from the SMEG database. Here, we assume that strains present in a sample, but absent in the database, can be assigned to a cluster using information on cluster-specific SNPs. Metagenomic reads are mapped to the representative strain coordinate map to identify clusters. A cluster is deemed present if the proportion of unique SNPs with coverage >0 exceeds a user-defined “cluster detection threshold,” which is set by the proportion of cluster-specific SNPs used to identify that cluster. For example, at a cluster detection threshold of 0.2, 20% of the unique SNPs in that cluster must have coverage >0 to be deemed present in a sample. The precision and recall of this threshold varies based on the allelic heterogeneity within a species, as more species with higher strain diversity will likely have more discriminatory SNPs, but those will likely have lower consensus quality scores to discriminate a cluster. Conversely, species with lower strain diversity will have fewer but more high consensus, discriminatory SNPs.
As an additional refinement step to increase numbers of discriminatory SNPs, a sample-specific, cluster-unique SNP profile is re-generated using only clusters identified in the sample (Fig. 1B), as we anticipate that most samples will not contain all clusters for a given species [gut metagenomes have been cited to have limited within-sample strain variation for different species (6)]. This step increases accuracy by removing nonrelevant clusters and increasing the number of discriminatory SNP sites for growth estimation. This is especially useful for clusters that had insufficient unique SNPs in the initial iterative SNP coordinate set creation (fig. S1B). Last, growth rate for each cluster is inferred by calculating nucleotide coverage at each cluster’s unique SNP sites across a variable sliding window, with the assumption that a rapidly growing strain would have higher coverage at SNP sites closer to the ori than the ter region (Fig. 1B).
As a complement, the reference-based approach assumes previous knowledge of strain composition in a dataset, which may have been determined using other means or tools such as StrainEst (7), metaSNV (8), or PathoScope (9). In this case, the user provides a list of strains, and SMEG subsequently calculates nucleotide coverage at SNP sites unique to each strain in a similar manner as the de novo approach (Fig. 1B). In contrast to the de novo approach, the reference-based approach is only applicable to strains already present in the database, which significantly limits its usability but has higher accuracy, allowing customization of the SNP coordinate set to only informative SNPs. However, this method is highly sensitive to the accurate determination of strain composition.
SMEG accurately detects subspecies clusters and predicts growth rate
Because the sensitivity and specificity of strain-level algorithms depend on genetic distance within a species, we tested SMEG’s sensitivity in silico using species with differing levels of within-species genetic diversity. For example, the phylogenetic distance between strains of the ubiquitous skin bacterium Cutibacterium acnes is relatively limited (10), which would provide a difficult scenario to assess our tool given the close relatedness of member strains. In contrast, skin commensal Staphylococcus epidermidis is characterized by substantial SNP and gene content diversity at the strain level (11). From 131 and 95 genomes, we generated 11 and 15 clusters for C. acnes and S. epidermidis, respectively. On the basis of biological estimates that the average number of strains for most species in an individual sample is estimated to be between one and five (6), we randomly selected one strain from eight different clusters of C. acnes and S. epidermidis and synthesized 30 mock metagenomic samples for each species, randomly varying relative abundances and genomic coverages to mimic a replicating strain in a mixed community.
First, we tested SMEG’s ability to correctly identify clusters in the dataset by calculating an F1 score, a metric that evaluates both maximal correctly detected clusters and minimal missing clusters (higher F1 ~ higher precision and recall). SMEG had a perfect F1 score for C. acnes and a near-perfect score for S. epidermidis at a cluster detection threshold of 0.2, reflecting the robustness of our de novo approach to correctly detect strains (Fig. 2A). SMEG had decreasing F1 score as a function of cluster detection threshold, because higher thresholds (e.g., 0.3 and 0.6) resulted in a higher false-negative rate, where fewer clusters are detectable in a sample as greater numbers of discriminatory SNPs are required to differentiate clusters (fig. S1C). In addition, SMEG lost accuracy at higher cluster detection thresholds for S. epidermidis, which is consistent with its strain-level genetic diversity: More diverse species have more potential discriminatory SNPs, but these are lower in consensus quality, and thus, recall is decreased, and fewer strains are identified by requiring more SNPs to identify a cluster (Fig. 2A). In less heterogeneous species, strains have fewer discriminatory but more consensus SNPs. Last, SMEG predictions were highly correlated with the expected growth rate across samples at different coverage cutoffs (Fig. 2A), indicating its ability to segregate rapidly growing and stationary strains in a sample.
Fig. 2. SMEG accurately detects strains and measures replication rate.
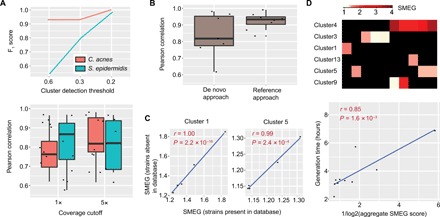
(A) F1 scores for both C. acnes and S. epidermidis, reflecting precision and recall, at several cluster detection thresholds from a 30-sample synthetic mock community containing eight strains of each species. The boxplot below shows the Pearson correlation between SMEG scores and expected PTR at 1× and 5× coverage cutoffs of strain abundance. (B) Pearson correlation between SMEG scores and expected PTR using both de novo and reference-based approach at a 5× coverage cutoff using the C. acnes dataset as in (A). (C) Pearson correlation between SMEG scores and expected PTR in a five-sample, high-complexity CAMI dataset spiked with synthetic C. acnes mock from two strains missing in the species database. (D) Heat map showing SMEG scores for S. aureus clusters. Each column in the heat map represents a sample, while black boxes indicate the absence of a cluster. The scatter plot below shows the Pearson correlation between S. aureus generation time and aggregate SMEG score.
In addition, we attempted to compare SMEG’s de novo strain profiler (i.e., cluster detection accuracy) with ConStrains (6), an algorithm that uses SNP patterns in a set of universal genes to reconstruct strains. We spiked our abovementioned mock reads into five high-complexity critical assessment of metagenome interpretation (CAMI) dataset (15 gigabase pairs each). ConStrains was unable to identify any strain as it complained of insufficient coverage despite our mock species having coverages >10 in all samples, which is above its minimum coverage requirement. On the other hand, SMEG detected clusters similar to pure mock samples, and growth predictions were highly correlated between pure mock samples and spiked-in complex dataset (fig. S2). We next evaluated the reference-based method using the same C. acnes mock metagenomes as above and observed improved accuracy in comparison to the de novo–based approach (Fig. 2B). Therefore, where a priori knowledge on strain composition is available or determined using other means or tools, we recommend using this option. Note that current tools for species-level growth rate inference from metagenomic samples (i.e., GRiD, iRep, and DEMIC) were unable to predict growth rate in the mock samples (fig. S3A and table S1).
We further examined the ability of SMEG to characterize the growth rate of strains whose genomes were absent from the database. We randomly excluded two C. acnes strains before database creation, synthesized a five-sample mock metagenomic dataset using a mixture of both strains, spiked reads into the high-complexity CAMI dataset (15 Gbp each), and estimated their growth rate. SMEG correctly determined the number of strains present in each sample, precisely assigned each strain to its cluster (i.e., F1 score = 1; fig. S4), and accurately estimated growth rates of the missing strains (Fig. 2C).
To determine whether SMEG could accurately estimate growth rate from real-world metagenomics samples, we used previously reported in situ generation times of Staphylococcus aureus in nasal samples (12) to verify the accuracy of SMEG. Because species-level growth rate represents the aggregate growth rate of individual strains within a population, we used average SMEG scores per sample for comparison and found strong correlation with reported generation times (Fig. 2D). We also evaluated SMEG parameters for microbes with differing levels of within-species genetic diversity, using S. aureus as an example of high strain heterogeneity (many discriminatory, but lower-quality SNPS) and C. acnes for low strain diversity (fewer, higher-quality SNPs). Our findings suggest that SMEG can detect clusters at up to 0.5× coverage, requires cluster coverage of 5× and 0.5× for microbes with high and low within-species genetic diversity, respectively, and requires at least 100 unique SNPs to accurately estimate growth rate (fig. S5). We recommend a 5× cutoff without a priori knowledge of the genomic characteristics of the species of interest.
We also explored the possibility of expanding SMEG to strains that may have been reconstituted de novo using DESMAN, an algorithm that identifies variants in core genes and uses co-occurrence across samples to link variants into haplotypes and abundance profiles (13). The major advantage to such an approach for strain reconstitution would be that the requirement for the creation of a species database using reference genomes might be circumvented and thus could be especially useful for species with few representatives in NCBI GenBank. We initially attempted to predict the correct order of core genes within a genome using coverage information across multiple samples, with the assumption that genes with high relative coverage are likely closer to the ori. However, in simulations of a S. epidermidis 30-sample mock metagenome simulating a single replicating strain, predicted gene order poorly correlated with the expected order (fig. S3B), which suggests that additional options for reordering genes are required for growth predictions with DESMAN-reconstituted haplotypes (e.g., reordering genes using the order in a closely related complete genome).
Next, we used DESMAN to predict strain variants in core genes from our previous C. acnes 30-sample mock metagenomic samples and estimated growth rate of haplotypes in the samples. However, because only a single haplotype (haplotype_5) was accurately resolved (i.e., phylogenetically similar to a reference test strain) (fig. S3C), we were unable to validate SMEG results for other haplotypes. Nevertheless, growth predictions using haplotype_5 and its phylogenetically similar reference strain (LRY_BL) were strongly correlated (fig. S3D). Therefore, when haplotypes are accurately reconstructed, SMEG’s reference-based approach can accommodate DESMAN strain predictions.
Replication rates of antibiotic-resistant and epidemiological outbreak strains
We next sought to examine SMEG’s versatility in uncovering new biological insights in real-world datasets. Metagenomic sequencing is increasingly used for epidemiological studies involving strain transmission in outbreaks (7, 14) to circumvent a dependence on large-scale isolate cultivation and sequencing and to provide microbial context to pathogenic characteristics. An expedient application of strain-level growth inferences would be to identify such strains, particularly those that are low abundance or uncultivatable or are opportunistic pathobionts that rise out of a mixed-strain consortia, e.g., catheter infections of S. epidermidis in the skin or Shiga toxin–producing Escherichia coli (STEC) infection in the gut.
As a first demonstration, we tested SMEG’s ability to identify antibiotic-resistant strains from a mixed strain culture in vitro. We grew two skin isolates of S. epidermidis [NIHLM001 and NIHLM023, 97% average nucleotide identity (ANI)], one of which (NIHLM001) was highly resistant to the bacteriostatic antibiotic erythromycin (Fig. 3A). We mixed both strains in a 1:1 ratio at an optical density at 600 (OD600) ~0.2, added erythromycin to the culture, and collected cells from three subsequent time points for metagenomic sequencing and analysis. SMEG accurately determined that NIHLM023’s growth was slowed after antibiotic treatment, whereas NIHLM001 was actively replicating (Fig. 3B). NIHLM001 grew more rapidly under antibiotics treatment, which suggested that it was inhibited by NIHLM023 in the absence of antibiotics. We later confirmed this assumption by determining the strain coverage at each time point (Fig. 3B). Note that OD600 alone was unable to determine whether the mixed cultures contained a susceptible strain (Fig. 3A) and relying solely on metagenomic data would require longitudinal sampling, whereas SMEG requires a single data point to identify actively dividing strains.
Fig. 3. In vitro antibiotic experiment for S. epidermidis strains.
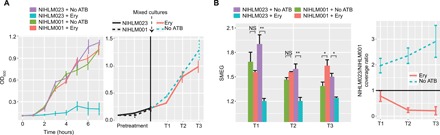
(A) In vitro growth curve of individual S. epidermidis strains NIHLM001 and NIHLM023 in the presence of the bacteriostatic antibiotic erythromycin (Ery). The figure on the right shows the growth curve when both cultures were combined and antibiotics were added to the mixture. T1, T2, and T3 represent hourly time points after antibiotic treatment. Experiments were conducted in triplicates, and error bars represent SEM. (B) SMEG output for strains NIHLM001 and NIHLM023 for samples collected every hour after antibiotic treatment (T1, T2, and T3). The figure on the right shows NIHLM023:NIHLM001 coverage ratios at time points after antibiotic treatment. Experiments were conducted in triplicates, and error bars represent SEM. NS, not significant; *P < 0.05; **P < 0.01, by one-way analysis of variance (ANOVA) analysis with Tukey’s multiple comparison test.
We next applied SMEG to 45 gut metagenomes from a 2011 outbreak caused by a STEC strain (2011C-3493) (15). SMEG correctly identified the outbreak cluster (cluster 22), which was actively replicating in 51% of the samples (Fig. 4A). All members of the outbreak cluster contained Shiga toxin (Stx) genes, validating the robustness of SMEG clustering. We further investigated potential microbiome characteristics that could suggest an underlying biological reasoning for lack of growth in some samples. Correlation analysis with the relative abundance of other bacterial species in the sample identified a likely antagonistic relationship between growth rate of cluster 22 and species belonging to the order Clostridiales and genus Bacteroides (Fig. 4B). In particular, we observed antagonism with Bacteroides thetaiotaomicron, in agreement with a previous study showing that the latter produces a 3-kDa molecule that represses the expression of Shiga toxin genes (16).
Fig. 4. SMEG validates known strain associations within an epidemiological outbreak.
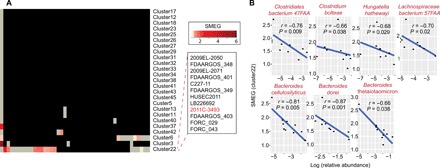
(A) The heat map shows the SMEG scores for E. coli clusters from a 2011 E. coli German outbreak (12), with a 5× coverage cutoff. Each column in the heat map represents a sample, while black boxes indicate the absence of a cluster. Gray boxes represent clusters that were present in a sample but had insufficient coverage and/or unique SNPs to accurately estimate growth rate. The predominant cluster (cluster 22) identified by SMEG correctly contained the outbreak strain 2011C-3493. (B) Spearman correlation between SMEG score of the outbreak cluster and relative abundance of commensal microbes.
Role of Akkermansia muciniphila strains in cancer immunotherapy
We also examined SMEG’s ability to uncover new biology. We analyzed a gut metagenomic dataset of lung and kidney cancer patients who had been treated with immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 axis (17). Previous analysis of this dataset showed a positive correlation between A. muciniphila abundance and positive patient outcomes (17). We thus examined whether the observed response is restricted to the growth of specific strains of A. muciniphila. Using SMEG’s de novo approach, we first determined cluster distributions in A. muciniphila–positive samples at baseline and identified three predominant clusters (clusters 1, 3, and 7) (Fig. 5A). Unexpectedly, cluster 1 was predominant in males, suggesting gender specificity (Fig. 5B). Similarly, cluster 1 was detectable in 31% of patients who responded to treatment, whereas it could only be detected in 15% of nonresponders (Fig. 5B), although its growth rate was not significantly associated with patient outcome. Note that while cluster 1 was predominant in males, the overall proportion of males who responded to treatment was similar to female responders (17). One interpretation of this data would be that female responsiveness may be independent of A. muciniphila. Strikingly, the growth rate of cluster 7, while less prevalent than cluster 1, was significantly associated with responsiveness (Fig. 5B).
Fig. 5. SMEG analysis of A. muciniphila strains in cancer patients treated with ICIs.
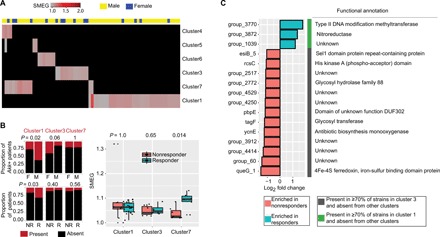
(A) Heat map displaying SMEG scores in A. muciniphila–positive samples at baseline. Each column in the heat map represents a sample, while black boxes indicate the absence of a cluster. Clusters were detected at 0.5× coverage, and gray boxes represent clusters that were present in a sample but had insufficient coverage (<1×) and/or unique SNPs (<100) to accurately estimate growth rate, with a 1× coverage cutoff optimized for A. muciniphila’s within-strain similarity. (B) Stacked barplots showing the distribution of clusters by gender and treatment outcome. P values were derived from two-sided Fisher’s exact test. The boxplot on the right shows SMEG scores at baseline. Significant differences between groups were computed with two-sided Wilcoxon rank sum test. M, male; F, female; R, responder; NR, nonresponder; Akk+, A. muciniphila positive. (C) Cluster-specific accessory genes for clusters 1 and 3 that were differentially enriched in responders and nonresponders at a log2-fold threshold of 1.
One unexpected observation was the competitive exclusion of clusters; i.e., we observed nearly no admixture of strains from different clusters within a patient. We investigated whether we could identify strain-specific genes whose presence or absence might be associated with treatment outcomes by examining the accessory gene content of clusters 1 and 7 versus cluster 3. Defining accessory genes as present in ≥70% of strains of a cluster (or present in two of the three strains of cluster 7) and absent in strains from other clusters, we found 39, 15, and 1006 cluster-specific accessory genes for clusters 1, 7, and 3, respectively, the latter of which reflected the higher gene content diversity specific to that cluster (fig. S6A). Accessory genes highly enriched in responders were mostly restricted to cluster 1 (none from 7), whereas highly enriched genes in nonresponders were restricted to cluster 3 (Fig. 5C). In addition, there was a strong correlation between cluster coverage and read counts for most genes, suggesting that genes were derived primarily from A. muciniphila and not from other species in the microbial community (fig. S6B). Because cluster 3 was associated with neither responsiveness nor nonresponsiveness (Fig. 5B), this suggests that the gene enrichment observed in nonresponders rather reflects a co-exclusion relationship between clusters 1 and 3.
Longitudinal growth rate of strains in response to antibiotic treatment
Last, we analyzed a longitudinal gut metagenomic dataset of 12 individuals examining the dynamics of antibiotic treatment, following microbial depletion and then repopulation over a 6-month period (18). We focused our analysis on strains of Faecalibacterium prausnitzii and Bacteroides vulgatus, which are two predominant species in gut metagenomes (19, 20). We identified two major states to describe strain-level dynamics for both species: At endpoint, subjects had either (i) lost strain diversity for a given species or (ii) maintained their original strain diversity (Fig. 6A), suggesting that strain mixtures may have stable states that are fixed a priori. Strains had posttreatment growth rates similar to the baseline time point (Fig. 6B), suggesting that these strains may have reached an optimal growth niche occupation.
Fig. 6. SMEG analysis of antibiotics-treated patients.
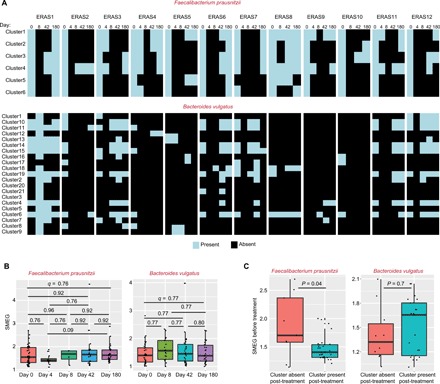
(A) Heat maps displaying the presence (light blue) and absence (black) of clusters for F. prausnitzii and B. vulgatus in 12 individuals treated with broad-spectrum antibiotics. Days 4 to 180 refer to time points after antibiotic use, while day 0 refers to baseline time point before antibiotics use. (B) Boxplots display SMEG scores (5× coverage cutoff) for clusters of F. prausnitzii and B. vulgatus at different time points after treatment. Day 0 refers to time point before antibiotics administration. Two-sided Wilcoxon rank sum tests were used to test for significant differences between groups, and false discovery rate (FDR) adjusted P values (q) are shown. (C) Boxplots display baseline SMEG scores for clusters absent (missing at day 42 or 180) and present (present at day 42 or 180) after treatment. Significant differences between groups were computed with two-sided Wilcoxon rank sum test.
Growth rate and metabolic state of bacteria have been shown separately to affect antibiotic efficacy; a more rapid growth rate (i.e., higher metabolic activity) has been associated with increased susceptibility to antibiotics (21). We tested this assumption and observed that strains of F. prausnitzii that were recovered after treatment significantly had lower SMEG scores before antibiotics in comparison to strains that were lost (Fig. 6C). However, it is likely that this phenomenon is species specific, as we did not observe this dynamic in B. vulgatus strains (Fig. 6C).
Limitations of SMEG
While we have demonstrated SMEG’s versatility and ability to uncover new biology in metagenomic datasets, we note several current limitations of SMEG. First, its deployment is on a species-by-species basis, analyzing growth rate of strains of a species of interest. Because of high computational requirements to build clusters and the susceptibility of accurate cluster formation to outliers, at this time, it cannot simultaneously estimate growth dynamics of strains from multiple species, such as our previous algorithm, GRiD. However, this allows a unique opportunity to optimize SMEG based on the unique characteristics of a species, such as genetic heterogeneity as we have demonstrated here, to increase sensitivity.
In addition, SMEG requires at least one strain with a pseudo-complete genome. However, in the absence of a complete reference genome, the draft genome with the least fragmentation can be reordered using a strain from a closely related species. It is important to properly reorient the contigs to the ori and ter to facilitate accurate coverage modeling in the representative strain. This reordering step can be performed using tools such as Mauve (22). Last, most parameters in SMEG can be user-defined to maximize flexibility for optimization. SMEG estimates growth rates using either a de novo or reference-based approach. A limitation with the de novo approach is it requires that clusters have sufficient unique SNPs, and in some cases, clusters can be composed of highly similar strain genomes. For instance, we were unable to generate unique SNP profiles for some clusters in E. coli. Therefore, if these missing clusters are present in a dataset, important biological information may be ignored. Although we note that decreasing the SNP assignment threshold would have enabled the generation of an SNP map for additional E. coli clusters, we avoided this option to ensure that only high-quality SNPs were used for analysis, with SNP quality defined as the proportion of strains harboring a unique SNP at a given position. Thus, in some cases, a user will need to choose between increased sensitivity and specificity when creating the species database. For this reason, the “auto” mode in SMEG outputs multiple SNP profiles using different parameters, in which case the user can optimize the levels based on their species and applications of interest.
In summary, our results with published and new metagenomic datasets highlight SMEG’s ability to accurately identify and predict the growth rate of strains associated with different disease conditions. Its high sensitivity to complex mixtures of closely related strains makes it a promising tool for outbreak epidemiology, infection dynamics, and understanding commensal strain diversity to guide future probiotics development. Last, as with most strain-level analyses tools, best practice remains to broadly survey different predictions and parameters to avoid misrepresentations of the underlying biology.
MATERIALS AND METHODS
SMEG algorithm
SMEG measures strain-specific bacterial growth rates from metagenomic samples. The pipeline consists of two steps (Fig. 1, A and B): (i) construction of species database and (ii) growth rate estimation. The species database is created using complete and draft genome sequences of a species of interest downloaded from NCBI GenBank. Genome sequences that have been annotated as plasmids are excluded. For draft-quality genomes, contigs are reordered using the software Mauve (22) by aligning the draft genome to a complete representative genome sequence of the same species. Next, a core genome phylogeny is constructed using FastTree (23) based on core genome alignments generated using Prokka (24) and Roary (25). The phylogeny is used to assign strains into clusters using dynamicTreeCut with deepSplit option of 4 (26). Outlier genomes, defined as having pairwise distances 30 times above the median, are excluded, as these genomes may have been misclassified taxonomically or may contain contaminant contigs.
For each cluster, SMEG identifies cluster-specific unique SNPs, i.e., SNPs that are shared between a given proportion of cluster members at a non-indel alignment position, but absent in all strains from other clusters (fig. S1A). This user-specified proportion is referred to as the SNP assignment threshold. For instance, setting the SNP assignment threshold to 0.8 indicates that a unique SNP will only be identified if it is shared between at least 80% of the cluster members and absent in strains from other clusters (fig. S1A). If the total number of unique SNPs of a given cluster is below the user-specified threshold, SMEG iteratively subclassifies the cluster and infers unique SNPs for each subcluster. SMEG repeats the subclassification step for a maximum of three iterations or until the threshold is met (Fig. 1A). This iterative subclassification process is especially useful in resolving large clusters, where the number of SNPs shared among cluster members is limited. Next, SMEG retrieves the coordinates of the cluster-specific SNPs in a representative genome, which is randomly selected from the cluster after favoring for genome completeness. While the optimal value of the SNP assignment threshold will vary depending on the species being analyzed (species with high strain-level genetic heterogeneity will have fewer discriminatory SNPs at high assignment thresholds), we implemented an auto option in SMEG, in which different threshold values are tested in parallel and output, giving the user the flexibility to select desired parameters and the associated database. Typically, a high threshold yielding sufficient number of unique SNPs is preferable. In addition, we provided precompiled databases for several selected bacterial species commonly encountered in human-associated metagenomes.
In the second step, growth rate is estimated using either a de novo or reference-based estimation approach (Fig. 1B). The de novo approach assumes no previous knowledge of strain composition in a sample. It allows the analysis of strains present in a sample, but missing in the SMEG database, by investigating features of phylogenetic clusters in the sample. To do this, metagenomic reads are mapped using bowtie2 (27) to the representative strain, and strains (whether or not characterized previously) of a cluster are considered present in a sample if the proportion of cluster-specific SNPs with read coverage >0 exceeds a cluster detection threshold. In scenarios where a sample does not contain all clusters in the species database, SMEG further generates a sample-specific SNP profile based solely on the identified clusters in a sample (Fig. 1B). This step increases the number of SNP sites for growth estimation by reducing the number of clusters compared and is especially useful for clusters lacking sufficient unique SNPs in the species database (fig. S1B). The reference-based approach requires the user to provide a list of genome names (if genomes already exist in the species database) or genome sequences. SMEG subsequently calculates nucleotide coverage at SNP sites unique to each strain and predicts the growth rates of the given strains in the metagenomic sample by mapping reads to each strain using bowtie2 (Fig. 1B) (27).
For both approaches, SMEG then generates an estimation of growth rate. First, SMEG computes the change in non-zero SNP sites for each cluster along the genome sequence using a moving window. The window size is proportional to the total number of unique SNPs, with a coefficient θ. Outlier SNP sites (i.e., sites with coverage below Q1 − 1.5 × IQR or above Q3 + 1.5 × IQR, where Q1 and Q3 are the first and third quartiles, respectively, and IQR is the interquartile range) resulting from GC bias or contamination are excluded. The SNPs are then fitted by a re-descending M estimator with Tukey’s biweight function (28). We have shown previously that this fit is resistant to noise arising from heterogeneous and low coverage strains (2). Then, SMEG computes the coverage ratio of the peak and trough of the curve (PTR), which is proportional to the rate of replication. SMEG outputs the median PTR based on three computations with θ = 0.02, 0.04, and 0.06. For clusters with insufficient cluster-specific SNPs (<200), θ = 0.01, 0.02, and 0.03 are used, which allows sufficient windows for growth estimation. For improved accuracy, we leverage information on the position of the chromosome initiator replication gene (dnaA) whenever possible. This is because dnaA is usually located in close proximity to the ori in most bacterial genomes (2). Therefore, when the position of dnaA is identified, SMEG ensures that the peak value is retrieved from the genomic quarter containing dnaA. Altogether, SMEG outputs four statistics for a given sample: (i) the median SMEG score, (ii) the coverage of phylogenetic clusters (in the de novo approach) or the user-provided strains (in the reference-based approach), (iii) the number of non-zero SNP sites used for the analysis, and (iv) the range of SMEG with different θ.
In silico C. acnes and S. epidermidis analyses
We downloaded 131 and 95 genomes of C. acnes and S. epidermidis, respectively, from NCBI GenBank (tables S2) to build the species database. Eleven clusters were identified using an SNP assignment threshold of 0.6 with iterative clustering for C. acnes [SNPs threshold for iterative clustering (−t parameter) set at 100 and strain KPA171202 as representative genome], while 15 clusters were identified for S. epidermidis at an SNP assignment threshold of 0.5 without iterative clustering and other parameters set at default. For both species, we selected a strain from eight different clusters and generated mock reads using the wgsim package included in samtools (29). Each of the eight strains was divided into 100-kb fragments, and varying amounts of reads were generated for each fragment to simulate the differential reads coverage observed for a replicating strain. To identify clusters in each sample, we used a cluster detection threshold of 0.2. We assessed the accuracy of SMEG by calculating F1 scores, which is a metric that evaluates both maximal correctly detected clusters and minimal missing clusters. Here, , where and . For spiked-in mock experiments, we retrieved high-complexity CAMI datasets from https://edwards.sdsu.edu/research/cami-challenge-datasets/.
S. aureus analysis
We retrieved 10 nasal samples from Szafrańska et al. (12), which had information on in situ generation times for S. aureus. Because of the large number of S. aureus strains present in NCBI, we built the SMEG database using strains annotated as “complete” or “chromosome” only (n = 396). We identified 13 clusters with iterative clustering at an SNP assignment threshold of 0.5 (tables S2). We filtered clusters having unique SNPs less than 10 in samples.
Determining the effect of coverage and unique SNP count on SMEG
We analyzed S. aureus clusters from 10 nasal samples obtained from Szafrańska et al. (12). For C. acnes, we retrieved 37 skin samples from back swabs from Oh et al. (30). Samples having at least a cluster with coverage >20× were subsampled to 10×, 5×, 2×, 1×, and 0.5× coverage to evaluate the effect of coverage on SMEG accuracy. We made the comparison between a cluster at >20× coverage and lower coverage. We fragmented reference strains (NCTC8325 and KPA171202 for S. aureus and C. acnes, respectively) into 100-kb fragments for GRiD and iRep analysis. Our accuracy evaluation was based on the widely used delta cutoff of 0.15 (2, 3).
To evaluate the effect of unique SNP count on SMEG score, we selected an S. aureus sample containing a cluster meeting the following requirements: (i) sufficient cluster coverage; (ii) high number of unique SNPs; and (iii) high PTR, which is indicative of high variation in SNP coverage across the genome. We subsampled unique SNPs count to 300, 200, 100, 75, and 50 SNPs. This subsampling step was repeated five times.
In vitro antibiotic experiment for S. epidermidis strains
S. epidermidis strains NIHLM001 and NIHLM023 (97% ANI) were grown in tryptic soy broth at 37°C to an OD600 of ~0.2. Both cultures were mixed, and erythromycin (125 μg/ml) was added (MICNIHLM001 = 4 mg/ml; MICNIHLM023 < 0.004 mg/ml). Growth continued for another 3 hours, and we collected samples every hour after antibiotic treatment for DNA extraction, Nextera XT library preparation, and metagenomic sequencing. We subsampled each sample to 2 million reads and performed SMEG analysis with the reference-based approach using reads mapping with a maximum of three mismatches.
SMEG analysis of E. coli outbreak samples
Metagenomic reads from 45 stool samples were downloaded from Loman et al. (15), 40 of which were positive for Shiga toxin E. coli (STEC). Because of the large number of E. coli strains present in NCBI, we only retrieved genomes annotated as complete or chromosome (n = 639) for building of the species database (table S2). We identified 30 clusters using iterative clustering with an SNP assignment threshold of 0.6. An additional 16 clusters (310 strains) had no cluster-unique SNPs identified under the given assignment threshold. We first conducted preliminary analysis using the 30 generated clusters and detected clusters in more than half of STEC samples. This suggested that the outbreak strain was contained in the 30 clusters. Consequently, we avoided the need to further lower the SNP assignment threshold, which would have generated cluster-specific SNPs for additional clusters without unique SNPs. We analyzed each metagenomic sample using a cluster detection threshold of 0.2 and detected clusters at 0.5× coverage cutoff. For improved accuracy, growth rates were only reported for clusters with non-zero SNP sites ≥100 and SNP coverage ≥5×. To determine non–E. coli species whose abundance was associated with the growth rate of the outbreak strain, we mapped reads to ReprDB database (31) and reassigned ambiguous reads using Pathoscope 2.0 (9). We filtered genomes having median coverage less than 0.001% across samples.
SMEG analysis of A. muciniphila strains in cancer patients treated with ICIs
We retrieved 219 gut metagenome samples from lung and kidney cancer patients who had been treated with ICIs targeting the PD-1/PD-L1 axis (17). These patients were sampled at baseline (before the commencement of treatment; T0) and 1 month (T1) and 2 months (T2) after treatment. We downloaded 64 strains of A. muciniphila from NCBI GenBank, constructed the species database using an SNP assignment threshold of 0.6 with iterative clustering, and detected seven clusters in samples using a cluster detection threshold of 0.2 (table S2). The association between strain growth rates and patient’s response to ICI were inferred based on T0 samples, or based on T1 samples when T0 samples were not available, as described previously (17). Because strains within A. muciniphila clusters or phylogroups are closely related and typically have an ANI of 97.2 to 100% (32), we detected clusters at a 0.5× coverage cutoff and reported growth rates for clusters with coverage ≥1× and non-zero SNP count ≥100 in a given sample. We retrieved the accessory gene content of clusters 1, 3, and 7 from Roary output (25) and identified cluster-specific genes, which we define as accessory genes present in ≥70% of strains of a cluster and absent in strains from other clusters. Because cluster 7 contains only three strains (table S2), we defined its accessory genes as genes present in at least two strains and absent from other clusters. This resulted in 39, 1006, and 15 cluster-specific accessory genes for clusters 1, 3, and 7, respectively. We used featureCounts (33) to estimate read counts for genes across samples, DESeq2 (34) to infer genes differentially enriched between responders and nonresponders, and eggNOG (35) for functional annotations. Last, we determined whether these genes were mainly contributed by A. muciniphila in the dataset by correlating cluster coverage and gene read count.
SMEG analysis of antibiotics-treated patients
Fifty-seven longitudinal gut metagenomic paired-end samples of pre- and post-antibiotic–treated patients were obtained from Palleja et al. (18). Forty-three and 83 strains of F. prausnitzii and B. vulgatus were retrieved from NCBI GenBank, respectively (tables S2). Species databases were constructed using an SNP assignment threshold of 0.8 with iterative clustering for F. prausnitzii, while an SNP assignment threshold of 0.5 without iterative clustering was used for B. vulgatus. SMEG identified 6 and 21 clusters for F. prausnitzii and B. vulgatus, respectively. For both species, one strain could not be assigned to a cluster. Phylogenetic clusters were detected in the metagenomic samples using a cluster detection threshold of 0.2. We detected clusters at 0.5× coverage cutoff and reported growth rates for only clusters with non-zero SNP sites ≥100 and SNP coverage ≥5×.
Supplementary Material
Acknowledgments
We would like to thank the Microbial Genomic Services and M. Adams of The Jackson Laboratory for their support in sequencing, and M. Adams and the Oh laboratory for critical reading of the manuscript. Funding: This work was funded by the NIH (DP2 GM126893-01 and K22 AI119231-01), the Mackenzie Foundation, and the Mark Foundation. J.O. is additionally supported by the NIH (1U54NS105539, 1 U19 AI142733, 1 R21 AR075174, and 1 R43 AR073562), the Department of Defense (W81XWH1810229), the NSF (1853071), the American Cancer Society, and Leo Foundation. A.E. is supported by The Jackson Laboratory Scholars Program and the Maximilian & Marion Hoffman Foundation Postdoctoral Fellow. Author contributions: A.E. and J.O. conceived the project. W.Z. contributed scripts to the development of SMEG. A.E. and J.O. analyzed data. A.E. and J.O. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Sequence reads generated for S. epidermidis strains NIHLM001 and NIHLM023 for in vitro growth analysis have been deposited in Sequence Reads Archive (PRJNA490375). Mock reads used for analysis can be retrieved from ftp://ftp.jax.org/ohlab/SMEG_mock_reads/. The S. aureus and C. acnes samples used for benchmark analysis are available from NCBI under BioProject accession numbers PRJEB27225 and PRJNA46333, respectively. The 2011 E. coli outbreak samples are available in the European Nucleotide Archive (ENA) under accession number ERP001956. Gut metagenomic samples from cancer patients treated with ICIs and the antibiotics-treated datasets are available from NCBI under BioProject accession numbers PRJEB22863 and PRJEB20800, respectively. The SMEG algorithm is available at https://github.com/ohlab/SMEG.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz2299/DC1
REFERENCES AND NOTES
- 1.Korem T., Zeevi D., Suez J., Weinberger A., Avnit-Sagi T., Pompan-Lotan M., Matot E., Jona G., Harmelin A., Cohen N., Sirota-Madi A., Thaiss C. A., Pevsner-Fischer M., Sorek R., Xavier R. J., Elinav E., Segal E., Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349, 1101–1106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emiola A., Oh J., High throughput in situ metagenomic measurement of bacterial replication at ultra-low sequencing coverage. Nat. Commun. 9, 4956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown C. T., Olm M. R., Thomas B. C., Banfield J. F., Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 34, 1256–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Y., Li H., Quantifying and comparing bacterial growth dynamics in multiple metagenomic samples. Nat. Methods 15, 1041–1044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd A. L., Byrd A. L., Deming C., Cassidy S. K. B., Harrison O. J., Ng W. I., Conlan S.; NISC Comparative Sequencing Program, Belkaid Y., Segre J. A., Kong H. H., Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med. 9, eaal4651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo C., Knight R., Siljander H., Knip M., Xavier R. J., Gevers D., ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol. 33, 1045–1052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albanese D., Donati C., Strain profiling and epidemiology of bacterial species from metagenomic sequencing. Nat. Commun. 8, 2260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costea P. I., Munch R., Coelho L. P., Paoli L., Sunagawa S., Bork P., metaSNV: A tool for metagenomic strain level analysis. PLOS ONE 12, e0182392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong C., Manimaran S., Shen Y., Perez-Rogers J. F., Byrd A. L., Castro-Nallar E., Crandall K. A., Johnson W. E., PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2, 33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholz C. F., Brüggemann H., Lomholt H. B., Tettelin H., Kilian M., Genome stability of Propionibacterium acnes: A comprehensive study of indels and homopolymeric tracts. Sci. Rep. 6, 20662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conlan S., Mijares L. A.; NISC Comparative Sequencing Program, Becker J., Blakesley R. W., Bouffard G. G., Brooks S., Coleman H., Gupta J., Gurson N., Park M., Schmidt B., Thomas P. J., Otto M., Kong H. H., Murray P. R., Segre J. A., Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol. 13, R64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szafrańska A. K., Junker V., Steglich M., Nübel U., Rapid cell division of Staphylococcus aureus during colonization of the human nose. BMC Genomics 20, 229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quince C., Delmont T. O., Raguideau S., Alneberg J., Darling A. E., Collins G., Eren A. M., DESMAN: A new tool for de novo extraction of strains from metagenomes. Genome Biol. 18, 181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholz M., Ward D. V., Pasolli E., Tolio T., Zolfo M., Asnicar F., Truong D. T., Tett A., Morrow A. L., Segata N., Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 13, 435–438 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Loman N. J., Constantinidou C., Christner M., Rohde H., Chan J. Z.-M., Quick J., Weir J. C., Quince C., Smith G. P., Betley J. R., Aepfelbacher M., Pallen M. J., A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104: H4. JAMA 309, 1502–1510 (2013). [DOI] [PubMed] [Google Scholar]
- 16.de Sablet T., Chassard C., Bernalier-Donadille A., Vareille M., Gobert A. P., Martin C., Human microbiota-secreted factors inhibit shiga toxin synthesis by enterohemorrhagic Escherichia coli O157: H7. Infect. Immun. 77, 783–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routy B., Le Chatelier E., Derosa L., Duong C. P. M., Alou M. T., Daillère R., Fluckiger A., Messaoudene M., Rauber C., Roberti M. P., Fidelle M., Flament C., Poirier-Colame V., Opolon P., Klein C., Iribarren K., Mondragón L., Jacquelot N., Qu B., Ferrere G., Clémenson C., Mezquita L., Masip J. R., Naltet C., Brosseau S., Kaderbhai C., Richard C., Rizvi H., Levenez F., Galleron N., Quinquis B., Pons N., Ryffel B., Minard-Colin V., Gonin P., Soria J.-C., Deutsch E., Loriot Y., Ghiringhelli F., Zalcman G., Goldwasser F., Escudier B., Hellmann M. D., Eggermont A., Raoult D., Albiges L., Kroemer G., Zitvogel L., Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Palleja A., Mikkelsen K. H., Forslund S. K., Kashani A., Allin K. H., Nielsen T., Hansen T. H., Liang S., Feng Q., Zhang C., Pyl P. T., Coelho L. P., Yang H., Wang J., Typas A., Nielsen M. F., Nielsen H. B., Bork P., Wang J., Vilsbøll T., Hansen T., Knop F. K., Arumugam M., Pedersen O., Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 3, 1255–1265 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Kraal L., Abubucker S., Kota K., Fischbach M. A., Mitreva M., The prevalence of species and strains in the human microbiome: A resource for experimental efforts. PLOS ONE 9, e97279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laursen M. F., Laursen M. F., Laursen R. P., Larnkjær A., Mølgaard C., Michaelsen K. F., Frøkiær H., Bahl M. I., Licht T. R., Faecalibacterium gut colonization is accelerated by presence of older siblings. mSphere 2, e00448-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopatkin A. J., Stokes J. M., Zheng E. J., Yang J. H., Takahashi M. K., You L., Collins J. J., Bacterial metabolic state more accurately predicts antibiotic lethality than growth rate. Nat. Microbiol. 4, 2109–2117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rissman A. I., Mau B., Biehl B. S., Darling A. E., Glasner J. D., Perna N. T., Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25, 2071–2073 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price M. N., Dehal P. S., Arkin A. P., FastTree 2—Approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seemann T., Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).24642063 [Google Scholar]
- 25.Page A. J., Cummins C. A., Hunt M., Wong V. K., Reuter S., Holden M. T. G., Fookes M., Falush D., Keane J. A., Parkhill J., Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langfelder P., Zhang B., Horvath S., Defining clusters from a hierarchical cluster tree: The dynamic tree cut package for R. Bioinformatics 24, 719–720 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kafadar K., The efficiency of the biweight as a robust estimator of location. J. Res. Natl. Bur. Stand. 88, 105–116 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh J., Byrd A. L., Park M.; NISC Comparative Sequencing Program, Kong H. H., Segre J. A., Temporal stability of the human skin microbiome. Cell 165, 854–866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou W., Gay N., Oh J., ReprDB and panDB: Minimalist databases with maximal microbial representation. Microbiome 6, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo X., Li S., Zhang J., Wu F., Li X., Wu D., Zhang M., Ou Z., Jie Z., Yan Q., Li P., Yi J., Peng Y., Genome sequencing of 39 Akkermansia muciniphila isolates reveals its population structure, genomic and functional diverisity, and global distribution in mammalian gut microbiotas. BMC Genomics 18, 800 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M. C., Rattei T., Mende D. R., Sunagawa S., Kuhn M., Jensen L. J., von Mering C., Bork P., eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.A. Rambaut, FigTree version 1.3.1 [computer program] (2009); http://tree.bio.ed.ac.uk/software/figtree/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaaz2299/DC1