

Abstract
Background
The human gut microbiome plays a critical role in the carcinogenesis of colorectal cancer (CRC). However, a comprehensive analysis of the interaction between the host and microbiome is still lacking.
Results
We found correlations between the change in abundance of microbial taxa, butyrate-related colonic metabolites, and methylation-associated host gene expression in colonic tumour mucosa tissues compared with the adjacent normal mucosa tissues. The increase of genus Fusobacterium abundance was correlated with a decrease in the level of 4-hydroxybutyric acid (4-HB) and expression of immune-related peptidase inhibitor 16 (PI16), Fc Receptor Like A (FCRLA) and Lymphocyte Specific Protein 1 (LSP1). The decrease in the abundance of another potentially 4-HB-associated genus, Prevotella 2, was also found to be correlated with the down-regulated expression of metallothionein 1 M (MT1M). Additionally, the increase of glutamic acid-related family Halomonadaceae was correlated with the decreased expression of reelin (RELN). The decreased abundance of genus Paeniclostridium and genus Enterococcus were correlated with increased lactic acid level, and were also linked to the expression change of Phospholipase C Beta 1 (PLCB1) and Immunoglobulin Superfamily Member 9 (IGSF9) respectively. Interestingly, 4-HB, glutamic acid and lactic acid are all butyrate precursors, which may modify gene expression by epigenetic regulation such as DNA methylation.
Conclusions
Our study identified associations between previously reported CRC-related microbial taxa, butyrate-related metabolites and DNA methylation-associated gene expression in tumour and normal colonic mucosa tissues from CRC patients, which uncovered a possible mechanism of the role of microbiome in the carcinogenesis of CRC. In addition, these findings offer insight into potential new biomarkers, therapeutic and/or prevention strategies for CRC.
Keywords: Colorectal cancer, Mucosal microbiome, Metabolome, Transcriptome, DNA methylation, Butyrate
Background
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and second leading cause of cancer death in both men and women globally, leading to an estimated 551,269 annual deaths worldwide [1]. Diet plays an important role in the initiation, promotion and progression of colon carcinogenesis [2]. Epidemiological surveys indicate that approximately 80% of CRC cases in Western countries are due to dietary factors [3].
The human intestine harbours a complex microbial community comprising more than 1014 microorganisms; it carries greater than 150 times the number of genes in the human genome [4]. Diet has been shown to have a dominant impact on the structure and composition of the gut microbiome, microbial generated metabolites and host metabolism [5, 6]. Many studies have demonstrated the effect of the gut microbiome on the pathogenesis of CRC, revealing potential pathogenic bacteria such as Fusobacterium as well as beneficial bacteria such as Lactobacillales [6–8].
Nevertheless, dietary factors have also been shown to induce epigenetic changes. The most extensively studied epigenetic mechanism is DNA methylation, which involves the transfer of methyl group to the cytosine of a CpG dinucleotide and is responsible for regulating the expression of genes related with critical pathological processes [9]. Carcinogenic gene silencing may occur due to aberrant gene-specific hypermethylation at the CpG rich region (CpG island) which is located in the promotors of the genes and interfere with transcription factor binding [10, 11].
One possible mechanism underlying the effects of diet and microbiome on CRC development is a potential interaction between the microbiome and the host, whereby the colonic metabolome is impacted, leading to a subsequent alteration in host epigenetic activity and host gene expression [12, 13]. Research on selected aspects of the microbiome, metabolome, host epigenome and host transcriptome have been carried out in human, animal and cell models [14]. For instance, the interaction between the microbiome and metabolome has been extensively studied, revealing various epigenome-modulation-related metabolites such as butyrate and folate [15, 16]. In addition, the contribution of commensal bacteria to epigenetic control in the host large intestine has been demonstrated by comparing conventional and germ-free mice [17]. Associations between the microbiome and differentially methylated genes have also been investigated in patients with ulcerative colitis [18]. The interplay between the microbiome, host transcripts related to adhesion molecules and fatty acid biosynthesis was strongly supported in one study of inflammatory bowel disease [19].
Despite the mounting evidence of a potential host-microbiome interaction, a comprehensive human study integrating all the aforementioned omics is still lacking. Thus, in this pilot study, we generated and analysed four types of omic data: the microbiome (16S rRNA sequencing; 36 pairs), the metabolome (untargeted GC/MS; 17 pairs), the host transcriptome (RNA-seq; 4 pairs) and the host epigenome (Infinium HumanMethylation850 BeadChip array; 4 pairs), as measured from paired tumour and adjacent normal colonic mucosa tissues samples obtained from CRC patients (details of the study design in Additional file 1: Figure S1).
Results
Comparison of microbial composition between tumour and adjacent normal tissues
Non-metric multidimensional scaling (NMDS) analysis based on the unweighted UniFrac distance on operational taxonomic units (OTUs) revealed that the microbial community composition of the cancerous tissues could be clearly distinguished from the non-cancerous tissues, which was confirmed by analysis of similarities (Anosim) (p-value = 0.002; Fig. 1a).
Fig. 1.

Microbial composition comparison between tumour tissues and paired normal tissues in CRC patients a. NMDS plot of microbiota based on unweighted UniFrac metric on OTUs. Each point represents one sample with colour indicating tumour tissues (red) and adjacent normal tissues (blue). b-d. Distribution of the top 10 most abundant taxa in the samples at phylum (b), family (c) and genus (d) level
The microbial population structure differed between tumour and healthy tissues at multiple levels (Fig. 1b-d). At the phylum level, lower abundance of Bacteroidetes, Firmicutes and Actinobacteria, and higher abundance of Proteobacteria and Fusobacteria, was observed in tumour tissues (p-value > 0.05). At the family level, there was an under-representation of Bacteroidaceae, Lachnospiraceae and Ruminococcaceae and predominance of Fusobacteriaceae in tumour tissues (p-value > 0.05). At the genus level, the most distinctive genera were Fusobacterium, Bacteroides and Faecalibacterium, with Fusobacterium higher in tumour tissues and the other two lower in tumour tissues (p-value > 0.05).
To further characterise the bacterial genera driving the difference between the paired samples, the paired zero-inflated Gaussian (ZIG) mixture model was used [20]. As shown in Fig. 2, 15 taxa were identified to be the most relevant to the differences between tumour and normal tissues (Additional file 2: Table. S1). We found that taxa in family Halomonadaceae, genus Halomonas, genus Shewanella, genus Fusobacterium, genus Fretibacterium, genus Peptostreptococcus and genus Klebsiella were highly enriched in cancer tissues (p-value< 0.05, Fig. 2a; details in Additional file 2: Table. S1), while genus Paeniclostridium, species Bacteroides eggerthii, genus Lactobacillus, genus Prevotella 2, Enterococcus, species Clostridium perfringens and genus Veillonella were less abundant in cancer tissues (p-value < 0.05, Fig. 2b; Additional file 2: Table. S1).
Fig. 2.

Boxplots of the differences in the mucosal microbiome between tumour tissues (red) and normal tissues (blue). Y axis: Relative counts normalized and log transformed by the default method in metagenomeSeq package. X axis: names of the significantly differentially abundant microbial taxa
Taken together, these results reveal the different microbial environments between the cancerous and normal colon mucosa.
Microbiota associated with CRC-related colonic metabolites
Seventeen pairs of samples were retained for colonic metabolome analysis, fifteen of which had matched microbiome data. In total, 309 metabolites were detected and quantified using the untargeted gas chromatography-mass spectrometry (GC-MS) approach. The score plot from orthogonal partial least squares discriminant analysis (OPLS-DA) revealed differences in metabolic features between the tissues (R2Y = 0.937; Q2 = 0.658; Fig. 3a). The reliability of the model was confirmed by seven-fold cross-validation and 200 response permutation tests on Fig. 3b, which has showed that all permutated values were lower than the original value. It indicated that the original classification model predicted the class better than the other newly calculated OPLS-DA models with randomly assigned class labels (Q2inter = − 0.377; Fig. 3b).
Fig. 3.
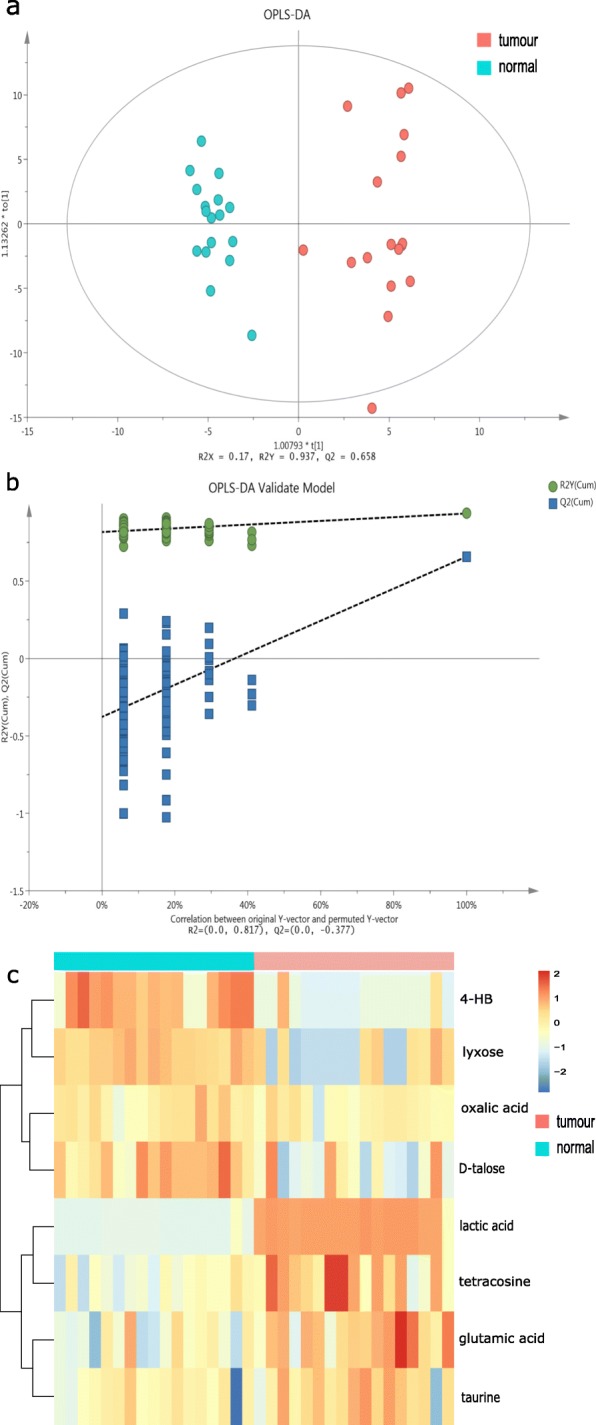
Comparison of the mucosal metabolome between tumour tissues and normal tissues in CRC patients a. OPLS-DA scores plot of metabolome between tissue samples. Each point represents one sample with colour indicating tumour tissues (red) and adjacent normal tissues (blue). b. OPLS-DA model validation based on seven-fold cross-validation and 200 permutation tests. Y axis: the R2 (green; goodness of fit) and Q2 (blue; predictability) of the all newly calculated and the original OPLS-DA models. X axis: correlation between the original Y observation and the permuted Y observation. R2inter and Q2inter are the intercept of the linear regression of R2 and Q2 from random models against those from original model. The negative Q2inter indicated the validity of the model. c. Heatmap visualizing the significantly differentially abundant metabolites between tumour (red) and normal tissues (blue) based on hierarchical clustering analysis. The rows demonstrate the metabolites and the columns display samples
Differential abundant metabolites were further identified based on Variable Influence on Projection (VIP) from the OPLS-DA model (threshold > 1) and significance level of 0.05 in the two-tailed paired t test. As shown in Fig. 3c, eight metabolites were selected, including lactic acid, 4-hydroxybutyric acid (4-HB), glutamic acid, taurine, lyxose, tetracosane, oxalic acid and D-talose. The heatmap revealed that the levels of 4-HB, lyxose, oxalic acid and D-talose were lower in tumour tissues compared to the paired normal tissues. On the other hand, the concentrations of lactic acid, glutamic acid, taurine and tetracosane were elevated in tumour tissues.
Pearson correlation analysis between changes in the level of the differentially abundant metabolites and the microbial genera in cancer and healthy tissues revealed several significant associations. The elevated abundance of genus Fusobacterium was significantly correlated with the decreased 4-HB level (r = 0.722, p-value = 0.002, degree of freedom(df) = 13; Table 1; details in Additional file 3: Figure S2). On the other hand, decrease in abundance of genus Prevotella 2 was correlated with the declined level of 4-HB in tumour tissues in comparison with matched normal tissues (r = − 0.593, p-value =0.02, df = 13; Table 1). In addition, the increase of family Halomonadaceae abundance exhibited a significant correlation with the increase in glutamic acid level between the tissues (r = 0.541, p-value = 0.037, df = 13; Table 1). The increase of lactic acid concentration in cancer tissues was also found to be significantly associated with the decrease in both genus Paeniclostridium level (r = − 0.537, p-value = 0.039, df = 13; Table 1) and genus Enterococcus level (r = − 0.527, p-value = 0.044, df = 13; Table 1).
Table 1.
Significant correlation of differentially abundant microbial taxa and metabolites (n = 15)
| Microbial taxa | Metabolites | r valuea | p-value |
|---|---|---|---|
|
Genus Fusobacterium (increased in tumour) |
4-HB (decreased in tumour) |
0.722 | 0.002 |
|
Genus Prevotella 2 (decreased in tumour) |
4-HB (decreased in tumour) |
−0.593 | 0.020 |
|
Family Halomonadaceae (increased in tumour) |
glutamic acid (increased in tumour) |
0.541 | 0.037 |
|
Genus Paeniclostridium (decreased in tumour) |
lactic acid (increased in tumour) |
−0.537 | 0.039 |
|
Genus Enterococcus (decreased in tumour) |
lactic acid (increased in tumour) |
−0.527 | 0.044 |
aFor the calculation of the correlations, the signed relative change of microbe abundance and metabolite level between paired tumour and normal tissues were used
Colonic metabolite-correlated microbial taxa associated with DNA methylation-related differential gene expression
In total, four tumour and normal tissue pairs were selected for transcriptome analysis using RNA-seq. After pre-processing and filtering, 15,832 gene transcripts were included in downstream analysis. Multi-dimensional scaling (MDS) was performed to visualise the differences in gene expression levels between cancer tissues and matched healthy tissues (Fig. 4a). After generalized linear model (GLM)-based paired differential comparison, 261 transcripts were found to be down-regulated in cancer tissues while 333 were found to be significantly up-regulated in tumour tissues (fold change (FC) > 2, p-value< 0.05; Fig. 4b).
Fig. 4.

Comparison of the mucosal transcriptome and DNA methylation profile between tumour tissues and normal tissues a. MDS plot of transcriptome between tissue samples. Each point represents one sample with colour indicating tumour tissues (red) and adjacent normal tissues (blue), shape indicating the patient. b. Volcano plot of the significant differences in the mucosal transcriptome between paired samples. The x-axis shows the gene expression log2FC between paired samples; the y-axis represents minus log10(FDR-adjusted p-value) calculated from GLM. Genes with more than two fold change in expression level are coloured in red (upregulated in tumour tissues) and green (downregulated in tumour tissues). c. MDS plot of DNA methylation for differentially expressed genes between paired tissue samples. Each point represents one sample with colour indicating tumour tissues (red) and adjacent normal tissues (blue), shape indicating the patient. d. Heatmap visualizing the DNA methylation related differentially expressed genes between tumour (red) and normal tissues (blue) based on hierarchical clustering analysis. The rows demonstrate the genes and the columns display samples
We then evaluated the methylation profile of four tumour and normal tissues pairs at the 594 differentially expressed genes, including all the probes that cover the promoters and gene body of these genes. After quality control, 14,108 probes were selected for subsequent analysis. As shown in Fig. 4c, the methylation patterns of these differentially expressed genes are clearly distinctive between the tumour and normal tissues. Of the 111 differentially methylated probes, 68 were highly methylated in cancer tissues (p-value< 0.05; Additional file 4: Table. S2). Due to the reported relationship of DNA hypermethylation located in gene promotor and gene transcription repression, genes with the opposite trend of DNA methylation at the promoter region and the corresponding gene expression level were considered as DNA methylation-related differentially expressed genes. This included sixteen genes: CHP2, EPHB1, FCRLA, FOLR2, FREM2, GAD1, GNG2, GPNMB, GRIN2D, IGSF9, LSP1, MT1M, PI16, PLCB1, RELN and SCG2 (Fig. 4d).
We further validated our hypothesis of a possible microbiome and host transcriptome interaction by analysing the correlation between the two profiles. The microbial taxa that were associated with the colonic metabolome were included. As a result, the increase in genus Fusobacterium abundance was found to be significantly associated with the down-regulated expression of PI16 (r = − 0.993, p-value = 0.008, df = 2; Table 2) and also significantly associated with the decreased expression of FCRLA (r = 0.956, p-value = 0.044, df = 2; Table 2) and LSP1 (r = 0.954, p-value = 0.046, df = 2; Table 2) in cancer tissues in comparison with the healthy tissues. The decrease in the abundance of genus Prevotella 2 was significantly associated with the decreased expression of MT1M in cancer tissues (r = − 0.999, p-value = 0.001, df = 2; Table 2); the positive abundance change of family Halomonadaceae was significantly associated with the reduction in the expression level of RELN (r = 0.957, p-value = 0.043, df = 2; Table 2). Nevertheless, the reduced abundance of genus Paeniclostridium and genus Enterococcus in cancer tissues also exhibited significant associations with the expression difference of PLCB1 (r = 0.995, p-value = 0.01, df = 2; Table 2) and IGSF9, respectively (r = − 0.997, p-value = 0.003, df = 2; Table 2).
Table 2.
Significant correlation of colonic metabolite-correlated microbial taxa and DNA methylation-related differential gene expression (n = 4)
| Microbial taxa | Genes | Coding protein | r valuea | p-value |
|---|---|---|---|---|
|
Genus Fusobacterium (increased in tumour) |
PI16 (decreased in tumour) |
Peptidase inhibitor 16 | −0.993 | 0.008 |
|
FCRLA (decreased in tumour) |
Fc Receptor Like A | 0.956 | 0.044 | |
|
LSP1 (decreased in tumour) |
Lymphocyte Specific Protein 1 | 0.954 | 0.046 | |
|
Genus Prevotella 2 (increased in tumour) |
MT1M (decreased in tumour) |
Metallothionein | −0.999 | 0.001 |
|
Family Halomonadaceae (increased in tumour) |
RELN (decreased in tumour) |
Reelin | 0.957 | 0.043 |
|
Genus Paeniclostridium (decreased in tumour) |
PLCB1 (increased in tumour) |
Phospholipase C beta 1 | 0.995 | 0.010 |
|
Genus Enterococcus (decreased in tumour) |
IGSF9 (decreased in tumour) |
Immunoglobulin Superfamily Member 9 | −0.997 | 0.003 |
aFor the calculation of the correlations, the signed relative change of microbe abundance and gene expression level between paired tumour and normal tissues were used
Discussion
In our present study, we comprehensively investigated the host-microbiome interaction in CRC by assessing multi-omics, including the mucosal microbiome, mucosal metabolome, host transcriptome and host DNA methylation profile.
We replicated several differentially abundant microorganisms as reported in previous CRC-related studies. At the genus level, potential pathogenic microorganisms such as genus Fusobacterium were found to be significantly enriched in tumour tissues compared with normal tissues in our samples. The genus Fusobacterium is a well-known potential pathogenic gut microorganism and enrichment of this genus has been reported to be associated with CRC in several studies [21–23]. The over-representation of genus Halomonas and genus Shewanella has also been revealed in tumour-associated microbiota in patients with rectal and distal colon cancers [24]. In addition, genus Peptostreptococcus has been shown to be associated with CRC in several studies [8, 25, 26]. Similarly, genus Klebsiella has been implicated in the progression of CRC [6, 27]. On the other hand, genus Lactobacillus and genus Enterococcus, were over-represented in healthy tissues in the current study, which have been long regarded as anti-inflammation and anti-tumorigenic probiotics [28, 29]. A recent study also reported that genus Lactobacillus was able to counterbalance the dysbiosis induced by CRC in human and mice [26, 30]. In an animal study, genus Enterococcus was shown to inhibit chemically-induced CRC [31].
The microbiota and its metabolic products have been reported to affect the health status of the host, especially in colon tissues [32]. In line with other metabolome studies in CRC, several metabolites were found to be significantly differentially abundant in cancer mucosal tissues in contrast to adjacent healthy tissues. Interestingly, the top three most distinguished differences between colon tissues in our study were lactic acid, 4-hydroxybutyric acid and glutamic acid. The elevation of lactic acid and glutamic acid in cancer tissues has been shown in multiple cohort studies of the CRC mucosa metabolome [33]. Additionally, taurine was found to be high in tumour tissues. Taurine is a sulphur amino acid, and studies have concluded that it is pro-inflammatory and genotoxic [34]. Oxalic acid has also been reported to be decreased in CRC samples [35]. Though tetracosane has been reported to be cytotoxic and potentially apoptotic, a high level of tetracosane in CRC tumour tissues has not yet been reported and warrants further study [36]. D-talose, which was found to be more abundant in normal tissues, is a rare sugar with possible anti-tumour application in the pharmaceutical industry [37, 38].
The key findings in our study were the associations between changes in microbial taxa, butyrate-related colonic metabolites, and DNA methylation-associated host gene expression between tumour and paired normal colon tissues. We identified a significant association between the increase of the pro-oncogenic genus Fusobacterium and decrease of 4-HB between the tissues. As a hydroxylated derivative of butyrate, 4-HB is involved in one of the four pathways of butyrate synthesis (the 4-aminobutyrate pathway) [39]. Although genus Fusobacterium, especially F. nucleatum, has been linked to colorectal tumorigenesis, the mechanism remains unclear [21–23]. Our finding suggests that, under tumour conditions, genus Fusobacterium might promote inflammatory response and carcinogenesis by interfering with butyrate synthesis in the tumour tissues. Butyrate is a short-chain fatty acid and has been revealed in several previous studies to be a potential anti-carcinogenic and anti-inflammatory metabolite [40, 41].
Butyrate is produced by the microbial fermentation of undigested polysaccharides that reach the colon [42]. In addition, butyrate also plays a crucial role in influencing gene expression in host colonic cells via epigenetic regulation [43, 44]. In human cells, there is evidence that butyrate has a direct effect on DNA methylation by regulating key enzymes, including methylcytosine dioxygenase (TET) and DNA methyltransferase 1 (DNMT1) [45, 46]. Butyrate has also been shown to modify DNA methylation and inhibit Wnt signalling in human gastric cancer cells, a pathway also known to be activated in CRC [47]. The potential association between Fusobacterium and DNA methylation-related carcinogenesis has been supported by a previous study [18]. It has also been demonstrated that Fusobacterium is enriched in CRC tissues, especially those with the methylation phenotype [22]. Butyrate may mediate the effect of Fusobacterium on host DNA methylation modifications.
In our study, the change in abundance of genus Fusobacterium was also correlated with DNA methylation-related expression differences in genes PI16, FCRLA and LSP1 in colon tissues. PI16 is a peptidase inhibitor; consistent with our findings, other studies have detected decreased expression of PI16 in colon cancer [48]. FCRLA is an unusual member of the lymphocyte receptor family and is associated with multiple Ig isotypes, including IgM, IgG and IgA [49, 50]. As a B cell-specific protein, FCRLA may play a crucial role in the mucosal immune response [51]. Down-regulation of LSP1 has been shown in several cancers and its over-expression is associated with cell apoptosis [52, 53].
Additionally, butyrate might also play a role in the effect of genus Prevotella 2 on CRC. The decrease of genus Prevotella 2 abundance and 4-HB concentration in tumour tissues showed a significant correlation. In terms of the correlation between genus Prevotella 2 and 4-HB, more detailed genomic analysis of its strains is warranted to identify the metabolic pathway. The change of genus Prevotella 2 abundance was also associated with the reduced expression of MT1M in cancer tissues compared with healthy tissues. A member of the metallothionein (MT) family, MT1M is a cysteine-rich metal-binding protein that is induced by inflammation and protects the cell against carcinogens [54].
Another observation in our study is the positive relationship between the increase of glutamic acid and microbial taxa in the potential pathogenic family Halomonadaceae in cancer tissues compared with normal tissues. In the large intestine, glutamic acid is partially originated from the digestion of alimentary and endogenous proteins by the colonic microbes [55]. Glutamic acid can also serve as butyrate precursor and at least five various pathways have been reported to be responsible for the fermentation of glutamic to butyrate by anaerobic bacteria [56]. Nonetheless, glutamic acid can also be metabolized to α-ketoglutarate (α-KG), which is co-factor of methylcytosine dioxygenase (TET) [57]. The link between Halomonadaceae and glutamic acid identified in our finding suggests that taxa in this family might involve in glutamic acid metabolism. Furthermore, the elevation of the taxa in family Halomonadaceae also correlated with the decreased expression of Reelin (RELN) in tumour tissues. The downregulation of Reelin has been found to be related to cell migration ability, tumour invasiveness and patients’ survival rates in several cancers, including gastric, breast, pancreatic and liver cancers [58–60]. Importantly, large CpG islands are located at Reelin promoter sites and its transcriptional silence has been shown to be strongly controlled by promoter hypermethylation [61]. Our finding might offer additional evidence of the mechanism of epigenetic regulation of RELN in colon cancer.
We also reported a significant association between the increase in lactic acid level and the decrease in abundance of genus Paeniclostridium as well as genus Enterococcus in cancer tissues, in contrast with normal tissues. This may in part be due to the excess lactic acid produced by cancer cells as a result of the high consumption of glucose in glycolysis, which is characteristic of tumour metabolism; this is termed the Warburg Effect [62]. Nonetheless, due to the synergic metabolism of lactate-producers such as genus Enterococcus and lactate-utilizing bacteria in the colon, less lactate might be disposed by lactate-utilizing bacteria in tumour tissues [63]. Lactate fermented by lactate-utilizing bacteria can be used to generate butyrate, which, as mentioned, might influence gene expression by modifying DNA methylation [64]. To explain the correlation between genus Paeniclostridium and lactic acid, further functional studies are required to explore their relationship. Interestingly, the reduced level of genus Enterococcus in tumour tissues was also found to be associated with the down-regulated expression of IGSF9 (Immunoglobulin Superfamily Member 9), which contributes to regulating immune cell activation [65]. Moreover, the reduced abundance of genus Paeniclostridium was related to the increased expression of Phospholipase C beta 1 (PLCB1). Phospholipase C (PLC) is critical for cell proliferation and apoptosis in several cell systems [66, 67]. The knockdown of PLCB1 is reported to initiate apoptosis, leading to strong growth arrest [68].
One limitation of our study is the use of 16sRNA sequencing technology; as such, we are unsure which specific species might distinguish tumour and normal tissues. However, the current shotgun metagenomic method is infeasible for mucosal microbiome study because of human DNA contamination [69]. Future development of sequencing technology would be critical for the identification of CRC-related microbes. Due to a relatively small sample size, the heterogeneity of CRC was also not investigated. However, our main aim was to conduct a pilot study to investigate the potential interaction between the host and microbiome from multiple “omics”. Future studies with larger sample sizes would help disentangle the complicated host-microbiome relationship in different stages and different types of CRC. Another issue is the causality of the reported relationship. Further animal intervention studies are needed to validate the correlation identified from our findings.
Apart from integrating multiple “omics”, another strength of our study is that we measured the microbiome from the mucosa sampled from CRC patients. The mucosal microbiome is different from the faecal microbiome, which is only an estimate of the former. The mucosal microbiome exhibits a more intimate and direct interaction with the host colonic epithelium; thus, it has a greater impact on the pathogenesis of disease [70].
Conclusions
In conclusion, our study identified associations between previously reported CRC-related microbial taxa, butyrate-related metabolties and DNA methylation-associated gene expression in tumour and paired normal colonic mucosa tissues from CRC patients, which uncovered a possible mechanism of the role of microbiome in the carcinogenesis of CRC. In addition, these findings offer insights into potential new biomarkers, therapeutic and/or prevention strategies for CRC.
Methods
Patient recruitment and DNA/RNA isolation
A total of 36 pairs of tumour tissues and adjacent healthy colon tissues were obtained from CRC patients in the Department of Colorectal and Anal Surgery, First Affiliated Hospital of Zhejiang University, China. Mucosa tissues were collected during surgery from both the tumour site and from an adjacent non-tumorous site that was more than 5 cm away from the tumour. The samples were flash-frozen in liquid nitrogen immediately after colonic resection and were stored at − 80 °C. Patients with diabetes or infectious diseases, those undergoing radiotherapy or chemotherapy, and those who had received antibiotics or probiotics within 4 weeks were excluded from the study. The study was approved by the Ethics Committee of the First Affiliated Hospital of the Medical School of Zhejiang University. Written informed consent was obtained from all subjects.
Total DNA and RNA were extracted from the tissue samples using AllPrep DNA/RNA mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions.
Mucosal microbiome analysis
After assessing DNA concentration and quality, PCR amplification of the V3-V4 regions of the 16S rRNA gene was performed. Then, the qualified amplicon libraries were sequenced on the Ion S5TM XL platform (Thermo Scientific) following the manufacturer’s protocol. The sequencing reads were processed using QIIME pipeline (v1.9.1) [71]. Quality filtering were performed under specific conditions according to the Cutadapt (Martin, 2011) [72]. Chimera sequences were identified using the UCHIME algorithm and were removed. After quality control and trimming, the reads were merged and clustered into operational taxonomic units (OTUs) using UPARSE software (v7.0.1001) with a 97% similarity threshold [73]. Taxonomy was assigned to each OTU cluster based on the representative reads using the RDP classifier based on the SILVA Database version 123 (confidence threshold: 80%) [74]. OTUs present in less than 25% of the total samples and with less than 3 reads were excluded. Non-metric multidimensional scaling (NMDS) was performed to visualise between-sample diversity based on unweighted UniFrac distances on OTUs using the vegan package with R software (v 2.15.3). Additionally, Analysis of similarities (Anosim) was conducted to statistically test the significance of the separation between samples. The default cumulative sum scaling (CSS) normalization procedure in metagenomeSeq R package and log transformation was used to normalize microbiome data. The paired zero-inflated Gaussian (ZIG) mixture model from the metagenomeSeq R package was applied to identify statistically significant and biologically relevant taxa in paired cancer and healthy colon mucosa samples [20].
Mucosal metabolomics analysis
50 mg of colon mucosa tissues was mixed with 800 μl ice-cold methanol and homogenized. After centrifugation, the supernatant was transferred to a new Eppendorf tube containing 20 μl of internal standard (1 mg/ml heptadecanoic). The remainder was extracted with 800 ul ice-cold methanol/chloroform (3:1, v/v) and the supernatant was then combined with the supernatant from the previous step. Next, the samples were dried under a nitrogen stream (Aosheng, Hangzhou, China). The dried residue was added to 80 μl of 20 mg/ml methoxylamine hydrochloride in anhydrous pyridine. The mixture was incubated at 37 °C for 24 h after vortexing. Then, the sample was mixed with 100 μl of N,O-bistrifluoroacetamide (BSTFA) [with 1% trimethylsilyl chloride (TMCS)] (Sigma-Aldrich, St. Louis, MO, USA) and derivatised (70 °C for 2 h). Untargeted metabolomics analysis was carried out by gas chromatography-mass spectrometry (GC-MS) with an Agilent 7890A GC and 5975C inert mass selective detector (MSD) system (Agilent Technologies, Santa Clara, CA, USA). The raw data from GC-MS was transformed by ChemStation software (version E.02.02.1431, Agilent, CA, USA) and pre-treated by ChromaTOF software (version 4.34, LECO, St. Joseph, MI, USA). The Fiehn database was referred to for identification of the metabolites. The GC-MS dataset was quantile normalized, auto-scaled and log transformed using MetaboAnalyst 4.0 (www.metaboanalyst.ca/). Subsequently, principal component analysis (PCA) and orthogonal partial least squares discriminant analysis (OPLS-DA) was performed using SIMCA (Umetrics, Umeå Sweden) to measure the differences in metabolites between the tissue samples. Model quality was summarized by R2Y and Q2 parameters, which measures the goodness of fit and the predictive ability of the model respectively (26). To measure the reliability of the OPLS-DA model from overfitting, we performed seven-fold cross-validation and 200 response permutation tests which randomly permutated the group attributes (Y) whereas the variables (X) remained same. Metabolites were considered statistically significant if the Variable Influence on Projection (VIP) value, a measure of the relative influence on the model, was greater than 1 and the p-value returned from the two-tailed Student’s t test on normalized data was less than 0.05.
Host transcriptome analysis
The RNA sequencing library was prepared as previously described [75] and sequenced on an Illumina Hiseq 2000 platform. After quality control, the paired-end clean reads were mapped to the reference genome hg19 using Hisat2 (v2.0.5). FeatureCounts (v1.5.0-p3) was used to count the number of reads mapped to genes [76]. Then, the gene expression value was quantified by the length of the gene and total read counts of the gene as count per million (CPM). The transcripts were subsequently filtered with the cut-off of CPM > 0.5 in at least 4 libraries. The bioconductor package edgeR was used to normalize and analyse differential gene expression levels between paired cancerous and non-cancerous tissues with a generalized linear model (GLM) [77]. Multi-dimensional scaling (MDS) plot was generated to visualize the difference between pairs of RNA samples. The resulting p-values were corrected using the Benjamini and Hochberg approach at False Discovery Rate (FDR) of 5% [78]. Genes with an adjusted p-value< 0.05 and fold change (FC) > 2 were considered differentially expressed between the groups.
Host methylation analysis
An EZ DNA Methylation Kit was used for bisulfite modification of the genomic DNA. Then, DNA methylation analysis was performed using the Infinium HumanMethylation850 BeadChip array (Illumina San Diego, CA, USA), which covers more than 850,000 CpG sites and 99% of the RefSeq genes. DNA methylation data analysis was performed following a cross-package Bioconductor workflow [79]. Methylation levels were quantified as beta value, which describes the proportion of methylation at each CpG locus. After background subtraction and data normalization, probes that were located at the region of the differentially expressed genes were selected for subsequent analysis. Differential DNA methylation between the groups was assessed by paired linear model using the limma R package and the p-values were adjusted for multiple testing using Benjamini-Hochberg method at FDR of 5% [78]. The probes with an adjusted p-value less than 0.05 were considered statistically significantly differentially methylated.
Interactions between omics
The correlations between change in the level of differentially abundant microbes and metabolites were estimated with Pearson correlation tests using the function cor.test in R (version 3.5.2). Due to the interdependence between paired tumour and normal tissues, the signed relative change of microbe abundance and metabolite concentration between paired tissues were used for the calculation of the correlations. Correlations were considered statistically significant if the p-value was less than 0.05. The same correlation tests were also conducted between the levels of differentially abundant microbes and differentially expressed genes that also exhibited opposite trend of differential methylation at the promoter region. Metabolites and methylation-related differentially expressed genes that were associated with the same microbe were reported.
Supplementary information
Additional file 1 Figure S1. Flow chart of study design.
Additional file 2 Table S1. Significantly differentially abundant microbial genera between tumour and normal colon tissues from CRC patients (n = 36).
Additional file 3 Figure S2. Illustration of correlation between microbial abundance and metabolite concentration.
Additional file 4 Table S2. Significantly differentially methylated probes that located within the differentially expressed genes between tumour and normal colon tissues from CRC patients (n = 4).
Acknowledgements
We acknowledge the support of the Victor Chang Cardiac Research Institute Innovation Centre, funded by the NSW Government.
About this supplement
This article has been published as part of BMC Microbiology Volume 20 Supplement 1, 2020: Proceedings of the Joint International GIW & ABACBS-2019 Conference: microbiology. The full contents of the supplement are available online at https://bmcmicrobiol.biomedcentral.com/articles/supplements/volume-20-supplement-1
Abbreviations
- CRC
Colorectal cancer
- NMDS
Non-metric multidimensional scaling
- OTU
Operational taxonomic unit
- Anosim
Analysis of similarities
- FDR
False discovery rate
- ZIG
Zero-inflated Gaussian
- GC-MS
Gas chromatography-mass spectrometry
- PCA
Principal component analysis
- OPLS-DA
Orthogonal partial least squares discriminant analysis
- VIP
Variable Influence on Projection
- MDS
Multi-dimensional scaling
- GLM
Generalized linear model
- FC
Fold change
- 4-HB
4-hydroxybutyric acid
- DNMT1
DNA methyltransferase 1
- TET
Methylcytosine dioxygenase
Authors’ contributions
QW, JZ.Y, DQ.F and LX.L designed and coordinated the study. JWKH, EG and LL supervised the project. QW and WR.W collected the samples. QW, DS, YT.L, LY.Y, XY.B and JJ.W carried out the experiments. XW.J, KC.W and QQ.W helped collect clinical data. MPH contributed to metabolome data analysis. QW, LT, JWKH and EG participated in the data analysis and result interpretation. QW wrote the manuscript. LT, MPH, JWKH, EG and LJ.L reviewed the manuscript. All authors have contributed and approved the final manuscript.
Funding
Funding for study design, data collection and data generation was provided by the National Natural Science Foundation of China (grant nos. 81790631, 81570512), the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (grant no. 81721091) and the National Key Research and Development Program of China (2018YFC2000500). JWKH participated in the data analysis and result interpretation and was supported by Career Development Fellowship by the National Health and Medical Research Council (1105271) and Future Leader Fellowship by the National Heart Foundation of Australia (100848). EG participated in the data analysis and result interpretation and is supported by the NSW Health Early-Mid Career Fellowship and the National Heart Foundation of Australia Future Leader Fellowship (101204). Publication costs are funded by the National Natural Science Foundation of China (grant no. 81790631).
Availability of data and materials
The datasets used and/or analysed during the current study are available from Sequence Read Archive (SRA) database (accession number: PRJNA599023). All data generated or analysed during this study are also available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All participants in this study provided written informed consent before sample collection. The research is in accordance with the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University (permit number: 2017–658-1).
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Eleni Giannoulatou, Email: e.giannoulatou@victorchang.edu.au.
Lanjuan Li, Email: ljli@zju.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12866-020-01762-2.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Raskov H, Pommergaard H-C, Burcharth J, Rosenberg J. Colorectal carcinogenesis-update and perspectives. World J Gastroenterol. 2014;20:18151. doi: 10.3748/wjg.v20.i48.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bingham SA. Diet and colorectal cancer prevention. Biochem Soc Trans. 2000;28:12–16. doi: 10.1042/bst0280012. [DOI] [PubMed] [Google Scholar]
- 4.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–329. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal Cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health: nutrition, epigenetics and health. Acta Physiol. 2011;202:103–118. doi: 10.1111/j.1748-1716.2011.02278.x. [DOI] [PubMed] [Google Scholar]
- 10.Sproul D, Meehan RR. Genomic insights into cancer-associated aberrant CpG island hypermethylation. Brief Funct Genomics. 2013;12:174–190. doi: 10.1093/bfgp/els063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mischke M, Plösch T. More than just a gut instinct-the potential interplay between a baby’s nutrition, its gut microbiome, and the epigenome. Am J Phys Regul Integr Comp Phys. 2013;304:R1065–R1069. doi: 10.1152/ajpregu.00551.2012. [DOI] [PubMed] [Google Scholar]
- 13.Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell. 2016;64:982–992. doi: 10.1016/j.molcel.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Q, Wang K, Wu W, Giannoulatou E, Ho JWK, Li L. Host and microbiome multi-omics integration: applications and methodologies. Biophys Rev. 2019;11:55–65. doi: 10.1007/s12551-018-0491-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 16.Paul B, Barnes S, Demark-Wahnefried W, Morrow C, Salvador C, Skibola C, et al. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin Epigenetics. 2015;7:112. doi: 10.1186/s13148-015-0144-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi K, Sugi Y, Nakano K, Tsuda M, Kurihara K, Hosono A, et al. Epigenetic control of the host gene by commensal Bacteria in large intestinal epithelial cells. J Biol Chem. 2011;286:35755–35762. doi: 10.1074/jbc.M111.271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahara T, Hirata I, Nakano N, Tahara S, Horiguchi N, Kawamura T, et al. Potential link between Fusobacterium enrichment and DNA methylation accumulation in the inflammatory colonic mucosa in ulcerative colitis. Oncotarget. 2017;8:61917–61926. doi: 10.18632/oncotarget.18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Häsler R, Sheibani-Tezerji R, Sinha A, Barann M, Rehman A, Esser D, et al. Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut. 2017;66(12):2087–2097. doi: 10.1136/gutjnl-2016-311651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–1202. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014;74:1311–1318. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–1980. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flemer B, Lynch DB, Brown JMR, Jeffery IB, Ryan FJ, Claesson MJ, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66:633–643. doi: 10.1136/gutjnl-2015-309595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology. 2017;152:1419–1433.e5. doi: 10.1053/j.gastro.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Hibberd AA, Lyra A, Ouwehand AC, Rolny P, Lindegren H, Cedgård L, et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterology. 2017;4:e000145. doi: 10.1136/bmjgast-2017-000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antonic V, Stojadinovic A, Kester KE, Weina PJ, Brücher BL, Protic M, et al. Significance of infectious agents in colorectal Cancer development. J Cancer. 2013;4:227–240. doi: 10.7150/jca.5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hove H, Nùrgaard H, Mortensen PB. Review lactic acid bacteria and the human gastrointestinal tract. Eur J Clin Nutr. 1999;53(5):339–350. doi: 10.1038/sj.ejcn.1600773. [DOI] [PubMed] [Google Scholar]
- 29.Zhong L, Zhang X, Covasa M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J Gastroenterol. 2014;20:7878–7886. doi: 10.3748/wjg.v20.i24.7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacouton E, Chain F, Sokol H, Langella P, Bermúdez-Humarán LG. Probiotic strain Lactobacillus casei BL23 prevents colitis-associated colorectal Cancer. Front Immunol. 2017;8:1553. doi: 10.3389/fimmu.2017.01553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sivieri K, Spinardi-Barbisan ALT, Barbisan LF, Bedani R, Pauly ND, Carlos IZ, et al. Probiotic Enterococcus faecium CRL 183 inhibit chemically induced colon cancer in male Wistar rats. Eur Food Res Technol. 2008;228:231–237. doi: 10.1007/s00217-008-0927-6. [DOI] [Google Scholar]
- 32.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian Colon. Cell Metab. 2011;13:517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu Y, Cai G, Zhou B, Li D, Zhao A, Xie G, et al. A distinct metabolic signature of human colorectal Cancer with prognostic potential. Clin Cancer Res. 2014;20:2136–2146. doi: 10.1158/1078-0432.CCR-13-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mal M, Koh PK, Cheah PY, Chan ECY. Metabotyping of human colorectal cancer using two-dimensional gas chromatography mass spectrometry. Anal Bioanal Chem. 2012;403:483–493. doi: 10.1007/s00216-012-5870-5. [DOI] [PubMed] [Google Scholar]
- 36.Uddin SJ, Grice D, Tiralongo E. Evaluation of cytotoxic activity of patriscabratine, tetracosane and various flavonoids isolated from the Bangladeshi medicinal plant Acrostichum aureum. Pharm Biol. 2012;50:1276–1280. doi: 10.3109/13880209.2012.673628. [DOI] [PubMed] [Google Scholar]
- 37.Sznaidman ML, Hecht SM. Studies on the total synthesis of tallysomycin. Synthesis of the threonylbithiazole moiety containing a structurally unique glycosylcarbinolamide. Org Lett. 2001;3:2811–2814. doi: 10.1021/ol0101178. [DOI] [PubMed] [Google Scholar]
- 38.Van Overtveldt S, Gevaert O, Cherlet M, Beerens K, Desmet T. Converting Galactose into the rare sugar Talose with Cellobiose 2-Epimerase as biocatalyst. Molecules. 2018;1:23(10). doi: 10.3390/molecules23102519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vital M, Howe AC, Tiedje JM. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)genomic Data. mBio. 2014;5(2):e00889. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. 2016;13:691–706. doi: 10.1038/nrgastro.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17:1519–1528. doi: 10.3748/wjg.v17.i12.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 43.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer R-J. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Fan C, Liang A, Fan X, Wang R, Li P, et al. Effects of SCFA on the DNA methylation pattern of adiponectin and resistin in high-fat-diet-induced obese male mice. Br J Nutr. 2018;120:385–392. doi: 10.1017/S0007114518001526. [DOI] [PubMed] [Google Scholar]
- 45.Wei YB, Melas PA, Wegener G, Mathé AA, Lavebratt C. Antidepressant-like effect of sodium butyrate is associated with an increase in TET1 and in 5-Hydroxymethylation levels in the Bdnf gene. Int J Neuropsychopharmacol. 2014;31:18(2). doi: 10.1093/ijnp/pyu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. Histone Deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011;31:2723–2732. [PubMed] [Google Scholar]
- 47.Shin H, Kim J-H, Lee YS, Lee YC. Change in gene expression profiles of secreted frizzled-related proteins (SFRPs) by sodium butyrate in gastric cancers: induction of promoter demethylation and histone modification causing inhibition of Wnt signaling. Int J Oncol. 2012;40:1533–1542. doi: 10.3892/ijo.2012.1327. [DOI] [PubMed] [Google Scholar]
- 48.Li B-Q, Huang T, Liu L, Cai Y-D, Chou K-C. Identification of colorectal Cancer related genes with mRMR and shortest path in protein-protein interaction network. PLoS One. 2012;7(4):e33393. doi: 10.1371/journal.pone.0033393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson TJ, Gilfillan S, Colonna M. Fc receptor-like a associates with intracellular IgG and IgM but is dispensable for antigen-specific immune responses. J Immunol. 2010;185:2960–2967. doi: 10.4049/jimmunol.1001428. [DOI] [PubMed] [Google Scholar]
- 50.Santiago T, Kulemzin SV, Reshetnikova ES, Chikaev NA, Volkova OY, Mechetina LV, et al. FCRLA is a resident endoplasmic reticulum protein that associates with intracellular Igs, IgM, IgG and IgA. Int Immunol. 2011;23:43–53. doi: 10.1093/intimm/dxq456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang W, Feng Q, Wang C, Zeng X, Du Y, Lin L, et al. Characterization of the B cell receptor repertoire in the intestinal mucosa and of tumor-infiltrating lymphocytes in colorectal adenoma and carcinoma. J Immunol. 2017;198:3719–3728. doi: 10.4049/jimmunol.1602039. [DOI] [PubMed] [Google Scholar]
- 52.Stone J, Thompson DJ, Dos Santos SI, Scott C, Tamimi RM, Lindstrom S, et al. Novel associations between common breast Cancer susceptibility variants and risk-predicting mammographic density measures. Cancer Res. 2015;75:2457–2467. doi: 10.1158/0008-5472.CAN-14-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S, Zheng Y, Ogundiran TO. Ojengbede O, Zheng W, Nathanson KL, et al. Association of Pancreatic Cancer Susceptibility Variants with risk of breast Cancer in women of European and African ancestry. Cancer Epidemiol Biomark Prev. 2018;27:116–118. doi: 10.1158/1055-9965.EPI-17-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bremner I. Nutritional and physiological significance of metallothionein. Experientia Suppl. 1987;52:81–107. doi: 10.1007/978-3-0348-6784-9_5. [DOI] [PubMed] [Google Scholar]
- 55.Blachier F, Boutry C, Bos C, Tomé D. Metabolism and functions of l-glutamate in the epithelial cells of the small and large intestines. Am J Clin Nutr. 2009;90:814S–821S. doi: 10.3945/ajcn.2009.27462S. [DOI] [PubMed] [Google Scholar]
- 56.Buckel W. Unusual enzymes involved in five pathways of glutamate fermentation. Appl Microbiol Biotechnol. 2001;57:263–273. doi: 10.1007/s002530100773. [DOI] [PubMed] [Google Scholar]
- 57.Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16:694–707. doi: 10.1038/nrc.2016.82. [DOI] [PubMed] [Google Scholar]
- 58.Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology. 2006;130:548–565. doi: 10.1053/j.gastro.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 59.Stein T, Cosimo E, Yu X, Smith PR, Simon R, Cottrell L, et al. Loss of Reelin expression in breast Cancer is epigenetically controlled and associated with poor prognosis. Am J Pathol. 2010;177:2323–2333. doi: 10.2353/ajpath.2010.100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okamura Y, Nomoto S, Kanda M, Hayashi M, Nishikawa Y, Fujii T, et al. Reduced expression of reelin (RELN) gene is associated with high recurrence rate of hepatocellular carcinoma. Ann Surg Oncol. 2011;18:572–579. doi: 10.1245/s10434-010-1273-z. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Sharma RP, Costa RH, Costa E, Grayson DR. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30:2930–2939. doi: 10.1093/nar/gkf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberti MV, Locasale JW. The Warburg effect: how does it benefit Cancer cells? Trends Biochem Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sato T, Matsumoto K, Okumura T, Yokoi W, Naito E, Yoshida Y, et al. Isolation of lactate-utilizing butyrate-producing bacteria from human feces and in vivo administration of Anaerostipes caccae strain L2 and galacto-oligosaccharides in a rat model. FEMS Microbiol Ecol. 2008;66:528–536. doi: 10.1111/j.1574-6941.2008.00528.x. [DOI] [PubMed] [Google Scholar]
- 64.Bourriaud C, Robins RJ, Martin L, Kozlowski F, Tenailleau E, Cherbut C, et al. Lactate is mainly fermented to butyrate by human intestinal microfloras but inter-individual variation is evident. J Appl Microbiol. 2005;99:201–212. doi: 10.1111/j.1365-2672.2005.02605.x. [DOI] [PubMed] [Google Scholar]
- 65.Hansen M, Walmod PS. IGSF9 family proteins. Neurochem Res. 2013;38:1236–1251. doi: 10.1007/s11064-013-0999-y. [DOI] [PubMed] [Google Scholar]
- 66.Faenza I, Fiume R, Piazzi M, Colantoni A, Cocco L. Nuclear inositide specific phospholipase C signalling - interactions and activity. FEBS J. 2013;280:6311–6321. doi: 10.1111/febs.12450. [DOI] [PubMed] [Google Scholar]
- 67.Li J, Zhao X, Wang D, He W, Zhang S, Cao W, et al. Up-regulated expression of phospholipase C, β1 is associated with tumor cell proliferation and poor prognosis in hepatocellular carcinoma. Onco Targets Ther. 2016;9:1697–1706. doi: 10.2147/OTT.S97189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fiume R, Huang X, Ramazzotti G, Mongiorgi S, Santi P, Somervaille T, et al. Phospholipase c beta 1 (PLCb1) in acute myeloid leukemia (AML): a novel potential therapeutic target. Ital J Anat Embryol. 2014;119:88. doi: 10.13128/IJAE-15925. [DOI] [Google Scholar]
- 69.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 70.Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A, et al. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J Gastroenterol. 2015;50:167–179. doi: 10.1007/s00535-014-0963-x. [DOI] [PubMed] [Google Scholar]
- 71.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJournal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 73.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 74.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haberman Y, Tickle TL, Dexheimer PJ, Kim M-O, Tang D, Karns R, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 77.Chen Y, Lun ATL, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438. doi: 10.12688/f1000research.8987.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 79.Maksimovic J, Phipson B, Oshlack A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Res. 2016;5:1281. doi: 10.12688/f1000research.8839.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Figure S1. Flow chart of study design.
Additional file 2 Table S1. Significantly differentially abundant microbial genera between tumour and normal colon tissues from CRC patients (n = 36).
Additional file 3 Figure S2. Illustration of correlation between microbial abundance and metabolite concentration.
Additional file 4 Table S2. Significantly differentially methylated probes that located within the differentially expressed genes between tumour and normal colon tissues from CRC patients (n = 4).
Data Availability Statement
The datasets used and/or analysed during the current study are available from Sequence Read Archive (SRA) database (accession number: PRJNA599023). All data generated or analysed during this study are also available from the corresponding author on reasonable request.