

Abstract
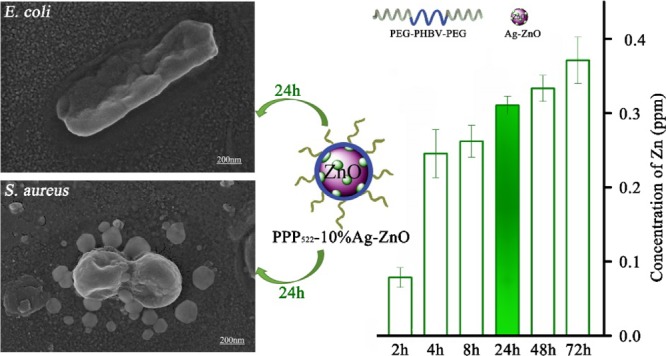
Antibacterial biomaterials with kill-resist dual functions by combining multiple active components have been constructed, with a final aim at decreasing the incidence of biomaterial-centered infection. Self-assemblies of bactericidal ZnO or Ag–ZnO nanoparticles (NPs) with triblock copolymers, poly(ethylene glycol)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate)–poly(ethylene glycol) (PEG–PHBV–PEG), showed a hydrophobic PHBV layer on NPs with PEG segments exposed outside via hydrogen bonding, resulting in long PEG (Mw = 2000) aggregation and short PEG (Mw = 1000) aggregation, respectively. These nanocomposite aggregations released ZnO or Ag–ZnO rapidly within initial few hours, and about 42–45% of NPs were left in the nanocomposites in deionized water for 16 d to improve the long-term antibacterial activity further. At the concentration below 50 μg/mL, the nanocomposite aggregation was cell-compatible with ATDC5 and showed sterilization rates over 91% against Escherichia coli and 98% against Staphylococcus aureus. Long PEG aggregation showed greater cell proliferation capacity than short PEG aggregation, as well as better bacterial resistance and bactericidal activity against both E. coli and S. aureus. The flexible self-assembling antibacterial NPs with antifouling block copolymers via adjusting the component ratio or the segment length have shown premise in the construction of the dual-function antibacterial materials.
1. Introduction
Biomaterial-centered infection has been posing a serious problem on human healthcare. Traditional clinical treatment is facing challenge because many bacterial strains have developed multiple resistance toward commonly used antibiotic drugs.1 Considerable efforts have recently been made on antibacterial surfaces such as bacteria-resistant surfaces and/or bactericidal surfaces to reduce the initial bacterial attachment and inhibit subsequent biofilm formation.2,3
Bactericidal surfaces can prevent the formation of viable biofilms by killing bacteria on surfaces and inhibit the proliferation of planktonic bacteria. A variety of metal and metal oxides such as Ag,4,5 Cu,6 ZnO,7 and TiO27,8 have been used as biocides. These nanoparticles (NPs) have strong and broad-spectrum antibacterial characteristics to damage the bacterial membrane as well as disrupt the function of bacterial enzymes and/or nucleic acid groups in cellular protein and DNA.9−12 Unfortunately, the concentration of these biocides decreases gradually during the release process, moreover, the surfaces will be contaminated by remaining dead bacteria to trigger immune responses or inflammation.13 Bacteria-resistant surfaces can prevent or reduce the initial bacterial attachment to interrupt biofilm formation. Hydrophilic polymers or oligomers are usually decorated on the bacteria-resistant surfaces, where a hydration layer prevents nonspecific interactions with proteins to reduce the adhesion of planktonic bacteria.14 Poly(ethylene glycol) (PEG) is the most commonly used bacteria-resistant material, and the bacterial resistance enhances as the number of ethylene glycol moieties increases.15,16 However, bacteria-resistant surfaces are unable to completely resist the adhesion of bacteria, these surfaces may be colonized by bacteria. Antibacterial surfaces with kill-resist dual functions via combining two or more active components into one composite have been designed to overcome these disadvantages.14,17,18
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) is a polyester produced by many strains of bacteria as an intracellular carbon and energy storage material. It has been widely utilized in biomedical areas because of biodegradable and biocompatible features.19−21 However, PHBV did not possess bacteria-resistant or bactericidal activity. Many efforts have therefore been focused on their composites with ZnO using electrospinning22 or a solvent casting technique,23,24 to fabricate bactericidal materials, where ZnO dispersed well in the matrix because of hydrogen bonding interactions. Ag NPs have better bactericidal activity against drug-sensitive and drug-resistant pathogenic bacteria than ZnO, but their cytotoxicity and genotoxicity to human normal cells have been unveiled continuously.25,26 Ag-doped ZnO is effective in reducing the amount of Ag NPs without sacrificing their antibacterial functions. Ag–ZnO hybrid NPs have attracted much interest because of the uniform distribution of Ag on the surface of ZnO without aggregation via a simply fabrication method.27,28 Once Ag–ZnO NPs release from surfaces, they can reduce bacterial colonization on surfaces and inhibit the proliferation of planktonic bacteria. Antifouling surfaces of PHB (or PHBV) via introducing hydrophilic PEG chains can prevent nonspecific interactions with proteins and reduce the adhesion of planktonic bacteria, but the exposed hydrophobic PHB chains in PHB/PEG blends are likely to be attacked by bacteria.29 Therefore, dual-function antibacterial surfaces with bacteria-resistant capacity and precise release of bactericidal agents may reduce the extent of initial bacterial attachment and thereby prevent the earliest stages of biofilm formation.
Blocking copolymers is a highly attractive option because of their versatility and flexibility to fabricate polymer chains aggregation.30 Polystyrene-b-poly(4-vinylpyridine) block copolymer membranes with highly ordered pore structures have been deposited with Ag NPs in situ to fabricate antibacterial surfaces against Pseudomonas aeruginosa.31 Synergistic interactions between self-organizing NPs and self-assembling polymeric matrices have been receiving much attention in developing functional hybrid materials. Our laboratory had previously synthesized PEG–PHBV–PEG amphiphilic block copolymers to introduce bacteria-resistant PEG with various chain lengths to obtain rod-shape polymeric aggregation where the hydrophilic PEG was distributed outside of the hydrophobic PHBV.32 These PEG–PHBV–PEG amphiphilic block copolymers may aggregate on ZnO or Ag–ZnO NPs to construct the composites with dual or multiple antibacterial components, where hydrophobic PHBV segments assemble on ZnO or Ag–ZnO to adjust the release of antibacterial agents, and hydrophilic PEG segments form a hydrate shell to resist bacterial adhesion. To the best of our knowledge, there are no studies on the self-assembly of Ag–ZnO NPs with PEG–PHBV–PEG triblock copolymers. In this work, amphiphilic block copolymers with different PEG chain lengths were employed to prepare composites with ZnO or Ag–ZnO bactericidal NPs. The effects of the PEG chain length on the antibacterial activity and cell compatibility were studied.
2. Materials and Methods
2.1. Materials
PEG (Mw = 1000 and 2000 g/mol, PEG1000 and PEG2000), isophorone diisocyanate (IPDI), diglyme, ethylene glycol, dibutyltin dilaurate, and anhydrous 1,2-dichloroethane were supplied by J&K Chemical (China). Chloroform, n-hexane, diethyl ether, ethanol, sodium hydroxide, silver nitrate, and zinc nitrate hexahydrate were obtained from Guangzhou Chemical Reagents (China) and used as received without further purification. PHBV with a content of 8 mol % 3-hydroxyvalerate was purchased from Sigma-Aldrich (USA). PHBV was purified by dissolution in CHCl3, filtration, and precipitation in n-hexane before use.
2.2. Synthesis of PEG–PHBV–PEG Block Copolymers
PEG–PHBV–PEG block copolymers (PPP) were synthesized as follows:32 briefly, 2 g of PHBV was dissolved in 20 mL diglyme at 140 °C under nitrogen, followed by the successive addition of 4 mL of ethylene glycol and 0.12 g of dibutyltin dilaurate. The solution was magnetically stirred for 7.5 or 9 h and precipitated in cold ethanol. The resulting telechelic-hydroxylated PHBV (PHBV-diol) showed Mw of 5000 or 3000 g/mol, measured by a Malvern (England) Viscotek Max VE 2001 gel permeation chromatography, denoted as PHBV-diol5000 and PHBV-diol3000, respectively. PHBV-diol (0.0001 mol), IPDI (0.0002 mol), and dibutyltin dilaurate (0.02 g) were added into 15 mL of anhydrous 1,2-dichloroethane at 75 °C. The mixture was stirred under nitrogen for 3 h and precipitated in n-hexane/ether (v/v, 1/1). In addition, 0.0001 mol of the above isocyanate terminated PHBV, and 0.0002 mol of PEG (PEG1000 or PEG2000) was dissolved in 15 mL of anhydrous 1,2-dichloroethane at 75 °C, followed by the addition of 1.0 wt % dibutyltin dilaurate. The mixture was magnetically stirred under nitrogen for 3 h and precipitated in n-hexane/ether (v/v, 1/1). Mw of PPP522 (PHBV-diol5000 and PEG2000) and PPP312 (PHBV-diol3000 and PEG1000) were 1.7 × 104 and 9.7 × 103 g/mol, respectively, and the molecular weight distribution was 1.4 and 2.7, respectively.
2.3. Preparation of PPP–ZnO and PPP–Ag–ZnO Nanocomposites
ZnO NPs were synthesized via a chemical precipitation method.33 Briefly, 7.425 g of zinc nitrate hexahydrate was dissolved in 50 mL of deionized water under constant stirring at room temperature, and 0.4 M sodium hydroxide was added dropwise to the solution until pH of 8–9 was reached. The mixture was stirred for 6 h, and the resulting ZnO NPs were purified thrice in deionized water via ultrasonication. ZnO NPs (0.25 g) and 5 mL of silver nitrate (50 mM) were added into 497.5 mL of deionized water. The suspension was ultrasonically treated, and 1 M of sodium hydroxide was added dropwise until pH of 8 was reached. After stirring at 80 °C for 5 h, the resulted Ag–ZnO NPs were washed with distilled water and ethanol for several times, then filtered and dried at 60 °C for 48 h.
PPP–ZnO and PPP–Ag–ZnO nanocomposites were prepared via a solution casting technique.34 PEG–PHBV–PEG (PPP522 or PPP312) (0.02 g) was first dissolved in 10 mL of chloroform. ZnO or Ag–ZnO NPs (5 or 10 wt %) was added into 5 mL of chloroform, ultrasonically treated for 4 h, and transferred into the above copolymer solution. The mixture was magnetically stirred for 12 h, ultrasonically treated for 2 h, and then cast onto a glass Petri dish. After maintained at room temperature for 8 h, the resulted composites were vacuum dried at 40 °C for 24 h. PPP–ZnO and PPP–Ag–ZnO nanocomposite aggregations were obtained as follows: 3.75 mg of the block copolymer was dissolved in 15 mL of CHCl3 and stirred magnetically at room temperature for 30 min. Ag–ZnO NPs (5 or 10 wt %) being suspended in deionized water were added to the copolymer solution dropwise. The mixture was magnetically stirred until the chloroform volatilized and freeze dried overnight for 48 h. The aggregation of the PPP522 or PPP312 triblock copolymer was also prepared in a similar way without the addition of the NPs.
2.4. Characterization
A Bruker (Germany) Vertex 70 Fourier transform infrared spectrometry (FTIR) was used to obtain the infrared analyses using a KBr pellet method. The spectra comprised 64 scans at a resolution of 1 cm–1 in the 4000–400 cm–1 spectral range. X-ray diffraction (XRD) analysis was performed using a Blagg MSAL-XD2 (Beijing, China) instrument with a Cu Kα radiation source (45 kV, 20 mA, and λ = 0.15406 nm). 2θ range of 10–80° was recorded in 0.02° steps at a rate of 2°/min. Transmission electron microscopy (TEM) was achieved on a transmission electron microscope (ZEISS, Tecnai-10) with an accelerating voltage of 20 kV to study the morphology of ZnO and Ag–ZnO NPs. Samples were mounted onto the Cu grid before observation. Scanning electron microscopy (SEM) was performed on a XL-30 scanning electron microscope (Philips) to study the morphology of the nanocomposites and their self-assemblies. A drop of aqueous aggregation was deposited onto a slide surface with a dimension of 5 mm × 5 mm, freeze dried overnight, and coated with gold.
Thermogravimetric analyses were carried out using a Netzsch (Germany) 209 F1 thermogravimetric analyzer. Approximately 10 mg of the sample was placed in a standard aluminum plate and referenced with an empty crucible as a background. Thermogravimetry analysis (TGA) curves and derivative thermogravimetry (DTG) curves were recorded from 35 to 800 °C under a nitrogen atmosphere at heating rates of 10 °C/min.
Inductively coupled plasma–optical emission spectroscopy (ICP, Optima 2000DV, PE company, America) was used to measure silver or zinc elemental contents released from Ag–ZnO NPs and PPP–Ag–ZnO aggregations. Briefly, 0.5 mg/mL of the sample suspension was filled into a dialysis bag (molecular weight cutoff = 106) and immersed in 500 mL of deionized water under low speed magnetically stirring at room temperature prior to quantification.
2.5. Cell Studies
2.5.1. Cell Culture
Cryo-preserved fifth-passage ATDC5 cells (P5) were thawed and cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Lifei Biotech, China) and 1% penicillin–streptomycin (Sigma) at 37 °C under a humidified atmosphere containing 5% CO2. At 80–90% confluence, the cells were rinsed in the phosphate buffered solution (PBS, pH 7.4, Gibco, USA) and then passaged by 0.25% trypsinase supplemented with 0.02% ethylene diamine-N,N-tetraacetic acid (Gibco, USA). The cell suspension was centrifuged at 1200 rpm for 5 min, and cell pellets (P6) were resuspended in DMEM supplemented with 10% FBS. Various nanocomposite aggregations were disinfected in 75% ethanol (vol %), followed by UV irradiation for 30 min before using.
2.5.2. Cell Viability
Cell viability was measured by an A1016-01 live/dead cell viability assay (Weikai Biotech, China), according to the manufacturer’s instruction. The cells (P6) were seeded onto a 24-well plate (3 × 103 cell/well) and incubated overnight at 37 °C for 6 h. Nanocomposite aggregations at concentrations of 10 or 50 μg/mL were added to the wells with Millicell culture supports (Millipore, Darmstadt, Germany). At 1 or 3 d, the cell complex with the released NPs was washed with PBS and stained with the working fluid for 30 min. Fluorescence images were taken by a Zeiss Axio scope A1 fluorescence microscope (Germany).
2.5.3. Cell Proliferation
Cell proliferation profiles were measured based on the reduction of tetrazolium salts in the medium using a cell-counting kit-8 (CCK-8, Beyotime Biotech, China). At 1, 3, or 5 d of culture, 300 μL of the CCK-8 solution (CCK-8/DMEM = 1:10) was added to the wells, and the cells were further incubated for 2 h. Media (100 μL) was transferred to a 96-well plate, and the absorbance was measured at 450 nm using a MULTISKAN MK3 microplate reader (Thermo Fisher, USA). The blank sample (only culture medium) was used as the control, and absorbance of each sample was the average of three separate wells.
2.6. Antibacterial Activity
2.6.1. Zone Inhibition Assay
Zone inhibition patterns on solid agar nutrient media plates against Escherichia coli and Staphylococcus aureus (Biological Tech, China) were performed as follows: briefly, 100 μL inoculums of each strain with a colony forming unit (CFU) (∼108/mL, OD600 = 0.1–0.4) were spread onto agar (Beyotime Biotech, China) and allowed to solidify for 5 min. Sterile filters of 10 mm in diameter were dropped with the nanocomposite aggregations, and the wet filter with distilled water was used as the negative control. The filters were dried at room temperature and placed in the Petri dishes containing S. aureus and E. coli, respectively. The plates were incubated at 37 °C for 24 h, and the diameters of antibacterial rings were the average of three separate measurements.
2.6.2. Antibacterial Rate Analysis
Activated bacteria (100 μL) at a final concentration of 105 to 106 CFU/mL were transferred to the nanocomposite aggregation suspension at a volume ratio of 1:100. The suspension was shaken at 37 °C for 24 h, 100 μL of which was pipetted onto the solid LB agar plates. The plates were then cultured at 37 °C for another 24 h, and the number of CFU was counted on the individual samples. Bacteria being incubated in the physiological saline were used as controls. Antibacterial rate (R) was finally calculated according to the following formula
| 1 |
where N0 and N are the average number of colonies in the control and experimental samples, respectively.
2.6.3. Live/Dead Assay
The viability of bacteria was assessed using live/dead BacLight bacterial viability kits (ThermoFisher Scientific, USA) to distinguish live bacteria with intact plasma membranes from dead bacteria with compromised membranes. Syto 9/PI reagents were used as the probes for live (green) and dead (red) stains, respectively. The diluted bacterial suspension (1 mL) (1 × 108 CFU/mL) was mixed with 9 mL of the nanocomposite suspension (50 mg/L), followed by oscillation at 37 °C for 24 h. Syto 9/PI working stains (150 μL) were added into the bacteria colonies and incubated at 37 °C for 15 min. The bacteria were then washed with 0.85% NaCl and centrifuged. A CLSM-TCS SP5 laser scanning confocal microscope (Germany) was used to take images of the bacteria treated with the nanocomposite aggregation. Untreated and alcohol-treated E. coli and S. aureus were used as controls of live and dead bacteria. The death rate of bacteria was calculated via ImageJ software.
2.6.4. Bacterial Morphology
PPP522-10% Ag–ZnO nanocomposites were treated with the diluted bacterial suspension (1 × 106 CFU/mL) at 37 °C for 24 h and washed with PBS. Samples were fixed with 2.5% glutaraldehyde for 30 min and washed with PBS three times. The bacteria were then dehydrated by slow water replacement using series of ethanol solutions (30, 50, 70, and 90%) for 10–15 min and deposited on glass, followed by coating with gold before SEM observation.
2.7. Statistical Analysis
Student t-test and one-way analysis of variance were used for the statistical analysis. p < 0.05 was considered to be statistically significant, and all data were represented as average ± SD.
3. Results and Discussion
3.1. Structure of the Nanocomposites
FTIR spectra of the nanocomposites are shown in Figure 1. The PPP522 block copolymer (Figure 1a) presented the C=O stretching vibration of PHBV at 1728.2 cm–1 and the absorption peak of the PEG crystal phase at 948.2 cm–1. The broad peak at 3435.8 cm–1 belonged to the distinct stretching vibration of −OH and N–H. The characteristic isocyanate groups at 2269.3 cm–1 disappeared in the block copolymer, confirming the chain extension reaction between PEG and isocyanate-terminated PHBV.32 The abovementioned absorption bands of PPP522 were shown in the spectra of the nanocomposites with ZnO and Ag–ZnO. The stretching bands in 400–600 cm–1 (Figure 1b–e) were the typical absorption peaks of ZnO NPs. Among these, the sharp peaks at about 520 and 420 cm–1 were attributed to the lattice vibration of ZnO.35 The additional peak at about 630 cm–1 was attributed to the interaction between Ag–ZnO of the nanocomposites (Figure S1, Supporting Information).11,36 The absorption peak at 2924.4 cm–1 was assigned to the symmetric stretching vibration of the methyl group of PPP522 and the stretching vibration of N–O. An intensity ratio was introduced as the peak height at half width of the peak at 2924.4 cm–1 against that of the absorption peak at 1728.2 cm–1 (the reference peak). It could be clearly seen that the intensity ratio of PPP522–Ag–ZnO was greater than that of PPP522 and increased with the increasing content of Ag–ZnO NPs. More importantly, carbonyl stretching vibration of PPP522-5% Ag–ZnO (Figure 1c) shifted to lower wavenumbers, by about 5 cm–1 in comparison to that of PPP522, suggesting the formation of hydrogen bonds with the hydroxyl moieties of ZnO NPs. Moreover, the lattice vibration of Zn–O (Figure 1d,e) appeared at 419.4 and 422.3 cm–1, nearly 10 cm–1 lower than that of ZnO (431.1 cm–1, Figure S1, Supporting Information), reconfirming the interactions between ZnO and triblock copolymers.
Figure 1.
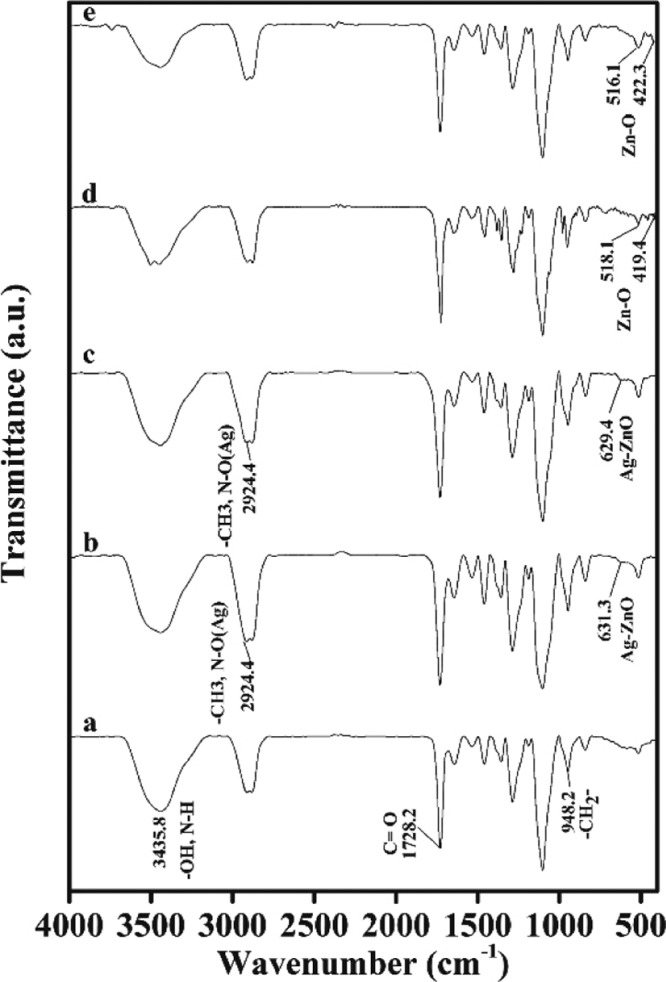
FTIR spectra of the nanocomposites of PPP522 (a), PPP522-10% Ag–ZnO (b), PPP522-5% Ag–ZnO (c), PPP522-10% ZnO (d), and PPP522-5% ZnO (e).
The diffractograms of the nanocomposites of PPP522 with ZnO or Ag–ZnO are shown in Figure 2(I) in which 2θ at 13.7, 17.0, 19.1, and 23.2° corresponded to the characteristic diffraction peak of PPP522,32 31.9, 34.6, 36.3, and 56.6° were assigned to the patterns of ZnO NPs,37 and the diffraction peaks at 38.6° were assigned to the patterns of Ag NPs (Figure S2, Supporting Information). The half width of PPP522-5% Ag–ZnO at 2θ of 36.3° was about 12.3% higher than that of PPP522-10% Ag–ZnO, and the value of PPP522-5% ZnO was about 33.3% higher than that of PPP522-10% ZnO. It is well known that the peak width is inversely proportional to the crystallite size according to the Scherrer formula. Therefore, it could be concluded that the average crystallite size of ZnO or Ag–ZnO NPs in the nanocomposites decreased with the decreasing loading percentage of NPs, perhaps owing to the restrain effect of the nanofiller–matrix interaction on the crystal growth. The microcrystal size of the PHBV phase in the nanocomposites being calculated from the (020) and (110) planes was slightly lower than that of the PPP522 triblock copolymer, indicating that ZnO NPs or Ag–ZnO NPs would have a limiting effect on the microcrystalline growth of the PHBV crystals. An interesting phenomenon was that the characteristic diffraction peaks of ZnO NPs and PHBV blocks in PPP312 nanocomposites (Figure 2(II)) were much stronger than the diffraction peaks in PPP522 nanocomposites, perhaps because the PEG1000 blocks in PPP312 would be in an amorphous state, and their restrain effects on PHBV blocks and ZnO NPs might be less than PPP522.
Figure 2.

XRD patterns of PPP–ZnO or PPP–Ag–ZnO nanocomposites: (I) PPP522 (a), PPP522-10% Ag–ZnO (b), PPP522-5% Ag–ZnO (c), PPP522-10% ZnO (d), and PPP522-5% ZnO (e); (II) PPP312 (a), PPP312-10% Ag–ZnO (b), PPP312-5% Ag–ZnO (c), PPP312-10% ZnO (d), and PPP312-5% ZnO (e).
TEM images (Figure 3a) illustrated the representative rod-like shape of ZnO NPs with a length of ∼183.3 nm and a width of ∼66.7 nm, which is in accordance with the diffraction results of the hexagonal Wurtzite crystals (Figure S2). The shape of Ag–ZnO NPs (Figure 3b) was basically similar to that of ZnO NPs. There were many small black spots on the surface of NPs, indicating the attachment of Ag NPs on the ZnO surface.38
Figure 3.

TEM and SEM images of ZnO NPs (a), Ag–ZnO NPs (b), and PPP522-10% Ag–ZnO and its enlargement (c,d).
Ag–ZnO NPs were also blended with PPP522 without using the assembly procedure. SEM images of the nanocomposites (Figure 3c,d) showed that rod Ag–ZnO NPs were homogeneously distributed throughout the triblock polymer matrix, with lengths and widths much greater than that of pure Ag–ZnO NPs because of their agglomeration effects. No obvious interface between the NPs and PPP522 could be seen, suggesting a strong physical crosslinking of the abundant hydroxyl groups on the ZnO surface with the matrix. The block copolymer chains might also form microcrystals via rearranging along the interface to improve the interface bonding.
3.2. Thermal Decomposition
The TGA and DTG plots of PPP312–ZnO and PPP312–Ag–ZnO with different contents of the NPs (Figure S3) and Table S1 (which summarized the corresponding parameters) are shown in the Supporting Information.
3.3. Self-Assemblies
The PPP522 copolymer formed rod assemblies with a length of ∼2 μm and a diameter of ∼1 μm (Figure 4a, enlargement). PHBV segments might be assembled into the hydrophobic core, and the hydrophilic PEG was distributed outside after solvent evaporation.32,39 The assemblies were not quite uniform because of various molecular weights and the segment composition of the triblock copolymers. The PPP522–Ag–ZnO nanocomposite assemblies showed different morphologies (Figure 4b), and NPs agglomeration increased the variation of shapes and sizes.
Figure 4.

SEM pictures of the PPP522 copolymer (a) and PPP522–Ag–ZnO nanocomposites (b).
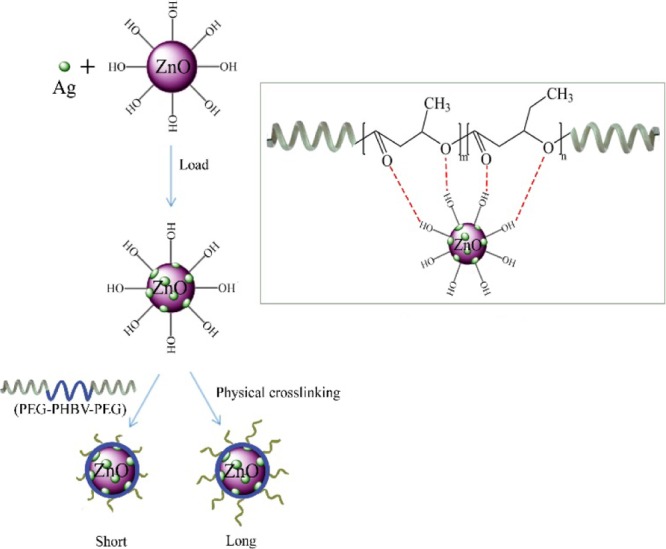
The nanocomposite aggregations might be derived via hydrogen bonding between ZnO NPs and PHBV segments (Scheme 1). As demonstrated above, the carbonyl stretching vibration of PHBV and the lattice vibration of Zn–O shifted to lower wavenumbers (Figure 1). Moreover, ZnO or Ag–ZnO NPs would limit the microcrystalline growth of PHBV (Figure 2), and the contents of the NPs influenced the thermal decomposition temperature of PHBV, but no obvious effect on PEG could be seen (Figurer S3). These strong ZnO–PHBV hydrogen bonds enhanced the physical crosslinking interaction between nanofillers and matrices (Figure 3d). Ag was also detected (Figure 1) in Ag–ZnO NPs and showed a little effect on the thermal degradation of the nanocomposites. Because Ag loading was very low (∼3.39% of mass fraction via ICP measurement), its involvement into the self-assembling process would be neglected. Therefore, PHBV segments would prefer to coat ZnO NPs and even rearrange regularly to form microcrystals induced by hydrogen bonds. The hydrophilic PEG segments would stretch outward from the PHBV–ZnO core to form a hydration shell. The triblock copolymer with long PEG chains would form a thicker hydrophilic layer (PPP522–Ag–ZnO), and the short PEG chains would present a thinner hydrophilic layer (PPP312–Ag–ZnO).
Scheme 1. Synthetic Schematic Diagram of PPP–Ag–ZnO Aggregations, Showing Long (PPP522–Ag–ZnO) or Short (PPP312–Ag–ZnO) Hydrophilic Chains on the Shell.
3.4. Release Profile of Antibacterial NPs
The sustained release of zinc from PPP522-10% Ag–ZnO and PPP312-10% Ag–ZnO nanocomposites at room temperature is presented in Figure 5. It was observed that the release rate of zinc from the nanocomposites was very fast within the first few hours and gradually slowed down after 2 d. Ag was not detected because of the very low loading in Ag–ZnO NPs, though it might be attached on the surface of ZnO crystals and released along with ZnO into the surrounding media. The concentration of Ag was calculated theoretically to be 0.0105 ppm based on the concentration of Zn (0.3100 ppm) at 1 d. The Zn concentration of PPP522-10% Ag–ZnO at 9d was 0.436 ppm, much more than that of PPP312-10% Ag–ZnO (0.344 ppm), indicating that long PEG aggregation released NPs faster than short PEG aggregation. ZnO–PHBV hydrogen bonds might be partially replaced by H2O–ZnO hydrogen bonds during the release period, thus Ag–ZnO NPs disassemble from the nanocomposite aggregations. Long PEG aggregation would provide a thick hydration layer surrounding Ag–ZnO NPs to promote the disassembly process in situ. At 16 d of release, about 44.3 and 42.0% of ZnO were left in the aggregations of PPP522-10% Ag–ZnO and PPP312-10% Ag–ZnO, respectively. These remaining NPs might be released along with the degradation of PHBV coating to extend the duration of antibacterial activity. Moreover, more antibacterial agents were allowed to be loaded in the nanocomposites because nearly half of these agents were covered by a biocompatible layer to enhance the cell compatibility.
Figure 5.
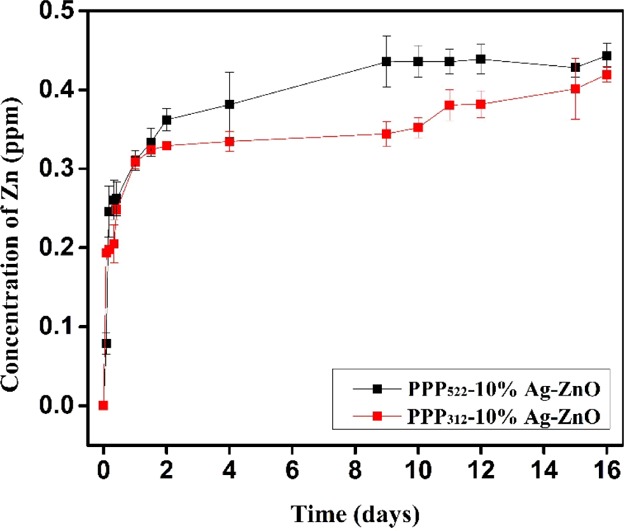
Release profile of Zn from PPP522-10% Ag–ZnO and PPP312-10% Ag–ZnO (n = 3).
3.5. ATDC5 Compatibility
ATDC5 being derived from a AT805 teratocarcinoma cell line was used to study the cell compatibility. Live/dead staining images of ATDC5 cells being incubated with various nanocomposite aggregations at concentrations of 10 and 50 μg/mL are shown in Figure 6. Cells did not present remarkable death when treated with PPP522 and PPP312, and no significant effect of the nanocomposites’ concentration on the viability of ATDC5 was shown. As the culture period increased, the amount of the live cells increased dramatically. However, more cells died while cultured with 10 μg/mL of PPP522–ZnO or PPP312–ZnO for 1 d. The amount of the dead cells increased with the culturing period, and the incorporation of Ag seemed to promote cell death. Because the nanocomposites were separated from the cells via the culture supports; therefore, the cell death would be attributed to the NPs released. Cells died remarkably when cocultured with 100 μg/mL of the nanocomposites (data not shown). Generally, ZnO and Ag–ZnO did not induce significant cell death when the concentration of the nanocomposites was below 50 μg/mL. However, the cells became mostly spindle-like with long pseudopodium when the concentration of the nanocomposites increased, showing the dedifferentiation morphology of ATDC5.
Figure 6.

Live/dead staining images of ATDC5 cells being incubated with various nanocomposites at 1 d and 3 d (I) (II).
CCK-8 results (Figure 7) show the proliferation of the cells treated with various nanocomposites, and the intensity ratio was defined as the intensity of the sample to control. Generally, the intensity ratios of triblock copolymers and their nanocomposites decreased as the culture period increased, indicating the growth inhibition effect on ATDC5 cells. The introduction of the NPs decreased the cell proliferation at some extent. Ag–ZnO containing nanocomposites showed lower intensity ratios than ZnO containing nanocomposites because of higher toxicity of Ag NPs, moreover, this growth inhibition effect was more significant at a longer culture period. Interestingly, long PEG nanocomposites presented greater intensity ratio than the short ones. Because there was no significant difference on pair comparison of samples at concentrations of 10 and 50 μg/mL, the amount of the released NPs was not the key factor though NPs were prone to release faster when self-assembled with long PEG triblock copolymers. In the nanocomposite aggregations, the hydrophobic PHBV segments assembled on the surface of nanoparticles to form a hydrophobic layer, and the hydrophilic PEG was distributed outside. Long PEG might present more hydrophilic domains to balance the hydrophobic domains because hydrophobic–hydrophilic balance was crucial to the cell–surface interaction.
Figure 7.

Cell proliferation of ATDC5 being incubated with various nanocomposites at the concentration of 10 (a) and 50 μg/mL (b). Intensity ratio was defined as the intensity of the sample to the control (n = 6, *p < 0.05, **p < 0.01).
3.6. Antibacterial Activity
3.6.1. Inhibition Zone
Figure 8 shows inhibition zones of the nanocomposites with different concentrations of ZnO and Ag–ZnO NPs against S. aureus and E. coli in the dark environment. PPP522 and PPP312 inhibited the bacterial growth because PEG segments had antifouling capability to prevent protein adsorption.40 The inhibition zone of PPP522 against E. coli was greater than that of PPP312 because long PEG segments of PPP522 would form an increased antifouling area against bacteria on the aggregation. However, there was no significant difference between both copolymers against S. aureus.
Figure 8.

Inhibition zones of various nanocomposites against E. coli (a,b) and S. aureus (c,d) (n = 3) (photograph courtesy of Qing Zhong. Copyright 2020).
The inhibition zone of PPP522-5% ZnO and PPP522-10% ZnO against E. coli were similar to PPP522, but the value increased about 52.28 and 56.06%, respectively, for PPP522-5% Ag–ZnO and PPP522-10% Ag–ZnO, showing the higher antibacterial capacity of Ag than ZnO NPs. As compared with that of PPP312, the inhibition zone of PPP312-5% ZnO, PPP312-10% ZnO, PPP312-5% Ag–ZnO, and PPP312-10% Ag–ZnO increased about 4.68, 21.36, 28.84, and 23.93%, respectively. The additional release of the antibacterial NPs (ZnO or Ag–ZnO) would result in much greater zone diameter of the nanocomposite aggregation than the triblock copolymer aggregation. The inhibition capability of the nanocomposites against S. aureus showed very similar tendency to that against E. coli. The zone diameter of PPP522-5% Ag–ZnO and PPP522-10% Ag–ZnO increased by about 43.44 and 40.60%, respectively, as compared with the PPP522 matrix. The introduction of the NPs to PPP312 increased the inhibition zone of PPP312-5% ZnO, PPP312-10% ZnO, PPP312-5% Ag–ZnO, and PPP312-10% Ag–ZnO by about 7.54, 25.15, 14.25, and 53.14%, respectively. Generally, the zone diameters of long PEG aggregation were greater than those of short PEG aggregation at the same content of NPs because the former possessed better antifouling capacity and much faster release rates of the NPs.
3.6.2. Antibacterial Rate
The bactericidal capacity of the nanocomposite aggregation was studied using the plate count method, as shown in Figure 9. The antibacterial rate of Ag–ZnO containing nanocomposites was quite higher than ZnO nanocomposites against E. coli and S. aureus. The value increased with the increasing concentration of Ag–ZnO NPs against E. coli, but no significant difference was shown for Ag–ZnO nanocomposites against S. aureus. The nanocomposites with higher ZnO concentrations presented greater antibacterial rate against S. aureus. At the concentration of 50 μg/mL, the nanocomposites showed antibacterial rates over 91% against E. coli and over 98% against S. aureus. Long PEG aggregation showed much greater antibacterial rate than short PEG aggregation, perhaps because of the higher accumulation of released NPs. Ag–ZnO nanocomposites presented better antibacterial rate than the Ag–ZnO control containing the same amount of NPs because these nanocomposites having the hydrophilic shell distributed well in the media to overcome the agglomeration of Ag–ZnO NPs. It should be noted that nearly half NPs were still left in the aggregations of PPP522-10% Ag–ZnO and PPP312-10% Ag–ZnO, which would be released further to increase the antibacterial interfaces.
Figure 9.
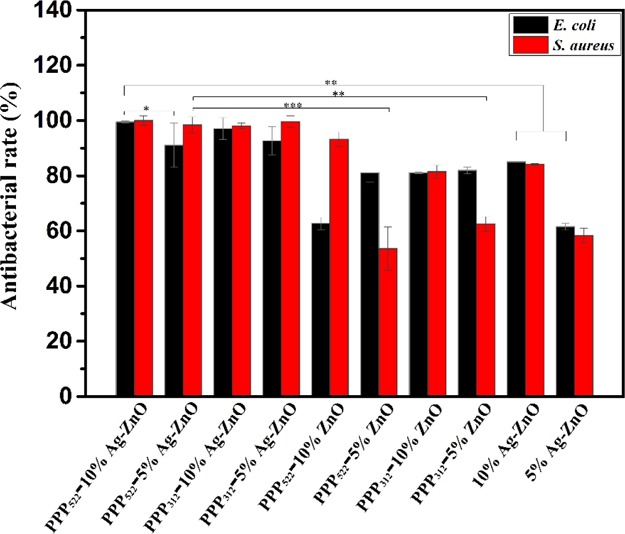
Antibacterial rates of E. coli and S. aureus treated by various nanocomposites at a concentration of 50 μg/mL for 24 h (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001).
3.6.3. Viability Assay
Figures 10 and 11 present the live/dead stained images of E. coli and S. aureus after incubating with the nanocomposites, where Syto 9 is a highly efficient cell permeable green fluorescent dye that binds to the DNA of living bacteria with intact cellular structures, and propidium iodide ) is a nonpermeable cell red fluorescent dye that can only stain DNA from dead bacteria with damage of the cell membrane.41,42 ZnO or Ag–ZnO nanocomposites increased the bactericidal capability compared with the controls. Dead bacteria of PPP522-10% Ag–ZnO was much greater than the other nanocomposites against E. coli and S. aureus. The bright yellow bacteria agglomeration of E. coli treated with PPP522-10% Ag–ZnO may be caused by the aggregation of dead bacteria and viable bacteria. Dead rates of PPP522-10% Ag–ZnO and PPP522-10% ZnO were 43.2 and 37.0% against E. coli, and these rates were 48.2 and 42.0% against S. aureus, respectively. It was obvious that Ag–ZnO nanocomposites presented better bactericidal capability than ZnO nanocomposites. Long PEG aggregation (PPP522-10% Ag–ZnO) also showed higher bactericidal capability than short PEG aggregation (PPP312-10% Ag–ZnO) against E. coli and S. aureus (34.4 and 44.0%), respectively.
Figure 10.

Live/dead staining images of E. coli incubated with various nanocomposites (top: live bacteria, green; middle: dead bacteria, red; bottom: combined).
Figure 11.

Live/dead staining images of S. aureus incubated with various nanocomposites (top: live bacteria, green; middle: dead bacteria, red; bottom: combined).
3.6.4. Bacterial Morphology
The SEM micrograph of E. coli and S. aureus being incubated with PPP522-10% Ag–ZnO for 24 h is shown in Figure 12. E. coli presented a rod shape with a length of approximately 1–2 μm. Bacterial cells are distorted with the ruptured cell membrane, indicative of the bactericidal effect of the nanocomposite aggregation (Figure 12a,b). Clusters of spherical S. aureus were rather small, and some ruptured membranes refused together (Figure 12c,d).
Figure 12.
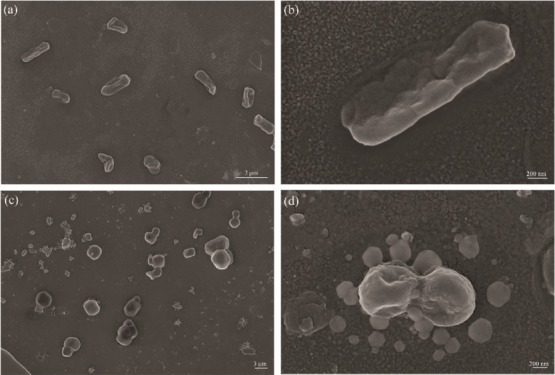
SEM images of E. coli (a,b) and S. aureus (c,d) treated by PPP522-10% Ag–ZnO.
The membrane rupture seemed as one main procedure to the death of bacteria. Ag and ZnO may interact with the membrane to change the bacterial permeability and further interact with the nucleic acids to inhibit the replication of bacteria; or produce reactive oxygen species upon microorganisms to oxidize the organic components of bacteria to carbon dioxide and water.43 Moreover, accumulation of Ag–ZnO NPs in the microbial membrane could result in the disintegration of the membrane and internalization into the microbial cell. Therefore, the antibacterial activity of the nanocomposites enhanced as the concentration of the NPs increased, and Ag–ZnO NPs presented stronger bacterial activity than ZnO.
Our result showed that E. coli was more sensitive than S. aureus for both long or short PEG aggregations. Membranes of both bacteria were negative with the surface charge of 2.56 × 10–16 g eq/cell and 0.24 × 10–16 g eq/cell, respectively, for S. aureus and E. coli.44 It seemed that the metal and metal oxide might be easier to combine with S. aureus, but E. coli had a complex outer membrane to restrain from the foreign substances. Once the antibacterial agents enter into the outer membrane, the complex mesh would also prevent them to escape. Therefore, the antibacterial agents might in situ destroy the cell membranes and enter into the bacterial cells.
An applicable concentration of bactericide is essential to the antibacterial composites because overdose of these NPs would lead to cell death significantly. Sustaining release of these NPs is also important for the surfaces with long-term antibacterial activity. The aggregations with dual functions resisted the bacterial adhesion and kill the bacteria efficiently upon rapid release of the antibacterial NPs. The remaining NPs that are protected by the biocompatible PHBV and PEG chains made high loading capacity of the nanocomposites available. As the hydrophobic PHBV segments self-assembled along the interface with the NPs, it allowed the hydrophilic PEG chains to be exposed outside the aggregations. Long PEG aggregations possessed better bacteria-resistance than short PEG aggregations, and released more NPs to present better bactericidal capacity. Even if PEG chains were contaminated by the dead bacteria, the released NPs would kill the bacteria continuously. It was very important for inhibiting the early formation of biofilms. The long-term antibacterial activity of the nanocomposite aggregation might further depend on the degradation of the block copolymer via controlling the composition of PHBV and PEG segments.
4. Conclusions
ZnO or Ag–ZnO self-assembly with PEG–PHBV–PEG block copolymers were successfully obtained, where a hydrophobic PHBV layer was introduced on the NPs with the PEG segments exposed outside to form a hydrophilic shell. These aggregations of NP-block copolymers decreased the thermal decomposition temperature of PHBV segments but did not influence PEG segments. At the concentration below 50 μg/mL, the nanocomposite aggregation was cell-compatible with ATDC5 cells. Long PEG aggregation showed greater cell proliferation capacity than short PEG aggregation, as well as better bacteria-resistance and bactericidal activity against both E. coli and S. aureus. These dual-function antibacterial materials indicated their great potential in decreasing the incidence of the biomaterial-centered infection.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (51572110, 51872124, and 21676116), the Natural Science Foundation of Guangdong Province of China (2016A030313085), the Science and Technology Projects of Guangdong Province (2015A020212025 and 2016A010103020), the Science and Technology Innovation Platform Project of Foshan City (2017AG100092), and the Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2017).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04086.
FTIR and XRD analyses of ZnO NPs and Ag–ZnO NPs and TGA and DTG plots of PPP312–ZnO and PPP312–Ag–ZnO with different contents of NPs (PDF)
Author Contributions
⊥ Q.Z. and W.H. contribute equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Khan A.; Miller W. R.; Arias C. A. Mechanisms of antimicrobial resistance among hospital-associated pathogens. Expert Rev. Anti-Infect. Ther. 2018, 16, 269–287. 10.1080/14787210.2018.1456919. [DOI] [PubMed] [Google Scholar]
- Salwiczek M.; Qu Y.; Gardiner J.; Strugnell R. A.; Lithgow T.; McLean K. M.; Thissen H. Emerging rules for effective antimicrobial coatings. Trends Biotechnol. 2014, 32, 82–90. 10.1016/j.tibtech.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Yang W. J.; Neoh K.-G.; Kang E.-T.; Teo S. L.-M.; Rittschof D. Polymer brush coatings for combating marine biofouling. Prog. Polym. Sci. 2014, 39, 1017–1042. 10.1016/j.progpolymsci.2014.02.002. [DOI] [Google Scholar]
- Durán N.; Durán M.; De Jesus M. B.; Seabra W. J.; Nakazato G.; Nakazato G. Silver nanoparticles: A new view on mechanistic aspects on antimicrobial activity. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 789–799. 10.1016/j.nano.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Mao C.; Xiang Y.; Liu X.; Cui Z.; Yang X.; Yeung K. W. K.; Pan H.; Wang X.; Chu P. K.; Wu S. Photo-inspired antibacterial activity and wound healing acceleration by hydrogel embedded with Ag/Ag@ AgCl/ZnO nanostructures. ACS Nano 2017, 11, 9010–9021. 10.1021/acsnano.7b03513. [DOI] [PubMed] [Google Scholar]
- Palza H. Antimicrobial Polymers with Metal Nanoparticles. Int. J. Mol. Sci. 2015, 16, 2099–2116. 10.3390/ijms16012099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.; Kim H.-K.; Wamer W. G.; Melka D.; Callahan J. H.; Yin J.-J. Photogenerated charge carriers and reactive oxygen species in ZnO/Au hybrid nanostructures with enhanced photocatalytic and antibacterial activity. J. Am. Chem. Soc. 2014, 136, 750–757. 10.1021/ja410800y. [DOI] [PubMed] [Google Scholar]
- Gupta K.; Singh R. P.; Pandey A.; Pandey A. Photocatalytic antibacterial performance of TiO2 and Ag-doped TiO2 against S. aureus. P. aeruginosa and E. coli. Beilstein J. Nanotechnol. 2013, 4, 345–351. 10.3762/bjnano.4.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambio-Jones C.; Hoek E. M. V. A review of the antibacterial effects of silver nanomaterials and potential implications for human health and the environment. J. Nanoparticle Res. 2010, 12, 1531–1551. 10.1007/s11051-010-9900-y. [DOI] [Google Scholar]
- Li J. H.; Hong R. Y.; Li M. Y.; Li H. Z.; Zheng Y.; Ding J. Effects of ZnO nanoparticles on the mechanical and antibacterial properties of polyurethane coatings. Prog. Org. Coat. 2009, 64, 504–509. 10.1016/j.porgcoat.2008.08.013. [DOI] [Google Scholar]
- Matai I.; Sachdev A.; Dubey P.; Uday Kumar S.; Bhushan B.; Gopinath P. Antibacterial activity and mechanism of Ag–ZnO nanocomposite on S. aureus and GFP-expressing antibiotic resistant E. coli. Colloids Surf., B 2014, 115, 359–367. 10.1016/j.colsurfb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wang T.; Zhao X.; Wang X.; Zhou L.; Yang Y.; Liao F.; Ju Y. Poly (lactic acid)/graphene oxide–ZnO nanocomposite films with good mechanical, dynamic mechanical, anti-UV and antibacterial properties. J. Chem. Technol. Biotechnol. 2015, 90, 1677–1684. 10.1002/jctb.4476. [DOI] [Google Scholar]
- Costerton J. W.; Stewart P. S.; Greenberg E. P. Bacterial biofilms: a common cause of persistent infections. Science 1999, 284, 1318–1322. 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Wu Z.; Chen H. Dual-function antibacterial surfaces for biomedical applications. Acta Biomater. 2015, 16, 1–13. 10.1016/j.actbio.2015.01.018. [DOI] [PubMed] [Google Scholar]
- Chapman R. G.; Ostuni E.; Liang M. N.; Meluleni G.; Kim E.; Yan L.; Pier G.; Warren H. S.; Whitesides G. M. Polymeric thin films that resist the adsorption of proteins and the adhesion of bacteria. Langmuir 2001, 17, 1225–1233. 10.1021/la001222d. [DOI] [Google Scholar]
- Ista L. K.; López G. P. Interfacial tension analysis of oligo (ethylene glycol)-terminated self-assembled monolayers and their resistance to bacterial attachment. Langmuir 2012, 28, 12844–12850. 10.1021/la302601x. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Ning C.; Zhou T.; Liu X.; Yeung K. W. K.; Zhang T.; Xu Z.; Wang X.; Wu S.; Chu P. K. Polymeric nanoarchitectures on Ti-based implants for antibacterial applications. ACS Appl. Mater. Interfaces 2014, 6, 17323–17345. 10.1021/am5045604. [DOI] [PubMed] [Google Scholar]
- Wei T.; Zhan W.; Cao L.; Hu C.; Qu Y.; Yu Q.; Chen H. Multifunctional and regenerable antibacterial surfaces fabricated by a universal strategy. ACS Appl. Mater. Interfaces 2016, 8, 30048–30057. 10.1021/acsami.6b11187. [DOI] [PubMed] [Google Scholar]
- Chen G.-Q.; Wu Q. The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials 2005, 26, 6565–6578. 10.1016/j.biomaterials.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Avella M.; Martuscelli E.; Raimo M. Review Properties of blends and composites based on poly (3-hydroxy) butyrate (PHB) and poly (3-hydroxybutyrate-hydroxyvalerate)(PHBV) copolymers. J. Mater. Sci. 2000, 35, 523–545. 10.1023/a:1004740522751. [DOI] [Google Scholar]
- Gong L.; Chase D. B.; Noda I.; Liu J.; Martin D. C.; Ni C.; Rabolt J. F. Discovery of β-form crystal structure in electrospun poly [(R)-3-hydroxybutyrate-co-(R)-3-hydroxyhexanoate](PHBHx) nanofibers: From fiber mats to single fibers. Macromolecules 2015, 48, 6197–6205. 10.1021/acs.macromol.5b00638. [DOI] [Google Scholar]
- Rodríguez-Tobías H.; Morales G.; Ledezma A.; Romero J.; Saldívar R.; Langlois V.; Renard E.; Grande D. Electrospinning and electrospraying techniques for designing novel antibacterial poly (3-hydroxybutyrate)/zinc oxide nanofibrous composites. J. Mater. Sci. 2016, 51, 8593–8609. 10.1007/s10853-016-0119-x. [DOI] [Google Scholar]
- Díez-Pascual A. M.; Díez-Vicente A. L. ZnO-reinforced poly (3-hydroxybutyrate-co-3-hydroxyvalerate) bionanocomposites with antimicrobial function for food packaging. ACS Appl. Mater. Interfaces 2014, 6, 9822–9834. 10.1021/am502261e. [DOI] [PubMed] [Google Scholar]
- Re Silva M. B.; Tavares M. I.; Junior A. W.; Neto R. P. Evaluation of Intermolecular Interactions in the PHB/ZnO Nanostructured Materials. J. Nanosci. Nanotechnol. 2016, 16, 7606–7610. 10.1166/jnn.2016.11760. [DOI] [Google Scholar]
- Lu Z.; Gao J.; He Q.; Wu J.; Liang D.; Yang H.; Chen R. Enhanced antibacterial and wound healing activities of microporous chitosan-Ag/ZnO composite dressing. Carbohydr. Polym. 2017, 156, 460–469. 10.1016/j.carbpol.2016.09.051. [DOI] [PubMed] [Google Scholar]
- AshaRani P. V.; Low Kah Mun G.; Hande M. P.; Valiyaveettil S. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano 2009, 3, 279–290. 10.1021/nn800596w. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Kim H.-I. Characterization and antibacterial properties of genipin-crosslinked chitosan/poly (ethylene glycol)/ZnO/Ag nanocomposites. Carbohydr. Polym. 2012, 89, 111–116. 10.1016/j.carbpol.2012.02.058. [DOI] [PubMed] [Google Scholar]
- Agnihotri S.; Bajaj G.; Mukherji S.; Mukherji S. Arginine-assisted immobilization of silver nanoparticles on ZnO nanorods: an enhanced and reusable antibacterial substrate without human cell cytotoxicity. Nanoscale 2015, 7, 7415–7429. 10.1039/c4nr06913g. [DOI] [PubMed] [Google Scholar]
- El-Khordagui L.; El-Sayed N.; Galal S.; El-Gowelli H.; Omar H.; Mohamed M. Photosensitizer-eluting nanofibers for enhanced photodynamic therapy of wounds: A preclinical study in immunocompromized rats. Int. J. Pharm. 2017, 520, 139–148. 10.1016/j.ijpharm.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Böker A.; He J.; Sill K.; Xiang H.; Abetz C.; Li X.; Wang J.; Emrick T.; Long S.; Wang Q.; Balazs A.; Russell T. P. Self-directed self-assembly of nanoparticle/copolymer mixtures. Nature 2005, 434, 55–59. 10.1038/nature03310. [DOI] [PubMed] [Google Scholar]
- Madhavan P.; Hong P.-Y.; Sougrat R.; Nunes S. P. Silver-enhanced block copolymer membranes with biocidal activity. ACS Appl. Mater. Interfaces 2014, 6, 18497–18501. 10.1021/am505594c. [DOI] [PubMed] [Google Scholar]
- Shi L.; Hu W.; He Y.; Ke Y.; Wu G.; Xiao M.; Huang L.; Tan S. Preparation and Characterization of Poly (ethylene glycol)-block-Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)-block-Poly (ethylene glycol) Triblock Copolymers. Macromol. Res. 2020, 10.1007/s13233-020-8005-4. [DOI] [Google Scholar]
- Shankar S.; Teng X.; Li G.; Rhim J.-W. Preparation, characterization, and antimicrobial activity of gelatin/ZnO nanocomposite films. Food Hydrocolloids 2015, 45, 264–271. 10.1016/j.foodhyd.2014.12.001. [DOI] [Google Scholar]
- Das B.; Khan M. I.; Jayabalan R.; Behera S. K.; Yun S.-I.; Tripathy S. K.; Mishra A. Understanding the antifungal mechanism of Ag@ ZnO core-shell nanocomposites against Candida krusei. Sci. Rep. 2016, 6, 36403. 10.1038/srep36403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaharia A.; Muşat V.; Pleşcan Ghisman V.; Baroiu N. Antimicrobial hybrid biocompatible materials based on acrylic copolymers modified with (Ag) ZnO/chitosan composite nanoparticles. Eur. Polym. J. 2016, 84, 550–564. 10.1016/j.eurpolymj.2016.09.018. [DOI] [Google Scholar]
- Sun D.; Zhang W.; Mou Z.; Chen Y.; Guo F.; Yang E.; Wang W. Transcriptome analysis reveals silver nanoparticle-decorated quercetin antibacterial molecular mechanism. ACS Appl. Mater. Interfaces 2017, 9, 10047–10060. 10.1021/acsami.7b02380. [DOI] [PubMed] [Google Scholar]
- Choi H.-J.; Choi S.-J.; Choo S.; Kim I.-D.; Lee H. Hierarchical ZnO nanowires-loaded Sb-doped SnO 2-ZnO micrograting pattern via direct imprinting-assisted hydrothermal growth and its selective detection of acetone molecules. Sci. Rep. 2016, 6, 18731. 10.1038/srep18731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alammar T.; Mudring A.-V. Facile preparation of Ag/ZnO nanoparticles via photoreduction. J. Mater. Sci. 2009, 44, 3218–3222. 10.1007/s10853-009-3429-4. [DOI] [Google Scholar]
- Cheng J.; Wang J. Syntheses of amphiphilic biodegradable copolymers of poly (ethyl ethylene phosphate) and poly (3-hydroxybutyrate) for drug delivery. Sci. China, Ser. B: Chem. 2009, 52, 961–968. 10.1007/s11426-009-0121-0. [DOI] [Google Scholar]
- Condat M.; Helary C.; Coradin T.; Dubot P.; Babinot J.; Faustini M.; Andaloussi S. A.; Renard E.; Langlois V.; Versace D.-L. Design of cytocompatible bacteria-repellent bio-based Polyester films via an aqueous photoactivated process. J. Mater. Chem. B 2016, 4, 2842–2850. 10.1039/c5tb02659h. [DOI] [PubMed] [Google Scholar]
- Loukanov A.; Filipov C.; Valcheva V.; Lecheva M.; Emin S. Growth stimulation of Bacillus cereus and Pseudomonas putida using nanostructured ZnO thin film as transducer element. J. Nanoparticle Res. 2015, 17, 196. 10.1007/s11051-015-3001-x. [DOI] [Google Scholar]
- Wei T.; Zhan W.; Yu Q.; Chen H. Smart biointerface with photoswitched functions between bactericidal activity and bacteria-releasing ability. ACS Appl. Mater. Interfaces 2017, 9, 25767–25774. 10.1021/acsami.7b06483. [DOI] [PubMed] [Google Scholar]
- Sinha R.; Karan R.; Sinha A.; Khare S. K. Interaction and nanotoxic effect of ZnO and Ag nanoparticles on mesophilic and halophilic bacterial cells. Bioresour. Technol. 2011, 102, 1516–1520. 10.1016/j.biortech.2010.07.117. [DOI] [PubMed] [Google Scholar]
- Kawabata N.; Ueno Y.; Torii K.; Matsumoto T. Capturing interaction between insoluble pyridinium-type polymer and bacterial cells. Agric. Biol. Chem. 1987, 51, 1085–1090. 10.1271/bbb1961.51.1085. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.