

Abstract
Although microRNA (miRNA) expression levels provide important information regarding disease states owing to their unique dysregulation patterns in tissues, translation of miRNA diagnostics into point-of-care (POC) settings has been limited by practical challenges. Here, we developed a hydrogel-based microfluidic platform for colorimetric profiling of miRNAs, without the use of complex external equipment for fluidics and imaging. For sensitive and reliable measurement without the risk of sequence bias, we employed a gold deposition-based signal amplification scheme and dark-field imaging, and seamlessly integrated a previously developed miRNA assay scheme into this platform. The assay demonstrated a limit of detection of 260 fM, along with multiplexing of small panels of miRNAs in healthy and cancer samples. We anticipate this versatile platform to facilitate a broad range of POC profiling of miRNAs in cancer-associated dysregulation with high-confidence by exploiting the unique features of hydrogel substrate in an on-chip format and colorimetric analysis.
Small noncoding RNAs, called microRNAs (miRNAs), have become increasingly important in disease diagnosis due to their abnormal expression in many diseases such as cancer, diabetes, neural, and heart diseases.1−4 Previous studies had demonstrated that miRNAs have high stability and can be detected in various biological fluids such as blood serum and plasma.5 However, clinicians still lack proper tools for high-confidence quantification of miRNA owing to their low abundance and sequence homology.6,7 Most challenges arise due to the grueling demands of an assay that could easily be integrated into a point-of-care (POC) clinical setting.8 An ideal platform ought to provide high sensitivity, high specificity, and multiplexing while minimizing the use of external equipment, and involving simple sample preparation and assay operation. While quantitative real-time polymerase chain reaction (qRT-PCR) is widely used as a gold standard for high sensitivity, it is limited in practical application, due to its requirement of substantial sample preparation steps, such as RNA extraction, expensive instruments, and intricately complex design of primers to avoid sequence bias arising from target-based amplification.9,10 To address these limitations of the conventional method, several techniques have now been developed for miRNA detection, fluorescent probes,11,12 isotachophoresis,13,14 and nanomaterial-based in vivo sensing.15,16 Despite advancements, the techniques do not satisfy all the requirements of POC regarding miRNAs, and the need for a more clinically feasible approach still remains.17
Hydrogel-based microfluidic approach can be advantageous in the development of reliable and affordable POC diagnostics.18−20 The superiority of nonfouling hydrogels has been previously demonstrated in comparison to surface-based systems for nucleic acid hybridization, especially from biologically complex samples.21 Better thermodynamics in the gel increases both sensitivity and specificity, and the solution-like environment of a hydrogel provides faster hybridization kinetics.22 Furthermore, it is possible to directly measure miRNAs in complex media such as cell lysate or serum, without RNA extraction and target amplification.12,23 In a previous gel particle-based approach, a universal labeling scheme was developed, which allowed the use of a single label for all captured miRNAs, and measured miRNA dysregulation patterns.24,25 Here, we adapted the same hydrogel chemistry and miRNA capture approach into an on-chip assay for POC applications.
POC diagnostics can facilitate fast and accurate identification of diseases, which leads to better treatment of patients. To fit the POC criteria of cost-effectiveness, portability, and accessibility, miRNA assays are commonly performed with nonfluorescent labeling molecules for visualization. Since such assay schemes offer only low to moderate detection sensitivity, miRNA detection methods often rely on signal boosting step, such as enzymatic and nucleic acid amplification.26−28 The Mirkin group has developed an assay using gold nanoparticles, with silver or gold deposition, which had been successfully applied to detect various biomarkers, including miRNAs.29−31 Their method provides robust signals without bleaching issues because of high photostability of gold-labeled conjugates, and very short amplification step with high sensitivity while minimizing equipment requirements.32 However, this microarray-based method typically requires a long assay time (overnight hybridization of target miRNAs) with complicated fluidic steps. Thus, we aim to integrate on-chip hydrogel assay and gold deposition scheme to achieve sensitive POC applications.
We leverage the advantages of microfluidic channels, and the chemical advantages of a polyethylene glycol (PEG) hydrogel scaffold for miRNA hybridization while optimizing an enzyme-free gold nanoparticle-based signal amplification scheme for POC diagnostics. Particularly, we sought to use dark-field imaging for high sensitivity, which had previously been demonstrated for ultrasensitive colorimetric nucleic acid assay.33 Dark-field imaging-based methods boost the scattered intensity of gold conjugates with white light illumination, thus minimizing the requirement of complex and expensive instrumentation for miRNA detection.34 With just a 15 min signal amplification step, we achieved a limit of detection of 260 fM and multiplexed miRNA quantification with minimal sample input. We expect this assay platform to be beneficial in a wide range of clinical samples, including cellular lysate and serum for POC applications.
Experimental Section
Microfluidic Chip Preparation
Commercial chips with straight channels (50 μm in height, 1 mm width, and 18 mm length) were purchased from Hilgenberg GmbH, Germany for performing all assays. Connection ports were fabricated in polydimethylsiloxane (PDMS) (Corning, Sylgard 184) with holes, which were punched with 15-gauge needles. All connection ports were bonded onto inlets and outlets of glass chips by oxygen plasma treatment (25 s on RF = high, Harrick Scientific, Pleasantville, NY). After a subsequent incubation at 80 °C for 20 min, a 2% (v/v) solution of 3-(trimethoxysilyl)propyl acrylate (Sigma) mixed in 24.5% (v/v) 1X PBS (phosphate buffered saline, Corning), and 73.5% (v/v) ethanol was filled inside the channels for tight adhesion of the gel pads on the glass walls. After 30 min, channels were rinsed with ethanol and dried with argon gas. After bioassays, chips were cleaned by soaking in 1 M NaOH for 1 h and then washed with DI water and ethanol. Next, they were dried with argon gas; chips were stored at 80 °C until the time of usage. These reusable glass chips were used for several assays (more than 10 times) by repeating the cleaning and activating procedures.
On-Chip Hydrogel Synthesis
All chemicals were purchased from Sigma-Aldrich (U.S.). Hydrogel posts were synthesized using projection lithography from a polyethylene glycol monomer mixture. The monomer mixture consisted of 20% (v/v) PEGDA 700 (poly(ethylene glycol) diacrylate, MW = 700 g/mol), 40% (v/v) PEG 600 (poly(ethylene glycol), MW = 600 g/mol), 5% (v/v) Darocur 1173 photoinitiator, and 35% (v/v) 3x TE (Tris-EDTA, USB Corporation) buffer with food coloring dye, which was previously optimized for the diffusion and reaction in bioassays.35,36 The monomer solution was diluted 9:1 with the acrydite-modified probes (Integrated DNA Technologies (IDT), Coralville, IA). The concentration of probe molecules was adjusted from the coarse rate matching as described in a previous study (miR-21:247 μM, miR-145, and let-7a: 50 μM).24 Probe-containing prepolymer solution was vortexed, centrifuged, and loaded into channels by pipetting. An inverted microscope (Zeiss Axio Observer A1) and a CCD camera (Andor Clara) were used for UV-initiated polymerization. With a desired photomask (Fineline Imaging) placed in the fieldstop, the polymerization was performed at 100 ms exposure (Lumen 200, Prior Scientific) using a 20× objective (Zeiss Plan-Neofluar) and a dichrioc filter for excitation at 365 nm. After polymerization, channels were rinsed with 1X TE buffer. For multiplexed assays, the subsequent monomer solution with a different probe was loaded in the same channel and polymerization was repeated as described in earlier. The posts containing different probe sequences were immobilized at spatially distinct locations in a single channel.
Fluidic Control
All assays were performed with gravity driven flow by appropriately inserting a 200 μL pipet tip into PDMS inlet port. The flow rate was observed to be 1–5 μL/min. Previously, we calculated the channel Péclet number (Pe) to be approximately 7000.25 With the high values of Pe (Pe ≫ 1), target depletion is assumed to be negligible.37 The solution was refilled in the tip of inlet port every 10 min to maintain height differences for a steady flow. Since our system is highly flexible and can be incorporated with various types of flows such as pressure-driven flows and syringe pump-driven flows, the manual iterative operation can be avoided by simply using an external equipment for fluidics.
MicroRNA Assay
Blocking solution of 3% (w/v) Pluronic F108 (Sigma-Aldrich) in nuclease-free water (Affymetrix) was filled into channels containing hydrogel posts. After 30 min, the process of target hybridization to specific probes was performed with the mixture of synthetic RNAs (IDT) or total RNA (BioChain, Newark, CA) in a TET buffer with a final concentration of 350 mM NaCl for 90 min. The sequence information on all the probes and targets used in this study was summarized in Supporting Information Table S1. Total RNA was stored at 100 ng/μL at −20 °C and 500 ng of total RNA dissolved in 200 μL was used for assay. Before performing the assay, the solution with total RNA was incubated at 95 °C for 5 min in a thermoshaker for disrupting secondary structures, and cooled down at room temperature. The hybridization mixture was loaded into the microchannel through the precut pipet tips in the injection port of the device. Target hybridization occurred at elevated temperature (55 °C) on a hot plate, and all subsequent steps were performed at room temperature. In previous works, the temperature in channels was validated to be constant based on a simple heat transfer calculation.25 For all assays, steady flows were maintained to deliver the molecules without depletion. In between assay steps, rinses were performed using 50 mM NaCl in TET for 30 s to impose sufficient stringency for high specific miRNA measurement. For ligation, the universal linker (IDT), T4 DNA ligase (800 U/mL), ATP (250 nM), and 10× NEB2 buffer (New England Biolabs, Ipswich, MA) were mixed in TET and incubated in the channels for 10 min as described in previous studies.24 After washing, Nanogold-Streptavidin (80 μg/mL, Nanoprobes, Inc., Yaphank, NY) diluted in 1× PBS (0.1% v/v) was loaded into channels for 30 min. For gold deposition, GoldEnhance (Nanoprobes, Inc.) was used, which consisted of Solution A (enhancer), Solution B (activator), Solution C (initiator), and Solution D (buffer). First, 200 μL of Solution A and B were mixed, and after 5 min, 200 μL of Solution C and D were added into the mixture. After vortexing, the mixture was delivered to the microchannels with posts for an optimal amplification time of 15 min.
Data Acquisition and Analysis
The hydrogel posts were imaged using a Zeiss Axio Observer A1 microscope with a 10× objective and a dark-field condenser. Each image was analyzed using ImageJ (National Institutes of Health).38 The color images we obtained were split to the respective red, green, and blue image components. The region of interest (ROI) was identified as a post circle with a defined diameter. For each ROI, the intensity of pixels in the red channel was only averaged to analyze the wine-red color of miRNA-gold conjugates. Background signal was calculated by averaging the red pixel intensities in a region outside the post, within the microfluidic chip. Target signal was computed by subtracting background signal from a raw signal with target incubation (Starget = Starget, raw – Sbackground). Control raw signal was obtained from the posts after incubation with 0 fM of synthetic targets. After subtracting background signal (Scontrol = Scontrol, raw – Sbackground), the control signal serves as the assay background, and the noise was defined as the standard deviation of the control signals (σcontrol). Control signal was subtracted from the desired target signal (Starget, net = Starget – Scontrol) to obtain a net signal. This makes Scontrol, net = 0. Finally, the signal-to-noise ratio (SNR) was determined as the ratio of net signal to assay noise (SNR = Starget, net/σcontrol). Limit of detection of the system was defined as the miRNA concentration at which the SNR equals 3, as per previously published protocols.25
Results and Discussion
Assay Development
For the development of a sensitive miRNA assay in point-of-care (POC) diagnostics, we sought to apply a rapid signal amplification scheme by gold deposition onto a hydrogel-based on-chip miRNA sensor. Previously, we had demonstrated ultrasensitive measurement of miRNAs using oil-isolated hydrogel chambers in microfluidic chip, with a limit of detection (LOD) of 22.6 fM.25 We had achieved high Péclet number in the previous work by using a gravity-driven flow to eliminate mass transfer limits that could arise from insufficient target molecule deliveries to the gel posts. Importantly, this strategy eliminated the need for complicated external flow controllers, making our system suitable for point of care applications. However, this assay was based on fluorescence measurements, which required expensive and complex imaging instruments. To overcome this issue, we used a colorimetric labeling approach based on gold deposition for signal amplification and dark-field imaging.
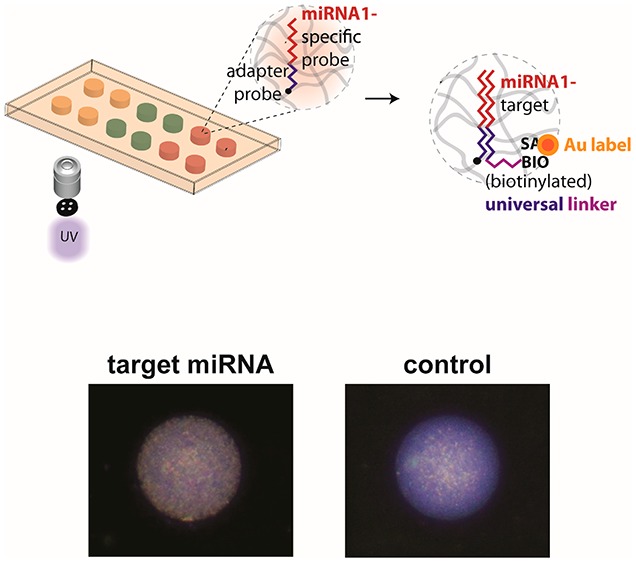
As done previously, we immobilized spatially encoded miRNA probe-bearing hydrogel posts in microfluidic channels using projection lithography to control their size and shape (Figure 1a). Multiplexing can be achieved with a spatial encoding scheme by exchanging monomer solutions in the device and polymerizing posts. The incorporated probes consist of two domains: a miRNA-specific domain and a universal linker domain. The linker used for labeling was the same for all measured miRNAs. A fully controlled steady gravity-driven flow was used for all assay steps, thus eliminating the need for expensive and cumbersome flow controllers. Target hybridization occurred at elevated temperatures (55 °C) on a hot plate, whereas all subsequent steps were performed at room temperature (Figure 1b). We ligated the biotinylated universal linker to the probe-target complex using the T4 DNA ligase for 10 min. Without target miRNA bound to the probe, the universal linker would be released during the stringent washing step with low salt (50 mM NaCl) buffer due to low binding affinity to probe cross-linked into the hydrogel. Next, the probe-target complex was labeled using a streptavidin-conjugated gold nanoparticle (SA-Au, 80 ng/mL, 30 min). Thereafter, the signal amplification scheme, based on gold ion deposition, was applied for 15 min.
Figure 1.

(A) Schematics of on-chip hydrogel post synthesis for multiplexing of small miRNA panels using projection lithography with the spatial encoding scheme. (B) miRNA assay scheme: target hybridization, universal linker ligation, gold nanoparticle labeling, and gold ion deposition-based signal amplification. (C) Dark-field images of posts after complete miRNA assay demonstrating the dose-dependent response of miRNAs. Scale bar represents 100 μm.
Both bright- and dark-field systems were considered for imaging the gold-labeled target miRNAs. For system characterization, we used immobilized gel posts functionalized with biotinylated DNA probe (5 nM). Although bright-field mode is widely used in colorimetric assay, its high background signals often generate large variations. More importantly, there was no difference between the presence and absence of probe after labeling streptavidin-conjugated gold nanoparticles and signal amplification with catalytic gold deposition (Supporting Information (SI) Figure S1). Conversely, with dark-field illumination we observed measurable signals from 5 nM probe. Thus, for sensitive measurement of surface plasmon resonance (SPR) scattering from gold nanoparticles, we used dark-field microscopy with the appearance of wine-red color. As shown in Figure 1c, in an initial trial with miRNA assay with the analysis using dark-field microscopy, the red dots were gradually increased as the concentration of target miRNA increased from 0 to 10 pM. Considering simplicity of the assay, further analysis was performed with red channel in RGB imaging.
Optimization and Assessment of Signal Amplification
Since previous studies had used the gold deposition scheme in a microarray format, we first needed to optimize the signal amplification step in the on-chip hydrogel-based assay. While previous studies used three rounds of 5 min gold deposition to maximize the signal-to-noise ratio (SNR) by increasing target-binding signal,31 we sought to run a single-step amplification to minimize the assay complexity for POC applications. We hypothesized that the solution-like and nonfouling environments of hydrogel posts would be superior to a microarray platform (rigid, planar surfaces) for gold deposition on targets without nonspecific background. For the optimization of the signal amplification step, we measured the SNR as a function of gold ion deposition time. Using immobilized gel posts functionalized with either biotinylated DNA (final concentration of 5 nM) or no biotin (serving as control), we loaded Nanogold-Streptavidin (80 ng/mL, PBS) for 30 min and, after washing, GoldEnhance mixture for 5–60 min. Then, we calculated SNR, which is defined as the net control-subtracted signal divided by the standard deviation of control measurements (assay-derived noise). As shown in Figure 2, SNR increased up to a time of 15 min, and then decreased due to high background signal. This indicated that a hydrogel-based system can achieve high sensitivity, simply with a single-step amplification without the need of multiple rounds.
Figure 2.
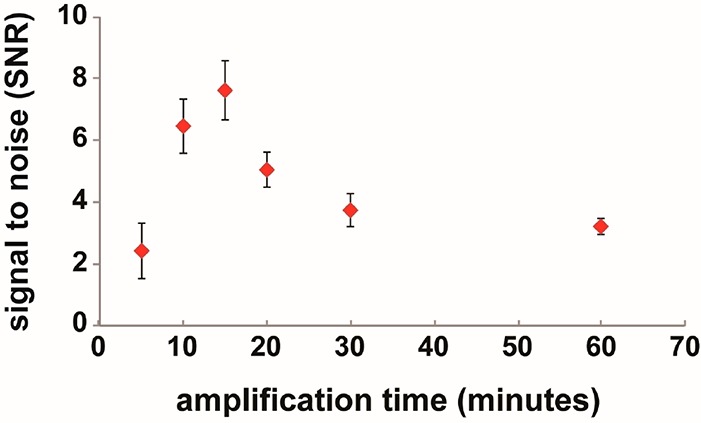
Optimization of amplification time. The signal-to-noise ratio (SNR) was continued to increase for up to 15 min of gold deposition-based signal amplification. After achieving a maximum value at 15 min, SNR decreased and then saturated due to nonspecific binding of gold ions. Error bars represent standard deviation (n = 8–15).
Next, we examined the assay stability, which is one of the important considerations for reliable measurement in POC diagnostics. An unstable signaling label would induce test errors. Also, sometimes assay platforms might need to be transferred from remote areas to a core facility for analysis by experts for accurate diagnostics. With 5 nM of the biotinylated probe, we analyzed the net signal from the gold-labeled conjugates over 24 h, after Nanogold-Streptavidin labeling and gold ion deposition steps. As described earlier, the net signal was determined as control (no biotinylated probe)-subtracted target signal. As expected, the net signal did not decrease, as shown in SI Figure S2. Importantly, it was noted that hydrogels and gold nanoparticles are stable for several months without degradation,21,39 which is expected to allow long-term storage and reimaging. Therefore, this assay platform would provide high stability in analysis, without photobleaching, and can be stored for a long time or be transported, if necessary, rendering it ideal for POC diagnostics.
Detection Sensitivity
Using the gold labeling scheme, we examined the signal changes in 10 pM spike-in of let-7a as a function of hybridization time. The target capture increased for 90 min and then saturated, as observed in previous studies (SI Figure S3).24,25 To minimize the RNA input requirement while retaining high sensitivity, we decided to use 90 min of hybridization. The 90 min hybridization was recommended due to the stringent buffer conditions (high temperature and low salt concentration), which we optimized earlier for specific miRNA measurements to distinguish between even 1–2 mismatches.24,25 If necessary, we could reduce the assay time, which would still give us a reasonable signal.
We next investigated the sensitivity of our hydrogel-based colorimetric detection scheme for detecting miRNAs. The synthetic miRNA spike-ins (from 100 fM to 10 pM let-7a) were incubated in each channel of a microfluidic chip. To account for the assay background, the net signal was considered–the control signal (0 fmol spike-in) was subtracted from the target signals. We show the dose-dependent responses from let-7a spike-ins in Figure 3. As shown in SI Figure S4, we then calculated the limit of detection (LOD), which was previously defined in Experimental Section. Our assay scheme provided a LOD of 260 fM, which is relatively sensitive compared to other colorimetric assays (pM to sub-pM).40−42 Although some recent studies on colorimetric miRNA assays show high sensitivity (fM to sub-fM),10,43,44 their methods rely on target-based amplification with the possible high risk of sequence bias. In addition, we might achieve better sensitivity if we run multiple rounds of signal amplification as a previous study optimized to perform the three rounds of gold deposition to maximize SNR.31
Figure 3.
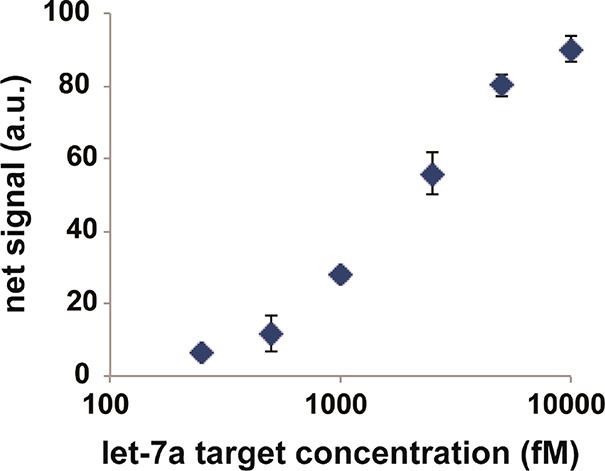
Assay sensitivity. The colorimetric miRNA measurements in microfluidics provided limit of detection (LOD) of 260 fM without expensive and complex instrument (see SI Figure S4 for SNR calculation). Error bars represent standard deviation (n = 5–14).
Additionally, the size and geometry of hydrogel posts can be optimized to improve the sensitivity of miRNA measurements. In a miRNA assay with the high concentration of targets such as 10 pM, we observed that more targets were attached to the edge of the post, rather than evenly distributed (Figure 1c). Previously, we optimized the pore size of gel scaffold to allow the fast diffusion of molecules. However, the rapid deposition of gold ions seems to affect the transport rate of labeling molecules, especially with the high level of probe-target conjugates. To consider flux into porous hydrogel posts, we could use ring structures instead of disk shapes as in a recent publication.20 By analyzing a ring-area around the edges, we expect that we could achieve higher mean signal and better sensitivity, which would be beneficial in POC diagnostics.
Multiplex Detection
After characterization, we measured the cross-reactivity of three microRNAs. Three clinical miRNA targets relevant in lung tumor were considered in this study: let-7a, miR-145, and miR-21. We immobilized three types of hydrogel posts, bearing each miRNA probe using the spatial encoding scheme. As shown in SI Figure S5, there was no significant interference among three different miRNA targets (∼20% of cross-reactivity). This minimal cross-reactivity of our hydrogel-based miRNA assay scheme enabled multiplexing analysis of small panels of miRNAs.
For multiplexing, each miRNA probe concentration was adjusted by coarse-rate matching (Experimental Section). All three miRNAs were expected to follow the same rate under the same hybridization condition (salt and temperature). To verify this hypothesis for the hydrogel-based colorimetric assay, we measured the detection limit for each microRNA target based on the calibration curve (SI Figure S4). As expected, all three microRNA targets showed similar LOD (SI Table S2).
As a proof of concept, we compared the miRNA expression in tumor and healthy tissue from total RNA samples using the colorimetric platform developed here. With the posts bearing three miRNA probes (let-7a, miR-145, and miR-21), we performed the multiplexed assay with total RNA samples, and we observed the dysregulation patterns of the three miRNA targets in healthy and tumor tissue (Figure 4). The patterns are consistent with prior studies the literature, since the expression of both let-7a and mir-145 are known to be decreased in lung tumor tissue, whereas that of miR-21 is elevated.24,25,45,46 Also, by using the same total RNA sample, signal from this colorimetric assay were comparable to those in previous studies25 using fluorescent labels (phycoerythrin-conjugated streptavidin reporter, SA-PE) (SI Figure S6), which were previously validated with qRT-PCR.24 This consistency with prior work demonstrates the high performance and reproducibility of our new miRNA measurement without using expensive fluorescence detection.
Figure 4.

Multiplexed measurements of three miRNA targets from total RNA derived from tissue. (A) The signal-to-noise ratio (SNR) of three miRNAs in tumor and healthy samples were plotted, and each of the tumor and healthy samples was measured 10 times. Error bars represent the standard deviation of targets normalized by assay noise (n = 10). (B) Dysregulation ratios of three miRNA targets in lung tumor versus healthy tissue were as expected from the previous studies, validating our assay multiplexing scheme. Error bars represent the standard deviation of miRNA expression measurements in tumor normalized by background-subtracted average miRNA signal in normal and by the ratio of tumor to normal miRNA expression.
Conclusions
Here, we present a technique for miRNA quantification in point-of-care (POC) diagnostics by using gold deposition-based signal amplification scheme with the on-chip hydrogel sensor platform. Unlike previous studies, the colorimetric assay developed here demonstrated highly stable and reliable miRNA measurement without the need of expensive instruments. This assay provided a limit of detection of 260 fM, which is relatively sensitive compared to other colorimetric assays. Moreover, our system enabled multiplex analysis of small panels of miRNAs with relatively simple assay steps. We successfully analyzed the dysregulation of miRNAs in lung tumor with respect to that in healthy tissues. In the future, it might be possible to integrate smartphone-based imaging system for immediate processing and analysis, with the help of smart algorithms. We envision that our system to have wide-ranging applications in POC clinical settings for various targets such as miRNAs, RNAs, DNA, and proteins.
Acknowledgments
We acknowledge the support from NIH-NIBIB Grant 5R21EB024101-02, the C1 Gas Refinery Program funded by the Ministry of Science and ICT (2018M3D3A1A01055732), National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018R1C1B6004086), and Korea Research Institute of Bioscience and Biotechnology Research Initiative Program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05043.
A list of reagents, additional supporting tables, and figures are available as noted in the text (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lu J.; Getz G.; Miska E. A.; Alvarez-Saavedra E.; Lamb J.; Peck D.; Sweet-Cordero A.; Ebert B. L.; Mak R. H.; Ferrando A. A.; Downing J. R.; Jacks T.; Horvitz H. R.; Golub T. R. Nature 2005, 435, 834–838. 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Leung S. W. Diabetologia 2015, 58, 900–911. 10.1007/s00125-015-3510-2. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Dezso Z.; MacKenzie C.; Oestreicher J.; Agoulnik S.; Byrne M.; Bernier F.; Yanagimachi M.; Aoshima K.; Oda Y. PLoS One 2013, 8, e69807 10.1371/journal.pone.0069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucharov C.; Bristow M. R.; Port J. D. J. Mol. Cell. Cardiol. 2008, 45, 185–192. 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard C. C.; Cheng H. H.; Tewari M. Nat. Rev. Genet. 2012, 13, 358–369. 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Nat. Methods 2010, 7, 687–692. 10.1038/nmeth0910-687. [DOI] [PubMed] [Google Scholar]
- Leshkowitz D.; Horn-Saban S.; Parmet Y.; Feldmesser E. RNA 2013, 19, 527–538. 10.1261/rna.036475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh P.; Dittmer D. P. Wiley Interdiscip Rev. RNA 2012, 3, 601–616. 10.1002/wrna.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P.; Hartmann N.; Baeriswyl L.; Andreasen D.; Bernard N.; Chen C.; Cheo D.; D’Andrade P.; DeMayo M.; Dennis L.; Derveaux S.; Feng Y.; Fulmer-Smentek S.; Gerstmayer B.; Gouffon J.; Grimley C.; Lader E.; Lee K. Y.; Luo S.; Mouritzen P.; et al. Nat. Methods 2014, 11, 809–815. 10.1038/nmeth.3014. [DOI] [PubMed] [Google Scholar]
- Feng C.; Mao X.; Shi H.; Bo B.; Chen X.; Chen T.; Zhu X.; Li G. Anal. Chem. 2017, 89, 6631–6636. 10.1021/acs.analchem.7b00850. [DOI] [PubMed] [Google Scholar]
- Ishihara R.; Hasegawa K.; Hosokawa K.; Maeda M. Anal. Sci. 2015, 31, 573–576. 10.2116/analsci.31.573. [DOI] [PubMed] [Google Scholar]
- Chapin S. C.; Doyle P. S. Anal. Chem. 2011, 83, 7179–7185. 10.1021/ac201618k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Schwarz G.; Santiago J. G. Angew. Chem., Int. Ed. 2013, 52, 11534–11537. 10.1002/anie.201305875. [DOI] [PubMed] [Google Scholar]
- Shintaku H.; Palko J. W.; Sanders G. M.; Santiago J. G. Angew. Chem., Int. Ed. 2014, 53, 13813–13816. 10.1002/anie.201408403. [DOI] [PubMed] [Google Scholar]
- Dong H.; Ding L.; Yan F.; Ji H.; Ju H. Biomaterials 2011, 32, 3875–3882. 10.1016/j.biomaterials.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Ryoo S. R.; Lee J.; Yeo J.; Na H. K.; Kim Y. K.; Jang H.; Lee J. H.; Han S. W.; Lee Y.; Kim V. N.; Min D. H. ACS Nano 2013, 7, 5882–5891. 10.1021/nn401183s. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Dong L.; Zhang J.; Zhao Y.; Li Z. Analyst 2018, 143, 1758–1774. 10.1039/C7AN02001E. [DOI] [PubMed] [Google Scholar]
- Yetisen A. K.; Butt H.; Volpatti L. R.; Pavlichenko I.; Humar M.; Kwok S. J.; Koo H.; Kim K. S.; Naydenova I.; Khademhosseini A.; Hahn S. K.; Yun S. H. Biotechnol. Adv. 2016, 34, 250–271. 10.1016/j.biotechadv.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Wei X.; Tian T.; Jia S.; Zhu Z.; Ma Y.; Sun J.; Lin Z.; Yang C. J. Anal. Chem. 2016, 88, 2345–2352. 10.1021/acs.analchem.5b04294. [DOI] [PubMed] [Google Scholar]
- Shapiro S. J.; Dendukuri D.; Doyle P. S. Anal. Chem. 2018, 90, 13572–13579. 10.1021/acs.analchem.8b03728. [DOI] [PubMed] [Google Scholar]
- Le Goff G. C.; Srinivas R. L.; Hill W. A.; Doyle P. S. Eur. Polym. J. 2015, 72, 386–412. 10.1016/j.eurpolymj.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pregibon D. C.; Doyle P. S. Anal. Chem. 2009, 81, 4873–4881. 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Shapiro S. J.; Chapin S. C.; Doyle P. S. Anal. Chem. 2016, 88, 3075–3081. 10.1021/acs.analchem.5b03902. [DOI] [PubMed] [Google Scholar]
- Chapin S. C.; Appleyard D. C.; Pregibon D. C.; Doyle P. S. Angew. Chem., Int. Ed. 2011, 50, 2289–2293. 10.1002/anie.201006523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Srinivas R. L.; Gupta A.; Doyle P. S. Angew. Chem., Int. Ed. 2015, 54, 2477–2481. 10.1002/anie.201409489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. M.; Martinez A. W.; Gong J.; Mace C. R.; Phillips S. T.; Carrilho E.; Mirica K. A.; Whitesides G. M. Angew. Chem., Int. Ed. 2010, 49, 4771–4774. 10.1002/anie.201001005. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Shi L.; Wang Q.; Mi L.; Li T. Anal. Chem. 2019, 91, 3652–3658. 10.1021/acs.analchem.8b05696. [DOI] [PubMed] [Google Scholar]
- Kim D.; Wei Q.; Kim D. H.; Tseng D.; Zhang J.; Pan E.; Garner O.; Ozcan A.; Di Carlo D. Anal. Chem. 2018, 90, 690–695. 10.1021/acs.analchem.7b03848. [DOI] [PubMed] [Google Scholar]
- Elghanian R.; Storhoff J. J.; Mucic R. C.; Letsinger R. L.; Mirkin C. A. Science 1997, 277, 1078–1081. 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- Kim D.; Daniel W. L.; Mirkin C. A. Anal. Chem. 2009, 81, 9183–9187. 10.1021/ac9018389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhasan A. H.; Kim D. Y.; Daniel W. L.; Watson E.; Meeks J. J.; Thaxton C. S.; Mirkin C. A. Anal. Chem. 2012, 84, 4153–4160. 10.1021/ac3004055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Li J. ACS Sens 2017, 2, 857–875. 10.1021/acssensors.7b00282. [DOI] [PubMed] [Google Scholar]
- Li J.; Kong C.; Liu Q.; Chen Z. Analyst 2018, 143, 4051–4056. 10.1039/C8AN00825F. [DOI] [PubMed] [Google Scholar]
- Hwu S.; Blickenstorfer Y.; Tiefenauer R. F.; Gonnelli C.; Schmidheini L.; Luchtefeld I.; Hoogenberg B. J.; Gisiger A. B.; Voros J. ACS Sens 2019, 4, 1950–1956. 10.1021/acssensors.9b00946. [DOI] [PubMed] [Google Scholar]
- Choi N. W.; Kim J.; Chapin S. C.; Duong T.; Donohue E.; Pandey P.; Broom W.; Hill W. A.; Doyle P. S. Anal. Chem. 2012, 84, 9370–9378. 10.1021/ac302128u. [DOI] [PubMed] [Google Scholar]
- Srinivas R. L.; Johnson S. D.; Doyle P. S. Anal. Chem. 2013, 85, 12099–12107. 10.1021/ac403201p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires T. M.; Messinger R. J.; Manalis S. R. Nat. Biotechnol. 2008, 26, 417–426. 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel M. C.; Astruc D. Chem. Rev. 2004, 104, 293–346. 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- Park J.; Yeo J. S. Chem. Commun. (Cambridge, U. K.) 2014, 50, 1366–1368. 10.1039/C3CC48154A. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Li R. D.; Yin B. C.; Ye B. C. Analyst 2015, 140, 6306–6312. 10.1039/C5AN01350J. [DOI] [PubMed] [Google Scholar]
- Miao J.; Wang J.; Guo J.; Gao H.; Han K.; Jiang C.; Miao P. Sci. Rep. 2016, 6, 32219. 10.1038/srep32219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Chen G.; Wang W.; Huang X.; Peng H.; Pu Q.; Du F.; Cui X.; Deng Y.; Tang Z. Anal. Chem. 2018, 90, 7107–7111. 10.1021/acs.analchem.8b01111. [DOI] [PubMed] [Google Scholar]
- Persano S.; Guevara M. L.; Wolfram J.; Blanco E.; Shen H.; Ferrari M.; Pompa P. P. ACS Omega 2016, 1, 448–455. 10.1021/acsomega.6b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J.; Konishi H.; Yanagisawa K.; Tomida S.; Osada H.; Endoh H.; Harano T.; Yatabe Y.; Nagino M.; Nimura Y.; Mitsudomi T.; Takahashi T. Cancer Res. 2004, 64, 3753–3756. 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Yanaihara N.; Caplen N.; Bowman E.; Seike M.; Kumamoto K.; Yi M.; Stephens R. M.; Okamoto A.; Yokota J.; Tanaka T.; Calin G. A.; Liu C. G.; Croce C. M.; Harris C. C. Cancer Cell 2006, 9, 189–198. 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.