

SUMMARY
Retinal degeneration is a form of neurodegenerative disease and is the leading cause of vision loss globally. The Toll-like receptors (TLRs) are primary components of the innate immune system involved in signal transduction. Here we show that TLR2 induces complement factors C3 and CFB, the common and rate-limiting factors of the alternative pathway in both retinal pigment epithelial (RPE) cells and mononuclear phagocytes. Neutralization of TLR2 reduces opsonizing fragments of C3 in the outer retina and protects photoreceptor neurons from oxidative stress-induced degeneration. TLR2 deficiency also preserves tight junction expression and promotes RPE resistance to fragmentation. Finally, oxidative stress-induced formation of the terminal complement membrane attack complex and Iba1+ cell infiltration are strikingly inhibited in the TLR2-deficient retina. Our data directly implicate TLR2 as a mediator of retinal degeneration in response to oxidative stress and present TLR2 as a bridge between oxidative damage and complement-mediated retinal pathology.
Graphical Abstract
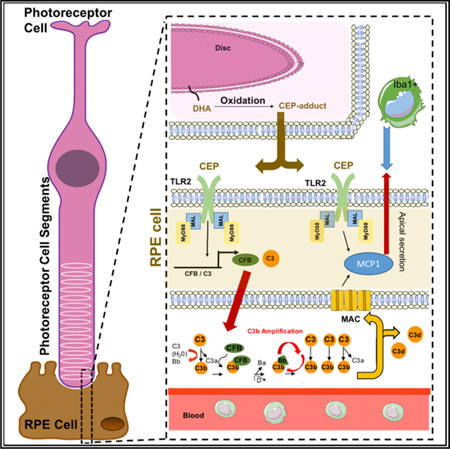
In Brief
Oxidative stress and complement deposition are common to many retinal degenerative diseases. Mulfaul et al. demonstrate that TLR2 blockade protects against photoreceptor neuronal cell death and RPE fragmentation in experimental models of oxidative stress-induced retinal degeneration and present TLR2 as a bridge between oxidative damage and complement-mediated retinal pathology.
INTRODUCTION
Toll-like receptors (TLRs) are a family of membrane-bound pattern recognition receptors (PRRs) located either on the cell surface or in endosomal compartments. These receptors are known to respond to host-molecules termed damage-associated molecular patterns (DAMPs) that have taken on the appearance of “non-self.” Sterile inflammation occurs in response to a growing list of DAMPs ranging from oxidized lipids or lipoproteins, to deposits of protein/lipid aggregates or particulate matter (Rock et al., 2010). As these stimuli are often not easily cleared, they can persist, causing over-activation of the immune system and contributing to disease pathogenesis. Ten human TLRs utilize four adaptor proteins to fine-tune the response required: MyD88, Mal/TIRAP, TRAM, and TRIF. Activation of TLRs leads to activation of a multitude of signaling pathways and transcription factors that determine the type and duration of the inflammatory response.
The retina is exposed to oxidative stress, which refers to cellular damage caused by reactive oxygen species (ROS), due to its high consumption of oxygen, its high proportion of polyunsaturated fatty acids, and its exposure to visible light. Excessive oxidative stress induces deleterious changes that result in visual impairment. Together, age-related macular degeneration (AMD), diabetic retinopathy (DR), and glaucoma are leading causes of visual impairment and involvement of oxidative stress has been reported for each disease (Nishimura et al., 2017). Furthermore, oxidative stress is thought to contribute to loss of cone photoreceptors in rare inherited retinopathies after degeneration of rod photoreceptors (Komeima et al., 2006). TLR2 heterodimerizes with either TLR1 or TLR6 and recognizes diacyl- and triacylated lipopeptides (Takeuchi et al., 2001, 2002). 2-(u-Carboxyethyl) pyrrole (CEP) is an oxidative-stress modification also recognized by TLR2 involved in promoting angiogenesis during wound healing (West et al., 2010; Wang et al., 2014). Excessive ROS can damage lipids through a mechanism known as lipid peroxidation and CEP modifications are generated by oxidation of docosahexaenoate (DHA)-containing lipids, which are found at high levels in the membrane of photoreceptor cells (Shindou et al., 2017). Of note, CEP-adducted proteins and CEP-ethanolamine phospholipids (CEP-EPs) are found in abundance in eyes and serum of patients with AMD compared with age-matched controls (Wang et al., 2014; Crabb et al., 2002; Gu et al., 2003) and are conceivably inducing activation of TLR2 in AMD and in other retinal diseases where ROS play a role in pathology.
TLR2 and TLR4 protect against infection in the anterior region of the eye (Kindzelskii et al., 2004; Kumar and Yu, 2006). However, investigations into roles for TLRs in outer retinal disease are sparse, and mainly confined to genetic investigations, including several contradicting reports of associations between various SNPs in TLRs and risk of AMD (Güven et al., 2016; Natoli et al., 2016b). AMD is the leading cause of central blindness in adults (Wong et al., 2014). End stage “dry” AMD is characterized by degeneration of the RPE, known as geographic atrophy (GA), resulting in photoreceptor cell degeneration. At present, there are no treatments for dry AMD. Genetic factors, age, diet, and smoking are risk factors for AMD. The common, coding variant Y402H in the complement factor H (CFH) gene is strongly associated with influencing susceptibility to AMD (Klein et al., 2005) and increasing severity of GA (Seddon et al., 2007). In fact, the role of complement in the retina is a topic of intense investigation, as it contributes to a variety of other retinal disease pathologies in addition to AMD (Sweigard et al., 2015; Sweigard et al., 2014; Kim et al., 2016). Variants in other complement-related genes are associated with AMD risk, including C3, CFI, and C9 (Yates et al., 2007; Despriet et al., 2009; van de Ven et al., 2013; Seddon et al., 2013); all result in an overly active complement system. Congruent with this, the deposition of C3, C5, and presence of membrane attack complex (MAC; Chirco et al., 2016), have been demonstrated in donor eyes with early AMD (Hageman et al., 2001; Anderson et al., 2010; Nozaki et al., 2006). Accordingly, SNPs in complement factors account for ~75% of the genetic risk for developing AMD. However, molecular triggers that initiate complement fixation in individuals with no apparent genetic risk remain unknown. Smoking is the largest modifiable risk factor for AMD; consequently, oxidative stress has been implicated in disease. Genotype and smoking have been independently related to AMD with multiplicative joint effects (Seddon et al., 2009), however, a tangible connection between the effects of oxidative stress and complement-associated pathology remains largely unidentified.
TLR signaling and the complement system have been linked in intestinal ischemia-reperfusion injury, where C3 deposition was markedly decreased in mice deficient in TLR4 (Pope et al., 2010), and in a renal transplant ischemia-reperfusion injury model, where inhibition of TLR2 led to a decrease in C3 deposition (Farrar et al., 2012). Furthermore, activation of TLR4 and TLR2 increases complement factor B (CFB) expression in macrophages (Kaczorowski et al., 2010; Zou et al., 2013). TLR function has not been explored in outer retinal degenerative disease and RPE pathology. Here we sought to explore whether TLR2 might act as a bridge between effects of oxidative stress and complement activation in the retina. In doing so, we discovered that TLR2 inhibition provides striking protection to the retina in response to oxidative stress. We show that oxidative stress activates TLR2 to trigger the proteolytic alternative pathway (AP) to completion with generation of the terminal complement complex, which forms sub-lytic MAC on the RPE and induces the pro-inflammatory chemokine MCP-1/CCL2. We demonstrate that inhibition of TLR2 reduces complement activation, C3 opsonization and MAC deposition; ameliorates RPE fragmentation; prevents Iba1+ve macrophage/microglial cell infiltration to the outer retina; and preserves photoreceptor cells in response to acute oxidative stress. Our data suggest that TLR2 signaling promotes an environment that drives a retinal degenerative phenotype, and presents TLR2 as a possible link between oxidative damage and excessive complement activation in retinal degenerative disease.
RESULTS
TLR2 Activation Induces AP Complement Factor Expression in Monocytes, Macrophages, and the RPE
To confirm a role for TLR2 in initiating the complement cascade, we measured gene expression levels of key AP components complement factor B (CFB) and complement factor 3 (C3) in response to TLR2 activation over time with a generic TLR2 ligand, Pam3Cys4–a synthetic triacylated lipopeptide PAMP. We observed upregulation of CFB and C3 in TLR2 activated bone-marrow–derived macrophages (BMDMs) (Figure 1A and 1B) and human monocytic cell lines (Figures 1C and 1D). Complement production and activation in the retina is reported to be distinct to systemic complement due to the physical barrier provided by Bruch’s membrane (BM) basolateral to the RPE (Khandhadia et al., 2013). In support of this, we observed a clear distinction in C3d staining (purple) in healthy aged donor eyes between the neural retina and the choroid, with the RPE acting as a border (Figure 1E). C3d is the final cleavage product of C3 and acts as an opsonin to mark dead and dying cells, or debris to be cleared by innate phagocytes. As C3d is relatively stable, it indicates historical complement activation. Higher magnifications show BM provided a distinct barrier separating the RPE and neural retina from systemic C3d staining in the choroid in healthy donor eyes (Figures 1F, S1, and S2; representative of n = 4). In these non-diseased donor eyes we observed C3d staining in the blood vessels of the choroid, but no C3d staining adjacent to the RPE/retina. In contrast, we observed C3d staining in the neural retina and in particular in photoreceptor segments in AMD eyes in both early (Figure 1G) and late (Figure 1H) disease. In contrast to healthy donor eyes, C3d staining in AMD donor eyes was apparent immediately subjacent to the RPE both in focused areas, and in a linear pattern immediately below the RPE (Figures 1I, S1, and S2; representative of n = 6). The different patterns of C3d staining between non-disease and AMD donor eyes suggests that either BM can no longer act effectively as a barrier between the outer retina and systemic factors and/or that local cells in the retina can produce complement. We next assessed whether the RPE itself could respond to TLR2 activation and induce complement. CFB and C3 were significantly upregulated in RPE cells in response to TLR2 activation (Figures 1J and 1K). By comparison, there were minor changes in several other complement proteins (Figure S2).
Figure 1. Oxidative Stress-Induced TLR2 Activation Induces AP Complement Factor Expression.

(A–D) qPCR of CFB (A and C) and C3 (B and D) expression in BMDMs or THP1s treated with 20 nM of Pam3Cys4.
(E–I) IHC of C3d (purple) in healthy non-disease donor (E and F) and AMD donor eyes (G–I).
(J and K) Black arrow and black asterisk denote C3d in CC and basal laminar deposits in AMD donor eye (representative of n = 4 non-disease donor eyes, n = 5 AMD donor eyes). qPCR of CFB (J) and C3 (K) in ARPE-19 cells treated with 20 nm of Pam3Cys4. Data shown are mean ± SD for a representative of three separate experiments. CC, choriocapillaris; RPE, retinal pigment epithelium; BM, Bruch’s membrane.
(L) Generation and chemical structure of CEP-adduct from DHA.
(M and N) qPCR of (M) C3 and (N) CFB in hfRPE cells treated with 0.1 µg of IgG or anti-TLR2 antibody prior to 10 µM of CEP-HSA for 24 h.
(O) IHC of TLR2 (purple) in a healthy donor. Bottom panel (black box) is photobleached to illustrate apical and basolateral RPE immunoreactivity (n = 4 non-disease donor eyes).
(P) Secreted CFB at 24 h and C3 at 48 h in hfRPE cells treated with 17.5 µM or 35 µM of CEP-HSA and 20 nM of Pam3Cys4, mean ± SD representative of three independent experiments: *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figures S1 and S2.
AMD-Associated Oxidative Stress Products Induce AP Complement Secretion from hfRPE Cells
We next assessed whether a physiologically relevant DAMP generated by oxidative stress could induce the same response in primary human fetal RPE (hfRPE) cells. The retina is one of the most highly metabolically active tissues in the body. This oxidative burden results in generation of lipid oxidation products such as CEP (Figure 1L). hfRPE cells were incubated with either a neutralizing antibody targeting TLR2 (anti-TLR2) or an immunoglobulin G (IgG) control prior to stimulation with CEP-adducted to human serum albumin (CEP-HSA). CEP-HSA induced C3 and CFB transcripts to similar levels observed for Pam3Cys4 in RPE cells and this was inhibited by the presence of anti-TLR2 neutralizing antibodies (Figures 1M and 1N). TLR2 localization was assayed in human donor eye tissue and observed both apically and basolaterally in the plasma membrane of the RPE (Figure 1O). Immunoblot analysis demonstrated TLR2 effect on transcript that resulted in a change at protein levels with secretion of both CFB and C3 protein into the supernatant of hfRPE cells in response to CEP-HSA or Pam3Cys4 (Figure 1P). Collectively, these data suggest that cells in the retina have the capacity to generate AP complement factors locally in response to TLR2 activation and oxidative stress product CEP.
Neutralization of TLR2 in a Photo-Oxidative Stress Model of Retinal Degeneration Decreases C3 Deposition and Promotes Survival of Photoreceptor Cells
Overexpressing C3 in the retina can promote many features of AMD (Cashman et al., 2011; Rohrer et al., 2007), while inhibiting various complement factors can protect against photoreceptor cell death in models of retinal degeneration (Sweigard et al., 2015; Rohrer et al., 2007). To define a role for TLR2 in bridging oxidative stress to complement activation and assessing its function in retinal degeneration, we utilized a well-characterized light-induced photo-oxidative stress model of retinal degeneration in which locally produced C3 is known to contribute causally to retinal degeneration (Natoli et al., 2017). In this model, there is a significant increase in C3+ macrophage/microglia in the photoreceptor layer and a decrease in outer nuclear layer (ONL) thickness (Natoli et al., 2016a). The ONL is made up of the nuclei of the rod and cone photoreceptors. A decrease in the ONL thickness is indicative of photoreceptor cell death and retinal degeneration. An anti-TLR2 neutralizing antibody or control anti-IgG was injected intravitreally (IVT) into both eyes of each animal. Animals were subsequently exposed to 100K lux light for 7 days continuously (Natoli et al., 2016a). We observed one to two more photoreceptor cell rows present in TLR2-neutralized retinas compared to IgG controls, indicating that TLR2 blockade confers protection from oxidative stress-induced photoreceptor cell loss in this model (Figures 2A–2C). The average number of ONL rows in a healthy wild-type C57BL/6 retina at the meridian analyzed is ~12, so a protection of one to two rows represents a meaningful preservation of photoreceptor numbers. C3 Immunohistochemistry (IHC) revealed that there was a significant reduction in the number of outer retinal C3+ cells/deposits observed in the outer segment (OS) layer of the photoreceptors in anti-TLR2-injected mice compared to IgG controls after photo-oxidative damage (Figures 2B–2D), despite the fact that DAPI+ cells are observed in the OS in the anti-TLR2 treated mice as well (Figure 2C, open arrows). In this model, C3 is deposited by macrophage/microglia that infiltrate the outer retina; therefore, these data indicate that, in vivo, TLR2 signaling in the retina is capable of inducing complement from macrophages/microglia in response to DAMPs generated by photo-oxidative stress, and that loss of TLR2 signaling reduces C3 deposition in the photoreceptor cells, enabling photoreceptor cell survival. Inhibition of Mal or MyD88 normally attenuates TLR2-dependent signaling. We utilized BMDMs deficient in TLR2 signaling pathway components TLR2, Mal, and MyD88, and measured AP induction in response to TLR2 activation. We observed an inhibition of C3 (Figure 2E) and CFB (Figure 2F) induction in response to TLR2 ligation in TLR2-, Mal-, and MyD88-deficient BMDMs. Conversely, overexpression of Mal or MyD88 through transient transfection leads to a dose-dependent activation of the C3-promoter in luciferase reporter assays (Figures 2G and 2H). Finally, to link back to our observations in the photo-oxidative stress model, we demonstrated that TLR2 activation of BMDMs and primary microglia (MG) isolated from mouse retina induced C3 secretion in response to TLR2 activation (Figures 2I and 2J).
Figure 2. Neutralization of TLR2 in a Photo-Oxidative Model of Retinal Degeneration and in Mononuclear Cells Decreases C3 Expression and Deposition.

(A–D) Three micrograms of anti-TLR2 or anti-IgG was injected IVT into C57Bl6 mice that were then exposed to 100K lux light for 7 days.
(A) Quantification of photoreceptor cell rows in anti-IgG versus anti-TLR2 groups.
(B and C) Immunofluorescence (IF) of C3 (green) and nuclear DAPI (blue) in mice injected with (B) anti-IgG versus (C) anti-TLR2 (scale bars represent 20 µm).
(D) Quantification of outer retinal C3 positive cells/deposits detected in ONL and subretinal space. Data shown are mean ± SEM. The p value was determined by nonparametric t test, p < 0.05. n = 9–10 per experiment, *p < 0.05.
(E and F) BMDMs from WT, TLR2–/–, Mal–/–, or MyD88–/–mice were treated for 3, 6, and 24 h with 20 nM of Pam3Cys4; expression of (E) C3 and (F) CFB was assayed by RT-PCR.
(G and H) HEK293-TLR2 cells were transfected for 24 h with C3 promoter-luciferase (100 ng), Renilla-luciferase (40 ng), and empty vector (EV) or plasmid expressing (G) Mal or (H) MyD88 at 10, 50, and 80 ng. Results are normalized for Renilla luciferase activity and represented as relative stimulation over the non-stimulated EV control, mean ± SD for triplicate determinations p value determined by one-way ANOVA and Tukey post test: *p < 0.05, **p < 0.01, ***p < 0.001.
(I and J) Secreted C3 expression in (I) BMDMs and (J) primary mouse microglia treated with 20 nM of Pam3Cys4 for 6 and 24 or 48 h.
These data support a model where, in response to light-induced photo-oxidative damage, endogenous DAMPs activate TLR2 on macrophages/microglia, driving gene induction of AP factors and C3 deposition in the outer retina, resulting in photoreceptor cell degeneration. This confirms a role for TLR2 in bridging oxidative stress to complement deposition in a photo-oxidative stress model. A key feature of the photo-oxidative stress model of retinal degeneration is that the primary cell types responding are the mononuclear phagocytic cells. Studies on the pharmacological NaIO3 model of retinal degeneration have demonstrated that the primary initial cell type to respond is the RPE cell (Kannan and Hinton, 2014).
Inhibiting Amplification of the AP Ameliorates RPE Degeneration in the NaIO3 Model of Retinal Degeneration
The NaIO3 mouse model of oxidative stress mimics some features of human retinal disease, albeit in an acute manner–notably complement deposition, RPE fragmentation, and photoreceptor cell degeneration (Li et al., 2006). In vitro, NaIO3 dose-dependently induced the expression of TLR2, CFB, and C3 in RPE cells (Figure S3), indicating the potential for activation of both TLR2 pathways and the AP in this model. Although there are potentially many DAMPs that drive TLR activation in response to oxidative damage, we tested for the appearance of CEP adducts. Figure S4 clearly demonstrates presence of CEP lipid oxidation product in the photoreceptor layer (third column) in NaIO3-treated animals, when compared to saline and IgG-control sections (first and second columns). CEP appears strongly in the central retina, with weaker staining in peripheral retina, a phenomena also observed by Rowan et al. (2017) in a high glycemic diet-induced model of retinal degeneration.
Surprisingly, evidence that AP activation has a role in promoting NaIO3-induced disease has not been reported. To assess whether activation of AP is pathological in this model, we administered sub-retinal injection of anti-CFD antibody, or anti-IgG control antibody, prior to IV NaIO3. Use of anti-CFD neutralizing antibody was chosen to prevent CFD cleaving CFB resulting in inhibition of the amplification of the AP in the retina. H&E staining of a cross section of a wild-type (WT) C57BL/6 mouse eye administered saline by tail-vein injection is provided for comparison (Figure S5A). Marked fragmentation of the RPE was observed in mice injected IV with NaIO3 and sub-retinal anti-IgG (Figure S5B). In contrast, administration of anti-CFD blocking antibody rescued rupture of the RPE monolayer. We also observed a difference in the numbers of pigmented cells in the outer segments (OSs) of the photoreceptor layers, with significantly fewer in the OS of eyes that had received anti-CFD treatment compared to anti-IgG controls (Figure S5C). To assess the effect of anti-CFD treatment on photoreceptor cell death, we counted the number of ONL rows for each treatment. Despite rescue of the RPE, we did not observe protection of rows of photoreceptors present in the ONL on neutralization of CFD (Figure S5D). However, it is possible that at a later time point this may be different, as TUNEL was significantly reduced in the anti-CFD blocking antibody group, compared with anti-IgG (Figures S5E and S5F). The localization of TUNEL was notable, as it appeared that in anti-CFD treated animals TUNEL was confined mainly to the inner layers of the ONL–suggesting that perhaps the antibody did not achieve full perfusion into the photoreceptor layer.
TLR2 Deficiency Protects against NaIO3-Induced RPE Fragmentation
Having established that oxidative stress-induced amplification of the AP is particularly damaging to the RPE in vivo, we next investigated whether TLR2 deficiency would modify this oxidative damage-induced RPE fragmentation using TLR2 knockout (TLR2–/–) mice. Marked degradation of the RPE was observed in the NaIO3 group compared with the vehicle NaCl group (Figure 3A). NaIO3-induced RPE fragmentation was similar in patterning to that observed in human tissue sections from AMD donor eyes (compare to Figure 1H). In contrast, TLR2 deficiency substantially protected RPE degeneration and retinal structure following NaIO3, although thinning of the RPE was still noted (Figure 3B). The RPE constitutes the outer blood retinal barrier (oBRB), functioning to separate the neural retina from blood-borne plasma proteins, white blood cells, and toxins. To quantify the protection observed in vivo, we selected the area of the RPE, shown in blue, and present representative images from WT and TLR2–/– mice 3 days post NaIO3 (Figure 3C, n > 5), and assessed area of the RPE/frame (Figure 3D), number of breaks in the RPE/frame (Figure 3E), and distance covered by RPE breaks/frame (Figure 3F). All methods of analysis demonstrated significant protection to the RPE provided by TLR2 deficiency under oxidizing conditions. In addition to analysis of cross sections of the RPE, we stained RPE flatmounts from WT or TLR2–/– mice for tight junction protein ZO-1, 8 h post NaIO3 treatment. ZO-1 was stained in the characteristic cobblestone pattern in TLR2-deficient mice (Figure 3G, right-hand panel). In direct contrast, ZO-1 staining in WT mice was patchy, with some areas demonstrating strong staining in a cobblestone pattern (Figure 3G, left-hand panel, closed arrows); however, there were also areas where no ZO-1 staining was apparent (Figure 3G, left-hand panel, asterisks) and many areas where ZO-1 appeared to be at cell–cell junctions in an inconsistent, non-uniform pattern (Figure 3G, left-hand panel, open arrows). Indeed, when hfRPE cells were treated with the TLR2 ligands CEP or Pam3Cys4 and then lysed and subjected to SDS-PAGE and immunoblotting, we observed a marked decrease in ZO-1 expression in all cases (Figure 3H). The data indicate that absence of TLR2 signaling results in delayed oBRB breakdown in the NaIO3 model of retinal degeneration. Interestingly, by 24 h after NaIO3 treatment, the cobblestone ZO-1 staining pattern is disrupted in both WT and TLR2–/– mice (data not shown), and yet the RPE monolayer appears to remain more intact in the TLR2–/– mice up to 3 days post NaIO3, indicating the existence of other TLR2-dependent mechanisms at play in promoting RPE degeneration.
Figure 3. TLR2–/– Mice Are Protected from Oxidative Stress-Induced RPE Damage.

(A–F) Histological H&E analysis of (A) WT and (B) TLR2–/– mice injected intravenously (i.v.), via tail vein, with NaCl or NaIO3 (50 mg/kg).
(C and D) Quantification of RPE area (in blue)/pixel (scale bars represent 20 µm) in (C) WT and (D) TLR2–/– mice. *p < 0.05, **p < 0.01, ***p < 0.001.
(E) Number of RPE breaks/frame.
(F) Distance between RPE monolayer breaks measured using the software ImageJ (n > 5 mice per genotype).
(G) IF of ZO-1 on RPE flatmounts from WT mice and TLR2–/– mice 8 h post NaIO3 (n > 5 per genotype; asterisks, ZO-1 loss; closed arrows, areas cobblestone patterning; open arrows, areas of discontinuous membrane staining).
(H) ZO-1 western blot in hfRPE cells treated with 17.5 µM or 35 µM of CEP-HSA and 20 nM of Pam3Cys4, 24 h. See also Figures S3–S5.
TLR2 Deficiency Regulates C3 Deposition and Protects against NaIO3-Induced Photoreceptor Cell Death
H&E staining of WT and TLR2–/– eyes post NaIO3 treatment indicated that the structure of the neural retina was better preserved in the TLR2–/– mice (Figures 3A and 3B). To assess the effect of TLR2 deficiency on photoreceptor cell death, we utilized TUNEL staining, which was significantly reduced in TLR2–/– mice compared with WT (Figures 4A amd 4B). Furthermore, on counting rows of nuclei in the ONL we found that TLR2 deficiency promoted survival of two to three rows of photoreceptor nuclei compared with WT mice (Figure 4C). This data indicated that in addition to preserving the RPE monolayer, genetic loss of TLR2 protected photoreceptor neurons from oxidative stress-induced cell death. While it is possible that this preservation of photoreceptor cells is a result of loss of photoreceptor-cell-intrinsic TLR2 signaling, we believed it more likely to be a secondary effect as a result of the improved preservation of the RPE and potentially a reduced C3 load in the outer retina. To investigate whether TLR2 deficiency effects C3 deposition in the NaIO3 model in an analogous manner to the photo-oxidative stress model, we stained for total C3 in WT and TLR2–/– mice (Figures 4D and 4E). C3 was observed in both WT and TLR2–/– retinas post NaIO3; however, C3 appeared to be differentially localized between WT and TLR2–/– mice with more C3 at areas of the RPE where cell junctions were separating in the WT retina, and immediately basolateral to the RPE, compared with TLR2–/– mice (Figures 4D and 4E, left-hand panels, insets, and right-hand top panels, arrows). We also noted C3 staining in the inner segments (ISs) of WT retina with rarer occurrences in the TLR2–/– retina. Pigmented cells appeared more frequently in the WT photoreceptor OS compared with the TLR2–/– retina, and these cells were C3+ (Figures 4D and 4E, right-hand lower panel); however, small focal points of C3 were observed in the OS of both WT and TLR2–/– retinas. To examine activation of C3 in response to NaIO3 we isolated RPE/choroid from WT or TLR2–/– mice injected IV with NaIO3 or NaCl and assayed for presence of the cleavage fragment iC3b. Upon activation, the thiol-ester bond in C3 is exposed, allowing covalent anchorage of C3b as well as its subsequent cleavage fragments to nearby molecules, and iC3b is formed when CFI cleaves C3b in such a way that C3b cannot associate with CFB and instead functions as an opsonin. In lysate isolated from saline, control WT mice C3α is observed, whereas iC3b is not present (Figure 4F, n = 2, lanes 1 and 2). In contrast, treatment with NaIO3 resulted in a clear reduction in C3α and the appearance of iC3b, indicative of C3 activation (Figure 4F, n = 3, lanes 3–5, quantified in Figure 4G). By comparison, there was no significant increase in iC3b formation in response to oxidative stress in the absence of TLR2 (Figures 4F and 4G), instead the data indicate that TLR2 may have a homeostatic role in regulating C3 activity, as levels of baseline C3α are lower and levels of iC3b are higher in TLR2–/– saline controls compared to saline controls in WT mice (Figure 4F, compare lanes 1 and 2 to lanes 6 and 7). Overall, the data indicate that DAMPs produced in response to oxidative stress engage TLR2 signaling to activate C3 cleavage and enhance opsonization of dying cells of the RPE.
Figure 4. TLR2 Deficiency Protects against NaIO3-Induced Photoreceptor Cell Death and Complement Factor C3 Deposition.

(A–C) WT and TLR2–/– mice were injected IV, with NaCl or NaIO3 (50 mg/kg) and eyes enucleated 72 h later.
(A) TUNEL (red) immunoreactivity (scale bars represent 20 µm) and (B) TUNEL quantification.
(C) Number of photoreceptor cell rows in the ONL. Data shown are mean ± SEM; p value determined by nonparametric t test.
(D and E) IF of C3 in (D) WT versus (E) TLR2–/– mice.
(F and G) Expression of C3 cleavage fragments in RPE/choroid tissue from WT and TLR2–/– mice injected i.v., with NaCl or NaIO3 (F) representative images and (G) quantification of iC3b.
*p < 0.05,**p < 0.01, ***p < 0.001.
Oxidative Product CEP-HSA Induces MAC Formation on the RPE
At this point, our data indicated that TLR2 plays a role in mediating C3 activation in response to oxidative stress, resulting in the deposition of C3 and its opsonizing cleavage products in the outer retina. We next investigated whether TLR2 can promote formation of the terminal complement complex MAC. CFB is the key rate-limiting AP complement factor, and, as such, small increases in CFB expression lead to formation of a C3 convertase (C3bBb) that amplifies the proteolytic cascade leading to C5 cleavage and ultimately formation of terminal complement MAC. During bacterial infection MAC usually leads to formation of a pore on the cell membrane, then lysis and death of the bacteria. However, MAC is rarely lytic for nucleated cells and is reported to induce signaling pathways resulting in pro-inflammatory and pro-angiogenic gene expression on the RPE. Of particular interest to us is that known consequences of sub-lytic MAC formation are the release of chemokine monocyte chemoattractant protein CCL2/MCP-1 and vascular endothelial growth factor (VEGF) (Lueck et al., 2011); both cytokines are believed to have fundamental roles in promotion of dry and wet AMD respectively. We suspected that the protective effect of TLR2 deficiency observed in the NaIO3 model of retinal degeneration may be partially attributed to blockade of sub-lytic MAC formation and signaling in the RPE. To determine whether the presence of CEP with provision of complete complement was sufficient to drive the proteolytic complement cascade to completion in vitro, we cultured hfRPE on transwell membranes for >4 weeks prior to stimulation with either HSA or CEP-HSA in the presence of heat-inactivated (Hi) or normal human serum (NHS) for provision of complete complement. Culture of hfRPE cells in the presence of 10% NHS and HSA resulted in the appearance of visible MAC (Figure 5A, white arrows, second panel), but culture of hfRPE cells in the presence of 10% NHS and CEP-HSA resulted in significantly more MAC being formed (Figures 5A, fourth and fifth panels, and 5B). hfRPE cells appeared to be less resistant to MAC formation in response to the presence of 10% NHS than ARPE-19 cells, as CEP-HSA in the presence of serum proteins was the only combination that induced MAC formation on the membrane in the cell line (Figures 5C–5E). Quantification of MAC by ELISA also confirms CEP-HSA can significantly drive MAC formation in the presence of NHS (Figure S6). Of note, soluble MAC was also detected in cells cultured in NHS alone in both ARPE-19 cells and primary hfRPE cells despite a lack and near-lack of membrane-associated MAC observed by confocal microscopy under these conditions. These data indicate that oxidative product CEP-HSA can promote the AP proteolytic cascade to completion with the embedding of MAC in the RPE membrane in vitro.
Figure 5. Oxidative Stress Products Drive AP Activation and MAC Formation in a TLR2-Dependent Manner.

(A–E) Polarized hfRPE cells (A and B) or ARPE-19 cells (C–E) were treated with 10% Hi-NHS or NHS in combination with HSA or CEP-HSA for 24 h, phase transmission, presence of MAC (red), Phalloidin (green), and DAPI (blue), with representative images from three separate experiments.
(B and E) Quantification of MAC+ specks in three 303 frames, data mean ± SD, one-way ANOVA followed by Tukey post-test to determine significance between groups; ***p < 0.001.
(F–I) IF of MAC (red), Phalloidin (green), and DAPI (blue) in ARPE-19 cells treated with (F and G) 0.1 µg of IgG or anti-TLR2 antibody for 1 h or (H and I) with DMSO or 40 µm of Mal peptide inhibitor for 2 h prior to CEP-HSA and 10% NHS for 24 h.
(G and I) quantification of MAC+ specks in four 20× frames, mean ± SD; p value determined by nonparametric t test; p < 0.05.
(J–O) WT and TLR2–/– mice injected IV, with NaIO3 (50 mg/kg).
(J) Quantification of MAC fluorescent intensity (scale bars represent 20 µm).
(K–O) Representative IF images of MAC at 72 h, (K and O) depicts MAC staining in the whole retina section, (L–N) show higher magnifications, (L) shows MAC in the IS and OS, (M) shows MAC in OS and on RPE, (N) shows MAC on RPE.
(P and R) Lysed tissue was assayed by western blot for expression of CFB(Bb). C9 and C9b at (P) 24 h and (R) 72 h.
(Q and S) Mean pixel density for C9b was quantified at (Q) 24 h and (S) 72 h using the software ImageJ.
See also Figures S6 and S7. *p < 0.05, **p < 0.01, ***p < 0.001.
Oxidative Stress-Induced TLR2 Activation Drives MAC Formation
We next wished to confirm a role for TLR2 in the recognition of CEP-HSA and promotion of MAC in the RPE. Our observation that ARPE-19 cells formed membrane-embedded MAC only in the presence of 10% NHS and CEP-HSA, with no visible membrane-embedded MAC formed in the presence of 10% NHS or 10% NHS + HSA alone (Figure 5C), allowed us to use these cells as a tool to examine CEP-HSA–inducible MAC specifically. RPE cells treated with NHS and CEP-HSA in the presence of a monoclonal neutralizing anti-TLR2 antibody or an isotype control (IgG) were assayed for MAC formation by confocal microscopy (Figures 5F and 5G). Neutralizing TLR2 attenuated the number of MAC formed in response to CEP-HSA by ~50% (Figures 5F and 5G). To further confirm the role of TLR2 in bridging oxidative stress to MAC formation, we investigated whether inhibiting TIR adaptor Mal would affect CEP-HSA induced MAC formation. RPE cells were cultured with NHS and CEP-HSA in the presence of a Mal inhibitor peptide or peptide control and MAC formation was assayed by confocal microscopy (Figures 5H and 5I). Inhibiting Mal significantly attenuated the number of MAC specks formed in response to CEP-HSA, in an analogous manner to TLR2 neutralization (Figures 5H and 5I). Together, these data confirm that TLR2 acts as a bridge between AMD-associated lipid oxidation product CEP and the induction of AP-driven MAC formation. However, given the inhibition of MAC formation following TLR2/Mal blockade is not complete, TLR2/Mal-independent signaling pathways are also implicated in contributing to CEP-induced MAC formation. MAC is known to be rapidly endocytosed from the RPE membrane in vitro and is rarely observed on the RPE in human tissue samples (Tan et al., 2016). In fact, outside of MAC-induced pathology of the choriocapillaris, a clear link between MAC deposition in the retina and retinal degeneration is not firmly delineated. To determine whether MAC formation may be involved in oxidative stress-induced retinal pathology, we examined MAC/C5b-9 by IHC in C5-deficient mice compared with WT mice 3 days after IV injection with NaIO3 (Figure S7). MAC/C5b-C9 was observed in the IS and OS of the photoreceptors in WT tissue sections (Figure S7, top-left panel), surrounding pigmented cells in the OS layer, and immediately adjacent to the RPE. MAC/C5b-C9 was not observed in the retina of C5-deficient mice (Figure S7, top-right panel) and H&E staining demonstrated that in addition to lacking MAC deposition, C5 deficiency also protected from oxidative stress-induced photoreceptor degeneration (Figure S7). Activation of C5 generates anaphylatoxin C5a and MAC-forming C5b products. Although the possibility exists that the protection observed could be due to loss of either C5a or C5b alone, as both products are generated simultaneously in parallel it is likely that loss of both contribute to the protection observed in response to oxidative stress, pointing to a role for MAC in this model of retinal degeneration.
We next examined the localization of MAC deposition 3 days post NaIO3 treatment in the WT and TLR2–/– mice (Figures 5J– 5O). MAC was observed in the IS and OS of the photoreceptors in WT tissue sections (Figure 5K), surrounding pigmented cells in the OS layer, and immediately adjacent to the RPE both apical and basolateral the RPE membrane (Figures 5I–5N, high magnification). In contrast, MAC was not observed in the retina of TLR2–/– mice (Figure 5O). (Note: this is a mouse IgG antibody, therefore the green fluorescence in the INL is indicative of the inner blood retinal vasculature.) As CFB-active fragment Bb is required to form the C5 convertase, we next investigated the appearance of active Bb and C9/C9b MAC products in retinal lysate from WT and TLR2–/– mice 1 day (Figures 5P and 5Q) or 3 days (Figures 5R and 5S) post NaIO3. Across both time points, Bb appeared more abundant in WT mice compared with TLR2–/– mice post NaIO3 treatment (Figures 5P and 5R, top panels). Zymogen C9 (70 kD) is proteolytically cleaved at a specific site to induce C9 polymerization (Tschopp et al., 1986). The 25 kD product, which is the carboxyl-terminal fragment of C9 capable of disturbing membrane potential, was found to a strikingly greater extent in the WT mice compared with the TLR2–/– mice at both 1 day (Figures 5P and 5Q) and 3 days (Figures 5R and 5S) post NaIO3 treatment. These data imply that TLR2 signaling drives the AP cascade to completion with formation of MAC in response to oxidative stress in vivo.
Oxidative Product CEP-HSA Induces Sub-lytic MAC Formation on the RPE and Secretion of Chemokine MCP-1
To interrogate whether CEP-HSA/NHS-induced MAC formation on the RPE was lytic or sub-lytic, we used a lactate dehydrogenase (LDH) assay as an indicator of cell death and an MCP-1/CCL2 ELISA as an indicator of sub-lytic MAC signaling. We harvested the RPE supernatant from our MAC-assay (Figure 5C) and observed no apparent cell death of RPE cells under conditions where MAC was forming (i.e., CEP-HSA + NHS) (Figure 6A); yet in the same samples we observed a significant increase in the secretion of MCP-1/CCL2 from cells with sub-lytic MAC formation (CEP-HSA + NHS), over cells treated with NHS or CEP-HSA alone (Figure 6B). Neutralization of TLR2 signaling using anti-TLR2 under the same conditions demonstrated a halving of CEP- and CEP/NHS-induced MCP-1/CCL2; in contrast, Pam3Cys4-induced MCP-1/CCL2 was abolished in the absence of TLR2 signaling (Figure 6C). The lack of complete penetrance of neutralizing TLR2 on CEP-induced MCP-1/CCL2 mirrors the incomplete inhibition of MAC formation observed with TLR2 neutralization (Figures 5F 5I), and implies the existence of both TLR2-dependent and -independent components to CEP signaling. However, collectively these data imply that TLR2 can act as a sensor for oxidative stressor CEP, and that CEP-TLR2 signaling synergizes with complement, resulting in sub-lytic MAC formation that functions to generate a chemokine signal from the RPE rather than cause cell lysis.
Figure 6. TLR2 Deficiency Reduces Oxidative Stress-Induced Iba1 Positive Mononuclear Cell Infiltration.

(A and B) ARPE-19 cells were treated with 10% NHS, CEP-HSA, or NHS plus CEP-HSA and (A) LDH activity.
(B) MCP-1 was assayed.
(C) MCP-1 secretion from ARPE-19 cells untreated or treated with anti-TLR2 0.1 mg for 1 h prior to 10% NHS, CEP, or both for 24 h.
(D–G) WT and TLR2–/– mice were injected IV, with 50 mg/kg of NaIO3 (n > 5). Retinal cryosections (D and F) were stained for IBA1 (green) and DAPI (blue) at 72 h (white arrows indicate iba1+cells subjacent to the RPE). Quantification of Iba1+ cells per 203 frame in the ONL (E) and photoreceptor OS (G), mean ± SEM; p value determined by nonparametric t test, minimum of eight frames counted/mouse (n = 10).
(H) THP-1 monocyte migration (after 120 min) from top chamber (8-µm pore transwells), toward fresh complete media, or conditioned media from RPE cells that were untreated, treated with CEP for 24 h, or treated with CEP + NHS, mean ± SEM for 4–5 counts; *p < 0.05.
(I) MCP_1 secreted from ARPE-19 cells treated with CEP-HSA or CEP-HSA in the presence of NHS for 24 h.
(J) CD86 MFI in CD45+CD66b-CD14+CD16+ monocytes treated with MCP-1 for 24 h. Experiments were carried out in triplicate and data are mean ± SE for three separate experiments. The p value was determined by nonparametric t test.
(K) Polarized ARPE-19 cells treated with NHS, CEP, or both NHS and CEP were assayed for secretion of MCP-1 by ELISA in both apical and basolateral supernatants. In all images, scale bars represent 20 µm.
TLR2 Deficiency Delays NaIO3-Induced Iba1+ Macrophage/Microglial Infiltration to the Outer Retina
TLR2 deficiency has been reported to reduce macrophage infiltration in the CNS in response to spinal nerve injury (Kim et al., 2011). The retina is an extension of the CNS and MCP-1/CCL2, is a potent chemoattractant, and is the major chemokine responsible for macrophage and microglial infiltration in the retina. The implication for our data is that TLR2 deficiency may result in reduced macrophage and microglial infiltration to the retina in response to oxidative stress. A recent report has demonstrated that photoreceptor cell death in the NaIO3 model is correlative with activated macrophage accumulation in the outer retina after RPE degeneration (Moriguchi et al., 2018). We next assessed the extent to which absence of TLR2 might influence macrophage/microglial cell migration into the outer retina, in response to oxidative stress. We carried out IHC for Iba1 72 h post NaIO3. Iba1 stains both macrophages and microglia and by 72 h, large Iba1+ cells were found both in the ONL (Figures 6D and 6E) and in among the OS of WT mice immediately apical to RPE (Figures 6D and 6G). Large Iba1+ cells were also observed appearing subjacent to RPE (Figure 6D, middle representations). There were significantly fewer Iba1+ cells in the ONL or OS of TLR2–/– mice (Figures 6D–6G, lower panels). These data indicate that oxidative stress-induced TLR2 signaling drives a chemokine gradient in vivo that has the potential to regulate both the migration of microglia from the inner to the outer retina and the migration and infiltration of myeloid cells from the choroid to the outer neural retina.
In vitro, we investigated whether conditioned media from RPE cells treated under CEP+NHS sub-lytic MAC conditions would affect monocyte migration across a transwell as a proxy for understanding whether blockade of sub-lytic MAC signaling in RPE cells in vivo might reduce macrophage/microglia cell infiltration analogous to what we observed in the NaIO3-treated TLR2–/–mice. We observed increased cell migration across a transwell membrane toward the chamber with media transferred from RPE cells when compared with controls (Figure 6H), MCP-1 levels in the transferred media are shown for context (Figure 6I). We next assessed the effect of MCP-1 on CD86 as a proxy for monocyte activation. MCP-1 treatment of peripheral blood mononuclear cells (PBMCs) increased CD86 membrane expression by ~1.4 fold (Figure 6J), indicating not only that sub-lytic MAC formation on RPE cells has potential to provide a chemokine gradient for monocytes but also that the monocytes may be further activated by the environment. Our IHC data indicated that, in addition to the decreased infiltration of Iba1+ve cells from the inner retina toward the outer retina, there is a deficit of macrophages infiltrating from the choroidal vasculature toward the outer retina also in TLR2–/– mice (Figures 6D and 6F, middle panels). RPE cells are polarized cells so we investigated the extent to which RPE cells secreted MCP-1/CCL2 apically versus basolaterally. Interestingly, we found that RPE cells cultured on transwells for >5weeks secrete MCP-1/CCL2 in a strikingly polarized manner when treated with CEP or CEP+NHS, with no significant secretion into the basolateral compartment compared with strong chemokine release into the apical compartment (Figure 6K). These data imply that while CEP and sub-lytic MAC signaling likely play a role in the infiltration of Iba1+ cells from the inner retina, there is likely an additional chemotactic signal that attracts macrophages from the choroid. The proteolytic complement cascade ending in MAC formation concomitantly generates soluble anaphylatoxins C3a and C5a. And despite being rapidly turned over, C5a in particular is a potent chemotactic agent for monocytes, macrophages, and microglia; therefore, it is possible that C5a is also involved in drawing Iba1+ cells into the retina in response to oxidative stress, although this remains to be tested.
Anti-TLR2 Therapy Protects against NaIO3-Induced RPE Degeneration and Photoreceptor Cell Death
Anti-TLR2-neutralizing antibodies had preserved photoreceptor degeneration in the focal photo-oxidative stress-induced model of retinal degeneration. We next investigated whether pharmacological blockade of TLR2 signaling, through use of the same anti-TLR2 neutralizing antibody, would rescue RPE fragmentation and photoreceptor degeneration in the NaIO3 model and could therefore broadly present TLR2 as a therapeutic target for oxidative stress-induced retinal degeneration. WT mice were injected IV with NaIO3 or with vehicle NaCl and with sub-retinal anti-IgG or anti-TLR2 antibodies and retinal histology was assessed 72 h after NaIO3. As expected, marked fragmentation of the RPE was observed in mice injected IV with NaIO3 and sub-retinal anti-IgG compared with mice injected IV with vehicle NaCl and sub-retinal anti-IgG (Figure 7A, top panels). In contrast, sub-retinal administration of anti-TLR2 blocking antibody at time of tail-vein NaIO3 administration rescued RPE degeneration and retinal structure (Figure 7A, top panels). To assess the effect of anti-TLR2 treatment on photoreceptor cell death, we utilized TUNEL staining. TUNEL staining was significantly reduced in mice that received anti-TLR2 blocking antibody, in comparison with those that received anti-IgG (Figures 7A, bottom panels, and 7B). Specifically, analogous to the TLR2–/– mice, we found that therapeutic neutralization of TLR2 also protected two to three rows of photoreceptors from oxidative stress-induced cell death (Figure 7C) and preserved the RPE monolayer post administration of NaIO3. Collectively, these data demonstrate that administration of a neutralizing anti-TLR2 antibody leads to a protection of the RPE, and a reduction in photoreceptor cell death under oxidative conditions.
Figure 7. Pharmacological Blockade of TLR2 Rescues NaIO3-Induced RPE and Photoreceptor Cell Degeneration.

(A–C) Wild-type mice were injected IV with NaCl or NaIO3 (50 mg/kg; n = 4 per treatment). At the same time, mice were injected IVT with 3 mg of anti-TLR2 antibody or anti-IgG. At 72 h later, eyes were prepared for (A, top row) H&E and (A, bottom row) TUNEL analysis. *p < 0.05.
(B) Quantification of TUNEL+ cells in NaIO3-injected WT mice in combination with anti-IgG versus anti-TLR2.
(C) Number of photoreceptor cell rows in IgG-injected versus anti-TLR2 ab-injected mice after NaIO3 treatment. Scale bars represent 20 µm.
DISCUSSION
With progressive age, increased oxidative damage occurs in many tissues, including the retina, and is thought to contribute to the progression of multiple forms of retinal degeneration most notably AMD. This is highlighted in a variety of experimental models where increased oxidative stress leads to a dry AMD-like pathology, including immunization with CEP, knockdown of SOD2, and NaIO3 injection (Hollyfield et al., 2008; Justilien et al., 2007; Kannan and Hinton, 2014). In addition to excessive oxidative stress, an accumulation of complement factors in the retina and choroid is a pathological hallmark of AMD (Hageman et al., 2005; Seth et al., 2008; Mullins et al., 2014). It has been suggested that products of photo-oxidation of bis-retinoid lipofuscin pigments could serve to activate complement (Zhou et al., 2006). However, the underlying mechanisms that trigger complement fixation in response to oxidative stress remain unknown. CEP has been shown to act as a ligand for TLR2, promoting angiogenesis in response to oxidative stress (West et al., 2010) and, indeed, blocking TLR2 signaling in two mouse models of choroidal neovascularization (CNV) was recently shown to be efficacious in reducing CNV lesion size (Marneros, 2016; Feng et al., 2017). This indicates that inhibitors of TLR2 have potential therapeutic utility for wet AMD.
We chose to study the effect of neutralizing TLR2 in two different experimental models of retinal degeneration. While both models utilized are oxidative stress-induced models of retinal degeneration known to deposit complement and result in loss of photoreceptor cells, the major cell types affected in each model differ (Natoli et al., 2017). In the photo-oxidative stress model, C3 is microglia-/macrophage-derived, deposited in the outer segments, and has been shown to contribute causally to photoreceptor loss. However, despite reports of C3 accumulation, no causative role for the AP had been implicated in retinal degeneration in the NaIO model. To determine whether AP activation was a driver of the pathology observed in the NaIO3 model or simply a bystander effect, we used an immunoprecipitating blocking antibody for complement factor D (CFD). CFD is a serine protease that cleaves CFB once bound to C3b, resulting in the assembly of the AP C3 convertase (Ricklin et al., 2010). An interesting observation relating to the use of anti-CFD in the NaIO3 model was the resulting protection of the RPE, with no significant loss of photoreceptor numbers. CFD binds to C3 only after it has bound CFB, at which point it cleaves CFB and enables amplification of the AP. For this reason, introduction of anti-CFD will block the amplification step of the AP, inhibiting MAC formation, but its inhibition of C3 cleavage into opsonizing fragments is less effective. With this in mind, our anti-CFD data indicate that photoreceptors are sensitive to C3 deposition/opsonization whereas the RPE may be more sensitive to the effects of amplifying the AP. Indeed, others have shown that complement regulators Cd55/Cd59 are reduced specifically in the photoreceptors in a model of retinal detachment, making photoreceptors especially sensitive to opsonization and complement-mediated death (Sweigard et al., 2015). By contrast, the fact that TLR2 deficiency protected both photoreceptor numbers and the RPE implies that, in response to oxidative stress, TLR2 signaling promotes both C3 opsonization and the amplification of the AP. Indeed, CFB is exclusive to the AP and is the key rate-limiting protein in AP activation (Nielsen et al., 1992). Simply increasing CFB expression can lead to the formation of the C3 convertase, activating the AP by cleaving C3. TLR2 activation consistently induced gene expression of both CFB and C3 to significant levels in all cell types tested, implying that TLR2 activation can universally activate the AP in vitro. Likewise, in vivo, we observed C3 opsonin fragment deposition in response to oxidative stress was lessened in the absence of TLR2. It is worth noting that CFH functions to inhibit the amplification of the AP by competing with CFB for binding with C3. In this way, variants in CFH that heighten risk for dry AMD and progression to GA are less efficient at preventing the amplification of the AP–again indicating a sensitivity of the RPE to the effects of the amplification of the AP. Amplification of the AP leads to terminal complement activation, whereupon its individual components C5b, C6, C7, C8, and C9 combine to form a lytic pore (C5b-9/MAC) on the surface of target cell membranes, capable of inducing cell lysis and inflammatory processes, as well as activating various cell signaling pathways (Lueck et al., 2011; Kunchithapautham and Rohrer, 2011; Triantafilou et al., 2013). In the human retina, the MAC complex is identified in Bruch’s membrane in eyes as young as 5 years of age (Mullins et al., 2014). The presence of MAC increases with normal aging, but it accumulates at higher levels in individuals with risk-associated AMD genotypes (Mullins et al., 2011) and has been identified in AMD patients within drusen in Bruch’s membrane (Seth et al., 2008) surrounding the choriocapillaris, and on RPE overlying drusen in vivo. The fact that the RPE is more intact in the absence of TLR2, despite being subjected to oxidative stress, implies that amplification of the AP has been inhibited due to the loss of TLR2 signaling. Indeed, the lack of active MAC formation in response to oxidative stress in the retina, in the absence of TLR2, was marked when compared to WT mice. Previous reports demonstrate that inhibition of TLR2 reduces C3 deposition in ischemia-reperfusion injury (Pope et al., 2010; Farrar et al., 2012; Stott and Korbelik, 2007); our data demonstrate that TLR2 can also directly trigger the proteolytic complement cascade to completion with formation of the terminal complement complex, MAC.
MAC activation on choroidal endothelial cells induces lysis (Zeng et al., 2016), but studies describe how RPE cells are resistant to MAC-mediated lysis, and efficiently remove MAC before lysis can take place; instead, sub-lytic MAC induces inflammatory signaling pathway activation (Lueck et al., 2011; Kunchithapautham and Rohrer, 2011; Georgiannakis et al., 2015). In support of these reports, we did not observe RPE cell death under TLR2-induced MAC-forming culture conditions in vitro, indicating that TLR2-induced MAC formation on RPE cells is sub-lytic. Sub-lytic MAC is characterized by secretion of MCP-1/CCL2, a key monocyte chemoattractant that also signals for monocyte differentiation into macrophages. In line with this characteristic, we observed a synergistic effect on MCP-1/CCL2 secretion under culture conditions where TLR2-induced MAC is formed above the induction observed in response to CEP alone, suggesting that MAC formed on RPE cells in response to TLR2 activation is sub-lytic and has the potential to create an environment that attracts phagocytes to the outer retina. Interestingly, MCP-1 secretion from the RPE was highly polarized, favoring a role for resident microglia activation in the neural retina. MCP-1/CCL2 has been shown to be increased in the outer retina in donor eyes with geographic atrophy, as is subretinal CCR2(+) inflammatory monocyte infiltration, and is considered to contribute to photoreceptor degeneration (Sennlaub et al., 2013). From a mechanistic stand point, support for MCP-1/CCL2 as a major factor in recruiting phagocytes in retinal degeneration comes from reports that genetic deletion of MCP-1/CCL2 prevents inflammatory monocyte recruitment, accumulation, and photoreceptor degeneration in vivo in mouse models (Sennlaub et al., 2013). The decreased Iba1+ staining and photoreceptor degeneration we observed in TLR2-deficient mice after treatment with NaIO3 supports the existence of a TLR2-driven chemokine gradient, attracting these cells to the outer retina and contributing to photoreceptor cell death, which may be a consequence of sub-lytic MAC signaling, although this remains to be definitively tested.
Our data indicate that TLR2 mediates complement deposition in response to oxidative stress that is pathological in nature, and that blocking TLR2 signaling preserves both photoreceptor and RPE integrity in vivo under conditions of acute oxidative stress. However, the respective contributions of the different cells in the retina that can respond to TLR2 and their individual contributions to oxidative stress-induced TLR2 promotion of retinal degeneration require further examination. We cannot yet distinguish between relative contributions to pathology made by TLR2-activated RPE and TLR2-activated mononuclear phagocytes. The observation that neutralizing TLR2 in the photo-oxidative damage model of retinal degeneration significantly reduced complement deposition and preserved photoreceptor cell layers indicates that, in addition to RPE-originating signals, blocking TLR2 signaling in the macrophage/microglia cells is also likely to contribute a significant aspect to the prevention of TLR2-mediated retinal degeneration (Natoli et al., 2017). Furthermore, given our data, and supporting literature, that MAC formation on the RPE is sub-lytic, it remains to be understood how oxidative stress results in RPE fragmentation in vivo and following this, why blocking TLR2 signaling in response to oxidative stress delays the RPE from this degeneration. Others have demonstrated that merely overexpressing C3 alone in vivo with C3-expressing adenovirus exhibited similarly significantly increased RPE death, in addition to loss of photoreceptor outer segments, and reactive gliosis (Cashman et al., 2011). So it appears that, in vivo, unregulated complement activation results in an environment that promotes RPE death, be it as a result of experimental AAV-inducible C3 overexpression (Cashman et al., 2011), or in our case, oxidative damage-induced TLR2-mediated C3/MAC activation. Importantly, during the course of our study, we also discovered TLR2 effects on the RPE that are independent of its role in inducing complement but undoubtedly contribute to RPE degeneration. Specifically, oxidative stress-induced TLR2 signaling can also reduce tight junction expression, likely contributing to weakening the RPE and consequently the outer blood retinal barrier. It is likely that additional TLR2-induced signaling pathways will be identified in further study. Finally, as we do not observe complete protection in either experimental model, other mechanisms must also be involved in oxidative stress-induced retinal degeneration.
In conclusion, we show that TLR2 deficiency reduces complement activation, delays oxidative damage-induced RPE fragmentation, delays migration of microglia/macrophages to the RPE and outer neural retina, and delays photoreceptor degeneration. Our data contribute toward understanding the mechanisms underlying oxidative stress-induced retinal degeneration and pinpoints TLR2 as a PRR bridging the detection of oxidative damage to activation of the complement response, providing new targets for the prevention of oxidative stress-induced pathology.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sarah Doyle (sarah.doyle@tcd.ie).
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal studies
NaIO3 model of retinal degeneration
All experiments were conducted in accordance with the ARVO Statement for Use of Animals in Ophthalmic and Vision Research, and approved by the Trinity College Dublin Animal Research Ethics Committee or the Australian National University (ANU) Animal Experimentation Ethics Committee. Mice used were C57BL/6J mice and Tlr2–/– (JAX stock #004650) at 8–12week old. DBA/2J C5-deficient mice (JAX stock #000671) were 10 months old and matched to 10 month old C57BL/6J mice. Comparator WT versus KO mice were sex matched, as sex has a measurable effect on the NaIO3 model.
RPE flatmounts
A single intravenous injection, via tail vein, of NaIO3 (50 mg/kg) was administered to C57BL/6J and TLR2–/– mice. Mice were euthanized 8 hours post injection, eyes fixed in ice-cold methanol for 15 minutes and the choroid/RPE dissected into flatmounts. Flatmounts were blocked and permeabilised in 5% NGS, 0.05% Triton X-100 for 1 hour and incubated with ZO-1 (1:100, Invitrogen) overnight at 4°C. Flatmounts were washed with PBS and incubated with goat anti-rabbit 488 for 2 hours at room temperature.
Iba1, C3, and MAC staining
A single intravenous injection, via tail vein, of NaIO3 (50 mg/kg) was administered to C57BL/6J and TLR2–/– mice. Mice were euthanized 72 hours post injection, eyes fixed in 4% paraformaldehyde for 90 minutes and after a 30% sucrose gradient, eyes were embedded in OCT. 12 µm sections were blocked and permeabilised in 5% NGS, 0.05% Triton X-100 for 1 hour and incubated with C3 (Abcam ab11887, 1:100), MAC (Biozol, FGI-10–1801, 1:100) or Iba1 (Wako 019–19741, 1:500) overnight at 4°C. Sections were washed with PBS and incubated with Alexa Fluor® goat anti-rabbit 488 or or Alexa Fluor® goat anti-mouse 488 1:500 in 5% NGS for 2 hours at room temperature and counterstained with Hoechst. To quantify the numbers of Iba1+ cells, a minimum of eight 20 x objective frames were counted per eye and counts were averaged per mouse. Images were cropped to only include the RPE and photoreceptor layers for MAC staining and the mean fluorescence intensity was measured.
CEP staining
A single intravenous injection, via tail vein, of NaIO3 (50 mg/kg) was administered to C57BL/6J mice. Mice were euthanized 24 hours post injection, eyes fixed in 4% paraformaldehyde for 90 minutes and after a 30% sucrose gradient, eyes were embedded in OCT. 12 µm sections were stained using anti-CEP ab 1:1000 (Kindly provided by Sheldon Rowan, Tufts University, USA) using the Vectastain ABC Kit following manufacturers protocol and detected using DAB (Vector Laboratories).
Inhibition by anti-CFD blocking antibody
A single intravenous injection was administered via the tail of NaIO3 (50 mg/kg) in NaCl. Control mice received NaCl. In tandem, mice received a single subretinal injection of either anti-CFD antibody (R&D Systems) antibody or IgG control (0.5 µg per eye). Mice were euthanized 72 h post injection. Eyes were fixed in Davidson’s fixative overnight, washed three times in PBS, and embedded in paraffin wax. Sections of 5 µm were cut with a microtome (Leica) and subject to xylene deparaffinizing and ethanol rehydration. For histology, slides were stained with hematoxylin and eosin. To detect cell death, sections were stained using an in situ Cell Death Detection kit with TMR Red (Roche) following the manufacturer’s protocols, and nuclei were counterstained with Hoechst.
Inhibition by anti-TLR2 blocking antibody
A single intravenous injection was administered via the tail of NaIO3 (50 mg/kg) in NaCl. Control mice received NaCl. In tandem, mice received a single subretinal injection of either anti-TLR2 blocking (Invivogen) antibody or IgG control (3 µg per eye). Mice were euthanized 3 days’ post injection eyes were fixed in Davidson’s fixative overnight, washed 3 times in PBS and embedded in paraffin wax. 5µm sections were cut with a microtome (Leica) and subject to xylene deparaffinising and ethanol rehydration. H&E
For histology slides were stained with hematoxylin and eosin.
TUNEL staining
To detect cell death, sections were stained using in situ Cell Death Detection kit, TMR red (Roche) for 1 hour at 37°C following manufacturer’s protocol and nuclei counterstained with Hoechst. The number of photoreceptor rows was calculated by counting the number of nuclei spanning the height of the ONL at three individual points per 20x frame (e.g., ~12 nuclei height in ONL of wild-type mice) and an average was taken per 20x frame. To quantify the numbers of ONL rows or TUNEL+ cells, a minimum of eight 20 x objective frames were counted per eye and counts were averaged per mouse.
Photo-oxidative stress model of retinal degeneration
C57BL/6J mice (8 weeks old) received a single intravitreal injection of either an anti-TLR2 antibody or IgG control (3 µg per eye). Animals were then exposed to 100Klux light for 7 days to induce photo-oxidative damage, as described previously (Natoli et al., 2016a). Following photo-oxidative damage, animals were euthanized and eyes collected for histological analysis. Retinal cryosections were stained with TUNEL (Roche) to detect photoreceptor cell death. C3 immunohistochemistry was performed using α-C3 antibody (1:100, Abcam), and C3+ cells/deposits in the outer retina (between the ONL and RPE) were counted per retinal section.
Human studies
Immunohistochemistry in human tissue
Human donor eyes obtained from the Iowa Lions Eye Bank (Iowa City, IA, USA) eyes were processed within 8 hours of death (Table S1). Macular punches which had been fixed in 4% paraformaldehyde and embedded in sucrose-optimal cutting medium were sectioned on a cryostat and stained for TLR2 (Abcam) and C3d (Dako) using VIP and Vectastain ABC Kit (Vector Laboratories). Characteristics of donor tissue used for immunohistochemistry are displayed in Table S1.
Cell lines
ARPE-19 cells (ATCC CRL 2302) 1:1 mixture of Dulbecco’s modified Eagle’s medium (DMEM)/nutrient mixture F-12 Ham with L-glutamine, 15 mM HEPES, sodium bicarbonate. THP-1 cells RPMI 1640 medium. Immortalized bone marrow derived macrophages wild-type MyD88–/– and Mal–/– mice (Kind gift Prof. Golenbock, UMass Medical School) DMEM. Peripheral blood mononuclear cells (PBMCs) were isolated from human blood RPMI 1640. All medium was supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin (Sigma-Aldrich). YFP+ Cx3cr1-expressing microglia were isolated from mouse retinas according to previously described methods (Fernando et al., 2016) and were sorted into a 48-well plate at 1500 cells per well. Isolated primary microglia were cultured for 3 weeks in DMEM-F12 supplemented with 10% FBS, 1% antibiotic-antimycotic (Thermo Fisher Scientific), 3% L-glutamine, 0.25ng/ml GM-CSF (Stem Cell Technologies) and 2.5 ng/ml M-CSF (Miltenyi Biotec) prior to TLR2 stimulation. Cells were maintained at 37°C, 5% CO2, 95% air.
Primary human fetal RPE culture
Cells, provided by Dr. Arvydas Maminishkis from the National Eye Institute (NEI), Bethesda, USA, were received as a confluent monolayer of P-0 cells assays were conducted using cells at P-1. Primary human fetal RPE cells were isolated from human donor eyes as previously described and cultured in MEM-a containing 5% FCS (Maminishkis et al., 2006).
METHOD DETAILS
Stimulation of cells
Bone marrow derived macrophages (BMDMs), human monocytic cell like THP1s, PBMCs ARPE-19 cells, primary human fetal RPE cells or primary retinal microglia were stimulated with the generic TLR2/1 ligand Pam3Cys4 (Invivogen) or with CEP-HSA (kindly provided by Prof. Robert G Salomon (Case Western Reserve University, Cleveland Ohio) at indicated concentrations. Where indicated RPE cells were pre-treated with 0.1 µg/ml TLR2 antibody for 1 hour (T2.5 Invivogen), corresponding IgG control 0.1 µg/ml (Invivogen), Mal peptide inhibitor resuspended in DMSO 40µM (Calbiochem) or an equal volume of DMSO was used in the control treatment.
Polarized ARPE-19 cell culture
0.4 µM polyester transwell inserts (VWR) were coated with 100 µg/ml Collagen IV (Sigma-Aldrich C5533) for 4 hours. ARPE-19 cells were seeded at a density of 1.7x105 cells per cm2 in DMEM F-12 Ham containing 10% (FBS). Two days later medium was replaced with complete medium containing 1% FBS and replenished twice weekly for 4–6 weeks.
Western blot
Antibodies for CFB (Atlas Antibodies Sigma) 1:250, C3 (MP Biomedicals-855444), C3d 1:1000 (Dako), ZO-1 (Invitrogen) 1:1000 and C5b-9 (Santa Cruz) 1:500 were incubated overnight at 4°C. Polyvinylidene fluoride (PVDF) membranes were wash 3 times with TBS-T and incubated with horseradish peroxidase conjugated anti-rabbit, anti-mouse or anti-goat 1:2000 (Sigma-Aldrich) for 1 hour at room temperature and developed using enhanced chemiluminescence. Densitometry was used to determine relative quantity of MAC and iC3b protein relative to actin loading control using ImageJ software. Scanned images were converted to 8-bit images. Each protein band was measured to obtain the area and mean value. The area was multiplied by the mean to obtain a measurement for each lane. The value obtained for the protein of interest was divided by the value obtained for the actin loading control for the corresponding well.
Quantitative RT-PCR
Total RNA was extracted from BMDMs, THP1s, ARPE-19 or hfRPE cells using Isolate II RNA Extraction Kit (Bioline) as per manufacturer’s instructions. RNA was reverse transcribed using MMLV Reverse Transcriptase (Promega). Target genes were amplified by real time PCR with SensiFast SYBR Green (Bioline) using the ABI 7900HT system (Applied Biosystems). The cycling threshold method was used for relative quantification after normalization to the ‘housekeeping’ gene βActin. The primers used were:
Human C3 forward 5′- CTGCCCAGTTTCGAGGTCAT-3′; reverse 5′ - CAATCGGAATGCGCTTGAGG-3′
Human CFB forward 5′- CAGGAAGGTGGCTCTTGGAG-3′; reverse 5′ – CCCATCCTCAGCATCGACTC-3′
Human TLR2 forward 5′- TGTAGCAACTGGCTTAGTTCA-3′; reverse 5′-TGGCCACAGAGGAGTCTCTTA-3′
βActin forward 5′-CGCGAGAAGATGACCCAGATC-3′; reverse 5′-GAGGCGTACAGGGATAGCAC-3′
Mouse C3 forward 5′ - AAGCATCAACACACCCAACA-3′; reverse 5′ - CTTGAGCTCCATTCGTGACA-3′
Mouse CFB forward 5′ - ATAGGCCCATCTGTCTCCCC-3′; reverse 5′ - CAGGTGGCTGTCTGAGGAA-3′
Measurement of MCP-1/CCL2 and MAC by ELISA
MCP-1 (TebuBio), and Soluble MAC (Abbexa) was detected in cell supernatants by sandwich ELISA according to manufacturer’s instructions. Absorbance was read at 450 nM on a 96 well plate spectrophotometer.
Measurement of membrane attack complex in vitro
Polarized ARPE-19 cells grown on transwell filters were maintained in serum free DMEM F-12 Ham for 48 hours. Cells were stimulated with 10% Normal Human Serum or heat inactivated Normal Human Serum (Hi) (56°C 30 minutes) either alone or with human serum albumin (HSA) or CEP-HSA for 24 hours. Where indicated cells were pre-treated for 1 hour with anti-TLR2 blocking antibody (T2.5 Invivogen), IgG control (0.1 µg/ml) or Mal peptide inhibitor (Calbiochem). Supernatants were harvested and assessed for soluble MAC formation by ELISA (Abbexa). Transwell inserts were fixed with 4% Paraformaldehyde for 10 minutes at room temperature, blocked with 5% bovine serum albumin (BSA) for 1 hour at room temperature and incubated with anti-mouse-C5b-9 1:25 (Santa cruz) overnight at 4°C. Transwells were washed 3 times in PBS and incubated with goat anti-mouse 647 1:500 (Invitrogen) and Phallodin 1:500 (Invitrogen) for 2 hours at room temperature. Cells were counted stained with Hoechst. Transwells inserts were carefully cut with a sterile blade and mounted on to polysine coated slides (Thermo Scientific) using Mowiol® 4–88. Staining was analyzed using a confocal laser scanning microscope Axio Observer Z1 Inverted Microscope equipped with a Zeiss LSM 700 T-PMT scanning unit and a 40x plan.
LDH viability assay
An LDH cytotoxicity kit (Pierce) was used to detect cell death following MAC formation as per manufacturer’s instructions absorbance was read at 490nm and background absorbance at 680nm.
Luciferase assay
HEK293-TLR2 cells were transfected for 24 hours with C3 promoter-luciferase (100 ng), Renilla-luciferase (40 ng) and empty vector (EV) or plasmid expressing Mal or MyD88 in increasing doses (10, 50 and 80 ng). Results are normalized for Renilla luciferase activity and represented as relative stimulation over the non-stimulated EV control and are expressed as mean ± SD for triplicate measurements.
Measurement of surface ICAM1 and CD86
PBMCs were labeled for the investigation of monocytes with the following fluorochrome-labeled antibodies: anti-CD16 (3G8) anti-CD86 (FM95) anti-CD45 (2D1); anti-CD66b (G10F5); CD14 (Tük4); CD80 (2D10); (Biolegend or Miltenyi). Each staining well contained 4 × 105 cells; cells were stained with LIVE/DEAD Aqua (Molecular Probes) followed by staining for 20 min on ice, washed, and analyzed by flow cytometry immediately. Flow cytometry was carried out on a BD LSR Fortessa cell analyzer and analyzed using FlowJo software (Tree Star).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was carried out using Prism Graphpad and details of each test used can be found in the figure legends.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets/code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TLR2 | Abcam | Cat# ab1655; RRID: AB_302428 |
| Anti-TLR2 neutralizing | InvivoGen | Clone T2.5; Cat# mab-mtlr2; RRID: AB_763722 |
| Mouse Control IgG1 | InvivoGen | Cat# mabg1-ctrlm; RRID: AB_11203233 |
| Anti-C3 | MP-Biomedicals | Cat# 855444 |
| Anti-C3 | Abcam | Cat# ab11887; RRID: AB_298669 |
| Anti-C3d | Agilent | Cat# A006302; RRID: AB_578478 |
| Anti-CFB | Atlas Antibodies; Sigma | Cat# HPA001817; RRID: AB_1078779 |
| Anti-CFD | R&D systems | Cat# MAB5430; RRID: AB_10640506 |
| Rat IgG1 Isotype Control | R&D systems | Cat# MAB005; RRID: AB_357348 |
| MAC/ anti-C5b-9 for Mouse IHC | Biozol | Cat# FGI-10–1801 |
| MAC/ anti-C5b-9 for human IHC | Santa Cruz Biotechnology | clone aE11; Cat# sc58935; RRID: AB_1119839 |
| MAC/ anti-C5b-9 for WB | Santa Cruz Biotechnology | Clone 2A1; Cat# sc66190; RRID: AB_1119840 |
| Anti-CEP | Dr. Rowan (Rowan et al., 2017) | NA |
| ZO-1 | Thermo Fisher Scientific | Cat# 40–2200; RRID: AB_2533456 |
| IBA1 | Wako | Cat# 019–19741; RRID: AB_839504 |
| Biotinylated Goat Anti-Rabbit IgG | Vector Laboratories | Cat# BA-1000; RRID: AB_2313606 |
| Biotinylated Rabbit Anti-Goat IgG | Vector Laboratories | Cat# BA-5000; RRID: AB_2336126 |
| Anti-human CD16 | BioLegend | Clone 3G8; Cat# 302006; RRID: AB_314206 |
| Anti-human CD86 | Miltenyi Biotec | Clone FM95; Cat# 130–094-877; RRID: AB_10839702 |
| Anti-human CD45 | BioLegend | Clone 2D1; Cat# 368506; RRID: AB_2566358 |
| Anti-CD66b | BioLegend | Clone G10F5; Cat# 305116; RRID: AB_2566605 |
| Anti-human CD14 | Miltenyi | Clone Tük4; Cat# 130–098-058; RRID: AB_2660173 |
| Anti-human CD80 | Miltenyi | Clone 2D10; Cat# 130–099-710; RRID: AB_2659260 |
| Alexa Fluor 488 Phalloidin | Thermo Fisher Scientific | Cat# A12379; RRID: AB_2315147 |
| Alexa Fluor 647 Donkey Anti-Mouse | Abcam | Cat# AB150107 |
| Alexa Fluor 488 Goat Anti-Rabbit | Thermo Fisher Scientific | Cat #A11034; RRID: AB_2576217 |
| Alexa Fluor 488 Goat anti-Mouse | Thermo Fisher Scientific | Cat #A11001; RRID: AB_2534069 |
| Biological Samples | ||
| Human donor eye tissue | The Iowa Lions Eye Bank | https://iowalionseyebank.org/ |
| Peripheral Blood Mononuclear Cells (PBMCs) | Isolated from healthy volunteers | NA |
| Primary Microglia | Isolated as per Fernando et al., 2016 | NA |
| Normal Human Serum | Sigma | Cat# H4522–20ml |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse M-CSF | Miltenyl Biotec | Cat# 130–094-129 |
| Mouse Recombinant GM-CSF | Stem Cell Technologies | Cat# 78017.1 |
| Pam3Cys4 | Invivogen | Cat# tlrl-pms |
| CEP-HSA | Prof. Salomon (Gu et al., 2003) | |
| Mal (TIRAP) Inhibitor Peptide | Calbiochem | Cat# 613570–1MG |
| Sodium Iodate | Sigma | Cat # S4007–100G |
| Hoechst | Sigma | Cat #B2261–25MG |
| Collagen IV | Sigma | Cat# C5533–5M |
| Critical Commercial Assays | ||
| In SITU Cell Death Detection Kit, TMR red | Roche | Cat# 12156792910 |
| MCP-1 ELISA | Tebubio | Cat# 900-K31 |
| MAC ELISA | Abbexa | Cat# abx350654 |
| Isolate II RNA Extraction Kit | Bioline | Cat# BIO-52073 |
| MMLV Reverse Transcriptase | Promega | Cat# M1705 |
| SensiFast SYBER Green | Bioline | Cat# BIO-92020 |
| Pierce LDH Viability Assay | Thermo Fisher Scientific | Cat# 13464269 |
| LIVE/DEAD Aqua | Thermo Fisher Scientific | Cat# L34957 |
| Mouse Vectastain ABC HRP Kit | Vector Laboratories | Cat#PK-4002; RRID: AB_2336811 |
| ImmPACT DAB Substrate | Vector Laboratories | Cat#SK-4105; RRID: AB_2336520 |
| Vectastain Elite ABC HRP kit | Vector Laboratories | Cat#PK-6200; RRID: AB_2336826 |
| Vector VIP Peroxidase (HRP) Substrate Kit | Vector Laboratories | Cat# SK-4600; RRID: AB_2336848 |
| Experimental Models: Cell Lines | ||
| ARPE19 | ATCC | CRL-2302 |
| Immortalized BMDMS | Prof. Golenbock, UMASS, USA | N/A |
| hfRPE | Dr. A. Maminishkis (Maminishkis et al., 2006) | N/A |
| THP-1 | ATCC | TIB-202 |
| HEK293-TLR2 | Dr. K. Fitzgerald, UMASS, USA | N/A |
| Experimental Models: Organisms/Strains | ||
| C57Bl6 | Jackson labs | |
| Tlr2–/– | Jackson labs | JAX stock #004650 |
| DBA/2J | Jackson labs | JAX stock #000671 |
| Oligonucleotides | ||
| Primers for SYBER qPCR, | This paper | N/A |
| Recombinant DNA | ||
| C3 luciferase (T1 del) | Addgene | |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com |
| ImageJ | https://imagej.nih.gov/ij/ | |
| FlowJo | Tree star | https://www.flowjo.com |
Highlights.
TLR2 activates the alternative complement pathway
TLR2 signaling triggers sub-lytic MAC formation on retinal pigment epithelial cells
TLR2 deficiency reduces oxidative stress-induced C3 and MAC in the outer retina
TLR2 blockade protects photoreceptors and RPE from oxidative stress-induced cell death
ACKNOWLEDGMENTS
This work was funded by the BrightFocus Foundation (M2016030), United States, Health Research Board Ireland (HRB-HRA/2013.290) and Science Foundation Ireland (SFI 15/CDA/3497), Royal Victoria Eye and Ear Hospital (RVEEH), Ireland, National Children’s Research Centre (NCRC), Ireland, Irish Research Council (IR-CLA/2017/295), Ireland, and NIH (grant R01EY016813), United States.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.064.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, et al. (2010). The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res 29, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman SM, Desai A, Ramo K, and Kumar-Singh R (2011). Expression of complement component 3 (C3) from an adenovirus leads to pathology in the murine retina. Invest. Ophthalmol. Vis. Sci 52, 3436–3445. [DOI] [PubMed] [Google Scholar]
- Chirco KR, Tucker BA, Stone EM, and Mullins RF (2016). Selective accumulation of the complement membrane attack complex in aging choriocapillaris. Exp. Eye Res 146, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, et al. (2002). Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 99, 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despriet DD, van Duijn CM, Oostra BA, Uitterlinden AG, Hofman A, Wright AF, ten Brink JB, Bakker A, de Jong PT, Vingerling JR, et al. (2009). Complement component C3 and risk of age-related macular degeneration. Ophthalmology 116, 474–480.e2. [DOI] [PubMed] [Google Scholar]
- Farrar CA, Keogh B, McCormack W, O’Shaughnessy A, Parker A, Reilly M, and Sacks SH (2012). Inhibition of TLR2 promotes graft function in a murine model of renal transplant ischemia-reperfusion injury. FASEB J 26, 799–807. [DOI] [PubMed] [Google Scholar]
- Feng L, Ju M, Lee KYV, Mackey A, Evangelista M, Iwata D, Adamson P, Lashkari K, Foxton R, Shima D, and Ng YS (2017). A proinflammatory function of toll-like receptor 2 in the retinal pigment epithelium as a novel target for reducing choroidal neovascularization in age-related macular degeneration. Am. J. Pathol 187, 2208–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando N, Natoli R, Valter K, Provis J, and Rutar M (2016). The broad-spectrum chemokine inhibitor NR58–3.14.3 modulates macrophage-mediated inflammation in the diseased retina. J. Neuroinflammation 13, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiannakis A, Burgoyne T, Lueck K, Futter C, Greenwood J, and Moss SE (2015). Retinal pigment epithelial cells mitigate the effects of complement attack by endocytosis of C5b-9. J. Immunol 195, 3382–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, and Salomon RG (2003). Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J. Biol. Chem 278, 42027–42035. [DOI] [PubMed] [Google Scholar]
- Güven M, Batar B, Mutlu T, Bostancı M, Mete M, Aras C, and Ünal M (2016). toll-like receptors 2 and 4 polymorphisms in age-related macular degeneration. Curr. Eye Res 41, 856–861. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, et al. (2005). A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 102, 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, and Mullins RF (2001). An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res 20, 705–732. [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, and Perez VL (2008). Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med 14, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justilien V, Pang J-J, Renganathan K, Zhan X, Crabb JW, Kim SR, Sparrow JR, Hauswirth WW, and Lewin AS (2007). SOD2 knockdown mouse model of early AMD. Invest. Ophthalmol. Vis. Sci 48, 4407–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski DJ, Afrazi A, Scott MJ, Kwak JH, Gill R, Edmonds RD, Liu Y, Fan J, and Billiar TR (2010). Pivotal advance: The pattern recognition receptor ligands lipopolysaccharide and polyinosine-polycytidylic acid stimulate factor B synthesis by the macrophage through distinct but overlapping mechanisms. J. Leukoc. Biol 88, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, and Hinton DR (2014). Sodium iodate induced retinal degeneration: New insights from an old model. Neural Regen. Res 9, 2044–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandhadia S, Hakobyan S, Heng LZ, Gibson J, Adams DH, Alexander GJ, Gibson JM, Martin KR, Menon G, Nash K, et al. (2013). Age-related macular degeneration and modification of systemic complement factor H production through liver transplantation. Ophthalmology 120, 1612–1618. [DOI] [PubMed] [Google Scholar]
- Kim C, Smith KE, Castillejos A, Diaz-Aguilar D, Saint-Geniez M, and Connor KM (2016). The alternative complement pathway aids in vascular regression during the early stages of a murine model of proliferative retinopathy. FASEB J 30, 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, You B, Lim H, and Lee SJ (2011). Toll-like receptor 2 contributes to chemokine gene expression and macrophage infiltration in the dorsal root ganglia after peripheral nerve injury. Mol. Pain 7, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindzelskii AL, Elner VM, Elner SG, Yang D, Hughes BA, and Petty HR (2004). Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J. Gen. Physiol 124, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, et al. (2005). Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeima K, Rogers BS, Lu L, and Campochiaro PA (2006). Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 103, 11300–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, and Yu F-SX (2006). Toll-like receptors and corneal innate immunity. Curr. Mol. Med 6, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunchithapautham K, and Rohrer B (2011). Sublytic membrane-attack-complex (MAC) activation alters regulated rather than constitutive vascular endothelial growth factor (VEGF) secretion in retinal pigment epithelium monolayers. J. Biol. Chem 286, 23717–23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Reca RG, Atmaca-Sonmez P, Ratajczak MZ, Ildstad ST, Kaplan HJ, and Enzmann V (2006). Retinal pigment epithelium damage enhances expression of chemoattractants and migration of bone marrow-derived stem cells. Invest. Ophthalmol. Vis. Sci 47, 1646–1652. [DOI] [PubMed] [Google Scholar]
- Lueck K, Wasmuth S, Williams J, Hughes TR, Morgan BP, Lommatzsch A, Greenwood J, Moss SE, and Pauleikhoff D (2011). Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye (Lond.) 25, 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maminishkis A, Chen S, Jalickee S, Banzon T, Shi G, Wang FE, Ehalt T, Hammer JA, and Miller SS (2006). Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest. Ophthalmol. Vis. Sci 47, 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marneros AG (2016). Increased VEGF-A promotes multiple distinct aging diseases of the eye through shared pathomechanisms. EMBO Mol. Med 8, 208–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi M, Nakamura S, Inoue Y, Nishinaka A, Nakamura M, Shimazawa M, and Hara H (2018). Irreversible photoreceptors and RPE cells damage by intravenous sodium iodate in mice is related to macrophage accumulation. Invest. Ophthalmol. Vis. Sci 59, 3476–3487. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, and Stone EM (2011). Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp. Eye Res 93, 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, Wang K, Tucker BA, and Stone EM (2014). The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol 184, 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli R, Fernando N, Jiao H, Racic T, Madigan M, Barnett NL, Chu-Tan JA, Valter K, Provis J, and Rutar M (2017). Retinal macrophages synthesize C3 and activate complement in AMD and in models of focal retinal degeneration. Invest. Ophthalmol. Vis. Sci 58, 2977–2990. [DOI] [PubMed] [Google Scholar]
- Natoli R, Jiao H, Barnett NL, Fernando N, Valter K, Provis JM, and Rutar M (2016a). A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp. Eye Res 147, 114–127. [DOI] [PubMed] [Google Scholar]
- Natoli R, Rutar M, Lu YZ, Chu-Tan JA, Chen Y, Saxena K, Madigan M, Valter K, and Provis JM (2016b). The role of pyruvate in protecting 661W photoreceptor-like cells against light-induced cell death. Curr. Eye Res 41, 1473–1481. [DOI] [PubMed] [Google Scholar]
- Nielsen HE, Larsen SO, and Vikingsdottir T (1992). Rate-limiting components and reaction steps in complement-mediated haemolysis. APMIS 100, 1053–1060. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Hara H, Kondo M, Hong S, and Matsugi T (2017). Oxidative stress in retinal diseases. Oxid. Med. Cell. Longev 2017, 4076518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, and Ambati J (2006). Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 103, 2328–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope MR, Hoffman SM, Tomlinson S, and Fleming SD (2010). Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Mol. Immunol 48, 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, and Lambris JD (2010). Complement: A key system for immune surveillance and homeostasis. Nat. Immunol 11, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Latz E, Ontiveros F, and Kono H (2010). The sterile inflammatory response. Annu. Rev. Immunol 28, 321–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Guo Y, Kunchithapautham K, and Gilkeson GS (2007). Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest. Ophthalmol. Vis. Sci 48, 5282–5289. [DOI] [PubMed] [Google Scholar]
- Rowan S, Jiang S, Korem T, Szymanski J, Chang ML, Szelog J, Cassalman C, Dasuri K, McGuire C, Nagai R, et al. (2017). Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc. Natl. Acad. Sci. USA 114, E4472–E4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Francis PJ, George S, Schultz DW, Rosner B, and Klein ML (2007). Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA 297, 1793–1800. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, and Rosner B (2009). Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest. Ophthalmol. Vis. Sci 50, 2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, Goldstein JI, Triebwasser M, Anderson HE, Zerbib J, et al. (2013). Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet 45, 1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E, et al. (2013). CCR2+ monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol. Med 5, 1775–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A, Cui J, To E, Kwee M, and Matsubara J (2008). Complement-associated deposits in the human retina. Invest. Ophthalmol. Vis. Sci 49, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou H, Koso H, Sasaki J, Nakanishi H, Sagara H, Nakagawa KM, Takahashi Y, Hishikawa D, Iizuka-Hishikawa Y, Tokumasu F, et al. (2017). Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem 292, 12054–12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott B, and Korbelik M (2007). Activation of complement C3, C5, and C9 genes in tumors treated by photodynamic therapy. Cancer Immunol. Immunother 56, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigard JH, Matsumoto H, Smith KE, Kim LA, Paschalis EI, Okonuki Y, Castillejos A, Kataoka K, Hasegawa E, Yanai R, et al. (2015). Inhibition of the alternative complement pathway preserves photoreceptors after retinal injury. Sci. Transl. Med 7, 297ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweigard JH, Yanai R, Gaissert P, Saint-Geniez M, Kataoka K, Thanos A, Stahl GL, Lambris JD, and Connor KM (2014). The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J 28, 3171–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Mühlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, and Akira S (2001). Discrimination of bacterial lipoproteins by toll-like receptor 6. Int. Immunol 13, 933–940. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, and Akira S (2002). Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol 169, 10–14. [DOI] [PubMed] [Google Scholar]
- Tan LX, Toops KA, and Lakkaraju A (2016). Protective responses to sub-lytic complement in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 113, 8789–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K, Hughes TR, Triantafilou M, and Morgan BP (2013). The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci 126, 2903–2913. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Amiguet P, and Schäfer S (1986). Increased hemolytic activity of the trypsin-cleaved ninth component of complement. Mol. Immunol 23, 57–62. [DOI] [PubMed] [Google Scholar]
- van de Ven JP, Nilsson SC, Tan PL, Buitendijk GH, Ristau T, Mohlin FC, Nabuurs SB, Schoenmaker-Koller FE, Smailhodzic D, Campochiaro PA, et al. (2013). A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet 45, 813–817. [DOI] [PubMed] [Google Scholar]
- Wang H, Guo J, West XZ, Bid HK, Lu L, Hong L, Jang GF, Zhang L, Crabb JW, Linetsky M, and Salomon RG; Clinical Genomic and Proteomic AMD Study Group (2014). Detection and biological activities of carboxyethylpyrrole ethanolamine phospholipids (CEP-EPs). Chem. Res. Toxicol 27, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, and Byzova TV (2010). Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, and Wong TY (2014). Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2, e106–e116. [DOI] [PubMed] [Google Scholar]
- Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, et al. ; Genetic Factors in AMD Study Group (2007). Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med 357, 553–561. [DOI] [PubMed] [Google Scholar]
- Zeng S, Whitmore SS, Sohn EH, Riker MJ, Wiley LA, Scheetz TE, Stone EM, Tucker BA, and Mullins RF (2016). Molecular response of chorioretinal endothelial cells to complement injury: Implications for macular degeneration. J. Pathol 238, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jang YP, Kim SR, and Sparrow JR (2006). Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 103, 16182–16187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Feng Y, Li Y, Zhang M, Chen C, Cai J, Gong Y, Wang L, Thurman JM, Wu X, et al. (2013). Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J. Immunol 191, 5625–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.