

Abstract
Amariin is an ellagitannin with two dehydrohexahydroxydiphenoyl (DHHDP) moieties connecting glucose 2,4- and 3,6-hydroxy groups. This tannin is predominant in the young leaves of Triadica sebifera and Carpinus japonica. However, as the leaves grow, the 3,6-DHHDP is converted to its reduced form, the hexahydroxydiphenoyl (HHDP) group, to generate geraniin, a predominant ellagitannin of the matured leaves. The purified amariin is unstable in aqueous solution, and the 3,6-(R)-DHHDP is spontaneously degraded to give HHDP, whereas 2,4-(R)-DHHDP is stable. The driving force of the selective reduction of the 3,6-DHHDP of amariin is shown to be the conformational change of glucose from O,3B to 1C4. Heating geraniin with pyridine affords 2,4-(R)-DHHDP reduction products. Furthermore, the acid hydrolysis of geraniin yields two equivalents of ellagic acid. Although the reaction mechanism is still ambiguous, these results propose an alternative biosynthetic route of the ellagitannin HHDP groups.
Keywords: dehydrohexahydroxydiphenic acid, hexahydroxydiphenic acid, amariin, geraniin, ellagitannin
1. Introduction
Ellagitannin is a group of polyphenols having hexahydroxydiphenoyl (HHDP) ester or its metabolites connected to mostly glucose [1,2]. Due to its high structural diversity and wide distribution in the plant kingdom, ellagitannins continue to attract intense interest within the natural product and organic chemistry communities [3,4]. The accumulation of ellagitannins in some plants suggests that these tannins play a significant role in plant defense systems [5,6,7], while the antiproliferative and antioxidative activities of ellagitannins in fruits and medicinal plants are well studied [8,9,10,11,12,13,14]. However, the biosynthesis of ellagitannins including the production mechanism of the HHDP group remains obscure. The HHDP group was supposed to be formed by oxidative coupling between two galloyl esters on glucopyranose, and this mechanism has been evidenced by the enzymatic preparation of ellagitannin from a gallotannin [15,16,17]. In addition, many successful total ellagitannin syntheses are also based on similar biomimetic strategies [18,19]. However, in 1993, Foo reported an important finding [20]. More specifically, that amariin, 1-O-galloyl-2,4;3,6-bis-(R)-dehydrohexahydroxydiphenoyl (DHHDP)-β-d-glucose (1), degraded spontaneously in aqueous ethanol to give the reduction product geraniin, 1-O-galloyl-2,4-(R)-DHHDP-3,6-(R)-HHDP-β-d-glucopyranose (2) as a major product [21,22]. Based on the implicit assumption that polyphenols are susceptible to oxidation, most natural product chemists supposed that DHHDP is generated by the oxidation of HHDP. However, Foo’s finding indicated that HHDP is possibly generated by the reduction of DHHDP. In this study, we investigated the reductive conversion of the DHHDP esters to HHDP esters.
2. Results and Discussion
2.1. Composition of Ellagitannins in Young and Matured Leaves.
In our continuous chemical study on ellagitannins, we found that similar reductive production of HHDP from DHHDP occurs in the fresh leaves of Triadica sebifera (syn. Sapium sebiferum). Amariin (1) was predominant in the small young leaves at the top of the twig collected in June; however, 1 was not detected in the larger and harder leaves of the same twig (Figure 1 and Supplementary Figure S1). Leaves of different sizes, ranging between the smallest leaf at the top and the large matured leaf were lyophilized, and the 60% acetonitrile extracts were analyzed by HPLC. The concentration of geraniin (2) per leaf increased proportionally with the leaf size and reached a plateau (Figure 2). In contrast, the concentration of 1 per leaf first increased as the leaves grew and then decreased as the leaves matured. These observations suggest that 2 is biosynthesized via 1. Similarly, 1 was predominant in the young leaves of Carpinus laxiflora (collected in April) (Figure 3) and decreased as the leaves grew; however, here 2 become predominant instead of 1 in the matured leaves in October [23]. A significant amount of elaeocarpsin, an ascorbic acid adduct of 2, was also detected in the matured leaves [24]. Furthermore, the young soft leaves of Elaeocarpus sylvestris var. ellipticus contain 1 as the major ellagitannin accompanied by less 2 content, and the matured, hard leaves contain 2 (Figure S2). These findings also suggest that 1 is a biosynthetic precursor of 2.
Figure 1.

HPLC profiles (310 nm) of extracts of the leaves of Triadica sebifera.
Figure 2.

Concentration of amariin (1) and geraniin (2) per leaf at different growing stages. A–D: see Figure 1.
Figure 3.

HPLC profiles of extracts of Carpinus laxiflora fresh leaves collected in April (A) and collected in October (B), and amariin (1) isolated from the April leaves (C).
A: young leaf at the top of a twig (leaf size about 1 cm in diameter), D: matured leaf of the same twig (about 8.5 cm in diameter), B (about 2 cm) and C (about 3.5 cm) are the leaves between A and D (50 mg of lyophilized leaf was extracted with 1.5 mL of 60% CH3CN, 0.1% TFA).
2.2. Structure of Isoamariin
In the young leaves of C. laxiflora, 1 was accompanied by a minor isomer named isoamariin (3). The molecular formula C41H28O28 was confirmed by high resolution fast atom bombardment mass spectrometry (HRFABMS) (m/z: 991.0656 [M + Na]+, calcd for C41H28O28Na, 991.0665). The 1H and 13C NMR spectra are closely related to 1. They indicated that the molecular parts of 3 are the same as those of 1. The NMR spectra exhibited duplicated signals caused by an equilibrium between the 6- and 5-membered ring hemiacetal structures of DHHDP group connected to the glucose 2,4-positions. In contrast to the 3,6-DHHDP of 1, the 3,6-DHHDP of 3 was almost fixed in the 6-membered ring form. In addition, the HMBC correlations (Figure 4) revealed that 3 is an isomer of 1, differing in the orientation of the 3,6-DHHDP on glucopyranose. The configuration of the DHHDP group was concluded to be R based on large negative and positive Cotton effects at 236 and 209 nm, respectively, in the electronic circular dichroism (ECD) spectrum. The structure of 3 is the same as that previously reported for didehydrogeraniin [25]; however, the 1H NMR chemical shifts of the didehydrogeraniin phenazine derivative in the literature coincided with those of the phenazine derivative of 1 and did not match the data for the phenazine derivative of 3 (Supplementary Table S1).
Figure 4.
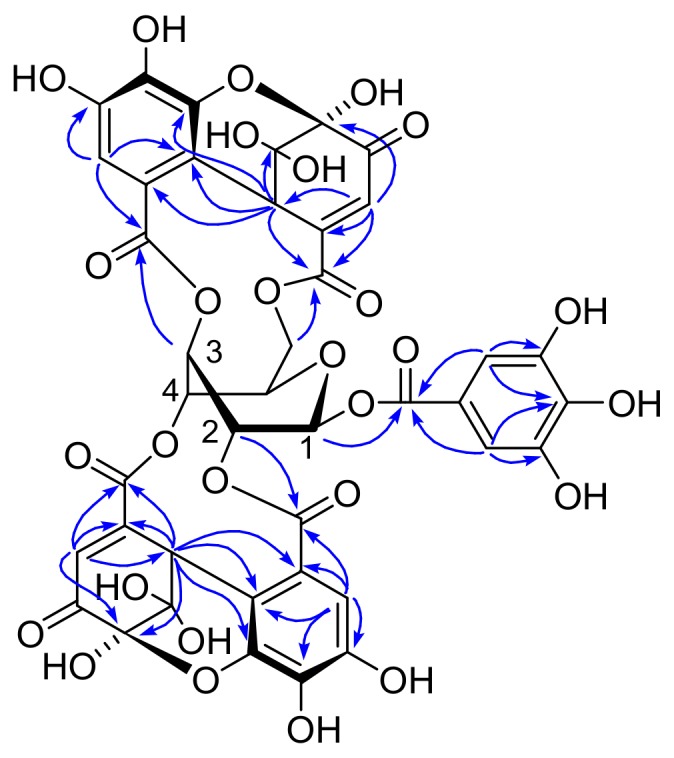
Selected HMBC correlations of major component of 3.
As reported by Foo [20], in aqueous solution 1 decomposed to give reduction product 2 as a major product (Scheme 1, Supplementary Figure S3). Similarly, isoamariin (3) was also decomposed under the same conditions to yield 2 (Supplementary Figure S4). Considering that reduction of 1 to 2 proceeded without any reductant, the reaction was presumed to be reduction-oxidation disproportionation. The isolation yield of 2 from 1 in citrate-phosphate buffer at pH 6 was 47%. Other products were identified to be 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucopyranose (furosin) [25], 1-O-galloyl-3,6-(R)-HHDP-β-d-glucopyranose (corilagin) [21], gallic acid, ellagic acid, and a new compound, 1-O-galloyl-3,6-(S)-HHDP-d-glucose (Supplementary Figure S5). The structure of the new compound was determined based on electrospray ionization (ESI)MS, HRFABMS, 1H, 13C NMR, 1H-1H correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond correlation (HMBC) and ECD spectra as shown in the Supplementary Materials (Figure S6). The 3,6-(S)-HHDP glucose is rare in nature; however, 1,2,4-tri-O-galloyl-3,6-(S)-HHDP-α-d-glucose is one of the major ellagitannin of Nuphar japonicum [26]. At the present time, we have not succeeded in identifying the oxidation products that are expected to be generated together with the reduction products.
Scheme 1.

Production of geraniin (2) from amariin (1) and isoamariin (3) in citrate-phosphate buffer (pH 6) at room temperature.
Only the 3,6-DHHDP of the two DHHDP groups of 1 selectively undergo the reduction; however, computational calculation indicated that the structures of the 2,4- and 3,6-DHHDP moieties are both ridged and essentially the same. The difference of the reactivity is thought to be caused by a difference in the strain from glucose to the DHHDP groups. The coupling constants of glucopyranose (J1,2 = 5.1 Hz, J2,3 = 1.3 Hz, J3,4 = 3.7 Hz, and J4,5 = 1.5 Hz) suggest that conformation of the glucopyranose of 1 is of the O,3B form. In contrast, X-ray crystallography confirmed that the glucopyranose core of 2 adopts the 1C4 conformation [27]. Due to the difficulty in analyzing the NMR signals caused by the equilibrium of the DHHDP groups, a bisacetonyl derivative 1a was prepared to fix the DHHDP groups in the 5-membered ring hemiacetal form [28], and DFT calculation was applied to 1a. The coupling constants observed for glucopyranose of 1a were essentially the same as those of 1, and they were also in agreement with those of 1a obtained by DFT calculation. The experimental NOEs observed for 1a agreed with the calculation results (Figure 5). Calculation also indicated that a conformer with 1C4-glucopyranose is less stable than in the O,3B form (ΔG = + 4.5 kcal/mol). In contrast, 2 prefers the 1C4 conformation, and 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucose is also shown to be adapted to 1C4 conformation based on the coupling constants and DFT calculation. These observations suggested that the difference in the reactivity of 3,6- and 2,4-DHHDP of 1 was due to the difference of strain present from the glucose to ester carbonyl groups.
Figure 5.

Selected NOE correlations and stereostructure obtained by DFT calculation of 1a.
DHHDP is regarded as a hydrated quinone dimer of galloyl groups. Similar pyrogallol-quinone dimers of epigallocatechin (4), that is, dehydrotheasinensins (5) are produced during the tea fermentation process in black tea manufacturing (Scheme 2) [29,30]. The catechin dimers are unstable and decompose during the drying process of black tea production to generate reduction products, theasinensins (6), as the major products. Its hexahydroxybiphenyl structure is structurally related to the HHDP group of ellagitannins. Experiments using pure dehydrotheasinensins showed that the reaction is reduction-oxidation disproportionation. However, only limited oxidation products, such as oolongtheanins (7) [31], were identified due to the complexity of the oxidation products. Theasinensins are catechin oxidation products characteristic of black tea, and the dimerization process seems to be related to the 3,6-HHDP formation of 2. The aforementioned production of the 3,6-(S)- and 3,6-(R)-HHDP glucoses from the (R)-DHHDP of 1 was also related to the production of theasinensins with R and S biphenyl bond, respectively, from (S)-dehydrotheasinensins.
Scheme 2.

Reduction-oxidation disproportionation of catechin quinone dimer during black tea production.
Previously, we reported that heating of 2 with pyridine in CH3CN yielded reduction products mallotusinin (8) with a characteristic dibenzofuran structure, 1-O-galloyl-2,4;3,6-bis-(R)-HHDP-β-d-glucose (10), and a decarboxylation product (11) [32]. The major product was 8 (30%, recovery of 2 was 15%)(Figure 6). In this study, an additional product 9 was isolated. The 1H and 13C NMR spectra indicated that 9 is a pyridine adduct of 8. The binding site of the pyridine was confirmed to be at the glucose C-4 side of the dibenzofuran moiety by HMBC correlations of pyridinium H-2,6 with the benzofuran 3’ carbon (Figure 7). This was supported by a large up field shift of the glucose H-4 (Δδ 0.67) as compared to that of 8. The production mechanism of 9 from 2 is proposed as shown in Scheme 3, and this may be related to the reduction of 2 to 8. The attack of hydride instead of pyridine produces 8.
Figure 6.

HPLC profiles of geraniin (2) (A) and reaction mixture after treatment with 4% pyridine in CH3CN (80 ˚C, 90 min) (B).
Figure 7.

Products obtained by treatment of 2 with pyridine in CH3CN (80 °C, 90 min) and HMBC correlations of 9.
Scheme 3.

Plausible production mechanism of 9 from 2.
In this experiment, we could not identify the expected oxidation products; however, oligomeric products (isolation yield 9.4%) may be candidates. The mixture of oligomers was detected at the origin during thin layer chromatography (TLC) and as a broad hump on the baseline of the HPLC (Figure 6 and Figure S7). The 13C NMR spectrum showed signals attributable to galloyl and HHDP esters together with glucose (Figure S8). Methylation and alkaline degradation of the oligomers confirmed the presence of galloyl, HHDP, and tetrahydroxy dibenzofuran moieties; however, we could not obtain evidence that the oligomers are oxidation products of 2.
Similar but contradictory results were obtained during acid hydrolysis. Foo reported that acid hydrolysis of amariin (1) gave ellagic acid [20], and Okuda et al. also indicated the production of excess ellagic acid upon acid hydrolysis of 2 [22]. These results suggested that reduction of the DHHDP esters also occurs during hydrolysis. Reexamined the acid hydrolysis of 2 with 2% H2SO4 at 100 ˚C afforded ellagic acid in the isolation yield of 180 mol%. Furthermore, the acid hydrolysis of 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucopyranose with 2% H2SO4 also yielded gallic acid and ellagic acid in the molar ratio of 1:0.73 (Supplementary Figure S9). These results suggested that the reduction of DHHDP to HHDP during the hydrolysis is not reduction-oxidation disproportionation. In this reaction, a possible electron donor candidate was glucose simultaneously generated by the hydrolysis; however, the 13C NMR spectrum and TLC of the sugar fraction obtained by hydrolysis confirmed the recovery of glucose.
3. Materials and Methods
3.1. General
Ultraviolet (UV) spectra were obtained on a JASCO V-560 UV/VIS spectrophotometer (JASCO, Tokyo, Japan). Optical rotations were measured with a JASCO DIP-370 digital polarimeter. The ECD spectra were measured with a JASCO J-725N spectrophotometer. 1H and 13C NMR spectra were recorded on a Varian Unity Plus 500 spectrometer (Agilent Technologies, Santa Clara, CA, USA) operating at 500 and 125 MHz for the 1H and 13C nuclei, respectively. NMR spectra were also recorded on a JEOL JNM-AL 400 spectrometer (JEOL Ltd., Tokyo, Japan) operating at 400 and 100 MHz for the 1H and 13C nuclei, respectively. ESIMS were obtained using a JEOL JMS-T100TD spectrometer. FABMS were recorded on a JMS700N spectrometer (JEOL Ltd.) using m-nitrobenzyl alcohol or glycerol as the matrix. Column chromatography was performed using Sephadex LH-20 (25–100 mm, GE Healthcare UK Ltd., Little Chalfont, UK), MCI-gel CHP20P (75–150 mm, Mitsubishi Chemical Co., Tokyo, Japan), Diaion HP20SS (Mitsubishi Chemical Co.), Toyopearl Butyl-650C (Tosoh Bioscience Japan, Tokyo, Japan), Cosmosil 75C18OPN (Nacalai Tesque Inc., Kyoto, Japan), and Chromatorex ODS (Fuji Silysia Chemical Ltd., Kasugai, Japan) columns. TLC was performed on precoated Kieselgel 60 F254 plates (0.2-mm thickness, Merck, Darmstadt, Germany), using toluene–ethyl formate–formic acid (1:7:1, v/v) and CHCl3–MeOH–H2O (7:3:0.5, v/v) mixtures as the eluents. The spots were detected using ultraviolet illumination and by spraying with 2% ethanolic FeCl3 solution (for phenolic compounds), 5% anisaldehyde in ethanolic 5% H2SO4 solution (for proanthocyanidins), or 5% H2SO4 solution followed by heating. Analytical HPLC was performed on a Cosmosil 5C18-ARII (Nacalai Tesque Inc., Kyoto, Japan) column (250 × 4.6 mm, i.d.) with a gradient elution of 4–30% (39 min) and 30–75% (15 min) CH3CN in 50 mM H3PO4 at 35 °C (flow rate, 0.8 mL/min; detection, JASCO photodiode array detector MD-2018 plus). Geraniin was obtained as yellow crystalline powder from previous our studies [33].
3.2. Plant Material
Fresh leaves of Triadica sebifera and Elaeocarpus sylvestris var. ellipticus were collected in Bunkyo campus of Nagasaki University. Fresh leaves of Carpinus laxiflora were collected Mt. Gokahara, Isahaya, Nagasaki prefecture.
3.3. Quantification of Ellagitannins in the Leaves of T. Sebifera
Fresh leaves of T. sebifera were lyophilized and powdered by grinding in a mortar. Aliquots (50 mg) of the powder (each 4 samples) were extracted with 60% CH3CN containing 0.1% trifluoroacetic acid (1.5 mL) at room temperature for 7 h. The extracts were analyzed by HPLC. Calibration curves were built using geraniin (320 nm) isolated in our previous study [24] and amariin (275 nm) isolated in this study. Average dried weights of the leaves in the Figure 1 were as follows: A, 0.007 g/leaf (fresh: 0.022 g/leaf); B, 0.029 g/leaf (fresh: 0.089 g/leaf); C, 0.065g/leaf (fresh: 0.20 g/leaf), and D, 0.195 g/leaf (fresh: 0.63 g/leaf).
3.4. Isolation of Amariin (1) and Isoamariin (3)
Fresh young leaves of Carpinus laxiflora (750 g) collected in April 30, 2014 were homogenized with 80% acetone containing 0.005% trifluoroacetic acid (TFA) (3 L) at room temperature 2 times. The extract was filtered, and the filtrate was concentrated using rotary evaporator. Resulting precipitates formed in the aqueous solution were removed by filtration. The filtrate (about 800 mL) was applied to Diaion HP20SS (8 cm i.d. × 37 cm) column with 0.005% TFA containing increasing proportions of MeOH [0% (500 mL), 20% (1 L), 30% (1 mL), 40% (1 L), 50% (1 L), 60% (1 L), 80% (1 L), and 100% (2 L)] to give 5 fractions. Fraction (Fr.) 3 (16.8 g) mainly containing 1 was subjected to Sephadex LH-20 column chromatography (5 cm i.d. × 22 cm) with 0%–100% MeOH in 0.005% TFA (20% stepwise, each 300 mL) to yield a crude sample of 1 (10.9 g). A portion (7.5 g) of the crude sample was purified by Diaion HP20SS column chromatography (4 cm i.d. × 27 cm, 0%–60% MeOH in 0.005% TFA, 5% stepwise, each 200 mL). Tubes containing 1 were combined and concentrated by rotary evaporator (40 ˚C) to give yellow precipitates of 1, which were collected by filtration (3.7 g). The filtrate (0.76 g) was separated by Chromatorex ODS (3 cm i.d. × 25 cm, 0%–50% MeOH in 0.005% TFA, 5% stepwise, each 100 mL) to yield 3 (121 mg).
Amariin (1): Yellow powder, 1H NMR (500 MHz, acetone-d6) δ: galloyl: 7.23, 7.22 (each s, galloyl-2,6); DHHDP (5- and 6-membered-ring hemiacetal structure): 7.26, 7.238, 7.236, 7.235, 7.233 (each s, H-3’), 6.71, 6.67, 6.60, 6.58 [each s, H(6)-3], 6.31, 6.26 [each d, J = 1.5 Hz, H(5)-3], 5.31. 5.29, 5.27, 5.26 [each s, H(6)-1], 4.90, 4.88, 4.86, 4.85 [each d, J = 1,5 Hz, H(5)-1]; glucose (two major isomers among 4 isomers), isomer A: 6.40 (d, J = 5.4 Hz, glc-H-1), 5.79 (dd, J = 1.5, 3.2 Hz, glc-3), 5.50 (br d, J = 3.2 Hz, glc-4), 5.47 (dt, J = 1.5, 5.4 Hz, glc-2), 5.23 (dd, J = 2.7, 13.3 Hz, glc-6), 4.77 (br s, glc-5), 4.39 (br d, J = 13.3 Hz, glc -6); isomer B: 6.35 (d, J = 5.1 Hz, glc-1), 5.65 (dt, J = 1.3, 3.7 Hz, glc-4), 5.62 (dt, J = 1.5, 5.1 Hz, glc-2), 5.53 (dd, J = 1.5, 3.7 Hz, glc-3), 5.33 (dd, J = 2.6, 13.3 Hz, glc-6), 4.83 (br s, glc-5), 4.39 (br d, J = 13.3 Hz, glc -6); 13C NMR (125 MHz, acetone-d6) δ: only signals of major isomers COO: 168.4, 168.1, 166.2, 165.1, 165.0, 164.8, 164.7, 164.6, 164.4; galloyl: 146.21, 146.18 (C-3,5), 139.9, 139.2 (C-4), 110.40, 110.39 (C-2,6); DHHDP: 194.2 [C(5)-4], 191.6, 191.2 [C(6)-4], 153.6, 152.5, 152.1 [C(6)-2], 148.6, 147.8. 147.3 [C(5)-6’], 143.5, 143.0, 142.9 [C(6)-6’], 129.2, 128.6 [C(6)-3], 125.7 [C(5)-3], 96.6, 96.2 [C(6)-5], 92.31, 92.27, 92.0 [C(5)-5, C(6)-6], 52.2, 51.1 [C(5)-1], 46.4, 45.0, 44.8 [C(6)-1]; other aromatic and sp2 carbons: 145.93, 145.86, 145.83, 139.2, 138.5, 138.4, 137.6, 120.3, 120.0, 119.9, 119.8, 119.7, 118.7, 117.0, 116.0, 115.0, 114.9, 113.9, 113.6, 113.1; glucose (two major isomers among 4 isomers): 95.7, 94.4 (glc-1), 78.1, 77.7 (glc-5), 73.7, 72.7 (glc-2), 70.4, 69.7 (glc-4), 67.6,66.1 (glc-3), 66.0, 65.8 (glc-6).
Isoamariin (3): Yellow powder, [α −136.8 (c 0.1, MeOH); FAB-MS m/z: 991 [M + Na]+; HR-FAB-MS m/z: 991.0656 [M + Na]+ (calcd. for C41H28O28Na, 991.0665); IR νmax cm−1: 3413, 1705, 1613, 1528, 1447, 1325, 1213; UV (MeOH) λmax (log ε): 279 (4.43), 221 (4.89); ECD (MeOH) λmax (Δε): 369 (−3.9), 326 (−1.4), 293 (−8.6), 270 (−2.1), 262 (−2.4), 257 (−2.1), 236 (−14.7), 228 (0), 209 (+36.4). 1H NMR (500 MHz, acetone-d6) δ: galloyl: 7.21, 7.20 (each s, galloyl-2,6); 2,4-DHHDP 5-membered-ring hemiacetal structure: 7.29 (s, H-3’), 6.26 (d, J = 1.4 Hz, H-3), 4.95 (d, J = 1.4 Hz, H-1); 2,4-DHHDP 6-membered-ring hemiacetal structure: 7.27 (s, H-3’), 6.60 (s, H-3), 5.35 (s, H-1); glucose with 5-membered-ring hemiacetal 2,4-DHHDP: 6.42 (d, J = 5.2 Hz, glc-1), 5.67 (dt, J = 1.3, 5.2 Hz, glc-2), 5.51 (dd, J = 1.3, 3.7 Hz, glc-3), 6.17 (dt, J = 1.3, 3.7 Hz, glc-4), 4.72 (br s, glc-5), 4.60 (dd, J = 3.1, 13.3 Hz, glc-6), 4.90 (dd, J = 1.7, 13.3 Hz, glc -6); glucose with 6-membered-ring hemiacetal 2,4-DHHDP: 6.36 (d, J = 4.7 Hz, glc-1), 5.78 (dt, J = 1.4, 4.7 Hz, glc-2), 5.36 (dd, J = 1.4, 3.7 Hz, glc-3), 6.28 (dt, J = 1.3, 3.5 Hz, glc-4), 4.76 (br s, glc-5), 4.62 (dd, J = 3.1, 12.3 Hz, glc-6), 4.93 (dd, J = 1.8, 12.3 Hz, glc-6); 13C NMR (125 MHz, acetone-d6) δ: galloyl: 119.7, 119.9 (galloyl-1), 110.3 (galloyl-2,6), 146.2 (galloyl-3,5), 139.8, 139.9 (galloyl-4), 164.8, 164.9 (galloyl-7); 2,4-DHHDP 5-membered-ring hemiacetal structure: 52.1 (C-1), 148.9 (C-2), 125.8 (C-3), 194.3 (C-4), 92.4 (C-5), 109.3 (C-6), 117.1 (C-1’), 120.3 (C-2’), 113.1 (C-3’), 147.8 (C-4’), 137.6 (C-5’), 147.3 (C-6’); 2,4-DHHDP 6-membered-ring hemiacetal structure: 46.4 (C-1), 154.0 (C-2), 129.3 (C-3), 191.7 (C-4), 96.2 (C-5), 92.4 (C-6); 116.1 (C-1’), 119.0 (C-2’), 113.6 (C-3’), 145.9 (C-4’), 139.2 (C-5’), 144.0 (C-6’); glucose with 5-membered-ring hemiacetal 2,4-DHHDP: 94.3 (glc-1), 74.2 (glc-2), 67.5 (glc-3), 69.6 (glc-4), 77.1 (glc-5), 68.0 (glc-6); glucose with 6-membered-ring hemiacetal 2,4-DHHDP: 95.4 (glc-1), 73.6 (glc-2), 69.0 (glc-3), 68.7 (glc-4), 76.5 (glc-5), 67.9 (glc-6).
3.5. Preparation of Acetonyl Derivative
Amariin (1) (130 mg) was dissolved in 80% acetone (20 mL) containing HCO2NH4 (130 mg) and stirred at r.t. for 13h. After removal of acetone by evaporation, the aqueous solution was subjected to Diaion HP20SS column chromatography (2 cm i.d. × 15 cm, 0%–60% MeOH in H2O, 5% stepwise, each 50 mL) to yield 1a (57.2 mg).
Bisacetonyl derivative (1a): off-white amorphous powder, [α −55.2 (c 0.1, MeOH); IRνmax cm−1: 3406, 1715, 1620, 1531, 1441; UV (MeOH) λmax (log ε): 282 (4.56), 221 (4.91). 1H NMR (500 MHz, acetone-d6) δ: 1H-NMR (acetone-d6, 500 MHz) δ: 2.19 (3H, s, 2,4-acyl-CH3), 2.23 (3H, s, 3,6-acyl-CH3), 2.96 (1H, d, J = 15.3 Hz, 3,6-acyl-CH2), 3.06 (1H, d, J = 15.6 Hz, 2,4-acyl-CH2), 3.46 (1H, d, J = 15.6 Hz, 2,4-acyl-CH2), 3.48 (1H, d, J = 15.3 Hz, 3,6-acyl-CH2), 4.21 (1H, dd, J = 1.2, 13.1 Hz, glc-6a), 4.83 (1H, d, J = 1.5 Hz, 3,6-acyl-1), 4.88 (1H, br. s, glc-5), 4.98 (1H, d, J = 1.5 Hz, 2,4-acyl-1’), 5.39 (1H, dt, J = 1.4, 3.8 Hz, glc-4), 5.47 (1H, dt, J = 1.5, 4.9 Hz, glc-2), 5.57 (1H, dd, J = 3.2, 13.1 Hz, glc-6b), 5.70 (1H, dd, J = 1.4, 3.8 Hz, glc-3), 6.35 (1H, d, J = 1.5 Hz, 2,4-acyl-3’), 6.44 (1H, d, J = 4.9 Hz, glc-1), 6.46 (1H, d, J = 1.5 Hz, 3,6-acyl-3), 7.20 (1H, s, 3,6-acyl-3’), 7.22 (1H, s, 2,4-acyl-3), 7.27 (2H, s, galloyl-H); 13C NMR (acetone-d6, 125 MHz) δ: 32.0 (2,4-acyl-CH3), 32.2 (3,6-acyl-CH3), 49.2 (3,6-acyl-CH2), 49.9 (2,4-acyl-CH2), 50.8 (3,6-acyl-1), 51.9 (2,4-acyl-1’), 64.5 (glc-6), 66.0 (glc-3), 69.7 (glc-4), 73.8 (glc-2), 78.1 (glc-5), 80.2 (3,6-acyl-5), 80.6 (2,4-acyl-5’), 94.3 (glc-1), 110.0 (2,4-acyl-6’), 110.4 (galloyl-2, 6), 110.6 (3,6-acyl-6), 113.0 (2,4-acyl-3, 3,6-acyl-3’), 116.9 (2,4-acyl-1), 117.9 (3,6-acyl-1’), 119.9 (3,6-acyl-2’), 120.1 (galloyl-1), 120.5 (2,4-acyl-2), 127.1 (3,6-acyl-3), 127.5 (2,4-acyl-3’), 136.5 (3,6-acyl-5’), 137.3 (2,4-acyl-5), 139.6 (galloyl-4), 144.6 (3,6-acyl-2), 144.9 (2,4-acyl-2’), 146.1 (galloyl-3, 5), 146.7 (3,6-acyl-6’), 147.0 (2,4-acyl-6), 147.4 (2,4-acyl-4, 3,6-acyl-4’), 164.3 (2,4-acyl-7), 164.9 (galloyl-7, 3,6-acyl-7), 166.1 (2,4-acyl-7’), 166.9 (3,6-acyl-7’), 196.9 (3,6-acyl-4), 197.2 (2,4-acyl-4’), 205.7 (2,4-acyl-CO), 205.9 (3,6-acyl-CO).
3.6. Preparation of Phenazine Derivative
Amariin (1) (50 mg) and o-phenylenediamine (10 mg) was dissolved in 10% AcOH in EtOH (2 mL) and heated at 50 ˚C for 2 h. The mixture was applied to Sephadex LH-20 (2 cm i.d. × 15 cm, 0-20% H2O in EtOH, 10% stepwise, each 100 mL) to yield 1b (30.2 mg). Phenazine derivative 3a was prepared from 3 in the same way.
Phenazinde derivative (1b): UV (MeOH) λmax (log ε) 445 (3.56), 378 (4.13), 281 (4.98), 205 (4.87); ECD (MeOH) Δε (nm) 377 (−1.48), 325 (+3.86), 284 (−67.5), 250 (+72.1), 221 (−42.0). 1H NMR (acetone-d6, 500 MHz) δ: 4.11 (1H, dd, J = 3.3, 12.0 Hz, glc-6a), 4.98 (1H, dd, J = 8.1, 12.0, glc-6b), 5.04 (1H, dd, J = 3.3, 8.1, glc-5), 5.40 (1H, d, J = 4.1, glc-4), 5.80 (1H, d, J = 4.1, glc-3), 5.86 (1H, d, J = 5.5, glc-2), 6.23 (1H, d, J = 5.5, glc-1), 7.01 (2H, s, galloyl-H), 7.03 (1H, s, 3,6-acyl-3’), 7.52 (1H, s, 2,4-acyl-3), 8.24 (1H, s, 3,6-acyl-3), 8.31 (1H, s, 2,4-acyl-3’); 13C-NMR (acetone-d6, 125 MHz) δ: 64.7 (glc-6), 67.7 (glc-3), 68.3 (glc-4), 76.2 (glc-2), 77.0 (glc-5), 91.7 (glc-1), 109.7 (2,4-acyl-3), 110.1 (galloyl-2, 6), 113.2 (3,6-acyl-3’), 115.2 (3,6-acyl-1’), 116.4 (2,4-acyl-1), 116.7 (3,6-acyl-1), 116.8 (2,4-acyl-1’), 119.8 (galloyl-1), 120.1 (2,4-acyl-3’), 120.6 (2,4-acyl-2), 120.7 (3,6-acyl-3), 123.1 (3,6-acyl-2’), 130.0, 130.4, 130.5 (2,4-acyl-4”, 5”, 3,6-acyl-4”, 5”), 132.2, 132.3 (2,4-acyl-3”, 6”, 3,6-acyl-3”, 6”), 136.0 (2,4-acyl-2’), 136.3 (3,6-acyl-2), 137.4 (3.6-acyl-5’), 138.0, 139.0 (2,4-acyl-5’, 3,6-acyl-5), 139.3 (2,4-acyl-5), 139.6 (galloyl-4), 142.8, 143.0, 143.1, 145.1, 145.3, 145.4, 145.6, 145.9 (2,4-acyl-6, 4’, 1”, 2”, 3,6-acyl-4, 4’, 6’, 1”, 2”), 145.3 (2,4-acyl-4), 146.0 (galloyl-3, 5), 152.1 (3,6-acyl-6), 152.3 (2,4-acyl-6’), 164.7 (galloyl-7), 165.9 (3,6-acyl-7), 166.6 (2,4-acyl-7’), 167.8 (3,6-acyl-7’), 167.9 (2,4-acyl-7).
Phenazine derivative (3a): Brown amorphous powder, [α −323.9 (c 0.06, MeOH); FAB-MS m/z: 1077 [M + H]+, 1099 [M + Na]+; HR-FAB-MS m/z: 1099.1403 [M + Na]+, (calcd. for C53H32O22N4Na, 1099.1406); IR vmax cm−1: 3336, 1732, 1613, 1509, 1463, 1355, 1335, 1308, 1189; UV (MeOH) λmax (log ε): 446 sh (3.51), 375 (4.15), 279 (5.00), 223 sh (4.86), 207 (4.89); ECD (MeOH) λmax (Δε): 381 (+3.8), 359 (+1.3), 304 (+16.9), 292 (0), 274 (−79.0), 257 (0), 247 (+29.4), 230 (0), 220 (−24.8), 212 (0); 1H NMR (acetone-d6, 500 MHz) δ: 4.12 (1H, t, J = 5.1, 11.6 Hz, glc-6a), 4.84 (1H, dd, J = 8.5, 11.6, glc-6b), 5.11 (1H, dd, J = 5.1, 8.5, glc-5), 5.48 (1H, d, J = 4.1, glc-3), 5.63 (1H, dd, J = 0.7, 4.1, glc-4), 5.71 (1H, d, J = 5.7, glc-2), 6.22 (1H, d, J = 5.7, glc-1), 6.99 (2H, s, galloyl-H), 7.16, 7.46, 7.86, 8.27 (each 1H, each s, aromatic) 7.93–8.01, 8.15–8.36 (each m, phenazine-H); 13C NMR (acetone-d6, 125 MHz) δ: 66.7, 68.2, 68.7, 76.2, 76.8 (glc-2~6), 91.7 (glc-1), 110.2 (galloyl-2,6), 113.5 (acyl-3), 115.7, 116.0, 116.6, 117.0, 118.5 (galloyl-1, acyl-1, 1’), 120.1, 120.2, 120.5, 122.7 (acyl-2, 3’), 130.2, 130.6, 130.7 (acyl-4”, 5”), 132.2, 132.4, 132.5 (acyl-3”, 6”), 136.1, 136.2 (acyl-2’), 138.2, 139.2, 139.5, 139.8, 139.9, 142.8, 142.9, 143.3, 143.5, 145.2, 145.3, 145.5, 145.7, 145.9 (galloyl-4, acyl-4, 5, 6, 4’, 5’, 1”, 2”), 146.2 (galloyl-3, 5), 152.3, 152.4 (acyl-6’), 164.9, 166.2, 166.6, 167.5, 167.9 (esters).
3.7. Treatment of 1 at pH 6
Amariin (1) (1.0 g) was dissolved in citrate-phosphate buffer (0.05 M, pH 6.0) and stirred at r.t. for 20 h. The mixture was acidified by addition of 5% TFA to pH 2 and applied to Sephadex LH-20 column chromatography (3 cm i.d. × 30 cm, 0%–100% MeOH in H2O, 20% stepwise, each 200 mL, followed by MeOH-acetone-H2O, 90:6:4, 80:12:8, 70:18:12, each 200 mL) to yield 11 fractions. Fr. 3 (23.5 mg) was identified as gallic acid. Fr. 4 (60.6 mg) was subjected to Chromatorex ODS column chromatography (2 cm i.d. × 17 cm, 0–70% MeOH in H2O, 5% stepwise, each 100 mL) to give 1-O-galloyl-3,6-(S)-HHDP-β-d-glucose (17.6 mg). Fr. 5 (43.7 mg) was purified by MCI gel CHP 20P column chromatography (2 cm i.d. × 17 cm, 0-50% MeOH in H2O, 5% stepwise, each 100 mL) to afford 1-O-galloyl-3,6-(R)-HHDP-β-d-glucose (corilagin, 24.0 mg). Similar separation of Fr. 7 (63.9 mg) yielded 1-O-galloyl-2,3-(R)-DHHDP-β-d-glucose (furosin, 34.4 mg). Fractions 9 and 10 were identified to be geraniin (2)(463.9 mg) and ellagic acid (48.0 mg), respectively.
1-O-Galloyl-3,6-(S)-HHDP-β-d-glucose: Brown amorphous powder; [α +20.7° (c 0.10, MeOH); UV (MeOH) λmax (log ε): 293 (4.36), 273 (4.49), 227 (4.80), 219 (4.88); IR νmax cm−1: 3378, 1709, 16213; ESI-MS (negative) m/z: 633 [M − H]−; HR-FAB-MS m/z: 657.0705 (calcd for C27H22O18Na, 657.0703); ECD (MeOH) Δε (nm): +3.38 (229), −2.16 (267), +1.45 (293); 1H NMR (acetone-d6, 500 MHz) δ: 7.21 (2H, s, galloyl-H-2,6), 7.23 (1H, s, HHDP-H-3), 7.01 (1H, s, HHDP-H-3’), 6.29 (1H, d, J = 9.0 Hz, glc-1), 5.10 (1H, dt, J = 1.1, 5.7 Hz, glc-3), 4.88 (1H, d, J = 12.3 Hz, glc-6), 4.25 (1H, br d, J = 2.4 Hz, glc-5), 3.96 (1H, dt, J = 5.7, 9.0 Hz, glc-2), 3.84 (1H, dd, J = 3.4, 12.3 Hz, glc-6), 3.59 (1H, br s, glc-4); 13C NMR (acetone-d6, 125 MHz) δ: 168.0 (HHDP-7), 167.7 (HHDP-7’), 166.1 (galloyl-7), 145.8 (galloyl-3,5), 144.6, 144.5 (HHDP-4,4’,6,6’), 139.1 (galloyl-4), 138.1 (HHDP-5), 136.4 (HHDP-5’), 126.4 (HHDP-2, 2’), 121.2 (galloyl-1), 117.5 (HHDP-1), 116.7 (HHDP-1’), 112.4 (HHDP-3’), 110.3 (galloyl-2,6), 108.1 (HHDP-3’), 92.3 (glc-1), 81.2 (glc-3), 80.1 (glc-5), 72.7 (glc-4), 70.2 (glc-2), 63.3 (glc-6).
3.8. Heating of 2 with Pyridine in CH3CN
A solution of 2 (2.0 g) in 4% pyridine in CH3CN (50 mL) was heated at 80 ˚C for 90 min. After cooling, the solution was mixed with 2% aqueous TFA (25 mL) and concentrated. Resulting aqueous solution was applied to Sephadex LH-20 column chromatography (3 cm i.d. × 25 cm, 60%–100% MeOH in H2O, 10% stepwise, each 100 mL, followed by MeOH-acetone-H2O, 80:10:10, 200 mL) to give 7 fractions. Fr. 3 (123.8 mg) was mainly composed of 1-O-galloyl-3,6-(R)-HHDP-β-d-glucose (corilagin) and product 9. Fr. 4 (78.7 mg) contained corilagin, 9, and ellagic acid. Fr. 5 (473.5 mg) was separated by Chromatorex ODS (4 cm i.d. × 25 cm, 0%–55% MeOH in H2O, 5% stepwise, each 100 mL) to give 2 (290.6 mg) and a mixture of 9 and 11, which were separated by Diaion HP20SS (3 cm i.d. × 17 cm, 10–60% MeOH in H2O, 5% stepwise, each 50 mL) followed by purification using Chromatorex ODS chromatography (3 cm i.d. × 28 cm) to yield 9 (23.2 mg) and 11 (39.3 mg). Fr. 6 was separated H2O soluble and insoluble parts, and the soluble fraction was purified by Avicel cellulose column chromatography (2 cm i.d. × 15 cm) with 2% AcOH to give 1-O-galoyl-2,3;4,6-bis-(R)-HHDP-β-d-glucose (10) (30.9 mg). The H2O insoluble precipitates were combined with Fr. 7 and subjected to size-exclusion column chromatography using Sephadex LH-20 (2 cm i.d. × 55 cm) with 7M urea:acetone (2:3, v/v, containing conc. HCl at 5 mL/L)[34] to give fractions containing oligomeric polyphenols and 8. The removal of urea by Diaion HP20SS column chromatography afforded 8 (595.8 mg) and oligomeric polyphenols (188.3 mg).
Pyridine adduct (9): A tan amorphous powder, [α −47.5 (c 0.1, MeOH); IR νmax cm−1: 3383, 1730, 1616, 1344; UV (MeOH) λmax (log ε): 349 (3.88), 280 (4.48), 217 (4.78). ESI-MS m/z: 996 [M + H]+, m/z: 995 [M − H]−; HRFABMS: m/z 996.1108 [M + H]+ (calcd for C46H30NO25, 996.1107); 1H NMR (acetone-d6, 500 MHz) δ: 9.53, 9.08 [each 1H, br d, J = 6.4 Hz, pyridine (py)-2,6], 8.62, 8.45 (each 1H, br t, J = 6.4 Hz, py-3,5), 7.37 [1H, s, benzofuran (BF)-3], 7.09 (2H, s, galloyl-2,6), 7.02 (1H, s, HHDP-3), 6.67 (1H, s, HHDP-3’), 6.39 (1H, br s, glc-3), 6.20 (1H, d, J = 3.1 Hz, glc-1), 5.39 (1H, br s, glc-2), 4.70 (1H, d, J = 3.8 Hz, glc-4), 4.41 (2H, m, glc-5, 6), 4.18 (1H, dd, J = 5.1, 10.6 Hz, glc-6); 13C NMR (acetone-d6, 125 MHz) δ: 168.3 (HHDP-7’), 166.7 (BF-7), 166.6 (HHDP-7), 165.2 (BF-7’), 165.1 (galloyl-7), 149.7, 149.2 (py-2,6), 147.9, 147.7, 147.5 (py-4, BF-4’,6’), 146.0 (galloyl-3,5), 145.9 (BF-6), 145.13, 145.06, 145.0, 144.6 (HHDP-4,4’,6,6’), 140.8 (BF-5’), 139.9 (galloyl-4), 137.3 (HHDP-5), 136.7 (HHDP-5’), 135.8 (BF-5), 134.6 (BF-2’), 129.8, 129.0 (py-3,5), 125.9 (BF-3’), 124.4, 123.8 (HHDP-2,2’), 119.6 (galloyl-1), 117.0 (BF-3), 116.7 (HHDP-1), 115.8 (BF-2), 115.7 (HHDP-1’), 115.1 (BF-1’), 114.3 (BF-1), 110.3 (galloyl-2,6), 109.5 (HHDP-3), 108.8 (HHDP-3’), 91.8 (glc-1), 74.7 (glc-5), 73.6 (glc-2), 69.3 (glc-4), 64.2 (glc-6), 62.1 (glc-3).
3.9. Methylation and Alkaline Degradation of Oligomeric Product
Oligomeric products (100 mg) in methanol (2 mL) was treated with etherial diazomethane for 2 days. The mixture was heated with 5% NaOH at 80 °C for 12 h, acidified with diluted HCl, and then partitioned with diethyl ether. The ether layer was concentrated and treated with diaomethane, and the products were separated by silica gel column chromatography (30%–100% acetone in hexane, 10% stepwise, each 50 mL) to yield methyl trimethoxybenzoate (5 mg), dimethyl (R)-hexamethoxydiphenate (2.0 mg) and dimethoxycarbonyl tetramethoxydibenzofuran (1.7 mg).
3.10. Acid Hydrolysis of DHHDP Esters
A portion (0.50 mL) of an acetone solution of 2 (10.0 mg/mL), 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucopyranose (6.0 mg/mL), and 1-O-galloyl-3,6-(R)-HHDP-β-d-glucopyranose (6.0 mg/mL) was dispensed into 5 mL screw-capped vials, and acetone was removed in vacuo. To each vial, 0.50 mL of 2% H2SO4 was added and heated at 100 °C for 7 h. The reaction mixture was mixed with DMSO (4.5 mL) to dissolve ellagic acid and analyzed by HPLC (Figure S9). Yield of the products was estimated based on the peak area and calibration curve obtained by using standard compounds: 4.76 μmol of gallic acid and 10.09 μmol of ellagic acid was produced from 5.25 μmol of 2. Similarly, 4.16 μmol of gallic acid and 3.05 μmol of ellagic acid was produced from 4.61 μmol of 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucopyranose, and 4.84 μmol of gallic acid and 5.59 μmol of ellagic acid was produced from 4.72 μmol of 1-O-galloyl-3,6-(R)-HHDP-β-d-glucopyranose.
3.11. Acid Hydrolysis of 2
Geraniin (2) (1.00 g, 1.05 mmol, dried over P4O10) was hydrolyzed with 2% H2SO4 (50 mL) under reflux for 2 days. Insoluble precipitates were collected by filtration and washed with MeOH to give ellagic acid (504.5 mg, 1.67 mmol). The filtrate was directly subjected to Diaion HP20SS column chromatography (3 cm i.d. × 25 cm, 0%–80% MeOH in H2O, 10% stepwise, each 100 mL) to yield gallic acid (206.2 mg, 1.21 mmol), flavogallonic acid (9.8 mg, 0.02 mmol), and ellagic acid (66.0 mg, 0.22 mol). Total isolation yield of ellagic acid was 1.89 mmol.
3.12. Computational Methods
A conformational search was performed using the Monte Carlo method and the MMFF94 force field with Spartan ′14 (Wavefunction, Irvine, CA, USA). The obtained low-energy conformers within 6 kcal/mol were optimized at the B3LYP/6-31G(d,p) level in acetone (PCM). The vibrational frequencies were also calculated at the same level to confirm their stability, and no imaginary frequencies were found. 1H NMR coupling constants of the low-energy conformers with Boltzmann populations greater than 1% were calculated at the B3LYP/6-31G(d,p)u + 1s (using only the Fermi contact term) level in acetone (PCM) and scaled by using the slope parameter 0.94 [35]. The calculated data for each conformer were averaged according to the Boltzmann distribution theory at 298 K based on their relative Gibbs free energies. All DFT calculations were performed using Gaussian 09 [36]. GaussView was used to draw the three-dimensional molecular structures [37].
4. Conclusions
The results presented and described in this paper strongly suggest that HHDP is produced by the reduction of DHHDP, at least in the case of 3,6-HHDP of geraniin (2) from amariin (1) and isoamariin (3). This may be the key mechanism in the ellagitannin biosynthesis, which otherwise remains unclear. The similarity to the reaction mechanism for the epigallocatechin B-B’ ring biphenyl bond formation during black tea production also supports our hypothesis. The difference in reactivity between 3,6- and 2,4-DHHDP is probably explained by tensile forces from glucose, which depend on the conformation of the pyranose ring. In our investigation on the production mechanism of HHDP from DHHDP, understanding the missing electron donor is the most critical issue. However, presently, we can only introduce the experimental results as described herein.
Acknowledgments
The authors are grateful to N. Tsuda, K. Chifuku, and H. Iwata at the Center for Industry, University and Government Cooperation, Nagasaki University, for recording the NMR and MS data, and M. Tanaka (Graduate School of Biomedical Sciences, Nagasaki University) for providing access to the ECD spectrometer. The computation was partly carried out using the computer facilities at the Research Institute for Information Technology, Kyushu University. This work was the result of using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting introduction of the new sharing system) Grant Number JPMXS0422500320.
Supplementary Materials
The following are available online. Figure S1. The leaves of Triadica sebifera used for the HPLC analysis, Figure S2. HPLC profiles of 60% CH3CN extracts of Elaeocarpus sylvestris var. ellipticus fresh leaves, Table S1 1H NMR data for phenazine derivatives. Figure S3. HPLC profiles of aqueous solution of amariin (1) in pH 6 citrate-phosphate buffer, Figure S4. HPLC profiles of isoamariin (3) in pH 6 citrate-phosphate buffer, Figure S5. Structures of minor products produced by degradation of 1, Figure S6. HMBC correlations and ECD spectra of 1-O-galloyl-3,6-(S)-HHDP-β-d-glucose. Figure S7. HPLC profile (A) and UV spectrum (B) of oligomeric polyphenol fraction obtained from 2, Figure S8. 13C NMR spectrum of oligomeric polyphenol fraction measured in acetone-d6. Figure S9. HPLC profiles of acid hydrolysis of 2, 1-O-galloyl-2,4-(R)-DHHDP-β-d-glucopyranose, and 1-O-galloyl-3,6-(R)-HHDP-β-d-glucopyranose. Figures S10–S31, 1D, 2D NMR spectra of compounds 1, 1a, 3, 3a, and 9, and computational methods.
Author Contributions
T.T. and Y.M. conceived and designed the experiments, and analyzed the data; M.E. performed the experiments and analyzed the data. Y.M. and M.E. performed the DFT calculation. M.E., Y.S. and T.T. discussed the conclusion and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant Nos. 17K08338 and 16K07741).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples are not available from the authors.
References
- 1.Okuda T., Yoshida T., Hatano T. Hydrolyzable tannins and related polyphenols. In: Herz W., Kirby G.W., Moore R.E., Steglich W., Tamm C., editors. Progress in the chemistry of organic natural products. Springer-Verlag; New York: 1995. pp. 1–117. [DOI] [PubMed] [Google Scholar]
- 2.Haslam E., Cai Y. Plant polyphenols (vegetable tannins): gallic acid metabolism. Nat. Prod. Rep. 1994;11:41–66. doi: 10.1039/np9941100041. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida T., Hatano T., Ito H., Okuda T. Structural diversity and antimicrobial activities of ellagitannins. In: Quideau S., editor. Chemistry and Biology of Ellagitannins, an Underestimated Class of Bioactive Plant Polyphenols. World Scientific Publishing; Singapore: 2009. pp. 55–93. [Google Scholar]
- 4.Quideau S., Feldman K.S. Ellagitannin chemistry. Chem. Rev. 1996;96:475–504. doi: 10.1021/cr940716a. [DOI] [PubMed] [Google Scholar]
- 5.Anstett D.N., Cheval I., D’Souza C., Salminen J., Johnson M.T.J. Ellagitannins from the Onagraceae decrease the performance of generalist and specialist herbivores. J. Chem. Ecol. 2019;45:86–94. doi: 10.1007/s10886-018-1038-x. [DOI] [PubMed] [Google Scholar]
- 6.Barbehenn R.V., Jones C.P., Hagerman A.E., Karonen M., Salminen J. Ellagitannins have greater oxidative activities than condensed tannins and galloyl glucoses at high pH: potential impact on caterpillars. J. Chem. Ecol. 2006;32:2253–2267. doi: 10.1007/s10886-006-9143-7. [DOI] [PubMed] [Google Scholar]
- 7.Salminen J., Roslin T., Karonen M., Sinkkonen J., Pihlaja K., Pulkkinen P. Seasonal variation in the content of hydrolyzable tannins, flavonoid glycosides, and proanthocyanidins in oak leaves. J. Chem. Ecol. 2004;30:1693–1711. doi: 10.1023/B:JOEC.0000042396.40756.b7. [DOI] [PubMed] [Google Scholar]
- 8.Londhe J.S., Devasagayam T.P.A., Foo L.Y., Shastry P., Ghaskadbi S.S. Geraniin and amariin, ellagitannins from Phyllanthus amarus, protect liver cells against ethanol induced cytotoxicity. Fitoterapia. 2012;83:1562–1568. doi: 10.1016/j.fitote.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Ross H.A., McDougall G.J., Stewart D. Antiproliferative activity is predominantly associated with ellagitannins in raspberry extracts. Phytochemistry. 2007;68:218–228. doi: 10.1016/j.phytochem.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Adams L.S., Zhang Y., Seeram N.P., Heber D., Chen S. Pomegranate ellagitannin–derived compounds exhibit antiproliferative and antiaromatase activity in breast cancer cells in vitro. Cancer Prev. Res. 2010;3:108–113. doi: 10.1158/1940-6207.CAPR-08-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parashar A., Gupta C., Gupta S.K., Kumar A. Antimicrobial ellagitannin from pomegranate (Punica granatum) Int. J. Fruit Sci. 2009;9:226–231. doi: 10.1080/15538360903241286. [DOI] [Google Scholar]
- 12.Ito H. Metabolites of the ellagitannin geraniin and their antioxidant activities. Planta Med. 2011;77:1110–1115. doi: 10.1055/s-0030-1270749. [DOI] [PubMed] [Google Scholar]
- 13.Landete J. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food Res. Int. 2011;44:1150–1160. doi: 10.1016/j.foodres.2011.04.027. [DOI] [Google Scholar]
- 14.Cerdá B., Hilary S., Espín J.C. Metabolism of Antioxidant and Chemopreventive Ellagitannins from Strawberries, Raspberries, Walnuts, and Oak-Aged Wine in Humans: Identification of Biomarkers and Individual Variability. J. Agric. Food Chem. 2005;53:227–235. doi: 10.1021/jf049144d. [DOI] [PubMed] [Google Scholar]
- 15.Gross G.G. Biosynthesis of ellagitannins: Old ideas and new solutions. In: Quideau S., editor. Chemistry and Biology of Ellagitannins, an Underestimated Class of Bioactive Plant Polyphenols. World Scientific Publishing; Singapore: 2009. pp. 94–118. [Google Scholar]
- 16.Niemetz R., Gross G.G. Enzymology of gallotannin and ellagitannin biosynthesis. Phytochemistry. 2005;66:2001–2011. doi: 10.1016/j.phytochem.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Niemetz R., Gross G.G. Ellagitannin biosynthesis: laccase-catalyzed dimerization of tellimagrandin II to cornusiin E in Tellima grandiflora. Phytochemistry. 2003;64:1197–1201. doi: 10.1016/j.phytochem.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Feldman K.S., Sahasrabudhe K., Quideau S., Hunter K.L., Lawlor M.D. Prospects and progress in ellagitannin synthesis. In: Gross G.G., Hemingway R.W., Yoshida T., editors. Plant Polyphenols 2: Chemistry, Biology, Pharmacognosy, Ecology. Kluwer Academic/Plenum Publishers; New York, NY, USA: 1999. pp. 101–125. [Google Scholar]
- 19.Khanbabaee K. Strategies for the synthesis of ellagitannins. In: Quideau S., editor. Chemistry and Biology of Ellagitannins, an Underestimated Class of Bioactive Plant Polyphenols. World Scientific Publishing; Singapore: 2009. pp. 152–202. [Google Scholar]
- 20.Foo L.Y. Amariin, a di-dehydrohexahydroxydiphenoyl hydrolysable tannin from Phyllanthus amarus. Phytochemistry. 1993;33:487–491. doi: 10.1016/0031-9422(93)85545-3. [DOI] [Google Scholar]
- 21.Okuda T., Yoshida T., Hatano T. Constituents of Geranium thunbergii Sieb. et Zucc. Part 12. Hydrated stereostructure and equibration of geraniin. J. Chem. Soc. Perkin Trans. 1. 1982:9–14. doi: 10.1039/p19820000009. [DOI] [Google Scholar]
- 22.Okuda T., Yoshida T., Nayeshiro H. Constituents of Geranium thunbergii Sieb. et Zucc. IV. Ellagitannins. (2). Structure of geraniin. Chem. Pharm. Bull. 1977;25:1862–1869. doi: 10.1248/cpb.25.1862. [DOI] [Google Scholar]
- 23.Nonaka G., Akazawa M., Nishioka I. Two new ellagitannin metabolites, carpinusin and carpinusin from Carpinus laxiflora. Heterocycles. 1992;33:597–606. doi: 10.3987/COM-91-S49. [DOI] [Google Scholar]
- 24.Tanaka T., Nonaka G., Nishioka I., Miyahara K., Kawasaki T. Tannins and related compounds. Part 37. Isolation and structure elucidation of elaeocarpusin, a novel ellagitannin from Elaeocarpus sylvestris var. Ellipticus. J. Chem. Soc. Perkin Trans. 1. 1986:369–376. doi: 10.1039/p19860000369. [DOI] [Google Scholar]
- 25.Yazaki K., Hatano T., Okuda T. Constituents of Geranium thunbergii Sieb. et Zucc. Part 14. Structures of didehydrogeraniin, furosinin, and furosin. J. Chem. Soc. Perkin Trans. 1. 1989:2289–2296. doi: 10.1039/p19890002289. [DOI] [Google Scholar]
- 26.Ishimatsu M., Tanaka T., Nonaka G., Nishioka I., Nishizawa M., Yamagishi T. Tannins and related compounds. LXXV. Isolation and characterization of novel diastereoisomeric ellagitannins, nupharins A and B, and their homologues from Nuphar japonicum DC. Chem. Pharm. Bull. 1989;37:129–134. doi: 10.1248/cpb.37.129. [DOI] [Google Scholar]
- 27.Luger P., Weber M., Kashino S., Amakura Y., Yoshida T., Beurskens G., Dauter Z. Structure of the tannin geraniin based on conventional X-ray data at 295 K and on synchrotron data at 293 and 120 K. Acta Cryst. 1998;B54:687–694. doi: 10.1107/S0108768198000081. [DOI] [Google Scholar]
- 28.Tanaka T., Fujisaki H., Nonaka G., Nishioka I. Tannins and related compounds. CXVIII. Structures, preparation, high-performance liquid chromatography and some reactions of dehydroellagitannin-acetone condensates. Chem. Pharm. Bull. 1992;40:2937–2944. doi: 10.1248/cpb.40.2937. [DOI] [Google Scholar]
- 29.Tanaka T., Mine C., Watarumi S., Fujioka T., Mihashi K., Zhang Y., Kouno I. Accumulation of epigallocatechin quinone dimers during tea fermentation and formation of theasinensins. J. Nat. Prod. 2002;65:1582–1587. doi: 10.1021/np020245k. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka T., Watarumi S., Matsuo Y., Kamei M., Kouno I. Production of theasinensins A and D, epigallocatechin gallate dimers of black tea, by oxidation-reduction dismutation of dehydrotheasinensin A. Tetrahedron. 2003;59:7939–7947. doi: 10.1016/j.tet.2003.08.025. [DOI] [Google Scholar]
- 31.Matsuo Y., Tadakuma F., Shii T., Saito Y., Tanaka T. Selective oxidation of pyrogallol-type catechins with unripe fruit homogenate of Citrus unshiu and structural revision of oolongtheanins. Tetrahedron. 2015;71:2540–2548. doi: 10.1016/j.tet.2015.03.016. [DOI] [Google Scholar]
- 32.Tanaka T., Nonaka G., Nishioka I. Tannins and related compounds. C. Reaction of dehydrohexahydroxydiphenic acid esters with bases, and its application to the structure determination of pomegranate tannins, granatins A and B. Chem. Pharm. Bull. 1990;38:2424–2428. doi: 10.1248/cpb.38.2424. [DOI] [Google Scholar]
- 33.Lee S.H., Tanaka T., Nonaka G., Nishioka I. Structure and biogenesis of jolkinin, a highly oxygenated ellagitannin from Euphorbia jolkinii. J. Nat. Prod. 2004;67:1018–1022. doi: 10.1021/np0400297. [DOI] [PubMed] [Google Scholar]
- 34.Yanagida A., Shoji T., Shibusawa Y. Separation of proanthocyanidins by degree of polymerization by means of size-exclusion chromatography and related techniques. J. Biochem. Biophysical Methods. 2003;56:311–322. doi: 10.1016/S0165-022X(03)00068-X. [DOI] [PubMed] [Google Scholar]
- 35.Bally T., Rablen P.R. Quantum-chemical simulation of 1H NMR spectra. 2. Comparison of DFT-based procedures for computing proton–proton coupling constants in organic molecules. J. Org. Chem. 2011;76:4818–4830. doi: 10.1021/jo200513q. [DOI] [PubMed] [Google Scholar]
- 36.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., et al. Gaussian 09. Gaussian, Inc.; Wallingford, CT, USA,: 2013. Revision D.01. [Google Scholar]
- 37.Dennington R., Keith T., Millam J. GaussView. Semichem Inc.; Shawnee Mission, KS, USA: 2009. Version 5.0.9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.