

Abstract
Improvements in early interventions after acute myocardial infarction (AMI), notably, the increased use of timely reperfusion therapy, have increased survival dramatically in recent decades. Despite this, maladaptive ventricular remodelling and subsequent heart failure (HF) following AMI remain a significant clinical challenge, particularly because several pre-clinical strategies to attenuate remodelling have failed to translate into clinical practice. Monocytes and macrophages, pleiotropic cells of the innate immune system, are integral in both the initial inflammatory response to injury and subsequent wound healing in many tissues, including the heart. However, maladaptive immune cell behaviour contributes to ventricular remodelling in mouse models, prompting experimental efforts to modulate the immune response to prevent the development of HF. Seminal work in macrophage biology defined macrophages as monocyte-derived cells that are comprised of two populations, pro-inflammatory M1 macrophages and reparative M2 macrophages, and initial investigations into cardiac macrophage populations following AMI suggested they aligned well to this model. However, more recent data, in the heart and other tissues, demonstrate remarkable heterogeneity and plasticity in macrophage development, phenotype, and function. These recent insights into macrophage biology may explain the failure of non-specific immunosuppressive strategies and offer novel opportunities for therapeutic targeting to prevent HF following AMI. Here, we summarize the traditional monocyte-macrophage paradigm, experimental evidence for the significance of these cells in HF after AMI, and the potential relevance of emerging evidence that refutes canonical models of monocyte and macrophage biology.
Keywords: Monocyte, Macrophage, Myocardial infarction
Graphical Abstract
Graphical Abstract.

1. Introduction
Emergency management of acute myocardial infarction (AMI) has been revolutionized by timely reperfusion therapy and, in particular, increasing access to primary percutaneous coronary intervention, leading to dramatic improvements in early survival.1–3 Despite this, AMI remains the commonest cause of heart failure (HF) and HF-related morbidity and mortality following AMI remain high.4–7 In view of the burden of HF following AMI, there is an unmet need for better understanding of the pathogenesis of ventricular remodelling and development of novel therapeutic targets.
In AMI, reduced blood flow to a region of myocardium results in infarction, mediated mainly through oncosis and necrosis.8 This necrotic myocardium becomes a region of mechanical weakness that requires scar deposition to prevent myocardial rupture and limit functional deterioration. This adaptive remodelling is necessary to prevent early mortality following AMI. However, excessive and progressive (maladaptive) ventricular remodelling, at both the infarct site and remote myocardium, alters ventricular size and function, culminating in the clinical syndrome of HF. Understanding the balance between adaptive and maladaptive remodelling is an emerging research area that has the potential to identify novel therapeutic targets for the prevention of HF after myocardial infarction (MI).
The innate immune response is an important regulator of this process and comprises three key phases: the inflammatory, proliferative, and maturation phases.8–10 Cell death triggers sterile inflammation, through exposure of endogenous damage-associated molecular patterns (DAMPs) to the innate immune system. Recognition of DAMPs by pattern recognition receptors on resident innate immune cells prompts a cascade of chemokine and pro-inflammatory cytokine release, recruiting and activating neutrophils, monocytes, and macrophages. Together, these cells degrade the extracellular matrix and phagocytose necrotic cells. This initial inflammatory phase, which peaks around Day 3 post-injury, is followed by a longer proliferative phase lasting around 10 days, also mediated by immune cells. This phase is characterized by anti-inflammatory signalling, fibroblast proliferation, and deposition of granulation tissue. The final, maturation phase occurs over subsequent months and is characterized by remodelling of the extracellular matrix, with few immune cells present at the site of injury. During this period following AMI, there are significant changes to ventricular size, shape, and function, with maladaptive remodelling leading to the development of HF.11
Monocytes and macrophages are implicated at all three stages of this response. Cardiac monocyte and macrophage numbers expand rapidly in the days following AMI.12,13 These initial, infiltrating populations demonstrate a pro-inflammatory phenotype which shifts over the ensuing days to a predominantly reparative phenotype, coordinating the deposition of scar tissue.14 By 2 weeks post-AMI, monocyte and macrophage populations at the site of infarction return to baseline, although macrophages persist for months after AMI in the remote, remodelling myocardium.15
Nonetheless, numerous trials of immunosuppressive agents have failed to mitigate HF following AMI, which is unsurprising given the complex interplay of reparative and deleterious processes, and demonstrates the need for a more detailed understanding of cellular and molecular pathways in ventricular remodelling to facilitate more targeted therapeutic interventions.16–18
The putative role of monocytes and macrophages in healing post-AMI, and the persistence of macrophages in remodelling ventricular myocardium, has led to interest in targeting these cells to prevent maladaptive remodelling. However, various strategies to target specific sub-populations have delivered seemingly contradictory results. This may relate to an overly constrictive traditional understanding and definitions of macrophage subsets, which are increasingly understood to be heterogeneous and plastic, impeding focused investigation of their role in ventricular remodelling after AMI.
Here, we discuss the canonical view of monocyte-derived macrophage differentiation and activation, and how strategies targeting monocyte and macrophage populations have impacted on ventricular remodelling in pre-clinical models. We then discuss emerging data on the complexity of macrophage development and phenotype that challenges the existing paradigm and may justify disparities in existing evidence. Finally, we discuss how recognition of macrophage heterogeneity and plasticity may benefit ongoing investigation and the development of new treatments in this important clinical area.
2. Traditional views of cardiac monocyte and macrophage populations
2.1 Monocyte and macrophage classification
The mononuclear phagocyte system is a family of myeloid cells comprising monocytes, macrophages, and dendritic cells. Monocytes are short-lived circulating cells that are implicated in inflammation through both direct effects and by differentiation into dendritic cells and macrophages. These cells develop from the common myeloid progenitor in the bone marrow and are released into the circulation, where they comprise two main subsets: conventionally known as classical and non-classical. These populations were originally described in humans, defined by CD16 (cluster of differentiation 16) expression, and later in mice. Phenotypic profiling suggests that these populations correspond, facilitating experimental characterization of populations in human studies.19,20
Classical monocytes, typically described as CD14+CD16neg in humans and Ly-6Chigh in mice, are implicated in inflammation.21,22 These cells are released from the bone marrow and extramedullary sites of haematopoiesis, such as the spleen, and traffic to sites of injury in a CCR2 [chemokine (C-C motif) receptor 2] dependent manner, and comprise over 90% of circulating monocytes.22 Upon extravasation, classical monocytes contribute to the innate immune response both via direct effects, such as tumour necrosis factor α (TNF-α) and nitric oxide production, and indirectly, by differentiation into macrophages and dendritic cells.23 More recently, these cells have been shown to play an important role in activating the adaptive immune response, through antigen presentation to T cells.23
In contrast, non-classical monocytes (CD14+CD16+/Ly-6Clow) are thought to persist in the circulation and their role in inflammation is less clear. In mice, these cells have been demonstrated to arise in an NR4A1 (nuclear receptor subfamily four group A member one) dependent manner, a transcription factor which is dispensable for classical monocyte development.24,25 Both murine and human non-classical monocytes may be differentiated from classical monocytes by high levels of CX3CR1 (C-X3-C motif chemokine receptor 1) expression and, in mice, this receptor has been demonstrated to be essential for their survival.22,26,27 These cells patrol the endothelium and may play a role in immune surveillance.28,29
On entry into tissues, monocytes give rise to dendritic cells and macrophages. Macrophages phenotypically differ from monocytes by increased expression of CD68 and MHCII (major histocompatibility 2), as well as F4/80 in mice, and reduced expression of CD14 in humans. Originally described as the prototypic phagocytic cells, macrophages are now recognized to mediate incredibly diverse processes, from cytokine production, phagocytosis, and co-ordination of the formation of granulation tissue in the context of inflammation, to organ-specific homoeostatic functions, the scope of which is still emerging in the heart.
Investigation of macrophage phenotype in vitro led to the widespread use of the M1 and M2 polarization model to describe macrophages in the context of inflammation.30 Macrophages stimulated with pro-inflammatory signals, such as LPS (lipopolysaccharide) and IFN-γ (interferon-gamma), demonstrate a stereotypic pro-inflammatory transcriptome and behaviour. These M1, or classically activated, macrophages demonstrate enhanced phagocytosis, antigen presentation on MHC II, and generation of reactive oxygen species. They also produce and release pro-inflammatory cytokines, such as IL-12 (interleukin 12), IL-23, IL-27, and TNF-α; chemokines, including CXCL9 (CXC motif chemokine ligand 9), CXCL10, CXCL11; and matrix metalloproteinases (MMP-1, 2, 7, 9, 12). Together these contribute to a pro-inflammatory micro-environment.
In contrast, macrophages stimulated with anti-inflammatory cytokines, such as IL-4 and IL-13, show expression of characteristic anti-inflammatory genes and a reparative phenotype.31 M2, or alternatively activated, macrophages produce anti-inflammatory cytokines (IL-10); chemokines, including CCL17 (C-C motif chemokine ligand 17),22,24 and growth factors, such as vascular endothelial growth factor and tumour growth factor beta. Together these mediators stimulate fibroblast-mediated production of the extracellular matrix, cell proliferation and angiogenesis, promoting tissue remodelling and repair.
Following these seminal studies, the M1–M2 activation paradigm continues to be widely used.
2.2 Cardiac monocyte and macrophage populations in health and disease
During homoeostasis, both classical and non-classical monocytes are found in the coronary vasculature. Intravital microscopy demonstrates classical monocytes circulate rapidly, whereas non-classical monocytes circulate more slowly, crawling along the endothelium.12,28,29 These patrolling monocytes appear to play in role in immuno-surveillance. Within the myocardium, macrophages are present in both the murine and human heart, comprising 6–8% of non-cardiomyocytes in the adult mouse.32,33 These cardiac macrophages play a role in cardiac development, immuno-surveillance and may contribute important specialized cardiac functions, such as conduction, though their exact functional significance is still emerging.10
Following AMI, monocyte and macrophage populations expand at the site of infarction and change their phenotype dramatically in the murine heart. Intravital microscopy demonstrates that monocyte recruitment to the infarct begins as early as 30 min following AMI, first from the vascular pool and later from the splenic reservoir.12 This initial recruitment is rapidly overtaken by neutrophil infiltration, and recent evidence suggests these sentinel infiltrating monocytes may play a role in neutrophil attraction.34
The infiltration of monocyte and macrophage populations into infarcted tissue occurs in two sequential phases.13,14 Ly-6Chigh monocytes are the predominant population early post-AMI, peaking between Days 3 and 5, and demonstrating a pro-inflammatory phenotype, including TNFα expression and high proteinase activity. Ly-6Chigh macrophages form the principal macrophage subset during this early phase, though they are less abundant than monocytes.35 As inflammation resolves, Ly-6Clow cells predominate, peaking in the infarct on Day 7 post-AMI.14 Initially described as a distinct wave of Ly-6Clow monocyte infiltration, more refined gating strategies and lineage tracing experiments have demonstrated these cells to be principally Ly-6Clow macrophages, which are derived from Ly-6Chigh monocytes.35
A similar pattern of sequential Ly-6Chigh and Ly-6Clow expansion is observed in the remote myocardium.13 However, Ly-6Clow numbers peak 5 days later than at the infarct site. Importantly, in contrast to the site of ischaemia, macrophages and Ly-6Chigh monocytes persist in the non-ischaemic myocardium (Figure 1).15
Figure 1.

Myocardial monocyte and macrophage populations after AMI. (A) Infarct site inflammatory cell populations expand rapidly following AMI with an initial expansion of Ly-6Chigh monocytes, peaking Day 3–5, followed by an expansion of Ly-6Clow macrophages, peaking on Day 7. (B) By 6 weeks, inflammatory cell infiltration at the infarct site has returned to baseline, leaving an acellular collagen based scar, whereas Ly-6Clow macrophages persist in the remote, remodelling myocardium.
These changes in monocyte and macrophage populations in the myocardium following AMI are accompanied by an expansion of circulating monocytes.14,36 The major source of circulating monocytes following AMI is the splenic reservoir, and these cells are generated both by seeding from the bone marrow and by local monocytopoiesis.36–38 The spleen contains myeloid precursors that expand rapidly after AMI; on adoptive transfer of splenic granulocyte-macrophage progenitors after AMI, these cells differentiate into splenic macrophages that mobilize to the healing myocardium.38 Monocytopoiesis at both medullary and extramedullary sites is regulated by sympathetic nervous signalling and IL-1, both in the acute and chronic phase following AMI.15,38,39
Human studies of cardiac populations following AMI demonstrate a similar pattern of infiltration to that observed in mice. Humans demonstrate peripheral blood monocytosis after AMI, with sequential peaks of CD14+CD16neg and CD14+CD16+ monocytes on Days 3 and 5, respectively, though CD14+CD16neg monocytes are more abundant throughout.40 This was confirmed in an analysis of post-mortem cardiac tissue, which showed a predominance of CD16− cells during the inflammatory phase, compared to comparable populations of CD16+ and CD16− cells during the proliferative phase.41 Both the bone marrow and splenic reservoir of monocytes (defined as CD14+ cells) were significantly depleted in this study.
3. Monocyte–macrophage phenotype and ventricular remodelling
3.1 Pro-inflammatory phenotypes correlate with ventricular dysfunction in mice and humans
The role of monocytes and macrophages in wound healing and the observation of two distinct waves of infiltration after AMI has led to the hypothesis that these cells may have a role in maladaptive ventricular remodelling and the development of HF. Observational studies in mice and humans demonstrate a correlation between classical monocytosis and degree of LV dysfunction after AMI. For example, the ApoEneg/neg (Apoprotein E) model demonstrates a chronically expanded pool of circulating Ly-6Chigh monocytes.42 Following coronary artery ligation, these mice show increased myocardial infiltration and persistence of Ly-6Chigh monocytes on Day 5 and reduced left ventricular ejection fraction (LVEF) at 3 weeks, compared to wild-type mice. Similarly, in patients following ST-segment elevation MI (STEMI), there is a negative correlation between peripheral blood monocytosis and recovery of ventricular function.43 In one study, peak circulating CD14+CD16neg levels following STEMI negatively correlated both with wound healing (assessed by the extent of myocardial salvage on cardiac magnetic resonance imaging at Day 7) and LVEF at 6 months, though the degree of HF at follow-up is likely to have been confounded by the greater initial infarct size.40
These findings have been corroborated when classical monocytes are defined as CD14+CD62L+ cells, with high levels associated with greater infarct size and regional systolic LV dysfunction at 4-month follow-up after STEMI, which remained significant even after stratification according to the extent of transmural infarction.44 An inverse trend was observed with the number of non-classical monocytes (CD14+CD62Lneg), with high levels correlating to improved LV function, though this was not statistically significant. However, crucially, these patients were taken from the HEBE trial, where subjects received intracoronary infusions of autologous peripheral blood mononuclear cells (PBMC) following AMI. Given the expansion of circulating classical monocyte populations following AMI, the outcomes at later time points are likely to be confounded by the intracoronary infusion of these pro-inflammatory skewed PBMC populations. The trial itself failed to show a significant effect of intracoronary infusion of autologous PBMC on left ventricular size or function at 4-month follow-up and, interestingly, revealed increased mortality and a reduction in recovery of left ventricular function at 24 months when compared with control.45,46
3.2 Targeting circulating monocytes and their reservoirs
To better understand, the specific contributions of monocyte and macrophage subsets on remodelling following AMI, studies have focused on either blocking monocyte recruitment to the myocardium or manipulating the phenotype of myocardial monocytes and macrophages (Figure 2).
Figure 2.
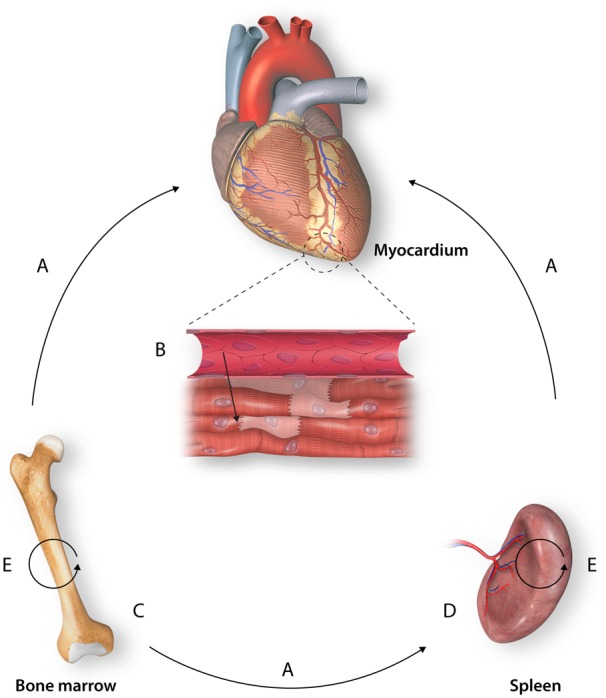
Targeting circulating monocytes and their reservoirs. Approaches depleting myocardial monocyte populations following AMI have focused on various stages of their trafficking upstream. (A) Depletion of all circulating monocytes (e.g. clodronate-loaded liposomes). (B) Inhibition of monocyte entry into the myocardium (e.g. Ccr2neg/neg). (C) Inhibition of monocyte release from the bone marrow (e.g. Ccr2neg/neg). (D) Depletion of the splenic reservoir or inhibition of splenic release (e.g. splenectomy, Agt1ar antagonism). (E) Inhibition of monocytopoiesis (e.g. beta-adrenergic blockade).
The correlation of monocytosis with poor outcomes led to the hypothesis that excessive monocyte infiltration is deleterious. However, treatment with clodronate-loaded liposomes after AMI, which deplete circulating monocytes (Figure 2A), either during the early pro-inflammatory phase or later proliferative phase, leads to impaired infarct repair.14 In both instances, reduced collagen deposition and angiogenesis are seen during the proliferative phase, whilst only depletion of early monocytes increases necrotic cells and debris, suggesting that both the pro-inflammatory early phase and later reparative phase are essential for infarct healing.
A number of studies have, therefore, sought to selectively target trafficking of Ly-6Chigh monocytes to the myocardium, taking advantage of the observation that Ly-6Clow monocytes traffic independently of CCR2 (Figure 2B). Following coronary artery ligation, Ccr2neg/negmice demonstrate reduced myocardial infiltration of Ly-6Chigh cells and siRNA knockdown of Ccr2 in wild-type mice reduces Ly-6Chigh monocyte infiltration of the myocardium and improves LVEF and left ventricular end-diastolic volume (LVEDV) in these mice.14,47 Notably, CCR2 is not only important for attraction of Ly-6Chigh monocytes to the myocardium but is also necessary for their mobilization from the bone marrow, and therefore, these models are likely to be exerting some of their effect via bone marrow mobilization and, in the case of constitutive knockout, prior seeding of the spleen by monocytes that originated in the bone marrow (Figure 2C).48
However, while targeting CCR2 can impede Ly-6Chigh monocyte trafficking, release of monocytes from the spleen occurs independently of CCR2 and splenic monocytes have been demonstrated to contribute to maladaptive ventricular remodelling (Figure 2D).36 Splenectomy in mice 8 weeks post-coronary artery ligation appears to improve ventricular function, as assessed by LVEDV and LVEF, whilst control mice that received sham abdominal surgery demonstrated ongoing deterioration of LV function.37 Similarly, adoptive transfer of splenic monocytes from mice with ventricular dysfunction post-AMI caused maladaptive ventricular remodelling in the recipients. In contrast, adoptive transfer of LPS-stimulated splenic monocytes did not induce ventricular remodelling, suggesting alterations specific to the splenic niche in the context of HF modulate monocyte phenotype, and that this phenotype is functionally distinct from that of in vitro derived pro-inflammatory monocytes. Interestingly, release of splenic monocytes has been shown to depend on the angiotensin II Type 1a receptor, and knockout of this receptor reduces both circulating and myocardial monocytes following AMI.36 It is possible that the effect of drugs that inhibit the renin–angiotensin system, which are in routine use in the prevention and treatment of HF following AMI, is partially mediated via this mechanism.
A final target to deplete myocardial monocyte populations is monocytopoiesis (Figure 2E). Targeting IL-1β, a cytokine that promotes monocytopoiesis following AMI, reduces circulating monocyte levels following coronary artery ligation and mitigated deterioration of LV function at 3 weeks.49 These findings are pertinent in light of the recent success of the monoclonal antibody to IL-1β, canakinumab, in prevention of cardiovascular events in patients with previous AMI.50 Sympathetic nervous system signalling has been demonstrated to drive monocytopoiesis and the routine use of β adrenergic blockade as cardio-protection following AMI may be exerting some effect via this mechanism.15 The impact on myocardial monocyte infiltration after AMI of inhibitors of both the renin–angiotensin system, implicated in splenocyte release, and the β adrenergic system, implicated in monocytopoiesis, remains to be characterized. Without such characterization, efforts to translate putative strategies targeting these cells may be confounded by the unknown impact of these commonly prescribed medications.
Other studies have hypothesized that Ly-6Clow monocytes may protect against ventricular remodelling by attenuating inflammation. Mice deficient for Nr4a1, an obligate transcription factor for Ly-6Clow monocyte development, showed absent circulating and myocardial Ly-6Clow monocytes and impaired LV function, with reduced LVEF and increased LVEDV at 21 days post-coronary artery ligation, compared to wild-type mice.35 However, rather than mediating its effects solely through depletion of Ly-6Clow monocytes, the authors demonstrated that Nr4a1 deletion also modulated the behaviours of Ly-6Chigh monocytes and both macrophage subsets. In particular, Ly-6Chigh monocytes demonstrated increased expression of CCR2 and infiltration of the infarct site was increased. Absolute numbers of Ly-6Chigh and Ly-6Clow macrophages were increased in the myocardium on Day 7, and global macrophage transcription was skewed towards a pro-inflammatory phenotype, with increased expression of IL-1βα, TNFα, and IL-6, and reduced expression of canonical M2 marker, CD206.
3.3 Targeting myocardial macrophage phenotype
NR4A1 is one of a number of identified molecular targets that links macrophage phenotype with ventricular remodelling. Initial studies showed that up-regulating canonical M2 genes in myocardial macrophage populations, using IL-4 or IL-10 treatment or GABAAR (gamma aminobutyric acid A receptor) agonism, following coronary artery ligation protects against ventricular remodelling.51–53 However, these approaches are non-specific as they also manipulate other cell types, including T-reg cells, and fail to demonstrate a causal role for macrophages or any particular subset.
Specific targeting of macrophage polarization, therefore, provides more convincing evidence of a causative role for macrophages in both early infarct repair and ventricular remodelling. Knockout of Trib1 (Tribbles pseudokinase 1) causes a selective depletion of CD206+ macrophages, a population described in this study as M2 macrophages.53 Following coronary artery ligation, Trib1 deficient mice demonstrate a nine-fold increase in cardiac rupture, and this phenotype is salvaged by adoptive transfer of CD206+ macrophages, providing evidence of a beneficial role for reparative macrophages early in the inflammatory phase. Trib1neg/neg demonstrated impaired LV function compared to wild type, implicating this macrophage population in protecting against ventricular remodelling. Similarly, in an Mmp28 knockout model, which shows reduced capacity of peritoneal macrophages to differentiate into M2 macrophages in vitro, ventricular function was impaired at 7 days compared to wild types after coronary artery ligation.54 Cardiac macrophage numbers were not significantly altered, but expression of canonical M1 markers was increased and M2 markers were reduced.
Efferocytosis, the phagocytosis of apoptotic cells, has been hypothesized as a cell-intrinsic mechanism by which macrophages might shift from a pro-inflammatory to reparative phenotype. Constitutive knockout of Mertk (Mer tyrosine protein kinase), a receptor tyrosine kinase required for effective efferocytosis, led to increased expression of canonical M1 genes in macrophages and exacerbated deterioration in LV function after coronary artery ligation.55
Conversely, blocking pathways implicated in M1 macrophage polarization was hypothesized to protect cardiac function after coronary artery ligation. siRNA targeting of Irf5 (interferon regulatory factor 5), a transcription factor implicated in M1 polarization, resulted in reduced expression of the canonical M1 markers TNF-α and IL-1β, but no change in M2 marker expression on Day 4 post-coronary artery ligation.56 These mice had similar infarct sizes but knockdown reduced LV dilatation at 3 weeks.
These observations led to the hypothesis that shifting the macrophage phenotype from the pro-inflammatory M1-like macrophage to an anti-inflammatory M2-like macrophage protects the heart from adverse remodelling following AMI. However, more recent evidence has challenged the use of the M1–M2 paradigm by demonstrating improved outcomes following knockout of Gata3 (GATA-binding factor-3), a transcription factor implicated in the differentiation of M2 macrophages.57 Selective knockout of Gata3 in myeloid cells depleted Ly-6Clow but not Ly-6Chigh macrophages following AMI and, surprisingly, the Ly-6Clow macrophage depleted mice demonstrated improved LV size and function at 2 months post-AMI. Additionally, absolute numbers of Ly-6Chigh macrophages increased in the knockout model, challenging the view that the presence of excessive early pro-inflammatory populations is deleterious.
To reconcile this study with the view that M2 macrophages are protective requires careful re-examination of the canonical M1–M2 macrophage model. The variety and limitations of methods to define macrophage populations means that cell populations described in various studies may not necessarily align to one another. Emerging analyses of the wide phenotypic spectrum of macrophages, as well as the existence of non-monocyte-derived macrophage subsets, supports this view.
4. Insights into the complexity of mononuclear cell populations following AMI
4.1 Macrophages in vitro and in vivo exhibit a network of stimulus-dependent phenotypes
The M1–M2 paradigm is based on the polarization of bone marrow-derived macrophages in response to LPS and IFN-γ or IL-4 and IL-13.30 However, when human macrophages are stimulated in vitro with a more diverse range of cytokines than originally described, many more distinct phenotypes result, with one study eliciting 299 distinct transcriptomes that do not map along a spectrum from M1 to M2.58 Similarly, the co-expression of canonical M1 and M2 markers in both mouse and human macrophages is well characterized, as is the ability of M1 or M2 macrophages to acquire canonical markers of the other subset in vitro.59,60 Accordingly, the M1–M2 paradigm is widely accepted as an over-simplification, and yet it continues to guide experimental study design in vivo.61
With this in mind, studies targeting particular transcriptional pathways are likely to be affecting only a subpopulation of M1 or M2 cells. For example, knockout of Gata3 depletes CCR2negLy-6Clow macrophages but does not eliminate them, suggesting that only a proportion of these cells are Gata3-dependent (Figure 3A). It is, therefore, misleading to conclude that M2 macrophages are responsible for the observed experimental phenotype. This also demonstrates the limitations in current definitions of macrophage subsets, as markers used to describe M1 and M2 populations vary between studies, and accordingly, the populations described are not comparable.
Figure 3.

Advantages of high throughput techniques to examine immune cell heterogeneity. (A) Flow cytometry is widely used to immunophenotype monocytes and macrophages. A common gating strategy in mice defines these cells as CD45+CD11b+Ly-6Glow. Based on Ly-6C and F4/80 expression these cells are commonly described as classical (Ly-6ChighF4/80low) and non-classical monocytes (Ly-6ClowF4/80low), and M1 ((Ly-6ChighF4/80high) and M2 (Ly-6ClowF4/80high). Cells expressing a particular transcription factor, termed X, may not align well with the M1/M2 paradigm, Knockout of this transcription factor may preferentially deplete a particular population, such as M2 macrophages, but is neither sensitive nor specific, failing to deplete all ‘M2’ macrophages, and also depleting cells of other subsets. (B) Novel high throughput techniques allow unbiased resolution of cells into distinct populations, based on gene expression profile (scRNA-seq) or large panels of differentially expressed proteins (mass cytometry). Such methods allow identification of candidate pathways for therapeutic targeting and ensure knockout models selectively deplete the population of interest. DC, dendritic cells; Mo, monocytes; Mϕ, macrophages; Nϕ, neutrophils.
Novel techniques that allow resolution of gene expression at the single-cell level, including single-cell RNA sequencing (scRNA-seq) and mass cytometry, may help overcome these issues (Figure 3B). scRNA-seq assesses the transcriptome at single-cell level and enables unbiased clustering of cells based on common expression profiles. scRNA-seq of murine aortic macrophages defined three clusters of macrophages: ‘Res-like’, which were present in healthy and atherosclerotic aortas, as well as inflammatory and Trem2high (triggering receptor expressed on myeloid cells 2) macrophages, which were only present in atherosclerosis.62 Whilst inflammatory and Res-like macrophages overlapped significantly with the transcriptomes of in vitro-derived M1 and M2 macrophages, respectively, inflammatory macrophages were enriched for Nrf2 (nuclear factor erythroid2 related factor 2), an anti-inflammatory transcription factor, and Mrc1 (mannose receptor C Type 1), which encodes CD206, a cell surface marker often used to define M2 macrophages. The newly described Trem2high subset did not align to either the M1 or M2 profile.
Interestingly, in contrast to the diversity of macrophage populations, application of scRNA-seq to circulating monocytes in mice suggests that these cells do align well to the classical and non-classical monocyte paradigm.63 Ly-6Chigh and Ly-6Clow cells have distinct and well-conserved transcriptomes. The authors of this particular study also address the transcriptional networks of a third population of monocytes. This third population, defined by intermediate expression of Ly-6C in mice and CD16 in humans, has long been recognized and is hypothesized to represent an intermediate developmental stage. The authors demonstrated heterogeneity in the transcriptional profiles of these Ly-6Cint cells, and argued that this lent further support to the hypothesis that these cells represent an intermediate developmental stage between classical and non-classical monocytes.
The resolution of scRNA-seq in distinguishing monocyte and macrophage populations is limited by significant overlap in transcriptional profiles between clusters, given their common lineage. Mass cytometry addresses this issue by using a large panel of heavy metal-conjugated antibodies targeting known or suspected differentially expressed proteins.64 Application of mass cytometry to the analysis of aortic macrophages described above was able to further subdivide phenotypes that had already been defined by scRNA-seq.65 This method of immunophenotyping allows simultaneous analysis of up to 40 proteins and more specific resolution of subsets compared to traditional flow cytometry. Comparison of populations defined using mass and flow cytometry has demonstrated a tendency to misclassify monocytes and macrophages and may account for some of the observed inter-study variation.14,35,66 Given the complex networks regulating gene expression at the translational and post-translational level, the combination of strategies to interrogate both RNA expression and protein expression is important to appreciate the relevant effector pathways in these cells.
Whilst mass cytometry is, to our knowledge, yet to be applied to cardiac macrophages, emerging data from scRNA-seq and novel experimental models of AMI illustrate macrophage diversity during homoeostasis and following AMI, novel mechanisms by which macrophage populations develop following AMI, and potential mechanism by which they modulate ventricular remodelling.
4.2 Cardiac mononuclear cells demonstrate diverse gene expression during homoeostasis
Tissue resident macrophages (TRM) are present in the myocardium during homoeostasis, comprising 7–8% of non-cardiomyocytes in the steady state.32 Initial fate mapping experiments in mice divided these cells according to CCR2 expression, demonstrating that CCR2+ cells are derived from and dependent on circulating CCR2+ monocytes, whereas CCR2neg macrophages are of mixed ontogeny, arising both from the embryonic yolk sac and from foetal liver haematopoietic stem cells. 67 This is in contrast to other organs, such as the brain, where TRM are entirely independent of circulating monocytes. These findings have been recapitulated in human hearts, where CCR2neg tissue resident populations have also been demonstrated to be maintained through proliferation, independent of circulating monocytes, whilst CCR2+ macrophages depend on circulating CCR2+ monocyte recruitment and proliferation.68
Use of scRNA-seq has demonstrated greater heterogeneity of myocardial TRMs than previously appreciated, identifying 11 clusters of mononuclear phagocytes with distinct gene expression profiles, comprising one monocyte cluster, four macrophage clusters, three dendritic cells clusters, one myeloid antigen presenting cell cluster, and two clusters of proliferating cells.69 Pathway analysis of these subsets suggests differing functions during homoeostasis. The first cluster is enriched for genes involved in reparative functions, such as efferocytosis and angiogenesis, including Timd4 (T-cell immunoglobulin and mucin containing domain-4), Lyve1 (lymphatic vessel endothelial hyaluronan receptor 1), and Igf1 (insulin-like growth factor 1). These cells are entirely maintained through local proliferation, and likely form a subset of the CCR2neg cells described before. A second cluster was unique in its expression of Ccr2, and was found to be enriched for genes involved in cell migration, hypoxic response, and the respiratory burst, suggesting a pro-inflammatory role. As previously described, these cells were entirely replaced by differentiation of circulating monocytes. The final populations were of mixed lineage, maintained both by local proliferation and by differentiation of circulating monocytes, and were characterized by enrichment of genes involved in antigen presentation and interferon-stimulated genes, respectively. This study demonstrates the utility of high throughput techniques in both resolving populations that are otherwise described homogenously and identifying novel target pathways.
In addition to these myocardial macrophages, the pericardial cavity, a serous cavity surrounding the heart, has long been known to contain macrophages.70,71 Whilst high throughput methods are yet to be applied to these macrophages, immunophenotyping by flow cytometry groups three distinct myeloid clusters, MHC II+ macrophages, GATA6+ macrophages, and dendritic cells, during homoeostasis.72 Bulk RNA-seq demonstrates that the transcriptional profile of GATA6+ pericardial cavity macrophages is more similar to that of other serous cavity macrophages, namely pleural and peritoneal, than to cardiac TRM, suggesting conserved functions for serous cavity macrophages across different organ systems. Pericardial macrophages were enriched for genes associated with homoeostasis and metabolism, whilst cardiac TRM were relatively enriched for genes involves in inflammation and, whilst this study did not take into account the newly recognized heterogeneity of cardiac TRM, it does illustrate the existence of a distinct populations of macrophages, whose contributions to cardiac homoeostasis and following injury has hitherto not been appreciated.
4.3 The heterogeneity and plasticity of macrophage transcriptomes following AMI
Comparison of macrophage populations before and after AMI offers insights into the mechanisms of macrophage differentiation and suggests candidate therapeutic targets to mitigate ventricular remodelling. scRNA-seq of mononuclear phagocytes in the infarcted myocardium 11 days after coronary artery ligation showed preservation of the four macrophage clusters described during homoeostasis, in addition to four novel macrophage populations.69 Pathways enriched in the AMI specific populations were involved in the inflammatory response, including TNF production, cell migration and adhesion, and hypoxic signalling.
Analysis of the preserved homoeostatic populations between non-infarcted and infarcted tissue, showed near identical gene expression, with the exception of a number of core TRM genes, including Timd4, Lyve1, and Igf1, which play important roles in angiogenesis and efferocytosis.69 It is well-established that TRM in the infarct zone undergo rapid cell death following AMI and that macrophages at the infarct site are derived from a rapid influx of circulating monocytes.32,38 These data, therefore, illustrate both the remarkable plasticity of circulating monocytes to differentiate into diverse macrophage populations, as well as a core signature of TRM genes involved in reparative functions that are lost following AMI and that may represent novel therapeutic targets.
Whilst TRM are depleted in the infarct zone following AMI, emerging data suggest these cells play critical roles in remodelling following AMI.73 Global depletion of TRM, by transient inducible knockout of Cx3cr1, results in increased recruitment of monocytes and macrophages to the remote myocardium and greater ventricular remodelling, with reduced LVEF, increased fibrosis and excess mortality 35 days following coronary artery ligation.69 However, selective depletion of Ccr2+ TRM results in reduced monocyte recruitment to the infarcted myocardium and improved ventricular size and function compared to wild types 28 days after an ischaemia–reperfusion (I/R) model of AMI, whilst selective depletion of CD169+ TRM resulted in worse ventricular outcomes, with reduced ejection fraction and end-diastolic volume, suggesting both critical and subset-specific roles for TRM populations in ventricular remodelling post-AMI.
To understand how TRM may be acting, it is important to consider the roles of macrophages in different regions of the myocardium following injury (Figure 4). Whilst TRM are rapidly depleted in the ischaemic zones following AMI, there is rapid expansion of Lyve1neg TRM specifically in the peri-infarct zone from Day 2 post-coronary artery ligation.69 Most studies investigating macrophages after AMI do not differentiate between regions (infarct, peri-infarct, and remote) of the myocardium, and those that do rarely investigate the peri-infarct or remote myocardium specifically, focusing instead on the infarct zone. In the remote myocardium, absolute numbers of macrophages in the remote myocardium expand and persist in the remote myocardium 8 weeks following AMI, and recent analyses suggests it is increased monocyte recruitment and differentiation, rather than TRM expansion, that accounts for this.15,69 Recognizing that macrophage populations may have different roles depending on their location following AMI is an important conceptual shift that may facilitate greater understanding of the mechanisms by which they contribute to ventricular remodelling.
Figure 4.
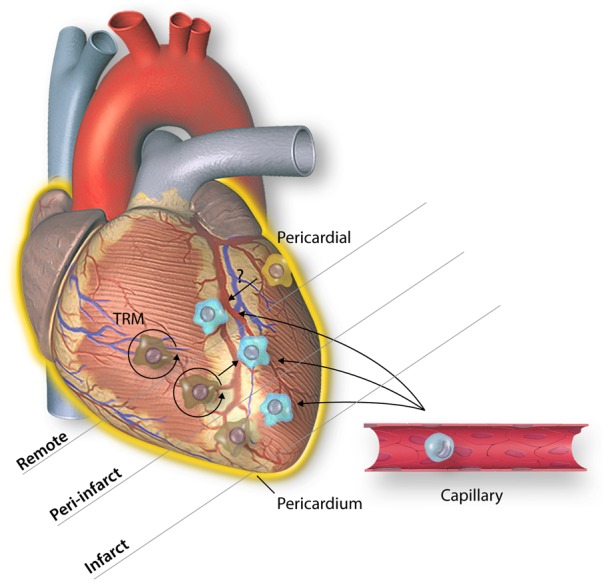
Novel macrophage populations appear to play spatially restricted roles in remodelling of the myocardium following AMI. Following AMI, at the infarct site, TRM undergo rapid cell death, and monocytes rapidly extravasate and differentiate into macrophages. In contrast, in the peri-infarct zone a subpopulation of TRM persist and rapidly proliferate following AMI, whilst monocyte-derived macrophages also contribute to rapid expansion of macrophage numbers at this site. Finally, in the remote myocardium, TRM persist but do not appear to contribute to the increased absolute numbers of macrophages, which is due to infiltration of circulating monocytes. A final, pericardial macrophage population has been identified following AMI. These cells do invade the myocardium but also attach to the epicardial surface and may mediate ventricular remodelling via paracrine signalling.
In addition to spatially restricted roles, there appears to be significant temporal variation in macrophage phenotype following AMI. Whilst scRNA-seq is yet to be applied to investigate temporal variation following AMI, bulk RNA-seq of cardiac macrophages illustrates a temporal shift in macrophage phenotype from pro-inflammatory to reparative to resident like.74,75 Whilst macrophages on Day 3 and Day 7 demonstrated predominantly pro-inflammatory and reparative gene expression profiles respectively, both populations showed mixed expression of canonical M1 and M2 genes, again supporting a more nuanced approach to description of M1-M2 phenotypes.75
Combination of scRNA-seq to identify candidate pathways and use of transient inhibitors offers a strategy to delineate temporally defined pathogenic pathways. For example, analysis of single-cell transcripts in cardiac macrophages on Day 4 following coronary artery ligation demonstrated the presence of an interferon inducible cell cluster, that was absent in Irf3neg/negmice.76 Such a cluster has also been identified during homoeostasis and following I/R injury, and, crucially, is distinct to the CCR2+ cluster, two populations that previously may have been described synonymously as M1 macrophages.69 To investigate the functional relevance of one such novel population, the investigators targeted one candidate pathway. Macrophages of this cluster were enriched for components of the IRF3-IFN (interferon regulatory transcription factor 3–interferon) pathway, which detects cytosolic DNA and initiates an interferon response. Deletion, using constitutive knockout models, of various components of the pathway protected against ventricular remodelling following coronary artery ligation. Crucially, of potential translational application, administration of IFNAR (interferon alpha receptor) neutralising antibody at 12 and 48 h post-AMI abrogated downstream gene expression, improved ventricular size, contractile function, and mouse survival.
The use of a technique to transiently block pathways allows better resolution of the role of particular macrophage phenotypes at different time points following AMI. For example, one might hypothesize that a pathway involved in stimulating fibroblast activity may be beneficial early following AMI but contribute to remodelling if persistently active days to weeks later. Constitutive knockouts are unable to delineate the relative significance of these pathways, whereas techniques to transiently target a pathway would offer translational potential by testing candidate therapies and demonstrating the ideal time points for intervention.
4.4 The impact of model systems on macrophage phenotype
The impact of both timing and location on macrophage phenotype suggests significant interplay between the microenvironment and macrophage plasticity. Permanent coronary artery ligation is widely used to model AMI because it causes significant necrosis and a reproducible inflammatory response. However, in the era of widespread clinical access to reperfusion therapy, important differences between permanent and transient coronary artery ligation models should be considered, including the infarct burden and contribution of reperfusion injury. For example, macrophage populations after I/R have a temporally shifted immune response, with macrophage populations at the infarct site peaking 4 days earlier than in the permanent ligation model and having almost completely resolved by 7 days.77 The study did not address changes in monocyte or macrophage phenotype, but this is an important outstanding research question. Without confirming the impact of therapeutic targets on ventricular remodelling using an I/R model, candidate studies risk identifying genes and pathways that are of limited translational relevance.
Moreover, recent insights into the role of serous cavity macrophages following visceral injury suggests a fundamental limitation of conventional coronary artery ligation and I/R models, as they require disruption of the parietal pericardium to access the coronary arteries and therefore may interfere with the response of pericardial macrophages to cardiac injury. GATA6+ serous cavity macrophages are found in the pericardial, pleural and peritoneal cavities during homoeostasis, and their function following injury is best characterized in the peritoneum, an analogous anatomical structure of the abdomen.78,79 These cells are rapidly recruited to the site of acute liver injury, via a non-vascular route that significantly outpaces extravasation of monocytes and contributes to tissue repair through disassembly of necrotic nuclei and promotion of angiogenesis.80 Modelling MI using a modified coronary artery ligation model, that leaves the parietal pericardium intact, produces a similar infarct size to conventional coronary artery ligation models but, crucially, mice with an intact pericardium show relatively preserved LV function at 4 weeks, suggesting a role for the pericardium in protecting against ischaemic remodelling.72 This cardio-protective effect of the pericardium was abrogated in mice lacking Gata6+pericardial macrophages. Specifically, mice lacking Gata6+macrophages showed increased fibrosis of the remote myocardium, impaired LV function and increased LV stiffness at 4 weeks following AMI, modelled using modified coronary artery ligation, despite similar initial infarct sizes, compared to wild-type controls. These cells do migrate into the myocardium but are localized predominantly around the epicardial surface, leading authors to postulate a possible paracrine mechanism of action. Accordingly, future strategies targeting macrophages may need to address not only cells present in the myocardium but also cells attached to the epicardial surface or in the pericardial space, which may contribute to modulation of remodelling via paracrine mechanisms.
5. Conclusions and future directions
Given the growing body of evidence demonstrating wide phenotypic variation of macrophages in vivo following AMI, understanding their contribution to pathological remodelling requires evolution from the established M1-M2 paradigm. This model assumes a continuum from M1 to M2, whereas in vivo evidence suggests a network of diverse macrophage behaviours, driven by a complex network of stimuli.
Interpretation of studies investigating monocyte and macrophage populations is limited by poor phenotypic resolution, and this may account for apparent inconsistencies in the literature. The emergence of unbiased, high throughput single-cell techniques will facilitate more specific identification of candidate macrophage populations and novel signalling pathways for therapeutic application. Furthermore, accounting for temporal and spatial variation will further contribute to our understanding of the regulation of monocyte and macrophage phenotype. These novel techniques will need to be complemented by mechanistic studies to demonstrate the functional significance of newly described phenotypes.
Despite compelling evidence for a key role for monocytes and macrophages in ventricular remodelling following AMI, targeted immunotherapeutic strategies are slow to emerge. Insights into macrophage biology in the context of atherosclerosis led to the first successful clinical trial of immunotherapy in cardiovascular disease (CANTOS) and this success provides a proof of concept for the utility of immunotherapy in this field. In light of the heterogeneity and plasticity of monocytes and macrophages in the heart following AMI, a conceptual shift from targeting arbitrarily and inconsistently defined myeloid cell populations to targeting candidate pathways identified in these cells may bridge the current gap between our current knowledge and therapeutic strategies.
Conflict of interest: none declared.
Funding
This work was supported by the King’s British Heart Foundation Centre for Excellence Award [RE/18/2/34213]. D.B. is supported by an Academy of Medical Sciences Starter Grant for Clinical Lecturers [SGL020\1087].
References
- 1. Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003;361:13–20. [DOI] [PubMed] [Google Scholar]
- 2. Nielsen PH, Maeng M, Busk M, Mortensen LS, Kristensen SD, Nielsen TT, Andersen HR.. Primary angioplasty versus fibrinolysis in acute myocardial infarction: long-term follow-up in the Danish acute myocardial infarction 2 trial. Circulation 2010;121:1484–1491. [DOI] [PubMed] [Google Scholar]
- 3. Zijlstra F, Hoorntje JCA, de Boer M-J, Reiffers S, Miedema K, Ottervanger JP, van 't Hof AWJ, Suryapranata H.. Long-term benefit of primary angioplasty as compared with thrombolytic therapy for acute myocardial infarction. N Engl J Med 1999;341:1413–1419. [DOI] [PubMed] [Google Scholar]
- 4. Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, Agostino RBD, Levy D, Kannel WB, Vasan RS.. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation 2008;118:2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen J, Hsieh AFC, Dharmarajan K, Masoudi FA, Krumholz HM.. National trends in heart failure hospitalization after acute myocardial infarction for Medicare beneficiaries 1998-2010. Circulation 2013;128:2577–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gerber Y, Weston SA, Berardi C, Mcnallan SM, Jiang R, Redfield MM, Roger VL.. Contemporary trends in heart failure with reduced and preserved ejection fraction after myocardial infarction: a community study. Am J Epidemiol 2013;178:1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ezekowitz JA, Kaul P, Bakal JA, Armstrong PW, Welsh RC, McAlister FA.. Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J Am Coll Cardiol 2009;53:13–20. [DOI] [PubMed] [Google Scholar]
- 8. Visan I. Myocardial infarct inflammation. Nat Immunol 2018;19:99.. [DOI] [PubMed] [Google Scholar]
- 9. Nahrendorf M. Myeloid cell contributions to cardiovascular health and disease. Nat Med 2018;24:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swirski FK, Nahrendorf M.. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol 2018;18:733–744. [DOI] [PubMed] [Google Scholar]
- 11. Sutton M, Sharpe N.. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 2000;101:2981–2988. [DOI] [PubMed] [Google Scholar]
- 12. Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, Nahrendorf M, Yun SH.. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res 2013;112:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee WW, Marinelli B, Laan AVD, Sena BF, Gorbatov R, Leuschner F, Dutta P, Iwamoto Y, Ueno T, Begieneman MPV, Niessen HWM, Piek JJ, Vinegoni C, Pittet MJ, Swirski FK, Tawakol A, Carli MD, Weissleder R, Nahrendorf M.. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol 2012;59:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, Libby P, Weissleder R, Pittet MJ.. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M.. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 2016;119:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saxena A, Russo I, Frangogiannis NG.. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res 2016;167:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE.. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol 2003;91:1055–1059. [DOI] [PubMed] [Google Scholar]
- 18. Silverman HS, Pfeifer MP.. Relation between use of anti- inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. Am J Cardiol 1987;59:363–364. [DOI] [PubMed] [Google Scholar]
- 19. Ziegler-Heitbrock H. Heterogeneity of human blood monocytes: the CD14+CD16+ subpopulation. Immunol Today 1996;17:424–428. [DOI] [PubMed] [Google Scholar]
- 20. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Andreas JR, Ziegler-Heitbrock L, Randolph GJ, Habenicht A.. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2014;115:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Passlick B, Flieger D, Ziegler-Heitbrock HW.. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989;74:2527–2534. [PubMed] [Google Scholar]
- 22. Geissmann F, Jung S, Littman DR.. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003;19:71–82. [DOI] [PubMed] [Google Scholar]
- 23. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, Rooijen NV, Grainger JR, Belkaid Y, Ma A, Riches DWH, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ.. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013;39:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC.. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C− monocytes. Nat Immunol 2011;12:778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomas GD, Hanna RN, Vasudevan NT, Jain MK, Glass CK, Hedrick CC, Function MG, Thomas GD, Hanna RN, Vasudevan NT, Hamers AA, Romanoski CE, Jain MK, Glass CK, Hedrick CC.. Deleting an Nr4a1 super-enhancer subdomain ablates Ly6C low monocytes while preserving article deleting an Nr4a1 super-enhancer subdomain ablates Ly6C low monocytes while preserving. Immunity 2016;45:975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D.. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 2003;197:1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Landsman L, Bar-On L, Zernecke A, Kim K, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S.. CX 3 CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 2009;113:963–973. [DOI] [PubMed] [Google Scholar]
- 28. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F.. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007;317:666–671. [DOI] [PubMed] [Google Scholar]
- 29. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F.. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013;153:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM.. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol 2000;164:6166–6173. [DOI] [PubMed] [Google Scholar]
- 31. Sica A, Erreni M, Allavena P, Porta C.. Macrophage polarization in pathology. Cell Mol Life Sci 2015;72:4111–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M.. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 2014;115:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Silva ND, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P.. Macrophages facilitate electrical conduction in the heart. Cell 2017;169:510–522.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W, Hsiao H-M, Higashikubo R, Saunders BT, Bharat A, Goldstein SR, Krupnick AS, Gelman AE, Lavine KJ, Kreisel D.. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016;1. pii: 87315. doi: 10.1172/jci.insight.87315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hilgendorf I, Gerhardt LMS, Tan TC, Winter C, Holderried TAW, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK.. Ly-6C high monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 2014;114:1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo J-L, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ.. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009;325:612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD.. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure critical importance of the cardiosplenic axis. Circ Res 2014;114:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M.. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 2012;209:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HWM, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M.. Myocardial infarction accelerates atherosclerosis. Nature 2012;487:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, Kitabata H, Okochi K, Arita Y, Ishibashi K, Komukai K, Kataiwa H, Nakamura N, Hirata K, Tanaka A, Akasaka T.. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol 2009;54:130–138. [DOI] [PubMed] [Google Scholar]
- 41. Laan AVD, Horst ET, Delewi R, Begieneman MPV, Krijnen PAJ, Hirsch A, Lavaei M, Nahrendorf M, Horrevoets AJ, Niessen HWM, Piek JJ.. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J 2014;35:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M.. Impaired infarct healing in atherosclerotic mice with Ly-6ChiMonocytosis. J Am Coll Cardiol 2010;55:1629–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maekawa Y, Anzai T, Yoshikawa T, Asakura Y, Takahashi T, Ishikawa S, Mitamura H, Ogawa S.. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J Am Coll Cardiol 2002;39:241–246. [DOI] [PubMed] [Google Scholar]
- 44. Laan AVD, Hirsch A, Robbers L, Nijveldt R, Lommerse I, Delewi R, Vleuten PVD, Biemond BJ, Zwaginga JJ, Giessen WVD, Zijlstra F, Rossum AV, Voermans C, Schoot CVD, Piek JJ.. A proinflammatory monocyte response is associated with myocardial injury and impaired functional outcome in patients with ST-segment elevation myocardial infarction: monocytes and myocardial infarction. Am Heart J 2012;163:57–65.e2. [DOI] [PubMed] [Google Scholar]
- 45. Hirsch A, Nijveldt R, Vleuten PVD, Tijssen JGP, Giessen WVD, Tio RA, Waltenberger J, Berg JT, Doevendans PA, Aengevaeren WRM, Zwaginga JJ, Biemond BJ, Rossum AV, Piek JJ, Zijlstra F.. Intracoronary infusion of mononuclear cells from bone marrow or peripheral blood compared with standard therapy in patients after acute myocardial infarction treated by primary percutaneous coronary intervention: results of the randomized controlled HEBE. Eur Heart J 2011;32:1736–1747. [DOI] [PubMed] [Google Scholar]
- 46. Delewi R, Laan AVD, Robbers L, Hirsch A, Nijveldt R, Vleuten PVD, Tijssen JGP, Tio RA, Waltenberger J, Berg JT, Doevendans PA, Gehlmann HR, Rossum AV, Piek JJ, Zijlstra F.. Long term outcome after mononuclear bone marrow or peripheral blood cells infusion after myocardial infarction. Heart 2015;101:363–368. [DOI] [PubMed] [Google Scholar]
- 47. Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, Gorbatov R, Iwamoto Y, Dutta P, Wojtkiewicz G, Courties G, Sebas M, Borodovsky A, Fitzgerald K, Nolte MW, Dickneite G, Chen JW, Anderson DG, Swirski FK, Weissleder R, Nahrendorf M.. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013;127:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsou C-L, Peters W, Si Y, Slaymaker S, Aslanian AM, Wesiberg SP, Mack M, Charo IF.. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 2007;117:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M, Reconstitution BM.. Targeting interleukin-1 β reduces leukocyte production after acute myocardial infarction. Circulation 2015;132:1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ridker PM, Dellborg M, Kastelein JJP, Rossi PRF, Pella D, Kobalava Z, Fonseca F, Everett BM, Troquay RPT, Glynn RJ, Genest J, Forster T, Cornel JH, Lorenzatti A, Thuren T, MacFadyen JG, Anker SD, Flather M, Cifkova R, Libby P, Vida-Simiti L, Koenig W, Shimokawa H, Nicolau J, Ogawa H, Ballantyne C, Chang WH, Pais P.. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 51. Wang Z, Huang S, Sheng Y, Peng X, Liu H, Jin N, Cai J, Shu Y, Li T, Li P, Fan C, Hu X, Zhang W, Long R, You Y, Huang C, Song Y, Xiang C, Wang J, Yang Y, Liu K.. Topiramate modulates post-infarction inflammation primarily by targeting monocytes or macrophages. Cardiovasc Res 2017;113:475–487. [DOI] [PubMed] [Google Scholar]
- 52. Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML.. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol 2017;112:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shintani Y, Yashiro K, Ishida H, Saba R, Suzuki K, Shiraishi M, Yamaguchi A, Adachi H, Shintani Y.. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 2016;126:2151–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin Y, Han H, Manicone AM, Lindsey ML.. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res 2012;78245:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, Thorp EB.. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 2013;113:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, Wojtkiewicz G, Dutta P, Sager HB, Borodovsky A, Novobrantseva T, Klebanov B, Fitzgerald K, Anderson DG, Libby P, Swirski FK, Weissleder R, Nahrendorf M.. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol 2014;63:1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang M, Song L, Wang L, Yukht A, Ruther H, Li F, Qin M, Ghiasi H, Sharifi BG, Shah PK.. Deficiency of GATA3-positive macrophages improves cardiac function following myocardial infarction or pressure overload hypertrophy. J Am Coll Cardiol 2018;72:885–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, DeNardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL.. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014;40:274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, Hacken NT, Cobos Jiménez V, Kootstra NA, Hamann J, Greaves DR, Locati M, Mantovani A, Gordon S.. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 2013;121:e57–e69. [DOI] [PubMed] [Google Scholar]
- 60. Czapla J, Matuszczak S, Wiśniewska E, Jarosz-Biej M, Smolarczyk R, Cichoń T, Głowala-Kosińska M, Śliwka J, Garbacz M, Szczypior M, Jaźwiec T, Langrzyk A, Zembala M, Szala S.. Human cardiac mesenchymal stromal cells with CD105+CD34- phenotype enhance the function of post-infarction heart in mice. PLoS One 2016;11:e0158745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nahrendorf M, Swirski FK.. Abandoning M1/M2 for a network model of macrophage function. Circ Res 2016;119:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A.. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 2018;122:1661–1674. [DOI] [PubMed] [Google Scholar]
- 63. Mildner A, Schönheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, Paul F, Chappell-Maor L, Priller J, Leutz A, Amit I, Jung S.. Genomic characterization of murine monocytes reveals C/EBPβ transcription factor dependence of Ly6C– cells. Immunity 2017;46:849–862. [DOI] [PubMed] [Google Scholar]
- 64. Spitzer MH, Nolan GP.. Mass cytometry: single cells, many features. Cell 2016;165:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, Zernecke A, Pramod AB, Ghosh AK, Michel NA, Hoppe N, Hilgendorf I, Zirlik A, Hedrick CC, Ley K, Wolf D.. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res 2018;122:1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thomas GD, Hamers AAJ, Nakao C, Marcovecchio P, Taylor AM, McSkimming C, Nguyen AT, McNamara CA, Hedrick CC.. Human blood monocyte subsets: a new gating strategy defined using cell surface markers identified by mass cytometry. Arterioscler Thromb Vasc Biol 2017;37:1548–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL.. Article embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ.. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 2018;24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epelman S.. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 2019;20:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ishihara T, Ferrans VJ, Jones M, Boyce SW, Kawanami O, Roberts WC.. Histologic and ultrastructural features of normal human parietal pericardium. Am J Cardiol 1980;46:744–753. [DOI] [PubMed] [Google Scholar]
- 71. Michailova KN, Usunoff KG.. Serosal membranes (pleura, pericardium, peritoneum). Normal structure, development and experimental pathology. Adv Anat Embryol Cell Biol 2006;183:ivii1–ivii144. [PubMed] [Google Scholar]
- 72. Deniset JF, Belke D, Lee W-Y, Jorch SK, Deppermann C, Hassanabad AF, Turnbull JD, Teng G, Rozich I, Hudspeth K, Kanno Y, Brooks SR, Hadjantonakis A-K, O’Shea JJ, Weber GF, Fedak PWM, Kubes P.. Gata6+ pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 2019;51:131–140.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao H-M, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ.. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 2019;124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML.. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 2018;113:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Walter W, Alonso-Herranz L, Trappetti V, Crespo I, Ibberson M, Cedenilla M, Karaszewska A, Núñez V, Xenarios I, Arroyo AG, Sánchez-Cabo F, Ricote M.. Deciphering the dynamic transcriptional and post-transcriptional networks of macrophages in the healthy heart and after myocardial injury. Cell Rep 2018;23:622–636. [DOI] [PubMed] [Google Scholar]
- 76. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, Kohler RH, Arlauckas SP, Yoshiko V, Savo A, Sadreyev RI, Kelly M, Fitzgibbons TP, Fitzgerald KA, Mitchison T, Libby P, Nahrendorf M, Weissleder R.. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 2017;23:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M.. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 78. Ghosn EEB, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA.. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A 2010;107:2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Okabe Y, Medzhitov R.. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014;157:832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang J, Kubes P.. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 2016;165:668–678. [DOI] [PubMed] [Google Scholar]