

Abstract
Synthetic protein-level circuits could enable engineering of powerful new cellular behaviors. Rational protein circuit design would be facilitated by a composable protein-protein regulation system in which individual protein components can regulate one another to create a variety of different circuit architectures. In this study, we show that engineered viral proteases can function as composable protein components, which can together implement a broad variety of circuit-level functions in mammalian cells. In this system, termed CHOMP (circuits of hacked orthogonal modular proteases), input proteases dock with and cleave target proteases to inhibit their function. These components can be connected to generate regulatory cascades, binary logic gates, and dynamic analog signal-processing functions. To demonstrate the utility of this system, we rationally designed a circuit that induces cell death in response to upstream activators of the Ras oncogene. Because CHOMP circuits can perform complex functions yet be encoded as single transcripts and delivered without genomic integration, they offer a scalable platform to facilitate protein circuit engineering for biotechnological applications.
Synthetic biology seeks to enable rational design of circuits that confer new functions in living cells. Most efforts thus far have centered on gene regulation because of the relative ease with which transcription factors and other nucleic acid–interacting proteins can be configured to regulate one another’s expression (1–10). However, many natural cellular functions are implemented by protein-level circuits, in which proteins specifically modify each other’s activity, localization, or stability. For example, caspase-mediated programmed cell death is regulated by a circuit of proteases that activate one another through cleavage (11). Synthetic protein circuits could provide advantages over gene regulation circuits, including faster operation, direct coupling to endogenous pathways, single-transcript delivery, and function without genomic integration (Fig. 1A).
Fig. 1. Design of composable protein circuit components.
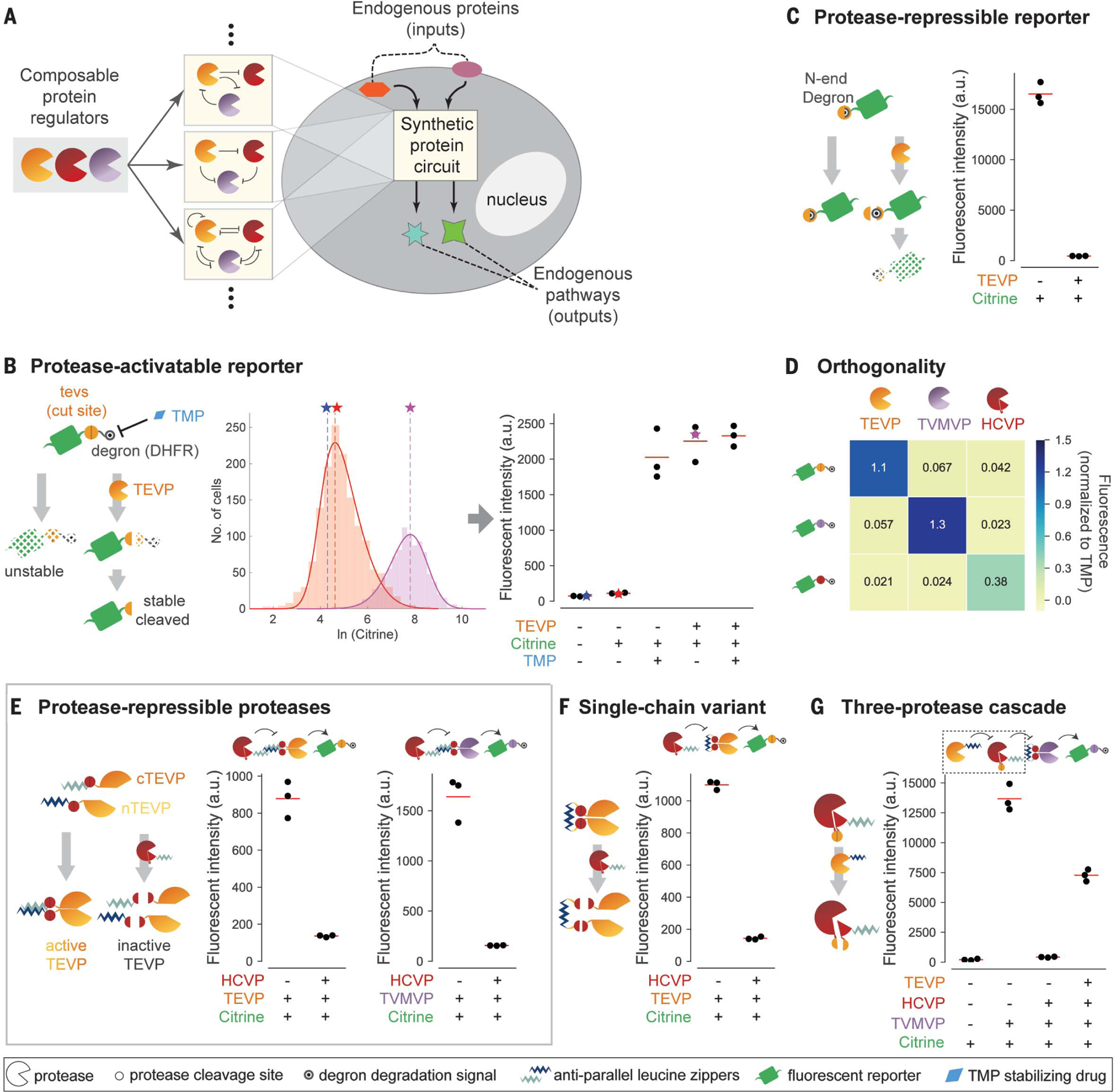
(A) Composable protein units (partial circles, left) can regulate one another in arbitrary configurations with diverse functions (middle). Protein-level circuits can interface directly with endogenous protein pathways and operate without modifying the genome or entering the nucleus (right). (B) (Left) The protease-activatable reporter (green) is stabilized by removal of a DHFR degron (black target) through protease (partial circle) cleavage of a corresponding target site (yellow circle).TMP (blue diamond) inhibits the degron and thus stabilizes the reporter. (Middle) Flow cytometry distributions of reporter fluorescence with (purple) or without (orange) TEVP. Distributions are limited to the gated area in fig. S1A. Solid curves indicate skew Gaussian fits. Vertical dashed lines and stars indicate distribution modes, which are plotted in subsequent figure panels. (Right) Analysis of reporter response to TMP and/or TEVP. Each dot represents one replicate. Stars indicate data from the middle panel. a.u., arbitrary units. (C) In the protease-repressible reporter, protease cleavage exposes an N-end degron (covered target) to destabilize the reporter. (D) Three proteases (columns) exhibit orthogonal regulation of three reporters (rows). Mean fluorescence intensity of three independent measurements is normalized to the TMP-stabilized value of the corresponding reporter. (E) Design for protease-repressible proteases. TEVP is split as indicated and then reconstituted through dimerizing leucine zippers (light and dark blue zig-zags). A leucine zipper–tagged HCVP (red partial circle) can dock with the target TEVP and cleave it to remove leucine zippers, effectively repressing TEVP.TVMVP (purple partial circle) can be regulated using the same design. (F) A single-chain variant of the HCV-repressible TEVP allows docking of and repressive cleavage by HCVP. (G) Protease regulation can propagate through a three-stage cascade. Repressible HCVP uses a variant design, in which TEVP cleavage separates core HCVP from its docking leucine zipper and activity-enhancing copeptide (small pie slice). In all panels, red lines indicate triplicate mean.
The key challenge is designing composable protein components whose inputs and outputs are of the same type, so that they can form a wide variety of protein circuits (12), much as a few electronic components can be wired to produce a variety of electronic circuits (Fig. 1A). Although natural protein domains have been combined to generate proteins with hybrid functions or to rewire cellular pathways for research (13–18) and biomedical applications (17, 19), the lack of composability has limited our ability to design protein-level function in living cells.
Viral proteases provide a promising basis for such a system (20, 21). Many of them exhibit strong specificity for short cognate target sites, which can be recognized and cleaved in various protein contexts (22–24). Natural viral diversity provides multiple proteases with distinct specificities (25). Viral proteases can be used with degrons to control protein stability (26–29). They can also activate transcription factors (30–32), synthetic intein zymogens (33), and other proteases in a purified protein system (20, 21).
We first focused on the well-characterized tobacco etch virus protease (TEVP) (34). To quantify TEVP activity, we designed a reporter in which a cognate cleavage site (tevs) is inserted between a citrine fluorescent protein and a dihydrofolate reductase (DHFR) degron, which can be inhibited by trimethoprim (TMP) as a positive control (35) (Fig. 1B). We transfected human embryonic kidney (HEK) 293 cells with plasmids expressing different combinations of TEVP, the reporter, and an mCherry cotransfection marker, and we analyzed cells by flow cytometry. We used the mCherry signal to select highly transfected cells, which showed the largest separation of basal reporter fluorescence from cellular autofluorescence to maximize the observable dynamic range of the reporter (materials and methods, Fig. 1B, and fig. S1A). Treating cells with TEVP strongly increased reporter abundance to levels similar to those obtained by TMP inhibition of the degron (Fig. 1B and fig. S1B, left). We also designed a complementary repressible reporter in which TEVP cleavage exposes a destabilizing N-terminal tyrosine residue (26, 36) (Fig. 1C and fig. S1B, right). These designs generalized in a straight-forward manner to the related tobacco vein mottling virus protease (TVMVP) (37) and, with some modifications, to the unrelated hepatitis C virus protease (HCVP) (24, 38) (fig. S1, C and D, and supplementary text). Furthermore, measuring activation of each reporter in response to each protease revealed limited cross-activation (Fig. 1D). Thus, three viral proteases can be used to orthogonally increase or decrease cognate reporters.
To enable the design of complex circuits, we next sought to achieve protease-protease regulation. The degron strategy used for the reporters failed to produce strong regulation, possibly because proteases can cleave degrons within the same protease molecule with relaxed specificity (28). Instead, we designed a scheme that regulates protease activity rather than abundance. We incorporated antiparallel heterodimerizing leucine zipper domains (39) to each half of a split TEVP (40) to reconstitute its activity (Fig. 1E, left). We also inserted HCVP cleavage sites between the leucine zippers and TEVP to allow HCVP to inhibit TEVP. Finally, we fused a leucine zipper (complementary to one of the zippers on split TEVP) to HCVP, thus enhancing its ability to dock with and inhibit its TEVP target (fig. S1E, left). This design successfully produced repression of TEVP by HCVP (Fig. 1E, left).
To generalize this design, we engineered a similar TEVP variant repressed by TVMVP (fig. S1E, right). On the basis of its sequence similarity to TEVP (fig. S1F), we also engineered TVMVP variants repressed by either HCVP (Fig.1E, right) or TEVP (fig. S1G). To make these designs more compact, we linked the two halves of each regulated protease with a single leucine zipper flanked by cleavage sites for the input protease, creating single-chain repressible proteases (Fig. 1F and fig. S1, H and I). Similar approaches enabled us to engineer protease regulation of the unrelated protease HCVP by using a different split strategy (supplementary text). In these constructs, cleavage by either TEVP or TVMVP strongly reduced HCVP activity, enabling signal propagation through three-stage protease cascades (Fig. 1G and fig. S1J). Together, this strategy established a composable protease regulation system.
Using this system, we designed core circuit functions, starting with Boolean logic. We identified three design principles that together would be sufficient to enable all eight two-input gates: First, incorporation of a consecutive pair of distinct cleavage sites between a degron and a target protein can implement OR logic, because cleavage of either site is sufficient to stabilize the protein (Fig. 2 and fig. S2A, OR). Second, to implement AND logic, we flanked the target protein with FKBP (41) and DHFR degrons on the N and C termini, respectively, each removable with a distinct cleavage site. On the N terminus, a leucine zipper was necessary to facilitate input protease docking. In this design, removal of both degrons was necessary to stabilize the protein (Fig. 2 and fig. S2A, AND). Third, to implement negation, we either used the N-end degron strategy (Fig. 1C) or propagated signals through an intermediate protease repression step (Fig. 1E). Cotransfection of each basic gate (OR, AND, and NOR as a specific case of negation) with varying concentrations of its inputs revealed the expected logic functions (Fig. 2 and fig. S2B). Further, varying the concentration of the reporter plasmid enabled tuning of output levels without disrupting the logical computation, facilitating matching of input and output levels in more complex circuits (fig. S2C). Finally, by utilizing the HCVP inhibitor asunaprevir (28) and a rapamycin-induced TEVP (40, 42), we found that these gates could also be controlled by small-molecule inputs (fig. S3A). These results thus show that three core gates exhibit robust and tunable operation across multiple input methods.
Fig. 2. CHOMP circuits implement binary logic gates.
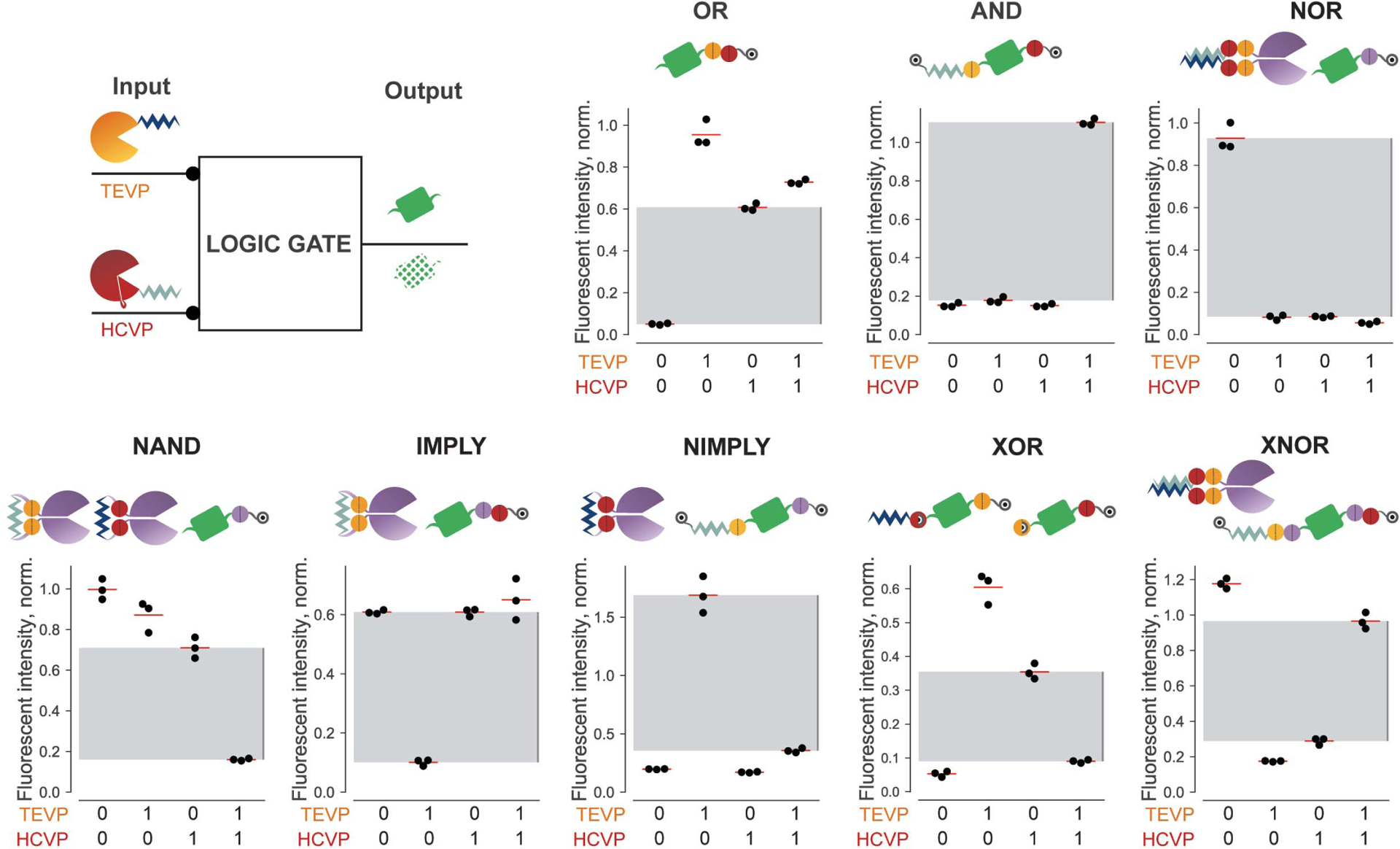
For each indicated gate, TEVP and HCVP serve as binary inputs, which are either included or excluded in transfections. Citrine fluorescence serves as gate output. The design and performance of each nontrivial two-input logic gate is shown for triplicate experiments (black dots). Fluorescence intensity in each panel is normalized to the corresponding reporter stabilized with TMP (for gates containing only C-terminal degrons) or Shield-1 plus TMP (for gates containing degrons at both termini). Gray regions indicate the range from maximum “OFF” value to minimum “ON” value for that gate.
Next, we combined these principles to design and validate the other two-input gates (Fig. 2 and fig. S2A). Furthermore, to test whether output from one gate could be directly used as input to a subsequent gate, we constructed a more complex nested NOR function by using additional orthogonal proteases from soybean mosaic virus (SMVP) (43) and herpes simplex virus (HSVP) (44) (fig. S3B). The output from this system was consistent with that expected from the logical function NOR[TEVP, NOR(SMVP, HSVP)] (fig. S3B).
Beyond Boolean logic, analog signal filtering can enable many cellular functions, such as the ability to selectively respond to specific input concentration ranges (45, 46). The incoherent feed-forward loop (IFFL) motif, in which an input both activates and inhibits the same target, provides a simple implementation for this function (47, 48). Inspired by the IFFL, we combined an activating arm, in which TEVP removes a C-terminal degron, with a repressing arm, in which TVMVP reveals a destabilizing N-end tyrosine (Fig. 3A). To tune the position and sharpness of the bandpass, we also introduced a positive-feedback loop based on reciprocal inhibition between HCVP and TVMVP on the repression arm, such that the amount of HCVP expression sets a threshold for TVMVP activity (Fig. 3A).
Fig. 3. Design of bandpass filtering and pulse-generation circuits.
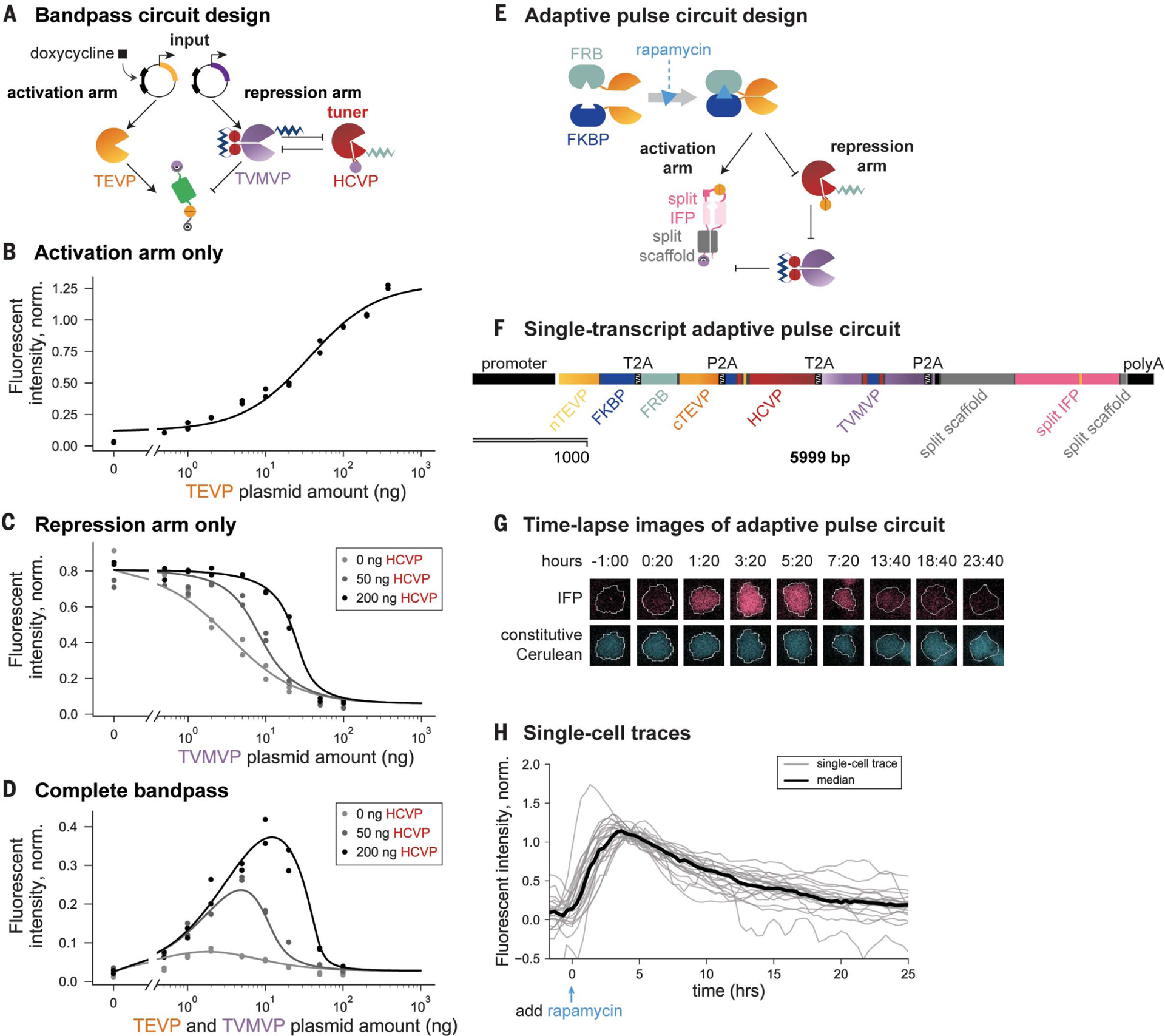
(A) For bandpass filtering, the expression of co-regulated inputs TEVP (yellow) and TVMVP (purple) are controlled by the amount of transfected DNA or by doxycycline (square) induction. The amount of HCVP (red) plasmid can be varied to tune the repression arm. (B) Input-output curve of the activation arm in the absence of TVMVP. Here and in subsequent panels, dots indicate duplicate measurements, and the curve is a model fit (materials and methods). (C) Input-output curve of the repression arm, in the presence of constant TEVP and increasing levels of HCVP (gray shades), which increases the repression threshold and sharpens the response. (D) Bandpass behavior of the complete circuit. Increasing HCVP expression (gray shades) shifts the position and increases the amplitude of the peak response. Data in (B) to (D) are normalized to the TMP-stabilized reporter. (E) Delayed repression can enable pulse generation. In this design, rapamycin-induced dimerization of FKBP and FRB domains reconstitutes TEVP. Cleavage of the reporter by TEVP allows maturation of far-red fluorescent protein (IFP, pink) (fig. S4D). (F) The pulse circuit was completely encoded on a single transcript, with protein components (indicated) separated by self-cleaving sequences (T2A and P2A) (47). (G) Filmstrips of a single cell stably incorporating both the pulse-generation circuit (pink) and a constitutive cerulean segmentation marker (blue). After rapamycin induction (t = 0), the output IFP signal (pink) increases and then decays, whereas the cerulean signal (blue) remains constant. (H) Traces of IFP fluorescence in 24 individual cells (gray lines). This analysis omits cells that exhibited phototoxicity or moved out of the field of view (see materials and methods). The black line indicates median fluorescence over all cells at each time point.
To characterize this bandpass circuit, we considered the abundance of TEVP and TVMVP as input and varied it through the concentration of transfected DNA, which correlated linearly with protein abundance (fig. S4A). The individual activating and repressing arms of the circuit generated increasing and decreasing responses, respectively, to increasing amounts of TEVP and TVMVP (Fig. 3, B and C). Addition of HCVP increased both the threshold and the sharpness of the response to TVMVP titration (Fig. 3C). Combining the two arms into a single circuit generated the anticipated bandpass behavior when we covaried TEVP and TVMVP expression through either different amounts of plasmid (Fig. 3D) or 4-epitetracycline induction (fig. S4B). Finally, varying the abundance of HCVP tuned the position and amplitude of the bandpass response (Fig. 3D and fig. S4B). These results demonstrate rational engineering of tunable analog bandpass filters.
Temporal signal processing, such as adaptation to a change in input, has a critical role in diverse biological systems (49) and has been demonstrated synthetically in bacteria at the gene regulation level (50). To engineer adaptation with the CHOMP (circuits of hacked orthogonal modular proteases) system, we designed an IFFL containing the three-step cascade (Fig. 1G) to introduce a delay in the repressing arm relative to the activating arm (Fig. 3E). To enable sudden induction, we adopted the rapamycin-induced TEVP used for the logic gates (figs. S3A and S4C). To facilitate dynamic readout of circuit output in individual cells, we used a far-red (infrared) fluorescent protein (IFP) that is synthesized in a nonfluorescent state but can be posttranslationally switched on by TEVP (51) (fig. S4D, left). We also added a conditional N-end degron to enable repression by TVMVP (Fig. 3E).
We encoded the entire pulse-generation circuit as a single open reading frame, with inter-leaved 2A self-cleaving peptides (52) to separate distinct protein components (Fig. 3F). This gene (encoding the single reading frame) was then stably incorporated in the genome (materials and methods). We used flow cytometry to analyze the response of the reporter in a single clone over time after rapamycin addition. Cells exhibited the expected adaptive dynamics, with a rise in fluorescence on a time scale of hours and a subsequent decay to baseline over ~1 day (fig. S4D, right). To obtain a direct view of dynamics in individual cells, we also analyzed the same cell line by time-lapse fluorescence microscopy (Fig. 3G and movie S1). Analysis of individual cells revealed similar adaptive dynamics, responding maximally at 269 ± 68 (mean ± SD) min after rapamycin addition, decaying to 50% of their peak values over the subsequent 491 ± 170 min, and eventually reaching fluorescence similar to that before induction (Fig. 3H). These results demonstrate the design of single-gene multicomponent circuits that generate dynamic signal responses.
By coupling directly to endogenous cellular outputs and inputs, protein-level circuits could act as programmable therapeutic devices. As a proof of principle for such a strategy, we designed a circuit to selectively kill cells with elevated activation of Ras, a protein whose activity is increased in many cancers (53, 54). More specifically, we designed a core circuit that responds to upstream activators of Ras, such as SOS and epidermal growth factor receptor (EGFR), by activating an engineered TEV protease, which in turn activates caspase-3 (Casp3) to induce cell death (11, 55) (Fig. 4A, core circuit). We then improved this circuit by incorporating additional proteases and interactions (Fig. 4A, full circuit).
Fig. 4. CHOMP circuit enables conditional activation of Casp3 in Ras-activating cells.
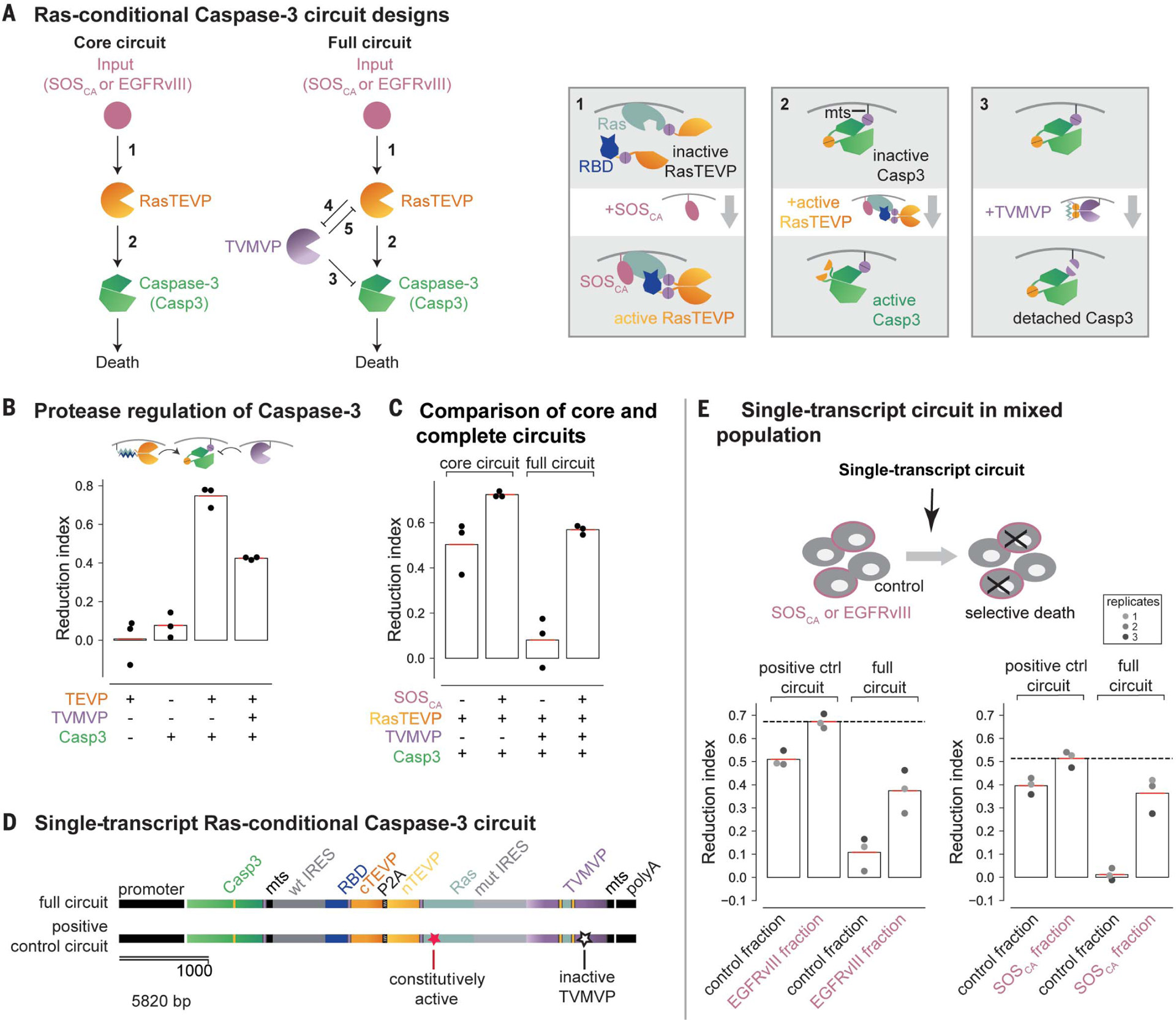
(A) The core circuit (left) links Ras activation by SOSCA or EGFRvIII to Casp3 activation. The full circuit (right) incorporates an additional TVMVP component to enhance selectivity. New regulatory features introduced in this circuit are explained schematically in the corresponding numbered boxes. Box 1: Input from upstream activators of Ras such as SOSCA and EGFRvIII (pink) activates Ras (light blue), causing it to bind RBD (dark blue), reconstituting RasTEVP. Box 2: Engineered Casp3 (green) tagged with a membrane localization sequence (mts) can be converted from an inactive to an active state by TEVP cleavage. Box 3: TVMVP cleavage detaches Casp3 from the membrane, reducing its ability to be activated by membrane-localized TEVP. (B) TEVP activates the engineered Casp3, whereas TVMVP inhibits this activation. Cells transfected with the indicated components were analyzed to determine the reduction index (percentage of cell number reduction compared to cells transfected with only a fluorescent marker; see materials and methods and fig. S5B). (C) The core circuit preferentially reduced cell number in the presence of ectopic SOSCA. The full circuit exhibited improved selectivity. (D) The full circuit (top) and a positive-control circuit incorporating a Gly12→Val mutation that makes Ras constitutively active and a Cys152→Ala mutation that abolishes TVMVP activity (bottom) were each encoded as a single transcript. (E) In a mixed population, the single-transcript circuit (D, top) conditionally reduced the number of EGFRvIII cells (left) and SOSCA cells (right) compared with that of cocultured control cells. The positive-control circuit (D, bottom) reduced the number of both fractions. The dashed line indicates the upper limit of the reduction index measured with the positive-control circuit. Dots from the same well are color matched.
To enable efficient protease-dependent induction of cell death at the plasma membrane, where Ras activation occurs, we membrane localized a TEVP-activated Casp3 variant (55) by incorporating the 20–amino acid membrane-targeting sequence (mts) from the C terminus of human H-Ras (56) (Fig. 4A, box 2). Using flow cytometry, we quantified the effect of this Casp3 variant on cell numbers in terms of a “reduction index” whose value measures the relative reduction in cell number compared with a control condition (materials and methods and fig. S5B). The membrane-targeted Casp3 decreased cell numbers when cotransfected with a similarly membrane-localized TEVP variant (Fig. 4B), with higher efficiency than the original cytoplasmic Casp3 variant (fig. S5D). Further, to allow bidirectional regulation by TEVP and TVMVP, we also incorporated a TVMVP cleavage site adjacent to the mts tag (Fig. 4A, box 3), enabling membrane-localized TVMVP to remove Casp3 from the membrane and thereby attenuate its activation by TEVP (Fig. 4B).
Next, to couple Ras-activating inputs to TEVP, we fused the N-terminal half of TEVP to Ras and its C-terminal half to the Ras-binding domain (RBD) of Raf, which binds to the active form of Ras (57, 58). In this design, upstream activators of Ras should reconstitute RasTEVP (Fig. 4A, core circuit and box 1, and fig. S5C) and thereby activate Casp3. To validate this design, we constructed a HEK293 cell line stably expressing a constitutively active SOS (SOSCA) variant with a membrane-localization myristoylation signal and no inhibitory C-terminal region (59). Transfection of the core circuit reduced cell numbers, both in this SOSCA cell line and in its parental control line that lacks ectopic SOSCA, but preferentially affected the SOSCA cells (Fig. 4C, core circuit, and fig. S5F). This selectivity required the regulated Ras-RBD interaction (fig. S5E and supplementary text). However, though this core circuit provided some selectivity, it also exhibited a relatively high background rate of Casp3 activation in the control cells.
To improve the circuit’s selectivity, we incorporated a TVMVP-TEVP reciprocal inhibition motif (fig. S5A, boxes 4 and 5) similar to the one used in the bandpass circuit, as well as feed-forward repression of Casp3 activation by TVMVP (Fig. 4A, box 3). In this full-circuit design, TVMVP should suppress activation of Casp3 in control cells, both directly and indirectly through TEVP. By contrast, in SOSCA cells, elevated activation of TEVP should override the inhibitory effects of TVMVP. The full circuit indeed improved selectivity (Fig.4C, figs.S5G and S6A, and supplementary text). More specifically, expressing TVMVP in amounts comparable to but lower than those of TEVP nearly abolished off-target effects in control cells while retaining most of the on-target reduction in cell number (Fig. 4C and fig. S5G).
To simulate a more biomedically relevant context, we encoded the full four-protein circuit on a single transcript, optimizing the relative abundance of components with internal ribosome entry site variants (60) (Fig. 4D, fig. S6B, and supplementary text), and transfected it into a mixed population of SOSCA and control cells. At its optimal concentration (fig. S6C), the single-transcript circuit reduced the number of SOSCA cells by ~40%, approaching the ~50% upper limit achieved by a positive-control circuit that constitutively activates Casp3 (Fig. 4D, and Fig. 4E, right). (The upper limit is constrained by gene delivery and expression efficiency.) Notably, it exhibited minimal effects on the control population (Fig. 4E, right). SOSCA-dependent killing could also be observed using annexin V staining as an independent readout of apoptosis (fig. S6D). Finally, to test the generality of the circuit, we considered the distinct and more biomedically relevant input EGFRvIII, an oncogenic EGFR mutant found in glioblastoma and other cancer types (61). The single-transcript full circuit also selectively killed EGFRvIII cells (Fig. 4E, left, and fig. S6D). Together, these results show that a CHOMP circuit can be engineered to detect and kill in response to upstream activators of Ras through rational iterative design optimization.
The considerable diversity of natural cellular behaviors stems from the flexibility with which regulatory components can form distinct circuits. Our results demonstrate how a set of composable protein regulators and circuit design principles can enable a broad range of protein-based circuits and functions. The use of a small number of composable components shifts the design problem, in part, from the level of the individual protein to the level of the protein circuit. Because the operation of CHOMP components does not depend on how they are expressed, they can be optimized through transient transfections, accelerating the overall design-build-test cycle. Although powerful, CHOMP could be improved with additional features. Protease-activating proteases would simplify some circuit designs and facilitate signal amplification. Protein design strategies to control the intrinsic nonlinearity (effective cooperativity) of input-output responses could enable the implementation of useful dynamics such as multistability (62) and oscillation (63, 64). With one exception, the circuits shown here were created with three proteases, but additional orthogonal proteases would allow larger and more complex circuits (25). Finally, future work could expand the range of CHOMP inputs and outputs, enabling direct sensing of the activities of Ras and other oncogenes, and allow for combinatorial sensing of multiple inputs.
CHOMP circuits could provide distinct capabilities compared with transcriptional systems. In terms of speed, proteases can respond rapidly to an increase in input protease activity (fig. S7 and supplementary text). CHOMP circuits can also operate in parallel at specific subcellular sites within a cell. Because CHOMP circuits have a relatively compact genetic design and do not require regulatory interactions with DNA, they could be introduced into differentiated and even postmitotic cells with gene therapy vectors or other viruses and could improve the specificity of oncolytic virotherapy (65). Synthetically, hybrid circuits combining transcriptional or translational regulation with engineered proteases could offer the programmability of base-pairing interactions together with protein-level operation. For example, existing cancer-detection circuits (66, 67) could conditionally express CHOMP components to increase specificity and couple to protein-mediated inputs and outputs. Integrating these capabilities, one can envision smart therapeutics or sentinels based on CHOMP circuits (68, 69).
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Markson, Y. Antebi, N. Nandagopal, and J. Ruan for technical assistance; A. Varshavsky, R. Deshaies, and P. Coffino for scientific input and advice; and R. Kishony, G. Seelig, J.G. Ojalvo, J. Markson, L. Potvin-Trottier, K. Frieda, R. Zhu, and A. Granados for critical feedback.
Funding: The research was funded by DARPA (HR0011-17-2-0008, M.B.E.), the Gordon and Betty Moore Foundation (GMBF2809, M.B.E.), NIH (T32 GM07616, L.S.C.), and the Helen Hay Whitney Foundation (F1047, X.J.G.). M.B.E is a Howard Hughes Medical Institute investigator.
Footnotes
Competing interests: All authors are inventors on U.S. patent application 62/619,001 (“A System for Programming Protein-level Circuits in Living Cells”) submitted by Caltech.
Data and materials availability: All DNA constructs are available from Addgene (www.addgene.org/depositing/75869/, and cell lines available from M.B.E. under a material transfer agreement with Caltech. The datasets generated and analyzed and the computer code used during the current study are available upon request from the corresponding author.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/361/6408/1252/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S7
Table S1
References (70–73)
Movie S1
REFERENCES AND NOTES
- 1.Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D, Science 340, 599–603 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Weinberg BH et al. , Nat. Biotechnol 35, 453–462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausländer S, Ausländer D, Müller M, Wieland M, Fussenegger M, Nature 487, 123–127 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Rinaudo K et al. , Nat. Biotechnol 25, 795–801 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Wroblewska L et al. , Nat. Biotechnol 33, 839–841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalil AS et al. , Cell 150, 647–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roquet N, Soleimany AP, Ferris AC, Aaronson S, Lu TK, Science 353, aad8559 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Nielsen AAK et al. , Science 352, aac7341 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Angelici B, Mailand E, Haefliger B, Benenson Y, Cell Reports 16, 2525–2537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohmueller JJ, Armel TZ, Silver PA, Nucleic Acids Res. 40, 5180–5187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X, Annu. Rev. Cell Dev. Biol 15, 269–290 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Marchisio MA, Stelling J, Bioinformatics 24, 1903–1910 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Yeh BJ, Rutigliano RJ, Deb A, Bar-Sagi D, Lim WA, Nature 447, 596–600 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Dueber JE, Yeh BJ, Chak K, Lim WA, Science 301, 1904–1908 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Park S-H, Zarrinpar A, Lim WA, Science 299, 1061–1064 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Howard PL, Chia MC, Del Rizzo S, Liu F-F, Pawson T, Proc. Natl. Acad. Sci. U.S.A 100, 11267–11272 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett DM, Singh N, Porter DL, Grupp SA, June CH, Annu. Rev. Med 65, 333–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morsut L et al. , Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roybal KT et al. , Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein V, Nabi M, Alexandrov K, ACS Synth. Biol 6, 1337–1342 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Stein V, Alexandrov K, Proc. Natl. Acad. Sci. U.S.A 111, 15934–15939 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrington JC, Dougherty WG, Proc. Natl. Acad. Sci. U.S.A 85, 3391–3395 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tözsér J et al. , FEBS J. 272, 514–523 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Bartenschlager R, J. Viral Hepat 6, 165–181 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Adams MJ, Antoniw JF, Beaudoin F, Mol. Plant Pathol 6, 471–487 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Taxis C, Stier G, Spadaccini R, Knop M, Mol. Syst. Biol 5, 267 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butko MT et al. , Nat. Neurosci 15, 1742–1751 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung HK et al. , Nat. Chem. Biol 11, 713–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Rodriguez J, Voigt CA, Nucleic Acids Res. 44, 6493–6502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kipniss NH et al. , Nat. Commun 8, 2212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnea G et al. , Proc. Natl. Acad. Sci. U.S.A 105, 64–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daringer NM, Dudek RM, Schwarz KA, Leonard JN, ACS Synth. Biol 3, 892–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gramespacher JA, Stevens AJ, Nguyen DP, Chin JW, Muir TW, J. Am. Chem. Soc 139, 8074–8077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waugh DS, Protein Expr. Purif 80, 283–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwamoto M, Björklund T, Lundberg C, Kirik D, Wandless TJ, Chem. Biol 17, 981–988 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varshavsky A, Proc. Natl. Acad. Sci. U.S.A 93, 12142–12149 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nallamsetty S et al. , Protein Expr. Purif 38, 108–115 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Taremi SS et al. , Protein Sci. 7, 2143–2149 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh I, Hamilton AD, Regan L, J. Am. Chem. Soc 122, 5658–5659 (2000). [Google Scholar]
- 40.Wehr MC et al. , Nat. Methods 3, 985–993 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL, Wandless TJ, Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi F, Charlton CA, Blau HM, Proc. Natl. Acad. Sci. U.S.A 94, 8405–8410 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghabrial SA, Smith HA, Parks TD, Dougherty WG, J. Gen. Virol 71, 1921–1927 (1990). [DOI] [PubMed] [Google Scholar]
- 44.Weinheimer SP et al. , J. Virol 67, 5813–5822 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart Y, Alon U, Mol. Cell 49, 213–221 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Porcher A, Dostatni N, Curr. Biol 20, R249–R254 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R, Nature 434, 1130–1134 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Greber D, Fussenegger M, Nucleic Acids Res. 38, e174 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma W, Trusina A, El-Samad H, Lim WA, Tang C, Cell 138, 760–773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basu S, Mehreja R, Thiberge S, Chen M-T, Weiss R, Proc. Natl. Acad. Sci. U.S.A 101, 6355–6360 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.To T-L et al. , Proc. Natl. Acad. Sci. U.S.A 112, 3338–3343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szymczak AL et al. , Nat. Biotechnol 22, 589–594 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ, Nat. Rev. Drug Discov 13, 828–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Downward J, Nat. Rev. Cancer 3, 11–22 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Gray DC, Mahrus S, Wells JA, Cell 142, 637–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hancock JF, Cadwallader K, Paterson H, Marshall CJ, EMBO J. 10, 4033–4039 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliveira AF, Yasuda R, PLOS ONE 8, e52874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yasuda R et al. , Nat. Neurosci 9, 283–291 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Aronheim A et al. , Cell 78, 949–961 (1994). [DOI] [PubMed] [Google Scholar]
- 60.Koh EYC et al. , PLOS ONE 8, e82100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wikstrand CJ, Reist CJ, Archer GE, Zalutsky MR, Bigner DD, J. Neurovirol 4, 148–158 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Gardner TS, Cantor CR, Collins JJ, Nature 403, 339–342 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Elowitz MB, Leibler S, Nature 403, 335–338 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Stricker J et al. , Nature 456, 516–519 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Russell SJ, Peng K-W, Bell JC, Nat. Biotechnol 30, 658–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nissim L, Bar-Ziv RH, Mol. Syst. Biol 6, 444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y, Science 333, 1307–1311 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Kojima R, Aubel D, Fussenegger M, Adv. Drug Deliv. Rev 105 (Pt A), 66–76 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Lienert F, Lohmueller JJ, Garg A, Silver PA, Nat. Rev. Mol. Cell Biol 15, 95–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.