

Graphic Abstract
Electronic supplementary material
The online version of this article (10.1007/s13659-020-00239-z) contains supplementary material, which is available to authorized users.
Keywords: Irpex lacteus, Meruliaceae, Irlactane, Tremulane, Sesquiterpene
Introduction
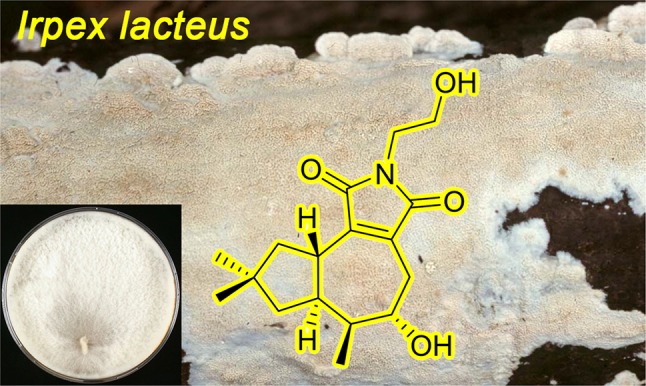
Medicinal fungi are dominate resources of natural products with diverse structural scaffolds and promising biological activities [1]. The wood-decaying fungus Irpex lacteus widely distributed throughout temperate areas globally is traditionally used as a medicinal fungus for diuretic, anti-bacterial, and anti-inflammatory function. The Yishenkang capsule made from the fermentated polysaccharide fraction of this fungus is clinically used as a remedy for chronic glomerulonephritis in China [2]. However, the fungus has not yet been fully chemically explored.
Tremulanes are a class of 5/7-ring-fused sesquiterpenoid which are typically found from fungi [1], and were originally isolated from the wood-decaying fungus Phellinus tremulae [3], and later from the medicinal fungus Phellinus igniarius, and the mushroom Conocybe siliginea. Recently, we reported ten tremulane/tremulane-related sesquiterpenoids, namely irlactins A–J from the cultures of the fungus I. lacteus, among which irlactins A–D featured by an unprecedented skeleton in sesquiterpenoid class [4–6]. Herein, as part of our ongoing research for secondary metabolites as promising drug leads from higher fungi, we reported fifteen previously undescribed irlactane/tremulanes, namely irlactin K (1), and irpexolactins A–N (2–15) (Fig. 1), along with nine known ones from the culture broth of the fungus I. lacteus.
Fig. 1.

Structures of compounds 1–24
Results and Discussion
Structure Elucidation of Previously Undescribed Fungal Metabolites (1–15)
Irlactin K (1) was isolated as colorless crystals. The 1H NMR spectroscopic data (Table 1) presented three methyl singlets (δH 0.88, 1.00, 1.23) and a methoxy group (δH 3.44). The 13C NMR spectrum (Table 4) showed sixteen carbon resonances ascribable to four methyls, four methylenes (one oxygenated), four methines (three oxygenated), and four quaternary carbons. All these data are reminiscent of those of irlactins C and D which were isolated from the cultures of the same fungus but different strains [4]. The presence of a methoxy group rather than a hydroxy group at C-12 in 1 as suggested by the HMBC correlation from the methoxy at δH 3.44 to the ketal carbon at δC 111.3 (C-12) discriminated its structure from those of irlactins C/D. The above assignments led to the determination of the planar structure of 1 as depicted in Fig. 2. The absolute configuration of 1 was determined to be 3R,5S,6R,7S,12R via X-ray single crystal diffraction analysis with the small Flack parameter 0.15(6) (Fig. 3).
Table 1.
1H NMR spectroscopic data of compounds 1–5 (600 MHz, δ in ppm)
| No | 1a | 2a | 3b | 4c | 5a |
|---|---|---|---|---|---|
| 1 | 2.83, overlapped | ||||
| 3 | 2.45, m | 2.66, dd (3.7, 3.7) | 2.98, dd (7.4, 7.4) | ||
| 4 |
1.99, ddd (11.6, 5.0, 3.6) 1.59, dd (11.6, 11.6) |
2.81, overlapped 2.65, ddd (17.3, 3.0, 3.0) |
1.98, ddd (14.5, 4.2, 2.5) 1.76, dd (14.5, 3.0) |
1.77, dddd (13.7, 13.7, 3.7, 3.7) 1.68, overlapped |
2.00, d (12.3) 1.83, dd (12.3, 7.4) |
| 5 | 3.76, dd (11.6, 3.6) | 3.63, overlapped |
2.44, dddd (14.4, 14.4, 3.3, 3.3) 1.74, m |
1.95, m 1.59, m |
|
| 6 | 1.98, m | 1.96, m | 2.10, m | 1.92, m | |
| 7 | 4.07, dd (11.2, 5.3) | 2.59, m | 3.59, m | 2.40, ddd (14.3, 6.6, 6.6) | 3.25, m |
| 8 |
1.53, dd (13.5, 11.2) 1.48, dd (13.5, 5.3) |
1.49, dd (14.2, 1.5) 1.45, d (14.2) |
1.65, br dd (12.5, 8.4) 1.45, dd (12.5, 11.0) |
1.67, overlapped 1.14, dd (12.5, 6.0) |
1.51, dd (12.1, 12.1) 1.46, dd (12.1, 8.5) |
| 10 |
1.96, br d (13.5) 1.78, br d (13.5) |
2.32, dd (12.9, 7.6) 1.54, dd (12.9, 10.9) |
3.25, d (17.8) 2.19, d (17.8) |
2.17, d (14.8) 1.65, d (14.8) |
2.30, br d (15.7) 1.99, br d (15.7) |
| 11 | 4.31, br s |
5.13, s 4.94, s |
4.07, d (12.1) 3.95, d (12.1) |
||
| 12 | 4.64, d (6.0) | 4.62, s |
4.32, dd (7.8, 7.4) 3.84, d (7.8) |
||
| 13 | 1.23, s | 1.03, d (7.0) | 0.84, d (7.0) | 0.89, d (7.0) | 0.87, d (7.0) |
| 14 | 0.88, s | 1.07, s | 0.89, s | 1.10, s | 1.11, s |
| 15 | 1.00, s | 1.07, s | 1.17, s |
3.39, d (10.0) 3.37, d (10.0) |
0.94, s |
| 1′ | 3.56, t (5.6) | 3.78, t (5.2) | |||
| 2′ | 3.63, t (5.6) | 3.83, t (5.2) | |||
| 2′-OH | 2.57, br s | ||||
| -OMe | 3.44, s | 3.19, s |
aRecorded in CD3OD
bRecorded in CDCl3
cRecorded in acetone-d6
Table 4.
13C NMR spectroscopic data of compounds 1–8 (150 MHz, δ in ppm)
| No | 1a | 2a | 3b | 4c | 5a | 6a,d | 7a,d | 8a |
|---|---|---|---|---|---|---|---|---|
| 1 | 133.1, C | 37.8, CH | 170.1, C | 93.7, C | 143.3, C | 135.39, C | 136.07, C | 140.6, C |
| 2 | 134.5, C | 145.5, C | 123.3, C | 156.1, C | 137.9, C | 131.28, C | 131.65, C | 126.8, C |
| 3 | 46.3, CH | 139.4, C | 74.4, C | 52.2, CH | 41.1, CH | 45.15, CH | 47.04, CH | 38.8, CH |
| 4 | 31.4, CH2 | 29.5, CH2 | 26.8, CH2 | 26.7, CH2 | 38.4, CH2 | 34.39, CH2 | 35.81, CH2 | 31.1, CH2 |
| 5 | 80.0, CH | 74.7, CH | 30.1, CH2 | 29.4, CH2 | 112.8, C | 76.79, CH | 76.48, CH | 73.6, CH |
| 6 | 44.4, C | 41.6, CH | 32.4, CH | 32.1, CH | 43.7, CH | 45.05, CH | 44.91, CH | 39.7, CH |
| 7 | 71.4, CH | 43.6, CH | 48.0, CH | 51.3, CH | 42.9, CH | 40.64, CH | 40.73, CH | 41.2, CH |
| 8 | 44.3, CH2 | 45.2, CH2 | 43.8, CH2 | 37.6, CH2 | 45.4, CH2 | 47.10, CH2 | 47.19, CH2 | 45.5, CH2 |
| 9 | 33.4, C | 37.3, C | 38.9, C | 41.5, C | 38.4, C | 38.59, C | 38.46, C | 39.6, C |
| 10 | 39.5, CH2 | 47.2, CH2 | 49.2, CH2 | 48.9, CH2 | 48.6, CH2 | 47.22, CH2 | 47.96, CH2 | 47.4, CH2 |
| 11 | 68.2, CH2 | 172.7, C | 168.6, C | 107.1, CH2 | 65.3, CH2 | 69.53, CH2 | 68.56, CH2 | 70.9, CH2 |
| 12 | 111.3, CH | 173.2, C | 179.8, C | 106.2, CH | 75.1, CH2 | 100.99, CH | 104.78, CH | 182.6, C |
| 13 | 18.0, CH3 | 12.6, CH3 | 11.6, CH3 | 14.7, CH3 | 12.7, CH3 | 14.96, CH3 | 14.81, CH3 | 12.1, CH3 |
| 14 | 25.6, CH3 | 32.0, CH3 | 27.3, CH3 | 26.1, CH3 | 29.3, CH3 | 29.78, CH3 | 29.70, CH3 | 27.5, CH3 |
| 15 | 32.5, CH3 | 31.6, CH3 | 28.5, CH3 | 71.1, CH2 | 27.3, CH3 | 28.24, CH3 | 28.16, CH3 | 29.0, CH3 |
| 1′ | 41.3, CH2 | 41.5, CH2 | ||||||
| 2′ | 60.4, CH2 | 61.0, CH2 | ||||||
| MeO- | 56.8, CH3 | 53.6, OCH3 |
aRecorded in CD3OD
bRecorded in CDCl3
cRecorded in acetone-d6
dAssignments were rounded to two decimal places to tell apart the close resonances
Fig. 2.

Key 1H-1H COSY and HMBC correlations of compounds 1, 2, 4, 6/7, 9, and 10
Fig. 3.
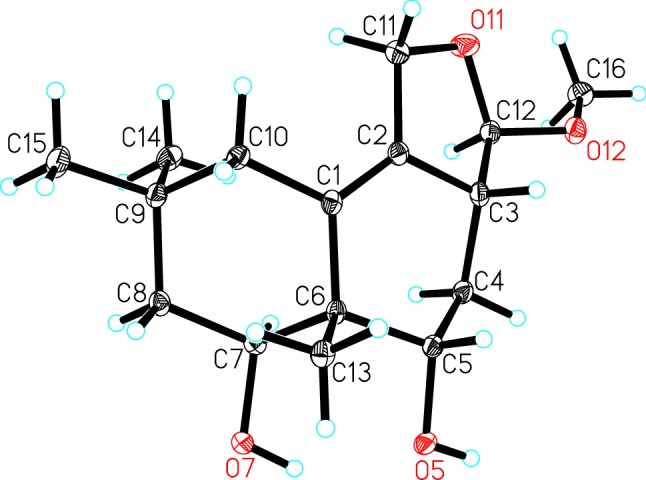
ORTEP drawing of compound 1
Irpexolactin A (2) was obtained as a pale-yellow oil. The molecular formula C17H25O4N with six degrees of unsaturation was deduced from the sodium adduct ion peak at m/z 330.1685 [M + Na]+ in the HRESIMS analysis (calcd for C17H25O4NNa, 330.1676). The 1H and 13C NMR data of 2 (Tables 1, 4) revealed the presence of three methyls (two singlets at δH 1.07, 1.07, one doublet at δH 1.03), five methylenes (one oxygenated at δC 60.4), four methines (one oxygenated at δC 74.7), and five quaternary carbons (two carbonyls at δC 172.7, C-11; 173.2, C-12) and a tetrasubstituted double bond at δC 145.5 (C-2), 139.4 (C-3), indicating three double bond equivalences. All these data bore striking similarity with those of coriolopsin A, a tremulane sesquiterpene isolated from the endophytic fungus Coriolopsis sp. J5 [7]. The structure of compound 2 was further corroborated by 2D NMR analysis, which suggested that the Δ1 double bond in coriolopsin A shifted to Δ2 in 2 by HMBC correlation from H-1 (δH 2.83), H2-4 (δH 2.81, 2.65) to C-2 and C-3 (Fig. 2). Furthermore, the oxygenated methine at δC 74.7 was assigned to C-5 by the HMBC correlation from H3-13 (δH 1.03) to C-5 and 1H-1H COSY correlation between H-5 (δH 3.63) and H-6 (δH 1.98) (Fig. 2). The remaining two methylenes was assigned to an aminoethanol group same as that harbored in vibralactamide A [8] according to the chemical shifts of C-1′ (δC 41.3), C-2′ (δC 60.4) and the 1H-1H COSY correlations between H-1′ (δH 3.56)/H-2′ (δH 3.63). The HMBC correlations from H-1′ to two carbonyls at C-11 and C-12, revealed a succinamide moiety.
Compound 2 possessed a 1-(2-hydroxyethyl)pyrrolidine-2,5-dione unit which is unusual in sesquiterpenoid family. The relative configuration of 2 was ascertained by the ROESY spectrum. The ROESY correlations of H-1/H3-13/H-5, and H-6/H-7 demonstrated that when 5-OH and H-7 occupied α orientation, H-1 and Me-13 would be β orientation (Fig. 4). All the above assignments successfully constructed the structure of 2 (Fig. 1) and irpexolactin A was given as the trivial name.
Fig. 4.

Key ROESY correlations of compounds 2, 4, 6/7, 9, and 10
The colorless oil 3 had a molecular formula of C17H25NO4 identical with that of 2 as suggested by HRESIMS result. The 13C NMR data of irlactam (22) [6] and 3 (Table 4) displayed remarkable disparities of C-1, C-2, and C-3, suggesting their main differences located at C-1 to C-3. The HMBC correlations from H-7 (δH 3.59), H-10 (δH 3.25 and 2.19) to C-1 (δC 170.1), C-2 (δC 123.3), and from H-4 (δH 1.98 and 1.76) to C-2, C-3 (δC 74.4) enabled the assignments of double bond between C-1 and C-2, and a hydroxy attached to C-3. The ROESY correlations between H-6 (δH 1.96) and H-7 revealed that the β orientation of Me-13 and α orientation of H-7. However, the configuration of 3-OH remained undetermined due to the shortage of sample for making any chemical derivatives. Thus, the structure of compound 3 was determined as shown in Fig. 1, and was named irpexolactin B.
Compound 4 was obtained as a colorless oil. The HRESIMS experiment gave a sodium adduct ion peak at m/z 289.1774 [M + Na]+, corresponding to a molecular formula of C16H26O3 (calcd for C16H26O3Na, 289.1774) with four double bond equivalences. The 1D NMR data (Tables 1, 4) displayed three methyls (one methoxy), six methylenes (one oxygenated, a terminal double bond), four methines (one acetal methine), and three quaternary carbons (one oxygenated, one embedded into a terminal double bond). All these data are reminiscent of the known compound 1β,12-epoxy-14-hydroxy-2(11)-tremulene [9]. The HMBC correlations from H-12 (δH 4.62) to C-1 (δC 93.7), C-2 (δC 156.1), C-3 (δC 52.2), and C-4 (δC 26.7), and from the methoxy at δH 3.19 to C-12 (δC 106.2) indicated that the methylene C-12 in 1β,12-epoxy-14-hydroxy-2(11)-tremulene was substituted by a methoxy to give 4 (Fig. 2). The ROESY correlations of H3-13 (δH 0.89)/H2-11 (δH 5.13, 4.94), H2-15 (δH 3.39, 3.37)/H-11a, and H3-14 (δH 1.10)/H-7 (δH 2.40) demonstrated the β orientations for Me-13 and 15-CH2OH (Fig. 4), which is different to that of 1β,12-epoxy-14-hydroxy-2(11)-tremulene. The key ROESY correlations of H-12 (δH 4.62)/H-3 (δH 2.66), H-12/H-4β (δH 1.68) (Fig. 4) helped to assign the relative configuration of C-12 as R* and α oriented. Thus, compound 4 was established as irpexolactin C.
Compounds 5 and 23 were inseparable mixture with a ratio of 1:1.6. HRESIMS results revealed that they had the same molecular formula as C15H24O3 (m/z 275.1624 [M + Na]+, calcd for C15H24O3Na, 275.1618). The NMR spectroscopic analysis of the mixture led to the identification of 11,12-dihydroxy-1-tremulen-5-one (23), a known tremulane sesquiterpenoid isolated from the mushroom Conocybe siliginea [10]. The remaining spectroscopic signals were ascribable to three methyls, five methylenes (two oxygenated), three methines, and four quaternary carbons (two olefinic, one ketal carbon) (Tables 1, 4). All these data showed resemblance with those of conocenol C (16) [11], except substituted patterns at C-5. In compound 5, a hydroxy group was attached to C-5, while in conocenol C a methoxy instead, which was deferred by HMBC correlation from H-12 (δH 4.32 and 3.84) to C-5 (δC 112.8) and the HRESIMS data. Notably, the inseparable mixture of 5 and 23 seems to be the interconversion of hemiketal form 5 and ketone form 23. Therefore, compound 5 was identified as irpexolactin D (shown in Fig. 1).
Compounds 6 and 7 were inseparable mixture with a ratio of 1.9:1 according to the integration of 1H NMR spectrum. The HRESIMS analysis demonstrated that they had the same molecular formula of C15H24O3 (m/z 275.1621 [M + Na]+, calcd for 275.1618). The doubled signals in the 1H and 13C NMR spectra and similar proton peak types indicated that the two compounds were epimers. The 1D NMR spectra of the mixture showed signals which were categorized into three methyls, four methylenes (one oxygenated), five methines (two oxygenated), and three quaternary carbons (two olefinic) (Tables 2, 4). The data showed a resemblance to those of ceriponol G [12], except an absence of a methoxy signal at C-12. Thus, the planar structures of 6 and 7 were elucidated as shown in Fig. 2. The inseparable feature of 6 and 7 implied that they were semiacetal tautomers. The 12-OH of 6 and 7 were assigned as α and β orientation, respectively, by the key ROESY cross peaks between H-3 (δH 2.54)/H-12 (δH 5.23) of 6, and the absence of ROESY signals between H-3 (δH 2.27) and H-12 (δH 4.74) of 7 (Fig. 4). The 5-OH of 6 and 7 were α orientation as suggested by the ROESY correlations between H3-13/H-5 (Fig. 4). Therefore, compounds 6 and 7 were identified as irpexolactins E and F.
Table 2.
1H NMR spectroscopic data of compounds 6–11 (600 MHz, δ in ppm)
| No | 6a | 7a | 8a | 9a | 10b | 11b |
|---|---|---|---|---|---|---|
| 1 | 2.84, ddd (13.3, 13.3, 10.5) | 2.78, ddd (13.3, 13.3, 10.5) | 2.76, ddd (13.3, 13.3, 10.5) | |||
| 3 | 2.54, m | 2.27, m | 3.64, br d (12.9) | |||
| 4 |
1.98, br dd (13.5, 6.3) 1.83, ddd (13.5, 13.5, 10.6) |
2.08, m 1.71, ddd (13.5, 13.5, 10.6) |
2.00, ddd (14.2, 5.0, 2.8) 1.84, ddd (14.2, 12.9, 2.0) |
2.53, ddd (17.2, 8.1, 2.5) 2.20, ddd (17.2, 10.6, 2.6) |
2.52, br d (17.0) 2.32, br dd (17.0, 2.6) |
2.51, ddd (17.0, 4.0, 4.0) 2.33, ddd (17.0, 12.0, 4.0) |
| 5 | 3.75, dd (10.6, 5.0) | 3.74, dd (10.6, 5.0) | 3.97, ddd (5.0, 5.0, 2.0) |
1.88, ddd (14.0, 8.1, 2.6) 1.69, ddd (14.0, 10.6, 2.5) |
1.75, m 1.68, m |
1.74, m 1.68, m |
| 6 | 1.93, m | 1.93, m | 1.87, m | 2.15, overlapped | 2.16, overlapped | |
| 7 | 3.38, m | 3.37, m | 3.45, m | 2.25, ddd (10.5, 10.5, 8.6) | 2.13, overlapped | 2.13, overlapped |
| 8 |
1.47, dd (12.3, 1.6) 1.44, d (12.3) |
1.47, dd (12.3, 1.6) 1.44, d (12.3) |
1.52, dd (13.6, 12.1) 1.48, d (13.6) |
1.73, dd (13.0, 8.6) 1.58, dd (13.0, 10.5) |
1.56, overlapped 1.45, dd (13.0, 7.5) |
1.55, dd (12.7, 12.7) 1.39, dd (12.7, 7.1) |
| 9 | ||||||
| 10 |
2.04, overlapped 1.92, overlapped |
2.05, overlapped 1.89, overlapped |
1.93, br s |
1.82, dd (12.2, 7.6) 1.50, dd (12.2,12.2) |
1.97, dd (12.7, 7.7) 1.33, dd (12.2, 12.2) |
2.02, dd (12.2, 7.6) 1.27, dd (12.2, 12.2) |
| 11 |
4.39, ddd (13.0, 5.0, 2.5) 4.18, ddd (13.0, 3.6, 2.2) |
4.32, ddd (13.0, 3.5, 2.0) 4.20, ddd (13.0, 4.5, 2.5) |
4.80, br d (13.5) 4.72, br d (13.5) |
4.77, d (18.0) 4.70, d (18.0) |
4.63, br d (17.0) 4.56, br d (17.0) |
4.64, d (16.9) 4.56, d (16.9) |
| 12 | 5.23, d (5.0) | 4.74, d (7.4) | ||||
| 13 | 0.91, d (7.6) | 0.92, d (7.6) | 0.88, d (7.0) | 1.24, s | 0.95, d (7.6) | 0.95, d (7.0) |
| 14 | 1.09, s | 1.09, s | 0.91, s | 1.09, s | 1.11, s | 1.09, s |
| 15 | 1.05, s | 1.03, s | 1.13, s | 1.08, s |
3.87, d (10.5) 3.81, d (10.5) |
3.40, d (10.7) 3.37, d (10.7) |
| AcO- | 2.09, s |
aRecorded in CD3OD
bRecorded in CDCl3
The colorless oil of compound 8 possessed a molecular formula of C15H22O3, suggested by HRESIMS analysis (m/z 273.1468 [M + Na]+, calcd for C15H22O3Na, 273.1461). The 1H and 13C NMR data of 8 (Tables 2, 4) are similar to those of 6/7, suggesting that 8 is congener of 6/7. Elucidating the 2D NMR spectra of 8 suggested that C-12 in 8 was substituted by a carbonyl as supported by the HMBC correlations from H-4 (δH 2.00 and 1.84) and H-11 (δH 4.80 and 4.72) to C-12 (δC 182.6). The relative configuration of 8 was consistent with that of 6/7 by the ROESY analysis. Thus, the chemical structure of compound 8 was established as shown in Fig. 1 and the name irpexolactin G.
Irpexolactin H (9) was isolated as a colorless oil. The 1D NMR spectra (Tables 2, 5) displayed three methyl singlets, five methylenes (one oxygenated), two methines, and five quaternary carbons (two sp3 ones, and three sp2 ones). The 1D NMR data of 9 showed similarity with those of ceriponol C [12], indicating they were structurally related. Interpreting of the 2D NMR spectra of 9 suggested that the hydroxy group was substituted at C-6 in 9 instead of C-8 in ceriponol C, which supported by HMBC correlations from H3-13 (δH 1.24) to C-5 (δC 43.0), C-6 (δC 75.0), and C-7 (δC 55.7), from H-4 (δH 2.53 and 2.20) and H-8 (δH 1.73 and 1.58) to C-6. The relative configuration of H-1, 6-OH, H-7 were assigned as β, α, α, respectively, based on the ROESY signals of H-1 (δH 2.84)/H3-13/H-8β (δH 1.58), and H-7 (δH 2.25)/H-8α (δH 1.73) (Fig. 4). All these assignments led to the establishment of the structure of 9 as shown in Fig. 3, which was consistent with the HRESIMS results (m/z 273.1465 [M + Na]+, calcd for C15H22O3Na, 273.1461). Thus, compound 9 was identified as 6α-hydroxy-tremul-2-en-12,11-olide.
Table 5.
13C NMR spectroscopic data of compounds 9–15 (150 MHz, δ in ppm)
| No | 9a | 10b | 11b | 12a | 13b | 14a | 15a |
|---|---|---|---|---|---|---|---|
| 1 | 41.3, CH | 40.4, CH | 39.1, CH | 146.9, C | 146.1, C | 144.8, C | 145.0, C |
| 2 | 168.1, C | 163.1, C | 163.5, C | 131.1, C | 131.4, C | 134.3, C | 133.9, C |
| 3 | 127.9, C | 128.1, C | 127.9, C | 52.8, CH | 40.7, CH | 45.8, CH | 40.3, CH |
| 4 | 20.3, CH2 | 19.9, CH2 | 20.0, CH2 | 67.5, CH | 21.5, CH2 | 22.2, CH2 | 25.4, CH2 |
| 5 | 43.0, CH2 | 32.1, CH2 | 33.5, CH2 | 42.6, CH2 | 31.9, CH2 | 33.2, CH2 | 42.2, CH2 |
| 6 | 75.0, C | 33.4, CH | 32.2, CH | 33.9, CH | 31.4, CH | 33.3, CH | 73.3, C |
| 7 | 55.7, CH | 48.6, CH | 48.6, CH | 46.6, CH | 46.2, CH | 47.1, CH | 53.5, CH |
| 8 | 42.9, CH2 | 39.78, CH2c | 40.1, CH2 | 46.5, CH2 | 45.3, CH2 | 41.4, CH2 | 44.0, CH2 |
| 9 | 37.3, C | 39.84, Cc | 41.6, C | 38.2, C | 37.1, C | 43.7, C | 37.1, C |
| 10 | 45.1, CH2 | 39.0, CH2 | 39.2, CH2 | 48.7, CH2 | 47.8, CH2 | 43.8, CH2 | 48.1, CH2 |
| 11 | 72.3, CH2 | 70.5, CH2 | 70.6, CH2 | 66.1, CH2 | 66.3, CH2 | 66.2, CH2 | 66.1. CH2 |
| 12 | 177.6, C | 175.4, C | 175.5, C | 60.2, CH2 | 63.7, CH2 | 61.8, CH2 | 63.9, CH2 |
| 13 | 21.1, CH3 | 11.5, CH3 | 11.6, CH3 | 12.9, CH3 | 11.6, CH3 | 12.1, CH3 | 19.9, CH3 |
| 14 | 31.4, CH3 | 26.9, CH3 | 26.5, CH3 | 29.0, CH3 | 28.5, CH3 | 22.8, CH3 | 29.0, CH3 |
| 15 | 31.1, CH3 | 72.2, CH2 | 71.4, CH2 | 27.3, CH3 | 26.8, CH3 | 71.6, CH2 | 27.2, CH3 |
| CH3CO- | 171.5, C | 171.1, C | 173.1, C | ||||
| CH3CO- | 21.2, CH3 | 21.1, CH3 | 21.0, CH3 |
aRecorded in CD3OD
bRecorded in CDCl3
cAssignments were rounded to two decimal places to tell apart the close resonances
The oily compounds irpexolactins I (10) and J (11) were determined to possess the molecular formulas of C17H24O4 and C15H22O3, respectively. The 1D NMR data of 10 (Tables 2, 5) resembled to those of the co-isolate tremulenolide D (17) [13], with the main difference located at C-14. The HMBC correlations from H3-14 (δH 1.11 for 10, 1.09 for 11) to C-15 (δC 72.2 for 10, 71.4 for 11), from H-15 (δH 3.87, 3.81 for 10) and a methyl singlet at δH 2.09 to a carbonyl at δC 171.5 suggested that H3-15 in tremulenolide D (17) was oxygenated into a hydroxymethyl to give 11, which was further acetylated to yield 10 (Fig. 1). The oxygen-bearing C-15 was assigned as β orientation which evidenced by the ROESY correlation between H-15 (δH 3.87, 3.81 for 10, 3.40, 3.37 for 11) and H-1 (δH 2.78 for 10, 2.76 for 11) (Fig. 4), and the relative configurations of Me-13 and H-7 were identical with those of compound 9. Therefore, the structures of 10 and 11 were established as shown in Fig. 1.
Irpexolactin K (12) was obtained as a colorless oil. It was assigned the molecular formula of C15H26O3 on the basis of HRESIMS (m/z 277.1781 [M + Na]+, calcd for C15H26O3Na, 277.1774). The NMR spectra of 12 (Tables 3, 5) exhibited similarities with those of the co-isolate tremulenediol A (18) [3]. The key difference was that a hydroxy group substituted at C-4 in 12 compared to 18, which in accordance with the chemical formula and further confirmed by 2D NMR spectra of 12. The 1H-1H COSY correlations between H-4 (δH 4.02) and H-3 (δH 2.80), H-5 (δH 1.98 and 1.85), and HMBC correlation from H-12 (δH 4.01, 3.84) to C-2 (δC 131.1), C-3 (δC 52.8), and C-4 (δC 67.5) corroborated the postulation. The relative configuration of 4-OH was determined as α based on the significant correlation between H-3/H-4. Hence, compound 12 was determined as shown in Fig. 1.
Table 3.
1H NMR spectroscopic data of compounds 12–15 (600 MHz, δ in ppm)
| No | 12a | 13b | 14a | 15a |
|---|---|---|---|---|
| 3 | 2.80, m | 2.74, m | 2.57, m | 2.79, m |
| 4 | 4.02, overlapped |
1.80, ddd (14.2, 2.6, 2.6) 1.64, overlapped |
1.81, overlapped 1.75, overlapped |
1.85, m 1.72, overlapped |
| 5 |
1.98, ddd (12.7, 12.7, 3.5) 1.85, overlapped |
1.89, ddd (13.8, 3.0, 3.0) 1.61, overlapped |
1.78, overlapped 1.62, m |
1.95, overlapped 1.74, overlapped |
| 6 | 1.88, overlapped | 1.77, m | 1.96, m | |
| 7 | 3.07, br dd (11.3, 8.9) | 3.04, br dd (9.8, 9.8) | 3.10, br dd (11.7, 8.5) | 3.09, dd (9.6, 9.6) |
| 8 |
1.55, dd (12.3, 8.9) 1.42, dd (12.3, 11.3) |
1.51, overlapped 1.38, dd (12.8, 11.8) |
1.55, dd (11.7, 11.7) 1.43, dd (11.7, 8.5) |
1.65, br dd (13.2, 8.3) 1.57, dd (13.2, 10.7) |
| 10 |
2.31, d (15.3) 1.93, d (15.3) |
2.24, d (15.1) 1.91, d (15.1) |
2.21, br d (15.2) 2.06, br d (15.2) |
2.31, d (14.8) 1.97, d (14.8) |
| 11 |
4.11, d (11.3) 3.93, d (11.3) |
4.11, dd (11.6, 6.5) 4.01, dd (11.6, 4.5) |
4.06, d (11.3) 3.95, d (11.3) |
4.03, d (12.0) 4.00, d (12.0) |
| 12 |
4.01, overlapped 3.84, dd (9.2, 9.2) |
4.29, dd (10.5, 7.4) 4.23, dd (10.5, 8.9) |
3.73, dd (10.5, 9.0) 3.70, dd (10.5, 7.4) |
4.34, dd (10.7, 10.7) 4.19, dd (10.7, 5.7) |
| 13 | 0.92, d (7.0) | 0.82, d (7.0) | 0.87, d (7.0) | 1.08, s |
| 14 | 1.08, s | 1.06, s | 0.89, s | 1.10, s |
| 15 | 0.88, s | 0.83, s |
3.38, d (10.0) 3.40, d (10.0) |
0.86, s |
| OAc | 2.06, s | 2.04, s | ||
| 11-OH | 1.54, dd (6.5, 4.5) |
aRecorded in CD3OD
bRecorded in CDCl3
The molecular formula of irpexolactin L (13) was assigned as C17H28O3 by HRESIMS (m/z 303.1941 [M + Na]+, calcd for C17H28O3Na, 303.1941), accounting for four degrees of unsaturation. The 1H and 13C NMR data of 13 (Tables 3, 5) showed features like those of tremulenediol A (18) [3], except the presence of an acetyl group (δH 2.06; δC 21.1 and 171.1). The HMBC correlation from H-12 (δH 4.29 and 4.23) to carbonyl of acetyl group, suggesting an acetoxy group substituted at C-12. Therefore, compound 13 was elucidated as shown in Fig. 1.
Compound 14 was obtained as a colorless oil and found to possess the molecular formula of C15H26O3 based on HRESIMS data (m/z 277.1778 [M + Na]+, calcd for C15H26O3Na, 277.1774). The 1H and 13C NMR data of 14 (Tables 3, 5) were highly similar to those of conocenol A [11]. The most significant deviation of the 13C NMR data between conocenol A and 14 located on the hydroxymethyl (δC 68.7 for conocenol A (C-15), δC 71.6 for 14 (C-14)) which were recorded in the same deuterium solvent (CD3OD), implying the only difference involved in the configuration of hydroxymethyl group attached to C-9. The crucial ROESY correlations of H-7/H-8α, H-15\H-8β helped to determine the β orientation of 15-CH2OH, while it was α in conocenol A when its relative configuration was drawn as same as 14. Thus, compound 14 was established as irpexolactin M.
Irpexolactin N (15), a colorless oil, showed a sodium adduct ion peak at m/z 319.1871 [M + Na]+ in the HRESIMS spectrum, indicating the molecular formula of C17H28O4 (calcd for C17H28O4Na, 319.1880). The 1D NMR (Tables 3, 5) spectra showed resonances of four methyl singlets, six methylenes (two oxygenated), two methines, and five quaternary carbons (Tables 3, 5). All these data showed similarity with those of (+)-(3S,6R,7R)-tremulene-6,11,12-triol (19) [14]. The presence of an additional acetyl group (δH 2.04; δC 21.0 and 173.1) in 15 as well as the HMBC correlation from H-12 (δH 4.34 and 4.19) to the acetyl carbonyl indicated that the 12-OH of 15 was acetylated compared to (+)-(3S,6R,7R)-tremulene-6,11,12-triol (19). Thus, compound 15 was identified as shown in Fig. 1.
The known sesquiterpenes were identified as conocenol C (16) [11], tremulenolide D (17) [13], tremulenediol A (18) [3], (+)-(3S,6R,7R)-tremulene-6,11,12-triol (19) [14, 15], conocenol B (20) [11], (−)-(3S,6S,7S,10S)-tremulene-10,11,12-triol (21) [14, 15], irlactam A (22) [6], 11,12-dihydroxy-1-tremulen-5-one (23) [10], and irlactin A (24) [4].
The absolute configuration of conocenol B (20) was determined as 3R,5R,6S,7R by single-crystal X-ray diffraction analysis (Fig. 5).
Fig. 5.
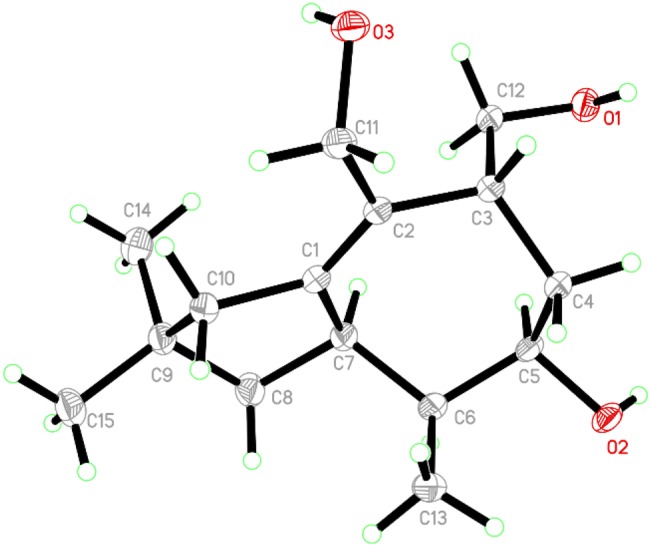
ORTEP drawing of compound 20
Biological Activities
Compounds 1, 2, 4, 12, and 22 were evaluated on vasorelaxant effect on KCl precontracted thoracic aorta rings. Nifedipine was used as the positive control. However, none of them showed significant vasorelaxant effect.
Conclusions
The chemical investigation on the culture broth of the medicinal fungus Irpex lacteus HFG1102 facilitated the isolation of 15 previously undescribed tremulane sesquiterpenes irlactin K (1), and irpexolactins A–N (2–15), and nine known ones, conocenol C (16), tremulenolide D (17), tremulenediol A (18), (+)-(3S,6R,7R)-tremulene-6,11,12-triol (19), conocenol B (20), (−)-(3S,6S,7S,10S)-tremulene-10,11,12-triol (21), irlactam A (22), 11,12-dihydroxy-1-tremulen-5-one (23), and irlactin A (24). Among the all isolates, irpexolactins A (2) and B (3) possessed an unusual 1-(2-hydroxyethyl)pyrrolidine-2,5-dione moiety. This research expands the structural diversity of mushroom derived tremulane-type sesquiterpenoids.
Experimental
General Experimental Procedures
Optical rotations were obtained on a JASCO P-1020 digital polarimeter (Horiba, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV-2401PC UV–visible recording spectrophotometer (Shimadzu, Kyoto, Japan). 1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer (Bruker Corporation, Karlsruhe, Germany). HRESIMS were recorded on an Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA, USA). HREIMS were recorded on a Waters Auto-Spec Premier P776 instrument (Waters, Milford, MA, USA). Single crystal X-ray diffraction was performed on Bruker APEX II DUO and D8 QUEST diffractometers (Bruker AXS GmbH, Karlsruhe, Germany). Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd) were used for column chromatography (CC). Medium Pressure Liquid Chromatography (MPLC) was performed on a Büchi Sepacore System equipped with pump manager C-615, pump modules C-605 and fraction collector C-660 (Büchi Labortechnik AG, Flawil, Switzerland), and columns packed with Chromatorex C-18 (dimensions 450 mm × i.d. 14 mm, particle size: 40–75 μm, Fuji Silysia Chemical Ltd., Kasugai, Japan). Preparative high performance liquid chromatography (prep. HPLC) were performed on an Agilent 1260 liquid chromatography system equipped with Zorbax SB-C18 columns (particle size 5 μm, dimensions 150 mm × i.d. 9.4 mm, flow rate 7 ml·min−1, respectively) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA).
Fungal Material
The fungus Irpex lacteus was collected from Changbai Mountain Nature Reserve in 2012, and was identified by Prof. Yu-Cheng Dai (Beijing Forestry University), an expert in mushroon taxonomy. The strain of I. lacteus in this study was isolated from the fresh fruiting bodies and kept on potato, dextrose, and agar (PDA) culture medium. A voucher specimen (No. CGBWSHFG1102) was deposited in the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences. The culture medium to ferment this fungus consisted of glucose (5%), peptone from porcine meat (0.15%), yeast powder (0.5%), KH2PO4 (0.05%) and MgSO4 (0.05%). Sixty Erlenmeyer flasks (500 ml) each containing 350 ml of above-mentioned culture medium were inoculated with I. lacteus strains, respectively. Fermentation were carried out on rotatory shakers at 25 °C and 150 rpm for 25 days in dark environment.
Extraction and Isolation
The culture broth (20 L) of I. lacteus HFG1102 was filtered and concentrated to 3 L followed by partitioned between EtOAc and water for four times to give an EtOAc layer. Meanwhile, the mycelia were extracted by EtOH (95%) for three times. The EtOAc layer together with the mycelium extract were concentrated under reduced pressure to afford a crude extract (16.0 g). This residue was separated by MPLC (ODS column) with MeOH/H2O (from 0:100 to 100:0) to give sixteen main fractions (A–P).
Subfraction E was fractionated by Sephadex LH-20 CC (MeOH) to afford five subfractions E1–E5. Compounds 12 (1.5 mg), 13 (2.4 mg), 14 (2.0 mg) were purified from E1 to E5, respectively, by Sephadex LH-20 CC using acetone as a mobile phase. Subfraction F was fractionated and purified by repeatedly Sephadex LH-20 (MeOH or acetone) to give compounds 16 (1.2 mg), 17 (1.8 mg), 19 (3.0 mg), 20 (2.5 mg).
Subfraction G was separated by Sephadex LH-20 (MeOH) to give four subfractions G1–G4. G4 was further separated by Sephadex LH-20 (acetone) to give three subfractions G4a–G4c. Compounds 10 (0.9 mg) and 15 (1.0 mg) were purified from G4a and G4b by prep-HPLC, respectively (10: MeCN/H2O, 30:70–54:46, 7 mL min−1, tR = 18.5 min; 15: MeCN/H2O, 25:75–45:55, 7 mL min−1, tR = 19.5 min). Subfraction G4c was purified by prep-HPLC (MeCN/H2O, 28:72–53:47, 7 mL min−1) to give compounds 5/23 (tR = 5.0 min, 1.2 mg), 6/7 (tR = 6.5 min, 2.2 mg).
Subfraction H was separated by Sephadex LH-20 (MeOH) to give two subfractions H1 and H2. H2 was further separated by Sephadex LH-20 (acetone) to give three subfractions H2a–H2c. H2b was separated by silica gel CC to give seven subfractions H2b1–H2b7. Subfraction H2b2 was purified by prep-HPLC to yield compound 11 (MeCN/H2O, 18:82–38:62, 7 mL min−1, tR = 17.5 min, 2.2 mg). Subfraction H2b7 was purified by prep-HPLC (MeCN/H2O, 23:77–43:57, 7 mL min−1) to yield compounds 2 (tR = 10.5 min, 1.8 mg), 22 (tR = 14.8 min, 2.3 mg), and 3 (tR = 18.2 min, 1.5 mg). and 21 (2.5 mg).
Subfraction I was separated by Sephadex LH-20 (MeOH) to give two subfractions I1 and I2. Subfraction I1 was further separated by Sephadex LH-20 (acetone) and prep-HPLC (MeCN/H2O, 28:72–48:52, 7 mL min−1) to give compound 8 (tR = 11.5 min, 3.2 mg). Subfraction I2 was further purified by prep-HPLC (MeCN/H2O, 30:70–50:50, 7 mL min−1) to give compound 4 (tR = 14.5 min, 3.0 mg).
Subfraction J was separated by Sephadex LH-20 (MeOH) and further purified by prep-HPLC (MeCN/H2O, 18:82–38:62, 7 mL min−1) to afford compound 9 (tR = 13.3 min, 2.1 mg).
Subfraction K was separated by Sephadex LH-20 (MeOH) to give two main subfractions K1 and K2. Subfraction K1 was purified by prep-HPLC (MeCN/H2O, 7:93–27:73, 7 ml min−1) to afford compound 18 (tR = 14.3 min, 1.1 mg) and 24 (tR = 16.0 mg, 1.5 mg). Subfraction K2 was purified by prep-HPLC (MeCN/H2O, 13:87–33:67, 7 mL min−1) to afford compound 1 (tR = 18.5 min, 0.9 mg).
Spectroscopic Data of Compounds
Irlactin K (1)
Colorless needles; UV (MeOH) λmax nm (log ε): 205 (2.54); 1H NMR (600 MHz, CDCl3) data, see Table 1; 13C NMR (150 MHz, CDCl3) data, see Table 4.
Irpexolactin A (2)
Pale-yellow oil; [α]26D + 79.2 (c 0.07, MeOH). UV (MeOH) λmax nm (log ε): 200 (3.69), 231 (3.96); IR (KBr) νmax 3426, 2927, 2859, 1701, 1632, 1406, 1030 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 1; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 330.1685 [M + Na]+ (calcd for C17H25NO4Na, 330.1676).
Irpexolactin B (3)
Colorless oil; [α]26D + 42.5 (c 0.10, MeOH). 1H NMR (600 MHz, CDCl3) data, see Table 1; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 330.1687 [M + Na]+ (calcd for C17H26NO4Na, 330.1676).
Irpexolactin C (4)
Colorless oil; [α]26D –95.9 (c 0.07, MeOH). UV (MeOH) λmax nm (log ε): 204 (3.64); IR (KBr) νmax 3428, 2926, 2858, 1632, 1384, 1031 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 1; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 289.1774 [M + Na]+ (calcd for C16H26O3Na, 289.1774).
Irpexolactin D (5) and 23 Mixture
Colorless oil; UV (MeOH) λmax nm (log ε): 206 (4.16), 244 (3.08); IR (KBr) νmax 3426, 2951, 2932, 2870, 1691, 1631, 1462, 1383, 1027 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 1; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 275.1624 [M + Na]+ (calcd for C15H24O3Na, 275.1618).
Irpexolactins E&F (6&7)
Yellow oil; UV (MeOH) λmax nm (log ε): 206 (3.66), 249 (3.36); IR (KBr) νmax 3426, 2953, 2928, 2861, 1632, 1385, 1029 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 2; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 275.1621 [M + Na]+ (calcd for C15H24O3Na, 275.1618).
Irpexolactin G (8)
Colorless oil; [α]26D + 17.1 (c 0.06, MeOH). UV (MeOH) λmax nm (log ε): 207 (3.66); IR (KBr) νmax 3421, 2953, 2928, 2869, 1755, 1629, 1381, 1171, 1029 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 2; 13C NMR (150 MHz, CDCl3) data, see Table 4; HRESIMS m/z 273.1468 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
Irpexolactin H (9)
Colorless oil; [α]26D + 5.7 (c 0.14, MeOH). UV (MeOH) λmax nm (log ε): 219 (3.71), 240.0 (3.27); IR (KBr) νmax 3424, 2928, 2859, 1726, 1631, 1384, 1029 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 2; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 273.1465 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
Irpexolactin I (10)
Colorless oil; [α]26D + 2.7 (c 0.10, MeOH). UV (MeOH) λmax nm (log ε): 210 (3.15); IR (KBr) νmax 3427, 2926, 2856, 1714, 1633, 1386, 1031 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 2; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 315.1560 [M + Na]+ (calcd for C17H24O4Na, 315.1567).
Irpexolactin J (11)
Colorless oil; [α]25D + 17.1 (c 0.08, MeOH). UV (MeOH) λmax nm (log ε): 219 (3.74); IR (KBr) νmax 3426, 2928, 2869, 1732, 1633, 1452, 1389, 1031 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 2; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 273.1467 [M + Na]+ (calcd for C15H22O3Na, 273.1461).
Irpexolactin K (12)
Colorless oil; [α]26D + 23.1 (c 0.08, MeOH). UV (MeOH) λmax nm (log ε): 208 (3.83); IR (KBr) νmax 3427, 2927, 2857, 1633, 1391, 1028 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 3; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 277.1781 [M + Na]+ (calcd for C15H26O3Na, 277.1774).
Irpexolactin L (13)
Colorless oil; [α]25D + 78.3 (c 0.10, MeOH). UV (MeOH) λmax nm (log ε): 210 (3.73); IR (KBr) νmax 3438, 2925, 2856, 1631, 1385, 1030 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 3; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 303.1941 [M + Na]+ (calcd for C17H28O3Na, 303.1941).
Irpexolactin M (14)
Colorless oil; [α]26D + 96.1 (c 0.10, MeOH). UV (MeOH) λmax nm (log ε): 207 (4.14), 252 (2.53); IR (KBr) νmax 3426, 2927, 2866, 1633, 1385, 1028 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 3; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 277.1778 [M + Na]+ (calcd for C15H26O3Na, 277.1774).
Irpexolactin N (15)
Colorless oil; [α]25D + 21.2 (c 0.04, MeOH). UV (MeOH) λmax nm (log ε): 208 (3.76), 250 (3.06); IR (KBr) νmax 3422, 2928, 2859, 1721, 1630, 1457, 1384, 1266, 1093, 1031 cm–1; 1H NMR (600 MHz, CDCl3) data, see Table 3; 13C NMR (150 MHz, CDCl3) data, see Table 5; HRESIMS m/z 319.1871 [M + Na]+ (calcd for C17H28O4Na, 319.1880).
Single-Crystal X-ray Diffraction Data of 1
Colorless crystals of 1 were obtained by crystallization from MeOH/H2O/petroleum ether. The crystal data were obtained from an APEX II DUO spectrophotometer (Bruker AXS GmbH, Karlsruhe, Germany) using graphite-monochromated Cu Ka radiation (λ = 1.54178 Å). The structures of 1 were solved by the direct method employing the SHELXS-97 program and then refined with full-matrix least-squares difference Fourier techniques. Crystallographic data of compound 1 was deposited to the Cambridge Crystallographic Data Centre (No. CCDC 1823228). These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/conts/retrieving.html. Crystal data for Cu_1_0m: C16H26O4, M = 282.37, a = 5.8192(2) Å, b = 9.6527(4) Å, c = 13.5674(6) Å, α = 90°, β = 93.526(2)°, γ = 90°, V = 760.65(5) Å3, T = 100(2) K, space group P21, Z = 2, μ(CuKα) = 0.702 mm−1, 6956 reflections measured, 2415 independent reflections (Rint = 0.0287). The final R1 values were 0.0300 (I > 2σ(I)). The final wR(F2) values were 0.0797 (I > 2σ(I)). The final R1 values were 0.0302 (all data). The final wR(F2) values were 0.0799 (all data). The goodness of fit on F2 was 1.070. Flack parameter = 0.15(6).
Single-Crystal X-ray Diffraction Data of 20
A light colorless platelet-like of C15H26O3, M = 254.36, approximate dimensions 0.095 × 0.116 × 0.263 mm3, was used for the X-ray crystallographic analysis on the BRUKER D8 QUEST diffractometer. The integration of the data using a monoclinic unit cell yielded a total of 12,363 reflections to a maximum θ angle of 79.29° (0.78 Å resolution), of which 3012 were independent (average redundancy 4.105, completeness = 98.1%, Rint = 5.36%, Rsig = 5.40%) and 2848 (94.56%) were greater than 2σ(F2). The final cell constants of a = 6.1670(3) Å, b = 7.7139(4) Å, c = 15.5645(8) Å, α = 90.00°, β = 93.047(2)°, γ = 90.00°, V = 739.38(6) Å3, T = 150.(2) K. Data were corrected for absorption effects using the Multi-Scan method (SADABS). The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P 1 21 1, with Z = 2, μ(CuKα) = 1.54178. The final anisotropic full-matrix least-squares refinement on F2 with 174 variables converged at R1 = 3.54%, for the observed data and ωR2 = 8.84% for all data. The goodness-of-fit was 1.009. The absolute configuration was determined by the Flack parameter = 0.01(9), which was determined using 1165 quotients [(I+) − (I−)]/[(I+) + (I−)]. Crystallographic data of compound 20 was deposited to the Cambridge Crystallographic Data Centre (No. CCDC 1977047). These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/conts/retrieving.html.
Vasorelaxant Effect Assay
Animals
Adult male Wistar rats (250–300 g) were kept in an animal room with a constant temperature of 22 ± 2 ℃, a humidity of 60 ± 5% and had free access to food and water. Experiments were performed in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Research Ethics Committee of the Kunming Institute of Botany, Chinese Academy of Sciences.
Methods
Vasorelaxant effects of 100 µmol/L of the compounds was evaluated on endothelium-intact thoracic aorta rings precontracted with KCl. Rat aortic rings were prepared according to that described [16, 17]. Nifedipine, a calcium channel blocker, was used as the positive control. Aortic rings were mounted on stainless steel hooks in organ baths containing 37 ℃ Krebs solution continuously bubbled with 95% O2 and 5% CO2, then equilibrated for 60 min under a resting tension of 1.5 g. After equilibration, the vessels were exposed to 1 μmol/l phenylephrine (PE), followed by 10 μmol/l acetylcholine (Ach) to check functional endothelial integrity, more than 80% relaxation of the rings was considered to be an endothelium-intact ring. Endothelium-intact rings precontracted with 60 mmol/l KCl were treated with different compounds, DMSO or nifedipine for 30 min or 60 min, the changes in tension of aortic rings were recorded. The vasorelaxant effect was calculated as a percentage of the relaxation in response to KCl on the aortic rings. Data were presented as mean ± SD and evaluated by one-way ANOVA followed by Bonferroni's test using SPSS 19.0. P < 0.05 was regarded to be statistically significant.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (Nos.81903512, 21961142008). We thank Analytical & Measuring Center, School of Pharmaceutical Sciences, South-Central University for Nationalities for MS and NMR spectra tests.
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Chen HP, Liu JK. Prog. Chem. Org. Nat. Prod. 2017;106:1–201. doi: 10.1007/978-3-319-59542-9_1. [DOI] [PubMed] [Google Scholar]
- 2.Dong XM, Song XH, Liu KB, Dong CH. Mycosystema. 2017;36:28–34. [Google Scholar]
- 3.Ayer WA, Cruz ER. J. Org. Chem. 1993;58:7529–7534. doi: 10.1021/jo00078a035. [DOI] [Google Scholar]
- 4.Ding JH, Feng T, Cui BK, Wei K, Li ZH, Liu JK. Tetrahedron Lett. 2013;54:2651–2654. doi: 10.1016/j.tetlet.2013.03.038. [DOI] [Google Scholar]
- 5.Ding JH, Li ZH, Feng T, Liu JK. Fitoterapia. 2018;125:245–248. doi: 10.1016/j.fitote.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Ding JH, Li ZH, Feng T, Liu JK. Nat. Prod. Res. 2019;33:316–320. doi: 10.1080/14786419.2018.1448816. [DOI] [PubMed] [Google Scholar]
- 7.Chen LL, Kong FD, Wang P, Yuan JZ, Guo ZK, Wang H, Dai HF, Mei WL. Chin. Chem. Lett. 2017;28:222–225. doi: 10.1016/j.cclet.2016.07.019. [DOI] [Google Scholar]
- 8.Chen HP, Jiang MY, Zhao ZZ, Feng T, Li ZH, Liu JK. Nat. Prod. Bioprospect. 2018;8:37–45. doi: 10.1007/s13659-017-0147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang XY, Feng T, Yin X, Li ZH, Zhang L, Liu JK. Chin. J. Chem. 2012;30:1231–1235. doi: 10.1002/cjoc.201200189. [DOI] [Google Scholar]
- 10.Zhou ZY, Tang JG, Wang F, Dong ZJ, Liu JK. J. Nat. Prod. 2008;71:1423–1426. doi: 10.1021/np8002657. [DOI] [PubMed] [Google Scholar]
- 11.Liu DZ, Wang F, Liu JK. J. Nat. Prod. 2007;70:1503–1506. doi: 10.1021/np070140n. [DOI] [PubMed] [Google Scholar]
- 12.Ying YM, Shan WG, Zhang LW, Zhan ZJ. Phytochemistry. 2013;95:360–367. doi: 10.1016/j.phytochem.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 13.Liu LY, Li ZH, Si J, Dong ZJ, Liu JK. J. Asian Nat. Prod. Res. 2013;15:300–304. doi: 10.1080/10286020.2013.763798. [DOI] [PubMed] [Google Scholar]
- 14.Wu XL, Lin S, Zhu CG, Yue ZG, Yu Y, Zhao F, Liu B, Dai JG, Shi JG. J. Nat. Prod. 2010;73:1294–1300. doi: 10.1021/np100216k. [DOI] [PubMed] [Google Scholar]
- 15.Yin RH, Zhao ZZ, Chen HP, Yin X, Ji X, Dong ZJ, Li ZH, Feng T, Liu JK. Phytochem. Lett. 2014;10:300–303. doi: 10.1016/j.phytol.2014.10.019. [DOI] [Google Scholar]
- 16.Chang GJ, Lin TP, Ko YS, Lin MS. Life Sci. 2010;86:869–879. doi: 10.1016/j.lfs.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Ji X, Trandafir CC, Wang AM, Kurahashi K. J. Stroke Cerebrovasc. 2013;22:1258–1262. doi: 10.1016/j.jstrokecerebrovasdis.2012.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.