

Pseudomonas aeruginosa is an opportunistic pathogen that is inherently resistant to many antibiotics and represents an increasing threat due to the emergence of drug-resistant strains. There is a pressing need to develop innovative antimicrobials against this pathogen. In this study, we identified the O-specific antigen (OSA) of P. aeruginosa serotype O6 as a novel target for therapeutic intervention. Binding of monoclonal antibodies and antigen-binding fragments therefrom to O6 OSA leads to rapid outer membrane destabilization and inhibition of cell growth.
KEYWORDS: lipopolysaccharide, LPS, O-specific antigen, antibacterial antibodies, outer membrane disruption, atomic force microscopy, AFM
ABSTRACT
Pseudomonas aeruginosa is an opportunistic pathogen that is inherently resistant to many antibiotics and represents an increasing threat due to the emergence of drug-resistant strains. There is a pressing need to develop innovative antimicrobials against this pathogen. In this study, we identified the O-specific antigen (OSA) of P. aeruginosa serotype O6 as a novel target for therapeutic intervention. Binding of monoclonal antibodies and antigen-binding fragments therefrom to O6 OSA leads to rapid outer membrane destabilization and inhibition of cell growth. The antimicrobial effect correlated directly with antibody affinity. Antibody binding to the O antigen of a second lipopolysaccharide (LPS) type present in P. aeruginosa or to the LPS core did not affect cell viability. Atomic force microscopy showed that antibody binding to OSA resulted in early flagellum loss, formation of membrane blebs, and eventually complete outer membrane loss. We hypothesize that antibody binding to OSA disrupts a key interaction in the P. aeruginosa outer membrane.
INTRODUCTION
Pseudomonas aeruginosa can cause life-threatening infections in immunocompromised individuals such as those suffering from AIDS, cancer, burn wounds, and cystic fibrosis (CF) (1, 2). In addition, P. aeruginosa is a leading cause of nosocomial infections, including pneumonia, urinary tract infections, surgical-site infections, bloodstream infections, and gastrointestinal infections (3, 4). Strains found in hospitals are becoming more difficult to treat as some are resistant to nearly all antibiotics, necessitating the search for more treatment options including biologic drugs (5–8). One of these biologics is a bispecific antibody targeting two virulence factors on the surface of P. aeruginosa cells (7). Prophylaxis or treatment with the bispecific antibody was protective in rabbit bloodstream and lung infection models (8).
A monoclonal antibody (MAb), S20, which is specific for the P. aeruginosa O6a, 6d O-specific antigen (OSA), a polysaccharide component of the outer membrane (OM) lipopolysaccharide (LPS), has an unusual antibacterial property that could open up new antibody treatment options for P. aeruginosa infections. It exhibits phagocytosis-independent and complement-independent antimicrobial activity against the O6a, 6d P. aeruginosa serotype with little or no antibacterial activity against other serotypes or other bacterial species and offers protection against infection in a leukopenic mouse model (9). S20 is a human IgG2 raised by immunizing immunoglobulin-knockout transgenic mice reconstituted with human immunoglobulin genes and has been shown to be opsonic for the killing of P. aeruginosa in the presence of human polymorphonuclear leukocytes (10). As for the direct antimicrobial activity of S20, propidium iodide penetration of S20-treated cells pointed to membrane damage, and scanning electron micrographs of S20-treated cells showed severe damage to the OM (9). The antimicrobial activity of S20 resides in the variable domains as the single-chain variable-fragment (scFv) exhibited antibacterial activity; however, it has not been confirmed that inhibitory activity is mediated by antigen binding (9). Surprisingly, MAb S20 was reported to exhibit antibacterial activity against P. aeruginosa PAO1, which is serotype 5 (9) and possesses a radically different OSA structure from that of O6a, 6d (Fig. 1a).
FIG 1.

Binding specificity and antibacterial activity of S20. (a) Structures of the OSAs of P. aeruginosa O5 (PAO1) and O6 (O6 IATS, PAK, O6a, 6d, and Fisher 1) strains. The unique β-linkage is shown in red. (b) Schematic showing representative P. aeruginosa LPS structures of OSA and CPA and the various antibodies used in this study. The schematic is not to scale. (c) ELISA results showing the high specificity of the S20 scFv to P. aeruginosa strain O6a, 6d compared with specificities of the other 20 IATS serotypes and PAO1. Data represent means of three replicates ± standard errors of the means. (d) Representative growth curve overlays of the IATS P. aeruginosa serotype O6a, 6d, PAO1, and O6 IATS in the absence (red lines) or presence (blue lines) of 10 μM S20 scFv monomer. The binding results shown in panel c correspond with antibacterial activity shown in panel d (see also Fig. S2 in the supplemental material) in that S20 exhibits antibacterial activity only toward the strain to which it binds.
The OSA of P. aeruginosa serotype O6 is composed of a four-sugar repeating unit comprised of l-rhamnose (l-Rha), 2-acetamido-2-deoxy-d-galacturonamide (d-GalNAcAN), 2-deoxy-2-formamido-d-galacturonic acid (d-GalNFoA), and 2-acetamido-2,6-dideoxy-d-glucose (d-QuiNAc) (11) (Fig. 1a). Subsets of this serotype, including O6a, 6d, differ in their O-antigen structures by the degree of d-GalNAcA amidation, by the presence/absence of 3-O-acetylation of GalNAcAN (3OAc), and by the linkages within their repeating units (α versus β and/or 1→2 versus 1→3) (12). The only unique structural difference between O6a, 6d and other O6 strains is the β-linkage between the d-QuiNAc and l-Rha residues of O6a, 6d (the other strains have an α-linkage) (12). Therefore, the exquisite specificity of the S20 antibody is likely in part due to interaction with this β-linkage.
In the present study we set out to further investigate the basis of the direct action of S20 on cells. We used a growth inhibition assay in addition to the plate count assay used previously (9) to verify the binding and antimicrobial activity of S20. The effect of S20 valency on antimicrobial activity was investigated with scFv monomer and dimer, and the effects of additional antibodies, with a range of specificities for P. aeruginosa LPS, on growth and cell viability were examined (Table 1). The MAb MF23-1 (13), specific for the OSAs of several Pseudomonas O6 strains, was tested for antibacterial activity. MF23-1, an IgG3, is one of a panel of MAbs isolated against 13 O-antigen serogroups (13) and has not been studied with regard to antibacterial activity. MF23-1 has a strong, but perhaps slightly less restrictive, preference for the α-linkage between the d-QuiNAc and l-Rha residues. We also examined the effects of MAb N1F10 (14), specific for the O chain of a second Pseudomonas LPS type in which the O chain is the Pseudomonas common polysaccharide antigen (CPA), and of MAb 7-4 (15), specific for the LPS inner core, on antibacterial activity (Fig. 1b). Finally, using atomic force microscopy (AFM), we examined the nature of outer membrane damage.
TABLE 1.
Summary of antibodies used in this study
| Antibodya | Formatb | Specificityc | Expression system | Reference or source |
|---|---|---|---|---|
| S20 | scFv | O6a, 6d OSA | P. pastoris | 9 |
| scFv dimer | O6a, 6d OSA | P. pastoris | This study | |
| MF23-1 | Murine IgG3 | O6 OSA | Hybridoma | 13 |
| Fab | O6 OSA | Papain digestion of MAb | This study | |
| N1F10 | Murine IgM | CPA | Hybridoma | 14 |
| scFv | CPA | P. pastoris | This study | |
| 7-4 | Murine IgG2b | Inner core LPS | Hybridoma | 15 |
| LPT3-1 | Murine IgG2a | Inner core LPS (N. meningitidis) | Hybridoma | 16 |
Unless specified, antibodies target the LPS of P. aeruginosa.
scFv, single-chain variable fragment; Fab, antigen binding fragment.
OSA, O-specific antigen; CPA, common polysaccharide antigen; LPS, lipopolysaccharide.
RESULTS
Binding and antibacterial specificity of S20.
Binding and growth inhibition assays were carried out using monomeric S20 scFv. Immobilized metal affinity chromatography (IMAC)-purified S20 scFv was subjected to size exclusion chromatography (SEC), which showed two prominent peaks representing S20 scFv dimer and S20 scFv monomer. Following SEC, reducing SDS-PAGE showed that the S20 scFv monomer and dimer were of high purity (see Fig. S1 in the supplemental material).
In addition to P. aeruginosa O6a, 6d, Xie et al. (9) reported that the S20 antibody displayed nonspecific growth inhibition of PAO1 (serotype O5), albeit at lower levels. To better understand the relationship between the binding and antibacterial specificities of S20, S20 scFv was screened for binding (Fig. 1c) and growth-inhibitory activity (Fig. 1d and Fig. S2) against all 20 International Antigenic Typing Scheme (IATS) serotypes of P. aeruginosa as well as PAO1 and O6a, 6d (Table S1). An enzyme-linked immunosorbent assay (ELISA) confirmed that the S20 antibody was highly specific for P. aeruginosa strain O6a, 6d and showed negligible binding to any of the 20 IATS serotypes or PAO1. A highly sensitive microtiter plate growth inhibition assay showed that S20 scFv delayed O6a, 6d growth. Western blotting indicated that S20 did not bind to P. aeruginosa O6a, 6d flagella (data not shown).
Binding and antibacterial specificity of MF23-1.
Like S20, MAb MF23-1 binds to P. aeruginosa O6a, 6d but also recognizes other O6 serotypes (13). Therefore, this MAb represents an ideal tool for testing if the antibacterial activity of S20 is unique or if antibody binding to OSA forms the basis of growth inhibition activity. Using agar plate assays, MAb MF23-1 and its Fab fragment significantly decreased the viability of all OSA positive (OSA+) O6 strains, including O6 IATS, Fisher 1, PAK, and O6a, 6d (Fig. 2). No inhibitory activity of MF23-1 was observed against O6 strains deficient in OSA (OSA−), including PAK wbpO (CPA+/OSA−) and PAK rmlC (CPA−/OSA−). LPT3-1, a MAb specific for Neisseria meningitidis inner core LPS (16), did not affect the growth of any of the P. aeruginosa strains tested. When strains were incubated for 30 min with MF23-1 MAb or Fab, the viability (in CFU counts per milliliter) of Fisher 1, O6 IATS, and PAK were all reduced more than 10-fold compared with a control treatment (phosphate-buffered saline [PBS]). This level of antibacterial activity was comparable to that observed for S20 and its fragments (Fab and scFv) against P. aeruginosa strain O6a, 6d. Although MF23-1 MAb and Fab reduced the viability of O6a, 6d, they were considerably less active against this strain than the other OSA+ O6 strains tested. The variability in MF23-1 activity against different O6 strains was proportional to the affinities of MF23-1 for their OSAs, with higher growth inhibition activity against strains displaying lower 50% effective concentrations (EC50s) in ELISAs (Fig. S3). Western blotting indicated that MF23-1 did not bind to P. aeruginosa O6a, 6d flagella (data not shown).
FIG 2.

Antibacterial activity of S20 scFv, MF23-1 MAb, and MF23-1 Fab against various P. aeruginosa O6 strains. The antibacterial activity was determined by counting CFU on agar plates following 30 min incubation of the bacteria (ca. 1 × 107 CFU ml−1) with the antibodies at 37°C. Untreated bacteria (PBS) and bacteria treated with the N. meningitidis LPS inner core-specific LPT3-1 MAb served as controls. Data represent means of three replicates ± standard errors of the means.
Binding kinetics and affinities of S20 and MF23-1.
The affinities of S20 scFv monomer and dimer for immobilized O6a, 6d LPS-containing liposomes were measured by surface plasmon resonance (SPR). Rate and affinity constants (Table 2) were determined by fitting the data to a 1:1 interaction model (Fig. S4). The good fit of the dimer data to the model indicated that the binding was homogeneous (i.e., all bivalent). These analyses revealed that the equilibrium dissociation constant (KD) of the S20 scFv monomer for O6a, 6d OSA was 560 nM, while the apparent affinity of the dimer was approximately 30-fold higher (KDapp of 21 nM) due to avidity.
TABLE 2.
Summary of the equilibrium dissociation and rate constants of S20 and MF23-1 antibodiesa
| Antibody | KD (M)b | kd (s−1) | ka (M−1 s−1) | Chi2 |
|---|---|---|---|---|
| S20 scFv monomer | ||||
| Immobilized LPS | 5.6 × 10−7 | 2.9 × 10−3 | 5.2 × 103 | 2.53 |
| 2 repeating units | 4.7 × 10−6 | 5.1 × 10−3 | 1.1 × 103 | 0.25 |
| 3 repeating units | 1.1 × 10−6 | 4.7 × 10−3 | 4.4 × 103 | 0.60 |
| S20 scFv dimer | ||||
| Immobilized LPS | 2.1 × 10−8c | 1.2 × 10−4 | 5.9 × 103 | 2.04 |
| 2 repeating units | ND | ND | ND | ND |
| 3 repeating units | ND | ND | ND | ND |
| MF23-1 MAb | ||||
| Immobilized LPS | ND | ND | ND | ND |
| 2 repeating units | 8.8 × 10−5d | 0.53 | ||
| 3 repeating units | 1.8 × 10−5d | 0.96 | ||
| MF23-1 Fab | ||||
| Immobilized LPS | ND | ND | ND | ND |
| 2 repeating units | ND | ND | ND | ND |
| 3 repeating units | 2.7 × 10−5d | 0.74 |
Antibodies were flowed over captured O6a, 6d LPS-containing liposomes and O6a, 6d repeating unit oligosaccharides (1, 2, or 3) were flowed over immobilized antibodies. No binding was detected for 1-repeating-unit oligosaccharides.
KD, equilibrium dissociation constant (KD = kd/ka, where kd is the dissociation rate constant and ka is the association rate constant). ND, not determined.
Apparent equilibrium dissociation constant, KDapp, due to avid binding.
Determined by steady-state affinity.
The OSA from P. aeruginosa strain O6a, 6d LPS was hydrolyzed, yielding 1-, 2-, and 3-repeating-unit oligosaccharides. The affinities of these three O6a, 6d oligosaccharides for immobilized S20 scFv monomer and MF23-1 MAb were measured by SPR (Fig. S5). Neither antibody bound the 1-repeating-unit oligosaccharide; however, both antibodies bound the 2- and 3-repeating-unit oligosaccharides. The data fit well to a 1:1 interaction model (Fig. S5). The affinity of S20 scFv was about 4-fold higher for the 3-repeating-unit oligosaccharide (KD of 1.1 μM) than that of the 2-repeating-unit oligosaccharide (KD of 4.7 μM) (Table 2). The affinity of S20 for the 3-repeating-unit oligosaccharide was slightly lower than that for intact LPS (KD of 560 nM), suggesting that 3 or 4 repeating units are required to stabilize the S20 epitope. Like S20 scFv, MF23-1 MAb had higher affinity for the 3-repeating-unit oligosaccharide (KD of 18 μM) than for the 2-repeating-unit oligosaccharide (KD of 88 μM) (Table 2). However, MF23-1 had about 20-fold weaker affinity than S20 for the oligosaccharides. Rate constants could not be determined due to fast off-rates, and affinities were determined by fitting the data to a steady-state model (Fig. S5).
Half-maximal effective concentrations (EC50s) of S20 and MF23-1.
Xie et al. (9) did not purify the S20 scFv by SEC, instead performing their experiments using a heterogeneous mixture of monomer, dimer, and higher-order aggregates. Therefore, it was unclear if the growth inhibition by S20 observed in previous assays was attributed to one or all of these scFv forms. We investigated these different formats to shed light on the nature of the antibacterial activity of S20; for example, if the monomer did not inhibit growth, this would suggest that cross-linking of surface LPS molecules is required for antibacterial activity.
The growth inhibition assay revealed that both the monomeric and dimeric forms of S20 scFv mediated direct antimicrobial activity against P. aeruginosa O6a, 6d (Fig. S6). The dimer more potently inhibited growth of O6a, 6d than the monomer. The lowest antibody concentrations that inhibited the growth of O6a, 6d over the entire growth curve were 78 nM and 10 nM for the monomer and dimer, respectively. The efficacy differences of the S20 scFv monomer and dimer were more clearly illustrated in turbidity assays (Fig. 3a). Interpolation of these data revealed that the monomer and dimer had EC50s of 450 nM and 60 nM, respectively. Thus, the antibacterial potency of the S20 scFv dimer was about 8-fold higher than that of the monomer (Fig. 3b).
FIG 3.
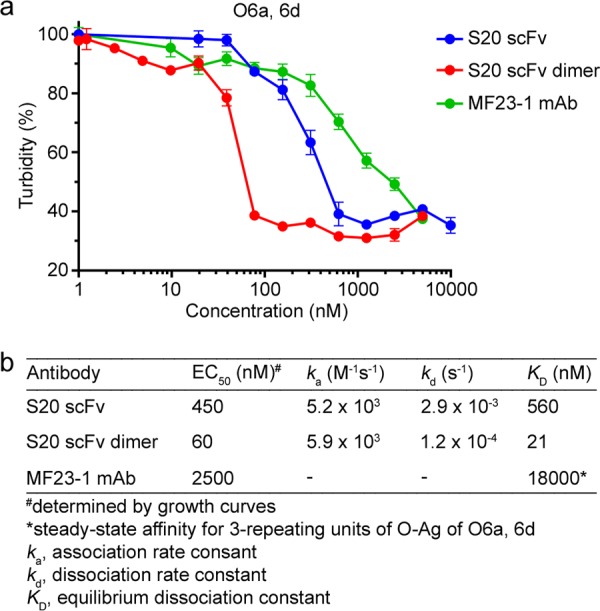
EC50s for S20 and MF23-1 inhibiting growth of P. aeruginosa O6a, 6d. (a) The data from the O6a, 6d growth curves generated from the experiment shown in Fig. S6 in the supplemental material were used to plot turbidity versus antibody concentration. The EC50 of cell turbidity was interpolated for each antibody. (b) Relationship between the EC50s of the antibodies and their SPR-derived binding affinities toward P. aeruginosa O6a, 6d. Growth inhibition was correlated with antibody affinity (R2 = 0.985).
As was observed for the S20 scFv monomer and dimer, the MF23-1 MAb showed antibacterial activity against its target strains (O6 IATS, PAK, O6a, 6d, and Fisher 1) in a concentration-dependent manner (Fig. S7). As revealed by turbidity assays (Fig. 4a), MF23-1 exhibited various degrees of antibacterial activity against these strains. EC50s were calculated as 40, 200, 230, and 2,500 nM for Fisher 1, PAK, O6 IATS, and O6a, 6d, respectively. As noted above, antibacterial activity against different O6 strains directly correlated with MF23-1 affinity for their OSAs (Fig. 4b and Fig. S3). The binding and antibacterial activities of MF23-1 against O6 IATS and PAK, which have the same OSA structures, were essentially identical.
FIG 4.
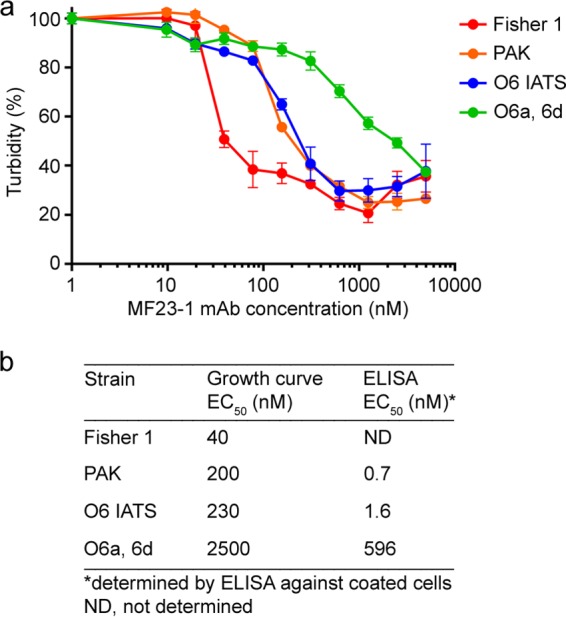
EC50s for MF23-1 inhibiting growth of various P. aeruginosa O6 strains. (a) Data from the growth curves generated from the experiment shown in Fig. S7 in the supplemental material were used to prepare a plot of turbidity versus MAb MF23-1 concentration. (b) Relationship of interpolated growth curve EC50 and ELISA-derived EC50 for each strain. Growth inhibition was correlated with antibody activity in ELISA (R2 = 0.999).
Antibodies against the LPS inner core are not antibacterial.
To ascertain if antibodies targeting other regions of the LPS molecule inhibited growth and in the hope of identifying an antibacterial epitope conserved among P. aeruginosa serotypes and present in clinical strains, we assessed the antibacterial activity of a MAb specific for the LPS inner core. MAb 7-4 (murine IgG2b), which was previously shown by Western blotting to bind the LPS inner core region of all P. aeruginosa serotypes (15), was chosen for this purpose. MAb 7-4 was screened for antibacterial activity against strains with smooth and rough phenotypes (Table S1). The smooth-phenotype strains included CPA+/OSA+ (O6a, 6d, PAK, and PAO1) and CPA+/OSA− (PAK wbpO) strains. The rough-phenotype strains were CPA−/OSA− (PAK rmlC, PAO1 ΔalgC::tet, and AK1012). The rough-phenotype strains were tested to exclude the possibility that long OSA chains could block access of MAb 7-4 to its target, preventing any antibacterial effect. As illustrated in Fig. 5a, MAb 7-4 had no effect on the viability of any of the strains with smooth or rough phenotypes. The only treatments that affected cell viability were the positive-control MAb MF23-1 against O6a, 6d (33% viability) and O6 IATS (9% viability). Although the affinity of MAb 7-4 for its epitope on the LPS inner core has not been determined, we assumed that it was unlikely to be dramatically lower than that of MF23-1 for O6a, 6d OSA (KD ≈ 20 μM). Thus, if MAb 7-4 had antibacterial activity, we would expect to detect it at the tested concentration of 8 μM since this concentration is considerably higher than any of the EC50 values determined for the anti-OSA antibodies (Fig. 3 and 4).
FIG 5.

Effect of inner core- and CPA-specific MAbs on P. aeruginosa viability. (a) MAb 7-4, an anti-inner core LPS antibody, does not show antibacterial activity toward various smooth and rough strains of P. aeruginosa serotypes O5 and O6 when tested at 8 μM. (b) MAb N1F10 (IgM), an anti-CPA antibody, and its scFv do not show antibacterial activity toward various strains of P. aeruginosa. Both MAbs were tested with the agar plate method. Untreated bacteria (PBS) and bacteria treated with the N. meningitidis LPS inner core-specific LPT3-1 antibody served as controls. MF23-1 MAb served as a positive control for serotype O6. Data represent means of three replicates ± standard errors of the means.
Antibodies against the CPA are not antibacterial.
In addition to producing LPS containing OSA, P. aeruginosa produces LPS containing CPA (11). We chose to target the CPA with N1F10 IgM (14) and scFv to determine if anti-CPA antibodies were also antibacterial. We screened N1F10 IgM and scFv against wild-type CPA+/OSA+ and CPA+/OSA− strains from serotypes O5 and O6 (Table S1). CPA+/OSA− strains were tested to provide better accessibility to the target antigen; OSA extends almost twice the length of CPA and could block antibody access to CPA (17, 18). Since N1F10 scFv is much smaller than the parent IgM, it should have better access to CPA. Growth inhibition assays demonstrated that N1F10 IgM and scFv had no effect on the viability of CPA+/OSA+ (PAK and PAO1) and CPA+/OSA− (PAK wbpO and AK1401) strains (Fig. 5b). Concentrations as high as 25 μM scFv were tested against PAK wbpO, and no effects were observed (data not shown). The only treatment that affected cell viability was the positive-control MAb MF23-1 against PAK.
AFM.
AFM images showed that untreated O6a, 6d cells had good structural integrity, with a well-defined periphery, a single flagellum at one pole, and a rough surface topography (Fig. 6a and b, top panels). The rough surface is believed to reflect the various lengths of O polysaccharides and corroborates other AFM studies of Gram-negative bacteria (11, 19). Natural outer membrane vesicles (nOMVs), which are known to be naturally released from the surfaces of virtually all Gram-negative bacteria, including P. aeruginosa (20, 21), were observed in the process of shedding and surrounding some untreated O6a, 6d cells (Fig. 6c). These nOMVs were very similar in morphology to those observed by Beveridge (20) in an electron micrograph of an unidentified Gram-negative bacterium.
FIG 6.

AFM images showing damage to P. aeruginosa O6a, 6d cells induced by treatment with various concentrations of MF23-1 MAb and S20 scFv at 37°C for 30 min. (a) AFM height (H), topographic (T), and phase-contrast (P) images of untreated O6a, 6d cells, of O6a, 6d cells treated with 10 μM MF23-1 MAb (4× EC50), and of O6a, 6d cells treated with 10 μM S20 scFv monomer (22× EC50), as indicated. (b) Enlargement of three-dimensional surface patches (400 by 400 nm) of untreated O6a, 6d cells, of O6a, 6d cells treated with 10 μM MF23-1 MAb, and of O6a, 6d cells treated with 10 μM S20 scFv monomer, as indicated. (c) Untreated O6a, 6d cell releasing natural OMVs (nOMVs) from the OM. (d) Topographic AFM images of O6a, 6d treated with 1 μM MF23-1 MAb (0.4× EC50) highlighting the release of irregularly shaped micelles and membrane components (top). Their release led to the appearance of pits 15 to 35 nm deep in the membrane (middle and bottom). Height (middle left) and derived three-dimensional (middle right) images, which were obtained by zooming into the smaller, red-boxed area (top), illustrate an area that was particularly affected by the appearance of pits. Cross-section (bottom) of the area indicated by the black line (middle left) shows pits measuring up to 35 nm in depth.
To capture early membranolytic effects, O6a, 6d cells were treated with 1 μM MF23-1 MAb, representing 0.4× EC50. Following treatment, cells were surrounded by large amounts of irregularly shaped micelles (Fig. 6d), which were morphologically distinct from nOMVs released from untreated cells (Fig. 6c). The surfaces of treated cells had microroughnesses comparable to those of untreated O6a, 6d cells although some had patchy areas with 15- to 30-nm-deep pits (Fig. 6d). Immediate and complete loss of motility was observed by phase-contrast microscopy following exposure to S20 or MF23-1 and suggested that flagellum loss was an early effect of antibody treatment.
When O6a, 6d cells were treated with a higher concentration of MF23-1 MAb (10 μM; 4× EC50), only a few micelle-like structures were observed, but shedding of quite large sections of OM was clearly apparent (Fig. 6a, middle). Topographical images showed that this treatment induced corrugated surfaces in cells (Fig. 6b, middle) rather than the microrough surfaces observed on untreated cells (Fig. 6b, top). Phase-contrast imaging detected two surface materials with different properties. Phase-contrast imaging uses a softness/hardness scale in which soft (elastic) surfaces are depicted as bright/white regions, and hard (stiff or rigid) surfaces are depicted as darker regions. We observed a lighter-colored layer being released from the cell surface, thereby revealing an underlying darker rigid layer, particularly at one end of the cell where the flagellum was possibly attached (Fig. 6a, middle). The softer layer is thought to represent the OM, and the underlying, more rigid layer represents peptidoglycan.
The surfaces of O6a, 6d cells treated with 22× EC50 (10 μM) S20 scFv monomer were smooth (Fig. 6a, bottom) and displayed none of the roughness observed on untreated cells. Phase-contrast images of cells treated with S20 scFv monomer (Fig. 6b, bottom) showed that their surfaces were almost completely devoid of the soft, light-colored material thought to correspond to OM (Fig. 6b, top).
Structural basis of S20 and MF23-1 OSA recognition and antibacterial activity.
To understand the molecular basis of S20 and MF23-1 OSA recognition, we determined and analyzed their variable region sequences. The S20 (XenoMouse-produced) heavy-chain variable (VH) region was rearranged from human IGHV3-33/IGHJ4 genes and had a 12-residue complementarity-determining region 3 (CDR-H3) loop (Table 3 and Fig. S8). Only a single residue of the VH region in proximity to CDR-H2 was mutated from the germ line (V55A). In contrast, the S20 light-chain variable (VL) region was rearranged from IGKV2D-29/IGKJ1 genes, and five residues were mutated from the germ line, four of which were within or flanked the CDR-L1 loop. The MF23-1 VH domain was rearranged from murine IGHV6-66/IGHJ3 genes and had a seven-residue CDR-H3 loop. Similar to the mutations in S20, only a single VH residue in CDR-H2 was mutated from the germ line (N48S). Unlike the VL region of S20, however, the MF23-1 VL domain, rearranged from IGKV9-124/IGKJ5 genes, was fully germ line. The VH domains of the anti-inner core and anti-CPA MAbs 7-4 and N1F10 were both significantly more divergent from the germ line (5 and 10 substitutions, respectively).
TABLE 3.
Characteristics of anti-P. aeruginosa LPS MAb variable region sequences
| Antibody | Isotype | IGHV and IGHJ genes | IGKV and IGKJ genes | Replacement mutation(s) by regionb |
|
|---|---|---|---|---|---|
| VH | VL | ||||
| S20 | IgG2(κ)a | IGHV3-33, IGHJ4 | IGKV2D-29, IGKJ1 | V55A | K24R, K36M, L39F, Y40S, S107N |
| MF23-1 | IgG3(κ) | IGHV6-6, IGHJ3 | IGKV9-124, IGKJ5 | N58S | |
| 7-4 | IgG2b(κ) | IGHV14-3, IGHJ3 | IGKV4-74, IGKJ1 | N29K, M39I, N64H, S83F, T99I | Y103S |
| N1F10 | IgM(κ) | IGHV1-9, IGHJ2 | IGKV15-103, IGKJ5 | L21I, G27D, T35S, G36S, S62I, G63Q, G74V, K75R, T96S, I101V | N48K |
Produced in XenoMouse.
IMGT numbering.
Homology models of the MAb S20 and MF23-1 paratopes were generated using RosettaAntibody3 (https://rosie.graylab.jhu.edu/antibody) (Fig. 7). Similar results were obtained using the Prediction of ImmunoGlobulin Structure (PIGS) server. Both MAbs had noticeably grooved paratopes suggestive of OSA-binding sites. Five of six replacement mutations in the S20 VH-VL domains lined or flanked the putative OSA-binding groove, as did the single MF23-1 VH replacement mutation. These models, in conjunction with the requirement of multiple oligosaccharide units for both MAbs to achieve full binding (Table 2 and Fig. S5), suggested that S20 and MF23-1 may have been selected to recognize the β- and α-linkages between the d-QuiNAc and l-Rha residues of O6a, 6d and O6 OSA, respectively. In the case of MAb S20, specificity for the O6a, 6d β-linkage may have arisen through somatic mutation and affinity maturation, while the MAb MF23-1 preference for the more common O6 α-linkage [especially the α-d-QuiNAc-(1→2)-α-l-Rha linkage of Fisher 1 strain OSA] appears to be germ line encoded.
FIG 7.

Homology models of the variable domains of MAb S20 (a) and MF23-1 (b). The Fv of each MAb is shown both as a ribbon diagram (left panels) and as a space-filling model (right panels). The side view (top rows) and top view (bottom rows) are shown. The VH and VL domains are shown in green and cyan, respectively. All CDRs are shown in white, and amino acid substitutions from the germ line are shown in red with side chains.
DISCUSSION
By investigating the effects of a panel of MAbs targeting P. aeruginosa LPS on cell viability and employing additional analytical tools and approaches, we have gained further insight into the antimicrobial properties of antibodies such as S20. Xie et al. (9) showed that the antimicrobial effect of S20 MAb and fragments therefrom was accompanied by membrane damage. This was established by a dye penetration assay, with EDTA and tobramycin serving as positive controls and with buffer and an anticancer MAb serving as negative controls. Scanning electron microscopy showed extensive outer membrane damage, as evidenced by blebbing, after treatment with 6.7 μM S20 scFv for 6 h at 37°C; control cells remained fully intact with no visible malformations. Our observation of an antibacterial effect of MF23-1, a MAb specific for all O6 serotypes, points to antibody binding to OSA being the critical event leading to growth inhibition of serogroup O6 cells. Significantly, the antibacterial activities of MF23-1 against two P. aeruginosa strains, O6 IATS and PAK, with identical OSA structures were essentially the same, and MAbs specific for the LPS core and CPA did not exhibit antibacterial activity. We have clearly established that antibody binding to OSA leads to OM disintegration and that antibacterial potency correlates with antibody affinity. We used three antibody concentrations with increasing EC50s and 30-min treatments to capture by AFM the fine detail of the various stages of OM damage. AFM showed that antibody binding leads to OM destabilization and shedding of the OM, exposing the underlying peptidoglycan.
It is highly improbable that antibody binding to other cell surface structures is responsible for the antibacterial effect observed here. Flagellin glycosylation on P. aeruginosa PAK (serogroup O6) requires one OSA biosynthesis enzyme, but two other enzymes in the biosynthetic pathway are dispensable, indicating that a complete OSA repeating unit is not needed for flagellin glycosylation (22). P. aeruginosa 1244 (serogroup O7) pilin is glycosylated with a single OSA repeating unit (23). Since S20 scFv and MF23-1 MAb require a minimum of two repeating units for binding (Table 2), they would not be expected to recognize these truncated OSA structures. Western blotting corroborated this hypothesis for O6a, 6d flagella.
The P. aeruginosa OM is unusual in several aspects. It is about 100-fold less permeable to small hydrophobic molecules (antibiotics and detergents) than the Escherichia coli OM (24). The lipidome of P. aeruginosa differs from that other enteric bacteria in that it contains phosphatidylcholine (25). The OM contains two types of LPS, one capped with OSA and a second capped with CPA; the latter is a neutrally charged, weakly immunogenic homopolymer of d-Rha (11). Biologically rare acidic and/or amino sugars, seldom found in other Gram-negative bacteria, are found in most OSAs (26). Pseudomonas spp. express a unique outer membrane protein (OprH) for which there is no homolog in E. coli; OprH interacts with negatively charged lipid A and LPS, contributing to the integrity of the OM (27, 28).
As demonstrated here and in earlier work (9, 10), P. aeruginosa OSA is potentially an alternative target for antimicrobial treatments. In support of this hypothesis is the observation that OSA, which is highly immunogenic, is reduced in amount or completely lost as a result of chronic selective pressure in clinical isolates, especially those from CF patients (29, 30). In this study, we have shown that anti-P. aeruginosa OSA MAbs and fragments therefrom only inhibited the growth of cells presenting their cognate antigens. In contrast, antibodies specific for P. aeruginosa CPA and for the LPS inner core were not antibacterial. Thus, antibodies targeting the OSA, but not the CPA, affect the viability of P. aeruginosa. These two types of LPS have several structural differences. Their O polysaccharides differ in their charges (OSA, negatively charged; CPA, neutral), their sugar compositions (OSA, rich in acidic and amino sugars and low in Rha; CPA, an Rha homopolymer), and, in many cases, their chain lengths (CPA chains of ≤23 sugars; OSA chains of ≤50 repeating units). Presumably, one or more of the unique features of OSA contribute to OM stability.
Only a few MAbs have been shown to exert antibacterial effects by obstructing the biological functions of outer membrane targets (9, 31–35). In addition to S20 and MF23-1, MAbs against four outer surface proteins of Borrelia burgdorferi, the cause of Lyme disease, and Borrelia hermsii, the cause of relapsing fever, exhibit concentration-dependent inhibitory and antimicrobial activity against their respective organisms (31–35). Some relevant observations with these MAbs include the following: (i) with one antigen, MAbs with complement-dependent and complement-independent antibacterial activity have been reported (36); (ii) the appearance of OM blebs is an early sign of cell damage (31–33, 35, 37); (iii) a MAb against one target induced destruction of the OM by formation of openings with diameters of 2.8 to 4.4 nm leading to osmotic lysis; (iv) expression of one target in the OM of E. coli did not confer susceptibility to its cognate MAb (37); (v) the antibacterial properties of anti-Borrelia MAbs reside in the variable domain as demonstrated by direct scFv growth inhibition (38); and (vi) the antimicrobial activity of one MAb required the presence of cholesterol and cholesterol glycolipids (39), which are, atypically for bacteria, present in Borrelia spp. (40). It may be that the common feature of antibacterial Borrelia MAbs is disruption of interactions between their protein antigens and cholesterol glycolipids.
There is evidence that, analogous to the cholesterol glycolipid-OM protein interactions in Borrelia spp., at least one of the P. aeruginosa OM proteins, OprH, interacts with LPS, albeit with the lipid A and core, not the OSA, as noted above (27, 28). However, the observation that OSA is reduced or completely lost in clinical isolates from CF patients (29, 30) suggests that it is unlikely that OSA interactions with OM proteins are required for membrane stability. Another more plausible hypothesis is that OSA interaction with divalent cations may play a role in P. aeruginosa OM stability. Divalent cations are known to neutralize and stabilize LPS in the OM through interactions with phosphate groups in the lipid A and core regions. Using supported lipid bilayer assemblies as model membranes, Adams et al. (41) showed that net-negative LPS-Na+ results in membrane destabilization through electrostatic repulsion between adjacent LPS molecules, which induces membrane curvature and leads to pore formation. Presumably, divalent cations also neutralize and stabilize OSA. Partial OSA neutralization by antibody binding could lead to OM destabilization in a manner analogous to that reported by Adams et al. (41). It should be noted that in this work we conducted our assays in tryptic soy broth (TSB), but use of a medium with a different cation concentration, such as cation-adjusted Mueller-Hinton broth, could have yielded differing results.
In summary, we demonstrated that binding of two MAbs to P. aeruginosa serotype O6 OSA, but not binding of MAbs to the LPS core or to CPA, was responsible for the antibacterial activity via OM destabilization. Antibacterial activity correlated directly with MAb specificity and affinity for the OSAs of different strains. Serotype O6 is the most prevalent serotype in nosocomial pneumonia, accounting for approximately 30% of the isolates, followed by O11 at about 25%, with numerous other serotypes and not typeable strains comprising the remainder (42). Most likely, the OSAs of other P. aeruginosa serotypes also represent targets for intervention in P. aeruginosa infections as all serotypes have the unusual OM features described above.
MATERIALS AND METHODS
Bacterial strains and culture.
Bacterial strains used in this study are listed in Table S1 in the supplemental material. P. aeruginosa O6a, 6d was provided by J. R. Schreiber, Tufts University. The complete set of International Antigenic Typing Scheme (IATS) P. aeruginosa strains, serotypes O1 to O20, was obtained from the American Type Culture Collection (ATCC). Fisher 1 (ATCC 27312, serotype O6, CPA+/OSA+), PAK (serotype O6, CPA+/OSA+), PAK wbpO (serotype O6, CPA+/OSA−, gentamicin resistant [Gmr, 200 μg ml−1]), PAK rmlC (serotype O6, CPA−/OSA−, Gmr [300 μg ml−1]), and PAO1 (serotype O5, CPA+/OSA+) were obtained from J. S. Lam, University of Guelph. PAO1 ΔalgC::tet (serotype O5, CPA−/OSA−, deficient in alginate exopolysaccharide; tetracycline resistant [100 μg ml−1]) was kindly provided by S. Müller-Loennies, Research Center Borstel, Germany. AK1012 (PAO1 derived, serotype O5, CPA−/OSA−) and AK1401 (PAO1 derived, serotype O5, CPA+/OSA−) were obtained from P. Fleming, National Research Council Canada.
All strains were cultured in TSB (Difco, Detroit, MI). Gentamicin sulfate (Sigma-Aldrich, Oakville, ON, Canada) and tetracycline (Sigma-Aldrich) were added for the growth of resistant strains. Bacteria from a single colony were picked from an agar plate and grown overnight (O/N) at 37°C and 220 rpm. Subcultures were inoculated with a 1:50 dilution from the O/N culture into 5 ml of broth and grown as described above until the optical density at 600 nm (OD600) reached 0.5 to 0.7. Bacteria were harvested by centrifugation (1,750 × g, 5 min, room temperature), washed three times with 5 ml of PBS, and then diluted to obtain the desired OD600 or number of CFU per milliliter. For ELISA, bacteria were heat killed at 60°C for 1 h and stored at –20°C.
Antibody production and purification.
Antibodies used in this study are listed in Table 1. Hybridomas MF23-1, 7-4, and N1F10 were provided by J. S. Lam, University of Guelph. MAb LPT3-1 was provided by Andrew Cox, National Research Council Canada. Hybridomas were grown in Dulbecco’s modified Eagle medium (Sigma-Aldrich) supplemented with 10% (vol/vol) Gibco fetal bovine serum (FBS) (ultralow IgG, <5 μg ml−1, U.S. origin; Invitrogen, Burlington, ON, Canada), 1% (vol/vol) antibiotic-antimycotic (Invitrogen), and 20 mM l-glutamine (Invitrogen). Immediately following partial thawing of one vial of a hybridoma clone in a 37°C water bath with swirling, the contents were transferred to 5 ml of culture medium and gently centrifuged at 20 × g for 3 min to remove dimethyl sulfoxide. The pelleted cells were gently resuspended in 5 ml of culture medium, transferred in a T25 tissue culture flask, and incubated at 37°C in a CO2 incubator. The hybridoma lines were split to obtain 106 cells ml−1 (1:4; every 2 to 3 days) and transferred to an appropriate tissue culture flask. Culture supernatants were collected, filtered through a 0.22-μm-pore-size Stericup filter unit (Millipore, Nepean, ON, Canada), and stored at 4°C until purification.
MF23-1 and LPT3-1 MAbs were purified from culture supernatants by standard protein G affinity chromatography, followed by buffer exchange to PBS (pH 7.4) using a HiPrep Desalting 26/10 column (GE Healthcare, Mississauga, ON, Canada). Purification of the 7-4 MAb required addition of 0.15 M NaCl to the elution buffer (0.1 M sodium citrate, 0.15 M NaCl, pH 3.6) and binding buffer (20 mM sodium phosphate, 0.15 M NaCl, pH 7.4) to prevent precipitation upon acidic elution from protein A/G columns; the MAb was purified using a 5-ml protein A column (GE Healthcare) and buffer exchanged to PBS (pH 7.4), as described above. N1F10 MAb was purified using a 1-ml HiTrap IgM Purification HP column (GE Healthcare) according to the manufacturer’s instructions and further purified by SEC using a HiLoad S200 16/60 column (GE Healthcare).
S20 scFv, which was previously cloned into the pPICZα-A vector (Invitrogen) (9), was expressed in Pichia pastoris strain X-33 essentially as described in the EasySelect Pichia expression kit manual (catalog no. K1740-01; Invitrogen) (43). S20 scFv, which has a C-terminal His6 tag, was purified by IMAC using a 5-ml HisTrap HP nickel affinity column (GE Healthcare) on an ÄKTAxpress system (GE Healthcare).
The VL (GenBank accession number U25104.1) and VH (GenBank accession number U25107.1) sequences of N1F10 were joined by a (Gly4Ser)3 linker and terminated with C-terminal tags (VL-linker-VH-c-Myc-His6). The coding sequence of the scFv was commercially synthesized, optimizing for P. pastoris codon utilization, and cloned into the P. pastoris expression vector, pD912-AKS, by DNA2.0 (Newark, CA). N1F10 scFv was expressed and purified as described for the S20 scFv. MF23-1 MAb was digested to release the Fab fragment using a protocol similar to that described by Parker et al. (44). The optimal digestion conditions were 0.02 mg ml−1 papain, 2 mg ml−1 MF23-1, and 4 h at 37°C. Antibody purity was assessed by SDS-PAGE under reducing conditions on precast 12% Mini-Protean TGX stain-free precast gels (Bio-Rad).
LPS and oligosaccharide preparation.
For LPS preparation, 24-liter P. aeruginosa strain O6a, 6d cultures were grown in Difco brain heart infusion broth in a 30-liter fermentor (newMBR, Zurich, Switzerland) at 37°C with dissolved oxygen controlled at 15% saturation. Cells were killed by the addition of phenol (2% final concentration), harvested, and lyophilized. Cells were washed with organic solvents to remove lipids and other lipophilic components (45). LPS was extracted from the air-dried cellular material using the hot-phenol/water method (46) essentially as previously outlined by St. Michael et al. (47).
For oligosaccharide preparation, O6a, 6d LPS (400 g) was heated with 1% acetic acid (100 ml, 100°C, 2 h). The lipid precipitate was removed by centrifugation, and the corresponding O polysaccharide (100 mg) as well as oligosaccharide fractions was isolated from the supernatants by Sephadex G-50 (GE Healthcare) SEC. The O6a, 6d O polysaccharide (100 mg) was treated with 10 ml of anhydrous hydrogen fluoride (HF; 10 ml) for 30 min at 0°C and then poured into a plastic petri dish for evaporation in a strongly ventilated hood. HF evaporated instantly, and the residue was dissolved in water and separated on a Biogel P6 (Bio-Rad) column in pyridine-acetic acid buffer, pH 4.5 (4 ml of pyridine and 10 ml of acetic acid [AcOH] in 1 liter of water), monitored with a refractive index detector. The main product was repeating-unit tetrasaccharide (30 mg); dimer (8 mg) and trimer (3 mg) of the repeating unit were also obtained. The products were analyzed by electrospray ionization-mass spectrometry (ESI-MS). The expected mass for 1 repeating unit was 811.7 (observed 812.7 m/z), and for 2 repeating units, it was 1,605.5 (observed 1607.7 m/z).
SPR.
Liposomes were prepared for analysis of antibody binding to LPS. Ten milligrams of lyophilized 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC; Sigma-Aldrich) was dissolved in 1 ml of chloroform. The lipids were placed on a heating block at low-medium heat and dried under a nitrogen stream, followed by 1 h under vacuum. One milliliter of purified LPS (from a 2 mg ml−1 solution in Milli-Q water) or PBS (for LPS-free negative-control liposomes) was added to the dried DMPC and incubated for 2 h at 37°C with shaking. The DMPC-LPS mixtures were then sonicated two to three times in a water bath for 30 s at 37°C. Liposomes were left for at least 16 h and as long as 48 h at 4°C, washed twice with 5 ml of SPR HBS-E running buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA) (GE Healthcare), and collected by ultracentrifugation for 1 h at 160,000 × g. The final liposome pellets were resuspended in 0.5 ml of HBS-E buffer and stored at 4°C until SPR analysis. Negative-control liposomes with incorporated LPS from Shigella flexneri serotype Y were prepared using the same protocol.
Immediately prior to analysis on a Biacore 3000 biosensor instrument (GE Healthcare), monomeric and dimeric S20 scFvs were separated by SEC. Control liposomes and liposomes containing O6a, 6d LPS were captured on L1 sensor chips (GE Healthcare) prepared as suggested by the manufacturer. Approximately 3,000 resonance units (RU) were captured at a flow rate of 5 μl min−1. Thereafter, the surfaces were stabilized by flowing HBS-E running buffer for 30 min at a flow rate of 100 μl min−1, followed by two injections of 5 μl of 10 mM NaOH at a flow rate of 50 μl min−1. Surfaces were blocked by injecting 10 μl of 0.1 mg ml−1 bovine serum albumin, followed by 5 μl of 10 mM NaOH. Analyses were performed in HBS-E running buffer at 25°C at a flow rate of 20 μl min−1. Surfaces were not regenerated between S20 scFv monomer injections in the same experiment; new surfaces were prepared between experiments or after each S20 scFv dimer injection. Prior to preparation of fresh surfaces, immobilized liposomes were stripped from the sensor chip by injecting three short pulses of 0.5% SDS and three short pulses of 50 mM NaOH-isopropanol (2:3, vol/vol) at a flow rate of 100 μl min−1. Rate and affinity constants were determined by locally fitting the data sets to a 1:1 interaction model using BIAevaluation, version 4.1, software (GE Healthcare).
The affinities of the S20 and MF23-1 antibodies for O6a, 6d repeating units were measured with a Biacore T200 instrument (GE Healthcare). S20 scFv (3,139 RU) and MF23-1 MAb (7,218 RU) were immobilized by amine coupling to a CM5 sensor chip (GE Healthcare) in 10 mM sodium acetate buffer (GE Healthcare) at pH 5.0 for S20 scFv and at pH 4.0 for MF23-1 MAb. The running buffer was HBS-EP+ (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% [vol/vol] surfactant P-20) (GE Healthcare). Repeating units were diluted in HBS-EP+ running buffer and flowed over the chip. All experiments were conducted at 25°C at a flow rate of 40 μl min−1, and the surfaces were not regenerated. The rate and affinity constants of the S20 scFv for the O6a, 6d oligosaccharides were determined by global fitting of the data set to a 1:1 interaction model. Since the rate constants of MF23-1 MAb were too fast for kinetic analysis, KD values were determined by fitting equilibrium data to a steady-state affinity model (Fig. S5). All analyses were performed using Biacore T200 evaluation software, version 1.0 (GE Healthcare).
Binding and growth inhibition assays.
ELISAs were performed on heat-killed cells as described by Xie et al. (9). Flagella purified from P. aeruginosa O6a, 6d grown overnight in L broth as previously described (48) were probed for antibody binding by Western blotting. Following SDS-PAGE and transfer to polyvinylidene difluoride membranes, membranes were blocked with 4% skim milk in PBS and incubated O/N with 10 μg of the desired MAb in 15 ml of PBS containing 0.1% Tween 20 (PBST) and 4% skim milk. The membranes were washed three times with PBST and probed with a rabbit anti-human IgG-alkaline phosphatase conjugate (Thermo Fisher, Waltham, MA) for the S20 IgG or with a goat anti-mouse F(ab′)2-alkaline phosphatase conjugate (Jackson ImmunoResearch, West Grove, PA) for MF23-1, both diluted 1:2,000.
Generally, antibodies were first screened for antibacterial activity using a microtiter plate method. For the microtiter plate method, antibodies in 50 μl of PBS at the desired concentrations were added to the wells of a 96-well flat-bottomed polystyrene plate (Corning, Corning, NY) containing 50 μl of the bacterial culture (OD600 of 0.024; ca. 5 × 107 CFU ml−1). The negative control contained sterile PBS instead of antibody. An N. meningitidis LPS inner core-specific MAb LPT3-1 was tested at the same time. Medium (50 μl) diluted with PBS (50 μl) served as a control for background subtraction. Three wells (triplicates) were used for each treatment. Microtiter plates were sealed with an adhesive film and incubated at 37°C and 50 rpm, using an automated Infinite 200 Pro microplate reader (Tecan, Mannedorf, Switzerland). Bacterial growth was evaluated by measuring the OD every 10 min for 16 h. The growth curves presented represent the average of three replicates. The second antibacterial assay using an agar plate method was performed as previously described by Xie et al. (9). Briefly, after 30 min of incubation with antibody at 37°C, mixtures were plated on growth medium, followed by colony enumeration after 18 h at 37°C.
EC50 determinations.
EC50s were determined using the microtiter plate method. Antibodies (S20 scFv monomer, S20 scFv dimer, and MAb MF23-1) were serially diluted 2-fold, starting at 20 μM for the S20 scFv monomer and 10 μM for the S20 scFv dimer and MAb MF23-1. The prepared antibody dilutions were added, in triplicate, to wells containing 50 μl of P. aeruginosa cells. The microtiter plates were incubated as described above. Midpoint growth curve data were used to prepare a plot of the percent turbidity (turbidity of antibody-treated cells/turbidity of the control cells × 100) versus antibody concentration (in nanomolar). The EC50 represents the interpolated antibody concentration that caused a 50% reduction in the turbidity relative to the level of the control.
AFM.
Purified MF23-1 MAb and S20 scFv monomer were diluted to the desired concentrations in 0.1 mM sodium phosphate buffer, pH 7.4, just before AFM experiments. O6a, 6d cells were treated with 1 μM MF23-1 MAb (0.4× EC50), 10 μM MF23-1 MAb (4× EC50), and 10 μM S20 scFv monomer (22× EC50).
A single colony of P. aeruginosa O6a, 6d from a TSB agar plate was used to inoculate 5 ml of TSB broth and grown for 16 h at 37°C and 220 rpm. To obtain healthy cells, a subculture was prepared by inoculating 50 ml of fresh TSB with 200 μl of the starter culture and grown as previously described until cells reached log phase (OD600 of 0.3 to 0.4). Cultures were centrifuged at 800 × g for 5 min, and pelleted cells were washed (four times with 25 ml) with decreasing concentrations of sodium phosphate buffer (5, 1, 0.5, and 0.1 mM sodium phosphate buffer, pH 7.4) to prevent osmolytic shock. The cell density was adjusted to an OD600 of 0.4 (ca. 6.4 × 108 CFU/ml) in 0.1 mM sodium phosphate buffer, pH 7.4. Cells (50 μl) were treated with antibody (50 μl; at concentrations listed above in 0.1 mM sodium phosphate buffer, pH 7.4) or 0.1 mM sodium phosphate buffer (50 μl; negative control) in a microcentrifuge tube for 30 min at 37°C with shaking at 220 rpm. After incubation, antibody-treated and untreated bacterial suspensions (25 μl) were spread onto poly-l-lysine-coated glass slides (Sigma) and allowed to immobilize for 5 to 10 min. Nonadherent bacteria were removed by rinsing the slides with 3 ml of 0.1 mM sodium phosphate buffer. Slides were air dried in a desiccator without vacuum for 16 h before AFM imaging.
AFM images were recorded using a JPK NanoWizard II BioAFM instrument (JPK Instruments, Berlin, Germany). Bacteria were mechanically probed with a NSC15/AlBS cantilever (spring constant [k] of 40 N/m; tip radius ca. of 10 nm [MikroMasch, Watsonville, CA]) in intermittent contact mode (tapping mode or AC mode), in air, using a resonance frequency of ca. 350 kHz and a scan rate of 0.3 to 0.4 Hz. After a large area (generally 500 by 500 μm) was scanned, six individual cells per treatment were randomly selected and scanned (<5 by 5 μm) to obtain higher-resolution images. Height and phase images were simultaneously collected. Height images were used to generate high-resolution topography/three-dimensional images. Images were processed with JPK Data Processing Software (JPK Instruments, Berlin, Germany).
Antibody sequence analyses and homology modeling.
The variable-region sequences of MAb S20 were determined by sequencing of the scFv coding regions in the vector pPICZα-A. The variable region coding sequences of MAbs MF23-1, 7-4, and N1F10 were PCR amplified from total hybridoma RNA using degenerate primers, cloned, and sequenced using Sanger’s method. The sequences of MAbs S20 and MF23-1 were compared with those previously published (10, 49). Antibody variable-region sequences were analyzed using IMGT V-QUEST (50). Homology modeling of the S20 and MF23-1 Fv fragments was conducted using either RosettaAntibody3 via the ROSIE server, as described below, or using the Prediction of ImmunoGlobulin Structure (PIGS) server (51) with default parameters.
For homology modeling of Fvs using Rosetta (52), templates were selected from the Protein Data Bank (PDB) database using BLAST independently for VH and VL framework regions and for CDR-H1, CDR-H2, CDR-L1, CDR-L2, and CDR-L3. Framework region sequences were threaded through the corresponding template matches (Table S2); CDR structures were grafted to build VH and VL domains, and the VH-VL orientation was modeled by multitemplate grafting. The structure of the CDR-H3 loop was modeled de novo using a Monte Carlo energy minimization algorithm. Following refinement, the lowest-energy structure was presented.
Supplementary Material
ACKNOWLEDGMENTS
We thank Maohui Chen and Linda Johnson for technical support on the use of the atomic force microscope. We thank Perry Fleming for preparing a large P. aeruginosa culture for LPS isolation and Susan Logan and Annie Aubry for preparing the flagella for Western blotting. We are grateful to Andrew Cox and Jim Richards for providing the LPT3-1 MAb and to Joe Lam for providing hybridomas MF23-1, 7-4, and N1F10. We thank Michael McLean, Joe Lam, Susan Logan, John Schreiber, and Sven Müller-Loennies for bacterial strains and/or valuable discussions.
This work was funded by contributions from NSERC and the Canada Research Chair Program to J.C.H. as well as support from the National Research Council Canada.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Govan JR, Deretic V. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60:539–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2:1051–1060. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 3.Spencer RC. 1996. Predominant pathogens found in the European Prevalence of Infection in Intensive Care Study. Eur J Clin Microbiol Infect Dis 15:281–285. doi: 10.1007/bf01695658. [DOI] [PubMed] [Google Scholar]
- 4.Koulenti D, EU-VAP/CAP Study Group, Lisboa T, Brun-Buisson C, Krueger W, Macor A, Sole-Violan J, Diaz E, Topeli A, DeWaele J, Carneiro A, Martin-Loeches I, Armaganidis A, Rello J. 2009. Spectrum of practice in the diagnosis of nosocomial pneumonia in patients requiring mechanical ventilation in European intensive care units. Crit Care Med 37:2360–2368. doi: 10.1097/CCM.0b013e3181a037ac. [DOI] [PubMed] [Google Scholar]
- 5.Wagner S, Sommer R, Hinsberger S, Lu C, Hartmann RW, Empting M, Titz A. 2016. Novel strategies for the treatment of Pseudomonas aeruginosa infections. J Med Chem 59:5929–5969. doi: 10.1021/acs.jmedchem.5b01698. [DOI] [PubMed] [Google Scholar]
- 6.Raz A, Serrano A, Hernandez A, Euler CW, Fischetti VA. 2019. Isolation of phage lysins that effectively kill Pseudomonas aeruginosa in mouse models of lung and skin infection. Antimicrob Agents Chemother 63. doi: 10.1128/AAC.00024-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, Bonnell J, Fleming R, Bezabeh B, Dimasi N, Sellman BR, Hilliard J, Guenther CM, Datta V, Zhao W, Gao C, Yu X-Q, Suzich JA, Stover CK. 2014. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med 6:262ra155. doi: 10.1126/scitranslmed.3009655. [DOI] [PubMed] [Google Scholar]
- 8.Le HN, Tran VG, Vu TTT, Gras E, Le VTM, Pinheiro MG, Aguiar-Alves F, Schneider-Smith E, Carter HC, Sellman BR, Stover CK, DiGiandomenico A, Diep BA. 2019. Treatment efficacy of MEDI3902 in Pseudomonas aeruginosa bloodstream infection and acute pneumonia rabbit models. Antimicrob Agents Chemother 63:e00710-19. doi: 10.1128/AAC.00710-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie X, McLean MD, Hall JC. 2010. Antibody-dependent cell-mediated cytotoxicity- and complement-dependent cytotoxicity-independent bactericidal activity of an IgG against Pseudomonas aeruginosa O6ad. J Immunol 184:3725–3733. doi: 10.4049/jimmunol.0902732. [DOI] [PubMed] [Google Scholar]
- 10.Hemachandra S, Kamboj K, Copfer J, Pier G, Green LL, Schreiber JR. 2001. Human monoclonal antibodies against Pseudomonas aeruginosa lipopolysaccharide derived from transgenic mice containing megabase human immunoglobulin loci are opsonic and protective against fatal pseudomonas sepsis. Infect Immun 69:2223–2229. doi: 10.1128/IAI.69.4.2223-2229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam JS, Taylor VL, Islam ST, Hao Y, Kocincova D. 2011. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front Microbiol 2:118. doi: 10.3389/fmicb.2011.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knirel YA, Bystrova OV, Kocharova NA, Zähringer U, Pier GB. 2006. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res 12:324–336. doi: 10.1179/096805106X118906. [DOI] [PubMed] [Google Scholar]
- 13.Lam JS, MacDonald LA, Lam MY, Duchesne LG, Southam GG. 1987. Production and characterization of monoclonal antibodies against serotype strains of Pseudomonas aeruginosa. Infect Immun 55:1051–1057. doi: 10.1128/IAI.55.5.1051-1057.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rocchetta HL, Lam JS. 1997. Identification and functional characterization of an ABC transport system involved in polysaccharide export of A-band lipopolysaccharide in Pseudomonas aeruginosa. J Bacteriol 179:4713–4724. doi: 10.1128/jb.179.15.4713-4724.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Kievit TR, Lam JS. 1994. Monoclonal antibodies that distinguish inner core, outer core, and lipid A regions of Pseudomonas aeruginosa lipopolysaccharide. J Bacteriol 176:7129–7139. doi: 10.1128/jb.176.23.7129-7139.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gidney MA, Plested JS, Lacelle S, Coull PA, Wright JC, Makepeace K, Brisson JR, Cox AD, Moxon ER, Richards JC. 2004. Development, characterization, and functional activity of a panel of specific monoclonal antibodies to inner core lipopolysaccharide epitopes in Neisseria meningitidis. Infect Immun 72:559–569. doi: 10.1128/iai.72.1.559-569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King JD, Kocincova D, Westman EL, Lam JS. 2009. Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun 15:261–312. doi: 10.1177/1753425909106436. [DOI] [PubMed] [Google Scholar]
- 18.Daniels C, Griffiths C, Cowles B, Lam JS. 2002. Pseudomonas aeruginosa O-antigen chain length is determined before ligation to lipid A core. Environ Microbiol 4:883–897. doi: 10.1046/j.1462-2920.2002.00288.x. [DOI] [PubMed] [Google Scholar]
- 19.Amro NA, Kotra LP, Wadu-Mesthrige A, Bulychev KA, Mobashery S, Liu G-y. 2000. High-resolution atomic force microscopy studies of the Escherichia coli out membrane: structural basis for permeability. Langmuir 16:2789–2796. doi: 10.1021/la991013x. [DOI] [Google Scholar]
- 20.Beveridge TJ. 1999. Structures of Gram-negative cell walls and their derived membrane vesicles. J Bacteriol 181:4725–4733. doi: 10.1128/JB.181.16.4725-4733.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadurugamuwa JL, Beveridge TJ. 1997. Natural release of virulence factors in membrane vesicles by Pseudomonas aeruginosa and the effect of aminoglycoside antibiotics on their release. J Antimicrob Chemother 40:615–621. doi: 10.1093/jac/40.5.615. [DOI] [PubMed] [Google Scholar]
- 22.Miller WL, Matewish MJ, McNally DJ, Ishiyama N, Anderson EM, Brewer D, Brisson JR, Berghuis AM, Lam JS. 2008. Flagellin glycosylation in Pseudomonas aeruginosa PAK requires the O-antigen biosynthesis enzyme WbpO. J Biol Chem 283:3507–3518. doi: 10.1074/jbc.M708894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castric P, Cassels FJ, Carlson RW. 2001. Structural characterization of the Pseudomonas aeruginosa 1244 pilin glycan. J Biol Chem 276:26479–26485. doi: 10.1074/jbc.M102685200. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimura F, Nikaido H. 1982. Permeability of Pseudomonas aeruginosa outer membrane to hydrophilic solutes. J Bacteriol 152:636–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilderman PJ, Vasil AI, Martin WE, Murphy RC, Vasil ML. 2002. Pseudomonas aeruginosa synthesizes phosphatidylcholine by use of the phosphatidylcholine synthase pathway. J Bacteriol 184:4792–4799. doi: 10.1128/jb.184.17.4792-4799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkinson SG. 1983. Composition and structure of lipopolysaccharides from Pseudomonas aeruginosa. Rev Infect Dis 5 Suppl 5:S941–S949. doi: 10.1093/clinids/5.supplement_5.s941. [DOI] [PubMed] [Google Scholar]
- 27.Kucharska I, Liang B, Ursini N, Tamm LK. 2016. Molecular interactions of lipopolysaccharide with an outer membrane protein from Pseudomonas aeruginosa probed by solution NMR. Biochemistry 55:5061–5072. doi: 10.1021/acs.biochem.6b00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, Patel DS, Kucharska I, Tamm LK, Im W. 2017. Refinement of OprH-LPS interactions by molecular simulations. Biophys J 112:346–355. doi: 10.1016/j.bpj.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam MY, McGroarty EJ, Kropinski AM, MacDonald LA, Pedersen SS, Hoiby N, Lam JS. 1989. Occurrence of a common lipopolysaccharide antigen in standard and clinical strains of Pseudomonas aeruginosa. J Clin Microbiol 27:962–967. doi: 10.1128/JCM.27.5.962-967.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hancock RE, Mutharia LM, Chan L, Darveau RP, Speert DP, Pier GB. 1983. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: a class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect Immun 42:170–177. doi: 10.1128/IAI.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman JL, Rogers RC, Benach JL. 1992. Selection of an escape variant of Borrelia burgdorferi by use of bactericidal monoclonal antibodies to OspB. Infect Immun 60:3098–3104. doi: 10.1128/IAI.60.8.3098-3104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Connolly SE, Thanassi DG, Benach JL. 2004. Generation of a complement-independent bactericidal IgM against a relapsing fever Borrelia. J Immunol 172:1191–1197. doi: 10.4049/jimmunol.172.2.1191. [DOI] [PubMed] [Google Scholar]
- 33.Šadžiene A, Jonsson M, Bergström S, Bright RK, Kennedy RC, Barbour AG. 1994. A bactericidal antibody to Borrelia burgdorferi is directed against a variable region of the OspB protein. Infect Immun 62:2037–2045. doi: 10.1128/IAI.62.5.2037-2045.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Šadžiene A, Thompson PA, Barbour AG. 1993. In vitro inhibition of Borrelia burgdorferi growth by antibodies. J Infect Dis 167:165–172. doi: 10.1093/infdis/167.1.165. [DOI] [PubMed] [Google Scholar]
- 35.Scriba M, Ebrahim JS, Schlott T, Eiffert H. 1993. The 39-kilodalton protein of Borrelia burgdorferi: a target for bactericidal human monoclonal antibodies. Infect Immun 61:4523–4526. doi: 10.1128/IAI.61.10.4523-4526.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma J, Gingrich-Baker C, Franchi PM, Bulger P, Coughlin RT. 1995. Molecular analysis of neutralizing epitopes on outer surface proteins A and B of Borrelia burgdorferi. Infect Immun 63:2221–2227. doi: 10.1128/IAI.63.6.2221-2227.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LaRocca TJ, Holthausen DJ, Hsieh C, Renken C, Mannella CA, Benach JL. 2009. The bactericidal effect of a complement-independent antibody is osmolytic and specific to Borrelia. Proc Natl Acad Sci U S A 106:10752–10757. doi: 10.1073/pnas.0901858106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaRocca TJ, Katona LI, Thanassi DG, Benach JL. 2008. Bactericidal action of a complement-independent antibody against relapsing fever Borrelia resides in its variable region. J Immunol 180:6222–6228. doi: 10.4049/jimmunol.180.9.6222. [DOI] [PubMed] [Google Scholar]
- 39.LaRocca TJ, Crowley JT, Cusack BJ, Pathak P, Benach J, London E, Garcia-Monco JC, Benach JL. 2010. Cholesterol lipids of Borrelia burgdorferi form lipid rafts and are required for the bactericidal activity of a complement-independent antibody. Cell Host Microbe 8:331–342. doi: 10.1016/j.chom.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Menachem G, Kubler-Kielb J, Coxon B, Yergey A, Schneerson R. 2003. A newly discovered cholesteryl galactoside from Borrelia burgdorferi. Proc Natl Acad Sci U S A 100:7913–7918. doi: 10.1073/pnas.1232451100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PG, Lamoureux L, Swingle KL, Mukundan H, Montano GA. 2014. Lipopolysaccharide-induced dynamic lipid membrane reorganization: tubules, perforations, and stacks. Biophys J 106:2395–2407. doi: 10.1016/j.bpj.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Q, Eggimann P, Luyt CE, Wolff M, Tamm M, Francois B, Mercier E, Garbino J, Laterre PF, Koch H, Gafner V, Rudolf MP, Mus E, Perez A, Lazar H, Chastre J, Rouby JJ. 2014. Pseudomonas aeruginosa serotypes in nosocomial pneumonia: prevalence and clinical outcomes. Crit Care 18:R17. doi: 10.1186/cc13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Invitrogen. 2010. EasySelect Pichia expression kit user manual. Invitrogen, Carlsbad, CA. [Google Scholar]
- 44.Parker MJ, Gomery K, Richard G, MacKenzie CR, Cox AD, Richards JC, Evans SV. 2014. Structural basis for selective cross-reactivity in a bactericidal antibody against inner core lipooligosaccharide from Neisseria meningitidis. Glycobiology 24:442–449. doi: 10.1093/glycob/cwu009. [DOI] [PubMed] [Google Scholar]
- 45.Masoud H, Moxon ER, Martin A, Krajcarski D, Richards JC. 1997. Structure of the variable and conserved lipopolysaccharide oligosaccharide epitopes expressed by Haemophilus influenzae serotype b strain Eagan. Biochemistry 36:2091–2103. doi: 10.1021/bi961989y. [DOI] [PubMed] [Google Scholar]
- 46.Westphal O. 1965. Bacterial lipopolysaccharide-extraction with phenol water and further application of procedure. Methods Carbohydr Chem 1:83–91. [Google Scholar]
- 47.St Michael F, Inzana TJ, Cox AD. 2006. Structural analysis of the lipooligosaccharide-derived oligosaccharide of Histophilus somni (Haemophilus somnus) strain 8025. Carbohydr Res 341:281–284. doi: 10.1016/j.carres.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 48.Totten PA, Lory S. 1990. Characterization of the type a flagellin gene from Pseudomonas aeruginosa PAK. J Bacteriol 172:7188–7199. doi: 10.1128/jb.172.12.7188-7199.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tout NL, Lam JS. 1997. Phage display and bacterial expression of a recombinant Fab specific for Pseudomonas aeruginosa serotype O6 lipopolysaccharide. Clin Diagn Lab Immunol 4:147–155. doi: 10.1128/CDLI.4.2.147-155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brochet X, Lefranc MP, Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36:W503–8. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marcatili P, Rosi A, Tramontano A. 2008. PIGS: automatic prediction of antibody structures. Bioinformatics 24:1953–1954. doi: 10.1093/bioinformatics/btn341. [DOI] [PubMed] [Google Scholar]
- 52.Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf-Bryfogle J, Biswas N, Dunbrack RL Jr, Gray JJ. 2017. Modeling and docking of antibody structures with Rosetta. Nat Protoc 12:401–416. doi: 10.1038/nprot.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.