

The rise of Gram-negative pathogens expressing metallo-β-lactamases (MBLs) is a growing concern, threatening the efficacy of β-lactam antibiotics, in particular, the carbapenems. There are no inhibitors of MBLs in current clinical use. Aspergillomarasmine A (AMA) is an MBL inhibitor isolated from Aspergillus versicolor with the ability to rescue meropenem activity in MBL-producing bacteria both in vitro and in vivo. Here, we systematically explored the pairing of AMA with six β-lactam antibiotic partners against 19 MBLs from three subclasses (B1, B2, and B3).
KEYWORDS: antibiotic resistance, aspergillomarasmine A, beta-lactams, carbapenems, cephems, metallo-beta-lactamases, penams
ABSTRACT
The rise of Gram-negative pathogens expressing metallo-β-lactamases (MBLs) is a growing concern, threatening the efficacy of β-lactam antibiotics, in particular, the carbapenems. There are no inhibitors of MBLs in current clinical use. Aspergillomarasmine A (AMA) is an MBL inhibitor isolated from Aspergillus versicolor with the ability to rescue meropenem activity in MBL-producing bacteria both in vitro and in vivo. Here, we systematically explored the pairing of AMA with six β-lactam antibiotic partners against 19 MBLs from three subclasses (B1, B2, and B3). Cell-based assays performed with Escherichia coli and Klebsiella pneumoniae showed that bacteria producing NDM-1 and VIM-2 of subclass B1 were the most susceptible to AMA inhibition, whereas bacteria producing CphA2 and AIM-1 of subclasses B2 and B3, respectively, were the least sensitive. Intracellular antibiotic accumulation assays and in vitro enzyme assays demonstrated that the efficacy of AMA/β-lactam combinations did not correlate with outer membrane permeability or drug efflux. We determined that the optimal β-lactam partners for AMA are the carbapenem antibiotics and that the efficacy of AMA is linked to the Zn2+ affinity of specific MBLs.
INTRODUCTION
β-Lactams are the most commonly prescribed family of antibiotics and are increasingly ineffective against many serious bacterial infections (1). Resistance to β-lactam antibiotics occurs predominantly through the production of β-lactamases. These enzymes are divided into four classes based on their structure and amino acid sequence. Classes A, C, and D are serine-β-lactamases (SBLs) that employ an active site serine residue to promote hydrolysis of the β-lactam ring (1). Class B enzymes are metallo-β-lactamases (MBLs) that require active site Zn2+ ions for their catalytic activity (2). MBLs are further organized into three subclasses (B1, B2, and B3). The B1 subclass is the largest and incorporates most of the clinically relevant MBLs, including the NDM, VIM, and IMP families (3–5). The enzymes of subclasses B2 (e.g., CphA2) and B3 (e.g., L1 and AIM-1) are less common in pathogens (6–8).
Several coformulations of β-lactam antibiotics with β-lactamase inhibitors, including avibactam, relebactam, vaborbactam, sulbactam, tazobactam, and clavulanic acid, are in clinical use (9, 10). These inhibitors are specific to SBLs and do not affect MBLs, revealing a growing therapeutic gap as MBL producers increase in frequency across the globe. Selective inhibition of MBLs has proven challenging as a result of their low sequence similarity, structurally fluid active sites, and poor selectivity over human metalloenzymes (11, 12). Furthermore, while many MBL inhibitors display potent activity in vitro, few exhibit comparable efficacy in whole-cell assays or in vivo animal models.
In a targeted screen for new MBL inhibitors, we discovered the fungal natural product aspergillomarasmine A (AMA), a potent inhibitor of the clinically important enzymes NDM-1 and VIM-2 (13). AMA restores the in vitro activity of meropenem in MBL-producing Enterobacteriaceae, Acinetobacter, and Pseudomonas isolates and was previously shown to be effective in rescuing meropenem activity against NDM-1-producing Klebsiella pneumoniae in a murine model of systemic infection (13). While these findings establish the potential of AMA as an MBL inhibitor, the effect of AMA against a broader panel of MBLs remains unknown.
In this study, we carried out a systematic analysis of the efficacy of AMA in combination with six β-lactam antibiotic partners from three subclasses (carbapenem, cephem, and penam). For susceptibility testing of the various antibiotic/inhibitor combinations, we selected 19 MBLs from three subclasses. The potency of each combination was evaluated using biochemical and cell-based assays, where individual MBLs were expressed in isogenic Escherichia coli and K. pneumoniae strains. The resulting data serve as a guide for the in vivo implementation of AMA and related MBL inhibitors.
RESULTS
The potency of AMA and meropenem combinations depends on the MBL subclass and allelic variant.
We previously demonstrated that AMA could inhibit two clinically relevant MBLs, NDM-1 and VIM-2, but was less effective toward IMP-7 (13). Consequently, we sought to rigorously establish the inhibitory spectrum of AMA against 19 MBLs from each subclass (B1, B2, B3). Using cell-based assays, we evaluated the efficacy of AMA in combination with meropenem. We scored efficacy based on the minimum concentration needed to restore the level of meropenem growth inhibition to its EUCAST (European Committee on Antimicrobial Susceptibility Testing) susceptibility breakpoint (2 μg/ml). To ensure consistency between the different enzymes tested, all MBL genes were identically cloned into the pGDP2 vector (low-copy-number plasmid with a Plac promoter) (14) and transformed into E. coli BW25113. The MIC values of meropenem against the wild-type and MBL-producing strains were measured in the absence of AMA to serve as a benchmark for resistance (see Table S1 in the supplemental material).
The results revealed that E. coli strains producing subclass B1 enzymes could be resensitized to meropenem (≤2 μg/ml) over AMA concentrations ranging from 4 to 32 μg/ml (Table 1; see also Table S4). Producers of NDM-1, VIM-1, VIM-2, VIM-7, CAM-1, and IND-1 from subclass B1 were the most susceptible to AMA (4 to 8 μg/ml). Cells producing NDM-4, NDM-5, NDM-6, NDM-7, IMP-1, IMP-7, and IMP-27 were moderately sensitive (12 to 16 μg/ml). E. coli producing B2 or B3 enzymes, except for L1, were the least sensitive to AMA (no rescue of meropenem at 2 μg/ml at AMA concentrations of ≥64 μg/ml) (Table 1; see also Table S4). The extent of meropenem potentiation by AMA was not necessarily related to the degree of antibiotic resistance conferred by the MBL. For example, while IMP-7 conferred a drug MIC of 16 μg/ml, it was less susceptible to AMA inhibition than other MBLs that conferred drug MIC values of 64 μg/ml.
TABLE 1.
Concentration of AMA needed to restore the activity of different β-lactam antibiotics to the level seen with their EUCAST susceptibility breakpoint concentration in MBL-producing E. coli BW25113a
| MBL | [AMA] at the susceptibility breakpoint of the antibiotic (μg/ml)b |
|||||
|---|---|---|---|---|---|---|
| Meropenem | Doripenem | Ertapenem | Imipenem | Cefotaxime | Ampicillin | |
| NDM-1 | 8 | 12 | 24 | 12 | 64 | 64 |
| NDM-4 | 16 | 16 | 64 | 16 | >64 | >64 |
| NDM-5 | 12 | 24 | 64 | 16 | >64 | >64 |
| NDM-6 | 16 | 16 | 64 | 16 | 64 | >64 |
| NDM-7 | 16 | 24 | 64 | 24 | >64 | >64 |
| VIM-1 | 8 | 8 | 12 | 12 | 24 | 64 |
| VIM-2 | 8 | 8 | 8 | 8 | 12 | 16 |
| VIM-7 | 8 | 8 | 8 | 8 | 8 | 16 |
| CAM-1 | 4 | 12 | 8 | 8 | 24 | 64 |
| DIM-1 | 12 | 12 | 16 | 8 | 24 | 64 |
| IND-1 | 8 | 8 | 12 | 12 | 24 | >64 |
| GIM-1 | 12 | 12 | 32 | 12 | >64 | >64 |
| IMP-1 | 16 | 12 | 64 | 24 | >64 | 64 |
| IMP-7 | 24 | 12 | >64 | 16 | >64 | 32 |
| IMP-27 | 32 | 24 | >64 | ≤0.5 | >64 | 12 |
| SPM-1 | 16 | 24 | 64 | 8 | >64 | >64 |
| CphA2 | >64 | 64 | >64 | >64 | ≤0.5 | ≤0.5 |
| L1 | 12 | 12 | 24 | 8 | ≤0.5 | >64 |
| AIM-1 | 64 | 64 | >64 | 24 | >64 | >64 |
All 19 MBL genes were cloned into the pGDP2 vector. All bioassays were conducted in duplicate. This table shows the results from replicate 1.
The EUCAST susceptibility breakpoint concentrations for meropenem, doripenem, ertapenem, imipenem, cefotaxime, and ampicillin are 2, 1, 0.5, 2, 1 and 8 μg/ml, respectively.
To examine whether the inhibitory potency of AMA was simply dependent on different levels of β-lactamase expression, a FLAG tag was engineered at the C termini of eight representative MBLs. These MBLs were specifically chosen because they covered a broad range of sensitivity to various combinations of AMA and meropenem. The level of MBL production was quantified using an anti-FLAG monoclonal antibody via Western blotting. The installation of the tag did not affect resistance. The relative protein levels of NDM-4, NDM-5, NDM-6, NDM-7, VIM-2, and IMP-7 were all within a 2-fold range (see Fig. S1 and S2 in the supplemental material). Although NDM-1 demonstrated the highest sensitivity to AMA inhibition, its relative expression level was ∼3-fold higher than that of any other enzyme. In contrast, IMP-7, which has a low sensitivity to AMA inhibition, showed lower protein levels. Together with our bioassay data, these results indicate that the MBL inhibition spectrum of AMA is broad but that it is more active toward class B1 enzymes.
The efficacy of AMA depends on the β-lactam antibiotic partner.
We partnered AMA with three different carbapenems (doripenem, ertapenem, and imipenem), a cephem (cefotaxime), and a penam (ampicillin) to explore the optimal AMA/antibiotic combination. We graded the efficacy of AMA based on its ability to restore the activity of each antibiotic to its EUCAST susceptibility breakpoint concentration (for doripenem, ertapenem, imipenem, cefotaxime, and ampicillin, 1, 0.5, 2, 1, and 8 μg/ml, respectively). As described above, the individual drug MIC values for each antibiotic against MBL-producing E. coli served as benchmarks for resistance (see Table S1). We found that the carbapenem and cephem susceptibilities of MBL-producing E. coli BW25113 were well (>7-fold) above the breakpoints. However, as this strain of E. coli encodes a chromosomal cephalosporinase (AmpC), it showed a baseline ampicillin MIC of 4 μg/ml and was much closer to the susceptibility breakpoint (see Table S1).
The levels of β-lactam potentiation by AMA were comparable for most of the carbapenems tested. Testing with AMA, the majority of strains producing MBLs showed either equivalent or 2-fold-increased levels of susceptibility for doripenem and imipenem relative to meropenem. Ertapenem, however, showed more variation. In particular, the IMP and NDM alleles were 3-fold to 5-fold less sensitive to AMA when paired with ertapenem relative to other carbapenems (Table 1; see also Table S4). This pattern of reduced susceptibility to AMA inhibition involving the VIM, IMP, and NDM enzymes was also evident when cefotaxime and ampicillin were the partner antibiotics (Table 1; see also Table S4). E. coli producing CphA2 was the most refractory to AMA combinations and showed high-level resistance against all carbapenem antibiotics (Table 1; see also Table S4).
β-Lactam potentiation by AMA is also MBL class dependent in K. pneumoniae.
To investigate whether the inhibitory potency of AMA with different β-lactam antibiotics was pathogen dependent, we transformed K. pneumoniae ATCC 33495 with plasmids carrying one of eight selected MBL genes. The MIC values of the different β-lactams against the wild-type and MBL-producing K. pneumoniae strains were measured in the absence of AMA to serve as a control (see Table S2). K. pneumoniae encodes a chromosomal penicillinase (SHV) and is consequently resistant to ampicillin with a drug MIC value of 64 μg/ml.
Similar to the results in E. coli, the potency of AMA combinations against MBLs produced in K. pneumoniae varied with the subclass of the β-lactam antibiotic partner. K. pneumoniae strains producing NDM-1 or VIM-2 were the most susceptible to carbapenems (Table 2; see also Table S5). However, K. pneumoniae producing other NDM variants demonstrated less sensitivity to AMA/carbapenem combinations, requiring higher AMA concentrations to achieve efficacy (Table 2; see also Table S5). Meropenem was the β-lactam partner most strongly potentiated by AMA. Concentrations of AMA ranging from 8 to 16 μg/ml reduced the MIC values of this carbapenem for most MBL-producing K. pneumoniae to ≤2 μg/ml (Table 2; see also Table S5). Since the MIC of meropenem with most MBL-producing K. pneumoniae strains was ≥32 μg/ml, this represents at least a 16-fold improvement of the antibiotic’s activity (see Table S2). The AMA and ampicillin pairing resulted in the poorest efficacy overall since all of the MBL-producing K. pneumoniae strains remained resistant to this antibiotic due to the production of an endogenous serine-dependent penicillinase (Table 2; see also Table S5). Like E. coli, CphA2 and AIM-1 were the least sensitive to AMA inhibition (Table 2; see also Table S5). These results suggest that the alteration of the potency of the AMA combinations contributed by the antibiotic partner was not pathogen dependent for two representative Enterobacteriaceae strains. Furthermore, the basal levels of β-lactam resistance shown by chromosomal β-lactamases play a significant role in determination of the appropriate β-lactam to be paired with AMA.
TABLE 2.
Concentration of AMA needed to restore the activity of different β-lactam antibiotics to the level seen with their EUCAST susceptibility breakpoint concentration in MBL-producing K. pneumoniae ATCC 33495a
| MBL | [AMA] at the susceptibility breakpoint of the antibiotic (μg/ml)b |
|||||
|---|---|---|---|---|---|---|
| Meropenem | Doripenem | Ertapenem | Imipenem | Cefotaxime | Ampicillin | |
| NDM-1 | 12 | 16 | 24 | 12 | 64 | >64 |
| NDM-4 | 16 | 24 | 64 | 24 | >64 | >64 |
| NDM-5 | 16 | 24 | 64 | 24 | >64 | >64 |
| NDM-6 | 12 | 16 | 64 | 24 | >64 | >64 |
| VIM-2 | 8 | 8 | 8 | 12 | 16 | >64 |
| IMP-7 | 24 | 24 | >64 | 64 | >64 | >64 |
| CphA2 | >64 | >64 | >64 | >64 | ≤0.5 | >64 |
| AIM-1 | 64 | 64 | >64 | >64 | >64 | >64 |
All 8 MBL genes were cloned into the pGDP2 vector. All bioassays were conducted in duplicate. This table shows the results from replicate 1.
The EUCAST susceptibility breakpoint concentrations for meropenem, doripenem, ertapenem, imipenem, cefotaxime, and ampicillin are 2, 1, 0.5, 2, 1 and 8 μg/ml, respectively.
Outer membrane permeability and efflux do not influence the activity of different β-lactam antibiotics against MBL-producing bacteria.
To probe whether the observed differences in the effects of β-lactam specificity on the efficacy of AMA were the result of differences in outer membrane penetration or efflux of these antibiotics, potentiation assays were conducted with hyperpermeable bamB/tolC deletion strains of E. coli BW25113 expressing eight different MBL genes. The BamB lipoprotein and the TolC outer membrane protein play essential roles in outer membrane permeability and the efflux of antibiotics, respectively (15, 16). We found that apart from ampicillin, for which the baseline MIC value decreased 8-fold, the carbapenem MIC values against the ΔbamB ΔtolC strain were similar to those seen with the wild type. Likewise, the levels of resistance shown by MBL-producing strains were comparable (see Table S3).
AMA treatment of the MBL-producing ΔbamB ΔtolC strains revealed that the extent of potentiation of each β-lactam was very similar to that seen with wild-type E. coli (Table 3; see also Table S6). We further probed the efficacy of the β-lactams by directly measuring the intracellular accumulation of AMA and each antibiotic in both the E. coli wild-type and ΔbamB ΔtolC strains. Although it was not possible to determine how much drug was lost to surface binding and periplasmic leeching, we reasoned that this would be a constant for each β-lactam in both the wild-type and ΔbamB ΔtolC strains. Any differences in accumulation should then be a result of increased penetration and/or decreased efflux (17). We found that AMA and the carbapenem antibiotics were detectable in cell extracts in wild-type bacteria but that the extent of accumulation varied with the subclass of the β-lactam antibiotic. Despite the drug MIC values being similar to those seen with the wild type, E. coli ΔbamB ΔtolC showed 5-fold, 2-fold, and 1.5-fold increases in accumulation for meropenem, doripenem, and imipenem, respectively (Fig. 1). Ertapenem was the only member of the carbapenems which showed no such change between the strains.
TABLE 3.
Concentration of AMA needed to restore the activity of different β-lactam antibiotics to the level seen with their EUCAST susceptibility breakpoint concentration in MBL-producing E. coli BW25113 ΔbamB ΔtolCa
| MBL | [AMA] at the susceptibility breakpoint of the antibiotic (μg/ml)b |
||
|---|---|---|---|
| Meropenem | Doripenem | Ampicillin | |
| NDM-1 | 8 | 12 | 32 |
| NDM-4 | 16 | 24 | >64 |
| NDM-5 | 16 | 24 | >64 |
| NDM-6 | 12 | 24 | >64 |
| VIM-2 | 8 | 8 | 24 |
| IMP-7 | 24 | 24 | >64 |
| CphA2 | 64 | >64 | ≤0.5 |
| AIM-1 | 64 | 64 | >64 |
All 8 MBL genes were cloned into the pGDP2 vector. All bioassays were conducted in duplicate. This table shows the results from replicate 1.
The EUCAST susceptibility breakpoint concentrations for meropenem, doripenem, and ampicillin are 2, 1 and 8 μg/ml, respectively.
FIG 1.
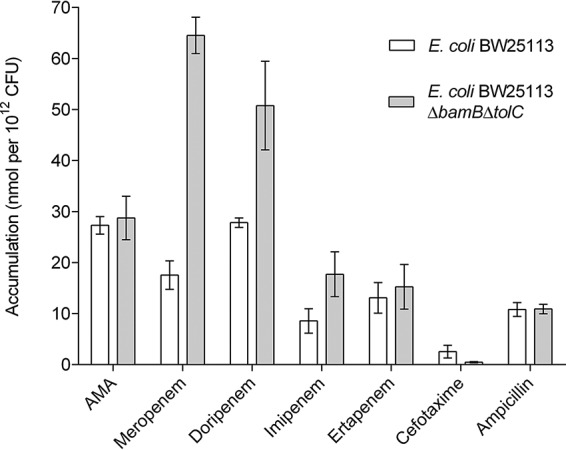
Accumulation assay data for different β-lactam antibiotics in E. coli BW25113 and E. coli BW25113 ΔbamB ΔtolC. All assays were performed in biological triplicate and technical duplicate. The error bars represent the standard deviations.
Our initial attempts to detect the accumulation of ampicillin failed and were likely hampered by susceptibility to endogenous AmpC. To circumvent AmpC-mediated hydrolysis, we cotreated the cells with avibactam. The treatment enabled the detection of ampicillin, which we found had accumulated similarly in the two strains, despite having a lower MIC in the hyperpermeable strain. It is possible that some of the ampicillin had been hydrolyzed, resulting in an underestimation of the precise amount present. We also faced challenges with cefotaxime since, despite several attempts, we could not demonstrate robust accumulation levels for this antibiotic.
β-Lactam inactivation is affected by substrate-specific zinc requirements of MBLs.
Previous work had shown that Zn2+ dissociates from MBLs during catalytic turnover of β-lactams and that this can lead to enzyme inactivation (18, 19). This phenomenon is particularly pronounced under conditions where excess Zn2+ is unavailable to replenish the enzyme. Since AMA binds to free Zn2+, it could potentially withhold Zn2+ from MBLs, and the β-lactam could promote inactivation. We therefore hypothesized that different β-lactam substrates could uniquely influence Zn2+ dissociation from MBLs and affect the efficacy of AMA.
We tested this hypothesis with a series of in vitro enzyme assays involving a subset of purified MBLs and the panel of β-lactam substrates used in the cell-based assays. In each enzyme stock (20 μM), a limited amount of AMA (100 nM) was included to sequester any residual Zn2+ in the Chelex-treated buffer. The reaction mixtures contained enzyme (4 to 10 nM), saturating amounts of substrate, and various concentrations of ZnSO4 (0.001 to 20 μM). The resulting reaction rates exhibited clear Zn2+ dependence for each substrate. From the progress curves of the initial reaction rates, the Zn2+ dissociation constant (Kd,Zn2) could be calculated as the concentration of ZnSO4 required to achieve half-maximal velocity. We presume on the basis of previous observations made by several different research groups (20) that metal dissociation occurs primarily from the second zinc site (Zn2) of the MBLs.
The levels of substrate-specific zinc dependence differed greatly among the MBLs and ranged from nanomolar to micromolar Kd,Zn2 values. In reaction mixtures containing ampicillin, cefotaxime, meropenem, and doripenem, NDM-1 showed the lowest average level of affinity for Zn2+ during catalysis, with substrate-specific Kd,Zn2 values that were mostly in the low micromolar range (0.9 to 3.2 μM) (Table 4; see also Fig. S3). This finding contrasted with the results seen with the other MBLs, for which nanomolar (140 to 950 nM) Zn2+ affinity was observed. Intriguingly, the Zn2+ dependence of imipenem and ertapenem hydrolysis did not follow this trend. For imipenem, the Kd,Zn2 values for NDM-1, VIM-2, IMP-7, and AIM-1 fell within a 2-fold range (400 to 950 nM), while NDM-4 and NDM-6 showed the highest affinity for Zn2+ during hydrolysis of this carbapenem (60 and 140 nM, respectively). Surprisingly, ertapenem showed low micromolar Zn2+ affinity (0.95 to 4.3 μM) for every MBL tested, which contrasted with the trends that we observed for the other substrates (Table 4; see also Fig. S3). Overall, these data allowed a general estimation of Zn2+ affinity for each MBL and showed that the specific choice of β-lactam influences the Kd,Zn2 values within a 3-fold range for each enzyme.
TABLE 4.
Zinc dependence of metallo-β-lactamase-catalyzed hydrolysis of β-lactam antibioticsa
| Substrate | Metallo-β-lactamase zinc dissociation constantb (Kd,Zn2) |
|||||
|---|---|---|---|---|---|---|
| NDM-1 | NDM-4 | NDM-6 | VIM-2 | IMP-7 | AIM-1 | |
| Ampicillin | 0.91 ± 0.07 | 0.15 ± 0.02 | 0.23 ± 0.02 | 0.45 ± 0.05 | 0.24 ± 0.04 | 0.19 ± 0.05 |
| Cefotaxime | 1.4 ± 0.3 | 0.17 ± 0.03 | 0.24 ± 0.02 | 0.21 ± 0.03 | 0.80 ± 0.4 | 0.33 ± 0.07 |
| Meropenem | 3.2 ± 0.5 | 0.3 ± 0.06 | 0.64 ± 0.08 | 0.63 ± 0.1 | 0.46 ± 0.08 | 0.52 ± 0.08 |
| Imipenem | 0.4 ± 0.04 | 0.06 ± 0.007 | 0.14 ± 0.01 | 0.34 ± 0.09 | 0.95 ± 0.3 | 0.59 ± 0.07 |
| Ertapenem | 2.8 ± 1 | 4.3 ± 2.6 | 2.0 ± 0.3 | 2.3 ± 1 | 2.0 ± 0.4 | 0.95 ± 0.2 |
| Doripenem | 1.1 ± 0.1 | 0.18 ± 0.02 | 0.14 ± 0.08 | 0.38 ± 0.03 | 0.082 ± 0.01 | 0.90 ± 0.1 |
Reactions were carried out with enzyme (4 to 10 nM) in Chelex-treated HEPES-NaOH, pH 7.5 (50 mM), supplemented with AMA (100 nM).
All constants are reported in micromolar (μM).
The Kd,Zn2 values failed to explain our observations where AMA differentially potentiated β-lactams. The results further support the hypothesis that the level of β-lactam resistance conferred by each MBL is the primary factor that dictates AMA susceptibility. However, the general level of Zn2+ affinity determined for each MBL was consistent with previous studies, where enzymes with higher affinity for Zn2+ were found to be better suited to withstanding its associated limitation in general (21). For example, NDM alleles, other than NDM-1, have evolved to increase metal affinity. This was evident in the current study as the concentrations of AMA needed to restore the activity of the antibiotics to their susceptibility breakpoints were generally lower for NDM-1 than for NDM-4, NDM-5, NDM-6, NDM-7, IMP-7, CphA2, and AIM-1. Interestingly, VIM-2 stands out as a particular case in that its low Kd,Zn2 values do not correlate well with the bioassay data, suggesting that this enzyme has an unknown property which increases its sensitivity to AMA.
DISCUSSION
AMA is an inhibitor of MBLs and potentiates the activity of meropenem against MBL-producing bacteria. Here, we explored the inhibitory activity of AMA paired with five other β-lactam antibiotics against 19 MBL enzymes produced in three bacterial strains.
Potentiation assays indicated that AMA achieved the highest inhibitory potency when combined with a carbapenem antibiotic. This was not influenced by the bacterial species or strain used during the bioassays (Tables 1 to 3). The results of the intracellular accumulation assays suggest that the inhibitory activity of AMA together with the different β-lactam antibiotics is not correlated with outer membrane permeability or drug efflux (Fig. 1). One possible explanation for the advantage of using AMA/carbapenem pairings may be related to the affinity of these drugs for their targets, the penicillin-binding proteins (PBPs). Kocaoglu and Carlson determined that each β-lactam antibiotic is selective for a subset of PBPs (22). While ampicillin targets a broader spectrum of PBPs, none of them are solely essential for bacterial growth, and the bactericidal effect is the result of inhibition of several of these enzymes.
On the other hand, meropenem, doripenem, and cefotaxime all inhibit at least one essential PBP and do so with high potency. Furthermore, unlike penams, which are prone to hydrolysis by class C SBLs (e.g., AmpC) and low-molecular-weight PBPs, carbapenems are not substrates for these enzymes. These observations are consistent with previous studies which demonstrated that meropenem, doripenem, and imipenem are the carbapenem antibiotics with the highest potencies and the broadest spectra of activity against different bacterial species (23), including extended-spectrum-β-lactamase (ESBL)-producing isolates of E. coli and K. pneumoniae (24).
Our work supports results of previous studies indicating that MBL-mediated carbapenem resistance is weaker than cefotaxime and ampicillin resistance (21, 25–27). Considering our data in conjunction with the current knowledge available for β-lactam efficacy and resistance, AMA is most likely to succeed when paired with a carbapenem, in particular, meropenem, doripenem, or imipenem.
Given our observations that the β-lactam substrate uniquely influences the AMA susceptibility of different MBLs, we wondered if its inhibitory activity was mechanistically dependent on substrate interactions. Substrate-induced Zn2+ dissociation (i.e., reduced Zn2+ affinity) was well documented in several MBL studies (18, 19, 28–30), and those results inspired us to develop the mechanistic basis for the following hypothesis: β-lactams promote the release of Zn2+ from MBLs (presumably by increasing the rate constant for dissociation), and then AMA acts as a recipient for the free metal. In effect, AMA would withhold Zn2+, leading to essentially irreversible inhibition. Precedents for this hypothesis can be found in the mechanism of inhibition proposed for d-penicillamine (d-Pen) toward human Zn2+-dependent carboxypeptidase A (ZnCPD). d-Pen directly catalyzes the removal of Zn2+ from ZnCPD through increased metal dissociation, but the high-affinity apoenzyme can rebind the metal and outcompete d-Pen. However, if a potent chelator such as EDTA is present, Zn2+ is sequestered, and ZnCPD is wholly inhibited (31, 32). In general, our data show that the level of Zn2+ affinity seen during β-lactam hydrolysis correlated well with the potency of AMA for numerous MBL/β-lactam pairs. This was consistent with several studies that have shown that increased affinity can overcome Zn2+ scarcity. For example, NDM variants contain single-point or multiple-point mutations which increase Zn2+ affinity. Furthermore, this increases the thermostability of these MBLs, which has been associated with an increase in resistance (21). This is also consistent with our observation that CphA2 is refractory to AMA, as this MBL is a mono-Zn2+ enzyme with a strong metal affinity (Kd,Zn value of 6 pM [33]).
Curiously, not all of the Kd,Zn2 values correlated with the biological activity of AMA and some of the results were counterintuitive. For example, the hydrolysis of ertapenem resulted in the highest Kd,Zn2 values for every MBL tested, which currently eludes explanation. Moreover, VIM-2 showed high Zn2+ affinity during hydrolysis of most β-lactams, although it was one of the most sensitive MBLs in our bioassays. Recently, it was shown that membrane anchoring stabilizes NDM variants when Zn2+ is scarce in the environment (34). All other MBLs contain cleavable signal peptides and are released into the periplasmic space and can be rapidly degraded in a manner depending on their stability. In the case of VIM-2, González et al. have shown that it is particularly susceptible to degradation (34), which could explain why, despite high Zn2+ affinity, VIM-producing bacteria are more susceptible to AMA.
Our results demonstrate that an AMA/carbapenem pairing would be the most effective combination for treating infections caused by MBL-producing bacteria in clinics, though higher concentrations of AMA are required to cover recently emerging NDM alleles.
MATERIALS AND METHODS
DNA manipulations and pGDP2 plasmid construction.
All oligonucleotide primers were purchased from IDT (Coralville, IA). The MBL genes used in this study were also purchased from IDT as gBlock gene fragments, except for blaNDM-4, blaNDM-5, blaNDM-6, and blaNDM-7. The gBlock sequences for each MBL gene were obtained from the Comprehensive Antibiotic Resistance Database (CARD; https://card.mcmaster.ca/). The sequence of blaAIM-1 from the CARD was codon optimized for E. coli K-12 to facilitate its expression in the E. coli and K. pneumoniae strains. Each MBL gene fragment was subsequently cloned into the pGDP2 vector. As blaNDM-4, blaNDM-5, blaNDM-6, and blaNDM-7 differ from blaNDM-1 by only a few point mutations, constructs of these variants were generated by site-directed mutagenesis of pGDP2:blaNDM-1 using the primers listed in Table S7 in the supplemental material. In comparison to NDM-1, the NDM-4 (M154L) and NDM-6 (A233V) variants required a single nucleotide substitution whereas the NDM-5 (M154L and V88L) and NDM-7 (D130N and M154L) variants needed two nucleotide substitutions. All MBL gene sequences were verified by Sanger sequencing. For antibiotic susceptibility and AMA potentiation assays, the purified plasmids were transformed into chemically competent E. coli BW25113 cells, E. coli BW25113 ΔbamB ΔtolC cells, or K. pneumoniae ATCC 33495 cells. The freeze-thaw transformation procedure used with the K. pneumoniae cells followed the protocol described in reference 35. These bacterial strains were chosen for the transformation because E. coli BW25113 and K. pneumoniae ATCC 33495 are carbapenem susceptible (see Table S1 to S3).
pE-SUMOstar and pET-28b plasmid construction.
For protein overproduction and purification, the MBL genes previously cloned into the pGDP2 vectors were used as templates for cloning into overexpression vectors using the primers listed in Table S8. SignalP (http://www.cbs.dtu.dk/services/SignalP/) was used to determine the sequence of the signal peptide, which was excluded from the final constructs to facilitate cytoplasmic accumulation. The VIM-2, IMP-7, and AIM-1 genes were ligated in a pET-28b vector in frame with a cleavable N-terminal 6×His tag. All NDM genes were ligated into a pE-SUMOstar vector in frame with an N-terminal 6×His-Smt3 tag. Smt3 is a ubiquitin-like protein from Saccharomyces cerevisiae. The purified plasmids were then transformed into chemically competent E. coli BL21(DE3) cells. Mature MBL gene sequences were verified by Sanger sequencing before overexpression.
Western blot analysis.
Engineering FLAG-tagged variants of each MBL was achieved through a PCR-based procedure. The MBL genes previously cloned into the pGDP2 vectors were used as templates for the addition of the protein tag by using the primers listed in Table S8. To allow the addition of the FLAG tag to the C terminus of the protein, the stop codon needed to be removed from the C terminus of the MBL gene. The tagged MBL genes were ligated into the pGDP2 vector and transformed into chemically competent E. coli BW25113 cells. Gene sequences were verified by Sanger sequencing before immunodetection.
Cation-adjusted Mueller-Hinton II broth (CAMHB) supplemented with kanamycin (50 μg/ml) was inoculated with E. coli BW25113 cells containing the FLAG-tagged MBL. The inoculated medium was incubated at 37°C until the optical density at 600 nm (OD600) reached 1.0. Cells (1 ml) were harvested by centrifugation (17,000 × g, 2 min, room temperature). The cell pellet was resuspended in 100 μl of double-distilled water (ddH2O) and mixed with 100 μl of 2× SDS running buffer. Proteins resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were transferred to a polyvinylidene difluoride membrane (PVDF) and probed with mouse-derived anti-FLAG monoclonal antibodies conjugated to horseradish peroxidase (HRP; GenScript, Piscataway, NJ) (1:5,000). Chemiluminescence signals were detected using a ChemiDoc MP imaging system (Bio-Rad, Hercules, CA). Following imaging, irreversible inhibition of HRP was conducted by incubating the Western blot with 30% hydrogen peroxide for 15 min at 37°C (36). The Western blot was then reprobed with mouse-derived anti-RpoA antibodies (BioLegend, San Diego, CA) (1:5,000) and anti-mouse IgG antibodies conjugated to HRP (Cell Signaling Technology, Danvers, MA) (1:5,000). Chemiluminescence signals were detected using a ChemiDoc MP imaging system. Protein band intensities were quantified using Image Lab software (Bio-Rad). These results were then plotted and analyzed using GraphPad Prism 8 (GraphPad, La Jolla, CA) to determine the relative protein expression levels. The antibodies targeting the alpha subunit of RNA polymerase (RpoA) served as loading controls for the analysis.
Protein purification.
For each MBL, LB medium supplemented with kanamycin (50 μg/ml) was inoculated with E. coli BL21(DE3) cells containing the appropriate plasmid. The inoculated medium was incubated at 37°C until the OD600 reached 0.6 to 0.8. Expression of the constructs was induced by the use of isopropyl-β-d-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. Cultures were then incubated at 16°C for 16 to 20 h. Cells were harvested by centrifugation (6,000 × g, 20 min, 4°C) using a Beckman Coulter Avanti J-25 centrifuge with a JLA 9.1000 rotor (Beckman Coulter, Fullerton, CA) and frozen at −20°C until required. For purification, the cell pellet was resuspended in lysis buffer (25 mM HEPES-NaOH, 300 mM NaCl, 10 mM imidazole, 100 μM ZnSO4, pH 7.5). Cells were then disrupted by sonication (8-s intervals for 8 min) using a Microson XL-2000 ultrasonic liquid processor (Qsonica, Newtown, CT) set at level 12. Unbroken cells were removed by centrifugation (40,000 × g, 20 min, 4°C) using a Beckman JLA 25.50 rotor (Beckman Coulter). His-tagged proteins were then bound to 2 ml of HisPur nickel-nitrilotriacetic acid (Ni-NTA) resin (Pierce, Rockford, IL) and applied to a gravity column. The resin was washed three times with lysis buffer (60 ml total volume), and the protein was then eluted with lysis buffer containing 300 mM imidazole. Fractions were analyzed using SDS-PAGE. All fractions shown to contain purified β-lactamase were combined and dialyzed for 16 to 20 h at 4°C in dialysis buffer (25 mM HEPES-NaOH, 150 mM NaCl, 100 μM ZnSO4, pH 7.5). Following dialysis, the 6×His tag was removed from the protein using thrombin (Sigma-Aldrich, St-Louis, MO). The 6×His tag and undigested protein were removed by passage through HisPur Ni-NTA resin equilibrated in dialysis buffer. Subsequently, thrombin was removed with p-aminobenzamidine-agarose resin. Protein samples were then analyzed by SDS-PAGE to confirm tag removal. The purified proteins were stored at 4°C or frozen at −20°C.
Purification of NDM-1 proceeded as described above, except for the addition of the Ulp-1 (ubiquitin-like protein-specific protease 1) to the dialysis buffer. The dialyzed protein was then applied to 2 ml of HisPur Ni-NTA resin to remove any uncleaved NDM-1 as well as Ulp-1. Fractions containing cleaved NDM-1, as assessed by SDS-PAGE, were pooled and stored at 4°C. NDM-4, NDM-5, and NDM-6 were purified using the same procedure as that used for NDM-1 purification.
AIM-1 was produced in an insoluble form and was acquired from the pellet obtained after centrifugation of the cell lysate. The AIM-1-containing protein pellet was solubilized using denaturation buffer (50 mM HEPES-NaOH, 150 mM NaCl, 6 M guanidine HCl, pH 7.5), followed by centrifugation (30,000 × g, 20 min, 4°C). The denatured protein sample (10 ml) was subsequently refolded in renaturation buffer (50 mM HEPES-NaOH, 150 mM NaCl, 50 mM l-arginine, 10 μM ZnSO4, pH 7.5) using a slow dialysis method. The dialysis membrane containing AIM-1 was subjected to buffer exchange in 50-ml volumes once per hour for a total of 8 h at room temperature. Precipitated protein was removed by centrifugation (30,000 × g, 10 min, 4°C), and the supernatant was then applied to HisPur Ni-NTA resin. The protein was eluted as described above, concentrated, and further purified to remove aggregates using a size exclusion column (HiLoad Superdex 200 16/600; GE Healthcare Bio-Sciences AB, Uppsala, Sweden) that was equilibrated and operated with dialysis buffer. SDS-PAGE-analyzed fractions containing monomeric protein were combined, and the His tag was removed as described above. The enzyme activity was tested and was comparable to previously published kinetic data (8).
β-Lactam potentiation assays.
Bacterial antibiotic susceptibility assays were conducted using AMA in combination with a β-lactam antibiotic based on the protocol described in reference 37. β-Lactam antibiotics were dissolved in water, and AMA was diluted in water containing ≤5% (vol/vol) ammonium hydroxide to ensure that the final pH was between 7.5 and 8.5. Compounds were subjected to filter sterilization. All assays were conducted in a 96-well format with 10 dilutions of AMA (0.5, 1, 2, 4, 8, 12, 16, 24, 32, and 64 μg/ml) being added to columns 1 to 10 while 8 2-fold dilutions of the β-lactam antibiotic (0.5–64 μg/ml) were added to rows A to H. The dilutions of the β-lactam antibiotics were also added to column 11 to confirm the MIC value of the β-lactam antibiotic with each different strain. The dilutions of AMA and the β-lactam antibiotic were conducted manually or with a Labcyte Echo 550 liquid dispenser (Labcyte, San Jose, CA) or with a Thermo Scientific Multidrop Combi nL reagent dispenser (Thermo Fisher Scientific, Waltham, MA). A bacterial inoculum was prepared from the bacterial cells of interest using colonies picked from overnight plates whose OD625 was adjusted to 0.08 to 0.10. Once the optimal OD625 was reached, a 200-fold dilution of the inoculum was conducted before its addition to the MIC plate. Dilution of the inoculum was performed using CAMHB. The inoculum was added to columns 1 through 11. The bacterial inoculum and the CAMHB were then added alternately to column 12 to serve as growth and sterility controls. The final assay volume was 100 μl when the assay was conducted in a 96-well round-base microtest plate (Sarstedt, Nümbrecht, Germany) or 50 μl when the assay was scaled to a 384-well format (Corning, Kennebunk, ME) (flat bottom, clear, tissue culture treated, polystyrene). Following a 20-h static incubation at 37°C, bioassay plates containing E. coli were shaken for 5 min to resuspend the bacterial cells. However, K. pneumoniae cells were resuspended manually using a pipette to minimize the formation of aerosols. The bioassay plates were read spectrophotometrically at a wavelength of 600 nm using a SpectraMax 384 Plus UV/Vis spectrophotometer/microplate reader (Molecular Devices, San Jose, CA). All potentiation assays were done with at least two replicates. The susceptibility breakpoints published by the European Committee on Antimicrobial Susceptibility Testing (EUCAST; http://www.eucast.org/clinical_breakpoints/) were used as a reference.
Accumulation assays.
Antibiotic accumulation was conducted as previously described using the E. coli wild-type and ΔbamB ΔtolC strains (38). Briefly, meropenem, doripenem, imipenem, ertapenem, cefotaxime, ampicillin, or AMA was added to cells in a volume of 875 μl ([β-lactam] = 50 μM and [AMA] = 100 μM). Avibactam (50 μM) was added to the assays containing either cefotaxime or ampicillin. Avibactam was used to inhibit the AmpC β-lactamase, which is chromosomally encoded by the E. coli strains and prevented the detection of these antibiotics. The antibiotics were incubated with the bacteria (10 min, 37°C), and 800 μl of the suspension was subsequently washed through 700 μl of ice-cold silicone oil (9:1 AR20/Sigma High Temperature) by centrifugation (12,000 × g, 2 min, room temperature). The cells were resuspended in 200 μl water and lysed by three freeze-thaw cycles. Cell debris was collected by centrifugation (17,000 × g, 2 min, room temperature) using a Fisher Scientific accuSpin Micro 17 microcentrifuge (Thermo Fisher Scientific), and the pellet was extracted with 100 μl methanol (MeOH). The cell extracts were pooled and quantitatively analyzed by the use of ultraperformance liquid chromatography (UPLC) coupled to a high-resolution quadrupole-time of flight (Q-TOF) 6550 mass spectrometer (Agilent, Santa Clara, CA). In order to detect AMA, samples were derivatized with benzoyl chloride (BzCl) based on the protocol described in reference 39. Briefly, samples were mixed with acetonitrile (1:1), 0.5 volumes of sodium carbonate (100 mM) and BzCl (2%) were added, and the mixture was subjected to vortex mixing. Any precipitated material present was removed by centrifugation (17,000 × g, 2 min, room temperature). Samples were loaded onto a C8 column (Agilent Eclipse XDB-C8) (100 mm by 2.1 mm; 3.5-μm pore size) that had previously been equilibrated with solvent A (water, 0.1% formic acid) and 5% solvent B (acetonitrile, 0.1% formic acid), and they were resolved using a linear gradient of 5% to 97% solvent B over 7 min, followed by a 1-min wash step at 97% solvent B at a flow rate of 0.4 ml/min. The Q-TOF system was operated in extended dynamic range positive-ion targeted tandem mass spectrometry (MS/MS) modes with an m/z range of 100 to 1,700 m/z and a capillary voltage of 0.5 kV. The collision energies and respective parent-daughter ion transitions used for AMA and each β-lactam antibiotic are listed in Table S9. Quantification was carried out with a calibration curve of each antibiotic using Agilent MassHunter Quantitative Analysis software. For each compound, biological and technical replicates were conducted in triplicate and duplicate, respectively.
In vitro enzyme assays.
MBL-catalyzed hydrolysis of β-lactam substrates was monitored spectrophotometrically by measuring the decrease in UV absorbance at 300 nm (meropenem, doripenem, imipenem, and ertapenem), 265 nm (cefotaxime), or 235 nm (ampicillin) in UV-grade 96-well flat-bottom microplates (Thermo Fisher Scientific). Before use in assays, enzymes (20 μM) were preincubated with AMA (0.5 mM; 16 h, 4°C) and subsequently dialyzed into reaction buffer (50 mM HEPES-NaOH [pH 7.5], 100 nM AMA, 4°C) to ensure that residual Zn2+ was removed from the enzyme preparation. HEPES-NaOH (1 M) buffer was pretreated with 5% (wt/vol) Chelex 100 prior to use (24 h, 25°C). Enzyme (4 to 10 nM) in reaction buffer was added to various concentrations (0.0007, 0.0015, 0.03, 0.15, 0.31, 0.62, 1.2, 2.5, 5, 10, and 20 μM) of ZnSO4 containing carbapenem (0.5 mM), ampicillin (0.5 mM), or cefotaxime (0.25 mM) substrate to initiate the reaction. The reactions were monitored using a BioTek Synergy H1 plate reader (Biotek, Winooski, VT) over 5 min at 25°C. To obtain Kd,Zn2 values, the initial rates of substrate hydrolysis at each ZnSO4 concentration were plotted and analyzed using nonlinear regression by fitting the data to equation 1 using GraphPad Prism 8. In cases where zinc inhibition was apparent, equation 2 was used. All enzyme assays were performed in duplicate.
| (1) |
| (2) |
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by a Canadian Institutes of Health Research grant (FRN-148463), a Canadian Institutes of Health Research Fellowship award (to D.S.), a Canada Research Chair in Antibiotic Biochemistry (to G.D.W.), and an Ontario Graduate Scholarship and Queen Elizabeth II Graduate Scholarship (to C.M.R.).
We thank Haley Zubyk for conducting the cloning and bacterial transformation of several MBL genes employed in this research and Susan McCusker for assistance with liquid handling instrumentation.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.King DT, Sobhanifar S, Strynadka N. 2016. One ring to rule them all: current trends in combating bacterial resistance to the β-lactams. Protein Sci 25:787–803. doi: 10.1002/pro.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bebrone C. 2007. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol 74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, Rao Z. 2011. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2:384–394. doi: 10.1007/s13238-011-1055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.García-Sáez I, Docquier J-D, Rossolini GM, Dideberg O. 2008. The three-dimensional structure of VIM-2, a Zn-β-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form. J Mol Biol 375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frère JM, Payne DJ, Bateson JH, Abdel-Meguid SS. 2000. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 6.Villadares MH, Galleni M, Frère J-M, Felici A, Perilli M, Franceschini N, Rossolini GM, Oratore A, Amicosante G. 1996. Overproduction and purification of the Aeromonas hydrophila CphA metallo-β-lactamase expressed in Escherichia coli. Microb Drug Resist 2:253–256. doi: 10.1089/mdr.1996.2.253. [DOI] [PubMed] [Google Scholar]
- 7.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. 1998. The crystal structure of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J Mol Biol 284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 8.Leiros H-K, Borra PS, Brandsdal BO, Edvardsen KSW, Spencer J, Walsh TR, Samuelsen Ø. 2012. Crystal structure of the mobile metallo-β-lactamase AIM-1 from Pseudomonas aeruginosa: insights into antibiotic binding and the role of Gln157. Antimicrob Agents Chemother 56:4341–4353. doi: 10.1128/AAC.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palzkill T. 2013. Metallo-β-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush K, Bradford PA. 2019. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 17:295–306. doi: 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- 11.Rotondo CM, Wright GD. 2017. Inhibitors of metallo-β-lactamases. Curr Opin Microbiol 39:96–105. doi: 10.1016/j.mib.2017.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Tehrani K, Martin NI. 2018. β-lactam/β-lactamase inhibitor combinations: an update. Medchemcomm 9:1439–1456. doi: 10.1039/c8md00342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD. 2014. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 510:503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox G, Sieron A, King AM, De Pascale G, Pawlowski AC, Koteva K, Wright GD. 2017. A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem Biol 24:98–109. doi: 10.1016/j.chembiol.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Namdari F, Hurtado-Escobar GA, Abed N, Trotereau J, Fardini Y, Giraud E, Velge P, Virlogeux-Payant I. 2012. Deciphering the roles of BamB and its interaction with BamA in outer membrane biogenesis, T3SS expression and virulence in Salmonella. PLoS One 7:e46050. doi: 10.1371/journal.pone.0046050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koronakis V, Eswaran J, Hughes C. 2004. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu Rev Biochem 73:467–489. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- 17.Li X-Z, Ma D, Livermore DM, Nikaido H. 1994. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: active efflux as a contributing factor to β-lactam resistance. Antimicrob Agents Chemother 38:1742–1752. doi: 10.1128/aac.38.8.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badarau A, Page MI. 2006. Enzyme deactivation due to metal-ion dissociation during turnover of the cobalt-β-lactamase catalyzed hydrolysis of β-lactams. Biochemistry 45:11012–11020. doi: 10.1021/bi0610146. [DOI] [PubMed] [Google Scholar]
- 19.Badarau A, Page MI. 2008. Loss of enzyme activity during turnover of the Bacillus cereus β-lactamase catalysed hydrolysis of β-lactams due to loss of zinc ion. J Biol Inorg Chem 13:919–928. doi: 10.1007/s00775-008-0379-2. [DOI] [PubMed] [Google Scholar]
- 20.Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, Fast W. 2011. Characterization of purified New Delhi metallo-β-lactamase-1. Biochemistry 50:10102–10113. doi: 10.1021/bi201449r. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Z, Thomas PW, Ju L, Bergstrom A, Mason K, Clayton D, Miller C, Bethel CR, VanPelt J, Tierney DL, Page RC, Bonomo RA, Fast W, Crowder MW. 2018. Evolution of New Delhi metallo-β-lactamase (NDM) in the clinic: effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J Biol Chem 293:12606–12618. doi: 10.1074/jbc.RA118.003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kocaoglu O, Carlson EE. 2015. Profiling of β-lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2. Antimicrob Agents Chemother 59:2785–2790. doi: 10.1128/AAC.04552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhanel GG, Wiebe R, Dilay L, Thomson K, Rubinstein E, Hoban DJ, Noreddin AM, Karlowsky JA. 2007. Comparative review of the carbapenems. Drugs 67:1027–1052. doi: 10.2165/00003495-200767070-00006. [DOI] [PubMed] [Google Scholar]
- 24.Jones RN, Sader HS, Fritsche TR. 2005. Comparative activity of doripenem and three other carbapenems tested against Gram-negative bacilli with various β-lactamase resistance mechanisms. Diagn Microbiol Infect Dis 52:71–74. doi: 10.1016/j.diagmicrobio.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Tada T, Miyoshi-Akiyama T, Shimada K, Shimojima M, Kirikae T. 2013. IMP-43 and IMP-44 metallo-β-lactamases with increased carbapenemase activities in multidrug-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:4427–4432. doi: 10.1128/AAC.00716-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poirel L, Naas T, Nicolas D, Collet L, Bellais S, Cavallo JD, Nordmann P. 2000. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob Agents Chemother 44:891–897. doi: 10.1128/aac.44.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yong D, Toleman MA, Bell J, Ritchie B, Pratt R, Ryley H, Walsh TR. 2012. Genetic and biochemical characterization of an acquired subgroup B3 metallo-β-lactamase gene, blaAIM-1, and its unique genetic context in Pseudomonas aeruginosa from Australia. Antimicrob Agents Chemother 56:6154–6159. doi: 10.1128/AAC.05654-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wommer S, Rival S, Heinz U, Galleni M, Frère J-M, Franceschini N, Amicosante G, Rasmussen B, Bauer R, Adolph H-W. 2002. Substrate-activated zinc binding of metallo-β-lactamases. J Biol Chem 277:24142–24147. doi: 10.1074/jbc.M202467200. [DOI] [PubMed] [Google Scholar]
- 29.Rasia RM, Vila AJ. 2002. Exploring the role and the binding affinity of a second zinc equivalent in B. cereus metallo-β-lactamase. Biochemistry 41:1853–1860. doi: 10.1021/bi010933n. [DOI] [PubMed] [Google Scholar]
- 30.Bounaga S, Laws AP, Galleni M, Page MI. 1998. The mechanism of catalysis and the inhibition of the Bacillus cereus zinc-dependent β-lactamase. Biochem J 331:703–711. doi: 10.1042/bj3310703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong CR, Auld DS. 2000. Inhibition of carboxypeptidase A by D-penicillamine: mechanism and implications for drug design. Biochemistry 39:7580–7588. doi: 10.1021/bi000101+. [DOI] [PubMed] [Google Scholar]
- 32.Chong CR, Auld DS. 2007. Catalysis of zinc transfer by D-penicillamine to secondary chelators. J Med Chem 50:5524–5527. doi: 10.1021/jm070803y. [DOI] [PubMed] [Google Scholar]
- 33.Hernandez Valladares M, Kiefer M, Heinz U, Soto RP, Meyer-Klaucke W, Nolting HF, Zeppezauer M, Galleni M, Frère JM, Rossolini GM, Amicosante G, Adolph HW. 2000. Kinetic and spectroscopic characterization of native and metal-substituted β-lactamase from Aeromonas hydrophila AE036. FEBS Lett 467:221–225. doi: 10.1016/S0014-5793(00)01102-9. [DOI] [PubMed] [Google Scholar]
- 34.González LJ, Bahr G, Nakashige TG, Nolan EM, Bonomo RA, Vila AJ. 2016. Membrane anchoring stabilizes and favors secretion of New Delhi metallo-β-lactamase. Nat Chem Biol 12:516–522. doi: 10.1038/nchembio.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merrick MJ, Gibbins JR, Postgate JR. 1987. A rapid and efficient method for plasmid transformation of Klebsiella pneumoniae and Escherichia coli. J Gen Microbiol 133:2053–2057. doi: 10.1099/00221287-133-8-2053. [DOI] [PubMed] [Google Scholar]
- 36.Sennepin AD, Charpentier S, Normand T, Sarré C, Legrand A, Mollet LM. 2009. Multiple reprobing of Western blots after inactivation of peroxidase activity by its substrate, hydrogen peroxide. Anal Biochem 393:129–131. doi: 10.1016/j.ab.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—ninth edition. CLSI, Wayne, PA. [Google Scholar]
- 38.Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ. 2017. Predictive rules for compound accumulation yield a broad-spectrum antibiotic. Nature 545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong JMT, Malec PA, Mabrouk OS, Ro J, Dus M, Kennedy RT. 2016. Benzoyl chloride derivatization with liquid chromatography–mass spectrometry for targeted metabolomics of neurochemicals in biological samples. J Chromatogr A 1446:78–90. doi: 10.1016/j.chroma.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.