

Abstract
The prevalence of psychiatric disorders has increased in recent years. Among existing mental disorders, major depressive disorder (MDD) has emerged as one of the leading causes of disability worldwide, affecting individuals throughout their lives. Currently, MDD affects 15% of adults in the Americas. Over the past 50 years, pharmacotherapy, psychotherapy, and brain stimulation have been used to treat MDD. The most common approach is still pharmacotherapy; however, studies show that about 40% of patients are refractory to existing treatments. Although the monoamine hypothesis has been widely accepted as a molecular mechanism to explain the etiology of depression, its relationship with other biochemical phenomena remains only partially understood. This is the case of the link between MDD and inflammation, mitochondrial dysfunction, and oxidative stress. Studies have found that depressive patients usually exhibit altered inflammatory markers, mitochondrial membrane depolarization, oxidized mitochondrial DNA, and thus high levels of both central and peripheral reactive oxygen species (ROS). The effect of antidepressants on these events remains unclear. Nevertheless, the effects of ROS on the brain are well known, including lipid peroxidation of neuronal membranes, accumulation of peroxidation products in neurons, protein and DNA damage, reduced antioxidant defenses, apoptosis induction, and neuroinflammation. Antioxidants such as ascorbic acid, tocopherols, and coenzyme Q have shown promise in some depressive patients, but without consensus on their efficacy. Hence, this paper provides a review of MDD and its association with inflammation, mitochondrial dysfunction, and oxidative stress and is aimed at thoroughly discussing the putative links between these events, which may contribute to the design and development of new therapeutic approaches for patients.
1. Introduction
Major depressive disorder (MDD) is a public health problem characterized as a mental disorder and is one of the leading causes of occupational or social disability worldwide. According to the World Health Organization [1], 322 million people are affected by this disorder, which is currently more predominant among women than men.
First-line treatment for depression includes talk therapies, antidepressant medications, or a combination of both. Patients suffering from mild depression are indicated for cognitive behavioral therapy, while for moderate to severe cases, antidepressants are indicated [1]. The full benefit of the medications occurs 4 to 6 weeks after initiation of administration [2].
Less than half of patients worldwide (in many countries, representing less than 10%) receive these treatments. In addition, other difficulties include lack of resources and/or skilled professionals, diversity of clinical manifestations, social stigma associated with mental disorders, and inaccurate assessment [1]. Despite the approaches available to treat MDD, only about one-third of depressed patients achieve remission upon receiving antidepressant treatment, and treatment response rates appear to drop with each subsequent retry [3, 4].
Currently available antidepressant therapies focus on modulating monoamine transmission, or they may limit it, as depression is a very broad disease and involves a sequence of events, and monoamine medications do not have a wide range of options. To assist the large number of refractory patients in recent years, the addition of atypical antipsychotics to antidepressants has been common and has some benefit [5]. Nevertheless, many patients continue to suffer from this disabling disease.
Treatment-resistant depression (TRD) is associated with increased functional impairment, mortality, morbidity, and long-term recurrent or chronic episodes [6, 7]. Therefore, an improved response to treatment by identifying predictive risk factors for nonresponse may help better disease prognosis [8].
Major depressive disorder has been associated with alterations in neurotransmitter biosynthesis, altered membrane receptor expression, alterations in cortical structure volume, and desensitization of the hypothalamic-adrenal-pituitary (HPA) axis [9]. HPA axis dysregulation causes excessive release of cortisol, a fundamental hormone for maintaining homeostasis, as it has numerous catabolic functions and anti-inflammatory action. However, its excessive production can suppress the immune system [10]; thus, inflammatory responses are triggered through the activation of macrophages and lymphocytes, as well as microglia and astrocytes [11]. The first studies on depression date back to the 1980s, and since then, the findings show that inflammation could play an important role in the pathophysiology of this disease [12–14].
In fact, several studies have shown changes in interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), and C-reactive protein (CRP) levels [15–17] in depressed patients. Some of these cytokines activate the enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO) from the kynurenine pathway, diverting tryptophan from its main route of serotonin production [18–20].
A growing body of evidence [21] indicates that inflammation may further cause deleterious changes in mitochondrial function, affecting oxidative phosphorylation and membrane polarity. These changes may lead to oxidative and nitrosative stress and apoptosis, events associated with the pathogenesis and pharmacological resistance of MDD [18, 21–23]. Thus, mitochondria should be considered a crucial target for the development of new antidepressant drugs, and specific forms of mitochondrial dysfunction can be identified as biomarkers to customize treatment and aid in early diagnosis [21]. In this paper, we discuss the involvement of inflammation, mitochondrial dysfunction, and oxidative stress in the etiology and pharmacological resistance of MDD as pathways for future therapeutic approaches.
2. Methodological Approach
Three hundred and seven (307) articles were selected from the search in the MEDLINE database. We used a combination of one or more of the following mesh-terms: treatment-resistant depression, major depressive disorder, redox imbalance, mitochondria, neuroinflammation, and antioxidants. The terms cited must appear in the title, keywords, or abstract of the article. After this search, we analyzed the abstract of each article and only those that contemplated the main scope of our discussion and could help us to describe the importance of oxidative stress and neuroinflammation on treatment-resistant depression were selected. Thus, 214 studies were included, which were analyzed and used to construct the rationale of our narrative review. Figure 1 illustrates the search methodology.
Figure 1.
Flowchart of the search methodology performed.
3. Inflammatory Hypothesis of Depression: HPA Axis Desensitization
Acute activation of the HPA axis plays a crucial role in responding appropriately to acute stress events. However, prolonged activation of the axis and a sustained increase of glucocorticoids are well documented and related to MDD [24]. Depressive individuals have an increase in plasma cortisol concentration and a decrease in HPA axis sensitivity to dexamethasone [25, 26]. Briefly, the alpha glucocorticoid receptor (GRα) mediates the negative feedback of the HPA axis, i.e., the ability of the cortisol to inhibit its secretion. In contrast, in depression and chronic stress situations, a decrease in GRα expression in the hypothalamus and pituitary leads to the desensitization of negative feedback, which in turn leads to HPA axis hyperactivity and a sustained increase in synthesis and secretion of glucocorticoids [27, 28].
The lack of adequate glucocorticoid-mediated inhibitory control promotes increased immune signaling, as demonstrated by increased levels of cytokines and proinflammatory cells activated by glucocorticoids [16, 29]. Lymphocytes from patients with MDD are also resistant to the suppressive effects of dexamethasone in vitro [30]. Glucocorticoid resistance and an intensification of proinflammatory signaling are found in about 85% of MDD studies [31]. Proinflammatory cytokines are also involved in glucocorticoid resistance by decreasing GR expression and function, leading to a pronounced increase in inflammatory responses [32]. These studies confirm that glucocorticoid resistance increases cortisol production and increased inflammatory signaling, which are coexisting biological responses and influence the therapeutic response in depression.
The antidepressant treatment enhances the synthesis of brain-derived neurotrophic factor (BNDF) and promotes neurogenesis. However, drug actions appear to depend on glutathione reductase (GR) activation, and glucocorticoid activation of GRα decreases neurogenesis. This contradiction may be related to the different effects of GRα due to its binding to different agonists and modifications of their phosphorylation state. Different antidepressants act on the glucocorticoid receptor and increase neurogenesis through mechanisms dependent on the activation of protein kinase A (PKA) and its signaling cascade [33]. However, cortisol decreases neurogenesis through upregulation of the kinase gene induced by serum and glucocorticoids (SGK1) [34]. This in vitro evidence demonstrates PKA antidepressant-induced positive regulation and cortisol-induced expression of SGK1 [34, 35]. These data indicate that persistent signaling of glucocorticoids and norepinephrine, in a situation of psychosocial stress, trauma, or persistent stressful events, contributes to exaggerated inflammation responses, desensitizing GRα and activating transcription of proinflammatory genes.
Peripherally released cytokines can reach the central nervous system (CNS) through three distinct pathways: the blood-brain barrier, the circumventricular organs, or the vagus nucleus of the solitary tract. When cytokines reach the CNS, they directly and indirectly affect the metabolism of neurotransmitters, which can stimulate apoptosis and decrease neurogenesis, affecting essential circuits for behavior maintenance. They alter the metabolism of serotonin, dopamine, and glutamate, which are neurotransmitters involved in mood regulation [16]. As serotonin levels decline, production of melatonin is impaired, which, in turn, disrupts the biological clock that controls neuronal physiological processes, including the sleep-wake cycle [36, 37]. This combination of factors accentuates depressive symptoms, as well as oxidative stress and inflammation in the CNS [38, 39]. A promising drug for treating circadian rhythm in psychiatric diseases is ramelteon (RMT), a melatonin receptor agonist [40]. Previous evidence indicates that RMT has neuroprotective, antioxidant, and anti-inflammatory activities [41].
Controlling inflammation in MDD patients is crucial. In response to stress, the innate immune system activates the “sterile inflammatory response” which releases molecules into the extracellular space. The Danger-Associated Molecular Pattern (DAMP) binds to Pattern Recognition Receptors (PRRs) expressed on the cytosol or innate immune cell membranes that may be NOD (NLR) or Toll (TLR) receptors. The cascade triggering of these PRRs leads to the activation of NLRP3 inflammasome and caspase-1 [42]. NLRP3 in turn activates IL-6, TNF-α, and IFN-γ, which may increase the activity of indoleamine 2,3-dioxygenase (IDO), an enzyme involved in the synthesis of kynurenine (KYN) from the amino acid tryptophan (TRP) [43, 44].
Tryptophan is an essential amino acid and is the primary precursor of serotonin. Activation of IDO reduces serotonin synthesis through a shift of tryptophan to the kynurenine pathway. In the brain, kynurenine is metabolized by the following cellular pathways: (1) neural progenitor cells and microglia, generating 3-hydroxykynurenine (3-HK) and quinolinic acid (QA), and (2) astrocytes, producing kynurenic acid (KA). The metabolite, 3-HK, is involved in oxidative stress. QA is an N-methyl-d-aspartate (NMDA) receptor agonist that promotes the increased release of glutamate and blocking its reuptake by astrocytes [45]. Increased glutamate is known to cause neurotoxicity because excess neurotransmitter triggers an influx of Ca2+ into the cell, which in turn can generate a potassium leak in the cell. This depolarizes the mitochondrial membrane, generating ROS and oxidized mitochondrial DNA (ox-mtDNA), which reactivates NLRP3 [46, 47]. Influx of Ca2+ may also activate other pathways that lead to mitochondrial dysfunction by reducing SIRT3 (Figure 2). This set of results can lead to excitotoxicity and neurodegeneration in essential brain areas of depressed individuals (Figure 2). In fact, an increase in QA concentration in the brain and cerebrospinal fluid of depressive individuals suffering from suicide was already found [48, 49]. In addition, increased QA concentration has been associated with oxidative stress and lipid peroxidation [50].
Figure 2.
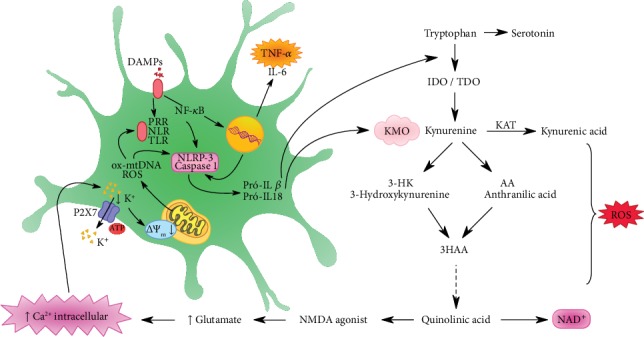
Danger-Associated Molecular Pattern (DAMP) binds to the Pattern Recognition Receptors (PRRs) expressed on the cytosol or in innate immune cell membranes. The cascade triggered by these PRRs leads to NLRP3 inflammasome and caspase-1 activation, which can activate IL-1β and IL-18. Oxidized mitochondrial DNA (ox-mtDNA) and mitochondrial reactive oxygen species (ROS) also activate the inflammasome. NF-κB, through the transcriptional activation pathway, generates tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). Proinflammatory cytokines IL-1β and IL-18 activate the enzymes IDO and TDO of the kynurenine pathway, degrading tryptophan into kynurenine. These two cytokines further activate KMO, which is the enzyme that directs kynurenine to be degraded to 3HK and quinolinic acid, both neurotoxic agents, over the kynurenic acid, a neuroprotective agent. Kynurenic acid is an NMDA receptor agonist and increases glutamate levels and consequently intracellular calcium. Excessive amounts of ROS are produced over the kynurenine pathway.
The reduction in serotonin synthesis by cytokines may also reduce dopamine synthesis. Injection of IFN-γ into rats resulted in a decreased concentration of tetrahydrobiopterin (BH4) and dopamine in the amygdala and raphe nuclei [51]. The BH4 is an essential cofactor for the regular activity of the enzyme tyrosine hydroxylase, which is the critical enzyme in the dopamine biosynthesis pathway [16]. The impact of cytokines on reducing synthesis and dopaminergic action in the brain can lead to a decreased motivation and pleasure (anhedonia), an essential and classic symptom of depressive behavior.
3.1. Neuroinflammation and the Role of Microglia
Some clinical studies suggest that microglia have a modified morphology and function in depressive individuals, with a less branched phenotype and less capacity for glutamate reuptake and maintenance of homeostasis [52, 53]. Moreover, in a study using positron emission tomography, individuals who presented a depressive episode demonstrated an increase in translocating protein (TSPO) labeling, which is a neuronal marker of inflammation [54]. Postmortem studies found an increase in the expression of cytokines and complement pathways in the prefrontal cortex and hippocampus of depressive individuals [55, 56], suggesting that neuroimmune dysregulation may represent a pathophysiological mechanism in depressive patients.
Changes in neuronal functions that occur concurrently with microglial activation imply reciprocal interactions between these two structures, and these responses may not only lead to neuroinflammation but also affect other essential functions of the CNS, such as neurotransmission. Morphological changes in microglia induced by neuroinflammation generally do not lead to acute neurotoxicity but may contribute to neuronal dystrophy after stress. Furthermore, exposure to psychosocial and environmental stress causes neuronal activation and release of glutamate and norepinephrine in corticolimbic brain regions, such as the prefrontal cortex, amygdala, and hippocampus [57]. This evidence suggests that dysfunctional neuronal activation associated with increased glucocorticoids leads to neuronal dystrophy in corticolimbic brain regions following chronic exposure to stress. For example, repeated stress caused dendritic atrophy and loss of synapses on pyramidal neurons in the prefrontal cortex of rats [58, 59], and these effects were also achieved after chronic corticosterone administration [60]. These neurobiological alterations contribute to the change in excitatory-inhibitory activity in the prefrontal cortex, a critical region for the maintenance and control of motivated behaviors, which is critically affected in depression [61, 62].
The increased release of DAMPs from the high mobility moiety (HMGB1) box 1 protein caused the activation of microglia and increased gene expression of proinflammatory cytokines [63]. Studies suggest that increased NLRP3 and IL-1β activation in the prefrontal cortex are mediated by the activation of microglia in chronic stress situations and that these responses are reversed under chronic treatment with fluoxetine [64].
Recent studies have also demonstrated that anxiolytics and antidepressants can block or reverse microglial activation. Administration of imipramine during a social defeat protocol reduced IL-6 expression in microglia and attenuated depressive-like behavior [65]. This result is similar to other studies, which found that selective serotonin reuptake inhibitors produce an anti-inflammatory response in microglia [66]. These findings raise important questions. (1) Do the interventions mentioned above aimed at normalizing monoamine neurotransmission exert their primary effects through neurotransmitter homeostasis? (2) Or can the inhibition of microglial activation be considered their primary mechanism of action? These results suggest that the pharmacological treatments currently used against depression may produce their therapeutic effects by enhancing neurotransmission and modulating microglial functions and inflammation. Understanding neuronal function and microglia and the distinction of molecular and cellular pathways that contribute to the maintenance of neuronal and microglial function after repeated exposure to stress may provide new insights into potential therapeutic targets.
Additional studies must be conducted to examine the relationships between microglia and neurons and, at the same time, to find out if the interaction between these cells contribute to the stress-induced inflammation. In addition, how can interventions involve these mechanisms to provide effective therapeutic benefits?
3.2. Inflammation and Treatment Resistance
The development of biomarkers capable of predicting the response to depression treatment is clinically important. These biomarkers could be critical for classifying patient subtypes and a good alternative for prescribing specific treatments if patients are resistant to the conventional antidepressant drug.
A recent review by Roman and Irwin [67] describes that proinflammatory cytokines (mainly IL-1β, IL-6, and TNF-α) may be useful biomarkers for investigating the presence or level of baseline inflammation during screening for depression. However, the choice of these immune biomarkers is limited due to the lack of standardization of assays for clinical application. The authors also cite a study in progress by Janssen Research & Development, LLC (ClinicalTrials.gov Identifier: NCT02902601), which is investigating the safety and tolerability of a new agent, JNJ 54175446, in patients with depression. JNJ 54175446 is a P2X7 purinergic receptor agonist capable of indirectly modulating inflammasome activation.
Despite decades of research about depression, we still lack a deeper understanding of the pathophysiology and mechanisms involved in drug resistance. In other areas, such as in diabetes or heart disease [68], a large genomic association between these illnesses and the molecular targets of marketed drugs is known, with high treatment efficacy. Unfortunately, the situation is entirely different in psychiatry, where none of the currently used drugs has a strong correlation with potential candidate biomarkers for Genome-wide Association Studies (GWAS) [69]. The genetic risk variants identified so far cover a broad spectrum of biological processes but are enriched for neurodevelopmental or synapse-related genes and are not directly related to the targets of current clinical medications. Taken together, these findings point to new avenues for antidepressant therapy, suggesting entirely new biology for these disorders and the urgent need to reconsider other factors involved with depression and drug resistance.
Many clinical factors have been related to the nontherapeutic response, including psychiatric comorbidities such as anxiety disorders, personality disorders, and bipolar disorder [70–72]. In addition, comorbidities of cardiovascular diseases, diabetes, and cancer have also been associated with an inefficient response to antidepressants [73]. Interestingly, these clinical conditions are associated with inflammation [16, 45, 74]. Investigation of the pathways involved with the pathophysiology of MDD has produced numerous promising therapeutic targets for treatment-resistant depression (TRD). Among these new markers, there is a particular interest in the inflammatory pathways and their link with oxidative and nitrosative stress [75–79].
We will now explore data about how inflammation and the outcomes that are related to it may influence the response to antidepressant treatment. A meta-analysis of 35 studies evaluating inflammatory markers before and after antidepressant treatment found that patients who were not responders were more likely to have a higher inflammatory profile at baseline and follow-up treatment than the responders [8]. In a study by Lindqvist et al., IL-6 decreased significantly in patients responding to selective serotonin reuptake inhibitors but did not reduce their concentrations in patients refractory to the treatment [80]. In a study of 241 depressive patients, Uhrer et al. indicated that the concentration of C-reactive protein (CRP) promoted a differentially predicted response to escitalopram and nortriptyline. Other studies found higher levels of CRP and other proinflammatory cytokines early in the study, and these inflammatory markers were associated with a weaker response to serotonin reuptake inhibitor antidepressants, including escitalopram, fluoxetine, and a low dosage of venlafaxine [81]. In a study by Haroon et al., patients who participated in multiple treatment trials and who failed symptom remission exhibited higher plasma concentrations of TNF-γ, sTNFR2, IL-6, and CRP compared to depressive individuals who responded efficiently. Following a body mass index (BMI) correction criterion, TNF-α, sTNF-R2, and IL-6 were the markers most associated with the number of failed treatment attempts [82].
Preclinical and clinical studies suggest that a decrease in NMDA receptor activity and an increase in AMPA receptors lead to favorable mood outcomes [83]. Increased stimulation of AMPA over NMDA receptors leads to an increase in calcium and sodium influx and a strengthening of intracellular signaling that enhances BDNF expression, leading to improved neuroplasticity and neuronal function [73, 77, 78]. Thus, increased AMPA activity and decreased NMDA activity may be essential outcomes for mood enhancement and cognition and promising targets for TRD [79, 83–85].
Using a preclinical model of inflammation, administration of lipopolysaccharide (LPS) has been shown to induce depressive-like behavior through NMDA receptor stimulation [86]. Briefly, LPS induces a depressive-like behavior by activating IDO [87], leading to an increase in the KYN/TRP ratio and an increase in the formation of QA [88], an agonist of the NMDA receptor. The discovery reinforced the importance of the NMDA receptor for depression influenced by inflammation in which ketamine improves the signs of distress by strengthening glutamatergic neurotransmission through AMPA receptors [86]. The use of ketamine for other purposes, including the treatment of MDD, has been studied [89]. Several studies have already shown an improvement in antidepressant response, including for patients with TRD. These findings will be discussed below in the section on new therapeutic perspectives.
The information above suggests that a portion of depressive patients has a high inflammatory profile and that such inflammation may be associated with failure in multiple treatment attempts. Thus, in patients with a history of failed antidepressant treatment, clinicians could evaluate and consider the use of therapies that act on the inflammatory profile or molecules that are related to inflammation.
4. Adjuvant Therapies
Many studies using TNF-α antagonists (infliximab); nonsteroidal anti-inflammatory drugs (NSAIDs); ketamine, natural anti-inflammatory agents, such as omega-3 polyunsaturated fatty acid and curcumin; and an NMDA receptor antagonist have gained attention. Among the aforementioned agents, infliximab has been the most evaluated for the treatment of TRD.
4.1. TNF-α Antagonist (Infliximab)
The TNF-α antagonists are used clinically in autoimmune disorders to prevent a systemic inflammatory response. The reduction in microglia activation with the use of TNF-α antagonists may be particularly relevant for TRD [90]. A recent meta-analysis (n = 2370) of seven randomized controlled trials using anticytokine agents (e.g., adalimumab, etanercept, tocilizumab, and infliximab) in conditions of chronic inflammation (e.g., rheumatoid arthritis) reported moderate antidepressant efficacy (standardized mean difference (SMD) = 0.40, 95% confidence interval (CI) = 0.22, 0.59) [91].
The antidepressive efficacy of infliximab alone has been investigated and reported as a primary outcome among adults with mood disorders. This randomized controlled clinical trial tested the efficacy of intravenous infliximab (5 mg/kg) administered at baseline and weeks 2, 4, and 6 of a 12-week protocol in a sample of 60 patients with TRD. The study found no difference between the treated and control groups; however, when patients were categorized for their inflammatory status based on (PCR) > 5 mg/L, a significant antidepressant effect was noted. Sixty-two percent of patients with TRD who received infliximab achieved a 50% reduction in HDRS scores, compared to only 33% of patients in the placebo group [92].
Although infliximab may be useful for a subset of patients with TRD, who also have increased inflammatory biomarkers, further studies are needed to confirm or refute this hypothesis. In addition, the number of adverse effects may be a limiting factor, as infliximab increases the risk of infection due to its potent anti-inflammatory effect.
4.2. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
Proinflammatory cytokines can trigger an inflammatory cascade in the brain, which includes increased activity of cyclooxygenase (COXs) that are critical enzymes in the production of prostaglandins [93]. Drugs targeting the cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) enzymes might have a beneficial effect on depressive patients with elevated levels of inflammatory cytokines. COX-2 expression is detected in synaptic dendrites and excitatory postsynaptic endings, especially on the cortex, hippocampus, and amygdala, whereas COX-1 is expressed in microglia and perivascular cells [94]. A study by Choi et al. [95] demonstrated that mice with COX-1 deficiency had a decrease in neuronal degeneration, microglial activation, and expression of proinflammatory cytokines and PGE2 after exposure to LPS.
However, COX-2 may play a neurotoxic or anti-inflammatory role, depending on the initial stimulus. The results of studies using animal models, especially with celecoxib treatment (a selective COX-2 inhibitor), are contradictory. In a chronic mild stress model in rats, treatment with celecoxib for 21 days reversed depressive behavior [96]. In another preclinical model of depressive-like behavior (bulbectomy), the use of celecoxib for 14 days reversed depressive behavior in treated rats. The concentration of the proinflammatory cytokines IL-1 and TNF-α in the prefrontal cortex and hypothalamus decreased, probably due to the reduction of PGE2 synthesis [97]. However, COX-2 may also have a neuroprotective function in response to an inflammatory challenge. Deletion of COX-2 may result in increased neuronal damage in the hippocampus and increased expression of TNF-α, IL-6, and IL-1β. Chronic administration of celecoxib for six weeks caused an increase in IL-1β levels in the brain of mice exposed to LPS [98].
NSAIDs, especially acetylsalicylic acid (ASA) and celecoxib, were also tested in an attempt to improve response in patients with TRD. Acetylsalicylic acid irreversibly inhibits COX-1 and COX-2, thus decreasing prostaglandin and thromboxane levels, and the production of TNF-α and IL-6 [99]. In 2013, Berk et al. published a systematic review that evaluated the role of ASA in the treatment of mental illness, evaluating studies from 1996 to 2012 [100]. Some evidence suggests beneficial effects for aspirin in mood disorders, through an improvement in the clinical response time to antidepressants [101]. That study found that patients who used fluoxetine associated with ASA had a higher rate of remission for depressive symptoms than the fluoxetine group. In another study with 70 depressive patients, administration of aspirin in combination with fluoxetine conferred a further reduction of oxidative stress parameters compared to fluoxetine monotherapy [102]. Despite the lack of significant clinical improvement and linear scientific data, NSAIDs act positively on many of the biochemical and molecular outcomes cited in the text. However, the use of NSAIDs chronically provides significant adverse effects to be considered and does not demonstrate a congruency in the results.
4.3. Ketamine: NMDA Antagonist
Interest in using ketamine to treat TRD has increased in the last decade. The long-term beneficial actions of ketamine on the central nervous system are very evident; however, there are not enough studies to discuss the long-term effects of a single ketamine administration on the functioning of neural circuits in MDD [103]. Previous evidence demonstrates that ketamine may increase the expression and synthesis of BDNF [103, 104]. The antidepressant effects of ketamine were inhibited in mice with BDNF deletion as well as after infusion of an anti-BDNF antibody into the prefrontal cortex [105]. Furthermore, preclinical studies show that ketamine exerts its antidepressant effects by increasing the expression of mammalian rapamycin target protein (mTOR), which modulates cell growth, proliferation, motility, survival, and protein synthesis [103, 106]. These observations provide insight into why the decreased levels of BDNF associated with depressive symptoms could be reversed through the rapid actions of ketamine. Several clinical studies have tested ketamine as an antidepressant agent for TRD.
In a recent meta-analysis [107], seven randomized controlled trials (RCTs) using intravenous infusion and intranasal ketamine evaluated its antidepressant effect in patients with MDD and bipolar depression. Ketamine was associated with higher clinical remission rates in the control groups (saline or midazolam) 24 hours (OR 7.06, NNT = 5), three days (OR 3.86, NNT = 6), and seven days (OR 4.00, NNT = 6) after remission. Ketamine is associated with transient psychotomimetic effects; however, these signs do not persist chronically [107].
In addition, several clinical trials were conducted to evaluate the role of ketamine in TRD. In a randomized, placebo-controlled, double-blind, crossover study, 18 patients with TRD received an infusion of ketamine (0.5 mg/kg) with a one-week interval between each administration. Patients who received ketamine obtain significant improvement in evaluations compared to placebo subjects. The positive effect was apparent within 110 minutes after injection and remained over the following days [108]. In other clinical studies, similar results have been observed [109, 110]. In two case-control studies, ketamine had an antisuicidal effect in the TRD subjects [111, 112].
Despite the promising effects of ketamine in patients with TRD, opinions about its use to treat MDD are still polarized, mainly because of its side effects. Some of the most common effects are dissociations related to depersonalization, as well as perceptual disturbances and mental confusion. The risk for the development of psychotic states, agitation, and dependence is also referred to as an adverse effect [113]. Nevertheless, ketamine is clearly a potent, rapid-acting antidepressant for TRD, which is entirely different from conventional antidepressants that take days to weeks before a crucial antidepressant effect is observed [114]. Therefore, ketamine may be a useful alternative for TRD. ESKETAMINE (ESK), its derivative, is a new NMDA drug recently approved by the FDA for intranasal use in TRD treatment. A recent systematic review reported that the intravenous infusion of ESK causes rapid and sustained antidepressant activity in refractory patients with MDD and TRD, as well as in patients with MDD at imminent risk of suicide [115]. However, despite its effectiveness, further preclinical and clinical studies are needed to investigate the long-term efficacy and safety of intranasal ESK. Another recent FDA-approved drug for treating TRD is Symbyax, which combines olanzapine (an atypical antipsychotic) and fluoxetine (a selective serotonin reuptake inhibitor) [116, 117]. A recent study of 25 patients treated with Symbyax for 8 weeks showed a decline in amygdala activity and right ventromedial prefrontal metabolism, and these events were correlated with improvement in depression after the intervention [118].
5. The Relation between Inflammation and Oxidative Stress
In the brain, oxidative stress and its related cellular damage are easily spread due to the physiological and physical characteristics of this organ, as well as the high metabolic rate of its cells, which make it highly dependent on an efficient mitochondrial oxidative phosphorylation system (OXPHOS). Mitochondria are intracellular organelles required for numerous cellular functions, including control of energy metabolism and regulation of ROS production, calcium homeostasis, and apoptosis. During the inflammatory process, released interleukins are capable of activating the KYN pathway, which generates catabolites, called TRYCATs that cause a high calcium influx inducing mitochondrial dysfunction along with an impairment in the cellular antioxidant system [119, 120]. This becomes a cycle as the mitochondrial dysfunction and potential losses of the mitochondrial membrane lead to a rapid increase in the production of mitochondrial reactive oxygen species (MROS), which are also activators of the inflammasome (NLRP3) [121] (Figure 2).
The macrophages involved during the inflammatory process have a P2X7 receptor (P2X7R), which is a cationic channel highly dependent on exogenous ATP. Activation of this channel causes potassium efflux, altering mitochondrial membrane potential and generating mtROS and oxidized mitochondrial DNA, which are released into the cytosol by directly activating NLRP3 [46]. Potassium efflux may also occur due to high levels of calcium from the kynurenine pathway (Figure 2).
The increase in ROS production is associated with a reduction in neuronal metabolism. This metabolic worsening may be associated with the reduced ability to convert external energy into substrates required for ATP biosynthesis, mainly due to mitochondrial dysfunction [122]. Metabolic stress and increased ROS production can lead to advanced glycation end products (AGEs) and lipid peroxidation end products in neurons [123].
A study by Anamika et al. reports that influx of Ca2+ activates neural nitric oxide synthase (nNOS) and nitric oxide release occurs. This nitric oxide can be combined with other types of ROS to form peroxynitrite (ONOO-), a highly unstable radical [124]. Excess nitric oxide may decrease SIRT3 activity and expression, causing mitochondrial dysfunction [125].
Sirtuins are NAD+-dependent histone deacetylases. Because they require NAD+ for their activity, the cellular level of sirtuins represents the redox status of cells and, thus, serves as metabolic stress sensors [125]. SIRT3 in particular is an isoform present in mitochondria, whose function is to modulate activity of several important mitochondrial proteins, to induce adaptive changes during bioenergetic deficits, and to regulate mitochondrial biogenesis and dynamics, ROS metabolism, ATP production, and maintenance of mitochondrial integrity [126, 127].
SIRT3 is also related to mitochondrial permeability transition pore (mPTP) blockade of the pores, preventing the release of cytochrome C and thus preventing apoptosis [125, 128]. All these changes can occur from the reduction of SIRT3, including low ATP production, which is very common in neurodegenerative diseases, including depressive patients [125].
With the reduction of SIRT3, the antioxidant defense mechanism also changes, since SIRT3 deacetylates the antioxidant enzyme MnSOD, and mitochondria are the main site of ROS generation, reducing antioxidant the immediate target of oxidation damage [129, 130]. The enzyme MnSOD catalyzes the conversion of superoxide anions (O−2) into hydrogen peroxide (H2O2) and oxygen (O2). Reduced MnSOD activity is the immediate target of oxidation damage [129, 130].
SIRT3 regulates the tricarboxylic acid cycle (TCA) by activating PDH (pyruvate dehydrogenase complex), the first enzyme that catalyzes the entry of pyruvate into the pathway of oxidative energy production [131]. In addition, SIRT3 can deacetylate mitochondrial complex I, specifically subcomplex 9 (NDUFA9) [132, 133]. SIRT3 is likely to regulate the expression of some complex IV subunits and is also related to the two subunits of ATP synthase F0-F1 [126, 134, 135]. Thus, inflammation generated by HPA axis imbalance leads to mitochondrial dysfunction, which in turn causes excessive ROS production.
A redox imbalance in the brain might be involved in the pathogenesis of depression beyond being related to other risk factors such as increased inflammation, impaired plasticity, and reducing neuron signaling [136]. Under physiological conditions, ROS and reactive nitrogen species (RNS) are regulated by antioxidant pathways, including enzymatic and nonenzymatic compounds. In excess or in situations where the antioxidant system is impaired, these species may damage lipids, proteins, and DNA. In depressed patients, the antioxidant system is also compromised because inflammation activates the KMO (kynurenine 3-monooxygenase) enzyme that degrades kynurenine into 3-hydroxykynurenine and other neurotoxic metabolites. The KMO enzyme is NADPH-dependent, so if the availability of this coenzyme is reduced, NADPH-dependent antioxidant defense systems are compromised, including glutathione (GSH) and catalase (CAT), which are key enzymes for reducing H2O2 in O2 and H2O [137] (Figure 3).
Figure 3.
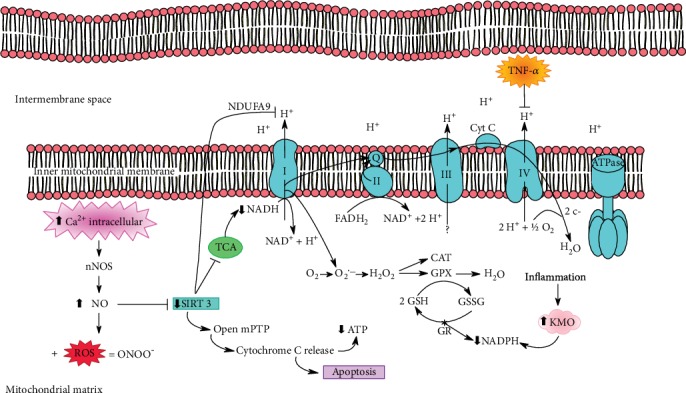
The increase of intracellular calcium activates neural nitric oxide synthase (nNOS), causing increased NO levels. This will decrease SIRT3, which acts as a key to control mitochondrial dysfunction. As SIRT3 activity decreases, mitochondrial permeability transition pores (mPTPs) open, which release cytochrome C, causing a decrease in ATP levels and inducing apoptosis. NO can also bind to ROS from the kynurenine pathway, generating peroxynitrite (ONOO), a highly unstable free radical. Reduction of SIRT3 deacetylates complex I NADH dehydrogenase, specifically in the NDUFA9 subunit, which interacts with two other ATP synthase subunits (F0 and F1). When SIRT3 is reduced, PDH activation is inadequate for the citric acid cycle, resulting in low levels of NADH and reduced activity of complex I. In addition, SIRT3 promotes deacetylation of MnSOD, an antioxidant enzyme that scavenges superoxide anion produced over the pathway. Another impaired antioxidant enzyme is GSH, because during the inflammatory process, the enzyme KMO, an enzyme dependent on NADPH, is activated, thus reducing the availability of this coenzyme for antioxidant defense systems. In parallel, TNF-α phosphorylates tyrosine 304 in subunit I of cytochrome C oxidase in complex IV, leading to further mitochondrial damage.
MDD is usually accompanied by a decrease in antioxidant enzyme activities and total antioxidant capacity (TAC). A recent meta-analysis identified lower TAC and some antioxidant parameters in acute episodes of depressed patients, as well as serum paraoxonase, uric acid, albumin, and zinc levels. On the other hand, higher oxidative damage products, including red blood cell (RBC) malondialdehyde, serum MDA and 8-F2-isoprostanes, and serum peroxide were found in MDD patients [138]. Table 1 summarizes oxidative/antioxidative markers in drug-naive patients with depression.
Table 1.
Markers of oxidative stress and antioxidants related to drug-naive patients.
| Study population | Outcome in DP | Reference |
|---|---|---|
| 247 DP and 248 HC | ↑ MDA | Islam et al. [139] |
| 77 adult DP and 47 HC | ↑ CP in DP at early stage; ↓ GPx activity at late stage | Diniz et al. [140] |
| 19 adolescent DP and 8HC | ↓ GSH | Freed et al. [141] |
| 50 aged DP and 55 HC | ↑ 8-OHdG | Lindqvist et al. [80] |
| 50 DP and 50 HC | ↑ MDA levels and ↓ SOD activity; no differences for CAT | Camkurt et al. [142] |
| 21 DP and 40 HC | ↑ CAT and SOD activities | Tsai and Huang [143] |
| 332 symptomatic patients, 141 DP and 622 HC | ↑ 8-OHdG | Black et al. [144] |
| 49 adult DP and 49 HC | ↑ TBARS and NO; ↓ SH; no differences for SOD | Kaufmann et al. [145] |
| 60 DP and 40 HC | ↑ MDA levels; ↓ SOD activity, nitrite and vitamin C levels | Bajpai et al. [146] |
| 322 aged DP | ↓ Vitamin C levels | Gariballa [147] |
| 15 DP and 19 HC | ↑ MDA and GR; ↓ SOD and GPx1 | Rybka et al. [148] |
| 82 adult DP and 94 HC | ↑ CP | Magalhães et al. [149] |
| 45 adult recurrent DP and 33 HC | ↑ NO | Talarowska et al. [150] |
| 70 aged DP and 35 HC | ↓ GPx and GSH levels; ↑ GR and SOD activities; no differences for CAT | Kodydková et al. [151] |
| 38 aged DP and 72 HC | ↑ 8-OHdG | Kupper et al. [152] |
| 35 DP and 35 HC | ↓ CoQ10 levels | Maes et al. [153] |
DP: depressive patients; HC: healthy controls; MDA: malondialdehyde; CP: carbonyl protein; GPx: glutathione peroxidase; SOD: superoxide dismutase; CAT: catalase; 8-OHdG: 8-hydroxydeguanosine; TBARS: thiobarbituric acid reactive species; NO: nitric oxide; SH: sulfhydryl; GR: glutathione reductase; GSH: glutathione; CoQ10: coenzyme Q10.
In the oxidative stress pathway, superoxide dismutase (SOD) is the primary antioxidant enzyme able to protect cells from damage caused by ROS. Disturbances in SOD activity are usually found in depressive patients, but the findings are still inconsistent with respect to the direction of this disruption [154]. Decreased SOD activity has been identified in MDD patients [148]. However, other studies reported increased SOD and catalase (CAT) activity in MDD patients [151]. Recently, Tsai and Huang found that serum SOD and CAT activities were significantly higher in the acute phase of MDD patients, suggesting that increased activities of both antioxidant enzymes might be indicators of acute depressive episodes on MDD [143].
Glutathione (GSH) is the most abundant low-molecular-weight thiol in the human body. It plays an important role in protecting cells and their components against ROS and represents a sensitive and consistently endogenous marker of oxidative stress [155]. The pathway that maintains intracellular GSH homeostasis comprises GSH redox cycling, which includes GSH oxidation by glutathione peroxidase (GPx) during detoxification of hydrogen peroxide (H2O2) or organic hydroperoxide and GSH reduction by glutathione reductase (GR) [155]. Peripheral measures in serum and plasma have already demonstrated a decrease in GPx activity in MDD patients [141]. In addition, negative correlations between GPx activity and severity of depressive symptoms were found, suggesting an impaired antioxidant protection in MDD [156]. Recently, a study using proton magnetic resonance spectroscopy to measure in vivo brain GSH in adolescents with MDD found low occipital GSH levels [141].
Production of H2O2 is balanced by catalytic action of antioxidant enzymes, such as CAT and GPx, and proteins with antioxidant function (peroxiredoxins) that act on H2O2 quickly and in synergy modulating the overall peroxide signal [157]. The transcription factor Nrf-2 (nuclear factor erythroid 2-related factor 2) is another target that seems to play an important role in redox homeostasis. At low oxidative stress levels, Nrf-2 is activated and stimulates the transcription of antioxidative genes, leading to the cytoprotective effects. Supporting the hypothesis that increased ROS plays a significant role in depression, Mellon et al. reported that genes regulated by Nrf-2 were elevated in MDD patients and decreased after effective antidepressant treatment [158]. In addition, a recent preclinical study with rodents found that vulnerability to depression resulted from a persistent state of oxidative stress, mediated by Nrf-2 dysfunction, which was reversed by treatment with antioxidants [159].
Several studies that evaluated lipid peroxidation in MDD patients suggest that increased serum levels of MDA, a product resulting from the reaction between ROS/RNS and lipids, are strongly associated with MDD [160]. A recent meta-analysis indicated that 8-hydroxy-2′-deoxyguanosine (8-OHdG) and F2-isoprostanes, which are measurements of oxidative DNA and lipid damage, respectively, are the two oxidative stress markers most consistently elevated in depression, usually with small to moderate effect sizes [144]. Increased MDA levels have also been found in patients with recurrent depression [148, 161, 162].
Increased levels of oxidative stress are also associated with larger cognitive impairment and one potential mechanism underlying neuroprogression and accelerated aging in mood disorders [163, 164]. Depression is known to be associated with accelerated brain aging [165]. One recent preclinical study that proposed a new model for aging brain found that oxidative stress at physiological levels may cause hippocampal dysfunction, specifically involving astrocytes, even before apoptosis detection [166]. Furthermore, a large body of evidence points to overproduction of ROS as one of the main causes of neuronal changes by inducing cell death and consequent atrophy of brain specific regions [167]. Smaller hippocampal volume has been associated with slower antidepressant response in late-life depression and increased peripheral oxidative stress in depressive patients [80, 168, 169]. All together, these findings may represent part of the interplay between oxidative stress, hippocampal volume, and treatment response.
The mechanisms of antidepressants against damage induced by excessive oxidative stress seem to be involved with remission of depressive symptoms and patients' recovery [136]. Evidence indicates that antidepressants may act to restore and normalize the activity of enzymes (such as SOD, GPx, GST, and GR) iNOS and xanthine oxidase (XO), increasing TAC levels, decreasing 8-OHdG and nitric oxide (NO) levels as well as lipid and protein oxidation and attenuating cell death induced by H2O2. Table 2 shows the effects of antidepressant drugs on redox metabolism in clinical, preclinical, and mammalian cell culture models.
Table 2.
Markers of oxidative stress and antioxidants related to antidepressant drugs (AD): preclinical and clinical studies.
| Study model | Antidepressant drugs/dose | Outcome | Reference |
|---|---|---|---|
| Clinical | SSRIs, TCAs, other antidepressants | ↓ 8-OHdG levels in antidepressant users | Black et al. [170] |
| Sertraline, fluoxetine, citalopram, and escitalopram | ↑ 8-OHdG levels in nonrespondent patients | Lindqvist et al. [80] | |
| Sertraline (25–100 mg) | ↓ O2·- and ·OH production; ↑ TRAP | Chang et al. [171] | |
| Escitalopram (20 mg) | ↓ SOD, CAT, MDA, and NO levels | Cimen et al. [172] | |
| Venlafaxine, paroxetine, escitalopram, sertraline, citalopram, milnacipran, fluoxetine, tianeptine, and moclobemide | ↓ SOD activity in red blood cells (24 weeks of treatment) (no difference exhibited after 6 and 12 weeks) | Kotan et al. [173] | |
| Fluoxetine (20 mg) | ↓ MDA levels; ↓ SOD1, CAT, and GSHP-x activities; ↑ TAS | Galecki et al. [102] | |
| Fluoxetine (20 mg) | ↑ ADA and SOD activities; ↓ NO and XO levels (no break down by medication group, reviewed meditations overall) | Herken et al. [174] | |
| Fluvoxamine (150 mg) | |||
| Sertraline (50 mg) | |||
| Citalopram (20 mg) | |||
| Fluoxetine and citalopram (20 mg) | ↓ MDA levels; ↓ SOD activity; ↑ ascorbic acid levels | Khanzode et al. [175] | |
| Citalopram and fluoxetine (20 mg) | ↓ MDA levels; ↓ GR, GPx, and SOD activities | Bilici et al. [176] | |
| Sertraline (50 mg) | |||
| Fluvoxamine (100 mg) | |||
|
| |||
| Preclinical animal model | Escitalopram (5 mg/kg) | ↓ ON in serum | Gammoh et al. [177] |
| Ketamine (5 mg/kg) | Ketamine induced antioxidant or proantioxidant effects dependent on antidepressant classes or brain area | Réus et al. [178] | |
| Fluoxetine (1.25 mg/kg) | |||
| Lamotrigine (5 mg/kg) | |||
| Quetiapine (5 mg/kg) | |||
| Escitalopram (5 mg/kg) | ↑ GSH in the brain | Matchkov et al. [179] | |
| Venlafaxine (5, 10, or 20 mg/kg) | ↓ MDA and NO plus ↑ GSH and TAC levels and GST activity in the hippocampus; ↓ 8-OHdG levels in serum and the hippocampus | Abdel-Wahab and Salama [180] | |
| Fluoxetine (5 mg/kg) | Restored SOD, CAT, and GSH levels in peripheral blood leucocytes | Novío et al. [181] | |
| Imipramine (10, 20, or 30 mg/kg) | ↑ SOD and CAT activities; ↓ MDA and carbonyl levels in the prefrontal cortex and hippocampus | Réus et al. [182] | |
| Fluoxetine (20 mg/kg) | Restored SOD, CAT, GST, and GR activities; ↑ GSH; ↓ MDA and carbonyl levels in the brain | Zafir et al. [183] | |
| Imipramine (10 mg/kg) | |||
| Venlafaxine (10 mg/kg) | |||
| Sertraline (5 or 10 mg/kg) | ↑ Glutathione levels in the brain | P. Kumar and A. Kumar [184] | |
|
| |||
| Preclinical cell culture | Clomipramine (15 μM) | ↓ NO production through attenuation of iNOS expression | Hwang et al. [185] |
| Imipramine (10 μM) | |||
| Desipramine (10−5, 10−6, or 10−7 M) | ↓ mRNA levels of SOD and CAT after treatment (2.5 h) ↑ mRNA levels of SOD, GST, and GR after treatment (24 h) |
Schmidt et al. [186] | |
| Imipramine (10−5, 10−6, or 10−7 M) | |||
| Maprotiline (10−5, 10−6, or 10−7 M) | |||
| Mirtazapine (10−5, 10−6, or 10−7 M) | |||
| Fluvoxamine (10 μM) | Inhibition of NO production | Hashioka et al. [187] | |
| Imipramine (50 μM) | |||
| Reboxetine (10 μM) | |||
| Amitriptyline (50 or 100 μmol/L) | Both agents attenuated cell death induced by H2O2; fluoxetine pretreatment ↑ SOD activity | Kolla et al. [188] | |
| Fluoxetine (50 μmol/L) | |||
8-OHdG: 8-hydroxydeguanosine; ADA: adenosine deaminase; CAT: catalase; GPx: glutathione peroxidase; GR: glutathione reductase; GSH: glutathione; GST: glutathione S-transferase; H2O2: hydrogen peroxide; iNOS: nitric oxide synthase; MDA: malondialdehyde; NO: nitric oxide; PON: paraoxonase; SOD: superoxide dismutase; SOD1: copper-zinc superoxide dismutase; SSRI: selective serotonin reuptake inhibitor; TAC: total antioxidant capacity; TAS: total antioxidant status; TBARS: thiobarbituric acid reactive species; TCA: tricyclic or tetracyclic antidepressant; XO: xanthine oxidase; TRAP: radical-trapping antioxidant parameter.
In general, antidepressants decrease oxidative damage markers in responsive patients, without altering this damage in nonresponsive patients (Table 2). However, there is no consensus about enzyme antioxidant defenses, as expected. Some studies found increased activity of antioxidant enzymes, while others observed a decrease. Most of these studies measured activity and nonexpression of enzymes, and this activity varies constantly and is not a static measure. Changes in enzyme activity are required to maintain or attempt to maintain cellular redox balance and occur in a matter of milliseconds. Studies with medicated depressive patients used different models, doses, and treatment times, which makes it difficult to compare their results. In addition, it remains unknown whether the effect of antidepressants on redox metabolism is direct or indirect or even a result of only improving the depressive state. Indirect effects can be mediated by different proteins, transcription factors, or anti-inflammatory effects. Thus, identifying the mechanisms responsible for the effects of antidepressants on redox metabolism is extremely important, because this is closely associated with the presence of symptoms in depressive patients.
Although many studies indicate increased lipid peroxidation in MDD, the first study was recently published that evaluated this parameter in TRD patients. Sowa-Kucma et al. found that TRD was accompanied by high levels of lipid peroxidation compared with non-TRD patients [189]. Another previous study investigated baseline OS markers (e.g., vitamin C and GPx) as predictors of antidepressant treatment response, finding negative results [190]. Other oxidative stress markers have been associated with poorer treatment response in MDD. Lindqvist et al. found higher baseline levels of F2-isoprostanes in nonresponders and a significant increase in 8-OHdG over the course of treatment in these patients [80].
Although antidepressant drugs have been shown to exert antioxidant effects in some study models (Table 2), it is unknown whether this is a direct or indirect effect of antidepressants. Together, lipid, protein, and DNA oxidative damage has been indicated as promising biomarkers for MDD patients under treatment; however, no clinical evidence for their use has yet been found. Nevertheless, there is evidence for the use of some antioxidants as a therapeutic approach for MDD. One promising candidate is N-acetylcysteine (NAC), a glutathione precursor that decreases inflammation and apoptosis, modulates levels of glutamate, promotes neurogenesis [191], and improves mitochondrial function [192]. These effects seem to be responsible for the remission of neurological symptoms in psychiatric diseases [193]. Berk et al. in a randomized carried 12-week study with a very expressive sampling (n = 269) reported antidepressant effects of NAC in patients with more severe depression (MADRS score of 25 or more) [194]. Furthermore, NAC exerts antioxidant effects in the anterior cingulate cortex of MDD patients in a multicenter RCT [195]. A recent study also described that chronic treatment with NAC improves depressive behavior and anxiety and spatial learning deficits as well as reverses pathological changes in the hippocampus in an animal model of neonatal depression [196].
Besides NAC, other antioxidants have been studied for their possible antidepressant effects. Recently, Abuelezz et al. reported that treatment with CoQ10 reversed depressive-like behavior, reduced lipid peroxidation, and restored GSSH and CAT levels in animals exposed to a chronic unpredictable mild stress (CUMS) protocol [197]. In addition, CoQ10 was able to recover the balance in kynurenine/serotonin levels by downregulating hippocampal IDO-1. Jahangard et al. found in a randomized study involving 89 patients with bipolar disorder, currently in a depressive episode, a significant increase in total thiol groups (TTG) and TAC, as well as a significant reduction in TNF-α, IL-10, and NO levels, after CoQ10 adjuvant therapy (200 mg/day) [198]. Corroborating with these findings, a decrease in CoQ10 levels was observed in TRD patients [153].
In turn, zinc (Zn) is an essential compound involved in several cellular processes and plays an important role against OS in the brain. Its neuroprotective properties include the blocking of excitotoxic Ca2+ influx and the upregulation of cellular antioxidant systems [199]. Clinical evidence points to an association of Zn deficiency and depressive behavior. According to the previous reports, Zn supplementation can improve mood in TRD patients [200]. Likewise, Zn administration increased BDNF expression and ameliorated the efficacy of antidepressant treatment, which can be relevant in the management of TRD [201, 202].
Curcumin, a potent anti-inflammatory plant metabolite, has been reported as a promising agent for the treatment of MDD [203–205]. Animal study models have already highlighted the potential of curcumin as an antidepressant [206, 207] with similar effects as fluoxetine and imipramine [208]. A meta-analysis that compared the use of curcumin with placebo demonstrated significant clinical efficacy of curcumin in improving depressive symptoms. Significant antianxiety effects were also reported [209]. Naqvi et al. demonstrated in a recent study using the CUMS protocol in rats that supplementation with curcumin (200 mg/kg) significantly attenuated the symptoms of depression and anxiety, reduced OS, and improved antioxidant status [210]. Curcumin also improved memory function and exhibited an inhibitory effect on acetylcholinesterase (AchE) activity. Similar results, demonstrated by Wang et al., showed that curcumin attenuated behavioral disorders associated with poststroke depression (PSD) in an animal model. These effects seem to be mediated by the control of Ca2+ levels in the CNS exhibited by curcumin [211].
Resveratrol (RES), another phenolic compound with recognized antioxidant activity, also has been studied as an antidepressant agent, but few studies have been conducted. Its beneficial effects on learning, memory, and anxiety [212] as well as on depressive-like behavior were reported [213]. Recently, in a study employing CUMS protocol, rats received RES (40 or 80 mg/kg/day) or fluoxetine (10 mg/kg/day) for four weeks. RES was able to reduce inflammation and apoptosis in the hippocampus and prefrontal cortex as well as expression of Akt/Akt and p-GSK3β/GSK3β proteins, achieving results similar to fluoxetine [214].
Considering all the evidence mentioned above, new investigations focusing on antidepressant effects of antioxidants or drugs with antioxidant activity should be performed, since the use of these compounds may emerge as a novel range of adjuvant therapy for resistant MDD management.
6. Limitations of the Current Research and Challenges Ahead
While this narrative review further supports that inflammation, mitochondrial dysfunction, and oxidative stress can be recognized as the central event in MDD, the question of why these events are still uncontrolled with conventional drug treatment remains unanswered. The few studies available on antidepressants and their clinical effects on inflammation/oxidation/mitochondrial dysfunction are often inconclusive or biased. Despite that, the combined evidence indicates that ATP depletion, oxidative stress, and inflammatory responses can activate apoptotic mechanisms that lead to neurodegeneration, a common feature in depressive patients. The data presented here demonstrate that MDD activates inflammation pathways through nuclear, cytosolic, and mitochondrial proteins, which can serve as a starting point for the screening of new drugs. However, the future challenge is to find effective new drugs that target inflammation and mitochondria, thus reducing cases of refractory patients. Finally, although the biochemistry mechanisms linked to the immune response in depression are gradually being elucidated, further research is needed to test these hypotheses in vivo.
Acknowledgments
Special thanks are due to L.A.B. for the technical assistance. This work was supported by the Coordenação de Apoio de Pessoal de Nível Superior (CAPES), the Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). A.P.V.V. is a fellowship of CAPES and M.S. of CNPq (88887.162616/2018-00 and 303383-2015-1, respectively).
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1.World Health Organization. Depression and other common mental disorders: global health estimates. World Health Organization; 2017. [Google Scholar]
- 2.Wharton R. N. In: Transtornos afetivos. Timothy A., editor. Merrit Tratado de Neurologia; 2011. [Google Scholar]
- 3.Holtzheimer P. E., Mayberg H. S. Stuck in a rut: rethinking depression and its treatment. Trends in Neurosciences. 2011;34(1):1–9. doi: 10.1016/j.tins.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Harbi K. S. treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Preference and Adherence. 2012;6:369–388. doi: 10.2147/PPA.S29716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson J. C., Papakostas G. I. Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials. American Journal of Psychiatry. 2009;166(9):980–991. doi: 10.1176/appi.ajp.2009.09030312. [DOI] [PubMed] [Google Scholar]
- 6.Fekadu A., Wooderson S. C., Markopoulo K., Donaldson C., Papadopoulos A., Cleare A. J. What happens to patients with treatment-resistant depression ? A systematic review of medium to long term outcome studies. Journal of Affective Disorders. 2009;116(1-2):4–11. doi: 10.1016/j.jad.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Fekadu A., Rane L. J., Wooderson S. C., Markopoulou K., Poon L., Cleare A. J. Prediction of longer-term outcome of treatment-resistant depression in tertiary care. The British Journal of Psychiatry. 2012;201(5):369–375. doi: 10.1192/bjp.bp.111.102665. [DOI] [PubMed] [Google Scholar]
- 8.Strawbridge R., Arnone D., Danese A., Papadopoulos A., Herane Vives A., Cleare A. J. Inflammation and clinical response to treatment in depression: a meta-analysis. European Neuropsychopharmacology. 2015;25(10):1532–1543. doi: 10.1016/j.euroneuro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Patel A. The role of inflammation in depression. Psychiatria Danubina. 2013;25(Supplement 2):216–223. [PubMed] [Google Scholar]
- 10.Vilela L. H. M., Juruena M. F. Avaliação do funcionamento do eixo HPA em deprimidos por meio de medidas basais: revisão sistemática da literatura e análise das metodologias utilizadas. Jornal Brasileiro de Psiquiatria. 2014;63(3):232–241. doi: 10.1590/0047-2085000000031. [DOI] [Google Scholar]
- 11.Felger J. C., Lotrich F. E. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013;246:199–229. doi: 10.1016/j.neuroscience.2013.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kronfol Z. Multiple sclerosis and depression. Archives of Neurology. 1985;42(4):310–310. doi: 10.1001/archneur.1985.04060040016004. [DOI] [PubMed] [Google Scholar]
- 13.Kronfol Z., Nasrallah H. A., Chapman S., House J. D. Depression, cortisol metabolism and lymphocytopenia. Journal of Affective Disorders. 1985;9(2):169–173. doi: 10.1016/0165-0327(85)90097-7. [DOI] [PubMed] [Google Scholar]
- 14.White J., Kivimäki M., Jokela M., Batty G. D. Association of inflammation with specific symptoms of depression in a general population of older people: The English Longitudinal Study of Ageing. Brain, Behavior, and Immunity. 2017;61:27–30. doi: 10.1016/j.bbi.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Blume J., Douglas S. D., Evans D. L. Immune suppression and immune activation in depression. Brain, Behavior, and Immunity. 2011;25(2):221–229. doi: 10.1016/j.bbi.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller A. H., Maletic V., Raison C. L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biological Psychiatry. 2009;65(9):732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon N. M., McNamara K., Chow C. W., et al. A detailed examination of cytokine abnormalities in major depressive disorder. European Neuropsychopharmacology. 2008;18(3):230–233. doi: 10.1016/j.euroneuro.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson G., Maes M. Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: treatment implications. Current Pharmaceutical Design. 2014;20(23):3812–3847. doi: 10.2174/13816128113196660738. [DOI] [PubMed] [Google Scholar]
- 19.Anderson G., Kubera M., Duda W., Lasoń W., Berk M., Maes M. Increased IL-6 trans-signaling in depression: focus on the tryptophan catabolite pathway, melatonin and neuroprogression. Pharmacological Reports. 2013;65(6):1647–1654. doi: 10.1016/S1734-1140(13)71526-3. [DOI] [PubMed] [Google Scholar]
- 20.Dowlati Y., Herrmann N., Swardfager W., et al. A meta-analysis of cytokines in major depression. Biological Psychiatry. 2010;67(5):446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Allen J., Romay-Tallon R., Brymer K. J., Caruncho H. J., Kalynchuk L. E. Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Frontiers in Neuroscience. 2018;12:p. 386. doi: 10.3389/fnins.2018.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner A., Boles R. G. Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(3):730–743. doi: 10.1016/j.pnpbp.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Alcocer-Gómez E., de Miguel M., Casas-Barquero N., et al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain, Behavior, and Immunity. 2014;36:111–117. doi: 10.1016/j.bbi.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Cohen S., Janicki-Deverts D., Miller G. E. Psychological stress and disease. JAMA. 2007;298(14):1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- 25.Hasler G., Drevets W. C., Manji H. K., Charney D. S. Discovering endophenotypes for major depression. Neuropsychopharmacology. 2004;29(10):1765–1781. doi: 10.1038/sj.npp.1300506. [DOI] [PubMed] [Google Scholar]
- 26.Ising M., Horstmann S., Kloiber S., et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression–a potential biomarker? Biological Psychiatry. 2007;62(1):47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 27.Pariante C. M., Lightman S. L. The HPA axis in major depression: classical theories and new developments. Trends in Neurosciences. 2008;31(9):464–468. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Pariante C. M., Miller A. H. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biological Psychiatry. 2001;49(5):391–404. doi: 10.1016/S0006-3223(00)01088-X. [DOI] [PubMed] [Google Scholar]
- 29.Cohen S., Janicki-Deverts D., Doyle W. J., et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(16):5995–5999. doi: 10.1073/pnas.1118355109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer M. E., Papadopoulos A., Poon L., et al. Dexamethasone-induced effects on lymphocyte distribution and expression of adhesion molecules in treatment-resistant depression. Psychiatry Research. 2002;113(1-2):1–15. doi: 10.1016/S0165-1781(02)00243-3. [DOI] [PubMed] [Google Scholar]
- 31.Horowitz M. A., Zunszain P. A. Neuroimmune and neuroendocrine abnormalities in depression: two sides of the same coin. Annals of the New York Academy of Sciences. 2015;1351(1):68–79. doi: 10.1111/nyas.12781. [DOI] [PubMed] [Google Scholar]
- 32.Pace T. W. W., Hu F., Miller A. H. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain, Behavior, and Immunity. 2007;21(1):9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anacker C., Zunszain P. A., Cattaneo A., et al. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Molecular Psychiatry. 2011;16(7):738–750. doi: 10.1038/mp.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anacker C., Cattaneo A., Musaelyan K., et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(21):8708–8713. doi: 10.1073/pnas.1300886110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anacker C., Cattaneo A., Luoni A., et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology. 2013;38(5):872–883. doi: 10.1038/npp.2012.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaki N. F. W., Spence D. W., BaHammam A. S., Pandi-Perumal S. R., Cardinali D. P., Brown G. M. Chronobiological theories of mood disorder. European Archives of Psychiatry and Clinical Neuroscience. 2018;268(2):107–118. doi: 10.1007/s00406-017-0835-5. [DOI] [PubMed] [Google Scholar]
- 37.Zaki N. F., Yousif M., BaHammam A. S., et al. Chronotherapeutics: Recognizing the importance of timing factors in the treatment of disease and sleep disorders. Clinical Neuropharmacology. 2019;42(3):80–87. doi: 10.1097/wnf.0000000000000341. [DOI] [PubMed] [Google Scholar]
- 38.Fındıklı E., Fatih Inci M., Gökçe M., Avni Fındıklı H., Altun H., Fatih Karaaslan M. Pineal gland volume in schizophrenia and mood disorders. Psychiatria Danubina. 2015;27(2) [PubMed] [Google Scholar]
- 39.Manchester L. C., Coto-Montes A., Boga J. A., et al. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. Journal of Pineal Research. 2015;59(4):403–419. doi: 10.1111/jpi.12267. [DOI] [PubMed] [Google Scholar]
- 40.Kashihara K., Nomura T., Maeda T., et al. Beneficial effects of ramelteon on rapid eye movement sleep behavior disorder associated with Parkinson’s disease - results of a multicenter open trial. Internal Medicine. 2016;55(3):231–236. doi: 10.2169/internalmedicine.55.5464. [DOI] [PubMed] [Google Scholar]
- 41.Satyanarayanan S. K., Su H., Lin Y. W., Su K. P. Circadian rhythm and melatonin in the treatment of depression. Current Pharmaceutical Design. 2018;24(22):2549–2555. doi: 10.2174/1381612824666180803112304. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 43.Dantzer R., O'Connor J. C., Freund G. G., Johnson R. W., Kelley K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nature Reviews Neuroscience. 2008;9(1):46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oxenkrug G. F. Genetic and hormonal regulation of tryptophan–kynurenine metabolism. Annals of the New York Academy of Sciences. 2007;1122(1):35–49. doi: 10.1196/annals.1403.003. [DOI] [PubMed] [Google Scholar]
- 45.Miller A. H., Raison C. L. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nature Reviews Immunology. 2016;16(1):22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinon F. Dangerous Liaisons: Mitochondrial DNA Meets the NLRP3 Inflammasome. Immunity. 2012;36(3):313–315. doi: 10.1016/j.immuni.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Chen H.-S. V., Lipton S. A. The chemical biology of clinically tolerated NMDA receptor antagonists. Journal of Neurochemistry. 2006;97(6):1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 48.Steiner J., Walter M., Gos T., et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? Journal of Neuroinflammation. 2011;8(1):p. 94. doi: 10.1186/1742-2094-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erhardt S., Lim C. K., Linderholm K. R., et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology. 2013;38(5):743–752. doi: 10.1038/npp.2012.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bansal Y., Singh R., Parhar I., Kuhad A., Soga T. Quinolinic acid and nuclear factor erythroid 2-related factor 2 in depression: role in neuroprogression. Frontiers in Pharmacology. 2019;10:p. 452. doi: 10.3389/fphar.2019.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamata M., Higuchi H., Yoshimoto M., Yoshida K., Shimizu T. Effect of single intracerebroventricular injection of α-interferon on monoamine concentrations in the rat brain. European Neuropsychopharmacology. 2000;10(2):129–132. doi: 10.1016/s0924-977x(99)00067-x. [DOI] [PubMed] [Google Scholar]
- 52.Torres-Platas S. G., Cruceanu C., Chen G. G., Turecki G., Mechawar N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain, Behavior, and Immunity. 2014;42, article S0889159114001299:50–59. doi: 10.1016/j.bbi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Steiner J., Bielau H., Brisch R., et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. Journal of Psychiatric Research. 2008;42(2):151–157. doi: 10.1016/j.jpsychires.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 54.Setiawan E., Wilson A. A., Mizrahi R., et al. Role of translocator protein Density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72(3):268–275. doi: 10.1001/jamapsychiatry.2014.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shelton R. C., Claiborne J., Sidoryk-Wegrzynowicz M., et al. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Molecular Psychiatry. 2011;16(7):751–762. doi: 10.1038/mp.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim Y.-K., Na K.-S., Myint A.-M., Leonard B. E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2016;64:277–284. doi: 10.1016/j.pnpbp.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Abdallah C. G., Averill L. A., Krystal J. H., Southwick S. M., Arnsten A. F. T. Glutamate and norepinephrine interaction: relevance to higher cognitive operations and psychopathology. The Behavioral and Brain Sciences. 2016;39, article e201 doi: 10.1017/S0140525X15001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Radley J. J., Sisti H. M., Hao J., et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125(1):1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Radley J. J., Rocher A. B., Rodriguez A., et al. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. Journal of Comparative Neurology. 2008;507(1):1141–1150. doi: 10.1002/cne.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wellman C. L. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. Journal of Neurobiology. 2001;49(3):245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- 61.Ressler K. J., Mayberg H. S. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nature Neuroscience. 2007;10(9):1116–1124. doi: 10.1038/nn1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price J. L., Drevets W. C. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber M. D., Frank M. G., Tracey K. J., Watkins L. R., Maier S. F. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats : a priming stimulus of microglia and the NLRP3 inflammasome. Journal of Neuroscience. 2015;35(1):316–324. doi: 10.1523/JNEUROSCI.3561-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan Y., Chen X. Y., Zhang Q. Y., Kong L. D. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain, Behavior, and Immunity. 2014;41:90–100. doi: 10.1016/j.bbi.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 65.Ramirez K., Sheridan J. F. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain, Behavior, and Immunity. 2016;57:293–303. doi: 10.1016/j.bbi.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tynan R. J., Weidenhofer J., Hinwood M., Cairns M. J., Day T. A., Walker F. R. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain, behavior, and Immunity. 2012;26(3):469–479. doi: 10.1016/j.bbi.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 67.Roman M., Irwin M. R. Novel neuroimmunologic therapeutics in depression: a clinical perspective on what we know so far. Brain, behavior, and immunity. 2020;83:7–21. doi: 10.1016/j.bbi.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins G. S., Mallett S., Omar O., Yu L. M. Developing risk prediction models for type 2 diabetes : a systematic review of methodology and reporting. BMC Medicine. 2011;9(1) doi: 10.1186/1741-7015-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leuchter A. F., Cook I. A., Hamilton S. P., et al. Biomarkers to predict antidepressant response. Current Psychiatry Reports. 2010;12(6):553–562. doi: 10.1007/s11920-010-0160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bock C., Bukh J. D., Vinberg M., Gether U., Kessing L. V. The influence of comorbid personality disorder and neuroticism on treatment Outcome in First Episode Depression. Psychopathology. 2010;43(3):197–204. doi: 10.1159/000304176. [DOI] [PubMed] [Google Scholar]
- 71.Corrêa H., Duval F., Claude M. M., et al. Noradrenergic dysfunction and antidepressant treatment response. European Neuropsychopharmacology. 2001;11(2):163–168. doi: 10.1016/S0924-977X(01)00079-7. [DOI] [PubMed] [Google Scholar]
- 72.Rush A. J., Siefert S. E. Clinical issues in considering vagus nerve stimulation for treatment-resistant depression. Experimental Neurology. 2009;219(1):36–43. doi: 10.1016/j.expneurol.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 73.Iosifescu D. V., Bankier B., Fava M. Impact of medical comorbid disease on antidepressant treatment of major depressive disorder. Current Psychiatry Reports. 2004;6(3):193–201. doi: 10.1007/s11920-004-0064-2. [DOI] [PubMed] [Google Scholar]
- 74.Couzin-Frankel J. Inflammation bares a dark side. Science. 2010;330(6011):p. 1621. doi: 10.1126/science.330.6011.1621. [DOI] [PubMed] [Google Scholar]
- 75.Carvalho A. F., Berk M., Hyphantis T. N., McIntyre R. S. The integrative management of treatment-resistant depression: a comprehensive review and perspectives. Psychotherapy and Psychosomatics. 2014;83(2):70–88. doi: 10.1159/000357500. [DOI] [PubMed] [Google Scholar]
- 76.Rosenblat J. D., Cha D. S., Mansur R. B., McIntyre R. S. Inflamed moods: a review of the interactions between inflammation and mood disorders. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014;53:23–34. doi: 10.1016/j.pnpbp.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 77.McIntyre R. S., Cha D. S., Soczynska J. K., et al. Cognitive deficits and functional outcomes in major depressive disorder: determinants, substrates, and treatment interventions. Depression and Anxiety. 2013;30(6):515–527. doi: 10.1002/da.22063. [DOI] [PubMed] [Google Scholar]
- 78.Hashimoto K. Inflammatory biomarkers as differential predictors of antidepressant response. International Journal of Molecular Sciences. 2015;16(12):7796–7801. doi: 10.3390/ijms16047796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hashimoto K. Role of the mTOR signaling pathway in the rapid antidepressant action of ketamine. Expert Review of Neurotherapeutics. 2011;11(1):33–36. doi: 10.1586/ern.10.176. [DOI] [PubMed] [Google Scholar]
- 80.Lindqvist D., Dhabhar F. S., James S. J., et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology. 2017;76:197–205. doi: 10.1016/j.psyneuen.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eller T., Vasar V., Shlik J., Maron E. Pro-inflammatory cytokines and treatment response to escitaloprsam in major depressive disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2008;32(2):445–450. doi: 10.1016/j.pnpbp.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 82.Haroon E., Chen X., Li Z., et al. Increased inflammation and brain glutamate define a subtype of depression with decreased regional homogeneity, impaired network integrity, and anhedonia. Translational Psychiatry. 2018;8(1):p. 189. doi: 10.1038/s41398-018-0241-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Research Reviews. 2009;61(2):105–123. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 84.Machado-Vieira R., Manji H. K., Zarate C. A. The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. The Neuroscientist. 2009;15(5):525–539. doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanacora G., Treccani G., Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62(1):63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walker A. K., Budac D. P., Bisulco S., et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology. 2013;38(9):1609–1616. doi: 10.1038/npp.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O'Connor J. C., Lawson M. A., André C., et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Molecular psychiatry. 2009;14(5):511–522. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saito K., Markey S. P., Heyes M. P. Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience. 1992;51(1):25–39. doi: 10.1016/0306-4522(92)90467-G. [DOI] [PubMed] [Google Scholar]
- 89.Gao M., Rejaei D., Liu H. Ketamine use in current clinical practice. Acta Pharmacologica Sinica. 2016;37(7):865–872. doi: 10.1038/aps.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ekdahl C. T. Microglial activation – tuning and pruning adult neurogenesis. Frontiers in Pharmacology. 2012;3(41) doi: 10.3389/fphar.2012.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kappelmann N., Lewis G., Dantzer R., Jones P. B., Khandaker G. M. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Molecular Psychiatry. 2018;23(2):335–343. doi: 10.1038/mp.2016.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Raison C. L., Rutherford R. E., Woolwine B. J., et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression. JAMA Psychiatry. 2013;70(1):31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harden J. L., Lewis S. M., Lish S. R., et al. The tryptophan metabolism enzyme L-kynureninase is a novel inflammatory factor in psoriasis and other inflammatory diseases. Journal of Allergy and Clinical Immunology. 2016;137(6):1830–1840. doi: 10.1016/j.jaci.2015.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maes M. Targeting cyclooxygenase-2 in depression is not a viable therapeutic approach and may even aggravate the pathophysiology underpinning depression. Metabolic Brain Disease. 2012;27(4):405–413. doi: 10.1007/s11011-012-9326-6. [DOI] [PubMed] [Google Scholar]
- 95.Choi S. K., Park Y. S., Choi D. K., Chang H. I. Effects of astaxanthin on the production of NO and the expression of COX-2 and iNOS in LPS-stimulated BV2 microglial cells. Journal Microbiollogy Biotechnology. 2008;18(12):1990–1996. [PubMed] [Google Scholar]
- 96.Guo J.-Y., Li C.-Y., Ruan Y.-P., et al. Chronic treatment with celecoxib reverses chronic unpredictable stress-induced depressive-like behavior via reducing cyclooxygenase-2 expression in rat brain. European Journal of Pharmacology. 2009;612(1-3):54–60. doi: 10.1016/j.ejphar.2009.03.076. [DOI] [PubMed] [Google Scholar]
- 97.Myint A.-M., Kim Y. K., Verkerk R., Scharpé S., Steinbusch H., Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. Journal of Affective Disorders. 2007;98(1-2):143–151. doi: 10.1016/j.jad.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 98.Aid S., Langenbach R., Bosetti F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. Journal of Neuroinflammation. 2008;5(1):p. 17. doi: 10.1186/1742-2094-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vane J. R., Botting R. M. The mechanism of action of aspirin. Thrombosis Research. 2003;110(5-6):255–258. doi: 10.1016/S0049-3848(03)00379-7. [DOI] [PubMed] [Google Scholar]
- 100.Berk M., Dean O., Drexhage H., et al. Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Medicine. 2013;11(1) doi: 10.1186/1741-7015-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mendlewicz J., Kriwin P., Oswald P., Souery D., Alboni S., Brunello N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. International Clinical Psychopharmacology. 2006;21(4):227–231. doi: 10.1097/00004850-200607000-00005. [DOI] [PubMed] [Google Scholar]
- 102.Gałecki P., Szemraj J., Bieńkiewicz M., Florkowski A., Gałecka E. Lipid peroxidation and antioxidant protection in patients during acute depressive episodes and in remission after fluoxetine treatment. Pharmacological Reports. 2009;61(3):436–447. doi: 10.1016/S1734-1140(09)70084-2. [DOI] [PubMed] [Google Scholar]
- 103.Salvadore G., Singh J. B. Ketamine as a fast acting antidepressant: current knowledge and open questions. CNS Neuroscience & Therapeutics. 2013;19(6):428–436. doi: 10.1111/cns.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wohleb E. S., Gerhard D., Thomas A., Duman R. S. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Current Neuropharmacology. 2017;15(1):11–20. doi: 10.2174/1570159x14666160309114549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lepack A. E., Fuchikami M., Dwyer J. M., Banasr M., Duman R. S. BDNF release is required for the behavioral actions of ketamine. International Journal of Neuropsychopharmacology. 2015;18(1) doi: 10.1093/ijnp/pyu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Abelaira H. M., Réus G. Z., Ignácio Z. M., et al. Effects of ketamine administration on mTOR and reticulum stress signaling pathways in the brain after the infusion of rapamycin into prefrontal cortex. Journal of Psychiatric Research. 2017;87:81–87. doi: 10.1016/j.jpsychires.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 107.McGirr A., Berlim M. T., Bond D. J., Fleck M. P., Yatham L. N., Lam R. W. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychological Medicine. 2015;45(4):693–704. doi: 10.1017/S0033291714001603. [DOI] [PubMed] [Google Scholar]
- 108.Zarate C. A., Singh J. B., Carlson P. J., et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 109.Diazgranados N., Ibrahim L., Brutsche N. E., et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Archives of General Psychiatry. 2010;67(8):793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ibrahim L., Diazgranados N., Luckenbaugh D. A., et al. Rapid decrease in depressive symptoms with an N-methyl-d-aspartate antagonist in ECT-resistant major depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(4):1155–1159. doi: 10.1016/j.pnpbp.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.DiazGranados N., Ibrahim L. A., Brutsche N. E., et al. Rapid resolution of suicidal ideation after a single infusion of anN-Methyl-D-Aspartate antagonist in patients With Treatment-Resistant Major Depressive Disorder. The Journal of Clinical Psychiatry. 2010;71(12):1605–1611. doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Price R. B., Nock M. K., Charney D. S., Mathew S. J. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biological Psychiatry. 2009;66(5):522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luckenbaugh D. A., Niciu M. J., Ionescu D. F., et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? Journal of Affective Disorders. 2014;159:56–61. doi: 10.1016/j.jad.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aleksandrova L. R., Phillips A. G., Wang Y. T. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. Journal of Psychiatry and Neuroscience. 2017;42(4):222–229. doi: 10.1503/jpn.160175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Bernardis D., Omasetti C., Pompili M., et al. ‘That’s the esketamine, baby, there’s nothing you can do about it!’ An update on glutamatergic system in suicidal depression. Current Topics in Medicinal Chemistry. 2020 doi: 10.2174/1568026620666200131100316. [DOI] [PubMed] [Google Scholar]
- 116.Chen A. T., Nasrallah H. A. Neuroprotective effects of the second generation antipsychotics. Schizophrenia Research. 2019;208:1–7. doi: 10.1016/j.schres.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 117.Thomas K., Saadabadi A. Olanzapine. Stat Pearls Publishing; 2020. [PubMed] [Google Scholar]
- 118.Pardo J. V., Sheikh S. A., Schwindt G., et al. A preliminary study of resting brain metabolism in treatment-resistant depression before and after treatment with olanzapine-fluoxetine combination. PLoS One. 2020;15(1) doi: 10.1371/journal.pone.0226486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shimada K., Crother T. R., Karlin J., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Culmsee C., Michels S., Scheu S., Arolt V., Dannlowski U., Alferink J. Mitochondria, microglia, and the immune system—how are they linked in affective disorders? Frontiers in Psychiatry. 2019;9 doi: 10.3389/fpsyt.2018.00739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yaron J. R., Gangaraju S., Rao M. Y., et al. K+ regulates Ca2+ to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death & Disease. 2015;6(10) doi: 10.1038/cddis.2015.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cunnane S., Nugent S., Roy M., et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. 2011;27(1):3–20. doi: 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xing Y., Zhang X., Song X., Lv Z., Hou L., Li F. Injury of cortical neurons is caused by the advanced glycation end products-mediated pathway. Neural Regeneration Research. 2013;8(10):909–915. doi: 10.3969/j.issn.1673-5374.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barreiros A. L. B. S., David J. M., David J. P. Estresse oxidativo: relação entre geração de espécies reativas e defesa do organismo. Química Nova. 2006;29(1):113–123. doi: 10.1590/S0100-40422006000100021. [DOI] [Google Scholar]
- 125.Anamika, Khanna A., Acharjee P., Acharjee A., Trigun S. K. Mitochondrial SIRT3 and neurodegenerative brain disorders. Journal of chemical neuroanatomy. 2019;95:43–53. doi: 10.1016/j.jchemneu.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 126.Bause A. S., Haigis M. C. SIRT3 regulation of mitochondrial oxidative stress. Experimental Gerontology. 2013;48(7):634–639. doi: 10.1016/j.exger.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 127.Verdin E., Hirschey M. D., Finley L. W. S., Haigis M. C. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in Biochemical Sciences. 2010;35(12):669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nicholls D. G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2009;1787(11):1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen Y., Fu L. L., Wen X., et al. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Disease. 2014;5(2):p. e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Qiu X., Brown K., Hirschey M. D., Verdin E., Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metabolism. 2010;12(6):662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 131.Jing E., O'Neill B. T., Rardin M. J., et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62(10):3404–3417. doi: 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ahn B. H., Kim H. S., Song S., et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lombard D. B., Tishkoff D. X., Bao J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Histone Deacetylases: the Biology and Clinical Implication. 2011;206:163–188. doi: 10.1007/978-3-642-21631-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Law I. K. M., Liu L., Xu A., et al. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the antiaging and metabolic effects of sirtuins. Proteomics. 2009;9(9):2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- 135.Duan W. Sirtuins: from metabolic regulation to brain aging. Frontiers in Aging Neuroscience. 2013;5 doi: 10.3389/fnagi.2013.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xu Y., Wang C., Klabnik J., O' Donnell J. Novel therapeutic targets in depression and anxiety: antioxidants as a candidate treatment. Current Neuropharmacology. 2014;12(2):108–119. doi: 10.2174/1570159X11666131120231448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Castellano-Gonzalez G., Jacobs K. R., Don E., et al. Kynurenine 3-monooxygenase activity in human primary neurons and effect on cellular bioenergetics identifies new neurotoxic mechanisms. Neurotoxicity Research. 2019;35(3):530–541. doi: 10.1007/s12640-019-9997-4. [DOI] [PubMed] [Google Scholar]
- 138.Liu T., Zhong S., Liao X., et al. A meta-analysis of oxidative stress markers in depression. PloS one. 2015;10(10) doi: 10.1371/journal.pone.0138904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Islam M. R., Islam M. R., Ahmed I., et al. Elevated serum levels of malondialdehyde and cortisol are associated with major depressive disorder: a case-control study. SAGE Open Medicine. 2018;6 doi: 10.1177/2050312118773953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Diniz B. S., Mendes-Silva A. P., Silva L. B., et al. Oxidative stress markers imbalance in late-life depression. Journal of Psychiatric Research. 2018;102:29–33. doi: 10.1016/j.jpsychires.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 141.Freed R. D., Hollenhorst C. N., Weiduschat N., et al. A pilot study of cortical glutathione in youth with depression. Psychiatry Research: Neuroimaging. 2017;270, article S0925492717300987:54–60. doi: 10.1016/j.pscychresns.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Camkurt M. A., Fındıklı E., İzci F., Kurutaş E. B., Tuman T. C. Evaluation of malondialdehyde, superoxide dismutase and catalase activity and their diagnostic value in drug naïve, first episode, non-smoker major depression patients and healthy controls. Psychiatry Research. 2016;238:81–85. doi: 10.1016/j.psychres.2016.01.075. [DOI] [PubMed] [Google Scholar]
- 143.Tsai M.-C., Huang T.-L. Increased activities of both superoxide dismutase and catalase were indicators of acute depressive episodes in patients with major depressive disorder. Psychiatry Research. 2016;235:38–42. doi: 10.1016/j.psychres.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 144.Black C. N., Bot M., Scheffer P. G., Cuijpers P., Penninx B. W. J. H. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology. 2015;51:164–175. doi: 10.1016/j.psyneuen.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 145.Kaufmann F. N., Gazal M., Mondin T. C., et al. Cognitive psychotherapy treatment decreases peripheral oxidative stress parameters associated with major depression disorder. Biological Psychology. 2015;110:175–181. doi: 10.1016/j.biopsycho.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 146.Bajpai A., Verma A. K., Srivastava M., Srivastava R. Oxidative stress and major depression. Journal of Clinical and Diagnostic Research: JCDR. 2014;8(12):CC04–CC07. doi: 10.7860/JCDR/2014/10258.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gariballa S. Poor vitamin C status is associated with increased depression symptoms following acute illness in older people. International Journal for Vitamin and Nutrition Research. 2014;84(1-2):12–17. doi: 10.1024/0300-9831/a000188. [DOI] [PubMed] [Google Scholar]
- 148.Rybka J., Kędziora-Kornatowska K., Banaś-Leżańska P., et al. Interplay between the pro-oxidant and antioxidant systems and proinflammatory cytokine levels, in relation to iron metabolism and the erythron in depression. Free Radical Biology and Medicine. 2013;63:187–194. doi: 10.1016/j.freeradbiomed.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 149.Magalhães P. V. S., Jansen K., Pinheiro R. T., et al. Peripheral oxidative damage in early-stage mood disorders: a nested population-based case-control study. International Journal of Neuropsychopharmacology. 2012;15(8):1043–1050. doi: 10.1017/S1461145711001532. [DOI] [PubMed] [Google Scholar]
- 150.Talarowska M., Gałecki P., Maes M., et al. Nitric oxide plasma concentration associated with cognitive impairment in patients with recurrent depressive disorder. Neuroscience Letters. 2012;510(2):127–131. doi: 10.1016/j.neulet.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 151.Kodydková J., Vávrová L., Zeman M., et al. Antioxidative enzymes and increased oxidative stress in depressive women. Clinical biochemistry. 2009;42(13-14):1368–1374. doi: 10.1016/j.clinbiochem.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 152.Kupper N., Gidron Y., Winter J., Denollet J. Association between type d personality, depression, and oxidative stress in patients with chronic heart failure. Psychosomatic Medicine. 2009;71(9):973–980. doi: 10.1097/PSY.0b013e3181bee6dc. [DOI] [PubMed] [Google Scholar]
- 153.Maes M., Mihaylova I., Kubera M., Uytterhoeven M., Vrydags N., Bosmans E. Lower plasma coenzyme Q10 in depression: a marker for treatment resistance and chronic fatigue in depression and a risk factor to cardiovascular disorder in that illness. Neuroendocrinology Letters. 2009;30(4):462–469. [PubMed] [Google Scholar]
- 154.Lopresti A. L., Maker G. L., Hood S. D., Drummond P. D. A review of peripheral biomarkers in major depression: the potential of inflammatory and oxidative stress biomarkers. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014;48:102–111. doi: 10.1016/j.pnpbp.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 155.Gu F., Chauhan V., Chauhan A. Glutathione redox imbalance in brain disorders. Current Opinion in Clinical Nutrition and Metabolic Care. 2015;18(1):89–95. doi: 10.1097/MCO.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 156.Maes M., Galecki P., Chang Y. S., Berk M. A review on the oxidative and nitrosative stress (O&NS;) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(3):676–692. doi: 10.1016/j.pnpbp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 157.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biology. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mellon S. H., Wolkowitz O. M., Schonemann M. D., et al. Alterations in leukocyte transcriptional control pathway activity associated with major depressive disorder and antidepressant treatment. Translational Psychiatry. 2016;6(5) doi: 10.1038/tp.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bouvier E., Brouillard F., Molet J., et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Molecular Psychiatry. 2017;22(12):1701–1713. doi: 10.1038/mp.2016.144. [DOI] [PubMed] [Google Scholar]
- 160.Lanctot K. L., Mazereeuw G., Herrmann N., Andreazza A., Khan M. A meta-analysis of lipid peroxidation markers in major depression. Neuropsychiatric Disease and Treatment. 2015;11:2479–2491. doi: 10.2147/NDT.S89922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Talarowska M., Gałecki P., Maes M., et al. Malondialdehyde plasma concentration correlates with declarative and working memory in patients with recurrent depressive disorder. Molecular Biology Reports. 2012;39(5):5359–5366. doi: 10.1007/s11033-011-1335-8. [DOI] [PubMed] [Google Scholar]
- 162.Wiener C., Rassier G. T., Kaster M. P., et al. Gender-based differences in oxidative stress parameters do not underlie the differences in mood disorders susceptibility between sexes. European Psychiatry. 2014;29(1):58–63. doi: 10.1016/j.eurpsy.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 163.Maurya P. K., Noto C., Rizzo L. B., et al. The role of oxidative and nitrosative stress in accelerated aging and major depressive disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2016;65:134–144. doi: 10.1016/j.pnpbp.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 164.Vieta E., Popovic D., Rosa A. R., et al. The clinical implications of cognitive impairment and allostatic load in bipolar disorder. European Psychiatry. 2013;28(1):21–29. doi: 10.1016/j.eurpsy.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 165.John A., Patel U., Rusted J., Richards M., Gaysina D. Affective problems and decline in cognitive state in older adults: a systematic review and meta-analysis. Psychological Medicine. 2019;49(3):353–365. doi: 10.1017/S0033291718001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ishii T., Takanashi Y., Sugita K., et al. Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: a new model for aging brain. Aging Cell. 2017;16(1):39–51. doi: 10.1111/acel.12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Michel T. M., Pulschen D., Thome J. The role of oxidative stress in depressive disorders. Current Pharmaceutical Design. 2012;18(36):5890–5899. doi: 10.2174/138161212803523554. [DOI] [PubMed] [Google Scholar]
- 168.Sheline Y. I., Disabato B. M., Hranilovich J., et al. Treatment course with antidepressant therapy in late-life depression. American Journal of Psychiatry. 2012;169(11):1185–1193. doi: 10.1176/appi.ajp.2012.12010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lindqvist D., Mueller S., Mellon S. H., et al. Peripheral antioxidant markers are associated with total hippocampal and CA3/dentate gyrus volume in MDD and healthy controls–preliminary findings. Psychiatry Research Neuroimaging. 2014;224(3):168–174. doi: 10.1016/j.pscychresns.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Black C. N., Bot M., Scheffer P. G., Penninx B. W. J. H. Oxidative stress in major depressive and anxiety disorders , and the association with antidepressant use; results from a large adult cohort. Psychological Medicine. 2017;47(5):936–948. doi: 10.1017/S0033291716002828. [DOI] [PubMed] [Google Scholar]
- 171.Chang C.-C., Lee C.-T., Lan T.-H., Ju P.-C., Hsieh Y.-H., Lai T.-J. Effects of antidepressant treatment on total antioxidant capacity and free radical levels in patients with major depressive disorder. Psychiatry Research. 2015;230(2):575–580. doi: 10.1016/j.psychres.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 172.Cimen B., Gumus C. B., Cetin I., Ozsoy S., Aydin M., Cimen L. The effects of escitalopram treatment on oxidative/antioxidative parameters in patients with depression. Klinik Psikofarmakoloji Bülteni-Bulletin of Clinical Psychopharmacology. 2015;25(3):272–279. doi: 10.5455/bcp.20150215102247. [DOI] [Google Scholar]
- 173.Kotan V. O., Sarandol E., Kirhan E., Ozkaya G., Kirli S. Effects of long-term antidepressant treatment on oxidative status in major depressive disorder: a 24-week follow-up study. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2011;35(5):1284–1290. doi: 10.1016/j.pnpbp.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 174.Herken H., Gurel A., Selek S., et al. Adenosine Deaminase, Nitric Oxide, Superoxide Dismutase, and Xanthine Oxidase in Patients with Major Depression: Impact of Antidepressant Treatment. Archives of Medical Research. 2007;38(2):247–252. doi: 10.1016/j.arcmed.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 175.Khanzode S. D., Dakhale G. N., Khanzode S. S., Saoji A., Palasodkar R. Oxidative damage and major depression: the potential antioxidant action of selective serotonin re-uptake inhibitors. Redox Report. 2003;8(6):365–370. doi: 10.1179/135100003225003393. [DOI] [PubMed] [Google Scholar]
- 176.Bilici M., Efe H., Köroğlu M. A., Uydu H. A., Bekaroğlu M., Değer O. Antioxidative enzyme activities and lipid peroxidation in major depression: alterations by antidepressant treatments. Journal of Affective Disorders. 2001;64(1):43–51. doi: 10.1016/S0165-0327(00)00199-3. [DOI] [PubMed] [Google Scholar]
- 177.Gammoh O., Mayyas F., Elhajji F. D. Chlorpheniramine and escitalopram: similar antidepressant and nitric oxide lowering roles in a mouse model of anxiety. Biomedical Reports. 2017;6(6):675–680. doi: 10.3892/br.2017.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Réus G. Z., Matias B. I., Maciel A. L., et al. Mechanism of synergistic action on behavior, oxidative stress and inflammation following co-treatment with ketamine and different antidepressant classes. Pharmacological Reports. 2017;69(5):1094–1102. doi: 10.1016/j.pharep.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 179.Matchkov V. V., Kravtsova V. V., Wiborg O., Aalkjaer C., Bouzinova E. V. Chronic selective serotonin reuptake inhibition modulates endothelial dysfunction and oxidative state in rat chronic mild stress model of depression. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2015;309(8):R814–R823. doi: 10.1152/ajpregu.00337.2014. [DOI] [PubMed] [Google Scholar]
- 180.Abdel-Wahab B. A., Salama R. H. Venlafaxine protects against stress-induced oxidative DNA damage in hippocampus during antidepressant testing in mice. Pharmacology Biochemistry and Behavior. 2011;100(1):59–65. doi: 10.1016/j.pbb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 181.Novío S., Núñez M. J., Amigo G., Freire-Garabal M. Effects of fluoxetine on the oxidative status of peripheral blood leucocytes of restraint-stressed mice. Basic & Clinical Pharmacology & Toxicology. 2011;109(5):365–371. doi: 10.1111/j.1742-7843.2011.00736.x. [DOI] [PubMed] [Google Scholar]
- 182.Réus G. Z., Stringari R. B., de Souza B., et al. Harmine and imipramine promote antioxidant activities in prefrontal cortex and hippocampus. Oxidative Medicine and Cellular Longevity. 2010;3(5):325–331. doi: 10.4161/oxim.3.5.13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zafir A., Ara A., Banu N. In vivo antioxidant status: A putative target of antidepressant action. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(2):220–228. doi: 10.1016/j.pnpbp.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 184.Kumar P., Kumar A. Protective role of sertraline against 3-nitropropionic acid-induced cognitive dysfunction and redox ratio in striatum, cortex and hippocampus of rat brain. Indian Journal of Experimental Biology. 2009;47(9):715–722. [PubMed] [Google Scholar]
- 185.Hwang J., Zheng L. T., Ock J., et al. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology. 2008;55(5):826–834. doi: 10.1016/j.neuropharm.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 186.Schmidt A. J., Heiser P., Hemmeter U. M., Krieg J. C., Vedder H. Effects of antidepressants on mRNA levels of antioxidant enzymes in human monocytic U-937 cells. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2008;32(6):1567–1573. doi: 10.1016/j.pnpbp.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 187.Hashioka S., Klegeris A., Monji A., et al. Antidepressants inhibit interferon-γ-induced microglial production of IL-6 and nitric oxide. Experimental Neurology. 2007;206(1):33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 188.Kolla N., Wei Z., Richardson J. S., Li X. M. Amitriptyline and fluoxetine protect PC12 cells from cell death induced by hydrogen peroxide. Journal of Psychiatry and Neuroscience. 2005;30(3):196–201. [PMC free article] [PubMed] [Google Scholar]
- 189.Sowa-Kućma M., Styczeń K., Siwek M., et al. Are there differences in lipid peroxidation and immune biomarkers between major depression and bipolar disorder: effects of melancholia, atypical depression, severity of illness, episode number, suicidal ideation and prior suicide attempts. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2018;81:372–383. doi: 10.1016/j.pnpbp.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 190.Sarandol A., Sarandol E., Eker S. S., Erdinc S., Vatansever E., Kirli S. Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative–antioxidative systems. Human Psychopharmacology: Clinical and Experimental. 2007;22(2):67–73. doi: 10.1002/hup.829. [DOI] [PubMed] [Google Scholar]
- 191.Berk M., Malhi G. S., Gray L. J., Dean O. M. The promise of N-acetylcysteine in neuropsychiatry. Trends in Pharmacological Sciences. 2013;34(3):167–177. doi: 10.1016/j.tips.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 192.Mocelin R., Marcon M., D’ambros S., et al. N-Acetylcysteine reverses anxiety and oxidative damage induced by unpredictable chronic stress in zebrafish. Molecular Neurobiology. 2019;56(2):1188–1195. doi: 10.1007/s12035-018-1165-y. [DOI] [PubMed] [Google Scholar]
- 193.Phensy A., Duzdabanian H. E., Brewer S., et al. Antioxidant treatment with N-acetyl cysteine prevents the development of cognitive and social behavioral deficits that result from perinatal ketamine treatment. Frontiers in Behavioral Neuroscience. 2017;11 doi: 10.3389/fnbeh.2017.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Berk M., Dean O. M., Cotton S. M., et al. The efficacy of AdjunctiveN-Acetylcysteine in major depressive Disorder. The Journal of Clinical Psychiatry. 2014;75(6):628–636. doi: 10.4088/JCP.13m08454. [DOI] [PubMed] [Google Scholar]
- 195.Das P., Tanious M., Fritz K., et al. Metabolite profiles in the anterior cingulate cortex of depressed patients differentiate those takingN-acetyl-cysteine versus placebo. Australian & New Zealand Journal of Psychiatry. 2013;47(4):347–354. doi: 10.1177/0004867412474074. [DOI] [PubMed] [Google Scholar]
- 196.Chakraborty S., Tripathi S. J., Srikumar B. N., Raju T. R., Shankaranarayana Rao B. S. N-acetyl cysteine ameliorates depression-induced cognitive deficits by restoring the volumes of hippocampal subfields and associated neurochemical changes. Neurochemistry International. 2020;132:p. 104605. doi: 10.1016/j.neuint.2019.104605. [DOI] [PubMed] [Google Scholar]
- 197.Abuelezz S. A., Hendawy N., Magdy Y. Targeting oxidative stress , cytokines and serotonin interactions via indoleamine 2, 3 dioxygenase by coenzyme Q10: role in suppressing depressive like behavior in rats. Journal of Neuroimmune Pharmacology. 2017;12(2):277–291. doi: 10.1007/s11481-016-9712-7. [DOI] [PubMed] [Google Scholar]
- 198.Jahangard L., Yasrebifar F., Haghighi M., Ranjbar A., Mehrpooya M. Influence of adjuvant coenzyme Q10 on inflammatory and oxidative stress biomarkers in patients with bipolar disorders during the depressive episode. Molecular Biology Reports. 2019;46(5):5333–5343. doi: 10.1007/s11033-019-04989-z. [DOI] [PubMed] [Google Scholar]
- 199.El-Hage W., Leman S., Camus V., Belzung C. Mechanisms of antidepressant resistance. Frontiers in Pharmacology. 2013;4 doi: 10.3389/fphar.2013.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Siwek M., Dudek D., Paul I. A., et al. Zinc supplementation augments efficacy of imipramine in treatment resistant patients: a double blind, placebo-controlled study. Journal of Affective Disorders. 2009;118(1-3):187–195. doi: 10.1016/j.jad.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 201.Rosenblat J. D., McIntyre R. S., Alves G. S., Fountoulakis K. N., Carvalho A. F. Beyond Monoamines-novel targets for treatment-resistant depression: a comprehensive review. Current Neuropharmacology. 2015;13(5):636–655. doi: 10.2174/1570159x13666150630175044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Swardfager W., Herrmann N., McIntyre R. S., et al. Potential roles of zinc in the pathophysiology and treatment of major depressive disorder. Neuroscience and Biobehavioral Reviews. 2013;37(5):911–929. doi: 10.1016/j.neubiorev.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 203.Ak T., Gülçin I. Antioxidant and radical scavenging properties of curcumin. Chemico-Biological Interactions. 2008;174(1):27–37. doi: 10.1016/j.cbi.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 204.Aggarwal B. B., Harikumar K. B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. The International Journal of Biochemistry & Cell Biology. 2009;41(1):40–59. doi: 10.1016/j.biocel.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dong S., Zeng Q., Mitchell E. S., et al. Curcumin enhances neurogenesis and cognition in aged rats: implications for transcriptional interactions related to growth and synaptic plasticity. PloS one. 2012;7(2):p. e31211. doi: 10.1371/journal.pone.0031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xu Y., Ku B. S., Yao H. Y., et al. The effects of curcumin on depressive-like behaviors in mice. European Journal of Pharmacology. 2005;518(1):40–46. doi: 10.1016/j.ejphar.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 207.Xu Y., Ku B.-S., Yao H.-Y., et al. Antidepressant effects of curcumin in the forced swim test and olfactory bulbectomy models of depression in rats. Pharmacology Biochemistry and Behavior. 2005;82(1):200–206. doi: 10.1016/j.pbb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 208.Li Y. C., Wang F. M., Pan Y., et al. Antidepressant-like effects of curcumin on serotonergic receptor-coupled AC-cAMP pathway in chronic unpredictable mild stress of rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(3):435–449. doi: 10.1016/j.pnpbp.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 209.Ng Q. X., Koh S. S. H., Chan H. W., Ho C. Y. X. Clinical use of curcumin in depression: a meta-analysis. Journal of the American Medical Directors Association. 2017;18(6):503–508. doi: 10.1016/j.jamda.2016.12.071. [DOI] [PubMed] [Google Scholar]
- 210.Naqvi F., Haider S., Naqvi F., Saleem S., Perveen T., Batool Z. A comparative study showing greater effects of curcumin compared to donepezil on memory function in rats. Pakistan Journal of Pharmaceutical Sciences. 2019;32(1) [PubMed] [Google Scholar]
- 211.Wang Z., Ren W., Zhao F., Han Y., Liu C., Jia K. Curcumin amends Ca2+ dysregulation in microglia by suppressing the activation of P2X7 receptor. Molecular and Cellular Biochemistry. 2020;465(1-2):65–73. doi: 10.1007/s11010-019-03668-8. [DOI] [PubMed] [Google Scholar]
- 212.de Oliveira M. R., Chenet A. L., Duarte A. R., Scaini G., Quevedo J. Molecular mechanisms underlying the anti-depressant effects of resveratrol: a review. Molecular Neurobiology. 2018;55(6):4543–4559. doi: 10.1007/s12035-017-0680-6. [DOI] [PubMed] [Google Scholar]
- 213.Moore A., Beidler J., Hong M. Resveratrol and depression in animal models: a systematic review of the biological mechanisms. Molecules. 2018;23(9):p. 2197. doi: 10.3390/molecules23092197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shen J., Qu C., Xu L., Sun H., Zhang J. Resveratrol exerts a protective effect in chronic unpredictable mild stress-induced depressive-like behavior: involvement of the AKT/GSK3β signaling pathway in hippocampus. Psychopharmacology. 2019;236(2):591–602. doi: 10.1007/s00213-018-5087-1. [DOI] [PubMed] [Google Scholar]