

ABSTRACT
Small extracellular vesicles (sEVs) present fairly distinctive lipid membrane features in the extracellular environment. These include high curvature, lipid-packing defects and a relative abundance in lipids such as phosphatidylserine and ceramide. sEV membrane could be then considered as a “universal” marker, alternative or complementary to traditional, characteristic, surface-associated proteins. Here, we introduce the use of membrane-sensing peptides as new, highly efficient ligands to directly integrate sEV capturing and analysis on a microarray platform. Samples were analysed by label-free, single-particle counting and sizing, and by fluorescence co-localisation immune staining with fluorescent anti-CD9/anti-CD63/anti-CD81 antibodies. Peptides performed as selective yet general sEV baits and showed a binding capacity higher than anti-tetraspanins antibodies. Insights into surface chemistry for optimal peptide performances are also discussed, as capturing efficiency is strictly bound to probes surface orientation effects. We anticipate that this new class of ligands, also due to the versatility and limited costs of synthetic peptides, may greatly enrich the molecular toolbox for EV analysis.
KEYWORDS: Extracellular vesicles, peptides, microarrays, membrane binding, membrane curvature
Introduction
Extracellular vesicles (EVs) are membranous micro- and nano-sized biological particles released by cells that play a major role in inter-cellular communication. EV shuttle an impressive amount of molecular information, including proteins and non-coding RNAs, thus representing a phenomenal source of circulating biomarkers [1]. As such, EVs are arising unparalleled expectations as the next-generation theranostic tools [2]. However, to fully realise EV potential, several challenges are yet to be overcome [3]. These are mainly related to the separation of specific EV populations from other bio-nanoparticles and contaminants commonly found in biological fluids (including protein aggregates, lipoproteins, viruses, organelles) that can plague the downstream analysis of vesicles with regards to their count, function and content [4]. Also the heterogeneity of cell-released EV accounts for this. Depending on their biogenesis pathway, EV can indeed be distinguished in endosome-origin exosomes (50–150 nm), plasma-membrane-derived microvesicles (50–1000 nm) (MVs) and apoptotic bodies (500–2000 nm). However, achieving such precise distinction remains extraordinarily difficult in routine practices since consensus has not yet emerged on specific markers of EV subtypes, for example, exosomes and MVs partially overlap in size and share many of the known biomarkers enriched in EVs. Therefore, ISEV guidelines encourage parallel classifications based on physical EV traits, including size or density; specifically, MISEV2018 [5] and recent literature [6–8] define as “small EVs” (sEVs) vesicles that are around 100 nm and in the range of 30–250 nm.
Analytical platforms for sEV high-throughput analysis that do not strictly rely on sample pre-treatment, limiting purification artefacts, are therefore highly desirable. In this scenario, EV microarrays have been introduced by Jørgensen and collaborators to phenotype EV on a protein microarray platform [9]. In this technique, antibodies are used for the selective capturing of EV by their most common surface-associated proteins (e.g. tetraspanins, MHC I and II, Annexin V, etc.), followed by fluorescence-based immune staining of characteristic trans-membrane proteins. This format has been extended to the analysis of antibody-captured vesicles in a label-free mode using surface plasmon resonance imaging (SPRi) [10] and single particle interferometric reflectance imaging sensor (SP-IRIS) [11]. However, targeting surface-exposed proteins still present several drawbacks: (i) the analysis can be biased by the presence of soluble antigens; (ii) the inherent variability of antibody specificity and affinity can impair EV capturing efficiency; (iii) protein markers relative abundance may be poor or subject to significant inter-individual fluctuations, thus reducing the value of comparative studies. The possibility to target a specific but “universal” EV marker such as the lipid membrane would therefore represent a paradigmatic shift, possibly expanding the available molecular tools towards an increased analytical consistence.
In this regard, sEV membrane is characterised by physical and chemical traits that are peculiar in the extracellular space [1]. sEVs have indeed highly curved membranes, whose outer leaflets typically contain a high amount of anionic, unsaturated phospholipids (e.g. phosphatidylserine) together with the presence of characteristic lipid-packing defects [12–14]. Of note, many proteins are physiologically involved in the dynamic modulation of membrane curvature that occurs during a multitude of cellular processes (including vesicles secretion); in addition, it is further worth highlighting that some of them are able to sense and bind with exquisite selectivity only highly curved membranes [15–18]. These include, among others, the Bin-Amphiphysin-Rvs (BAR) domain of ampiphysin [19], the ArfGAP1 lipid-packing sensor (ALPS) proteins [20], the C2B domain of synaptotagmin-I and the effector domain of the myristoylated alanine-rich C-kinase substrate protein (MARCKS-ED) [21]. Accordingly, peptides derived from membrane-sensing proteins have emerged as convenient, easy-to-synthetise novel molecular probes for targeting highly curved membranes [14,22–24]. In this frame, proposed mechanisms of membrane curvature sensing by protein domains and peptides can be multiple and co-operative (Figure 1). In many cases, the early events of membrane recognition and binding are based on complementary electrostatic interactions between the peptide/protein effector domain and the phospholipids on the outer membrane leaflet, that subsequently can lead to the insertion of the sensing effector into the membrane defects that characterise highly curved membranes [16,21,24]. This mechanism is characteristic of amphipathic peptides. Other recognition pathways were also described [25].
Figure 1.
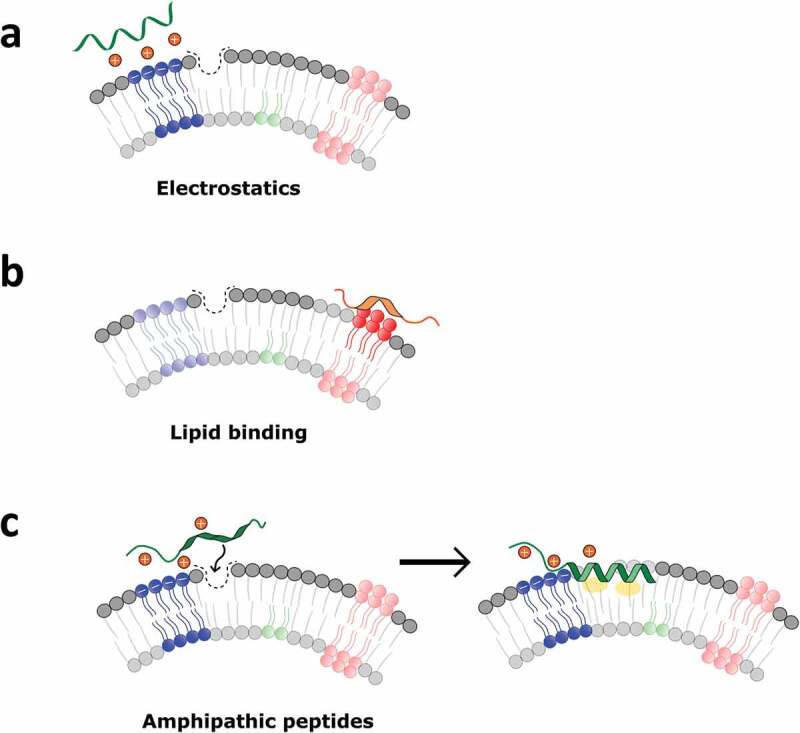
Common mechanisms involved in membrane recognition by curvature-sensing peptides. (a) Purely electrostatic interactions are typical of cationic peptides; (b) specific binding to lipids particularly abundant in small vesicles (e.g. phosphatidylserine) can drive the interaction; (c) amphipathic peptides usually approach highly curved membranes through electrostatics, and subsequently insert into lipid-packing defects. Binding can be stabilised by peptide folding within the membrane, facilitated by the presence of hydrophobic groups.
Here, we introduce for the first time the use of membrane-sensing peptide ligands as molecular baits for sEV and we demonstrate their use in a microarray platform as valuable alternative/complement to antibodies. The peptide ligands were able to capture sEV from purified samples and from untreated human serum with high specificity and binding capacity.
To the best of our knowledge, this is the first application of membrane-sensing peptides to sEV profiling and the first-reported example of sEV peptide microarrays.
Results and discussion
Peptide design and microarray
Among the set of reported curvature-sensing peptides, we decided to focus on a short amino acid sequence (RPPGFSPFR, BK) derived from bradykinin that was reported to bind in solution to highly curved lipid nanovesicles, including sEV [26]. An electrostatics-dominated recognition mechanism further stabilised by hydrophobic interactions has been proposed to be responsible for this peculiar affinity and, of note, selectivity towards small nanovesicles is consistently amplified by multimeric and multivalent peptide display, leading to very limited affinity towards larger (ca. 400 nm) vesicles. Based on these premises, we adapted the reported sequence to fit our previously developed microarray platform for site-selective oriented peptide immobilisation [27]. Briefly, the membrane-sensing sequence was extended with a short PEG spacer terminating with a propargyl group for click-based bioconjugation to azide copolymer (MCP-6)-coated analytical surfaces [28]. We realised three different peptide baits, including a linear (BP), a branched (BPb) and a tandem (BPt) derivatives, to assess the possible contribution of probes multivalency to sEV capturing (Scheme 1). Finally, given the key role of electrostatic interactions in initiating the complex membrane-sensing mechanism reported for BK, we also synthesised a negative control peptide where arginine residues were mutated to (oppositely charged) glutamic acid residues (BPn).
Scheme 1.
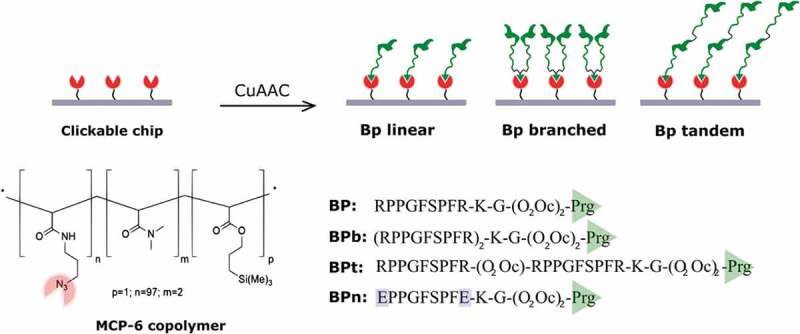
Membrane-sensing peptides are used to capture EV on sensing surfaces. Peptidic probes are immobilised on chips through chemoselective click-type reaction between azido groups provided by MCP-6 surface coating and propargyl-glycine-terminated peptides. Peptidic probes are synthesised in a linear form (BP) and in two multivalent presentation: branched (BPb) and tandem (BPt). As a negative control (BPn), a peptide where arginine residues were mutated to (oppositely charged) glutamic acid residues was synthesised.
sEV capturing on peptide microarray
Patterned silicon chips with 80 nm SiO2 layer, suitable for SP-IRIS by the ExoView™platform, were arrayed with five replicated spots of peptides via copper catalysed azide-alkyne cycloaddition (CuAAC) mediated chemoselective immobilisation (Scheme 1). Microchips were first characterised by quantifying the immobilisation density of peptides; according to our previous findings [27,29], the average binding yield of each spotted peptide was 2.0–3.5 ng/mm2. As a first experimental set to probe peptide-based EV capturing, EVs were isolated from HEK cells by ultracentrifugation (UC) and characterised according to MISEV2018 guidelines [5], by nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM) and Western blotting (WB) to demonstrate the presence of EV membrane and luminal proteins. Results confirmed the presence of vesicles with size, morphology and protein content compatible with EV as shown in Figure 2(a–c).
Figure 2.
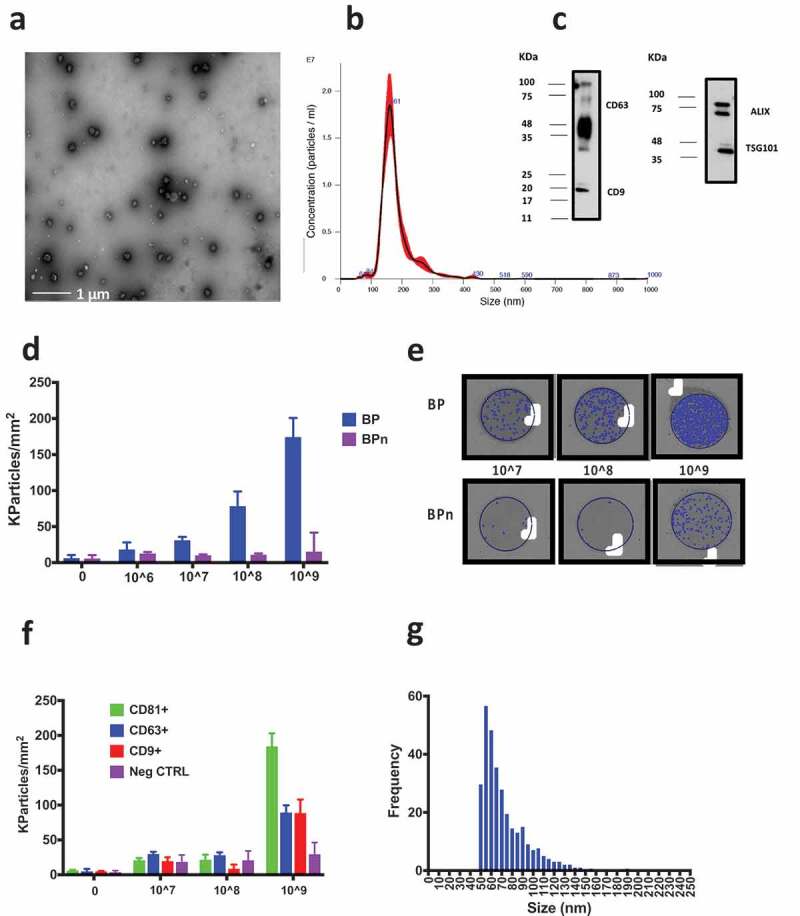
Characterisation of UC-isolated EVs from HEK cell-line culture performed by TEM, NTA and WB. (a) TEM imaging of the bulk UC-isolated HEK EVs after negative staining by phosphotungstic acid. (b) Results of the analysis by NTA providing a mean particle size of 180 ± 1 nm and a concentration of 1.2 × 1012 particles/mL. (c) The presence of transmembrane protein CD63 and CD9 and luminal proteins ALIX and TSG101 was assessed by Western blotting. The UC preparation resulted positive to all the four proteins. (d) HEK UC particle density per mm2 detected on BP and BPn peptide spots in a blank sample (filtered PBS) and in 1 × 106–1 × 109 particles/mL concentrations range. A clear dose-response effect is visible. Signal on BPn peptide is negligible. (e) Representative images of BP and BPn peptide spots incubated with 1 × 107–1 × 109 particles/mL: blue dots indicate detected particles. (f) HEK UC particle density per mm2 detected on antibody microarray (anti CD81/CD63/CD9). Only 1 × 109 particles/mL concentration provides on CD antibodies spots a signal distinguishable from that on the negative control antibody. (g) Observed size distribution of captured particles reported as the number of counts detected in each 5 nm bin. Representative peptide spot images are reported in the supplementary information (Figure S1).
Based on NTA quantifications, serial dilutions of the HEK UC sample in the 1 × 106–1 × 109 particles/mL range were prepared in PBS and incubated for 150 min on peptide chips. sEV capturing from the HEK UC sample on peptide microarray was analysed by SP-IRIS on the ExoView™ platform that digitally count, size and image individual, low-refractive index and ≥ 50 nm nanovesicles based on an interferometric principle [11] (Scheme 2); net values of detected EV on BP and BPn from five spot replicates were averaged (Figure 2(d)).
Scheme 2.
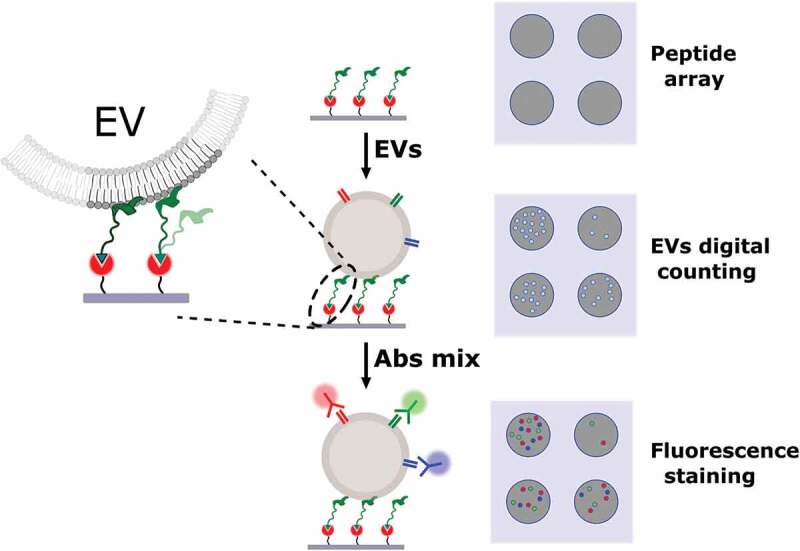
Scheme of the assay for label-free and fluorescence detection of EV captured on microarray chips. A silicon chip is arrayed with spots of capturing peptides and incubated with the EV sample. SP-IRIS platform images the chip and provides a label-free counting and sizing of the captured EV. The same chip can then be further incubated with fluorescent antibodies for immune staining of EV-associated proteins and three-colour fluorescence based co-localisation of EV surface markers.
Remarkably, we could immediately assess that a significant amount of EV (blue dots) could be detected within the area of BP spots (Figure 2(e)), whilst negligible binding was observed for the negative control peptide BPn. Moreover, microchips incubation with different sample concentrations highlighted a dose-response effect, with detected particle signal on BP being clearly distinguishable down to 1 × 107 particles/mL concentration (Figure 2(d)). It is worth noting that, when the same serial dilutions of the HEK UC sample (1 × 106–1 × 109 particles/mL) were incubated on an antibody microarray chip spotted with anti-tetraspanins IgG (anti CD81/CD63/CD9), only the highest concentration (1 × 109 particles/mL) provided a distinguishable counting from the non-specific signals detected on the negative antibody control (Figure 2(f)). Of note, the average particle counting detected on the antibody spots was lower than that provided by BP for each tested HEK UC sample. As we could expect based on our probe selection, the size distribution observed for captured particles (Figure 2(g)) suggested the preferential affinity of BP peptide for small vesicles.
Size dependency of BP capturing
Liposomes of three different sizes (150, 250, 450 nm) made of the same lipidic composition (supplementary information) were used in a model experiment to investigate the size dependency of vesicle capturing by BP peptide. The binding of liposomes to peptides was analysed by SPRi on chips spotted with the set of linear (BP), branched (BPb) and a tandem (BPt) peptides, immobilised on chip by same click-based strategy [27]. Sensorgrams were recorded after injection of liposomes at the concentration of 2.5 × 109 particles/mL (Figure S2) showing a clear binding for the small liposomes (150 nm) whereas negligible signal was observed for the larger (250 and 450 nm) liposomes. Notably, these data are in accordance with previously reported results on bradykinin-derived peptides that guided our probe selection [26].
We also compared BP binding of sEVs-enriched vs MVs-enriched samples prepared from non-filtered, fresh, platelet-free plasma, by centrifugation at 100.000 g for 2 h and 10.000 g for 1 h, respectively. Figure 3(a–c) reports NTA measurements and WB analysis of the two samples. The peptide capture from the two preparations was similar and limited to the vesicles in the 50–120 nm size present in both samples (Figure 3(d,e)). Interestingly, the size distribution of vesicles captured by a tetraspanin antibody chip was broader (Figure 3(f,g)), especially for MVs-enriched sample on CD41, a platelet-derived MVs-associated biomarker [30]. Overall, these data suggest that BP peptides can be used as generic baits for both MVs and sEV within their overlapping size range (50–120 nm).
Figure 3.
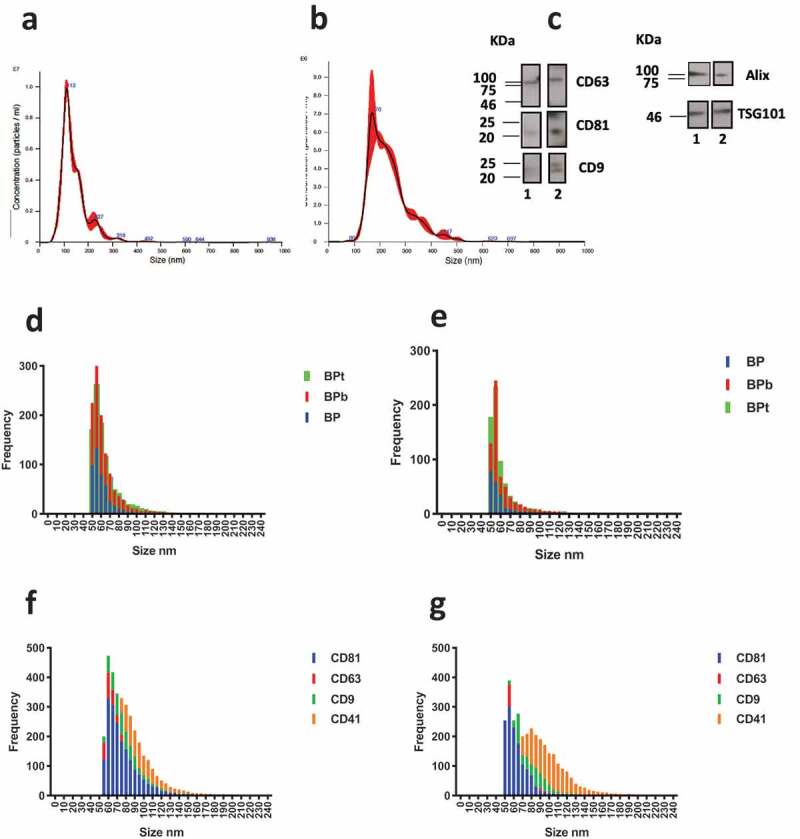
NTA analysis of (a) sEV-enriched sample obtained by centrifugation at 100.000 g for 2 h (mean particle diameter 143 ± 1 nm) and (b) MVs-enriched sample obtained at 10.000 g for 1 h (mean particle diameter 236 ± 5 nm). (c) Western blotting analysis for sEVs (lane 1) and MVs-enriched sample (lane 2). CD63, CD9 and CD81 markers are confirmed for both samples. Similarly, luminal proteins Alix and TSG101 are detected for both sEVs and MVs samples. (d) Observed size distribution on peptide chips for captured vesicles from the sEVs and (e) MVs-enriched sample obtained at 10.000 g. The size is reported as the number of counts detected in each 5 nm bin. (f) Size distribution on tetraspanins antibody chip for the sEVS sample and (g) MVs sample.
To confirm the EV nature of the peptide-captured vesicles, we performed a three-colour fluorescence detection of EV transmembrane proteins (CD81, CD63, CD9) by the use of a secondary antibody staining with a cocktail of anti-CD81, anti-CD9 and anti-CD63 antibodies labelled with Cy3, AF488 and Cy5, respectively. When HEK UC sample at the concentration of 2 × 109 particles/mL was incubated on peptide chip, label-free counting (Figure 4(a)) and bright fluorescence signals were detected on particles captured by BP peptides in the three fluorescence channels (Figure 4(b,c)). As for the multivalent peptide forms BPb and BPt, we observed only a slight increase in vesicle binding (Figure 4(a)). In this sense, a surface multivalency effect due to peptides co-operative interaction in nanovesicles binding is likely to account for the observed high capturing efficiency (Scheme 2). In accordance to label-free data, the negative control BPn showed negligible non-specific binding (Figure 3(a,b)).
Figure 4.
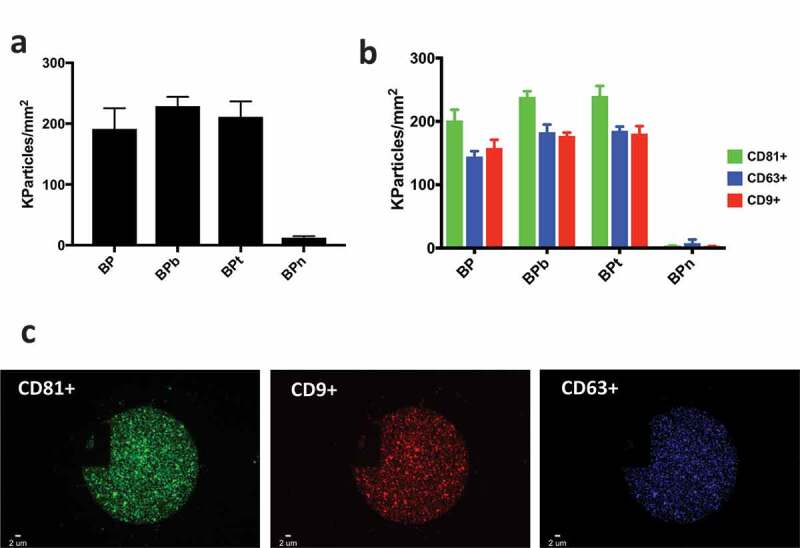
(a) EV density after incubation with HEK UC sample at the concentration of 1 × 109 particles/mL label free detected on BP peptides; (b) correspondent EV density detected by fluorescence on BP peptides; (c) representative BP spot and fluorescence immune staining. Images were acquired on the three different fluorescence channels: green particles are vesicles captured by BP and positive for CD81; blue particles are vesicles captured by BP and positive for CD63 whereas red particles are vesicles captured by BP and positive for CD9.
EVs from human serum
In order to test our peptide arrays with samples of increasing complexity, capturing of EVs from human serum by membrane-sensing peptides was assessed with vesicles derived from a pool of serum samples. Nowadays, EV purification from blood relies on techniques that use biophysical or biochemical features of EVs, such as size, density and surface molecules [3]. However, the specificity of each isolation protocol is constrained by the overlap of biophysical and biochemical characteristics of targeted EVs with other biological components that prevail in that particular body fluid. In blood for example, EVs overlap in size with protein aggregates, VLDLs, LDLs and chylomicrons which make isolation from blood plasma or serum a highly challenging task [31–33]. EV isolation from complex biological samples thus often requires a combination of multiple procedures such as density cushion with size exclusion chromatography (SEC) [34] in plasma or UC and SEC in human synovial fluid [35].
Here, we isolated EVs from human serum by UC followed by a combined polymer-based precipitation and SEC. The serum-derived EVs were then characterised by TEM and NTA (Figure 5(a,b)). The analysis by WB (Figure 5(c)) confirmed the presence of transmembrane proteins CD63, CD81 and CD9 and luminal proteins ALIX and TSG101 in the purified EVs (lane 2); assessment of apolipoprotein A I (Apo AI) by WB confirmed a remarkable decrease of lipoprotein contamination in the purified EV sample (lane 2).
Figure 5.
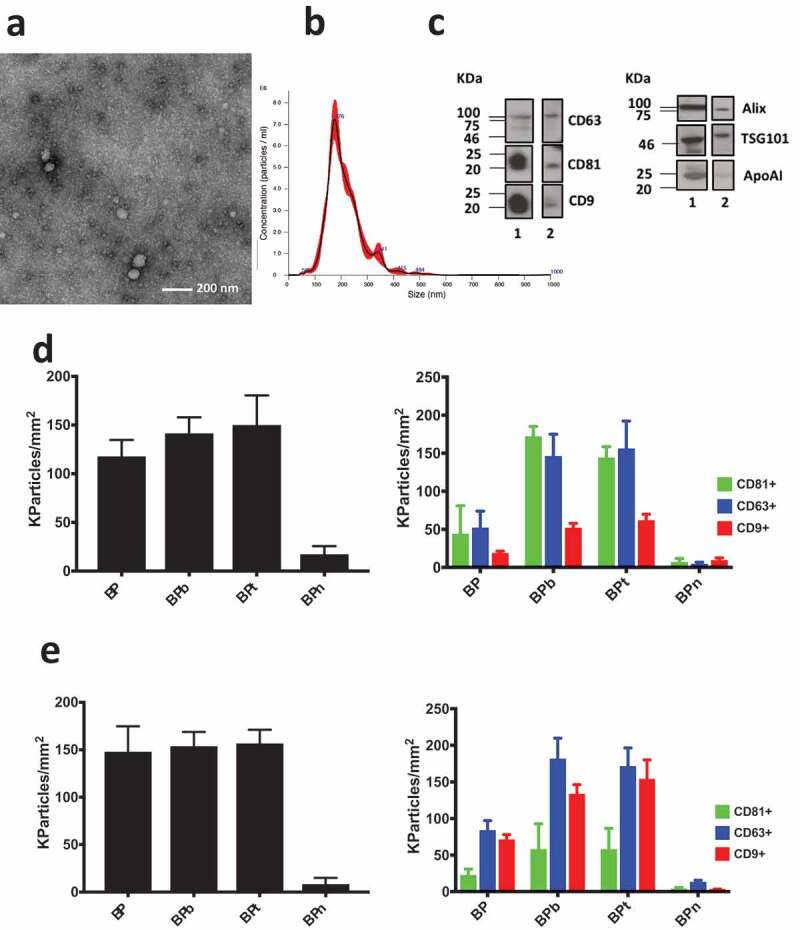
(a) TEM imaging of EVs obtained from human serum by UC followed by combined polymer precipitation and SEC showed results comparable with analogous analysis reported elsewhere [36]; (b) NTA analysis provided a mean particle size of 203 ± 3 nm and a concentration of 8.2 × 1011 particles/mL; (c) the presence of transmembrane protein CD63, CD81 and CD9 and luminal proteins ALIX and TSG101 was assessed by Western blotting of serum (lane 1) and after combined isolation of EVs. Contamination by lipoproteins is assessed by WB of Apo AI in serum (lane 1) and in the purified EVs (lane 2). (d) Analysis of EV isolated by ultracentrifugation, polymer precipitation and SEC from human serum incubated on peptide microarrays at 1 × 109 particles/mL concentration. Density of particles captured by BP peptides (left panel) is confirmed by fluorescence staining using CD81/CD/63/CD9 fluorescent antibodies (right panel). (e) Analysis performed on unpurified human serum diluted 1:8. Density of particles captured by BP peptides is detected label free (left panel) and by fluorescence staining using CD81/CD/63/CD9 fluorescent antibodies (right panel). Observed size distribution of captured EV is reported in the supplementary information (Figure S3).
Label-free counting on BP peptides and fluorescence co-localisation of tetraspanins were performed (Figure 5(d)). Differently from what observed with the HEK-derived vesicles, the multivalent BPb and BPt peptides showed an improved capturing capacity of serum-derived vesicles in comparison to linear BP. Importantly, co-localisation detection of EV transmembrane proteins (CD81, CD63, CD9) confirm the capturing of EV by the entire set of peptides except for the negative control and show the same trend of increased binding yield for multivalent peptides observed by label-free counting. Given the relevance of isolation-free workflows to obtain unbiased EV analysis, in addition to purified vesicles, peptide ligands were tested with pure serum (Figure 5(e)). A pooled sera sample was filtered by a 0.2 µm syringe filter, diluted 1:8 in PBS and incubated on a peptide arrayed chip for 150 min. Particles from untreated serum were label free detected with similar efficiency on all peptides except for the negative control BPn (Figure 5(e)). The observed size distribution range of captured EVs is shown in Figure S3. Remarkably, fluorescence staining with anti-CD9/CD63/CD81 antibodies revealed a clear effect of peptide multivalency in capturing a higher number of tetraspanin positive particles compared to linear BP (Figure 5(e)).
Specific sEV binding on BP peptides directly from untreated serum was further demonstrated by a widely used and independent detection technique using a SPRi assay. To this aim, unpurified serum diluted 1:10 was injected on an SPRi chip arrayed with the set of BP peptides according to previously devised protocols [27] and as described in the experimental section. EV binding was detected label free and then the effective immobilisation of sEV on the surface of SPRi chip was confirmed by injecting a mixture of anti-CD9/CD63/CD81 antibodies that interact with surfaces of EV detected on BP peptides (Figure S4).
The role of surface chemistry in vesicles binding
To assess the role of peptide spatial exposure upon chip surface on the efficiency of sEV capturing, we compared a non-specific immobilisation on nucleophile-reactive polymer-coated chips vs a click-type peptides binding strategy able to provide pre-determined probes orientation. Of note, both strategies rely on the same polymer for chips coating (MCP-2) [37], that can be differently functionalised to introduce azido reactive handles (MCP-6) [28]. SEC-isolated EVs from serum (see NTA, WB and TEM characterisation in Figure 7) were incubated on peptide microarrays at the concentration of 1 × 109 particles/mL. Strikingly, for the entire set of BP peptides, the EV binding capacity observable for site-selectively immobilised peptides was totally abolished when peptides were randomly bound onto the surface (Figure 6(a,b)). To provide additional insights on the general feasibility of our approach, peptide surface orientation was also pursued by means of the well-known biotin-streptavidin system, that though less efficient than click-type immobilisation, maintained EV binding capacity (supplementary information Figure S5).
Figure 7.
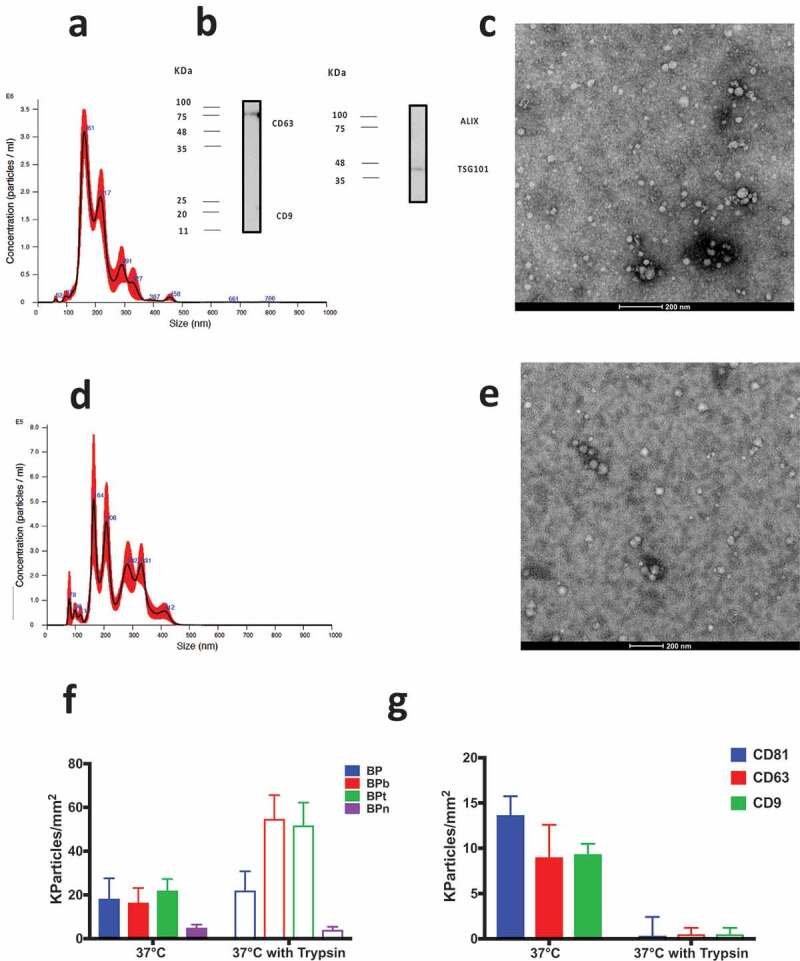
(a) NTA analysis of the SEC-isolated EVs. Mean particle size: 208 ± 3 nm. (b) WB analysis of SEC-isolated EVs from human serum. The presence of the transmembrane proteins CD9 and CD63 and the luminal proteins ALIX and TSG101 is checked and positivity confirmed for CD63 and TSG101; (c) TEM imaging after negative staining showed results in line with analogous analysis reported elsewhere [36,39]; (d) NTA analysis of SEC-isolated EVs after 6 h incubation at 37°C in presence of trypsin mean particle size: 247 ± 3 nm; (e) TEM imaging after trypsin treatment. (f) Particle density of EV from human serum incubated at the concentration of 1 × 109 particles/mL. EV capturing by BP peptides is not affected by trypsin treatment. (g) EV binding on CD81/CD63/CD9 antibody chip is abolished by surface protein digestion using trypsin.
Figure 6.
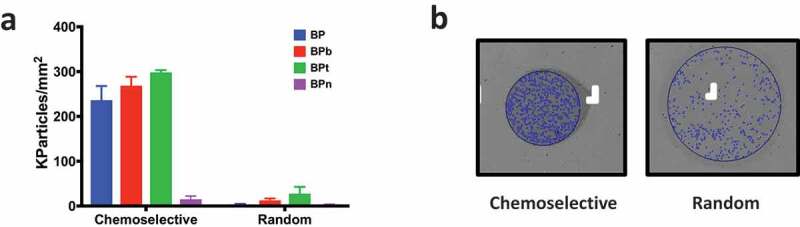
(a) EV density on BP peptides immobilised either chemoselectively on MCP-6 or randomly on MCP-2. EV capturing capacity is abolished when peptides are not oriented on the microarray surface. (b) Comparison of representative images of BP spots either chemoselectively or randomly immobilised. Spot size is smaller on MCP-6, edges well defined and particle counting after incubation higher than on the random bound peptide.
Vesicles binding on BP peptides is not mediated by surface-associated proteins
In order to get insights into the EV-peptide binding mechanism and verify whether it is directly mediated by the lipid membrane or influenced by its associated proteins, serum EV isolated by SEC were subjected to trypsin digestion [38] for 6 h to ensure complete proteolysis and binding on peptide and antibody chips was investigated. Figure 7 reports EV characterisation before trypsin incubation (A, B, C) and after proteolytic treatment (D and E). After treatment, EVs were incubated at the concentration of 1 × 109 particles/mL on peptide microarrays (Figure 7(f)). Particle density of the trypsin-treated sample detected on BP spots compared to that of a sample subjected to analogous incubation in absence of proteolytic enzyme, demonstrated that membrane protein digestion does not affect vesicles binding. Indeed, the binding in some cases (namely with the multivalent peptides) is even enhanced by proteolysis thus not being directly mediated by surface associated proteins. Oppositely, when the same samples were incubated on an antibody (CD81/CD63/CD9) microarray, as expected, no binding was detectable with the trypsin digested sample likely due to proteolysis of EV surface tetraspanins (Figure 7(g)).
Discussion
The use of peptidic ligands to capture EVs is a promising approach which could complement the traditional use of other affinity probes such as antibodies [40]. Peptides are indeed attractive due to low costs, stability, synthetic versatility and ease of tunability of functional groups. Inspired by the work by Yin and co-workers [26], we here tested the ability of membrane curvature-sensing peptides to capture extracellular vesicles onto analytical surfaces, with a particular focus on microarray chips. Compared to current approaches in EV on-chip analysis [9] and to the use of antibody probes directed against EV surface markers, our approach markedly differs in that a general yet sEV-specific trait, i.e. the highly curved membrane, is targeted. In principle, this approach should enable a more efficient EV capturing, as membrane-exposed protein biomarkers are inherently limited in number, whereas the binding of the lipid membrane is not affected by low availability of surface markers. We indeed observed that peptides have a binding capacity higher than anti-tetraspanin antibodies, resulting in a lower limit of detection of vesicles. Arguably, we ascribe this result to the co-operative interactions (surface multivalency) occurring at the chip surface between peptides and EV membranes over a broad contact area. Of note, the efficiency of binding is strictly connected to the correct and controlled peptide display on the surface, which maximises the probability for interaction (Figure 6). The preserved EV binding even upon trypsin treatment (Figure 7) supports the idea that BP peptides binding to EV is indeed not mediated by membrane-associated proteins, i.e. it is unbiased by differential surface protein expression.
Most importantly, the size dependency of captured EV showed that BP peptides have preferential affinity towards vesicles in the 50–150 nm size (sEV), as we demonstrated by using either synthetic liposomes of different sizes (150–400 nm range) and by using EV obtained from samples enriched with sEVs or MVs (different UC fractions). These pieces of evidence are in full agreement with previous reports on the striking size selectivity that curvature-sensing peptides are able to display [16,17,20,41]. The combined efficiency and selectivity of sEV binding were also demonstrated by the on-chip capture of EV isolated from serum after multi-step purification (see Figure 5) as well as directly from untreated serum. In complex samples such as untreated serum, the use of multivalent peptides showed to be particularly convenient to increase affinity and capturing specificity, in agreement with previous reports on the favourable role of multivalency in peptide microarrays [27].
Materials and methods
Reagents
Reagents for peptide synthesis were from Iris Biotech (Marktredwitz, Germany). Other chemicals were from Sigma-Aldrich (St. Louis, MO, the USA) if not stated otherwise. Bare silicon chips and Tetraspanin Kits (chips spotted with anti-CD9/CD63/CD81 antibodies) were provided by NanoView Biosciences (Bostom MA). MCP-6 azido copolymer was obtained from Lucidant Polymers (Sunnyvale, CA, the USA). EVsingle Size Exclusion Column kit was from Izon Science (Oxford, the UK).
Isolation of HEK EV by ultracentrifugation
Three-day-conditioned media from HEK cells were harvested and centrifuged at 500 g for 25 min. Supernatants were filtered with 0.22 mm filters (Merck Millipore) and centrifuged in a SorvallTM WX Ultracentrifuge (ThermoFisher Scientific, WX Ultra 100 #75,000,100) at 150.000 g for 90 min at 4°C with a SureSpinTM 630 swinging bucket rotor (ThermoFisher Scientific) to pellet EV. After supernatant was carefully removed, EV-containing pellet was resuspended in PBS and stored at −80°C until use.
Serum separation
A total of 4 mL of blood were collected in BD VACUTAINER (clot activator tube). Serum samples were separated after centrifugation within 2 h from blood collection at 1900 g for 10 min at 4°C. Serum samples from five healthy controls were pooled and frozen at −20°C until use.
Plasma separation
A total of 6 mL of peripheral blood were collected in EDTA vacutainer tubes. Blood was immediately iced and processed within 2 h from venesection. After centrifuging at 1600 g for 10 min at 4°C, the plasma (supernatant) was carefully removed and re-centrifuged at 100.000 g for 2 h or at 10.000 g for 1 h.
Isolation of serum EV by ultracentrifugation
A total of 1 mL of serum pool was filtered with 0.22 mm filters (Merck Millipore) diluted 1:1 with PBS and centrifuged in a Optima™ TLX Preparative Ultracentrifuge, Beckman CoulterTM at 150.000 g for 120 min at 4°C with a TLA-55 Rotor (Beckman CoulterTM) to pellet EV. After supernatant was carefully removed, EV-containing pellet was stored at −80°C until use.
Isolation of EVs by combined polymer-based precipitation and SEC
Serum EVs isolated by UC as described above were further subjected to the Exo-Spin™ (Cell Guidance Systems, the UK) purification kit according to the manufacturer instructions.
EV isolation by size exclusion chromatography
SEC was performed according to the manufacturer’s instructions of the qEV single column (Izon, Christchurch, New Zealand). Briefly, after column equilibration with PBS, 150 µL of the pool of sera were loaded, 200 µL were collected; the first five fractions (F1–F5) were discarded. The seventh fraction (F7) that according to the manufacturer represents the fraction with the highest amount of vesicles was used for analysis and trypsin treatment.
Trypsin treatment
F7 SEC sample was incubated with 25 µg/mL trypsin for 6 h at 37°C in a Eppendorf Thermomixer according to published protocols [38,42].
Nanoparticle tracking analysis
NTA was performed according to manufacturer’s instructions using a NanoSight NS300 system (Malvern Technologies, Malvern, UK) configured with 532 nm laser. All samples were diluted in filtered PBS to a final volume of 1 mL. Ideal measurement concentrations were found by pre-testing the ideal particle per frame value (20–100 particles/frame). Following settings were set according to the manufacturer’s software manual. A syringe pump with constant flow injection was used and three videos of 60 s were captured and analysed with NTA software version 3.2. From each video, the mean, mode and median EV size was used to calculate samples concentration expressed in nanoparticles/mL.
Transmission electron microscopy
Isolated EVs were absorbed on glow discharged carbon-coated formvar copper grids, washed with water, contrasted with 2% uranyl acetate and air dried. Grids were observed with a Zeiss LEO 512 transmission electron microscope. Images were acquired by a 2 k × 2 k bottom-mounted slow-scan Proscan camera controlled by EsivisionPro 3.2 software.
Western blot analysis
Purified EVs were resuspended in not reducing Laemmli buffer for the detection of CD9 and CD63, in reducing buffer for ALIX and TSG101 and boiled for 5 min at 95°C. Proteins were resolved by SDS-PAGE and electro-transferred onto a nitrocellulose membrane. Non-specific sites were blocked with 5% (w/v) skimmed milk in T-TBS (tris-buffered saline: 150 mM NaCl, 20 mM TrisHCl, pH 7.4, and 0.5% Tween 20). Membranes were incubated overnight at 4°C with the following antibodies: mouse anti-CD9 (1:5000, BD Pharmingen, #555,370, San Jose, CA, the USA), mouse anti-CD63 (1:20,000; BD Pharmingen, #556,019, San Jose, CA, the USA), mouse anti-ALIX (1:500, Santa Cruz, #sc-271,975, Santa Cruz, CA, the USA), mouse anti-CD81 (1:5000; BD Pharmingen #555,675, San Jose, California, the USA), mouse anti-Apo AI (1:1000; AbCam #ab17278, Abcam Inc. Cambridge, the UK) and mouse anti-TSG101 (1:500, Novus Bio, #NB200-112, Littleton, CO, the USA). After washing with T-TBS, membranes were incubated with goat anti-mouse (1:10,000–1:50,000) IgG conjugated to horse-radish peroxidase for 45 min. Positive immunoreactive bands were detected by the enhanced chemiluminescence method (ImmobilonTM HRP substrate, #WBKLS0500, Millipore Corp., Billerica, MA, the USA).
Coating of microarray silicon chips with MCP-6
Silicon chips were coated according to the protocol described in [25,27]. Briefly, chips were immersed in a MCP-6 solution (1% w/v in 0.9 M (NH4)2SO4), filtered at 200 μm, for 30 min. The chips were then rinsed with Milli-Q water and cured for 15 min at 80°C.
Peptide synthesis and characterisation
Peptides were assembled by stepwise microwave-assisted Fmoc-SPPS on a Biotage ALSTRA Initiator+ peptide synthesiser, operating in a 0.05 mmol scale. Activation of entering Fmoc-protected amino acids (0.3 M solution in DMF) was performed using 0.5 M Oxyma in DMF/0.5 M DIC in DMF (1:1:1 molar ratio), with a five equivalent excess over the initial resin loading. For the linear form BP and BPn coupling steps were performed for 20 min at 50°C. For the branched and tandem forms (BPb and BPt) coupling steps were performed for 45 min at 50°C. Chain ramification for the branched BPb was introduced by coupling Fmoc-Lys(Fmoc)-OH to the growing peptide chain. Capping steps were performed by treatment with a 0.3 M Ac2O/0.3 M DIEA solution in DMF (1 × 5 min). Fmoc-deprotection steps were performed by treatment with a 20% piperidine solution in DMF at room temperature (1 × 10 min). Following each coupling, capping or deprotection step, peptidyl-resin was washed with DMF (3 × 3.5 mL). Upon complete chain assembly, resin was washed with DCM (5 × 3.5 mL) and gently dried under nitrogen flow. Resin-bound peptide was treated with an ice-cold TFA, TIS, water, thioanisole mixture (90:5:2.5:2.5 v/v/v/v, 3 mL). After gently shaking the resin for 2 h at room temperature, the resin was filtered and washed with neat TFA (2 × 4 mL). Cleavage mixture was concentrated under nitrogen stream and then added dropwise to ice-cold diethyl ether (40 mL) to precipitate the crude peptide. The crude peptide was collected by centrifugation and washed with further cold diethyl ether to remove scavengers. Peptide was then dissolved in 0.1% TFA aqueous buffer (with minimal addition of ACN to aid dissolution, if necessary). Residual diethyl ether was removed by a gentle nitrogen stream and the crude peptide was purified by RP-HPLC and pure fractions combined and analysed by ESI-MS.
Peptide microarrays
Microarrays were arrayed on MCP-6-coated patterned silicon chips, with 80 nm oxide layer thickness, using a non-contact S12 Spotter (Scienion Co., Berlin, Germany), depositing one drop for each spot. Peptides were first dissolved in DMSO to 1 mM stock solution and then diluted to the final spotting concentration (100 μM) into the printing buffer for CuACC conjugation on MCP-6-coated surfaces (5 mM Na/Acetate pH 4.8, 50 mM Trehalose, 100 μM CuSO4, 400 μM THPTA and 6.25 mM ascorbic acid).
Printed chips were placed in a humid chamber and incubated overnight at room temperature. The following day chips were immersed in a filtered 2 mM EDTA water solution for 1 h, then washed with Milli-Q water and dried.
Probe density for each immobilised peptide on the array was assessed using label-free film thickness measurement using on the Nanoview platform by nanoQC2.3.2-IT software (NanoView Biosciences, Boston, MA) and previously determined calibration factors for IRIS [43,44]. The average amount of each immobilised peptide was 1.5–3.5 ng/mm2.
EV analyses with ExoView
EV samples were diluted in filtered PBS and incubated for 2 h and 30 min in static conditions on the printed chips in a humid chamber. EV label-free analysis was carried out using the ExoView R100 reader (NanoView Biosciences, Boston, MA). The reader automatically acquires interferometric images of the microarray. NanoViewer 2.6.0 software counts nanoparticles captured on the peptide spots within a user-defined particle contrast.
For fluorescence immune staining on antibody chips, samples were diluted in incubation buffer (NanoView Biosciences). The samples were incubated on the ExoView Tetraspanin Chip (EV-TC-TTS-01) placed in a sealed 24-well plate for 16 h at room temperature. The chips were then washed three times in 750 µL of incubation buffer for 3 min each on an orbital shaker. Then, chips were incubated with ExoView Tetraspanin Labelling ABs (EV-TC-AB-01) that consist of anti-CD81 Alexa-555, anti-CD63 Alexa-488 and anti-CD9 Alexa-647. The antibodies were diluted 1:5000 in incubation buffer with 2% BSA. The chips were incubated with 500 µL of the labelling solution for 2 h. The chips were then washed once in incubation buffer, three times in wash buffer followed by a rinse in filtered DI water and dried. The chips were then imaged with the ExoView R100 reader using the nScan 2.8.4 acquisition software. The data were then analysed using NanoViewer 2.8.4. On peptide chips the previous protocol was followed using a mixture of tetraspanin antibodies (monoclonal anti-CD81, anti-CD63, anti-CD9 from Adipogen Lifescience) in house labelled by Cyanine3 NHS ester, AF-488 NHS ester and Cyanine5 NHS ester from Lumiprobe (Hannover, Germany).
SPRi
Peptide microarrays were also prepared on gold SPRi chips, purchased from Horiba Scientific SAS (SPRi-Biochip), following the same protocol described above. XelPleX instrument (Horiba Scientific SAS) was firstly calibrated with a solution of 3 mg/mL of sucrose and then 500 µL of serum sample (diluted 1:10 in running buffer) were injected on the surface of the chip with a flow rate of 10 µL/min. Subsequently, 200 µL of a mixture of anti-human CD9/CD63/CD81 antibodies (500 nM each), purchased from Ancell Corporation, were injected at 25 µL/min in order to confirm the presence of EV immobilised on the chip. EzSuite and OriginLab softwares were used to analyse the SPRi signals related to each injection.
Liposomes
Liposomes were purchased by Nanovex Biotechnologies (Spain). Pronanosome Lipo-N was used to obtain liposomes, details in the supplementary information.
We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV190066) (Van Deun J, et al. EV-TRACK: transparent reporting and centralising knowledge in extracellular vesicle research. Nature methods. 2017;14(3):228–32).
Conclusions
We here provided a proof-of-concept demonstration of the use of membrane-sensing peptides as a novel class of molecular ligands for integrated sEV isolation and analysis, reporting for the first time on peptide microarrays for extracellular vesicles. Given their affinity to the membrane of sEV, these molecules can serve as general baits, enabling vesicles capturing unbiased by differential surface protein expression. We showed that highly efficient sEV capturing can be obtained, even from unpurified and complex samples such as serum, provided the surface chemistry of peptidic probes presentation is accurately tailored. These new class of molecular probes may be nicely integrated with the use of protein markers towards improved sEV isolation and characterisation. Of note, compared to proteins and antibodies, peptides are characterised by low cost of preparation, remarkable stability and ease of chemical manipulation, offering virtually unlimited possibilities for experimental design. Overall, these features may further prompt the implementation of membrane-sensing peptides into EV affinity-based isolation techniques, enriching the analytical and pre-analytical EV toolbox. We therefore envision these molecular tools to find broad applications within the extracellular vesicles scenario in the near future.
Supplementary Material
Acknowledgements
HYDROGEX (Regione Lombardia & Fondazione Cariplo, Grant no. 2018-1720) and INDEX (European Union H2020 Grant Agreement no. 7664669) are acknowledged for partial financial support to A.G., M.C. and M.C. We thank Prof. Vincenza Dolo for TEM imaging of HEK cells EVs and for helpful discussions.
Disclosure statement
Alessandro Gori, Marina Cretich and Marcella Chiari have filed a US Provisional Patent Application (62/897,042) on the bases of these data.
Supplementary data
The supplementary data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- [1].van Niel G, D’Angelo G, Raposo G.. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–16. [DOI] [PubMed] [Google Scholar]
- [2].Andaloussi SEL, Mäger I, Breakefield XO, et al. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. [DOI] [PubMed] [Google Scholar]
- [3].Konoshenko MY, Lekchnov EA, Vlassov AV, et al. Isolation of extracellular vesicles: general methodologies and latest trends. Biomed Res Int. 2018;2018:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Merchant ML, Rood IM, Deegens JKJ, et al. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. 2017;13:731–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018. DOI: 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Busatto S, Zendrini A, Radeghieri A, et al. The nanostructured secretome. Biomater Sci. 2020;8:39–63. [DOI] [PubMed] [Google Scholar]
- [7].Yekula A, Minciacchi VR, Morello M, et al. Large and small extracellular vesicles released by glioma cells in vitro and in vivo. J Extracell Vesicles. 2020;9:1689784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Matsumoto A, Takahashi Y, Chang H-Y, et al. Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. J Extracell Vesicles. 2020;9:1696517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jørgensen M, Bæk R, Pedersen S, et al. Extracellular Vesicle (EV) Array: microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. J Extracell Vesicles. 2013;2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rojalin T, Phong B, Koster HJ, et al. Nanoplasmonic approaches for sensitive detection and molecular characterization of extracellular vesicles. Front Chem. 2019;7 DOI: 10.3389/fchem.2019.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Daaboul GG, Gagni P, Benussi L, et al. Digital detection of exosomes by interferometric imaging. Sci Rep. 2016;6:37246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].György B, Szabó TG, Pásztói M, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hugel B, Martínez MC, Kunzelmann C, et al. Membrane microparticles: two sides of the coin. Physiology. 2005;20:22–27. [DOI] [PubMed] [Google Scholar]
- [14].Kastelowitz N, Yin H.. Exosomes and microvesicles: identification and targeting by particle size and lipid chemical probes. ChemBioChem. 2014;15:923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008. DOI: 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- [16].Antonny B. Mechanisms of membrane curvature sensing. Annu Rev Biochem. 2011. DOI: 10.1146/annurev-biochem-052809-155121 [DOI] [PubMed] [Google Scholar]
- [17].Cui H, Lyman E, Voth GA. Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys J. 2011. DOI: 10.1016/j.bpj.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hatzakis NS, Bhatia VK, Larsen J, et al. How curved membranes recruit amphipathic helices and protein anchoring motifs. Nat Chem Biol. 2009. DOI: 10.1038/nchembio.213 [DOI] [PubMed] [Google Scholar]
- [19].Bhatia VK, Madsen KL, Bolinger PY, et al. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. Embo J. 2009. DOI: 10.1038/emboj.2009.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bigay J, Casella JF, Drin G, et al. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. Embo J. 2005. DOI: 10.1038/sj.emboj.7600714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].De Jesus AJ, White OR, Flynn AD, et al. Determinants of curvature-sensing behavior for MARCKS-fragment peptides. Biophys J. 2016;110:1980–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Flynn AD, Yin H. Lipid-targeting peptide probes for extracellular vesicles. J Cell Physiol. 2016;231:2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gómez-Llobregat J, Elías-Wolff F, Lindén M. Anisotropic membrane curvature sensing by amphipathic peptides. Biophys J. 2016;110:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Jesus AJ, Yin H. Computational design of membrane curvature-sensing peptides In Methods in Molecular Biology, Humana Press, New York. Vol. 1529 2017. p. 417–437. [DOI] [PubMed] [Google Scholar]
- [25].Zeno WF, Thatte AS, Wang L, et al. Molecular mechanisms of membrane curvature sensing by a disordered protein. J Am Chem Soc. 2019;141:10361–10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saludes JP, Morton LA, Coulup SK, et al. Multivalency amplifies the selection and affinity of bradykinin-derived peptides for lipid nanovesicles. Mol Biosyst. 2013;9:2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gori A, Cretich M, Vanna R, et al. Multiple epitope presentation and surface density control enabled by chemoselective immobilization lead to enhanced performance in IgE-binding fingerprinting on peptide microarrays. Anal Chim Acta. 2017;983:189–197. [DOI] [PubMed] [Google Scholar]
- [28].Sola L, Damin F, Gagni P, et al. Synthesis of clickable coating polymers by postpolymerization modification: applications in microarray technology. Langmuir. 2016;32:10284–10295. [DOI] [PubMed] [Google Scholar]
- [29].Gori A, Sola L, Gagni P, et al. Screening complex biological samples with peptide microarrays: the favorable impact of probe orientation via chemoselective immobilization strategies on clickable polymeric coatings. Bioconjug Chem. 2016;27:2669–2677. [DOI] [PubMed] [Google Scholar]
- [30].Brisson AR, Tan S, Gounou C, et al. Extracellular vesicles from activated platelets: A quantitative cryoelectron microscopy and immuno-gold labelling study. J Extracell Vesicles. 2017;6:4. [DOI] [PubMed] [Google Scholar]
- [31].Yuana Y, Levels J, Grootemaat A, et al. Co-isolation of extracellular vesicles and high-density lipoproteins using density gradient ultracentrifugation. J Extracell Vesicles. 2014;3:23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hong C-S, Funk S, Muller L, et al. Isolation of biologically active and morphologically intact exosomes from plasma of patients with cancer. J Extracell Vesicles. 2016;5:29289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lobb RJ, Becker M, Wen Wen S, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. 2015;4:27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Karimi N, Cvjetkovic A, Jang SC, et al. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell Mol Life Sci. 2018. DOI: 10.1007/s00018-018-2773-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Foers AD, Chatfield S, Dagley LF, et al. Enrichment of extracellular vesicles from human synovial fluid using size exclusion chromatography. J Extracell Vesicles. 2018. DOI: 10.1080/20013078.2018.1490145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tian Y, Gong M, Hu Y, et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J Extracell Vesicles. 2020;9:1697028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pirri G, Damin F, Chiari M, et al. Characterization of a polymeric adsorbed coating for DNA microarray glass slides. Anal Chem. 2004;76:1352–1358. [DOI] [PubMed] [Google Scholar]
- [38].Cvjetkovic A, Jang SC, Konečná B, et al. Detailed analysis of protein topology of extracellular vesicles-evidence of unconventional membrane protein orientation. Sci Rep. 2016. DOI: 10.1038/srep36338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Buschmann D, Kirchner B, Hermann S, et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J Extracell Vesicles. 2018;7:1481321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gaillard M, Thuaire A, Nonglaton G, et al. Biosensing extracellular vesicles: contribution of biomolecules in affinity-based methods for detection and isolation. Analyst. 2020. DOI: 10.1039/C9AN01949A [DOI] [PubMed] [Google Scholar]
- [41].Drin G, Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010. DOI: 10.1016/j.febslet.2009.10.022 [DOI] [PubMed] [Google Scholar]
- [42].Skliar M, Chernyshev VS, Belnap DM, et al. Membrane proteins significantly restrict exosome mobility. Biochem Biophys Res Commun. 2018;501:1055–1059. [DOI] [PubMed] [Google Scholar]
- [43].Ozkumur E, Yalçin A, Cretich M, et al. Quantification of DNA and protein adsorption by optical phase shift. Biosens Bioelectron. 2009;25:167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cretich M, Monroe MR, Reddington A, et al. Interferometric silicon biochips for label and label-free DNA and protein microarrays. Proteomics. 2012;12:2963–2977. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.