

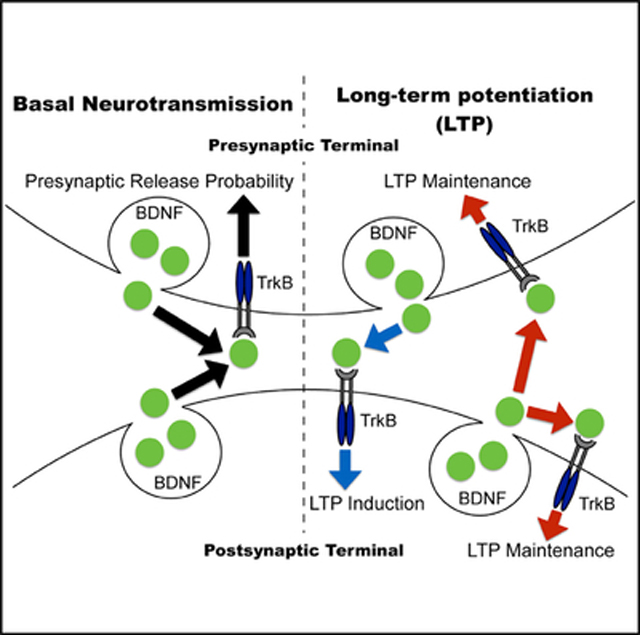
SUMMARY
Brain-derived neurotrophic factor (BDNF) and its high-affinity receptor, tropomyosin receptor kinase B (TrkB), regulate long-term potentiation (LTP) in the hippocampus, although the sites of BDNF-TrkB receptors in this process are controversial. We used a viral-mediated approach to delete BDNF or TrkB specifically in CA1 and CA3 regions of the Schaffer collateral pathway. Deletion of BDNF in CA3 or CA1 revealed that presynaptic BDNF is involved in LTP induction, while postsynaptic BDNF contributes to LTP maintenance. Similarly, loss of presynaptic or postsynaptic TrkB receptors leads to distinct LTP deficits, with presynaptic TrkB required to maintain LTP, while postsynaptic TrkB is essential for LTP formation. In addition, loss of TrkB in CA3 significantly diminishes release probability, uncovering a role for presynaptic TrkB receptors in basal neurotransmission. Taken together, this direct comparison of presynaptic and postsynaptic BDNF-TrkB reveals insight into BDNF release and TrkB activation sites in hippocampal LTP.
In Brief
Lin et al. directly compare a role for presynaptic and postsynaptic BDNF and TrkB receptors in hippocampal LTP. They find that LTP induction is mediated by anterograde BDNF-TrkB signaling, while both anterograde and retrograde BDNFTrkB signaling persists presynaptically and postsynaptically for LTP maintenance.
Graphical Abstract
INTRODUCTION
Brain-derived neurotrophic factor (BDNF) and its high-affinity receptor, tropomyosin receptor kinase B (TrkB), regulate certain forms of synaptic plasticity including long-term potentiation (LTP) and effect synaptic transmission (Hohn et al., 1990; Park and Poo, 2013; Thoenen, 1995). The induction of LTP refers to the transient events serving to trigger the formation of LTP. The maintenance of LTP refers to the persisting biochemical signal that lasts in the cell (Sweatt, 1999). Early studies have shown that homozygous and heterozygous BDNF knockout mice exhibit significant deficits in LTP at CA1 hippocampal synapses (Korte et al., 1995; Patterson et al., 1996), which could be rescued with exogenous BDNF (Patterson et al., 1996) or viral expression of BDNF (Korte et al., 1996). These lines of evidence suggest that the LTP deficits are not due to developmental defects but rather point to a functional role of BDNF in hippocampal LTP (Korte et al., 1996). To specifically dissect a role of BDNF in LTP of adult brain, we previously generated an inducible BDNF knockout mouse line in which BDNF is selectively deleted in adult forebrain and found significant impairments in hippocampal LTP as well as learning and memory deficits (Monteggia et al., 2004). While loss of BDNF in hippocampus impairs hippocampal LTP, the effects on basal synaptic transmission have not been as consistent (Korte et al., 1995; Patterson et al., 1996). Homozygous BDNF-null mice have been reported to have impaired induction of high-frequency transmission coupled with a significant reduction in the number of docked vesicles at active zones in the CA1 region (Pozzo-Miller et al., 1999). Previous studies have also shown that loss of TrkB function in hippocampus impacts LTP. For example, blocking TrkB activation with TrkB antiserum (TrkB-Ab) has been shown to attenuate LTP in the hippocampus (Kang et al., 1997). Characterization of conditional TrkB knockout mice, in which the gene was deleted during postnatal development in forebrain, revealed deficits in hippocampal LTP (Minichiello et al., 1999). While these data established critical roles of BDNF-TrkB in hippocampal LTP, the origin of BDNF release and the site of TrkB activation for hippocampal LTP have been controversial.
BDNF and TrkB are expressed in presynaptic CA3 and postsynaptic CA1 neurons (Baj et al., 2012; Minichiello, 2009). Previous work using mutant mice that lack BDNF in CA3-CA1, or selectively in CA1, revealed that high-frequency stimulation (HFS)- and theta burst stimulation (TBS)-induced LTP require presynaptic BDNF release from CA3 (Zakharenko et al., 2003). However, similar LTP induction protocols failed to evoke LTP in a BDNF mutant mouse in which dendritic BDNF was absent from CA1 dendritic localization (An et al., 2008). BDNF can enhance basal synaptic strength through TrkB activation potentially via presynaptic and postsynaptic mechanisms, although there have been conflicting data on how this occurs (Kang and Schuman, 1995a; Levine et al., 1995; Magby et al., 2006; Minichiello, 2009; Park and Poo, 2013). In regard to TrkB, concurrent blockade of presynaptic and postsynaptic of TrkB-mediated PLCγ signaling produces a similar impairment in LTP (Gärtner et al., 2006), as exhibited by conditional TrkB knockout mice (Minichiello et al., 1999). Nevertheless, blockade of either presynaptic or postsynaptic TrkB-mediated PLCg signaling alone did not interfere with LTP (Gärtner et al., 2006).
While there is evidence for both presynaptic and postsynaptic release for BDNF (Dieni et al., 2012; Matsuda et al., 2009) coupled with a concurrent presynaptic and postsynaptic role for TrkB (Alder et al., 2005; Chen et al., 1999) in LTP, there has not been a direct comparison examining the selective loss of BDNF in the CA3 or CA1 region with mice lacking TrkB in either presynaptic or postsynaptic neurons. In this study, we use a viral-mediated approach to genetically delete BDNF or TrkB selectively in either the CA3 or CA1 and examine its impact on synaptic plasticity and basal neurotransmission. This approach allows for the identification of the origin of BDNF release and its site of TrkB action in hippocampal LTP.
RESULTS
Localized Region-Specific Deletion of BDNF and TrkB in Subregions of Hippocampus
To examine the site of BDNF release in the hippocampus during LTP and basal synaptic transmission, and to identify the role of presynaptic and postsynaptic TrkB receptors during these processes, we performed stereotaxic injection of adeno-associated virus (AAV) expressing Cre recombinase tagged to GFP (AAV-GFP-Cre) bilaterally into either the CA3 (presynaptic neurons) or the Placements of the stereotaxic injections were verified in each mouse at the completion of experiments by checking GFP expression in hippocampal slices (Figures 1A–1C and S1), and BDNF or TrkB protein expression in CA3 or CA1 via western blotting. Only mice with targeted CA3 or CA1 placements were included in the analysis. Western blot analysis showed that BDNF protein expression was significantly reduced to 30.1% ± 4.7% or 26.5% ± 4.9% in Bdnffl/fl mice injected with AAV-GFP-Cre in the CA3 or CA1, compared to that of their respective AAV-GFP-injected controls (Figure 1D). The Ntrk2fl/fl mice injected with AAVGFP-Cre displayed a significant reduction to 22.2% ± 7.0% or 18.3% ± 5.5% in TrkB protein expression in the CA3 or CA1 region as compared to that of the respective AAV-GFP-injected mice (Figure 1E). CA1 (postsynaptic neurons) of the Schaffer collateral pathway in the dorsal hippocampus of 12-week-old Bdnffl/fl mice (Rios et al., 2001) or Ntrk2fl/fl mice (Luikart et al., 2005) (Figures 1A–1C). Littermate Bdnffl/fl or Ntrk2fl/fl mice injected with AAV-GFP bilaterally into either the CA3 or CA1 regions served as controls. Animals were utilized 3 weeks after surgery for electrophysiology and behavioral experiments, a time point in which Cre-mediated recombination occurs in brain (Adachietal.,2017;Bertonetal.,2006).Using this method, we specifically deleted BDNF or TrkB bilaterally in either the CA3 or CA1 subregion of adult hippocampus to precisely target the endogenous gene and avoid the potential confound of developmental defects.
Figure 1. Localized Deletion of BDNF or TrkB in the Hippocampus.

(A and B) Representative images of GFP-expressing neurons in the (A) CA3 (presynaptic terminal of Schaffer collateral pathway) or (B) CA1 (postsynaptic area of Schaffer collateral pathway) of Bdnffl/fl mice or Ntrk2fl/fl mice.
(C) Schematic diagram of the approximate injection site in the CA1 and CA3 regions.
(D and E) Representative western blot images of mature BDNF and TrkB expression in Bdnffl/fl or Ntrk2fl/fl mice injected with AAV-GFP or AAV-GFP-Cre. (D) BDNF protein expression in CA1 (n = 8) or CA3 (n = 7) of AAV-GFP-Cre-injected Bdnffl/fl mice was significantly reduced to 26.5% ± 4.9% and 30.1% ± 4.7% of AAV-GFP-injected Bdnffl/fl mice (n = 8 and n = 9, respectively). (E) TrkB protein expression in CA1 (n = 9) or CA3 (n = 8) of AAV-GF-PCre-injected Ntrk2fl/fl mice was significantly reduced to 18.3% ± 5.5% and 22.2% ± 7.0% of AAV-GFP-injected Ntrk2fl/fl mice (n = 8 for each).
Data are mean ± SEM.
Impact of Localized Deletion of BDNF or TrkB on LTP
To assess LTP and basal synaptic transmission, the CA3-CA1 Schaffer collateral afferents were stimulated and field excitatory postsynaptic potentials (fEPSPs) were recorded from hippocampal sliced of the CA1 region in mice 3 weeks after stereotaxic surgery (Figure 2A). In mice with deletion of BDNF in the CA3 region, HFS induced a stable induction of LTP in hippocampal slices with a significant decrease in the magnitude of LTP compared to GFP-injected control slices (Figure 2B). In contrast, selective BDNF deletion in the CA1 region resulted in post-stimulus potentiation indistinguishable from that of GFP-injected mice; however, it was not maintained following the induction phase (Figure 2C). There were no differences in fEPSPs slopes induced by each pulse during HFS from mice with BDNF deletion in either the CA3 or CA1 region compared to GFP-injected mice, demonstrating that the deficits in LTP were not associated with altered glutamatergic neurotransmission (Figure S2). Taken together, these data suggest presynaptic (CA3) BDNF contributes to the induction of LTP while postsynaptic (CA1) BDNF is required for the maintenance of LTP at CA3-CA1 synapses.
Figure 2. Region-Specific Deletion of BDNF Impairs LTP.

(A) Experimental protocol for recording field excitatory postsynaptic potential (fEPSP) from CA1 while stimulating the CA3-CA1 Schaffer collateral afferents. Baseline responses were collected for 20min.Pairedpulse stimulation was applied to harvest the ratio of the response to the second stimulus over response to the first stimulus (P2/P1 in Figures 4 and 5). Paired-pulse stimulation was applied before baseline recording and after 1-hr LTP recording.
(B) LTP induced by HFS (filled triangle) in hippocampal slices (n = 7) with deletion of BDNF in the CA3 region was stable but significantly smaller than in hippocampal slices (n = 9) of AAV-GFP-injected Bdnffl/fl mice (two-way ANOVA: F(153, 2,156) = 26.43, p < 0.0001 for group, Sidak’s post hoc tests for least 10 min: p < 0.001). Inset, representative waveforms from AAV-GFP- and AAV-GFP-Cre-injected slices recorded baseline (1) and after HFS (2).
(C) HFS induced a similar level of post-tetanic potentiation in hippocampal slices (n = 8) with CA1 BDNF deletion immediately following HFS, compared to AAV-GFP-injected Bdnffl/fl mice, but responses decreased drastically in the following 10 min, and potentiation was not maintained during the rest of the 40- to 50-min recording (two-way ANOVA: F(143, 2,011) = 15.91, p < 0.0001 for group, Sidak’s post hoc tests for last 10 min: p < 0.001). Inset, representative waveforms from AAV-GFP-and AAV-GFP-Cre-injected slices recorded baseline (1) and after HFS (2).
Data are mean ± SEM.
To examine the function of presynaptic or postsynaptic TrkB receptors in hippocampal LTP, we performed similar electrophysiological analysis in hippocampal slices from stereotaxic injected mice in which the CA3-CA1 afferents were stimulated and fEPSPs recorded from the CA1 region (Figure 3A). Mice with selective deletion of TrkB in the CA3 region displayed an initial induction of LTP but a failure to maintain it compared to GFP-injected mice (Figure 3B). In contrast, selective deletion of TrkB in the CA1 region resulted in an attenuated induction and maintenance of LTP compared to GFP control-injected mice (Figure 3C). Similar to the BDNF findings, selective deletion of TrkB from either the CA3 or CA1 subregion did not alter the slope of fEPSPs induced by each pulse during HFS of hippocampal slices compared to GFP-injected mice, suggesting that the LTP deficits were not due to alterations in baseline glutamatergic neurotransmission (Figure S3). Collectively, these data show that presynaptic (CA3) TrkB is required for LTP maintenance, while postsynaptic (CA1) TrkB signaling is essential to fully induce LTP.
Figure 3. Region-Specific Deletion of TrkB Affects LTP Formation.

(A) Experimental protocol.
(B) HFS (filled triangle) initiated the same level of potentiation in hippocampal slices (n = 8) with deletion of TrkB in the CA3, compared to Ntrk2fl/fl mice injected with AAV-GFP (n = 8). However, this potentiation was not maintained. Responses were gradually decreased after HFS with a significant difference in the responses in the last 10 min of recording between hippocampal slices from AAV-GFP and AAV-GFP-Cre CA3-injected Ntrk2fl/fl mice (two-way ANOVA: F(150, 2,114) = 18.93, p < 0.0001 for group; Sidak’s post hoc tests for least 10 min: p < 0.001). Inset, representative waveforms from AAV-GFP- and AAV-GFP-Cre-injected slices recorded baseline (1) and after HFS (2).
(C) Compared to LTP induced in hippocampal slices (n = 8) of AAV-GFP-injected Ntrk2fl/fl mice (GFP), deletion of TrkB from CA1 (n = 9) nearly blocked LTP formation (two-way ANOVA, F(154, 2,325) = 14.45 for group, p < 0.0001, Sidak’s post hoc tests for the last 10 min: p < 0.001). Inset, representative waveforms from AAV-GFP- and AAV-GFP-Cre-injected slices recorded baseline (1) and after HFS (2).
Data are mean ± SEM.
Loss of Presynaptic TrkB Results in Decreased Neurotransmitter Release Probability
To assess basal neurotransmission, we applied paired-pulse stimulation to hippocampal slices from mice with a selective deletion of BDNF or TrkB in either the CA3 or CA1 region and plotted paired-pulse ratios (response to the second stimulus/ response to the first stimulus [P2/P1]) (Figure 4A). Synaptic responses to a pair of closely spaced stimuli can show facilitation or depression depending on the initial neurotransmitter release probability. We found mice with selective deletion of BDNF in either the CA3 or CA1 had indistinguishable paired-pulse ratios (PPRs) compared to the GFP-injected controls (Figure 4B). Previous work reported that BDNF homozygous and heterozygous knockout mice display reductions in maximal fEPSP slope and paired-pulse facilitation (PPF), suggesting that loss of BDNF impairs basal neurotransmission (Patterson et al., 1996). To reconcile our current findings with the previous literature, we applied paired-pulse stimulation to hippocampal slices from an inducible Bdnf knockout mouse line in which BDNF is reduced ~70% throughout the hippocampus and cortex (Monteggia et al., 2004). The inducible Bdnf knockout (KO) mice exhibited a significantly higher PPR than littermate controls (Figure 4C) suggestive of a decrease in release probability, which is in agreement with prior work with homozygous and heterozygous BDNF KO mice (Patterson et al., 1996). To examine whether the findings obtained with the inducible Bdnf KO mice were impacted by developmental compensation, we injected AAV-GFP-Cre in both the CA1 and CA3 regions of adult Bdnffl/fl mice and found enhanced PPR compared to GFP-injected mice, indicating a reduction of presynaptic release probability (Figures 4D and S4). Collectively, these data demonstrate that loss of BDNF in both presynaptic and postsynaptic neurons results in alteration in release probability while sparing BDNF expression in either presynaptic or postsynaptic neurons compensates for the regulation of basal neurotransmission.
Figure 4. Deletion of Presynaptic TrkB Alters Release Probability.

(A) Experimental protocol.
(B) Compared to GFP controls, paired-pulse ratio (PPR) (P2/P1) before LTP was not altered by deletion of BDNF in either CA1 or CA3 (two-way ANOVA: F(2,192) = 12.21, p = 0.3474 for group; F(7,192) = 22.1, p < 0.0001 for interstimulus interval).
(C) PPR was increased in hippocampal slices (n = 7) of Bdnf knockout (KO) mice, compared to littermate CTLs (n = 7) (two-way ANOVA: F(1,88) = 24.78, p < 0.0001 for group; F(7,208) = 12.81, p < 0.0001 for interstimulus interval; Sidak’s post hoc tests: CTL versus KO, 20 ms, p = 0.0388; 30 ms, p = 0.0006; 50 s, p = 0.0054; 100 ms, p = 0.0491).
(D) Deletion of BDNF in both CA1 and CA3 regions increased PPR (two-way ANOVA: F(1,184) = 40.97, p < 0.0001 for group; F(7,184) = 22.23, p < 0.0001 for interstimulus interval: Sidak’s post hoc tests: GFP versus GFP-Cre, 20 ms, p < 0.0001; 30 ms, p = 0.0046; 50 ms, p = 0.0194) before LTP. Representative traces show pulse 1 following by pulse 2 with 20-ms interstimulus interval.
(E) Compared to GFP controls, deletion of TrkB in CA1 had no impact on PPR before LTP; however, selectively deleting TrkB from CA3 enhanced PPR (two-wayANOVA: F(2,208) = 57.4, p < 0.0001 for group; F(7,208) = 35.64, p < 0.0001 for interstimulus interval; Sidak’s post hoc tests: CTL versus GFP-Cre Ntrk2fl/fl -CA3, 20 ms, p = 0.0003; 30 ms, p = 0.0001; 50 ms, p < 0.0001; 100 ms, p < 0.0001; 200 s, p = 0.0008) before LTP. Representative traces show pulse 1 following by pulse 2 with 20-ms interstimulus interval.
(F) Top, To measure release probability, NMDA-fEPSP was recorded from the CA3-CA1 Schaffer collateral pathway, using 0.1-Hz stimulation, before and afterMK-801 bath application. Traces show responses of the 1st and the 15th stimulus. NMDA-fEPSP amplitudes evoked every 10 s in the presence of MK-801 decayed slower in slices with deletion of TrkB in CA3 (n = 7) compared to GFP (n = 8).
(G) Rate constant of NMDA-fEPSP amplitudes in the presence of MK-801 was significantly lower in slices with deletion of TrkB in CA3 than GFP (t(13) = 3.631, p = 0.003).
*p < 0.05. Data are mean ± SEM.
We next applied paired-pulse stimulation to mice with a localized deletion of TrkB in the CA3 or CA1 of the hippocampus. The selective loss of TrkB in the CA3 region resulted in a significant enhancement in PPR, suggesting a decrease in presynaptic release probability (Figure 4E). In contrast, the loss of TrkB in the CA1 region did not alter the PPR compared to GFP-injected mice (Figure 4D). To explore a potential role for presynaptic TrkB receptors in neurotransmitter release, we perfused MK-801, a use-dependent blocker of NMDA (N-methyl-D-aspartate) receptors, onto hippocampal slices and measured NMDA receptor-mediated fEPSPs (Figure 4F). In this experimental paradigm, a lower release probability would be expected to lead to a slower block of NMDA receptor-mediated responses by MK-801 (Hessler et al., 1993). We found a selective loss of TrkB from the CA3 region resulted in a significant reduction in the rate of NMDA-fEPSP block by MK-801 (Figure 4G). Collectively, data from the paired-pulse experiments and the MK-801 studies demonstrate that presynaptic TrkB receptors are required to maintain normal presynaptic release probability.
Deletion of Presynaptic TrkB Unveiled a Presynaptic Component of LTP
We next examined potential changes in neurotransmitter release before and after LTP by applying paired-pulse stimulation before and after LTP and analyzing the PPRs (Figure 5A). Changes in PPR suggest the involvement of presynaptic components in LTP (Schulz et al., 1994). GFP-injected mice in either the CA3 or CA1 subregion showed no alterations in the PPRs before or after LTP, showing that this experimental protocol does not shift baseline synaptic transmission (Figures 5B and 5E). PPR was also not altered after LTP induction in CA3 or CA1 BDNF deletion hippocampal slices compared to the ratios before LTP (Figures 5C and 5D). In contrast, after LTP, the selective deletion of TrkB in the CA3 region significantly reduced the PPRs (Figure 5F), which was elevated during basal conditions (Figure 4D). Thus, while our data show that loss of presynaptic TrkB leads to a decrease in baseline presynaptic release probability, the induction of LTP under the same conditions appears to augment release and partially rescue this phenotype. In mice with a selective deletion of TrkB in the CA1, there was no difference in PPRs before or after LTP, further corroborating the regional differences of presynaptic and postsynaptic TrkB in LTP (Figure 5G).
Figure 5. Deletion of Presynaptic TrkB Unmasks a Presynaptic Component of LTP.

(A) Experimental protocol.
(B) PPF was unaltered before and after LTP in hippocampal slices of AAV-GFP injected Bdnffl/fl mice (F(1,88) = 0.3372, p = 0.5625).
(C and D) No difference was found before and after LTP in the hippocampal slices of either (C) AAV-GFP-Cre CA3-injected Bdnffl/fl mice (F(1,56) = 3.332, p = 0.0703) or (D) AAV-GFP-Cre CA1-injected Bdnffl/fl mice (F(1,48) = 1.471, p = 0.2276).
(E and G) There was no significant difference between PPR before and after LTP in (E) AAV-GFP-injected Ntrk2fl/fl controls (F(1,88) = 0.2463, p = 0.6203) or (G) AAVGFP-Cre CA1-injected Ntrk2fl/fl mice (F(1,64) = 0.2967, p = 0.5867).
(F) However, a significant reduction of PPR after LTP was observed in hippocampal slices of AAV-GFP-Cre CA3-injected Ntrk2fl/fl mice (F(1,56) = 28.56, p < 0.0001, post hoc tests: before versus after LTP, 30 ms, p = 0.0418; 100 ms, p = 0.0136; 200 ms, p = 0.0361). Representative traces show pulse 1 following by pulse 2 with 20-ms interstimulus interval.
*p < 0.05. Data are mean ± SEM.
Deletion of TrkB from CA3 or CA1 Results in Impaired Hippocampal-Dependent Memory
Our data to this point identified a rather surprising role of presynaptic TrkB receptors in synaptic transmission. Therefore, we examined whether the deletion of TrkB selectively in CA3 impacts hippocampal-dependent learning and memory. We also tested mice with a selective deletion of TrkB in CA1, given the importance of postsynaptic TrkB in LTP induction. Mice were assessed in behavior 3 weeks following surgery. Locomotor activity was assessed as the total number of horizontal beam breaks during a 60-min testing period. There was no difference in ambulation across the 60-min testing period between the mice with CA3 or CA1 TrkB deletion and controls with GFP injection (inset, Figure 6A). We further analyzed the locomotor activity data in 5-min time intervals and found no significant difference in the number of beam breaks at any of the time points examined (Figure 6A). To assess hippocampal-dependent learning and memory, we tested mice in the fear conditioning paradigm and the novel object recognition task. In the fear conditioning paradigm, mice lacking TrkB in CA3 or CA1 displayed similar baseline freezing compared to GFP-injected mice (data not shown). Twenty-four hours after training, we examined context-dependent fear-conditioned response and found no significant difference in freezing behavior in mice with a selective loss of TrkB in CA3 or CA1 compared with GFP-injected mice (Figure 6B). Similarly, cue-dependent fear-conditioned responses assessed 4 hr later in a novel context with tone only was not altered across different groups (Figure 6B). The deletion of TrkB in CA3 or CA1 did not alter responses to footshock intensities compared to GFP-injected mice (data not shown). We also assessed mice in the novel object recognition task, a single-trial memory test that examines the ability to discriminate a novel object from a familiar object (Bevins and Besheer, 2006). There was no difference in the total amount of time spent with the objects during the familiarization period for all groups of mice (data not shown). The following day, one of the familiar objects was replaced with a novel object. Deletion of TrkB in CA3 or CA1 resulted in less preference (lower discrimination index) for the novel object relative to the familiar object compared to GFP-injected mice, suggesting a deficit in novel object recognition (Figures 6C and 6D).
Figure 6. Mice with Deletion of TrkB from CA1 or CA3 Exhibit Deficits in Hippocampal-Dependent Memory.

(A) Deletion of TrkB from CA1 or CA3 did not alter locomotor activity. The total number of beam breaks (inset) was similar between Ntrk2fl/fl mice receiving AAV-GFP (CTL, n = 10) or AAV-GFP-Cre into either CA1 (n = 9) or CA3 (n = 10) (F(2,26) = 0.07179, p = 0.9310). There was also no significant difference in ambulation measured over the 1-hr testing period.
(B) Context-dependent fear conditioning was tested 24 hr after training, and cue-dependent fear was assessed 4 hr after contextual fear conditioning. Deletion of TrkB from CA1 or CA3 did not result in significant deficits (F(2,52) = 1.781, p = 0.1777).
(C) Ntrk2fl/fl mice that received AAV-GFP-Cre into either CA1 or CA3 spent less time exploring the novel object compared to GFP controls in the novel object recognition test.
(D) A significant reduction of discrimination index([time spent on novel object – time spent on familiar object]/total time) was found in the mice with deletion of TrkB in either CA1 or CA3 compared to GFP controls (F(2,26) = 4.66, p = 0.0186; post hoc tests: CTL versus CA1, p = 0.0383; CTL versus CA3, p = 0.0345).
*p < 0.05. Data are mean ± SEM.
DISCUSSION
In this study, we selectively deleted BDNF or TrkB from the CA3 or CA1 subregions of the hippocampus of adult mice to examine the role of presynaptic and postsynaptic BDNF or TrkB in synaptic plasticity. It is well established that BDNF-TrkB signaling regulates synaptic plasticity (Waterhouse and Xu, 2009); nevertheless, the site of BDNF release and the location of TrkB receptors in the process remain controversial. Stereotaxic injections of AAV expressing GFP-Cre recombinase into either the CA3 or CA1 resulted in a robust reduction of BDNF or TrkB protein expression specifically in these hippocampal subregions. The deletion of BDNF in CA3 or CA1 led to differing LTP deficits in hippocampal slices, suggesting that presynaptic and postsynaptic BDNF are both required, yet contribute separately in LTP. These data reveal that presynaptic BDNF is involved in the induction events regulating the strength of LTP while postsynaptic BDNF contributes to LTP maintenance. Moreover, the loss of presynaptic or postsynaptic TrkB receptors also leads to distinct deficits in LTP in which presynaptic TrkB is required to maintain LTP while postsynaptic TrkB is essential to fully induce LTP.
These results reveal a potential induction mechanism of LTP mediated by anterograde BDNF-TrkB signaling, and suggest that after induction, both anterograde and retrograde BDNF-TrkB signaling persist in presynapses and postsynapses for LTP maintenance. The findings from this study also uncovered a presynaptic role for TrkB in basal neurotransmission. The loss of presynaptic TrkB significantly diminishes release probability under basal conditions similar to the phenotype observed in inducible BDNF KO hippocampal slices, suggesting that BDNF signaling augments basal neurotransmission through presynaptic TrkB receptors. Additionally, the deletion of presynaptic TrkB resulted in increased release probability following LTP, suggesting the potential involvement of a presynaptic TrkB component in LTP. To determine whether these changes impact hippocampal-dependent learning and memory, we tested mice in the novel object recognition test and found TrkB deletion in either CA3 or CA1 resulted in significant deficits. Taken together, these data provide further information on the role of presynaptic and postsynaptic effects of BDNF and TrkB in synaptic plasticity and basal neurotransmission.
Previous studies have established that BDNF-TrkB signaling plays a role in LTP and measures of synaptic plasticity. However, little is known about whether BDNF is secreted from presynaptic or postsynaptic terminals and how presynaptic or postsynaptic TrkB are individually involved in LTP formation. The disparity in the literature stems from differing patterns of synaptic activity used in individual studies and differences of in vivo or in vitro preparations without a direct comparison for the roles of BDNF and TrkB in this process. Our study was designed to directly compare the contributions of presynaptic and postsynaptic BDNF and TrkB in hippocampal LTP. The choice of a viral-mediated approach to delete BDNF or TrkB in adult animals and then examine LTP 3 weeks later was to avoid potential developmental confounds; however, this may not fully reflect the acute action of BDNF-TrkB.
We found that the absence of presynaptic or postsynaptic BDNF resulted in differing LTP deficits, demonstrating that the site of BDNF release confers specific function in HFS-induced LTP formation. Our data are in agreement with a previous study that reported deletion of BDNF in the CA3 region, the presynaptic side, impaired the presynaptic component of LTP (Zakharenko et al., 2003). A potential role for BDNF in postsynaptic neurons has also been shown in a prior study that reported tetanic stimulation leading to postsynaptic depolarization in hippocampal cultures triggers BDNF release from postsynaptic terminals (Hartmann et al., 2001). Another study has shown induction and maintenance stages of LTP require BDNF-TrkB signaling (Pang et al., 2016), although this work did not address presynaptic or postsynaptic BDNF release or the role of presynaptic or postsynaptic TrkB receptors in the process. A separate paper reported a role for perisynaptic glia in the perirhinal cortex in recycling and release of BDNF to activate TrkB receptors in the maintenance phase of LTP (Vignoli et al., 2016). We examined the parallel comparison of presynaptic and postsynaptic BDNF-TrkB signaling in LTP in the Schaffer collateral pathway, in which circuitry organization and cell composition could be different from that in perirhinal cortex. Our findings regarding a role for postsynaptic BDNF in LTP maintenance is similar to data from a BDNF mutant mouse in which dendritic BDNF was absent and the impairments in LTP were found at the Schaffer collateral-CA1 synapses (An et al., 2008). Additionally, a recent study showed that postsynaptic TrkB activation during structural LTP depends on autocrine postsynaptic BDNF; however, additional mechanisms include other sources of BDNF such as presynaptic and paracrine secretions (Harward et al., 2016). In our study, deleting presynaptic BDNF reduces the strength of LTP (Figure 2B), and lack of postsynaptic BDNF impacts the maintenance of LTP but preserves a similar level of post-stimulation potentiation as in control slices (Figure 2C). These results suggest HFS-induced BDNF release from the presynaptic terminal produces post-stimulation potentiation and controls the initial strength of LTP while BDNF release from postsynaptic terminal induced by HFS acts to prolong LTP maintenance.
While there have been several studies examining the role of BDNF in LTP, there has been little focus on the role of TrkB in this process. Rather, many studies have extrapolated inferences on TrkB signaling from BDNF findings. However, BDNF is a secreted protein and its release site may be different from the site of TrkB receptor signaling. The current study allowed the individual involvement of presynaptic or postsynaptic TrkB in LTP induction to be assessed and provided direct evidence of distinct roles of TrkB receptors in the CA3 or CA1 region. We found that presynaptic (CA3) TrkB is required for LTP maintenance while postsynaptic (CA1) TrkB is essential for LTP formation. Prior work has shown that mice with a selective deletion of TrkB in the CA1 subregion show no alteration in postsynaptic depolarization induced LTP, suggesting that presynaptic BDNF-TrkB signaling in CA3 afferents or interneurons mediates LTP (Xu et al., 2000). However, BDNF facilitates spike-timing-dependent LTP (tLTP) (Lu et al., 2013) via binding to postsynaptic TrkB receptors (Edelmann et al., 2015), suggesting the involvement of postsynaptic TrkB in long-term synaptic changes. Moreover, previous studies suggested that a restricted time window is critical for BDNF-TrkB action on LTP maintenance. A prior study showed that endogenous BDNF release for 8–12 min after stimulation is required for LTP maintenance in perirhinal cortex (Aicardi et al., 2004). Another study reported that blockade of TrkB phosphorylation by NMPP1 from 1 to 40 min after stimulation prevented late LTP formation in the CA1 region (Lu et al., 2011). In our work, when deleting BDNF from the CA1 region, HFS-induced potentiation dropped rapidly in the first 10 min post-stimulation and no late LTP formed 60 min after HFS (Figure 2C); additionally, deletion of TrkB from CA1 region blocked LTP formation (Figure 3C), suggesting that postsynaptic BDNF-TrkB action with a restricted temporal expression is essential for long-term synaptic plasticity.
Changes in BDNF-TrkB signaling can lead to alterations in basal synaptic transmission. Prior work demonstrated that applying BDNF to neuronal cultures enhances synaptic strength through TrkB (Kang and Schuman, 1995a; Levine et al., 1995; Magby et al., 2006). Intriguingly, TrkB KO mice exhibit normal basal neurotransmission as assessed with PPRs (Minichiello et al., 1999). In our study, hippocampal slices from inducible BDNF KO mice, which have a broad forebrain deletion, and mice in which we selectively deleted BDNF from both CA1 and CA3 regions, showed a significant increase in the PPR, supporting previous studies that suggested BDNF regulates basal neurotransmission and synaptic efficacy (Kang and Schuman, 1995a; Patterson et al., 1996). However, selective deletion of presynaptic or postsynaptic BDNF did not alter the PPR or input-output curves (data not shown), indicating that BDNF secreted either presynaptically or postsynaptically triggers adaptations that maintain basal synaptic strength.
BDNF homozygous and heterozygous KO mice have reduced PPR as well as a decrease in the maximum fEPSP response slope (Patterson et al., 1996), suggesting that reduced BDNF expression alters basal neurotransmission. Interestingly, conditional TrkB KO mice exhibit no alterations in PPR and fEPSP slope (Minichiello et al., 2002), suggesting there is a network regulation rather than an oversimplified dichotomy between BDNF and TrkB signaling. We found that the deletion of presynaptic TrkB resulted in increased PPR (Figure 4D) and reduced the decay of NMDA activity as assessed by application of MK-801 (Figures 4E and 4F). The implication that TrkB signaling mediates a presynaptic modification during plasticity (Kang and Schuman, 1995b) was corroborated by our results showing that presynaptic TrkB regulates presynaptic release probability. Our results unmasked a presynaptic component mediating LTP formation in slice when TrkB is deleted presynaptically; however, this potentiation gradually depressed over time, suggesting a regulatory pathway between presynaptic TrkB signaling and postsynaptic changes that sustain long-lasting LTP.
Conditional TrkB KO mice have been reported to exhibit impairments in hippocampal-dependent long-term memory (Minichiello et al., 1999); however, the individual impact of presynaptic or postsynaptic TrkB deletion in learning and memory has not been previously explored. While deficits in LTP appeared in hippocampal slices for all TrkB gene manipulations, there were significant deficits in novel object recognition with no effect on fear memory. In our work, the regions of BDNF or TrkB deletion are restricted, implying that BDNF or TrkB expression in the hippocampus may be required to reach a minimum reduction threshold in CA1 or CA3 to achieve impairment in fear memory at the behavioral level. Alternatively, the coordinates used in our stereotaxic surgeries specifically targeted to dorsal CA1 or CA3 subregions of the hippocampus; therefore, it is possible that dorsal and ventral expressions of BDNF or TrkB may be redundant for fear memory performance. It is known that amygdala and hippocampus contribute differently in cue and contextual fear conditioning, in which amygdala is required for both cue and contextual fear conditioning, and hippocampus is involved in contextual fear conditioning (Phillips and LeDoux, 1992); furthermore, BDNF-TrkB signaling in amygdala is critical for fear conditioning (Penzo et al., 2015). In contrast, mice displayed significant deficits in novel object recognition, which is a more dorsal hippocampal-dependent memory (Goulart et al., 2010). During the novel object recognition test, animals distinguish a novel object from a familiar object, which indicates “recognition” of the previously explored object, as well as detection of differences between the objects. This recognition, by definition, requires intact memory of the previously explored object. Thus, this task is a useful tool for assessing the behavioral and neural processes mediating storage and/or subsequent recall of a long-term memory (Bevins and Besheer, 2006). The mechanism underlying these processes may require long-lasting LTP. The correlation between impaired LTP maintenance, which is commonly observed in hippocampal slices with TrkB deletion in either CA1 or CA3, and the recognition deficit of the novel object 24 hr after training in mice in the absence of CA1 or CA3 TrkB, supports the physiological relevance of presynaptic TrkB or postsynaptic TrkB in memory encoding. However, considering the complex role of presynaptic or postsynaptic TrkB in LTP and basal neurotransmission, further behavioral experiments will be necessary to precisely dissect their function in various learning and memory tests.
In this study, we identified distinct roles of presynaptic and postsynaptic BDNF and TrkB in LTP. We also report a role for presynaptic TrkB in release probability, highlighting a role for presynaptic TrkB receptors in basal neurotransmission. Moreover, the loss of presynaptic or postsynaptic TrkB impacted performance in the novel object recognition task, suggesting a link to learning and memory. Taken together, these results provide insight into the role of BDNF and TrkB receptors in hippocampal synaptic plasticity processes.
EXPERIMENTAL PROCEDURES
Animals
Mice were housed in a vivarium on a 12-hr light/dark cycle with access to food and water ad libitum. The Bdnffl/fl (Rios et al., 2001) and Ntrk2fl/fl (Luikart et al., 2005) mice were generated as previously described and maintained as homozygous crosses. Since LTP varies across the estrous cycle (Warren et al., 1995), 3-month-old male mice were utilized. Electrophysiological recordings and behavior were performed 3 weeks after surgery. Inducible Bdnf KO mice were generated as previously described (Monteggia et al., 2004). All animal procedures were approved by the Institutional Animal Care and Use Committee at The University of Texas Southwestern Medical Center in compliance with US Public Health Service guidelines.
AAV
AAV expressing Cre recombinase tagged to GFP (AAV-GFP-Cre) and the control virus AAV-GFP were obtained from the Penn Vector Core; AAV-GFP-Cre and AAV-GFP are AAV2/1.CMV.HI.GFP-Cre.SV40 and AAV2/ 1.CMV.PI.EGFP.WPRE.bGH, respectively. Previous work using AAV-GFP-Cre has shown that GFP does not interfere with Cre recombinase activity (Adachi et al., 2008).
Stereotaxic Surgery
The Bdnffl/fl and Ntrk2fl/fl mice were anesthetized with ketamine (100 mg/kg, intraperitoneally [i.p.])/xylazine (10 mg/kg, i.p.) and then mounted on a stereotaxic apparatus. Using a drill, bilateral holes were made above the target injection sites. The coordinates relative to bregma for CA1 and CA3 were as follows: CA1, anteroposterior, −2.4 mm; lateral, −1.5 mm; dorsoventral, −1.7 mm; and CA3, anteroposterior, −1.7 mm; lateral, −3.5 mm; dorsoventral, −1.9 mm at a 10 angle. A total of 1 μL of virus was bilaterally infused with a 33G Hamilton syringe over a 4-min period. The syringe was left in for an additional 5 min to ensure diffusion of the virus. Mice were injected with AAV-GFP or AAV-GFP-Cre. The use of GFP allowed the visualization of the infected neurons.
Hippocampal Slice Electrophysiology
Three weeks after the stereotaxic surgery, mice were anesthetized with isoflurane and decapitated. Brains were removed and immersed in ice-cold dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose, and 10 glucose for 2–3 min. Hippocampi were dissected and then cut with a vibratome into 400-mm-thick transverse sections in ice-cold dissection buffer continuously aerated with 95% O2 and 5% CO2. Area CA3 was surgically removed and collected for western blot analysis from each slice immediately after sectioning. Sections were recovered in oxygenated artificial cerebrospinal fluid (ACSF) containing the following (in mM): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 glucose, pH 7.4 (continuously equilibrated with 95% O2 and 5% CO2) for 2–3 hr at 30C. Hippocampal slices were transferred to the recording chamber and perfused with ACSF at a rate of 2–3 mL/min at 30C. fEPSPs were evoked by inserting a concentric bipolar stimulating electrode (FHC) to Schaffer collateral/commissural afferents. Extracellular recording electrodes filled with ACSF (resistance, 1–2 MΩ) were inserted into the CA1 area proximally below the molecular layer. Baseline responses were collected every 30 s using an input stimulus intensity that induced 30%–40% of the maximum response. PPF was elicited by paired-pulse stimulations at decreasing interstimulus intervals (ISIs) of 500, 400, 200, 100, 50, 30, and 20 ms and analyzed by dividing the fEPSP slope of pulse 2 (P2) by pulse 1 (P1). Paired-pulse stimulation was applied before and after LTP. LTP was induced by HFS composed of four trains at 100-Hz pulses for 1 s. The initial slopes of the fEPSPs were expressed as percentages of the preconditioning baseline average; the time-matched, normalized data were averaged across experiments. After recording, the CA1 region was dissected from slices for western blot analysis.
To examine release probability, NMDA receptor (NMDAR) field potentials (NMDA-fEPSPs) were recorded in solution containing the following: 124 mM NaCl, 2 mM KCl, 3 mM CaCl2, 0.1 mM MgCl2, 10 μM glucose, 1.2 μM NaH2PO4, 26 mM NaHCO3, 10 mM glycine, 20 μM DNQX (6,7-dinitroquinoxaline-2,3-dione), and 50 μM picrotoxin. After a 20-min stable baseline recording, stimulation was paused for 10 min to equilibrate while 20 μM (+)-MK-801 was added to the bath. One hundred stimuli at 0.1 Hz produced a decay curve of NMDA-fEPSP amplitudes with (+)-MK-801 perfusion.
Western Blot Analysis
CA1 and CA3 tissue were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 1% NP-40, 0.1% SDS, 0.5% Na deoxycholate, 4 mM EDTA, 150 mM NaCl, protease and phosphatase inhibitors [cOmplete mini tablets (Roche)], 10 mM sodium pyrophosphate, 50 mM NaF, and 2 mM sodium orthovanadate). Total protein concentration was quantified by BCA (bicinchoninic acid) protein assay. Twenty micrograms of total protein per well was loaded on SDS-PAGE gels and transferred to nitrocellulose blots, and then placed in blocking solution for 60 min at room temperature. Blots were incubated in anti-BDNF (1:2,000; Abcam), anti-TrkB (1:2,500; Abcam), or anti-GAPDH (1:50,000; Cell Signaling) antibodies at 4°C overnight. HRP-conjugated anti-mouse secondary antibody was used for BDNF (1:2,000), and anti-rabbit secondary antibody for TrkB (1:2,000) and GAPDH (1:10,000). Bands were developed with enzymatic chemiluminescence (ECL) and detected by UVP Biospectrum imaging system. Image files were analyzed with ImageJ. BDNF or TrkB signals were normalized to their respective GAPDH signals and expressed as percentage to the control group.
Behavioral Overview
Mice recovered for 3 weeks following stereotaxic surgeries before behavioral testing, a time point sufficient to induce gene recombination (Adachi et al., 2008). Mice were habituated to the behavior rooms for at least 1 hr prior to testing, which was conducted during the light cycle. Behavioral tasks were performed such that mice were tested in one paradigm per day, from least to most stressful (locomotor activity, novel object recognition, and fear conditioning). In all experiments, mice were age matched and groups balanced by genotype, and testing performed by an individual blind to group/treatment assignment.
Locomotor Activity
Mice were placed in a new home cage under red light for 1 hr, and ambulation was assessed by the number of photocell beam breaks, with signals digitized by PAS software (San Diego Instruments).
Novel Object Recognition
Mice were placed in a 44 × 22-cm2 open field under dim lighting for 10 min to habituate to the testing arena. On day 2, mice were reintroduced to the arena containing two of the same objects (familiar objects). Mice interacted with the familiar objects for 30 s, or after a 10-min exposure in the testing arena, whichever took place first, were returned to their home cage. On day 3, one familiar object was switched to a new object (novel object), and mice were allowed to explore in the arena for 10 min. An experimenter blind to treatment scored the time the animal spent interacting with the familiar and novel object. The discrimination index was calculated as the difference between time spent exploring the novel object and the familiar object divided by the total exposure time exploring both two objects ([time spent on novel object time spent on familiar object]/total time).
Fear Conditioning
Mice were placed in individual fear conditioning chambers (Med Associates). During the acquisition phase, mice were habituated to the chamber for 2 min, and then received three trains of a 30-s loud tone (90 dB) followed immediately by a 0.5-mA footshock for 2 s with a 1-min intertrain interval. Mice remained in the chamber for an additional 1 min before being returned to their home cages. For context-dependent fear conditioning, 24 hr later mice were returned to the chambers for 5 min without a tone or shock, and the amount of time spent freezing was determined. Freezing behavior was defined as no movement except for respiration. Cue-dependent fear conditioning was assessed 4 hr later. Mice were habituated in a novel environment scented with vanilla without any tone or shock for 3 min followed by a 3-min tone. The amount of time spent freezing during the 3-min tone period was determined. The following day, the animals’ response to shock was assessed by exposing the mice to increasing footshock intensities (0.05–0.45 mA).
Statistical Analysis
Electrophysiological data are presented as mean ± SEM and analyzed using two-way ANOVA. Sidak’s multiple-comparisons test were used for post hoc analyses if significant interaction effects were found. The decay rate constant of NMDA-fEPSP amplitudes was analyzed using Student’s t test.
Behavioral data are presented as mean ± SEM. Statistical significance was determined by one-way ANOVA. Turkey’s multiple-comparisons test was used for post hoc analysis. In all experiments, p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Presynaptic BDNF and postsynaptic TrkB are involved in LTP induction
Postsynaptic BDNF, as well as both presynaptic and postsynaptic TrkB, contributes to LTP maintenance
BDNF in both presynaptic and postsynaptic terminals modulates basal neurotransmission
Presynaptic TrkB regulates presynaptic release probability
ACKNOWLEDGMENTS
We are grateful to members of the Monteggia and Kavalali labs for helpful discussion on the manuscript. We thank Drs. L. Parada for the Ntrk2fl/fl mice, R. Jaenisch for the Bdnffl/fl mice, and Z. Ma for valuable advice and confocal image support. This work was supported by National Institutes of Health Grants MH070727 (L.M.M.) and MH066198 (E.T.K.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.020.
REFERENCES
- Adachi M, Barrot M, Autry AE, Theobald D, and Monteggia LM (2008). Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry 63, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Autry AE, Mahgoub M, Suzuki K, and Monteggia LM (2017). TrkB signaling in dorsal raphe nucleus is essential for antidepressant efficacy and normal aggression behavior. Neuropsychopharmacology 42, 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, and Canossa M. (2004). Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 101, 15788–15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder J, Thakker-Varia S, Crozier RA, Shaheen A, Plummer MR, and Black IB (2005). Early presynaptic and late postsynaptic components contribute independently to brain-derived neurotrophic factor-induced synaptic plasticity. J. Neurosci 25, 3080–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, and Xu B. (2008). Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baj G, D’Alessandro V, Musazzi L, Mallei A, Sartori CR, Sciancalepore M, Tardito D, Langone F, Popoli M, and Tongiorgi E. (2012). Physical exercise and antidepressants enhance BDNF targeting in hippocampal CA3 dendrites: further evidence of a spatial code for BDNF splice variants. Neuropsychopharmacology 37, 1600–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, et al. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311, 864–868. [DOI] [PubMed] [Google Scholar]
- Bevins RA, and Besheer J. (2006). Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.”. Nat. Protoc 1, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, and Kossel A. (1999). Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J. Neurosci 19, 7983–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, and Barde YA (2012). BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol 196, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann E, Cepeda-Prado E, Franck M, Lichtenecker P, Brigadski T, and Leßmann V. (2015). Theta burst firing recruits BDNF release and signaling in postsynaptic CA1 neurons in spike-timing-dependent LTP. Neuron 86, 1041–1054. [DOI] [PubMed] [Google Scholar]
- Gärtner A, Polnau DG, Staiger V, Sciarretta C, Minichiello L, Thoenen H, Bonhoeffer T, and Korte M. (2006). Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J. Neurosci 26, 3496–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulart BK, de Lima MN, de Farias CB, Reolon GK, Almeida VR, Quevedo J, Kapczinski F, Schrö der N, and Roesler R. (2010). Ketamine impairs recognition memory consolidation and prevents learning-induced increase in hippocampal brain-derived neurotrophic factor levels. Neuroscience 167, 969–973. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, and Lessmann V. (2001). Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 20, 5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, and McNamara JO (2016). Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, and Malinow R. (1993). The probability of transmitter release at a mammalian central synapse. Nature 366, 569–572. [DOI] [PubMed] [Google Scholar]
- Hohn A, Leibrock J, Bailey K, and Barde YA (1990). Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature 344, 339–341. [DOI] [PubMed] [Google Scholar]
- Kang H, and Schuman EM (1995a). Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662. [DOI] [PubMed] [Google Scholar]
- Kang HJ, and Schuman EM (1995b). Neurotrophin-induced modulation of synaptic transmission in the adult hippocampus. J. Physiol. Paris 89, 11–22. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, and Schuman EM (1997). Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19, 653–664. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, and Bonhoeffer T. (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, and Bonhoeffer T. (1996). Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA 93, 12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, and Plummer MR (1995). Brainderived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc. Natl. Acad.Sci. USA 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Ji Y, Ganesan S, Schloesser R, Martinowich K, Sun M, Mei F, Chao MV, and Lu B. (2011). TrkB as a potential synaptic and behavioral tag. J. Neurosci 31, 11762–11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Park H, and Poo MM (2013). Spike-timing-dependent BDNF secretion and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, and Parada LF (2005). TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J. Neurosci 25, 3774–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magby JP, Bi C, Chen ZY, Lee FS, and Plummer MR (2006). Singlecell characterization of retrograde signaling by brain-derived neurotrophic factor. J. Neurosci 26, 13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, and Poo MM (2009). Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci 29, 14185–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci 10, 850–860. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Ku€hn R, Unsicker K, Cestari V, RossiArnaud C, Lipp HP, Bonhoeffer T, and Klein R. (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24, 401–414. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, and Korte M. (2002). Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36, 121–137. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, and Nestler EJ (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. USA 101, 10827–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Nagappan G, Guo W, and Lu B. (2016). Extracellular and intracellular cleavages of proBDNF required at two distinct stages of late-phase LTP. NPJ Sci. Learn 1, 16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, and Poo MM (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, and Kandel ER (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137–1145. [DOI] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, and LeDoux JE (1992). Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav. Neurosci 106, 274–285. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, and Lu B. (1999). Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci 19, 4972–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, and Jaenisch R. (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol. Endocrinol 15, 1748–1757. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, and Johnston D. (1994). Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J. Neurosci 14, 5325–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (1999). Toward a molecular explanation for long-term potentiation. Learn. Mem 6, 399–416. [DOI] [PubMed] [Google Scholar]
- Thoenen H. (1995). Neurotrophins and neuronal plasticity. Science 270, 593–598. [DOI] [PubMed] [Google Scholar]
- Vignoli B, Battistini G, Melani R, Blum R, Santi S, Berardi N, and Canossa M. (2016). Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. Neuron 92, 873–887. [DOI] [PubMed] [Google Scholar]
- Warren SG, Humphreys AG, Juraska JM, and Greenough WT (1995). LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res 703, 26–30. [DOI] [PubMed] [Google Scholar]
- Waterhouse EG, and Xu B. (2009). New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol. Cell. Neurosci 42, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, and Reichardt LF (2000). The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci 20, 6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, and Morozov A. (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39, 975–990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.