

Impaired FOXO1 expression in CD4 T cell responses from old adults causes expanded multivesicular bodies sequestering GSK3β.
Abstract
T cell differentiation involves the dynamic regulation of FOXO1 expression, which rapidly declines after activation and is subsequently restored. Reexpression is impaired in naïve CD4+ T cell responses from older individuals. Here, we show that FOXO1 promotes lysosome function through the induction of the key transcription factor for lysosomal proteins, TFEB. Subdued FOXO1 reexpression in activated CD4+ T cells impairs lysosomal activity, causing an expansion of multivesicular bodies (MVBs). Expansion of the MVB compartment induces the sequestration of glycogen synthase kinase 3β (GSK3β), thereby suppressing protein turnover and enhancing glycolytic activity. As a consequence, older activated CD4+ T cells develop features reminiscent of senescent cells. They acquire an increased cell mass, preferentially differentiate into short-lived effector T cells, and secrete exosomes that harm cells in the local environment through the release of granzyme B.
INTRODUCTION
Aging is associated with a decline in adaptive immunity, resulting in increased susceptibility to infection and the decreased efficacy of vaccination (1, 2). Studies in humans have so far failed to identify a single major defect. Decline in T cell generation due to thymic involution is, in part, compensated by homeostatic proliferation that maintains a sizable and diverse naïve CD4+ T cell compartment. The CD8+ T cell compartment is more affected by aging with a diminution of naïve and central memory subsets and an increase in effector T cells (3, 4). End-differentiated effector CD8+ CD45RA+ CD28− T cells accumulate that have lost replicative potential and exhibit an expression pattern of cytokines and the cytotoxic proteins perforin and granzyme B, reminiscent of innate cell function and the senescence-associated secretory phenotype (SASP) (5, 6). Less frequently, CD4+ CD45RA+ CD28− effector T cells accumulate in older individuals. These end-differentiated T cells also exhibit reduced proliferative capacity and potent effector functions (7). With the exception of these effector T cells that are frequently specific for latent viruses, unstimulated circulating naïve and memory CD4+ and CD8+ T cells do not show evidence of cellular senescence that could account for age-associated immune defects (8). Rather than senescence-associated defects, epigenetic signatures and microRNA profiles induced by T cell aging are indicative of activated differentiation pathways that favor development into short-lived effector cells (9).
In addition to cellular senescence and epigenetic changes, many cellular pathways contribute to aging (10). One of the generic aging hallmarks is loss of protein homeostasis (proteostasis), i.e., a defect in degradation of misfolded or damaged proteins by lysosomes or proteasomes. FOXO1, a conserved longevity factor, is important in maintaining proteostasis in many cell types by promoting the expression of genes involved in autophagy and the ubiquitin-proteasome system (11). In quiescent T cells, FOXO1 is located in the nucleus, regulating transcriptional activity. T cell activation induces AKT-mediated phosphorylation of FOXO1, nuclear exclusion, ubiquitin-proteasome degradation, and hence termination of transcriptional regulatory activity (12). In our previous studies, naïve CD4+ T cells from older individuals exhibited sustained AKT activity after activation due to miR-21–mediated reduced PTEN expression, resulting in the preferential differentiation of effector over memory T cells (13). How sustained AKT activation affects the kinetics of FOXO1 activity in a T cell response is unknown but likely has consequences for proteostasis and differentiation.
Here, we show that the dynamic regulation of FOXO1 expression after T cell activation affects proteostasis through the regulation of lysosomal activity and exosome secretion. FOXO1 induces the transcription of TFEB, the master transcription factor for lysosome biogenesis (14). Responses of older naïve CD4+ T cells are characterized by a failure to restore FOXO1 expression after the initial activation-induced decline, resulting in impaired lysosomal proteolytic activity. Instead, the formation of multivesicular bodies (MVBs), an acidic organelle that has no proteolytic activity and is the major source of exosomes, is enhanced (15). Glycogen synthase kinase 3β (GSK3β) is sequestered into MVBs, resulting in reduced intracellular GSK3β activity in older activated naïve CD4+ T cells. The sustained FOXO1 repression and the associated GSK3β sequestration induce a senescence-like defect in protein homeostasis manifesting as increased cell size and exosome disposal; glycolytic capacity and glycogen synthesis are increased, and T cell function is biased toward the production of pro-inflammatory cytokines and expression of granzyme B that is secreted in the exosomes.
RESULTS
Age-associated failure in FOXO1 reexpression impairs lysosomal function in aged naïve CD4+ T cell responses
In our previous studies, we found that naïve CD4+ T cells from older individuals have sustained activation of the phosphatidylinositol 3-kinase (PI3K)–AKT–mechanistic target of rapamycin complex 1 (mTORC1) axis after activation, resulting in the preferential differentiation into effector T cells (13). FOXO1 is known to be dynamically regulated in T cell responses, in part due to AKT-mediated phosphorylation and subsequent degradation (12). Upon stimulation with anti-CD3 and anti-CD28 antibody–coated beads in vitro, FOXO1 protein expression in young naïve CD4+ T cells markedly declined to a very low level on day 3 to then partially recover by day 5 (Fig. 1A). Pre-activation levels and the activation-induced decline of FOXO1 were not influenced by age. In contrast, recovery was subdued in naïve CD4+ T cells from older (65 to 85 years) compared to young (20 to 35 years) healthy adults; FOXO1 protein expression in aged cells was reduced by 35% on day 5 after stimulation (Fig. 1A and fig. S1A) and continued to be lower over subsequent days (fig. S1, B and C). Treatment with the AKT inhibitor (MK-2206 2HCl) restored FOXO1 protein expression by day 5–activated naïve CD4+ T cells from older individuals (fig. S1D). FOXO1 transcripts were also lower by 50% in cells from older compared to young individuals (fig. S1E), likely reflecting that FOXO1 regulates its own transcription (fig. S1F) (16). Global gene expression profiles of day 5–activated CD4+ T cells obtained by RNA sequencing (RNA-seq) supported the notion that the reduced reexpression of FOXO1 is functionally important. Gene set enrichment analysis (GSEA) showed that age-associated transcriptional signatures of day 5–activated naïve CD4+ T cells were concordant with those down-regulated in mouse Foxo1 knockout T cells compared with wild-type cells (Fig. 1B). In contrast, an age-dependent FOXO1-dependent signature was not observed in unstimulated naïve CD4+ T cells (fig. S1G).
Fig. 1. Age-associated failure in FOXO1 reexpression impairs lysosomal function in naïve CD4+ T cell responses.
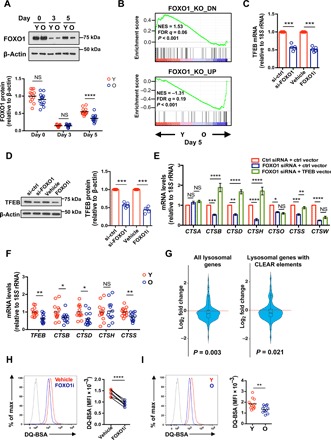
(A) Naïve CD4+ T cells were activated with anti-CD3/anti-CD28 beads. FOXO1 protein expression was determined by Western blotting. Representative Western blots of cells from one young (Y) and one old (O) adult and summary data from 13 young (20 to 35 years old) and 13 old (65 to 85 years old) healthy individuals. Intensities of FOXO1 protein expression were normalized to β-actin and are shown relative to the mean of unstimulated naïve CD4+ T cells from young individuals. The horizontal lines represent mean values; comparison by two-tailed unpaired t test. NS: not significant. (B) GSEA comparing fold transcript differences in young compared with old naïve CD4+ T cells on day 5 after stimulation (accession number: SRA: SRP158502) (19) with that of mouse Foxo1 knockout (KO) unstimulated T cells (46). NES, normalized enrichment score; FDR, false discovery rate. (C) Naïve CD4+ T cells from a young healthy adult were activated with anti-CD3/anti-CD28 beads, transfected with FOXO1 or control siRNA on day 2, and then cultured on plates coated with anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) antibodies. Alternatively, naïve CD4+ T cells were activated with anti-CD3/anti-CD28 beads; vehicle or 50 nM FOXO1 inhibitor (AS1842856) was added on day 3. TFEB mRNA was quantified by reverse transcription polymerase chain reaction (RT-PCR) on day 5 of culture. Results are normalized to control-silenced or vehicle-treated cells; mean of five experiments; two-tailed paired t test. (D) TFEB protein expression in cells treated as described in (C) was determined by Western blotting. Representative blots (left) and relative intensities, normalized to control samples (right), are shown; mean of five experiments, two-tailed paired t test. (E) Naïve CD4+ T cells were activated as described in (C) and transfected with indicated siRNA and plasmid. Lysosomal cathepsin gene expressions were quantified by RT-PCR. Results are normalized to control samples; mean ± SEM of four to six experiments; comparison by two-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. (F) TFEB and cathepsin transcripts from day 5–activated naïve CD4+ T cells from thirteen 20- to 35-year-old and thirteen 65- to 85-year-old healthy adults. Results are expressed relative to the mean of cells from young individuals. Horizontal lines represent mean values; comparison by two-tailed unpaired t test. (G) Transcriptome data from naïve CD4+ T cells from three young and three old healthy individuals stimulated with anti-CD3 and anti-CD28 beads for 5 days (accession number: SRA: SRP158502) (19). Violin plots and boxplots showing the fold gene expression differences of lysosomal genes, comparing the transcriptome of old to young T cells. Lysosomal gene sets were drawn from published experiments in human-derived cell lines (14, 18). Statistical analysis by Wilcoxon rank sum tests. (H and I) Naïve CD4+ T cells from a young healthy individual were stimulated and treated with FOXO1 inhibitor as in (C). Cells were then treated with DQ-BSA (5 μg/ml) for 5 hours. Fluorescence of cleaved DQ-BSA was analyzed by flow cytometry. (H) Representative histograms (left) and results of paired samples from seven young healthy individuals (right); two-tailed paired t test. (I) Representative histograms (left) and cumulative data from 12 young and 12 old healthy individuals; two-tailed unpaired t test. The gray histogram represents untreated naïve CD4+ T cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. MFI: mean fluorescence intensity.
FOXO1 plays a pivotal role in cellular quality control, which is critical in preventing age-related diseases (11). TFEB, a master transcription factor of the lysosomal gene network (14), was recently identified as a FOXO1 target gene in adipocytes (17). To determine whether FOXO1 regulates lysosomal gene expression in T cell responses by controlling TFEB transcription, we inhibited the recovery of FOXO1 activity either by small interfering RNA (siRNA) silencing (fig. S2A) or by pharmacological inhibition by adding a FOXO1 inhibitor (AS1842856) on day 3 after activation. Both TFEB transcripts and protein expression were down-regulated by either intervention (Fig. 1, C and D). Moreover, FOXO1 silencing and inhibition resulted in reduced transcription of six of seven cathepsin genes (Fig. 1E). Expression of four (CTSB, CTSD, CTSH, and CTSS) of these six cathepsins could be rescued by TFEB overexpression (Fig. 1E and fig. S2, B and C). Together, FOXO1 reexpression after T cell activation is required for the expression of lysosomal genes, at least in part, by inducing TFEB transcription. Because FOXO1 reexpression is impaired in older activated T cells, we examined the influence of age on gene expression of TFEB and those four cathepsin genes that were rescued by TFEB overexpression. Results from day 5–activated CD4+ T cells from 13 young and 13 old healthy adults are shown in Fig. 1F. TFEB as well as CTSB, CTSD, and CTSS transcripts were reduced in older activated naïve CD4+ T cells, while CTSH expression varied widely. The influence of age on lysosomal gene expression was confirmed by analyzing two gene sets, one of 95 lysosomal genes expressed in T cells and one of 62 lysosomal genes that have CLEAR (coordinated lysosomal expression and regulation) elements (TFEB-binding motif) (14, 18), in a previously published RNA-seq dataset from activated naïve CD4+ T cells [accession number: SRA (Sequence Read Archive): SRP158502] (19). Expressions of lysosomal genes from both sets were significantly reduced with age (Fig. 1G). As expected, we did not find any age-associated changes in lysosomal gene expressions in unstimulated naïve CD4+ T cells (fig. S3).
To determine whether FOXO1 regulates lysosomal proteolytic activities, we used de-quenched bovine serum albumin (DQ-BSA) labeling. DQ-BSA is self-quenched, as a result of heavy labeling by BODIPY (boron-dipyrromethene) dyes. When added to culture medium, DQ-BSA is incorporated into the liquid-phase cellular endosomal compartment by non–receptor-mediated endocytosis. Upon fusion with lysosomes, DQ-BSA is digested into smaller fragments, relieving self-quenching and generating a fluorescent signal that correlates with the proteolytic activities (18). FOXO1 inhibition decreased lysosomal proteolytic activities by 35% (Fig. 1H). Consistent with reduced FOXO activity, we found less lysosomal proteolytic activities in cells from older individuals compared with those from young adults (Fig. 1I). Together, lysosomal proteolytic activity is dependent on the FOXO1-dependent transcription of lysosomal genes and is therefore impaired in older naïve CD4+ T cells that fail to appropriately reexpress FOXO1 after activation.
Failure in FOXO1 reexpression promotes MVB expansion in and exosome secretion by old naïve CD4+ T cells
Inhibition of lysosome has been shown to lead to intracellular accumulation of LAMP1-positive compartments (20) that represent late endosomes or MVBs but not functional lysosomes (20). Treatment with the lysosome inhibitors leupeptin or bafilomycin A1 up-regulated LAMP1 in activated naïve CD4+ T cells (fig. S4). Likewise, we observed a threefold increase in LAMP1 protein expression upon silencing or pharmacological inhibition of FOXO1 in activated naïve CD4+ T cells (Fig. 2A), indicating that sustained FOXO1 deficiency in T cell responses leads to the expansion of MVBs. In support of this notion, CD63, a tetraspanin protein that marks the intraluminal vesicles (ILVs) of MVBs, was twofold increased upon FOXO1 inhibition (Fig. 2B). LysoTracker staining showed an increased number of acidic organelles in cells treated with the FOXO1 inhibitor (Fig. 2C). Given the defective lysosomal proteolytic activities in these cells (Fig. 1H), these acidic organelles are likely MVBs and not functional lysosomes. Immunofluorescence studies showed accumulation of enlarged, partially colocalizing LAMP1- and CD63-positive structures after FOXO1 inhibition, consistent with the expansion of MVBs (Fig. 2D).
Fig. 2. Failure in FOXO1 reexpression promotes MVB expansion and exosome secretion in old naïve CD4+ T cells.
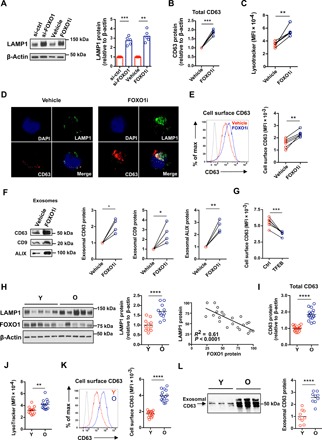
(A to E) Naïve CD4+ T cells were activated, silenced for FOXO1 expression, or treated with FOXO inhibitor as described in Fig. 1C. (A) LAMP1 protein expression was determined by Western blotting. Representative Western blots (left) and results relative to those of cells from control-silenced or vehicle-treated samples (right) (n = 5, mean, two-tailed paired t test). (B) Western blot results for CD63, expressed relative to vehicle-treated cells (n = 5, two-tailed paired t test). (C) Flow cytometry of LysoTracker staining. MFI from five experiments (two-tailed paired t test). (D) Cells were stained with anti-LAMP1 (green) and anti-CD63 (red). Confocal images representative of three independent experiments show accumulation of LAMP1 and CD63 in FOXO1-inhibited naïve CD4+ T cells. DAPI, 4′,6-diamidino-2-phenylindole. (E) Flow cytometry of cell surface CD63 expression. Representative histograms (left) and results from five experiments (right; two-tailed paired t test). The shaded histogram represents isotype control. (F) Naïve CD4+ T cells were cultured for the past 24 hours of the 5-day culture in exosome-depleted medium. Exosomes were isolated and assessed for exosomal markers (CD63, CD9, and ALIX) by immunoblotting. Representative Western blots and results from four experiments (expressed relative to vehicle-treated cells, two-tailed paired t test). (G) TFEB expression was restored in old naïve CD4+ T cells by transfection with a TFEB construct (fig. S5). CD63 cell surface expression in control- and TFEB-transfected activated CD4+ T cells was compared (n = 6, two-tailed paired t test). (H to L) Naïve CD4+ T cells from 20- to 35- and 65- to 85-year-old healthy individuals were stimulated with anti-CD3/anti-CD28 beads for 5 days. (H) LAMP1 and FOXO1 protein expression were determined by Western blotting. Representative Western blots and cumulative data from 11 young and 11 old healthy individuals (two-tailed unpaired t test, left); Pearson correlation plot of FOXO1 and LAMP1 (right). (I) CD63 Western blots from experiments of 16 young and 16 old individuals (two-tailed unpaired t test). (J) Acidic organelles were quantified as in (D). Cumulative data from 13 young and 13 old healthy individuals (two-tailed unpaired t test). (K) Flow cytometry of cell surface CD63. Representative histograms (left) and cumulative data from 15 young and 15 old healthy individuals (right; two-tailed unpaired t test). The shaded histogram represents isotype control. (L) Western blots for CD63 in exosomes isolated as described in (F). Representative Western blots (left) and results from nine young and nine old individuals (right; two-tailed unpaired t test). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Fusion of MVB with the plasma membrane results in the release of ILVs as exosomes (21); secretion of exosomes has been shown to increase after lysosome inhibition (21). This prompted us to test whether lysosome deficiency and MVB expansion caused by FOXO1 inhibition promote exosome secretion by activated naïve CD4+ T cells. FOXO1 inhibition increased cell surface expression of CD63, a surrogate marker of MVB fusion with the plasma membrane (Fig. 2E) (22). Exosomes in cell culture supernatants from FOXO1-inhibited compared to control CD4+ T cells were twofold enriched as determined by Western blotting for the MVB markers CD63, CD9, and ALIX (Fig. 2F) (21). Conversely, overexpression of the FOXO1-dependent, transcriptional regulator of lysosomal genes, TFEB, reduced CD63 expression (Fig. 2G).
Next, we explored whether the reduced FOXO1 expression in activated naïve CD4+ T cells from older adults had functional consequences for MVB content and exosome secretion. LAMP1 and CD63 protein expression levels in older naïve CD4+ T cells were twofold higher compared to those in young cells (Fig. 2, H and I). The protein expression level of LAMP1 was inversely correlated with that of FOXO1 (Fig. 2H). LysoTracker staining was increased, indicating the expansion of MVBs (Fig. 2J). Cell surface CD63 expression (Fig. 2K) and exosomal CD63 (Fig. 2L) were even more increased, indicating an enhanced activation of MVB and exosome secretion by older T cells. Together, these data suggest that the age-associated reduced FOXO1 expression in activated naïve CD4+ T cells promotes MVB expansion, MVB/plasma membrane fusion, and exosome secretion.
MVB expansion induces sequestration of GSK3β
Because MVBs have been reported to be important regulators of GSK3β activity by sequestering the kinase (23), we examined whether FOXO1 inhibition reduces GSK3β activity in T cells. We found high protein expression of GSK3β in exosomes isolated from day 5–stimulated naïve CD4+ T cells (Fig. 3A), indicating that T cells regulate intracellular GSK3β activity by releasing GSK3β-containing exosomes into extracellular fluid. Naïve CD4+ T cells activated for 5 days and treated with the FOXO1 inhibitor for the past 24 hours of culture showed a more than twofold increase in secretion of exosomal GSK3β protein compared with control cells, leading to the decrease of intracellular GSK3β protein (Fig. 3B). Of note, the protein size of exosomal GSK3β was lower than that of intracellular GSK3β (Fig. 3B).
Fig. 3. Sequestration of GSK3β by expanded MVBs induces increased cell size in old activated naïve CD4+ T cells.
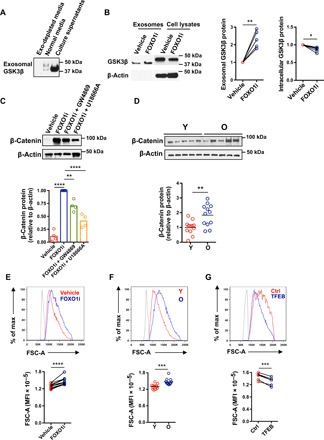
(A) Naïve CD4+ T cells were activated; exosomes were isolated on day 5 as described in Fig. 2F and probed for GSK3β. Western blots are representative of four experiments. (B) Exosomal and cytoplasmic GSK3β from naïve CD4+ T cells cultured in the presence of vehicle or FOXO1 inhibitor for the past 24 hours of 5-day culture after activation. Representative Western blots and results from five experiments (expressed relative to vehicle-treated cells, two-tailed paired t test). (C) Naïve CD4+ T cells from young healthy individuals were activated with anti-CD3/anti-CD28 beads for 5 days, the past 2 days in the presence of vehicle or indicated inhibitors. β-Catenin expression was determined by Western blotting. Representative Western blots (top) and results from four experiments (expressed relative to FOXO1 inhibitor–treated cells, bottom; one-way ANOVA followed by Tukey’s multiple comparisons test). (D) β-Catenin expression in CD4+ naïve T cells from twelve 20- to 35-year-old and twelve 65- to 85-year-old healthy individuals on day 5 after stimulation. Representative Western blots (top) and cumulative results (expressed relative to young individuals, bottom; two-tailed unpaired t test). (E) FSC-A of activated naïve CD4+ T cells cultured in the presence or absence of a FOXO1 inhibitor for the past 48 hours was determined by flow cytometry. Representative histograms (top) and results of paired samples from 10 young healthy individuals (bottom; two-tailed paired t test). The shaded histogram represents unstimulated naïve CD4+ T cells. (F) Representative FSC-A histograms (top) and data from day 5–activated naïve CD4+ T cells from 15 young and 15 old healthy individuals (bottom; two-tailed unpaired t test). The shaded histogram represents unstimulated naïve CD4+ T cells. The horizontal lines represent mean values. (G) TFEB expression was restored in activated naïve CD4+ T cells from old individuals by transfection with a TFEB construct (fig. S5). FSC-A in control and TFEB-transfected activated CD4+ T cells were compared (n = 6, two-tailed paired t test). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The increased exosomal secretion of GSK3β protein in FOXO1-inhibited cells prompted us to test whether intracellular GSK3β activity was deficient in these cells. We found a 4- to 10-fold increase of β-catenin expression, suggesting that reduced GSK3β activity in these cells prevented β-catenin degradation (Fig. 3C). The exosome formation and release inhibitor GW4869 and the MVB inhibitor U18666A partially prevented the increase of β-catenin in FOXO1-inhibited cells (Fig. 3C). Treatment with these inhibitors was not toxic to the cells (fig. S6A). Consistent with the increase in MVBs, β-catenin protein levels were higher in day 5–activated naïve CD4+ T cells from old healthy individuals (Fig. 3D). This up-regulation of β-catenin was posttranscriptional, and transcript levels were not different (fig. S6B).
Maintained FOXO1 activity after T cell activation has been shown to prevent biomass accumulation and suppress the increase in cell size (24). Conversely, GSK3β activity has been reported to reduce cell mass (18, 23) and cell size (25). Naïve CD4+ T cells treated with a GSK3β inhibitor (BIO) had increased cell size as indicated by increased forward scatter area (FSC-A) (fig. S7A). Accordingly, FSC-A in FOXO1-inhibited naïve CD4+ T cells was higher compared with control cells (Fig. 3E). Consistent with the lower expression of FOXO1, old naïve CD4+ T cells exhibited increased cell size compared to young cells (Fig. 3F). Conversely, TFEB reduced FSC-A (Fig. 3G). Together, these data suggest that loss of FOXO1 promotes MVB sequestration and exosomal secretion of GSK3β, thereby affecting protein turnover and increasing the cell size of old naïve CD4+ T cells.
MVB sequestration of GSK3β regulates glucose homeostasis in old naïve CD4+ T cells
GSK3β is an important metabolic regulator in macrophages (26) and B cells (25); its role in the metabolic regulation of T cells is less studied. GSK3β inhibits glycogen synthesis by phosphorylating glycogen synthase (27). We found a decrease in phosphorylated glycogen synthase in FOXO1-inhibited naïve CD4+ T cells, which was reversed by the treatment with an MVB inhibitor (U18666A) (Fig. 4A). Accordingly, FOXO1 inhibition raised intracellular glycogen levels by more than threefold (Fig. 4B). Again, these functional consequences of FOXO1 and GSK3β activity deficiency were pertinent for T cell aging. Phosphorylated glycogen synthase in activated naïve CD4+ T cells from old healthy individuals was lower (Fig. 4C), and intracellular glycogen storage was higher compared to cells from young adults, consistent with the observed reduced GSK3β activity (Fig. 4D).
Fig. 4. MVB sequestration of GSK3β regulates glucose homeostasis in old naïve CD4+ T cells.
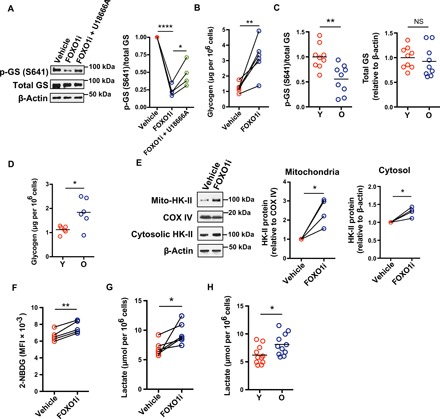
(A) Naïve CD4+ T cells from a young healthy individual were activated with anti-CD3/anti-CD28 beads for 3 days and then treated with vehicle or inhibitors for 2 days. Phosphorylated glycogen synthase (p-GS) and total glycogen synthase (GS) were determined by Western blotting. Representative Western blots (left) and results from four experiments shown as the ratios of p-GS and total GS relative to vehicle-treated cells (right; one-way ANOVA followed by Tukey’s multiple comparisons test). (B) Intracellular glycogen was quantified in naïve CD4+ T cells from six young individuals activated by anti-CD3/anti-CD28 beads and treated with vehicle control or FOXO inhibitors for the past 24 hours of culture; comparison by two-tailed paired t test. (C) p-GS and total GS in CD4+ naïve T cells on day 5 after stimulation were determined by Western blotting. Results of the ratios of p-GS (left) and total GS (right) from nine young and nine old healthy individuals (two-tailed unpaired t test). (D) Intracellular glycogen levels of CD4+ naïve T cells from six young and six old healthy individuals on day 5 after stimulation (two-tailed unpaired t test). (E) Naïve CD4+ T cells were activated and treated with FOXO1 inhibitor (AS1842856) as in (A). Mitochondria and cell cytosol were isolated. Mito-HK-II and cytosolic HK-II were assessed by Western blotting. Representative Western blots of cells from one healthy individual (left) and results from four experiments for mito-HK-II (middle) and for cytosolic HK-II (right), expressed relative to vehicle-treated cells (two-tailed paired t test). (F and G) Naïve CD4+ T cells were activated and treated with FOXO1 inhibitor (AS1842856) as in (B). Glucose uptake was determined by flow cytometry of 2-NBDG uptake. Results from cells of five young healthy individuals; two-tailed paired t test (F). Lactate concentrations were measured in culture supernatants of cells from six young healthy individuals; two-tailed paired t test (G). (H) Lactate concentrations from supernatants of activated naïve CD4+ T cells from 11 young and 11 old individuals (two-tailed unpaired t test). The horizontal lines represent mean values. *P < 0.05, **P < 0.01, ***P < 0.001.
GSK3β inhibition induces an accumulation of hexokinase II (HK-II) at mitochondria (28), thereby enhancing glycolysis (29). We found a twofold increase of mitochondrial and a lesser increase of cytoplasmic HK-II (mito-HK-II) in FOXO1-inhibited naïve CD4+ T cells (Fig. 4E) as well as an increased glucose uptake and lactate secretion compared to control cells (Fig. 4, F and G). Again, a similar increased lactate production was seen for day 5–activated naïve CD4+ T cells from older individuals (Fig. 4H).
Reduced FOXO1 reexpression in old individuals favors differentiation into granzyme B–expressing effector cells and promotes secretion of B cell cytotoxic exosomes
Metabolic reprogramming with enhanced glycolysis favors the induction of effector functions including granzyme B expression (30, 31). We found a threefold increase of inducible granzyme B production when FOXO1 activity was inhibited throughout differentiation (Fig. 5A); this increase was prevented by treatment with the glycolysis inhibitor 2-deoxy-d-glucose (2DG) (fig. S7B). Transcript studies confirmed a biased differentiation into effector CD4+ T cells upon FOXO1 inhibition, with increased expression of GZMB, PRF1, and PRDM1, but no significant difference in RUNX3 (Fig. 5B). Consistent with their FOXO1 deficiency, old activated naïve CD4+ T cells had enhanced granzyme B production compared to young cells (Fig. 5C).
Fig. 5. Reduced FOXO1 reexpression in old individuals favors differentiation into granzyme B–expressing effector cells and promotes secretion of B cell cytotoxic exosomes.
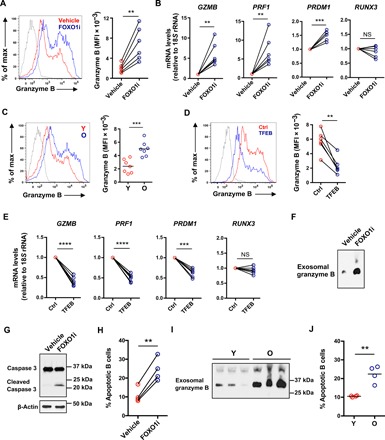
(A and B) Naïve CD4+ T cells from young healthy individuals were activated with anti-CD3/anti-CD28 beads for 3 days followed by 2-day culture with vehicle or 50 nM FOXO1 inhibitor (AS1842856). Representative histograms (left) of intracellular granzyme B after restimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin (A). The shaded histogram represents isotype control. Data (right) from six young healthy individuals (two-tailed paired t test). Expression of GZMB, PRF1, PRDM1, and RUNX3 determined by RT-PCR (B). Results are presented relative to vehicle-treated cells (two-tailed paired t test). (C) Naïve CD4+ T cells were activated with anti-CD3/anti-CD28 beads for 5 days. Representative histograms (left) of intracellular granzyme B after restimulation with PMA and ionomycin and cumulative data (right) in cells from seven 20- to 35-year-old and seven 65- to 85-year-old healthy individuals (two-tailed unpaired t test). (D and E) Unstimulated naïve CD4+ T cells from six 65- to 85-year-old healthy individuals were transfected with a TFEB expression vector or a control vector and then stimulated with anti-CD3/CD28 beads for 5 days. Representative histograms (left) of and cumulative data (right) of intracellular granzyme B after restimulation with PMA and ionomycin (two-tailed paired t test) (D). The shaded histogram represents isotype control. Gene expression was measured by RT-PCR (E). Results are expressed relative to control vector–transfected cells (n = 6; two-tailed paired t test). **P < 0.01, ***P < 0.001, ****P < 0.0001. (F) Naïve CD4+ T cells from young individuals were cultured for the past 24 hours of the 5-day culture in exosome-depleted medium with vehicle or 50 nM FOXO1 inhibitor (AS1842856). Exosomes were isolated and assessed for granzyme B by immunoblotting. Western blots representative of three experiments. (G and H) Freshly isolated peripheral blood B cells were treated with exosomes isolated as in (F) for 24 hours. Caspase-3 activity in B cells was determined by immunoblotting using an antibody recognizing both uncleaved and cleaved caspase-3. Western blots representative of two experiments (G). Relative frequencies of Annexin V+ 7-AAD+ B cells (H); n = 3, two-tailed paired t test. (I and J) Exosomes from young and old naïve CD4+ T cells were isolated as in (F). Exosomal granzyme B was detected by immunoblotting. Representative Western blots of cells from three young and three old individuals (I) and frequencies of apoptotic B cells induced by exosomes isolated from four young and four old individuals (J); two-tailed unpaired t test, **P < 0.01.
In support of the notion that the increased granzyme B production by FOXO1-inhibited naïve CD4+ T cells was due to sequestration of GSK3β, GSK3β inhibition during T cell differentiation reproduced enhanced granzyme B expression (fig. S7C), whereas addition of an MVB or an exosome inhibitor prevented differentiation into granzyme-producing effector T cells (fig. S7D). We could reverse the aging-related phenotype of biased effector cell differentiation by genetically overexpressing TFEB. Granzyme B production (Fig. 5D) and GZMB, PRF1, and PRDM1 transcription in activated naïve CD4+ T cells from old individuals (Fig. 5E) were significantly reduced by TFEB overexpression compared to control vector–transfected cells. Conversely, the biased differentiation into effector T cells was linked to FOXO1 expression kinetics. While FOXO1 inhibition reduced TFEB expression and induced the effector cell program, in part by up-regulating glycolysis, 2DG treatment or TFEB overexpression increased FOXO1 protein expression (fig. S7E). These data indicate a positive feedback loop that stabilizes the effector cell differentiation program after FOXO1 inhibition.
The co-occurrence of high exosome secretion and increased production of granzyme B in FOXO1-inhibited cells and in T cells from older individuals prompted us to investigate whether these exosomes exhibit cytotoxicity to neighboring cells. As shown in Fig. 5F, exosomes contained granzyme B, and exosomal granzyme B was enriched in supernatants of FOXO1-inhibited cells compared to that from vehicle-treated cells. In a coculture system with B cells, granzyme B–containing exosomes following FOXO1 inhibition induced B cell apoptosis as shown by the increase of cleaved caspase-3 (Fig. 5G) and the increased frequencies of Annexin V+ 7-AAD+ B cells (Fig. 5H). Again, the findings were pertinent for T cell aging; old activated naïve CD4+ T cells secreted more exosomal granzyme B than young cells (Fig. 5I), and exosomes derived from older T cells were more cytotoxic, resulting in increased frequencies of apoptotic B cells (Fig. 5J).
Tfeb deficiency reproduces human aged phenotypes in a mouse model in vivo
Our in vitro studies on human naïve CD4+ T cell responses demonstrated that TFEB deficiency due to reduced FOXO1 up-regulation is one of the major defects in older CD4+ T cell responses, manifesting as MVB expansion, increased cell size, and enhanced CD4+ effector T cell differentiation. To examine the functional consequences of TFEB deficiency in vivo, we retrovirally transduced SMARTA CD4+ T cells with short hairpin RNA (shRNA) specific for Tfeb (shTfeb) or control shRNA (shCtrl) and transferred the cells into B6 mice followed by lymphocytic choriomeningitis virus (LCMV) infection. Reduced Tfeb expression in shTfeb-transduced SMARTA CD4+ T cells was confirmed by Western blotting (Fig. 6A). Tfeb silencing reproduced the age-associated phenotypes without affecting proliferative expansion (Fig. 6B). Tfeb-silenced SMARTA CD4+ T cells expressed more CD63 (Fig. 6C), exhibited increased cell size (Fig. 6D), and were biased toward effector cell differentiation with increased frequencies of granzyme B–producing SMARTA cells (Fig. 6E).
Fig. 6. Tfeb deficiency reproduces human aged phenotypes in a mouse model in vivo.
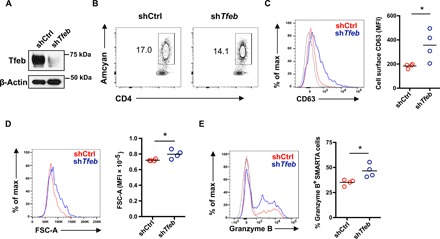
Tfeb shRNA retrovirally transduced Amcyan+ LCMV-specific naïve SMARTA transgenic CD4+ T cells (2 × 105) were adoptively transferred into CD45.2+ naïve recipients. Mice were infected with 2 × 105 plaque-forming units of LCMV Armstrong. On day 5 after infection, spleens were harvested and analyzed. Fluorescence-activated cell sorting plots are gated on CD4+ Amcyan+ SMARTA cells. (A) Tfeb silencing in sorted CD4+ Amcyan+ SMARTA cells was confirmed by Western blotting. (B) Contour plot showing percentage of retrovirally transduced Amcyan+ SMARTA cells on day 5 after LCMV infection. (C) Representative histogram of CD63 cell surface expression on transduced SMARTA cells on day 5 after infection (left) and cumulative data from four mice in each group (right). (D) Representative histogram of FSC-A (left) and cumulative data from four mice in each group (right). (E) Representative histogram of intracellular granzyme B (left) and cumulative data of percentage of granzyme B+ cells in Amcyan+ SMARTA cells (right) from four mice in each group. Data are representative of one of two independent experiments each with n = 4 mice. *P < 0.05.
DISCUSSION
Here, we show that FOXO1 regulates organelle and protein homeostasis by promoting lysosome function through the induction of the key transcription factor for lysosomal proteins, TFEB. This pathway is pertinent for T cell responses, because T cells undergo a rapid decline of FOXO1 after activation followed by a slow recovery. Reexpression is impaired in activated CD4+ T cells of older individuals, resulting in reduced lysosomal activity and expansion of the MVB compartment. Expansion of MVBs increases sequestration of GSK3β that influences T cell function and differentiation by regulating protein turnover and glycolytic activity. As a consequence, older T cells have an increased cell mass and preferentially differentiate into short-lived effector T cells characterized by high granzyme B production. MVBs in these T cells default to exosome secretion that harms the local environment through the release of granzyme B.
T cell aging is a complex process, involving changes in epigenetic states, T cell activation pathways, as well as T cell differentiation and proliferation (8, 10). While defects in FOXO1 activity cannot account for all age-related defects, it was striking to see how well FOXO inhibition can reproduce many of the age-associated changes observed after T cell activation. One of the most consistent findings in older T cell responses is their bias to differentiate into short-lived effector T cells. Data presented in this study attributed this bias to reduced FOXO1 activities and downstream GSK inhibition. We observed not only qualitative but also quantitative correlation between FOXO inhibition in young T cell activation and age-associated changes in several readout systems, such as increased LAMP1 (Fig. 2A) and β-catenin protein expression (Fig. 3C), emphasizing the relevance of this pathway.
The endolysosomal system is a dynamic membrane compartment crucial to the maintenance of proteostasis. It regulates signaling, secretion, recycling, and degradation of cellular protein and lipid components. After recyclable components are sorted back to the cell membrane or the trans-Golgi network, cargo destined for degradation is delivered to late endosomes. These proteins are incorporated into ILVs that bud from the limiting membrane to form multivesicular endosomes or MVBs. Stable lysosomal membrane proteins such as LAMP1 remain on the limiting membrane, a factor distinguishing MVBs from other organelles with internal membranes, such as autophagic bodies or multilamellar lysosomes (15). MVBs are acidic organelles that, in contrast to lysosomes, do not have proteolytic activity. They function as temporary storage tanks for proteins that are sorted either into exosomal release pathway or into lysosomal degradation pathway (15). Fusion of MVBs with lysosomes initiates the degradation of ILVs and their contents (15). If lysosomal function is inhibited, MVBs will expand (20) and enter the exosomal release pathway (21). Consistently, our data showed that lysosomal defects due to FOXO1 deficiency induce an expansion of MVBs, resulting in accumulation of LAMP1 and CD63 proteins (Fig. 2, A, B, and D). LysoTracker staining intensity reflecting the quantity of acidic organelles in cells is also increased after FOXO1 inhibition (Fig. 2C), indicating again the expansion of MVBs. Exosome secretion is enhanced following MVB expansion in FOXO1-inhibited cells (Fig. 2, E and F). These exosomes contain GSK3β (Fig. 3A). Consistent with their reduced FOXO1 activity, activated CD4+ T cells from old individuals resembled young cells treated with a FOXO1 inhibitor to prevent recovery of FOXO1 activity subsequent to activation.
In examining the functional consequences of MVB expansion, we focused on their ability to sequester GSK3β. GSK3 forms a destruction complex with Axin and adenomatous polyposis coli (APC) that cleaves β-catenin. In canonical Wnt signaling, this destruction complex is disassembled and β-catenin is released to accumulate and function as transcriptional regulator (32). Studies by De Robertis et al. (23) have shown that sequestration of GSK3β inside MVBs separates the kinase from its many cytosolic substrates including β-catenin and is required for Wnt signaling. Expansion of MVBs induced by lysosome inhibition resulted in a more efficient sequestration of GSK3 and increased Wnt responsiveness (20). Accordingly, FOXO1-inhibited T cells or T cells from old individuals with low FOXO1 activity had reduced intracellular GSK3β activity (Fig. 3B) due to the increase of MVB sequestration and exosomal release of GSK3β. Reduced GSK3β activity increased β-catenin stability but did not induce the transcriptional signatures of Wnt signaling in old activated naïve CD4+ T cells (Fig. 3, C and D) (13). A possible explanation is that FOXO1-deficient day 5–activated CD4+ naïve T cells preferentially expressed the Wnt inhibitory truncated instead of the Wnt stimulatory full-length TCF1 protein (fig. S8, A and B).
We have previously described that naïve CD4+ T cells from older individuals have a higher propensity to differentiate into short-lived effector T cells rather than memory or follicular helper cells (13). Our results here suggest that the reduced reexpression of FOXO1 and the associated increased sequestration of GSK3β contribute to this differentiation preference. Inhibition of FOXO1 or GSK3β activity in naïve CD4+ T cells from young individuals resulted in the preferential expression of PRDM1, perforin, and granzyme B (Fig. 5B and fig. S7C). Consistent with our findings in human CD4+ T cells, granzyme B production and cytotoxicity in CD8+ T and NK cells are increased following GSK3 inhibition under both in vitro and in vivo conditions (33, 34). GSK3β-mediated metabolic control may contribute to this differentiation bias. Effector T cells undergo a metabolic switch from oxidative phosphorylation to glycolysis, in part, to produce precursor molecules important for proliferation (35). In contrast, generation of long-lived memory T requires fatty acid oxidation. We observed an increase of glycolytic flux in old naïve CD4+ T cells after activation compared to that in young cells (Fig. 4H). This was, at least in part, due to the increased mitochondria translocation of HK-II induced by a loss in GSK3β activity (Fig. 4E). Our findings on preferential effector T cell differentiation are consistent with a previous study showing that impaired lysosomal function diverts T cell differentiation toward pro-inflammatory subsets and exacerbates the in vivo inflammatory response (36). However, our data appear to be in contrast to the observation that GSK3β inhibition favors the generation of CD8+ stem-like memory T cells (37). The GSK3 effect on differentiation is likely depending on the context in which GSK3 inhibition occurs. For inducing stem-like memory T cells, GSK3 was inhibited at the time of T cell activation, while we inhibited GSK3 at a later time point to mimic the increased expansion of MVBs occurring in activated old naïve T cells. Moreover, a recent study claimed that generation of memory stem cells is not due to the up-regulation of Wnt-dependent genes, indicating the existence of posttranscriptional mechanisms of regulation or of alternative signaling pathways other than GSK3/β-catenin pathway (38).
With increased cell size (Fig. 3F), increased glycogen storage and glycolytic flux (Fig. 4, D and H), and increased exosome production and secretion of pro-inflammatory and cytotoxic molecules (Fig. 5, C and I), activated older naïve CD4+ T cells with reduced GSK3 activity exhibit features reminiscent of senescent cells. Cell size is normally controlled by the balance between cell growth (mass accumulation) and proliferation (cell division) (39). Senescent cells become enlarged when they have arrested cell cycle but maintain protein synthesis. In contrast to truly senescent cells, the old naïve CD4+ T cells described here do not decrease cell division rate (fig. S9), nor do old naïve CD8+ T cells (40). Moreover, Tfeb-silenced SMARTA cells that mimic aged T cells also exhibit increased cell size (Fig. 6D) but comparable expansion after LCMV infection under in vivo (Fig. 6B). Loss of proteostasis following autophagy-lysosome inhibition alone has been shown to induce increased cell size in nonsenescent cells (41). Likewise, GSK3β-deficient old T cells undergo mass accumulation and become larger. GSK3 reduces the half-life of about 20% of total cellular proteins playing an important role in proteostasis. GSK3-mediated phosphorylation promotes protein ubiquitination and subsequent proteasomal degradation (23). It is likely that the increased cellular protein stability in GSK3β-deficient old CD4+ T cells leads to not only increased cell size but also the accumulation of damaged proteins that eventually induce cellular senescence. Previous reports have linked GSK3β inhibition to senescence-associated growth suppression (42); moreover, GSK3 inhibition–mediated glycogenesis is involved in cell senescence (43).
In addition to the defects in proteostasis, FOXO1-deficient old activated CD4+ T cells also share with senescent cells the increased production of exosomes. Production of pro-inflammatory cytokines and chemokines, as well as growth factors and proteases by senescent cells, a phenotype termed SASP (44), has been implicated in causing nonspecific cytotoxicity to the surrounding tissues as well as contributing to oncogenesis. Removal of senescent cells is currently explored to improve systemic symptoms associated with the aging process including SASP and to promote healthy life span (45). Similar to senescent cells, activated naïve CD4+ T cells from older individuals exert increased nonspecific cytotoxicity as shown in Fig. 5 as the example of B cells. This cytotoxicity might be of particular importance in lymph nodes where it could contribute to the defective B cell response with aging. This age-associated T cell defect is unlikely to be responsive to classical senolytics but could be targeted by restoring FOXO1 activity.
In summary, we identified a FOXO1/lysosome/MVB/GSK3β pathway that acts as an important regulator for the maintenance of proteostasis and the control of effector T cell differentiation. Aged naïve CD4+ T cells fail to restore FOXO1 activity after activation, resulting in the expansion of MVBs and sequestration of GSK3β. As a consequence, activated T cells acquire a senescence-like phenotype, including the secretion of granzyme B–containing exosomes causing nonspecific cytotoxicity. Targeting the FOXO1/lysosome/MVB/GSK3β pathway might serve as a strategy to counteract the defects associated with immune aging.
MATERIALS AND METHODS
Study population and cell isolation
Peripheral blood samples were obtained from 52 young (20 to 35 years old) and 49 old (65 to 85 years old) healthy individuals with no history of autoimmune disease or cancer and no uncontrolled renal disease, diabetes mellitus, or cardiovascular disease. Sixty-two of them (31 young-old pairs) were deidentified samples purchased from the Stanford Blood Center (Palo Alto, CA, USA) from donors younger than 35 years or older than 65 years. Thirty-nine samples were from individuals recruited from the Palo Alto area. The study was in accordance with the Declaration of Helsinki, approved by the Stanford Institutional Review Board, and all participants gave informed written consent. Naïve CD4+ T cells were purified with human CD4+ T cell enrichment cocktail (15062, STEMCELL Technologies), followed by negative selection with anti-CD45RO magnetic beads (19555, STEMCELL Technologies). Peripheral blood B cells were purified by positive selection with anti-CD19 microbeads (130-050-301, Miltenyi Biotec).
Cell culture
Isolated naïve CD4+ T cells were activated with Dynabeads Human T-Activator CD3/CD28 (11132D, Thermo Fisher Scientific) in RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) and penicillin and streptomycin (100 U/ml) (Thermo Fisher Scientific). For exosome-related experiments, cells were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% exosome-depleted FBS (A2720803, Thermo Fisher Scientific).
Western blotting
Cells were lysed in radioimmunoprecipitation assay buffer containing phenylmethylsulfonyl fluoride and protease and phosphatase inhibitors (sc-24948, Santa Cruz Biotechnology) for 30 min on ice. Proteins were separated on denaturing 4 to 15% SDS–polyacrylamide gel electrophoresis (4561086, Bio-Rad), transferred onto nitrocellulose membrane (1704270, Bio-Rad), and probed with antibodies to FOXO1 (2880, Cell Signaling Technology), TFEB (anti-human: ab220695, Abcam; anti-mouse: 32361, Cell Signaling Technology), LAMP1 (9091, Cell Signaling Technology), CD63 (ab68418, Abcam), CD9 (13403), ALIX (92880), GSK3β (12456), β-catenin (8480), phospho-glycogen synthase (Ser641) (3891), total glycogen synthase (3886), HK-II (2867), TCF1 (2206), granzyme B (17215), caspase-3 (14220), and β-actin (4970, all from Cell Signaling Technology). Membranes were developed using horseradish peroxidase–conjugated secondary antibodies (Cell Signaling Technology) and Chemiluminescent Western Blot Detection Substrate (Thermo Fisher Scientific).
Flow cytometry
For cell surface staining, cells were incubated with fluorescently conjugated antibodies in phosphate-buffered saline (PBS) containing 2% FBS at 4°C for 30 min. For intracellular cytokine assays, cells were restimulated with phorbol 12-myristate 13-acetate (PMA) (25 ng/ml; Sigma-Aldrich) and ionomycin (1 μg/ml; Sigma-Aldrich) in the presence of brefeldin A (GolgiPlug, BD Biosciences) for 4 hours at 37°C. Cells were then incubated with cell surface antibodies, permeabilized with Cytofix/Cytoperm kit (BD Biosciences), and stained with fluorescently labeled antibodies specific for the indicated cytokines. Annexin V apoptosis detection kit (BD Biosciences) was used to detect apoptotic cells. Dead cells were excluded from the analysis using LIVE/DEAD Fixable Aqua (eBioscience). The following fluorochrome-conjugated antibodies were used for flow cytometry: anti-CD4 (anti-human: RPA-T4; anti-mouse: RM4-5; BD Biosciences), anti-CD63 (anti-human: H5C6; anti-mouse: NVG-2; BD Biosciences), anti–granzyme B (GB11; BD Biosciences), and anti-FOXO1 (14262, Cell Signaling Technology). Cells were analyzed on an LSRII or LSRFortessa (BD Biosciences). Flow cytometry data were analyzed using FlowJo (TreeStar).
RNA isolation and quantitative RT-PCR
Total RNA was isolated using the RNeasy Plus Mini Kit (74134, Qiagen) and converted to complementary DNA (cDNA) using the SuperScript VILO cDNA Synthesis Kit (11754, Invitrogen). Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed on the ABI 7900HT System (Applied Biosystems) using Power SYBR Green PCR Master Mix (4368706, Thermo Fisher Scientific) according to the manufacturer’s instructions. Oligonucleotide primer sets are shown in table S1.
Gene set enrichment analysis
GSEA software from the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp) was used to determine the enrichment of gene sets in young (20 to 35 years old) or old (65 to 85 years old) T cells. The datasets describing age-associated differences in activated CD4+ T cells were obtained from the SRA database under accession number SRP158502 (19). Gene sets caused by Foxo1 deficiency were from GSE15037 (46).
Transfection
For FOXO1 silencing and TFEB rescue, naïve CD4+ T cells activated with Dynabeads for 48 hours were washed and cotransfected with either SMARTpool negative control siRNA (Dharmacon) and negative control DNA vector (pcDNA3.1), SMARTpool FOXO1 siRNA (Dharmacon) and negative control DNA vector, or SMARTpool FOXO1 siRNA and TFEB expression vector (99955, Addgene) using the Amaxa Nucleofector System and P3 Primary Cell Nucleofector Kit (Lonza). Cells were rested for 24 hours and cultured on plates coated with anti-CD3 (5 μg/ml) and anti-CD28 antibody (5 μg/ml) (eBioscience) for 2 days. Cells were then harvested and analyzed. For TFEB overexpression, resting naïve CD4+ T cells were transfected with a TFEB expression vector or a negative control vector using the Amaxa Nucleofector System and P3 Primary Cell Nucleofector Kit (Lonza). Cells were resting for 24 hours and stimulated with anti-CD3 and anti-CD28 beads for 5 days. Cells were then harvested and analyzed.
Pharmacologic inhibitors
A FOXO1 inhibitor (AS1842856, EMD Millipore) was used at a dose of 50 nM for all experiments. The AKT inhibitor MK-2206 2HCl was used at a dose of 0.5 μM. The lysosome inhibitors leupeptin (14026, Cayman Chemical) and bafilomycin A1 (11038, Cayman Chemical) were used at doses of 50 μM and 10 nM, respectively. An MVB inhibitor (U18666A, Cayman Chemical) was used at a dose of 2 μM. An exosome inhibitor (GW4869, Cayman Chemical) was used at a dose of 1 μM. The GSK3β inhibitors BIO (Cayman Chemical) and SB216763 (Sigma-Aldrich) were used at doses of 0.5 and 5 μM, respectively. 2DG (Sigma-Aldrich) was used at a dose of 2 mM. A β-catenin inhibitor (FH535, Cayman Chemical) was used at a dose of 10 μM.
DQ-BSA lysosomal activity assay and LysoTracker staining
For studying lysosomal proteolysis, activated naïve CD4+ T cells were treated with DQ-BSA (5 μg/ml; D12050, Thermo Fisher Scientific) diluted in prewarmed medium and incubated at 37°C for 5 hours. Cells were then briefly washed once with ice-cold PBS containing 2% FBS and kept on ice. Fluorescence of cleaved BSA-DQ was analyzed by flow cytometry. For quantitatively analyzing LysoTracker staining, naïve CD4+ T cells were treated with 100 nM LysoTracker (L7528, Thermo Fisher Scientific) diluted in prewarmed medium, incubated for 2 min at 37°C, and then washed with ice-cold PBS containing 2% FBS and kept on ice. LysoTracker fluorescence was analyzed and quantified by flow cytometry.
Confocal microscopy
Cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and incubated with primary antibodies to LAMP1 (9091, Cell Signaling Technology) and CD63 (556019, BD Pharmingen) at 4°C for overnight. Incubation with secondary antibodies was performed at room temperature for 2 hours using Alexa Fluor 488–conjugated AffiniPure Donkey anti-Rabbit immunoglobulin G (IgG) or Cy3-conjugated AffiniPure Donkey anti-Mouse IgG (Jackson ImmunoResearch Laboratories). The images were analyzed using an LSM 710 microscope system with the ZEN 2010 software (Carl Zeiss) and a 63× oil immersion objective (Carl Zeiss).
Exosome isolation
Naïve CD4+ T cells were activated with anti-CD3 and anti-CD28 beads for 4 days. After bead removal and washing, 1 million cells were cultured in 1-ml exosome-depleted medium for 24 hours. One-milliliter cell culture supernatant or 1-ml control medium containing 10% FBS was centrifuged at 2000g for 30 min to remove cells and debris. Exosomes were isolated from supernatants by the Total Exosome Isolation Reagent (4478359, Thermo Fisher Scientific) according to the manufacturer’s instructions. Exosomal protein contents were quantified by the Bradford assay.
In vitro B cell cytotoxic assays
Exosomes, collected from supernatants of 24-hour culture of 1 million day 4–activated naïve CD4+ T cells, were used to treat 1 million of freshly purified B cells. After 24 hours of treatment, B cells were collected and washed, and apoptotic B cells were detected by an Annexin V apoptosis detection kit (BD Biosciences). Caspase-3 activity was determined by immunoblotting using anti–caspase-3 (14220, Cell Signaling Technology) that recognizes both uncleaved and cleaved caspase-3.
Metabolic assays
Intracellular glycogen levels were measured using the Glycogen Assay Kit (700480, Cayman Chemical) according to the manufacturer’s instructions. To determine glucose uptake and lactate production, naïve CD4+ T cells were activated with anti-CD3 and anti-CD28 beads for 4 days, and beads were removed. After washing, 1 million cells were cultured in 1-ml fresh normal medium for 24 hours and lactate production was determined using the Lactate Colorimetric/Fluorometric Assay Kit (K607, BioVision). Glucose uptake in activated CD4+ T cells was determined by incubating cells in glucose-free medium containing 200 μM 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-d-glucose (2-NBDG) (11046, Cayman Chemical) for 2 hours at 37°C, followed by flow cytometry–based analysis.
Mitochondria and cytosol fractions
Twenty million cells were used to isolate mitochondria using the Mitochondria Isolation Kit for Cultured Cells (89874, Thermo Fisher Scientific) according to the manufacturer’s instructions. Cytosol fraction was obtained by collecting the supernatants after mitochondria pelleting.
Mice and adoptive transfers
Naïve CD4+ T cells specific to the GP66–77 epitope of LCMV obtained from SMARTA T cell receptor transgenic mice (CD45.1, a gift from R. Ahmed at the Emory University) were activated in plates coated with anti-CD3 antibody (8 μg/ml; 16-0032-82, eBioscience) and anti-CD28 antibody (8 μg/ml; 16-0281-82, eBioscience). Cells were transduced with a retroviral vector expressing either scrambled control RNA or Tfeb shRNA (5′-CGGCAGTACTATGACTATGAT-3′) on days 1 and 2 after activation. At day 6 after activation, retrovirus-transduced Amcyan+ SMARTA cells were isolated, and 2 × 105 transduced cells were intravenously transferred to naïve C57BL/6 (CD45.2) mice (The Jackson Laboratory). At day 3 after transfer, mice were infected intraperitoneally with 2 × 105 plaque-forming units of LCMV Armstrong. At day 5 after infection, spleens were harvested and analyzed. Experiments were approved by the Stanford Administrative Panel on Laboratory Animal Care Committee.
Statistical analysis
Statistical analysis was performed using the Prism 6.0 software (GraphPad Software Inc.). Paired or unpaired two-tailed Student’s t tests were used for comparing two groups. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was used for multigroup comparisons. Two-way ANOVA with Tukey’s post hoc test was used for multigroup comparisons when studying gene expressions of different cathepsins in three different transfection groups. A two-tailed Pearson’s correlation test was used for correlation analysis. P < 0.05 was considered statistically significant. Statistical details and significance levels can be found in the figure legends.
Supplementary Material
Acknowledgments
Funding: This work was supported by the NIH (R01 AR042527, R01 HL117913, R01 AI108906, R01 HL142068, and P01 HL129941 to C.M.W. and R01 AI108891, R01 AG045779, U19 AI057266, and R01 AI129191 to J.J.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Author contributions: J.J., C.M.W., and J.J.G. designed the study. J.J., X.L., B.H., C.K., W.C., and H.Z. performed the experiments. J.J., C.M.W., and J.J.G. analyzed and interpreted the data. J.J. and J.J.G. wrote the manuscript with all authors providing feedback. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaba1808/DC1
REFERENCES AND NOTES
- 1.Nikolich-Žugich J., The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Haynes L., Swain S. L., Why aging T cells fail: Implications for vaccination. Immunity 24, 663–666 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Czesnikiewicz-Guzik M., Lee W. W., Cui D., Hiruma Y., Lamar D. L., Yang Z. Z., Ouslander J. G., Weyand C. M., Goronzy J. J., T cell subset-specific susceptibility to aging. Clin. Immunol. 127, 107–118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wertheimer A. M., Bennett M. S., Park B., Uhrlaub J. L., Martinez C., Pulko V., Currier N. L., Nikolich-Žugich D., Kaye J., Nikolich-Žugich J., Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J. Immunol. 192, 2143–2155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akbar A. N., Henson S. M., Lanna A., Senescence of T Lymphocytes: Implications for enhancing human immunity. Trends Immunol. 37, 866–876 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Callender L. A., Carroll E. C., Beal R. W. J., Chambers E. S., Nourshargh S., Akbar A. N., Henson S. M., Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 17, e12675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallejo A. N., CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 205, 158–169 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Goronzy J. J., Weyand C. M., Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 19, 573–583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goronzy J. J., Hu B., Kim C., Jadhav R. R., Weyand C. M., Epigenetics of T cell aging. J. Leukoc. Biol. 104, 691–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.López-Otin C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webb A. E., Brunet A., FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedrick S. M., Hess Michelini R., Doedens A. L., Goldrath A. W., Stone E. L., FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 12, 649–661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim C., Hu B., Jadhav R. R., Jin J., Zhang H., Cavanagh M. M., Akondy R. S., Ahmed R., Weyand C. M., Goronzy J. J., Activation of miR-21-regulated pathways in immune aging selects against signatures characteristic of memory T cells. Cell Rep. 25, 2148–2162.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., Ballabio A., A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Piper R. C., Katzmann D. J., Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23, 519–547 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Essaghir A., Dif N., Marbehant C. Y., Coffer P. J., Demoulin J.-B., The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J. Biol. Chem. 284, 10334–10342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L., Tao Z., Zheng L. D., Brooke J. P., Smith C. M., Liu D., Long Y. C., Cheng Z., FoxO1 interacts with transcription factor EB and differentially regulates mitochondrial uncoupling proteins via autophagy in adipocytes. Cell Death Discov. 2, 16066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ploper D., Taelman V. F., Robert L., Perez B. S., Titz B., Chen H.W., Graeber T. G., von Euw E., Ribas A., De Robertis E. M., MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. U.S.A. 112, E420–E429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu B., Li G., Ye Z., Gustafson C. E., Tian L., Weyand C. M., Goronzy J. J., Transcription factor networks in aged naïve CD4 T cells bias lineage differentiation. Aging Cell 18, e12957 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobrowolski R., Vick P., Ploper D., Gumper I., Snitkin H., Sabatini D. D., de Robertis E. M., Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2, 1316–1328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eitan E., Suire C., Zhang S., Mattson M. P., Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 32, 65–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verweij F. J., Bebelman M. P., Jimenez C. R., Garcia-Vallejo J. J., Janssen H., Neefjes J., Knol J. C., de Goeij-de Haas R., Piersma S. R., Baglio S. R., Verhage M., Middeldorp J. M., Zomer A., van Rheenen J., Coppolino M. G., Hurbain I., Raposo G., Smit M. J., Toonen R. F. G., van Niel G., Pegtel D. M., Quantifying exosome secretion from single cells reveals a modulatory role for GPCR signaling. J. Cell Biol. 217, 1129–1142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taelman V. F., Dobrowolski R., Plouhinec J. L., Fuentealba L. C., Vorwald P. P., Gumper I., Sabatini D. D., De Robertis E. M., Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newton R. H., Shrestha S., Sullivan J. M., Yates K. B., Compeer E. B., Ron-Harel N., Blazar B. R., Bensinger S. J., Haining W. N., Dustin M. L., Campbell D. J., Chi H., Turka L. A., Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat. Immunol. 19, 838–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jellusova J., Cato M. H., Apgar J. R., Ramezani-Rad P., Leung C. R., Chen C., Richardson A. D., Conner E. M., Benschop R. J., Woodgett J. R., Rickert R. C., Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 18, 303–312 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeisbrich M., Yanes R. E., Zhang H., Watanabe R., Li Y., Brosig L., Hong J., Wallis B. B., Giacomini J. C., Assimes T. L., Goronzy J. J., Weyand C. M., Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann. Rheum. Dis. 77, 1053–1062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oreña S. J., Torchia A. J., Garofalo R. S., Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J. Biol. Chem. 275, 15765–15772 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Pastorino J. G., Hoek J. B., Shulga N., Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Mathupala S. P., Ko Y. H., Pedersen P. L., Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 19, 17–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sukumar M., Liu J., Ji Y., Subramanian M., Crompton J. G., Yu Z., Roychoudhuri R., Palmer D. C., Muranski P., Karoly E. D., Mohney R. P., Klebanoff C. A., Lal A., Finkel T., Restifo N. P., Gattinoni L., Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 123, 4479–4488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buck M. D., O’Sullivan D., Klein Geltink R. I., Curtis J. D., Chang C.H., Sanin D. E., Qiu J., Kretz O., Braas D., van der Windt G. J. W., Chen Q., Huang S. C.-C., O’Neill C. M., Edelson B. T., Pearce E. J., Sesaki H., Huber T. B., Rambold A. S., Pearce E. L., Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nusse R., Clevers H., Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Taylor A., Harker J. A., Chanthong K., Stevenson P. G., Zuniga E. I., Rudd C. E., Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of Co-receptor PD-1 to enhance CD8+ cytolytic T cell responses. Immunity 44, 274–286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cichocki F., Valamehr B., Bjordahl R., Zhang B., Rezner B., Rogers P., Gaidarova S., Moreno S., Tuininga K., Dougherty P., McCullar V., Howard P., Sarhan D., Taras E., Schlums H., Abbot S., Shoemaker D., Bryceson Y. T., Blazar B. R., Wolchko S., Cooley S., Miller J. S., GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 77, 5664–5675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman N. M., Boothby M. R., Chi H., Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 20, 55–70 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Baixauli F., Acín-Pérez R., Villarroya-Beltrí C., Mazzeo C., Nuñez-Andrade N., Gabandé-Rodriguez E., Ledesma M. D., Blázquez A., Martin M. A., Falcón-Pérez J. M., Redondo J. M., Enríquez J. A., Mittelbrunn M., Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gattinoni L., Zhong X. S., Palmer D. C., Ji Y., Hinrichs C. S., Yu Z., Wrzesinski C., Boni A., Cassard L., Garvin L. M., Paulos C. M., Muranski P., Restifo N. P., Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cieri N., Camisa B., Cocchiarella F., Forcato M., Oliveira G., Provasi E., Bondanza A., Bordignon C., Peccatori J., Ciceri F., Lupo-Stanghellini M. T., Mavilio F., Mondino A., Bicciato S., Recchia A., Bonini C., IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121, 573–584 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Lloyd A. C., The regulation of cell size. Cell 154, 1194–1205 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Quinn K. M., Fox A., Harland K. L., Russ B. E., Li J., Nguyen T. H. O., Loh L., Olshanksy M., Naeem H., Tsyganov K., Wiede F., Webster R., Blyth C., Sng X. Y. X., Tiganis T., Powell D., Doherty P. C., Turner S. J., Kedzierska K., La Gruta N. L., Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 23, 3512–3524 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Miettinen T. P., Björklund M., Mevalonate pathway regulates cell size homeostasis and proteostasis through autophagy. Cell Rep. 13, 2610–2620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y. M., Song I., Seo Y. H., Yoon G., Glycogen synthase kinase 3 inactivation induces cell senescence through sterol regulatory element binding protein 1-mediated lipogenesis in chang cells. Endocrinol. Metab. 28, 297–308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo Y.-H., Jung H.-J., Shin H.-T., Kim Y.-M., Yim H., Chung H.-Y., Lim I. K., Yoon G., Enhanced glycogenesis is involved in cellular senescence via GSK3/GS modulation. Aging Cell 7, 894–907 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Herranz N., Gil J., Mechanisms and functions of cellular senescence. J. Clin. Invest. 128, 1238–1246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Deursen J. M., The role of senescent cells in ageing. Nature 509, 439–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouyang W., Beckett O., Flavell R. A., Li M. O., An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 30, 358–371 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/17/eaba1808/DC1