

Abstract
Aims
We previously reported that increased protein O-GlcNAcylation in diabetic mice led to vascular rarefaction in the heart. In this study, we aimed to investigate whether and how coronary endothelial cell (EC) apoptosis is enhanced by protein O-GlcNAcylation and thus induces coronary microvascular disease (CMD) and subsequent cardiac dysfunction in diabetes. We hypothesize that excessive protein O-GlcNAcylation increases p53 that leads to CMD and reduced cardiac contractility.
Methods and results
We conducted in vivo functional experiments in control mice, TALLYHO/Jng (TH) mice, a polygenic type 2 diabetic (T2D) model, and EC-specific O-GlcNAcase (OGA, an enzyme that catalyzes the removal of O-GlcNAc from proteins)-overexpressing TH mice, as well as in vitro experiments in isolated ECs from these mice. TH mice exhibited a significant increase in coronary EC apoptosis and reduction of coronary flow velocity reserve (CFVR), an assessment of coronary microvascular function, in comparison to wild-type mice. The decreased CFVR, due at least partially to EC apoptosis, was associated with decreased cardiac contractility in TH mice. Western blot experiments showed that p53 protein level was significantly higher in coronary ECs from TH mice and T2D patients than in control ECs. High glucose treatment also increased p53 protein level in control ECs. Furthermore, overexpression of OGA decreased protein O-GlcNAcylation and down-regulated p53 in coronary ECs, and conferred a protective effect on cardiac function in TH mice. Inhibition of p53 with pifithrin-α attenuated coronary EC apoptosis and restored CFVR and cardiac contractility in TH mice.
Conclusions
The data from this study indicate that inhibition of p53 or down-regulation of p53 by OGA overexpression attenuates coronary EC apoptosis and improves CFVR and cardiac function in diabetes. Lowering coronary endothelial p53 levels via OGA overexpression could be a potential therapeutic approach for CMD in diabetes.
Keywords: Coronary microcirculation, Coronary blood flow, Apoptosis, Capillaries, Cardiovascular disease
Graphical Abstract
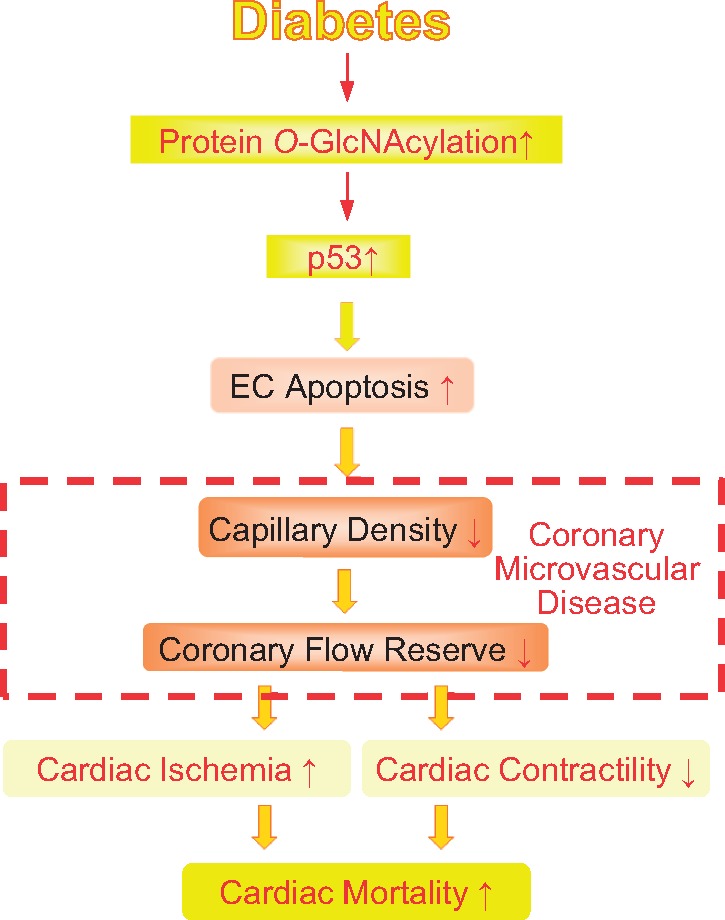
Graphical Abstract.
1. Introduction
The increasing prevalence of diabetes is a global phenomenon and many people with diabetes suffer from and die of heart disease or stroke. Diabetic heart disease includes diabetic cardiomyopathy and coronary artery disease (CAD). CAD is classified into two groups: obstructive CAD and coronary microvascular disease (CMD, also known as non-obstructive CAD). Recent reports emphasize the importance of CMD in diabetes as a risk factor for cardiac mortality.1,2 The mechanisms responsible for CMD include microvascular rarefaction, small vascular remodelling, and attenuated vasodilatation in coronary small arteries.3,4 Coronary vascular endothelial injury and dysfunction are implicated in the development and progression of CMD; however, the detailed sequence of events remains unclear.
Mutation of transcription factor p53 is associated with 50% of known cancers and mutant p53 promotes cancer cell proliferation and survival. p53 is known to activate cell apoptosis,5 and overexpression of p53 is one of the causes for cell death during cardiac ischaemia.6,7 p53 levels are significantly increased in normal endothelial cells (ECs) following high glucose (HG) treatment,8 and in diabetic hearts.9 In this study, we will examine whether and how p53 is up-regulated in coronary ECs in diabetes and if increased p53 alters coronary microvascular function and subsequently changes cardiac contractility in diabetes.
Protein O-GlcNAcylation is a post-translational modification that involves the addition of N-acetylglucosamine (GlcNAc) to serine or threonine residues of proteins. Two enzymes regulate protein O-GlcNAc modification: O-GlcNAc transferase that catalyzes the addition of O-GlcNAc and O-GlcNAcase (OGA) that removes O-GlcNAc from proteins.10 Excessive protein O-GlcNAcylation has been detected in diabetes and it contributes to the functional changes in many tissues including the endothelium.11–15 The activity of endothelial nitric oxide synthase is decreased by its O-GlcNAcylation, through inhibition of Ser1177 phosphorylation.12 HG treatment increases protein O-GlcNAc modification and decreases angiogenic capacity in ECs, due in part to decreased Akt phosphorylation.13 Excess protein O-GlcNAcylation in ECs increases ICAM-1 levels through Sp1 up-regulation.14 Yao et al.15 reported that up-regulation of ICAM-1 and VCAM1 by HG treatment was mediated by increased angiopoietin-2 transcription through O-GlcNAcylation of Sp3. We have demonstrated that O-GlcNAcylation of connexin 40, a component of gap junctions, attenuates intercellular communication between ECs, and decreases endothelium-derived hyperpolarization-dependent relaxation in coronary artery of type 1 diabetic (T1D) mice.11 Despite the extensive studies, few reports have examined the role of endothelial OGA (or protein O-GlcNAcylation) in coronary microvascular dysfunction in type 2 diabetes.
In this study, we report that hyperglycaemia leads to p53 overexpression in coronary ECs via excessive protein O-GlcNAcylation. OGA overexpression in ECs down-regulates p53 expression and subsequently restores coronary blood flow (CBF) and cardiac function in type 2 diabetic (T2D) mice.
2. Methods
2.1 Animals
All experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Arizona (UA) and the University of California, San Diego (UCSD) and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The universities have been certified by Public Health Service with Animal Welfare Assurance number A3248-01 (UA) and A3033-01 (UCSD); the approved IACUC protocol numbers for this study are 14-520 (at UA) and S18185 (at UCSD). The laboratory personnel who conducted following experiments took all trainings required for animal handling and were certified by the IACUC.
Male TALLYHO/Jng (TH) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred in our animal facility. The TH mice are a polygenic T2D model and male TH mice exhibit hyperglycaemia, hyperinsulinaemia, hyperlipidaemia, and obesity.16,17 We used age-matched male C57BL/6 mice as wild type (Wt) controls according to the Jackson Laboratory guideline. All mice were fed with a normal chow diet. TH and Wt mice were used for the experiments at age of 17–24 weeks. Pifithrin-α (PFT) is a chemical developed as a specific blocker of p53 transcriptional activity and it is widely used to inhibit the downstream signalling of p53.18–20 We purchased it from Cayman Chemical (Ann Arbor, MI, USA) and administered to mice by intraperitoneal (i.p.) injection (q.o.d.) at 2.2 mg/kg for 4 weeks.21,22 0.22% DMSO (vehicle) was injected as a vehicle control. Oral glucose tolerance test (OGTT) and measurements of plasma cholesterol, high density lipoprotein, triglyceride, and insulin levels were performed as described previously.23
Mice were anesthetized with a mixture of ketamine (100 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.) before heart dissection (one time injection per mouse). All efforts were made to minimize suffering. Mice that underwent ischaemia/reperfusion (I/R) surgery were given buprenorphine SR (1.0 mg/kg, s.c., once) following anesthaesia (1% isoflurane, constant inhalation during surgery, Henry Schein, Melville, NY, USA) to minimize pain during the recovery phase. Animals were monitored daily following all surgical procedures, and evaluated for signs of pain, distress, or morbidity.
2.2 EC-specific OGA overexpression in TH mice
EC-specific, tetracycline-inducible OGA overexpressing mice were generated in our laboratory.11 The background strain for OGA overexpressing mice is C57BL/6. OGA overexpressing mice were crossed with TH mice and the resulting mice, termed THOGA, were used for OGA overexpression in the T2D model. We performed OGTT at 7 weeks of age, and THOGA mice with a diabetic phenotype (Dia-THOGA) were then randomly separated into two groups: one group with doxycycline (DOX) treatment (+DOX) and the other without DOX treatment (−DOX). Mice with the glucose level >150 mg/dL at time 0 and >250 mg/dL at time 60 min by OGTT were classified as diabetic mice. THOGA mice that did not develop T2D still exhibited an obese phenotype and were excluded from these studies. OGA overexpression was achieved by DOX administration in drinking water (0.2 mg/mL) for 6 weeks (DOX was started at 10 weeks of age and mice were used for the experiments at 16 weeks of age 6 weeks after DOX administration).
2.3 Coronary flow velocity reserve measurement
Coronary flow velocity reserve (CFVR), instead of coronary flow reserve (CFR), was used to assess coronary microvascular function because of the difficulty in precisely measuring coronary arterial diameter in mice.24 Coronary blood flow velocity (CFV) was measured using a Vevo 2100 system (FUJIFILM Visual Sonics, Inc., Toronto, Canada). Mice were anesthetized with isoflurane (1%) and the resting level of CFV was obtained. CFVR was defined as the ratio of the maximal hyperaemic CFV (induced by 2.5% isoflurane) divided by the resting CFV (1% isoflurane).25
2.4 Analysis of EC apoptosis and capillary densities in the left ventricular myocardium
The evaluation of EC apoptosis and capillary density in the left ventricle (LV) were conducted as described previously.11,26–28
2.5 Myocardial I/R-induced infarction
We performed in vivo I/R on mice under anesthaesia with isoflurane. The left anterior descending coronary artery (LAD) was ligated for 30 min, followed by reperfusion for 48 h.29 The LAD was then reoccluded and 2% Evans Blue dye was injected through the descending aorta to determine the area at risk (AAR, the area without blue colour). The hearts were then dissected, frozen, and cut into 1 mm-thick transverse sections. The tissue sections were stained with 1% 2,3,5-triphenyltetrazolium chloride for 10 min at 37°C to determine the infarcted area (white colour). Percentage of infarct size divided by AAR was compared between groups.
2.6 Haemodynamic studies
In mice anesthetized with isoflurane, a pressure-volume loop catheter (PVR-1030, Millar Instruments, Houston, TX, USA) was inserted into the right carotid artery and advanced into the LV.29 Haemodynamic parameters were measured and characterized using an MPVS Ultra system (Millar Instruments) and recorded by Lab Chart Pro 8 (AD Instruments Inc., Colorado Springs, CO, USA).
2.7 Isolation of mouse coronary ECs
Mouse coronary ECs (MCECs) were isolated using a method described previously.11,23,26
2.8 Western blot analysis
Cells were lysed using NP40-based lysis buffer with protease inhibitor cocktail, phosphatase inhibitor, and PugNAc. Protein levels were analysed using SDS-PAGE separation and electrophoretic transfer to nitrocellulose membranes. Primary antibodies used in this study are listed in Supplementary material online, Table S1.
2.9 Human coronary ECs
Human coronary endothelial cells (HCECs) from five control and four T2D patients were purchased from commercial suppliers (Supplementary material online, Table S2) and cultured in EC media composed of M199 supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 20 μg/mL endothelial cell growth supplement (ECGS), and 16 U/mL heparin.
2.10 HG treatment and OGA gene transduction in HCECs
For HG treatment, 20 mmol/L glucose was added to growth media (final glucose concentration: 25 mmol/L). In normal glucose (NG) control cells, 20 mmol/L mannitol was added to exclude the potential effect of changes in osmolality (NG; glucose concentration: 5 mmol/L). Cells were cultured for 96 h before experiments. Adenovirus (Adv) encoding human OGA (OGA-Adv) was used to introduce the OGA transgene into HCECs.11 Adv with empty vector (Control-Adv) was used as a control. Cells were transduced at a concentration of 100 pfu/cell 1 day before initiating HG treatment.
2.11 Tube formation assay in HCECs
HCECs (4 × 104) were seeded on Matrigel26 and treated with PFT (20 µmol/L) or vehicle (0.008% DMSO).18 Twenty-four hours after plating cells, four microscopic fields, selected at random, were photographed using an EVOS FL Auto Cell Imaging System with 4× objective lens (Thermo Fisher Scientific Inc., Rockford, IL, USA). Total tube lengths and capillary network density were analysed using ImageJ 1.51k software.
2.12 Real-time PCR
mRNA levels were measured by real time PCR.11,23 18S ribosomal RNA (18S) was used as an endogenous reference gene. Primers sequences are listed in Supplementary material online, Table S4.
2.13 Data and statistical analysis
Data collection and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology. We conducted data analysis in a blinded fashion wherever possible and set proper controls for every experimental approach. The mouse numbers and independent experiment numbers are described in the figure legend.
Statistical analysis was performed using GraphPad Prism 7.04 (La Jolla, CA, USA). Data are presented as mean ± S.E. Two-tailed Student’s t-test was used for comparison of two groups, and one-way ANOVA was used for comparison of multiple groups. Statistical comparison between dose-response curves was made by two-way ANOVA with Bonferroni post hoc test. Differences were considered to be statistically significant when P < 0.05.
2.14 Materials
Medium 199 (M199), streptomycin/penicillin, trypsin/EDTA, ECGS, FBS, Matrigel, and Dynabeads® Sheep Anti-Rat IgG were obtained from Thermo Fisher Scientific. Collagenase II and dispase II were purchased from Worthington Biochemical Corporation (Lakewood, NJ, USA). Rat anti-mouse CD31 was obtained from BD Biosciences (San Jose, CA, USA). Ketamine, xylazine, and buprenorphine SR were purchased from Henry Schein. All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA).
3. Result
3.1 Metabolic characteristics
Blood samples were collected from Wt mice and TH mice to measure metabolic characteristics.23 TH mice exhibited increased body weight, heart weight, hyperglycaemia, hyperinsulinaemia, and hyperlipidaemia (Table 1). Furthermore, TH mice showed impaired glucose tolerance in comparison to Wt mice (Figure 1A).
Table 1.
Pharmacological and haemodynamic parameters of Wt and TH mice with or without I/R
| Without I/R |
||||
|---|---|---|---|---|
| Wt (n = 8) | TH (n = 6) | |||
| Body weight (g) | 27.4 ± 0.4 | 32.4 ± 1.0* | ||
| Blood glucose (mg/dL) | 156.3 ± 13.2 | 492.0 ± 37.9* | ||
| Insulin (ng/mL) | 1.9 ± 0.2 | 3.4 ± 0.2* | ||
| TC (mg/dL) | 121.3 ± 21.8 | 182.5 ± 9.0* | ||
| HDL (mg/dL) | 45.7 ± 3.6 | 91.3 ± 9.8* | ||
| TG (mg/dL) | 32.5 ± 7.7 | 264.6 ± 49.1* | ||
|
| ||||
|
Without I/R
|
With I/R
|
|||
| WT (n = 6) | TH (n = 6) | WT (n = 6) | TH (n = 6) | |
|
| ||||
| Body weight (g) | 27.3 ± 0.5 | 31.5 ± 1.3* | 25.4 ± 1.0 | 32.9 ± 1.5* |
| Heart weight (mg) | 108.7 ± 1.9 | 127.5 ± 3.7* | 105.8 ± 1.6 | 117.8 ± 1.9* |
| Heart weight/body weight | 4.0 ± 0.1 | 4.1 ± 0.3 | 4.2 ± 0.1 | 3.6 ± 0.2* |
| Blood glucose (mg/dL) | 164.5 ± 12.8 | 521.5 ± 42.1* | 163.0 ± 8.5 | 545.2 ± 10.6* |
| Mean arterial pressure (mmHg) | 117.2 ± 3.3 | 123.5 ± 2.1 | 116.0 ± 2.4 | 126.0 ± 3.8 |
| Heart rate (bpm) | 530.3 ± 14.4 | 514.2 ± 8.2 | 527.3 ± 10.3 | 520.2 ± 8.1 |
| Cardiac output (µL/min) | 12 031 ± 1248 | 8471 ± 616* | 11 243 ±629 | 7730 ±747* |
| Stroke volume (µL) | 23.2 ± 2.5 | 17.2 ± 1.2 | 21.9 ± 0.7 | 15.1 ± 1.2* |
| LVSP (mmHg) | 104.3 ± 6.6 | 111.8 ± 5.3 | 105.8 ± 6.9 | 87.1 ± 2.1* |
| dP/dt max (mmHg/s) | 7399 ± 384 | 5746 ± 445* | 5981 ± 182 | 3580 ± 202* |
| dP/dt min (mmHg/s) | −5456 ± 350 | −4055 ± 349* | −4308 ± 190 | −2378 ± 144* |
| Ejection fraction (%) | 48.2 ± 1.8 | 34.0 ± 1.1* | 38.8 ± 1.1 | 26.2 ± 1.0* |
| ESPVR (mmHg/µL) | 7.1 ± 0.4 | 3.2 ± 0.3* | 3.9 ± 0.1 | 2.1 ± 0.1* |
ESPVR, end-systolic pressure–volume relation; HDL, plasma high density lipoprotein; LVSP, left ventricle systolic pressure; TC, plasma total cholesterol; TG: plasma triglyceride.
N shows the number of mice. Data are mean ± S.E.
P < 0.05 vs. Wt.
Figure 1.

Characterization of Tallyho (TH) mice. (A) Oral glucose tolerance test (OGTT). Wild-type mice (Wt, nmice = 7); TH mice (TH, nmice = 7). (B) Coronary flow velocity reserve (CFVR). Wt, nmice = 6; TH, nmice = 5. (C) Representative photographs showing the capillary density and EC apoptosis. ECs were stained by BS-lectin-FITC (a marker of ECs) and apoptotic cells were detected by TUNEL staining (red). An arrow indicates co-stained cells (apoptotic ECs, orange). Bar = 50µm. (D) Averaged data showing the capillary density in Wt (nmice = 5) and TH (nmice = 5). (E) Summarized data of the percentage of apoptotic ECs (the number of apoptotic ECs divided by total number of ECs). Nmice = 5 for each group. (F) Representative images of hearts after I/R. The area at risk (AAR) is shown in non-blue (= white + red) and the infarcted area is shown in white. Five cross sections (each 1 mm thick) from the ligation site to the apex. Averaged data of infarcted area in the AAR (G) and the AAR in the ventricle (H). Nmice = 6 per group. (I) Protein O-GlcNAcylation in MCECs was detected by the anti-O-GlcNAc antibody (RL2). Arrow indicates a representative band of proteins showing increased O-GlcNAcylation. Actin was used as a loading control. (J) Summarized data of the intensity of the band indicated by the arrow (left) and total RL2 signals (right) (nmice = 7 per group). Data are mean ± S.E. *P < 0.05 vs. Wt. Statistical comparison between time-response curves was made by two-way ANOVA with Bonferroni post hoc test. Unpaired Student’s t-test was used for comparisons of two experimental groups.
3.2 Reduced CFVR, decreased capillary density, and increased endothelial apoptosis in the LV of TH mice
CFVR was measured to assess coronary microvascular function in mice.24Figure 1B demonstrates that CFVR was significantly decreased in TH mice compared to Wt mice. One of the causes for reduced CFVR is vascular rarefaction in the heart. We found that TH mice exhibited decreased capillary density (Figure 1D) and increased EC apoptosis (Figure 1E) in the LV compared to Wt mice.
3.3 Decreased cardiac contractility and increased susceptibility to I/R in TH mice
It has been shown that sustained reduction of CBF attenuates cardiac function.30,31Table 1 demonstrates that TH mice exhibit attenuated cardiac contractility compared to Wt mice. Decreased capillary density also exacerbates cardiac damage following I/R. We therefore tested the effect of 30-min occlusion and 48-h reperfusion of the LAD on infarct size in Wt and TH mice. Figure 1F–H shows that infarct size after myocardial I/R was significantly larger in TH mice than in Wt mice.
3.4 Increased protein O-GlcNAc modification in coronary ECs isolated from TH mice
Hyperglycaemia and cellular stresses increase pathophysiological protein O-GlcNAcylation.11–13 We isolated mouse coronary ECs (MCECs) from Wt and TH mice and compared the levels of protein O-GlcNAc modification. O-GlcNAc modification was detected with RL2 (an anti-O-GlcNAc antibody).11 As shown in Figure 1I and J, protein O-GlcNAcylation was significantly increased in MCECs of TH mice compared to Wt mice.
3.5 EC-specific OGA overexpression inhibits protein O-GlcNAcylation in MCECs and restores endothelial and cardiac functions in TH mice
DOX administration successfully increased OGA protein levels and decreased protein O-GlcNAc modification in MCECs of Dia-THOGA mice (Figure 2A and B). OGA overexpression in ECs did not alter the glucose tolerance (Figure 2C), nor the body weight, heart weight, hyperglycaemia, hyperinsulinaemia, or dyslipidaemia seen in Dia-THOGA mice (Table 2). However, OGA overexpression in Dia-THOGA mice fully restored CFVR with partial, but significant, improvement of capillary density and EC apoptosis in the LV towards the level seen in Wt mice (Figure 2D–F). Furthermore, attenuated cardiac contractility in Dia-THOGA mice was improved by OGA overexpression in ECs (Table 2).
Figure 2.

Characterization of Dia-THOGA mice. (A) Western blots showing OGA and Actin protein levels in MCECs (left panel). Right dot plot shows OGA protein level normalized to Actin. Nmice = 5 per group. (B) Protein O-GlcNAcylation in MCECs detected with RL2 antibody (left). Dot plots show the summarized data of the intensity of the band indicated by the arrow (A and B) and total RL2 signals (right). Nmice = 5 per group. (C) OGTT. Wt, nmice = 7; Dia-THOGA w/o DOX, nmice = 11; Dia-THOGA + DOX, nmice = 9. (D) CFVR. Wt, nmice = 6; Dia-THOGA w/o DOX, nmice = 8; Dia-THOGA + DOX, nmice = 6. (E) Capillary density. Wt, nmice = 8; Dia-THOGA w/o DOX, nmice = 6; Dia-THOGA + DOX, nmice = 8. (F) Apoptotic ECs. Wt, nmice = 8; Dia-THOGA w/o DOX, nmice = 6; Dia-THOGA + DOX, nmice = 8. Data are mean ± S.E. *P < 0.05 vs. Wt, #P < 0.05 vs. Dia-THOGA + DOX. Statistical comparison between time-response curves was made by two-way ANOVA with Bonferroni post hoc test. Statistical comparison between three groups was made by one-way ANOVA with Bonferroni post hoc test.
Table 2.
Pharmacological and haemodynamic parameters of Wt, Dia-THOGA − DOX, and Dia-THOGA + DOX
| Wt | Dia-THOGA − DOX | Dia-THOGA + DOX | |
|---|---|---|---|
| Pharmacological parameters | N = 6 | N = 9 | N = 9 |
| Body weight (g) | 26.7 ± 1.2 | 48.5 ± 1.9* | 52.2 ± 1.3* |
| Blood glucose (mg/dL) | 125.8 ± 7.6 | 398.4 ± 44.9* | 389.3 ± 47.3* |
| Insulin (ng/mL) | 1.3 ± 0.5 | 3.6 ± 0.1* | 3.5 ± 0.1* |
| TC (mg/dL) | 96.8 ± 16.2 | 183.0 ± 22.7* | 194.8 ± 15.3* |
| HDL (mg/dL) | 67.7 ± 14.7 | 88.8 ± 11.7 | 124.2 ± 6.8* |
| TG (mg/dL) | 44.8 ± 10.7 | 106.2 ± 35.4 | 126.2 ± 39.1 |
| Haemodynamic parameters | N = 8 | N = 6 | N = 8 |
| Body weight (g) | 24.7 ± 1.3 | 48.9 ± 3.0* | 47.1 ± 1.4* |
| Heart weight (mg) | 108.3 ± 3.0 | 168.9 ± 6.9* | 164.6 ± 8.0* |
| Heart weight/body weight | 4.9 ± 0.4 | 3.5 ± 0.3* | 3.5 ± 0.1* |
| Blood glucose (mg/dL) | 120.0 ± 6.0 | 177.2 ± 13.1* | 210.1 ± 16.7* |
| Mean arterial pressure (mmHg) | 119.5 ± 3.2 | 115.3 ± 5.5 | 118.1 ± 4.0 |
| Heart rate (bpm) | 519.0 ± 11.2 | 533.3 ± 5.5 | 521.3 ± 9.3 |
| Cardiac output (µL/min) | 9050 ± 825 | 6250 ± 424* | 8226 ± 665 |
| Stroke volume (µL) | 19.8 ± 1.3 | 13.4 ± 1.0* | 16.4 ± 1.0 |
| LVSP (mmHg) | 100.0 ± 1.7 | 89.8 ± 4.5 | 98.0 ± 2.7 |
| dP/dt max (mmHg/s) | 5941 ± 224 | 3128 ± 358* | 4509 ± 239*,# |
| dP/dt min (mmHg/s) | −4587 ± 136 | −2706 ± 129* | −3373 ± 224* |
| Ejection fraction (%) | 46.9 ± 1.1 | 24.8 ± 1.1* | 32.6 ± 0.9*,# |
| ESPVR (mmHg/µL) | 5.5 ± 0.4 | 3.1 ± 0.3* | 4.7 ± 0.3# |
N shows the number of mice. Data are mean ± S.E.
P < 0.05 vs. Wt.
P < 0.05 vs. Dia-THOGA − DOX.
3.6 Exposure to HG up-regulates p53 protein levels in HCECs and OGA overexpression down-regulates p53 protein levels in HG-treated HCECs
There is increasing evidence showing that p53 protein levels are regulated by O-GlcNAcylation.32 To examine whether excess protein O-GlcNAcylation is involved in the p53 overexpression observed in HG-treated HCECs, we first tested whether OGA overexpression altered p53 levels in coronary ECs. To induce O-GlcNAcylation, HCECs were treated with HG for 4 days. As shown in Figure 3C, protein O-GlcNAcylation was significantly increased by HG treatment. OGA-Adv transduction increased OGA protein levels (Figure 3B) and decreased protein O-GlcNAcylation (Figure 3C). Furthermore, we found that OGA overexpression significantly decreased p53 levels in HG-treated HCECs (Figure 3D).
Figure 3.

Effect of hyperglycaemia and OGA overexpression on protein O-GlcNAcylation, p53 protein expression, and tube formation. (A) Representative Western blot image of OGA, O-GlcNAcylation (RL2 antibody), and Actin. (B) OGA protein levels in HCECs treated with normal glucose (NG, 5 mmol/L glucose + 20 mmol/L Mannitol, 4 days) or high glucose (HG, 25 mmol/L glucose, 4 days) with control adenovirus (Cont-Adv) or OGA-Adv transduction. Nexperiments = 6 per group. *P < 0.05 vs. NG + Cont-Adv. (C) Protein O-GlcNAcylation detected by RL2 antibody. Nexperiments = 6 per group. *P < 0.05 vs. NG + Cont-Adv, #P < 0.05 vs. HG + Cont-Adv. (D) p53 protein levels detected by Western blot. Nexperiments = 8 per group. *P < 0.05 vs. NG + Cont-Adv, #P < 0.05 vs. HG + Cont-Adv. (E) p53 protein levels in HCECs from control and diabetic patients detected by Western blot. Control patients (C, npatients = 5); and T2D patients (D, npatients = 4). *P < 0.05 vs. Cont. (F) p53 protein levels in MCECs freshly isolated from Wt and TH mice detected by Western blot. Nmice = 15 per group. *P < 0.05 vs. Wt. (G) p53 protein levels in MCECs freshly isolated from Wt, Dia-THOGA w/o DOX and Dia-THOGA + DOX detected by Western blot. Nmice = 6 per group. *P < 0.05 vs. Wt. (H) Representative photograph image of tube formation. (I) Summarized data of total length, branch numbers, and junction numbers in control HCECs with vehicle (0.008% DMSO), diabetic HCECs with vehicle, and diabetic HCECs with PFT treatment (20 µmol/L, 24 h). Nexperiments = 6 per group. *P < 0.05 vs. Cont + vehicle, #P < 0.05 vs. Dia + vehicle. Data are mean ± S.E. Unpaired Student’s t-test was used for comparisons of two experimental groups. Statistical comparison between three groups was made by one-way ANOVA with Bonferroni post hoc test.
3.7 p53 protein levels are increased in coronary ECs from diabetic patients and mice, and in vivo OGA overexpression decreases p53 protein levels in diabetic mice
Consistent with the results from HG-treated HCECs, HCECs from T2D patients (Figure 3E) and MCECs from TH mice (Figure 3F) both exhibited a significant increase in p53 protein levels. Furthermore, increased p53 levels in MCECs from Dia-THOGA mice were diminished by OGA overexpression towards the level seen in Wt mice (Figure 3G).
3.8 Inhibition of p53 restores tube formation in HCECs from diabetic patients
Capillary density in vivo is regulated by endothelial proliferation, migration, and apoptosis. To assess the angiogenic capability of ECs, an ex vivo tube formation assay was performed using HCECs from control and diabetic patients. Figure 3H and I shows that HCECs from diabetic patients exhibited significantly less tube formation than cells from control patients, while treatment with PFT18 (a p53 inhibitor) in HCECs from diabetic patients restored the level of tube formation towards that in control HCECs.
3.9 Inhibition of p53 restores the levels of CFVR, capillary density, EC apoptosis, and decreases the cardiac damage after I/R, in TH mice
Four-week administration of PFT did not change the pharmacological parameters in TH mice (Figure 4A and Table 3), but increased CFVR (Figure 4B) and capillary density in the LV (Figure 4C), and decreased coronary EC apoptosis (Figure 4D) in TH mice. In addition, cardiac contractility was enhanced and mean arterial pressure was decreased by chronic PFT administration in TH mice (Table 3). The infarct size after I/R was partially, but significantly, decreased after PFT administration in TH mice (Figure 4E).
Figure 4.

Effect of chronic PFT treatment in TH mice on haemodynamics and mRNA levels of genes involved with p53 signalling. (A) OGTT. Wt + vehicle, nmice = 10; TH + vehicle, nmice = 11; TH + PFT, nmice = 10. (B) CFVR. Wt + vehicle, nmice = 8; TH + vehicle, nmice = 9; TH + PFT, nmice = 9. (C) Capillary density. Wt + vehicle, nmice = 5; TH + vehicle, nmice = 5; TH + PFT, nmice = 6. (D) Apoptotic ECs. Wt + vehicle, nmice = 5; TH + vehicle, nmice = 5; TH + PFT, nmice = 6. (E and F) Infarcted area in the AAR (E) and the AAR in the ventricle (F). Nmice = 6 per group. (G) p53 mRNA levels in MCECs. Wt + vehicle, nmice = 8; TH + vehicle, nmice = 8; TH + PFT, nmice = 8. (H) Tnf mRNA levels in MCECs. Wt + vehicle, nmice = 7; TH + vehicle, nmice = 6; TH + PFT, nmice = 6. (I) Bax mRNA levels in MCECs. Wt + vehicle, nmice = 7; TH + vehicle, nmice = 8; TH + PFT, nmice = 7. (J) Bbc3 mRNA levels in MCECs. Wt + vehicle, nmice = 7; TH + vehicle, nmice = 7; TH + PFT, nmice = 7. (K) Casp2 mRNA levels in MCECs. Wt + vehicle, nmice = 7; TH + vehicle, nmice = 6; TH + PFT, nmice = 6. Data are mean ± S.E. *P < 0.05 vs. Wt + vehicle, #P < 0.05 vs. TH + vehicle. Statistical comparison between time-response curves was made by two-way ANOVA with Bonferroni post hoc test. Statistical comparison between three groups was made by one-way ANOVA with Bonferroni post hoc test.
Table 3.
Pharmacological and haemodynamic parameters of Wt + vehicle, TH + vehicle, and TH + PFT
| Wt + vehicle | TH + vehicle | TH + PFT | |
|---|---|---|---|
| Pharmacological parameters | N = 6 | N = 10 | N = 8 |
| Body weight (g) | 31.1 ± 0.7 | 36.1 ± 1.1 | 36.1 ± 1.7 |
| Blood glucose (mg/dL) | 132.2 ± 9.8 | 354.0 ± 49.2* | 376.6 ± 25.2* |
| Insulin (ng/mL) | 1.1 ± 0.3 | 3.6 ± 0.1* | 3.7 ± 0.1* |
| TC (mg/dL) | 88.2 ± 3.8 | 209.9 ± 22.5* | 200.6 ± 30.8* |
| HDL (mg/dL) | 56.6 ± 6.1 | 81.4 ± 7.4 | 86.6 ± 8.3* |
| TG (mg/dL) | 24.0 ± 5.4 | 210.2 ± 26.5* | 189.9 ± 34.0* |
| Haemodynamic parameters | N = 5 | N = 5 | N = 6 |
| Body weight (g) | 31.3 ± 0.6 | 38.5 ± 3.2* | 36.3 ± 1.7 |
| Heart weight (mg) | 110.8 ± 3.9 | 132.2 ± 2.6* | 132.3 ± 2.0* |
| Heart weight/body weight | 3.5 ± 0.1 | 3.4 ± 0.1 | 3.7 ± 0.2 |
| Blood glucose (mg/dL) | 118.2 ± 4.5 | 461.0 ± 14.6* | 401.5 ± 25.1* |
| Mean arterial pressure (mmHg) | 117.8 ± 4.9 | 140.6 ± 4.5* | 120.8 ± 2.4# |
| Heart rate (bpm) | 496.1 ± 3.1 | 526.5 ± 21.5 | 522.6 ± 11.3 |
| Cardiac output (µL/min) | 9813 ± 1010 | 7986 ± 474 | 8881 ± 866 |
| Stroke volume (µL) | 21.7 ± 1.6 | 15.4 ± 0.6* | 17.9 ± 1.1 |
| LVSP (mmHg) | 110.5 ± 3.0 | 105.2 ± 7.2 | 101.2 ± 5.1 |
| dP/dt max (mmHg/s) | 6374 ± 105 | 3146 ± 87* | 4112 ± 133*,# |
| dP/dt min (mmHg/s) | −4352 ± 195 | −2326 ± 191* | −3169 ±181*,# |
| Ejection fraction (%) | 47.1 ± 2.4 | 31.3 ± 1.5* | 37.2 ± 0.2*,# |
| ESPVR (mmHg/µL) | 4.3 ± 0.7 | 2.1 ± 0.1* | 3.1 ± 0.1 |
N shows the number of mice. Data are mean ± S.E.
P < 0.05 vs. Wt + vehicle.
P < 0.05 vs. TH + vehicle.
3.10 Identification of genes that are regulated by p53 in TH mice
MCECs were freshly isolated from Wt mice, TH mice, and TH mice administered PFT for 4 weeks. We first used a Mouse p53 Signalling Pathway PCR Array (QIAGEN) and screened mRNAs. Four out of 84 mRNAs were selected for validation by real-time PCR based on the following criteria: (i) the level is significantly altered in TH mice and is restored by PFT treatment and (ii) the altered level is consistent with the increased cell apoptosis observed in TH mice. Based on these criteria, we focused on four mRNAs: tumour necrosis factor (Tnf), Bcl2-associated X protein (Bax), Bcl-2-binding component 3 (Bbc3), and caspase 2 (Casp2) (Supplementary material online, Table S3). Figure 4H–K shows that Tnf, Bax, and Bbc3 mRNAs were significantly increased in TH mice, while PFT treatment decreased the expression level of these genes to a level similar to that in Wt mice. There was, however, no statistically significant difference in Casp2 mRNA level among the three groups. We also found that PFT treatment decreased p53 mRNA level in TH mice (Figure 4G).
4. Discussion
CMD leads to cardiac ischaemia and heart attack.30,31 The major difference between obstructive CAD and CMD is the cause of the disease. Obstructive CAD is primarily initiated by plaque development in coronary arteries and the blockage or narrowing of the vessels that leads to ischaemia. On the other hand, CMD is caused by the loss of capillaries or increased small vessel contraction. The angiograph of patients with chest pain due to CMD (without CAD) is normal and therefore often misdiagnosed as another disease such as myocarditis. CFR is commonly used as an indicator of CMD in patients without obstructive CAD4,33,34 and CFR is decreased in patients with diabetes.35–38 Coronary microvascular rarefaction, as a result of endothelial apoptosis, decreases CBF and exacerbates ischaemia-induced cardiac damage due to a shortage of vascular supply relative to cardiac demand.3,4 Sustained reduction of CBF also leads to cardiac dysfunction.30,31 We and other investigators show that capillary density is decreased in diabetic hearts;26–28,39–41 however, the molecular mechanisms that lead to lowered capillary density in the diabetic heart are still incompletely understood. In this study, we examined the role of p53 and protein O-GlcNAcylation in the regulation of coronary EC apoptosis, CBF, and cardiac contractility in diabetic mice.
TH mice exhibited hyperglycaemia, dyslipidaemia, hyperinsulinaemia, and abnormal glucose tolerance (Figure 1A and Table 1). The phenotype of this strain appeared to be very similar to that observed in T2D patients.17 CFVR is a well-accepted assessment of CMD in mice without obstructive CAD.24 TH mice do not develop atherosclerotic plaques; therefore, CFVR reflects the occurrence of coronary microvascular dysfunction. TH mice exhibited impaired CFVR (Figure 1B), accompanied by decreased capillary density and increased EC apoptosis in the LV (Figure 1C–E). Since the number of capillary density positively correlates with CFR,42–44 our data suggest that vascular rarefaction due to enhanced coronary EC apoptosis likely contributes to the development of CMD in TH mice. It has been reported that db/db mice (a monogenic T2D model) exhibit attenuated CFVR due to coronary arterial remodelling45 or attenuated coronary vasodilatation.46 We reported that endothelium-dependent relaxation (EDR) in coronary arteries was significantly attenuated in inducible T1D and T2D mice11,23,26 and our preliminary data showed that TH mice also exhibited impaired EDR in coronary arteries compared to Wt mice (Supplementary material online, Figure S1). The physiological and pharmacological properties of blood vessels with different size (or different order or different branch) are quite different; ECs from different sized vessels (e.g. capillary ECs and arterial ECs) play a very different role in the regulation of vascular functions.47,48 We will conduct more studies to define detailed molecular mechanisms by which EDR is attenuated in coronary arteries of TH mice and whether attenuated EDR contributes to reduced CFVR in TH mice in the future. TH mice exhibited larger infarct size following myocardial I/R and significantly reduced cardiac contractility, while myocardial I/R led to further attenuation of cardiac function in TH (Figure 1F–H and Table 1). The impaired cardiac function observed in TH mice might result not only from reduced CFVR, but also from a direct effect of chronic hyperglycaemia on the function of cardiac myocytes. We therefore used endothelium-specific OGA overexpressing TH mice in our experiments to define the specific role of endothelial dysfunction in the regulation of cardiac function in diabetes.
DOX treatment in Dia-THOGA successfully increased OGA expression and decreased protein O-GlcNAc modification in coronary ECs (Figure 2A and B), while diabetic characteristics (e.g. glucose tolerance, plasma insulin level) were not altered by DOX treatment (Figure 2C and Table 2). It has been reported that the inhibition of OGA increases protein O-GlcNAcylation and decreases insulin secretion from β-cells,49 whereas prolonged overexpression of OGA in β-cells increases serum insulin levels in non-diabetic mice.50 OGA overexpression in the liver decreases gluconeogenesis and plasma glucose concentration in diabetes.51 By overexpressing OGA ubiquitously in TH mice, we expect to observe improved metabolic characteristics through activation of insulin secretion from β-cells and attenuation of gluconeogenesis of hepatocytes.
OGA overexpression significantly restored CFVR accompanied with decreased coronary EC apoptosis in Dia-THOGA mice (Figure 2D–F). These observations suggest that excess protein O-GlcNAcylation increases EC apoptosis, decreases capillary density, and results in attenuated CFVR. We also found that OGA overexpression in only ECs augmented cardiac contractility in Dia-THOGA mice (Table 2), indicating that restoration of CFVR, via repairing EC function, can be a therapeutic target for cardiac dysfunction in diabetes. We realize that the magnitude of CFVR recovery after OGA overexpression is more prominent than the recovery rate of capillary density and EC apoptosis; therefore, we speculate that OGA overexpression might have also restored EDR in the coronary arteries of Dia-THOGA mice. We also observed that cardiac function was significantly restored by OGA overexpression; however, function did not reach the level in control hearts, suggesting that the defect in cardiac myocytes is caused not only by sustained reduction of CFVR, but also by direct effects of hyperglycaemia on cardiac myocyte function. Hu et al.52 reported that OGA overexpression restored Ca2+ transients and contractility of cardiac myocytes isolated from T1D mice by restoring the level of sarcoplasmic reticulum Ca2+-ATPase, indicating that pathophysiological protein O-GlcNAcylation may also affect the function of cardiac myocytes.
Pro-angiogenic therapy is one of the approaches used to restore heart damage caused by cardiac ischaemia.53 Successful data were obtained by VEGF treatment in rodent models; however, clinical trial results failed to show that VEGF gene therapy has a beneficial effect on increasing coronary flow.54 There are still ongoing projects to increase capillary density by pro-angiogenic therapy using VEGF as well as other growth factors.53 In the meantime, some investigators have started to focus on preventing the loss of capillary networks.55,56 Among various pro-apoptotic factors, p53 is a well-studied protein whose gene expression and activation are modified by protein O-GlcNAcylation; O-GlcNAcylation at Ser149 of p53 inhibits phosphorylation of p53 at Thr155 and subsequently decreases ubiquitination of p53 and stabilizes p53.32 HG treatment of coronary ECs significantly increased p53 protein levels while OGA overexpression in these cells decreased p53 to the level similar to those measured in normal-glucose-treated ECs (Figure 3D). HCECs from diabetic patients and MCECs from TH mice exhibited significantly increased p53 protein levels compared to controls (Figure 3E and F), while OGA overexpression markedly decreased p53 levels in Dia-THOGA mice (Figure 3G). These data suggest that excessive protein O-GlcNAcylation resulting from hyperglycaemia leads to increased endothelial p53 protein levels in vitro and in vivo. While we focused on p53 as a downstream target of protein O-GlcNAcylation in this study, we acknowledge that O-GlcNAcylation might also affect the expression of other proteins that regulate EC apoptosis. Therefore, we will examine other candidates regulated by protein O-GlcNAcylation and test the role of these proteins on coronary microvascular dysfunction in diabetes in future studies.
To elucidate the role of p53 on the angiogenic properties of ECs, we conducted the tube formation assay using HCECs from control or T2D diabetic patients. Figure 3H and I shows that HCECs from diabetic patients exhibited attenuated angiogenic capacity and that PFT treatment of diabetic HCECs significantly increased angiogenic capacity towards the level seen in control HCEC. In addition, Supplementary material online, Figure S2 shows that inhibition of p53 by p53 siRNA significantly increased tube formation in diabetic HCECs. Those data suggest that increased p53 protein level might lead to decreased capillary density in the diabetic heart. In line with the ex vivo study, we found that p53 inhibition in vivo exerts beneficial effects on CVFR, capillary density in the LV, and coronary EC apoptosis in diabetes (Figure 4B–D) and reduces infarct size after I/R (Figure 4E and F and Table 3). We applied PFT systemically; therefore, PFT not only affects coronary endothelial function, but also influences the function in other organs such as cardiac myocytes.57 The restoration of cardiac contractility shown in Table 3 is thus due not only to the recovery of CFVR, but also to the direct PFT effect on cardiac myocytes. Interestingly, mean arterial pressure was significantly reduced by PFT treatment (Table 3). The main vasculature that contributes to regulating arterial pressure is the resistance arteries in the mesenteric and renal circulation. If PFT can decrease vascular resistance in those vascular beds, the restoration of mean arterial pressure by PFT treatment could be expected. Pfaff et al.58 demonstrated that p53 deletion enhanced new capillary formation in the mesenteric arterial bed after arterial ligation. Kim et al.59 showed that overexpression of p53 decreased endothelium-dependent vascular relaxation, although these data were obtained in aorta. These observations imply that PFT treatment in TH mice might be able to decrease arterial pressure via reducing vascular resistance. Further experimentation is necessary to confirm our hypothesis.
Although OGA overexpression in HG-treated HCECs and in MCECs from diabetic mice decreased p53 levels towards those in the control mice, there might be other signalling cascades involved in hyperglycaemia-induced p53 overexpression. Overexpression of Sirtuin 1 (NAD+-dependent histone deacetylase) decreases p53 levels in aorta of diabetic mice.60 HG treatment decreases murine double minute 2 (an E3 ubiquitin ligase which degrades p53) level and increases p53 protein level.61 Hyperglycaemia-induced p53 overexpression is decreased by the treatment of glucagon-like peptide-1 receptor agonist (exendin-4) in retinal epithelial cells.62 Therefore, further experiments are required to identify the molecular mechanisms by which hyperglycaemia induces p53 overexpression in coronary ECs.
p53-mediated cell apoptosis involves many genes. After conducting a p53-focused PCR array followed by real-time PCR validation in freshly isolated MCECs, we identified three mRNAs with increased levels in diabetes and restored levels following PFT treatment (Figure 4H–K). This is very useful information for the next step to study p53 signalling cascades in the development of CMD in diabetes. In line with other investigators,63,64 we found that PFT decreased p53 transcript levels (Figure 4G); however, the precise mechanisms by which PFT inhibits p53 signalling is still unknown.
We previously demonstrated that reactive oxygen species (ROS) is increased in coronary ECs of T1D and T2D mice23,27,28,65 and increased ROS leads to cell apoptosis.27 It has been well documented that overexpression of p53 increases ROS production;66,67 therefore, the p53-mediated EC apoptosis may partially result from excess ROS production in diabetic mice.
The data from this study demonstrated that increased coronary EC apoptosis in TH mice is one of the causes for decreased capillary density in the heart. TH mice had significantly increased heart weights; therefore, the increased cardiac mass cannot be ignored when determining capillary density. Here we analysed and reported the capillary metrics per mm2 of tissue, where a smaller number of capillaries per mm2 is equal to an increased distance between the capillaries. Both increased EC apoptosis and increased cardiac mass would increase the distance between capillaries and decrease the diffusion rate of O2 for cardiac myocytes, thereby increasing the risk of cardiac ischaemia. Neither overexpression of OGA nor inhibition of p53 had an effect on heart weight, but both interventions did increase capillary density. These results suggest that no matter how capillary density is decreased in TH mice, OGA overexpression and p53 inhibition can increase capillary density and O2 diffusion, and thereby mitigate the risk of cardiac ischaemia.
In summary, the observations from this study suggest that protein O-GlcNAcylation leads to coronary microvascular dysfunction in diabetes due, at least in part, to p53 overexpression in coronary ECs. Overexpression of OGA reduced coronary EC apoptosis, increased capillary density in the LV, and restored CFVR in the diabetic heart. In addition, repairing endothelial function has a beneficial effect on decreased cardiac contractility in diabetes. Therefore, we here propose that inhibition of protein O-GlcNAcylation in ECs via OGA overexpression may be a novel therapeutic approach for CMD in diabetes.
Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL142214 and HL146764 to A.M.).
Supplementary Material
Time for primary review: 23 days
Translational perspective
Coronary microvascular disease (CMD) is one of the causes of cardiac ischaemia and heart attack in patients with diabetes; however, the molecular mechanisms which chronic hyperglycaemia leads to CMD is not well understood. Our study demonstrated that lowering p53 (an apoptosis inducer) via overexpression of OGA (an enzyme to reduce protein O-GlcNAcylation) inhibited coronary endothelial apoptosis, increased coronary flow, and improved cardiac contractility in diabetes. Therefore, inhibiting p53 is a potential therapeutic approach for CMD in diabetes.
References
- 1. Murthy VL, Naya M, Foster CR, Gaber M, Hainer J, Klein J, Dorbala S, Blankstein R, Di Carli MF.. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 2012;126:1858–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Erdogan D, Yucel H, Uysal BA, Ersoy IH, Icli A, Akcay S, Arslan A, Aksoy F, Ozaydin M, Tamer MN.. Effects of prediabetes and diabetes on left ventricular and coronary microvascular functions. Metabolism 2013;62:1123–1130. [DOI] [PubMed] [Google Scholar]
- 3. Camici PG, d'Amati G, Rimoldi O.. Coronary microvascular dysfunction: mechanisms and functional assessment. Nat Rev Cardiol 2015;12:48–62. [DOI] [PubMed] [Google Scholar]
- 4. Dean J, Cruz SD, Mehta PK, Merz CN.. Coronary microvascular dysfunction: sex-specific risk, diagnosis, and therapy. Nat Rev Cardiol 2015;12:406–414. [DOI] [PubMed] [Google Scholar]
- 5. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A.. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 2018;25:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, Ponnusamy M, Shan C, Xu S, Wang Q, Zhang YH, Zhang J, Wang K.. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun 2018;9:29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forini F, Kusmic C, Nicolini G, Mariani L, Zucchi R, Matteucci M, Iervasi G, Pitto L.. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014;155:4581–4590. [DOI] [PubMed] [Google Scholar]
- 8. Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I.. Protective role of SIRT1 in diabetic vascular dysfunction. ATVB 2009;29:889–894. [DOI] [PubMed] [Google Scholar]
- 9. Rawal S, Munasinghe PE, Nagesh PT, Lew JKS, Jones GT, Williams MJA, Davis P, Bunton D, Galvin IF, Manning P, Lamberts RR, Katare R.. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the type 2 diabetic human and mouse heart. Clin Sci 2017;131:847–863. [DOI] [PubMed] [Google Scholar]
- 10. Hart GW, Housley MP, Slawson C.. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007;446:1017–1022. [DOI] [PubMed] [Google Scholar]
- 11. Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, Scott BT, Wang H, Dillmann WH.. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol 2015;309:C593–C599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R.. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 2002;106:466–472. [DOI] [PubMed] [Google Scholar]
- 13. Luo B, Soesanto Y, McClain DA.. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. ATVB 2008;28:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Qu Y, Niu T, Wang H, Liu K.. O-GlcNAc modification of Sp1 mediates hyperglycaemia-induced ICAM-1 up-regulation in endothelial cells. Biochem Biophys Res Commun 2017;484:79–84. [DOI] [PubMed] [Google Scholar]
- 15. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M.. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem 2007;282:31038–31045. [DOI] [PubMed] [Google Scholar]
- 16. Clee SM, Attie AD.. The genetic landscape of type 2 diabetes in mice. Endocr Rev 2007;28:48–83. [DOI] [PubMed] [Google Scholar]
- 17. Kim JH, Saxton AM.. The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol Biol 2012;933:75–87. [DOI] [PubMed] [Google Scholar]
- 18. Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV.. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999;285:1733–1737. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Ibrahim YF, Das D, Zungu-Edmondson M, Shults NV, Suzuki YJ.. Carfilzomib reverses pulmonary arterial hypertension. Cardiovasc Res 2016;110:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park AM, Nagase H, Liu L, Vinod Kumar S, Szwergold N, Wong CM, Suzuki YJ.. Mechanism of anthracycline-mediated down-regulation of GATA4 in the heart. Cardiovasc Res 2011;90:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR.. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol 2013;58:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu H, Menendez S, Schlegelberger B, Bae N, Aplan PD, Gohring G, Deblasio TR, Nimer SD.. Loss of p53 accelerates the complications of myelodysplastic syndrome in a NUP98-HOXD13-driven mouse model. Blood 2012;120:3089–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho YE, Basu A, Dai A, Heldak M, Makino A.. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am J Physiol Cell Physiol 2013;305:C1033–C1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wikstrom J, Gronros J, Gan LM.. Adenosine induces dilation of epicardial coronary arteries in mice: relationship between coronary flow velocity reserve and coronary flow reserve in vivo using transthoracic echocardiography. Ultrasound Med Biol 2008;34:1053–1062. [DOI] [PubMed] [Google Scholar]
- 25. You J, Wu J, Ge J, Zou Y.. Comparison between adenosine and isoflurane for assessing the coronary flow reserve in mouse models of left ventricular pressure and volume overload. Am J Physiol Heart Circ Physiol 2012;303:H1199–H1207. [DOI] [PubMed] [Google Scholar]
- 26. Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH.. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol 2008;295:C221–C230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sasaki K, Donthamsetty R, Heldak M, Cho YE, Scott BT, Makino A.. VDAC: old protein with new roles in diabetes. Am J Physiol Cell Physiol 2012;303:C1055–C1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan M, Han Y, Basu A, Dai A, Si R, Willson C, Balistrieri A, Scott BT, Makino A.. Overexpression of hexokinase 2 reduces mitochondrial calcium overload in coronary endothelial cells of type 2 diabetic mice. Am J Physiol Cell Physiol 2018;314:C732–C740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suarez J, Wang H, Scott BT, Ling H, Makino A, Swanson E, Brown JH, Suarez JA, Feinstein S, Diaz-Juarez J, Dillmann WH.. In vivo selective expression of thyroid hormone receptor α1 in endothelial cells attenuates myocardial injury in experimental myocardial infarction in mice. Am J Physiol Regul Integr Comp Physiol 2014;307:R340–R346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selthofer-Relatic K, Mihalj M, Kibel A, Stupin A, Stupin M, Jukic I, Koller A, Drenjancevic I.. Coronary microcirculatory dysfunction in human cardiomyopathies: a pathologic and pathophysiologic review. Cardiol Rev 2017;25:165–178. [DOI] [PubMed] [Google Scholar]
- 31. Spoladore R, Fisicaro A, Faccini A, Camici PG.. Coronary microvascular dysfunction in primary cardiomyopathies. Heart 2014;100:806–813. [DOI] [PubMed] [Google Scholar]
- 32. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, Cho JW.. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol 2006;8:1074–1083. [DOI] [PubMed] [Google Scholar]
- 33. Kuruvilla S, Kramer CM.. Coronary microvascular dysfunction in women: an overview of diagnostic strategies. Expert Rev Cardiovasc Ther 2013;11:1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marinescu MA, Loffler AI, Ouellette M, Smith L, Kramer CM, Bourque JM.. Coronary microvascular dysfunction, microvascular angina, and treatment strategies. JACC Cardiovasc Imaging 2015;8:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nahser PJ Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD.. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation 1995;91:635–640. [DOI] [PubMed] [Google Scholar]
- 36. Strauer BE, Motz W, Vogt M, Schwartzkopff B.. Impaired coronary flow reserve in NIDDM: a possible role for diabetic cardiopathy in humans. Diabetes 1997;46(Suppl 2):S119–S124. [DOI] [PubMed] [Google Scholar]
- 37. Pitkanen OP, Nuutila P, Raitakari OT, Ronnemaa T, Koskinen PJ, Iida H, Lehtimaki TJ, Laine HK, Takala T, Viikari JS, Knuuti J.. Coronary flow reserve is reduced in young men with IDDM. Diabetes 1998;47:248–254. [DOI] [PubMed] [Google Scholar]
- 38. Nitenberg A, Valensi P, Sachs R, Dali M, Aptecar E, Attali JR.. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes 1993;42:1017–1025. [DOI] [PubMed] [Google Scholar]
- 39. Chung AW, Hsiang YN, Matzke LA, McManus BM, van Breemen C, Okon EB.. Reduced expression of vascular endothelial growth factor paralleled with the increased angiostatin expression resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in human type 2 diabetic arterial vasculature. Circ Res 2006;99:140–148. [DOI] [PubMed] [Google Scholar]
- 40. Teng X, Ji C, Zhong H, Zheng D, Ni R, Hill DJ, Xiong S, Fan GC, Greer PA, Shen Z, Peng T.. Selective deletion of endothelial cell calpain in mice reduces diabetic cardiomyopathy by improving angiogenesis. Diabetologia 2019;62:860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hinkel R, Howe A, Renner S, Ng J, Lee S, Klett K, Kaczmarek V, Moretti A, Laugwitz KL, Skroblin P, Mayr M, Milting H, Dendorfer A, Reichart B, Wolf E, Kupatt C.. Diabetes mellitus-induced microvascular destabilization in the myocardium. J Am Coll Cardiol 2017;69:131–143. [DOI] [PubMed] [Google Scholar]
- 42. Tsagalou EP, Anastasiou-Nana M, Agapitos E, Gika A, Drakos SG, Terrovitis JV, Ntalianis A, Nanas JN.. Depressed coronary flow reserve is associated with decreased myocardial capillary density in patients with heart failure due to idiopathic dilated cardiomyopathy. J Am Coll Cardiol 2008;52:1391–1398. [DOI] [PubMed] [Google Scholar]
- 43. Kofflard MJ, Michels M, Krams R, Kliffen M, Geleijnse ML, Ten Cate FJ, Serruys PW.. Coronary flow reserve in hypertrophic cardiomyopathy: relation with microvascular dysfunction and pathophysiological characteristics. Neth Heart J 2007;15:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaul S, Jayaweera AR.. Myocardial capillaries and coronary flow reserve. J Am Coll Cardiol 2008;52:1399–1401. [DOI] [PubMed] [Google Scholar]
- 45. Husarek KE, Katz PS, Trask AJ, Galantowicz ML, Cismowski MJ, Lucchesi PA.. The angiotensin receptor blocker losartan reduces coronary arteriole remodeling in type 2 diabetic mice. Vascul Pharmacol 2016;76:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Przygodzki T, Talar M, Przygodzka P, Watala C.. Inhibition of cyclooxygenase-2 causes a decrease in coronary flow in diabetic mice. The possible role of PGE2 and dysfunctional vasodilation mediated by prostacyclin receptor. J Physiol Biochem 2015;71:351–358. [DOI] [PubMed] [Google Scholar]
- 47. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 2007;100:174–190. [DOI] [PubMed] [Google Scholar]
- 48. Castro PR, Barbosa AS, Pereira JM, Ranfley H, Felipetto M, Goncalves CAX, Paiva IR, Berg BB, Barcelos LS.. Cellular and molecular heterogeneity associated with vessel formation processes. Biomed Res Int 2018;2018:6740408.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akimoto Y, Hart GW, Wells L, Vosseller K, Yamamoto K, Munetomo E, Ohara-Imaizumi M, Nishiwaki C, Nagamatsu S, Hirano H, Kawakami H.. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology 2007;17:127–140. [DOI] [PubMed] [Google Scholar]
- 50. Soesanto Y, Luo B, Parker G, Jones D, Cooksey RC, McClain DA.. Pleiotropic and age-dependent effects of decreased protein modification by O-linked N-acetylglucosamine on pancreatic β-cell function and vascularization. J Biol Chem 2011;286:26118–26126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dentin R, Hedrick S, Xie J, Yates J, Montminy M.. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008;319:1402–1405. [DOI] [PubMed] [Google Scholar]
- 52. Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH.. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res 2005;96:1006–1013. [DOI] [PubMed] [Google Scholar]
- 53. Lavu M, Gundewar S, Lefer DJ.. Gene therapy for ischemic heart disease. J Mol Cell Cardiol 2011;50:742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taimeh Z, Loughran J, Birks EJ, Bolli R.. Vascular endothelial growth factor in heart failure. Nat Rev Cardiol 2013;10:519–530. [DOI] [PubMed] [Google Scholar]
- 55. Gogiraju R, Xu X, Bochenek ML, Steinbrecher JH, Lehnart SE, Wenzel P, Kessel M, Zeisberg EM, Dobbelstein M, Schafer K.. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc 2015;4:e001770.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nako H, Kataoka K, Koibuchi N, Dong YF, Toyama K, Yamamoto E, Yasuda O, Ichijo H, Ogawa H, Kim-Mitsuyama S.. Novel mechanism of angiotensin II-induced cardiac injury in hypertensive rats: the critical role of ASK1 and VEGF. Hypertens Res 2012;35:194–200. [DOI] [PubMed] [Google Scholar]
- 57. Gu J, Wang S, Guo H, Tan Y, Liang Y, Feng A, Liu Q, Damodaran C, Zhang Z, Keller BB, Zhang C, Cai L.. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis 2018;9:82.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pfaff MJ, Mukhopadhyay S, Hoofnagle M, Chabasse C, Sarkar R.. Tumor suppressor protein p53 negatively regulates ischemia-induced angiogenesis and arteriogenesis. J Vasc Surg 2018;68:222S–233S.e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, Cole MP, Jeon BH, Irani K.. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res 2008;103:1441–1450. [DOI] [PubMed] [Google Scholar]
- 60. Chen H, Wan Y, Zhou S, Lu Y, Zhang Z, Zhang R, Chen F, Hao D, Zhao X, Guo Z, Liu D, Liang C.. Endothelium-specific SIRT1 overexpression inhibits hyperglycemia-induced upregulation of vascular cell senescence. Sci China Life Sci 2012;55:467–473. [DOI] [PubMed] [Google Scholar]
- 61. Barzalobre-Geronimo R, Flores-Lopez LA, Baiza-Gutman LA, Cruz M, Garcia-Macedo R, Avalos-Rodriguez A, Contreras-Ramos A, Diaz-Flores A, Ortega-Camarillo C.. Hyperglycemia promotes p53-Mdm2 interaction but reduces p53 ubiquitination in RINm5F cells. Mol Cell Biochem 2015;405:257–264. [DOI] [PubMed] [Google Scholar]
- 62. Kim DI, Park MJ, Choi JH, Lim SK, Choi HJ, Park SH.. Hyperglycemia-induced GLP-1R downregulation causes RPE cell apoptosis. Int J Biochem Cell Biol 2015;59:41–51. [DOI] [PubMed] [Google Scholar]
- 63. Bragado P, Armesilla A, Silva A, Porras A.. Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 2007;12:1733–1742. [DOI] [PubMed] [Google Scholar]
- 64. Proietti De Santis L, Balajee AS, Lorenti Garcia C, Pepe G, Worboys AM, Palitti F.. Inhibition of p53, p21 and Bax by pifithrin-α does not affect UV induced apoptotic response in CS-B cells. DNA Repair (Amst) 2003;2:891–900. [DOI] [PubMed] [Google Scholar]
- 65. Makino A, Scott BT, Dillmann WH.. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010;53:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu B, Chen Y, St Clair DK.. ROS and p53: a versatile partnership. Free Radic Biol Med 2008;44:1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simabuco FM, Morale MG, Pavan ICB, Morelli AP, Silva FR, Tamura RE.. p53 and metabolism: from mechanism to therapeutics. Oncotarget 2018;9:23780–23823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.