

Enterococcus faecalis and Enterococcus faecium are commensals of the gastrointestinal tract of most terrestrial organisms, including humans, and are major causes of health care-associated infections. Such infections are difficult or impossible to treat, as the enterococcal strains responsible are often resistant to multiple antibiotics. One intrinsic resistance trait that is conserved among E. faecalis and E. faecium is cephalosporin resistance, and prior exposure to cephalosporins is one of the most well-known risk factors for acquisition of an enterococcal infection.
KEYWORDS: beta-lactam resistance, cephalosporins, enterococci, penicillin-binding proteins, peptidoglycan synthesis
ABSTRACT
Enterococcus faecalis and Enterococcus faecium are commensals of the gastrointestinal tract of most terrestrial organisms, including humans, and are major causes of health care-associated infections. Such infections are difficult or impossible to treat, as the enterococcal strains responsible are often resistant to multiple antibiotics. One intrinsic resistance trait that is conserved among E. faecalis and E. faecium is cephalosporin resistance, and prior exposure to cephalosporins is one of the most well-known risk factors for acquisition of an enterococcal infection. Cephalosporins inhibit peptidoglycan biosynthesis by acylating the active-site serine of penicillin-binding proteins (PBPs) to prevent the PBPs from catalyzing cross-linking during peptidoglycan synthesis. For decades, a specific PBP (known as Pbp4 or Pbp5) that exhibits low reactivity toward cephalosporins has been thought to be the primary PBP required for cephalosporin resistance. We analyzed other PBPs and report that in both E. faecalis and E. faecium, a second PBP, PbpA(2b), is also required for resistance; notably, the cephalosporin ceftriaxone exhibits a lethal effect on the ΔpbpA mutant. Strikingly, PbpA(2b) exhibits low intrinsic reactivity with cephalosporins in vivo and in vitro. Unlike the Δpbp5 mutant, the ΔpbpA mutant exhibits a variety of phenotypic defects in growth kinetics, cell wall integrity, and cellular morphology, indicating that PbpA(2b) and Pbp5(4) are not functionally redundant and that PbpA(2b) plays a more central role in peptidoglycan synthesis. Collectively, our results shift the current understanding of enterococcal cephalosporin resistance and suggest a model in which PbpA(2b) and Pbp5(4) cooperate to coordinately mediate peptidoglycan cross-linking in the presence of cephalosporins.
INTRODUCTION
Enterococci are commensal residents of the gastrointestinal tract of most terrestrial organisms, including humans. However, some species of enterococci, in particular Enterococcus faecalis and Enterococcus faecium, are also major causes of health care-associated infections (1, 2). Such infections can be difficult or sometimes impossible to treat, as the enterococcal strains responsible are often resistant to multiple antibiotics (3–5). This antibiotic resistance includes acquired resistance traits (e.g., vancomycin resistance) as well as intrinsic resistance traits exhibited by all members of the species. One such intrinsic resistance trait that is conserved among E. faecalis and E. faecium is that of cephalosporin resistance (4, 6, 7). Importantly, prior exposure to cephalosporins is one of the most well-known risk factors for acquisition of an enterococcal infection (8, 9), because enterococci proliferate to abnormally high densities in the intestinal tract of patients receiving cephalosporins and subsequently escape to cause infection elsewhere (10–12).
Cephalosporins belong to the beta-lactam family of antibiotics that inhibit peptidoglycan (PG) biosynthesis by acylating the active-site serine of penicillin-binding proteins (PBPs) to prevent the PBPs from performing the cross-linking reaction of PG synthesis (13). High-molecular-weight PBPs responsible for PG synthesis can be classified as class A (capable of performing both PG transglycosylation and cross-linking steps of PG synthesis; referred to herein as “aPBPs”) or class B (capable of performing only cross-linking; “bPBPs”) (14). Most bacteria encode multiple PBP homologs representing both PBP types. The genomes of E. faecalis and E. faecium, for example, each encode 3 aPBPs and 3 bPBPs (15, 16). In general, beta-lactam antibiotics are capable of acylating both aPBPs and bPBPs to prevent their cross-linking activity, although the extent of reactivity of different PBPs toward a given beta-lactam antibiotic can vary. Hence, the effectiveness of a given beta-lactam antibiotic in preventing PG cross-linking will reflect the reactivity of that beta-lactam with the entire repertoire of PBPs expressed by a bacterium. A subset of specific PBPs exhibit particularly low beta-lactam reactivities (often referred to as “low affinity” PBPs) and therefore confer beta-lactam resistance to their bacterial hosts by enabling PG cross-linking to continue in the presence of beta-lactams when other PBPs have been acylated. One common example is the staphylococcal bPBP Pbp2a, which provides beta-lactam resistance to methicillin-resistant Staphylococcus aureus (17, 18).
A specific enterococcal bPBP, referred to as either Pbp5 or Pbp4 in the literature, has been known for decades to be required for cephalosporin resistance (15, 19). This bPBP [herein referred to as Pbp5(4) for consistency with the nomenclature of both Arbeloa et al. (15) and Sauvage et al. (14)] exhibits intrinsically low reactivity toward cephalosporins, thereby enabling growth in the presence of cephalosporins. Given these findings, it has long been tacitly assumed that Pbp5(4) was the primary enterococcal PBP responsible for PG cross-linking to mediate cephalosporin resistance. However, Pbp5(4) has not been demonstrated to be solely sufficient to provide all PG cross-linking required for growth in the presence of cephalosporins. Previous studies investigated potential requirements for enterococcal aPBPs in cephalosporin resistance, finding that no individual aPBP is strictly required (15, 16). In those studies, either of 2 aPBPs (PonA or PbpF) were required for cephalosporin resistance, although their PG cross-linking activity was not required for this function. These findings were interpreted to mean that the PG glycosyltransferase activity of either PonA or PbpF was (at least partly) functionally redundant with the other and was required for PG synthesis in cooperation with Pbp5(4) to mediate cephalosporin resistance. To the best of our knowledge, no studies have specifically investigated enterococcal bPBPs other than Pbp5(4) for a role in cephalosporin resistance. Given the importance of PBPs in PG synthesis and their roles as targets for beta-lactam antibiotics, we sought to determine if other enterococcal PBPs were also required for cephalosporin resistance.
Here, we report that in both E. faecalis and E. faecium, a second bPBP [known as PbpA or Pbp2b, herein referred to as PbpA(2b)] is required for cephalosporin resistance in addition to Pbp5(4). Surprisingly, the cephalosporin ceftriaxone exhibits a marked lethality against the E. faecalis ΔpbpA mutant that is not observed in other strains. PbpA(2b) exhibits strikingly low intrinsic reactivity with cephalosporins in vivo and in vitro, consistent with a critical role for PbpA(2b) in mediating cephalosporin resistance. Unlike the Δpbp5 mutant, the ΔpbpA mutant exhibits a variety of phenotypic defects in growth kinetics, cell wall integrity, PG synthesis, and cellular morphology, indicating that PbpA(2b) and Pbp5(4) are not functionally redundant and that PbpA(2b) plays a more central role in PG synthesis. Collectively, our results point to a model in which PbpA(2b) and Pbp5(4) cooperate to coordinately mediate PG cross-linking in the presence of cephalosporins and identify an attractive new extracellular target for small-molecule therapeutics designed to disable intrinsic cephalosporin resistance in all clinically relevant enterococci.
RESULTS
bPBPs are functionally distinct in E. faecalis.
Three bPBPs are encoded in the enterococcal genome [Pbp5(4), PbpA(2b), and PbpB(2)] (15). These 3 bPBPs from E. faecalis OG1RF are ∼22 to 27% identical to each other at the primary amino acid sequence level (determined using MUSCLE with default parameters at https://www.ebi.ac.uk/Tools/msa/muscle/), illustrating that their primary sequences are quite divergent from each other despite the fact that they catalyze the same chemical reactions. Pbp5(4) (OG1RF_11907; GenBank accession number AEA94594.1) has long been known to be required for cephalosporin resistance, but virtually nothing is known about the functions of enterococcal PbpA(2b) (OG1RF_12158; GenBank accession number AEA94845.1) or PbpB [also known as Pbp2; herein referred to as PbpB(2)] (OG1RF_10724; GenBank accession number AEA93411.1). PbpB(2) is encoded in a gene cluster including genes involved in cell division (such as ftsQ, ftsA, ftsZ, and divIVA), suggesting that PbpB(2) participates in septal PG synthesis during cell division and might therefore be essential for viability. Consistent with this, despite multiple attempts, we were unable to construct an E. faecalis mutant lacking pbpB.
In contrast, we successfully constructed a mutant of E. faecalis lacking pbpA and compared it to our previously constructed Δpbp5 mutant (20). Unlike the Δpbp5 mutant, the ΔpbpA mutant exhibited a significant growth defect compared to wild-type E. faecalis (see Fig. S1 in the supplemental material), the first indication that Pbp5(4) and PbpA(2b) are not functionally redundant. To determine if PbpA(2b) contributes to cell wall integrity, we used a previously described method to assess the susceptibility of intact bacteria to lysis (destruction of the membrane) by SDS (21). In this approach, wild-type E. faecalis with intact PG experiences minimal lysis and releases relatively little protein upon SDS treatment due to the intact PG sacculus, while mutant cells with impaired cell wall integrity (i.e., improperly formed or partially un-cross-linked sacculi) release much more protein upon SDS treatment due to considerable cell lysis. For mutant cells with significantly impaired PG, the extent of protein release can be comparable to wild-type cells for which the PG was predigested with lysozyme. To analyze our PBP mutants using this approach, exponentially growing bacteria were collected, exposed (or not) to lysozyme prior to treatment with 2% SDS in Laemmli SDS loading buffer, and subjected to electrophoresis to assess protein release (Fig. S2). As expected, all lysozyme-treated samples lysed efficiently upon treatment with SDS. Relatively little lysis was detected with wild-type or Δpbp5 cells, indicating that the cell wall of these strains was predominantly intact. In contrast, the ΔpbpA mutant lysed extensively without lysozyme pretreatment, consistent with the hypothesis that PbpA(2b)-mediated peptidoglycan cross-linking is essential for cell wall integrity. To confirm that the phenotypes of the ΔpbpA mutant were indeed due to loss of PbpA(2b), we performed complementation analysis by introducing a copy of pbpA at an ectopic location in the genome. PbpA(2b) expression was restored to levels comparable to those of the wild type in the complemented strain (Fig. S3), and we found that both the growth defect and the cell wall integrity defect were also restored upon ectopic PbpA(2b) production (Fig. S1 and S2).
Because PbpA(2b) is required for cell wall integrity, we hypothesized that synthesis of PG would be impaired in the absence of PbpA(2b). To test this, we used a previously described assay that monitors incorporation of [14C]GlcNAc into SDS-insoluble peptidoglycan of exponentially growing cells (22, 23). Pulse labeling of exponentially growing wild-type E. faecalis cells with [14C]GlcNAc led to incorporation of label (Fig. 1), as reported previously (22). To validate label incorporation as a specific measure of PG synthesis, we analyzed the incorporation of label into wild-type E. faecalis treated with fosfomycin, an antibiotic that inhibits the first committed step of PG synthesis in the cytoplasm, revealing that fosfomycin blocked incorporation of label as expected (Fig. 1). We conclude that under our experimental conditions, label incorporation reflects synthesis of PG and not incorporation into any other polymers. Therefore, we analyzed [14C]GlcNAc incorporation by the ΔpbpA mutant, revealing an ∼51% reduction in the rate of incorporation compared to the wild type, normalized to total biomass (Fig. 1), consistent with the hypothesis that PbpA(2b) is critical for PG synthesis in enterococci.
FIG 1.
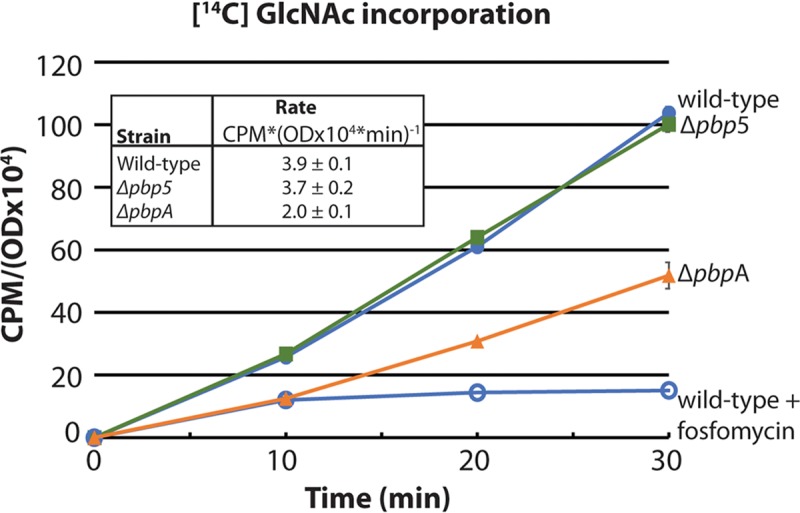
The ΔpbpA mutant is defective at peptidoglycan synthesis. Exponentially growing E. faecalis cells were pulse-labeled with [14C] GlcNAc to monitor incorporation into SDS-insoluble peptidoglycan. For wild-type cells, an aliquot was also treated with fosfomycin to inhibit peptidoglycan synthesis. At intervals, aliquots were treated with SDS, and insoluble peptidoglycan was subjected to scintillation counting. Incorporated radioactivity (CPM) was normalized to total biomass (OD600) and expressed as CPM/(OD600 × 104). Data represent the mean ± standard error from two independent experiments. Strains were wild-type, OG1; ΔpbpA, JL632; and Δpbp5, JL339.
Examination of the morphology of E. faecalis bPBP mutants revealed a marked difference between mutants lacking Pbp5(4) and PbpA(2b). The Δpbp5 mutant was indistinguishable from the wild type when live cells were examined using differential inference contrast (DIC) microscopy or transmission electron microscopy (Fig. 2). In contrast, the ΔpbpA mutant exhibited a marked morphological defect, growing as clumps of irregularly shaped cells rather than the typical diplococcal arrangement of ovoid cells characteristic of enterococci. Transmission electron microscopy (TEM) revealed that ΔpbpA mutant cells exhibited aberrant placement of the cell division septa (Fig. 2), suggesting a loss of coordination between peripheral and septal PG synthesis machineries. The morphological abnormalities were complemented by ectopic production of PbpA(2b).
FIG 2.
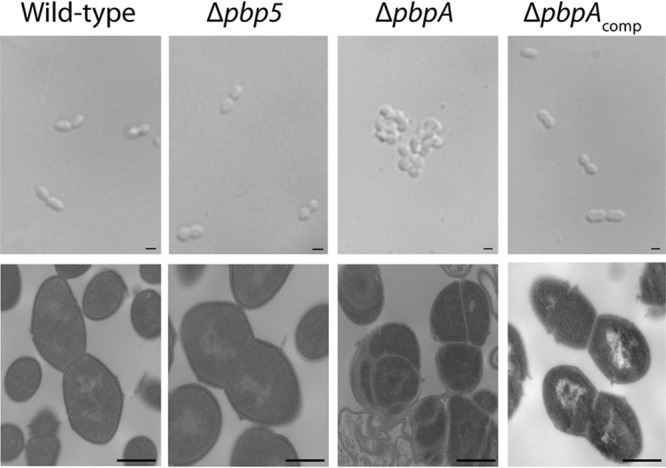
The ΔpbpA mutant exhibits aberrant cellular morphology. E. faecalis strains growing exponentially in MHB were examined using differential interference contrast microscopy (top panels, scale bars = 1 μm) and by transmission electron microscopy (bottom panels, scale bars = 0.5 μm). Strains were wild-type, OG1; ΔpbpA, JL632; Δpbp5, JL339; and ΔpbpAcomp, DDJ241.
PbpA(2b) is required for enterococcal cephalosporin resistance.
To test for a role of PbpA(2b) in enterococcal antimicrobial resistance, we determined MICs against a panel of antibiotics using broth microdilution assays. As expected (15), the Δpbp5 mutant exhibited a substantial loss of resistance to cephalosporins, including representative 2nd-, 3rd-, 4th-, and 5th-generation cephalosporins (cefuroxime, ceftriaxone, cefepime, and ceftaroline, respectively; Table 1). Similar defects were observed for the Δpbp5 mutant of a diverse E. faecalis lineage (Table 1). No defects were observed for the beta-lactam ampicillin or for bacitracin (which impairs cell wall synthesis by preventing bactoprenol recycling) or gentamicin (which impairs protein synthesis) in the absence of Pbp5(4). A 2-fold reduction in resistance to the carbapenems meropenem and imipenem was observed, although such 2-fold changes are generally considered to be within the error of broth microdilution measurements. Overall, these observations are consistent with the known role for Pbp5(4) in mediating enterococcal cephalosporin resistance and extend previous observations to multiple generations of cephalosporins.
TABLE 1.
Resistance of PBP mutants to antimicrobialsa
| Antimicrobial | MIC (μg/ml) E. faecalis OG1 |
MIC (μg/ml) E. faecalis CK221 |
|||||
|---|---|---|---|---|---|---|---|
| Wild typeb | Δpbp5 | ΔpbpA | pbpAcomp | Wild-type | Δpbp5 | ΔpbpA | |
| Cefuroxime | 128 | 0.5 | 0.5 | 256 | 512 | 1 | 0.5 |
| Ceftriaxone | 64 | 1 | 1 | 128 | 512 | 1 | 0.25 |
| Cefepime | 16 | 4 | 2 | 16 | 32 | 4 | 4 |
| Ceftaroline | 32 | 2 | 2 | 16 | 32 | 2 | 2 |
| Ampicillin | 0.5 | 0.5 | 0.5 | NDc | 0.5 | 0.5 | 0.5 |
| Meropenem | 2 | 1 | 8 | 4 | ND | ND | ND |
| Imipenem | 1 | 0.5 | 0.5 | ND | ND | ND | ND |
| Bacitracin | 64 | 64 | 32 | ND | 32 | 32 | 16 |
| Gentamicin | 32 | 32 | 16 | ND | ND | ND | ND |
Median MICs determined from a minimum of 3 independent replicates.
Derivatives of E. faecalis OG1 were Δpbp5, JL339; ΔpbpA, JL632; and pbpAcomp DDJ241. Derivatives of E. faecalis CK221 were Δpbp5, JL640; and ΔpbpA, JL639.
ND, not determined.
Analysis of the ΔpbpA mutants from both lineages of E. faecalis revealed a substantial loss of resistance to representative 2nd-, 3rd-, 4th-, and 5th-generation cephalosporins, similar to the phenotype of the Δpbp5 mutant, that was complemented by ectopic expression of pbpA (Table 1). Immunoblotting verified that the defect of the ΔpbpA mutant is not due to loss of Pbp5(4) expression in the ΔpbpA mutant (Fig. S4), indicating that both bPBPs are therefore required for enterococcal cephalosporin resistance. As with the Δpbp5 mutant, the defects of the ΔpbpA mutant were largely specific to cephalosporins, as no changes in resistance were observed for ampicillin, bacitracin, or gentamicin. Hence, the growth defects of the ΔpbpA mutant per se do not explain its loss of cephalosporin resistance. Additionally, the ΔpbpA mutant exhibited an increase in resistance to meropenem. This phenomenon was not universal among carbapenems (imipenem resistance did not increase; Table 1). Although the mechanistic basis for the increase in meropenem resistance remains unexplained, this increase also indicates that a general growth defect does not explain the specific loss of cephalosporin resistance. Plasmid-based overexpression of PbpA(2b) (Fig. S3) drove enhanced resistance to ceftriaxone (Table 2). Moreover, killing assays revealed that the viability of E. faecalis ΔpbpA cells declines substantially after treatment with ceftriaxone concomitant with cell lysis (Fig. 3). This effect is also unique to the ΔpbpA mutant, as the Δpbp5 mutant is not killed by ceftriaxone, nor does it lyse upon ceftriaxone treatment (Fig. 3), despite being susceptible to ceftriaxone as determined by MIC assays. Together, these findings represent the first evidence that a bPBP other than Pbp5(4) drives cephalosporin resistance in enterococci.
TABLE 2.
Resistance of plasmid-carrying strains to ceftriaxone
| Strainb | MIC (μg/ml)a |
|---|---|
| E. faecalis | |
| Wild-type + vector | 64 |
| ΔpbpA + vector | 1 |
| ΔpbpA + P-pbpA-his6 | >128 |
| E. faecium | |
| Wild-type | 64 |
| pbpA knockdown | 8 |
Median MICs for ceftriaxone were determined from a minimum of 3 independent replicates.
Strains were E. faecalis OG1/pJRG9, E. faecalis JL632/pJRG9, E. faecalis JL632/pEAW9, E. faecium 1141733/pJLL105, and E. faecium 1141733/pJLL264.
FIG 3.

The ΔpbpA mutant is susceptible to killing by ceftriaxone. E. faecalis cells growing exponentially in MHB were treated with ceftriaxone. Samples were removed and enumerated for viability (A) and the OD600 determined (B) at intervals. Data represent the mean ± standard error from two independent experiments. Strains were wild-type, OG1; ΔpbpA, JL632; Δpbp5, JL339; and ΔpbpAcomp, DDJ241.
The PbpA(2b) homolog from E. faecium is ∼55% identical to PbpA(2b) of E. faecalis at the primary amino acid sequence level. To test if PbpA(2b) is required for cephalosporin resistance in E. faecium as well, we attempted to construct a pbpA deletion mutant in the E. faecium 1141733 clinical isolate but were unable to do so despite multiple attempts, suggesting that PbpA(2b) might be even more critical for viability in E. faecium. Inspection of the pbpA gene neighborhood suggested that pbpA was likely to be encoded on a monocistronic transcript. Therefore, as an alternative to genetic deletion, we used a plasmid containing an inducible promoter to express an antisense RNA directed against E. faecium pbpA to knock down PbpA(2b) expression. Antimicrobial susceptibility assays on antisense-expressing E. faecium revealed that PbpA(2b) also promotes cephalosporin resistance in E. faecium (Table 2). Hence, PbpA(2b) drives cephalosporin resistance in both E. faecalis and E. faecium, the two enterococcal species of greatest clinical significance.
PbpA(2b) exhibits low reactivity with ceftriaxone.
Pbp5(4) has been known for decades to exhibit low reactivity toward cephalosporins, a property that enables Pbp5(4) to carry out PG cross-linking and thereby promote growth despite the presence of cephalosporins in the environment. Given that PbpA(2b) is also required for cephalosporin resistance, we hypothesized that PbpA(2b) would similarly exhibit low reactivity toward cephalosporins. To test this, we used the activity-based probe Bocillin FL (24) to covalently label enterococcal PBPs on live cells. Both Bocillin FL and cephalosporins (like all beta-lactams) acylate PBPs at their active-site serine, thereby preventing PBP transpeptidase activity. Therefore, we used an experimental competition format in which growing cells are first exposed to a cephalosporin, followed by Bocillin FL to label any remaining unacylated PBPs. Any PBPs that are reactive with the cephalosporin will be acylated and unable to be subsequently labeled by Bocillin FL, whereas PBPs that are not reactive with the cephalosporin (i.e., not acylated) will subsequently be labeled upon exposure to excess Bocillin FL (and therefore be fluorescent). Exposure of exponentially growing E. faecalis cells to ceftriaxone resulted in ceftriaxone-mediated acylation of multiple PBPs (missing bands in Fig. 4); however, 3 PBPs were essentially not acylated by ceftriaxone, enabling subsequent acylation by Bocillin FL. This pattern was conserved in both evolutionarily diverse E. faecalis strains we examined. To determine which specific PBPs were unreactive with ceftriaxone, we analyzed Bocillin FL-mediated PBP labeling of a panel of E. faecalis PBP deletion mutants (no cephalosporin treatment), which enabled us to assign the identities of each of the 6 labeled bands resolved under our experimental conditions (Fig. S5). With this information, we identified the non-ceftriaxone-reactive PBPs as Pbp5(4), PonA, and PbpA(2b). These same PBPs [including PbpA(2b)] were also not reactive with additional 2nd- and 3rd-generation cephalosporins (cefuroxime and ceftazidime; Fig. S6). The 4th-generation cephalosporin cefepime largely acylated all PBPs except for PbpA(2b), and the 5th-generation cephalosporin ceftaroline also did not acylate PbpA(2b) (Fig. S6). Together, these results reveal that even cephalosporins capable of acylating the low-reactivity Pbp5(4) cannot acylate PbpA(2b). Thus, PbpA(2b) is required for enterococcal cephalosporin resistance and is not reactive with cephalosporins in vivo, a property that presumably enables PbpA(2b) to carry out PG cross-linking in the presence of cephalosporins to promote growth.
FIG 4.
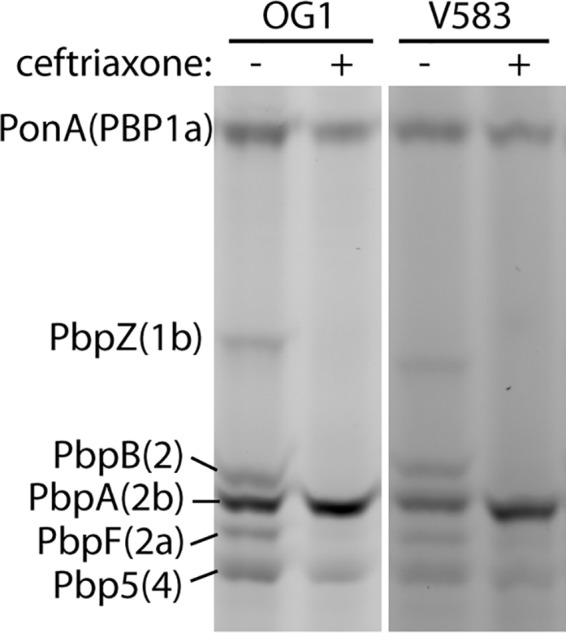
Ceftriaxone acylates only a subset of E. faecalis PBPs on growing cells. Two evolutionarily diverse E. faecalis strains (OG1 and an erythromycin-sensitive derivative of the clinical isolate V583) were cultured in MHB to the exponential phase and treated with 2 mg/ml ceftriaxone (or not) as indicated. Cells were collected after 20 min and treated with Bocillin FL to acylate any PBPs that had not been previously acylated by ceftriaxone. Total cell lysates were subjected to SDS-PAGE and scanned to visualize fluorescently labeled PBPs. Data are representative of more than 3 independent experiments.
To determine if low cephalosporin reactivity is an inherent property of PbpA(2b) itself or if the cephalosporin reactivity of PbpA(2b) is modulated by other factors in vivo, we expressed and purified a soluble recombinant E. faecalis PbpA(2b) lacking its N-terminal transmembrane anchor [PbpA(2b) ΔTM]. This strategy has been used previously to investigate structure and function of other enterococcal PBPs (25–28). Antibiotic competition assays were performed in vitro by incubating the purified protein with various concentrations of antibiotic for 20 min, followed by labeling any remaining unacylated PBP with excess Bocillin FL. We found that although PbpA(2b) ΔTM was reactive with ampicillin (50% inhibitory concentration [IC50] estimated to be ∼3 to 10 μM), it was virtually completely unreactive with ceftriaxone at any concentration up to 30,000 μM (equivalent to 19.8 mg/ml, the highest concentration tested; Fig. 5). As a control, ceftriaxone reacted robustly with recombinant PbpB(2) ΔTM under our experimental conditions (IC50 estimated to be ∼1 to 3 μM), consistent with the robust ceftriaxone reactivity of PbpB(2) observed in vivo (Fig. 4; Fig. S6 and S7). As expected, recombinant Pbp5(4) ΔTM also exhibited low ceftriaxone reactivity (IC50 estimated to be ∼3,000 μM; Fig. S7), though Pbp5(4) was modestly more reactive with ceftriaxone than PbpA(2b). If low reactivity toward cephalosporins is functionally important for cephalosporin resistance, we hypothesized that it would be conserved for PbpA(2b) of E. faecium as well. To test this, we performed competition experiments with recombinant E. faecium PbpA(2b) ΔTM. The results (Fig. S8) revealed that, despite being only ∼55% identical to PbpA(2b) of E. faecalis, E. faecium PbpA(2b) also exhibits inherently low reactivity with ceftriaxone (IC50 estimated to be ∼10,000 μM), as well as ampicillin. Collectively these results establish that PbpA(2b) of clinically relevant enterococci exhibits inherently low reactivity with cephalosporins that is similar to, or even less reactive than, the enterococcal PBP [Pbp5(4)] known to exhibit low-cephalosporin reactivity.
FIG 5.

PbpA(2b) exhibits intrinsically low reactivity for ceftriaxone in vitro. Purified, recombinant E. faecalis His6-PbpA(2b) ΔTM (3 μM) was incubated with the indicated concentrations of ceftriaxone (A) or ampicillin (B) for 20 min and then treated with excess Bocillin FL. The reactions were subjected to SDS-PAGE and scanned to visualize fluorescently labeled PbpA(2b), followed by Coomassie staining for total protein. Data are representative of 3 independent experiments.
DISCUSSION
A specific enterococcal bPBP, historically referred to in the literature as either Pbp4 or Pbp5, has been known for decades to be required for beta-lactam resistance. Pbp5(4) contains a MecA_N (pfam05223) domain, which—although its specific molecular function is not well understood—is also encoded in the staphylococcal Pbp2a protein known to be responsible for beta-lactam resistance in methicillin-resistant Staphylococcus aureus. Given its low reactivity with beta-lactam antibiotics, enterococcal Pbp5(4) has been thought to be the primary PBP responsible (i.e., necessary and sufficient) for resistance to beta-lactams, including cephalosporins, in enterococci. This is consistent with studies demonstrating loss of cephalosporin resistance by a Δpbp5 mutant (15). However, a potential role in cephalosporin resistance for the other 2 bPBPs [PbpB(2) and PbpA(2b)] encoded by the clinically relevant enterococci E. faecalis and E. faecium has never been excluded, or—to the best of our knowledge—explicitly tested.
To determine if PbpB(2) or PbpA(2b) is important for enterococcal cephalosporin resistance, we attempted to construct deletion mutants lacking the corresponding genes. We have thus far been unable to construct an E. faecalis mutant lacking PbpB(2), suggesting that it may be essential for viability. Two bits of circumstantial evidence suggest a direct role for PbpB(2) in cell division; first, the gene neighborhood in which pbpB is found contains many cooriented genes whose products are involved in cell division, and second, PbpB(2) contains two C-terminal penicillin-and-serine/threonine-associated (PASTA) domains, and the homologous PASTA-containing PBP in Streptococcus pneumoniae (Pbp2x) has been implicated in septal PG synthesis during cell division (29, 30). In any case, given our inability to construct the ΔpbpB mutant, we have not yet been able to directly interrogate PbpB(2) for a role in cephalosporin resistance via genetic approaches. However, our PBP acylation data indicate that PbpB(2) is highly reactive with cephalosporins both in vivo and in vitro at concentrations below 2 μg/ml, which means that PbpB(2) cannot drive PG cross-linking in the presence of cephalosporins.
In contrast to PbpB(2), we successfully constructed mutants of 2 diverse lineages of E. faecalis lacking PbpA(2b) and compared them to mutants lacking Pbp5(4). The ΔpbpA mutants exhibited a substantial loss of resistance to cephalosporins, comparable in magnitude and scope to that of the Δpbp5 mutants, despite the fact that Pbp5(4) was expressed in the absence of PbpA(2b). In addition, ceftriaxone exhibited a marked bactericidal effect on the ΔpbpA mutant, in contrast to wild-type or Δpbp5 mutant cells. Together, these results shift the paradigm for enterococcal cephalosporin resistance by demonstrating that Pbp5(4) is not sufficient for resistance and that a second class B PBP, PbpA(2b), is specifically required in addition to Pbp5(4). Expression of antisense RNA directed at transcripts encoding PbpA(2b) of E. faecium also led to a reduction of cephalosporin resistance, indicating that PbpA(2b) function is evolutionarily conserved and is required for cephalosporin resistance in both enterococcal species of greatest clinical significance. In line with this essential requirement for PbpA(2b) to grow in the presence of cephalosporins, PbpA(2b) from both E. faecalis and E. faecium exhibits intrinsically low reactivity toward cephalosporins. As with Pbp5(4), this intrinsically low reactivity enables PbpA(2b) to avoid acylation (i.e., inactivation) by cephalosporins, preserving its ability to catalyze PG cross-linking despite the presence of cephalosporins in the environment. It is worth noting that the Bocillin FL-mediated acylation of PbpA(2b) observed in Fig. 4 and 5 could also occur if PbpA(2b) was initially acylated by ceftriaxone but subsequently underwent rapid hydrolysis. Such a hypothesis predicts that the concentration of remaining intact, active ceftriaxone would be reduced after exposure to PbpA(2b) due to PbpA(2b)-mediated hydrolysis. We tested for such an effect (Fig. S9) and did not observe any change in the activity of ceftriaxone. Thus, these results support the hypothesis that PbpA(2b) is unreactive with cephalosporins and, as a result, remains available to be acylated by Bocillin FL in the context of PBP labeling experiments. Of note, although PbpA(2b) is a class B PBP, PbpA(2b) does not contain a MecA_N domain as found in Pbp5(4) (Fig. S10). Hence, the MecA_N domain is not required for bPBPs to evolve intrinsically low reactivity toward beta-lactam antibiotics.
The ΔpbpA mutant also exhibited a variety of other phenotypic defects, including reduced growth kinetics, impaired cell wall integrity, reduced PG synthesis, and aberrant cellular morphology, that were not shared with the Δpbp5 mutant. Thus, although both PbpA(2b) and Pbp5(4) are required for cephalosporin resistance, they perform distinct functions in enterococcal cells. Collectively the phenotypes suggest that the activity of Pbp5(4) is important primarily to enable growth in the presence of cephalosporins, but the activity of PbpA(2b) plays a more central role in overall PG synthesis, wall integrity, and cellular morphology under all conditions that extends beyond its critical requirement for cephalosporin resistance.
Recent work on PG synthesis in several bacterial species has revealed that SEDS-family (shape, elongation, division, sporulation) integral membrane proteins such as RodA and FtsW function as PG glycosyltransferases and, in complex with a cognate bPBP, act as multiprotein machines (known as “synthases” or polymerization complexes) that mediate PG synthesis at distinct cellular locations (31–34). In recent models, one synthase is localized to the site of cell division (the “divisome”) and builds septal PG during cell division, while a separate synthase (the “elongasome”) builds peripheral PG. Coordinated activity of both synthases is required for proper growth and division. Based on our results, we speculate that PbpA(2b) and Pbp5(4) contribute to PG synthesis as essential components of distinct PG synthase machines, which catalyze PG synthesis at different cellular locations, and that proper function of both PbpA(2b)- and Pbp5(4)-containing synthases is simultaneously required for growth in the presence of cephalosporins. Such a model is consistent with our observations that inactivation of either Pbp5(4) or PbpA(2b) alone leads to loss of cephalosporin resistance. Moreover, the relative lack of phenotypes exhibited by the Δpbp5 mutant in the absence of cephalosporins suggests that the synthase to which Pbp5(4) belongs also contains one or more additional PBPs capable of providing PG cross-linking during normal growth. Such PBPs are likely to exhibit high reactivity with cephalosporins, rendering Pbp5(4) essential for PG cross-linking by its synthase when cephalosporins are present in the environment. Conversely, the diverse phenotypes of the ΔpbpA mutant suggest that other PBPs, including Pbp5(4), cannot functionally compensate for PG cross-linking in its synthase.
One possibility is that Pbp5(4) is a member of the divisome PG synthase together with PbpB(2). Given that PbpB(2) is highly reactive with cephalosporins, Pbp5(4) could provide PG cross-linking activity to build septal PG as part of the divisome when PbpB(2) is inactivated by cephalosporins, while PbpA(2b) catalyzes peripheral PG synthesis as part of the elongasome, together enabling growth and cephalosporin resistance (Fig. S11). Such a model would be consistent with the relative lack of phenotypes exhibited by the Δpbp5 mutant in the absence of cephalosporins [when PbpB(2) remains active] and with our observation that the ΔpbpA mutant remains capable of building division septa (albeit aberrantly). However, this model is likely oversimplified, as it predicts that Pbp5(4) should be able to substitute for the complete absence of PbpB(2), which is not consistent with our inability to construct the pbpB deletion mutant. A caveat to that observation, though, is that we have not yet formally established that the underlying reason we have been unable to genetically delete pbpB is, in fact, due to the loss of the PbpB protein itself; it remains possible that the pbpB deletion impacts expression of other downstream genes critical for cell division. In addition, it is unclear at present how the aPBPs PonA and PbpF would fit into this model, although aPBPs have been found to be associated with the bPBP-containing synthases in other bacteria and therefore could very well be a part of the PbpA(2b)- and/or Pbp5(4)-containing synthases. Future work will focus on defining the mechanisms by which PbpA(2b) and Pbp5(4) coordinate with each other to catalyze PG synthesis in the presence of cephalosporins.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1. Enterococci were propagated on Mueller-Hinton broth (MHB) or agar (prepared according to the manufacturer’s instructions; Difco) at 30°C unless indicated otherwise. Chloramphenicol was included at 10 μg/ml when required for maintenance of plasmids.
Genetic manipulation of enterococci.
Mutant strains were constructed using the temperature-sensitive, counterselectable allelic exchange plasmid pJH086 as previously described (35). In addition to p-Cl-Phe, counterselection plates contained 20% sucrose. Fragments of genomic DNA from upstream and downstream of the gene of interest were amplified by PCR and introduced into pJH086 via isothermal assembly (36). Deletion alleles were in-frame and retained a variable number of codons at the beginning and end of each gene, in an effort to avoid perturbing the expression of adjacent genes. Table S1 contains the specific details of each deletion allele. All mutant or complemented strains were constructed independently at least twice and analyzed to ensure that their phenotypes were concordant. The inserts of all recombinant plasmids were sequenced in their entirety to ensure the absence of any undesired mutations.
Complementation of the E. faecalis ΔpbpA mutant was achieved by integrating a single copy of the pbpA gene along with 400 bp of upstream DNA (presumably containing the pbpA promoter) into an ectopic locus in the chromosome of the ΔpbpA mutant, using a homologous recombination-based allelic exchange strategy as described previously (37). PCR analysis confirmed the presence of the original ΔpbpA deletion in the complemented strain. Immunoblot analysis on the complemented strain verified restoration of near wild-type levels of PbpA(2b) production (Fig. S3).
Antisense RNA for knockdown of E. faecium PbpA(2b).
To generate an inducible antisense construct to knock down expression of E. faecium pbpA, we used an approach analogous to that described previously to knock down expression of genes in Staphylococcus aureus (38–40). Our construct included the entire E. faecium pbpA open reading frame (ORF) plus 58 nucleotides upstream of the ORF in plasmid pBK2, cloned in the reverse orientation such that transcription from the inducible plasmid-encoded promoter would lead to production of the antisense transcript. The resulting plasmid (pJLL264) was introduced into wild-type E. faecium by electroporation. Expression from the plasmid-encoded promoter was induced by addition of cCF10 peptide when desired, as we have done previously (35).
Antimicrobial susceptibility assays.
MICs were determined using a broth microdilution method. Briefly, bacteria from stationary-phase cultures were inoculated into wells containing 2-fold serial dilutions of antibiotics in MHB (including chloramphenicol when required for maintenance of plasmids) at a normalized optical density at 600 nm (OD600) of 4 × 10−5 (∼1 × 105 CFU for the wild type). Plates were incubated in a Bioscreen C plate reader at 37°C for 24 h with brief shaking before each measurement. The OD600 was determined every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC. For time-kill analysis, stationary-phase cultures were diluted to an OD600 of 0.01 in MHB and incubated at 37°C with shaking until the early exponential phase (OD600, ∼0.13), at which time aliquots were treated with 2 mg/ml ceftriaxone. Samples were removed at intervals (0, 1, 4, and 24 h) for determination of OD600, and viable bacteria were enumerated on MH agar. We note that because of the cell division defect and clumping phenotype exhibited by the ΔpbpA mutant, CFU counts are not an accurate representation of the number of cells in a culture. Instead, we normalized the inocula based on OD600 values, which provide a more representative estimation of the amount of biomass present (see Supplementary Data for the analysis of protein levels in cultures normalized by OD600).
Analysis of cell wall integrity.
Stationary-phase cultures of strains grown at 30°C were diluted to an OD600 of 0.01 in MH broth and incubated at 37°C with shaking until the exponential phase (OD600, ∼0.2). Bacteria were harvested by centrifugation, resuspended in 50 μl of lysozyme solution (10 mM Tris [pH 8], 50 mM NaCl, 20% sucrose), and normalized to an equivalent OD600. Cell suspensions were split into two equal aliquots, one of which was subsequently treated with lysozyme (5 mg/ml). All samples were incubated for 5 min at 37°C. Laemmli SDS buffer was added, and samples were boiled (5 min) and subjected to SDS-PAGE followed by staining for total protein using GelCode blue stain (Pierce) according to the manufacturer’s instructions.
Immunoblot analysis.
Bacteria were grown to the exponential phase in MHB at 37°C as described above. Cultures were mixed with an equal volume of EtOH/acetone (1:1), collected by centrifugation, and washed once with water. To prepare total cell lysates, bacteria were suspended in lysozyme buffer (10 mM Tris [pH 8], 50 mM NaCl, 20% sucrose), normalized to an equivalent OD600, treated with lysozyme (5 mg/ml) for 30 min at 37°C, and mixed with Laemmli SDS sample buffer. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride (PVDF; Bio-Rad) and probed with rabbit anti-PbpA(2b), rabbit anti-Pbp5(4), and rabbit anti-RpoA antisera (custom polyclonal antisera). Detection was performed with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Invitrogen).
Peptidoglycan synthesis assays.
Peptidoglycan synthesis was monitored by uptake of [14C]GlcNAc into SDS-insoluble peptidoglycan as described previously (22, 23) with minor modifications. Bacteria were grown to the exponential phase in MHB at 37°C as described above. Cultures were diluted 6-fold into prewarmed MHB supplemented with 0.4 μCi/ml [14C]GlcNAc. At intervals, aliquots were mixed with equal volumes of 0.2% SDS. The cells were collected by centrifugation and suspended in 150 μl water and then transferred into 5 ml Econo-Safe scintillation fluid for scintillation counting. Optical density measurements were done in triplicate by transferring 200 μl from parallel label-free cultures to a 96-well plate and measuring the OD600 using a SpectraMax5 plate reader (Molecular Devices). Data were expressed as counts per minute (CPM)/(OD600 × 104).
Microscopy.
Bacteria were grown to the exponential phase in MHB at 37°C as described above. For transmission electron microscopy, cultures were directly mixed with an equal volume of 4% glutaraldehyde in 0.1 M sodium cacodylate buffer and fixed at room temperature for 30 min. Bacteria were washed 3 times with buffer and postfixed for 1 h on ice with 1% osmium tetroxide. Following fixation, cells were rinsed with buffer and dehydrated through a graded methanol series (50%, 20 min; 70%, 20 min; 95%, 20 min; 100% 3×, 20 min) followed by acetonitrile and then 60 min in a 50/50 mixture of acetonitrile and epoxy resin (EMbed-812; Electron Microscopy Sciences, Hatfield, PA). Following 2 changes (3 h each) of epoxy resin, the samples were centrifuged to pellet in Eppendorf tubes and polymerized at 70°C overnight. Thin (60 nm) sections were cut, and the sections were stained with uranyl acetate and lead citrate and then imaged using a Hitachi H600 transmission electron microscope, and the images were recorded using a Hamamatsu side entry charge-coupled-device (CCD) camera and AMT imaging software. For light microscopy, bacteria were fixed using 1% formaldehyde and examined with differential interference contrast microscopy using a Nikon Eclipse Ti-U inverted microscope. Examination of live cells confirmed that fixation did not alter their morphology.
PBP acylation in vivo.
Bacteria were grown to the exponential phase in MHB at 37°C as described above. Cultures were split and treated (or not) with a cephalosporin for 20 min at 37°C. Cells were chilled in an ice-water bath, collected by centrifugation, suspended in buffer (25 mM Tris [pH 7.5], 150 mM NaCl, 100 μg/ml chloramphenicol), and normalized to an equivalent OD600 on ice. Bocillin FL was added to 40 μM and incubated at 37°C for 20 min. Cells were washed once with buffer and then suspended in lysozyme buffer for digestion with 5 mg/ml lysozyme for 15 min at 37°C to make whole-cell lysates as described above. Lysates were subjected to 8% SDS-PAGE on 20-cm gels at constant amperage for ∼16 h and scanned on a Typhoon Trio with excitation at 488 nm.
Expression and purification of recombinant PBPs.
PBPs were expressed from pET28b in Escherichia coli BL21(DE3) host cells. All PBPs were cloned and expressed without their N-terminal membrane anchors as indicated in Table S1. BL21(DE3) carrying a pET28b derivative was cultured to an OD600 of 0.6, induced with 1 mM IPTG, and incubated for 4 h at 37°C for induction. Cells were broken via a French press, and recombinant His-tagged protein was purified via IMAC on Ni-NTA resin prior to dialysis into 1× PBS containing 10% glycerol.
PBP acylation in vitro.
PBPs were acylated in vitro as previously described (41) with minor modifications. Reaction mixtures containing a given purified recombinant PBP (3 μM) and various concentrations of antibiotics were prepared in 1× PBS and incubated at 37°C for 20 min, at which time Bocillin FL was added to 30× molar excess (91 μM) and incubated for an additional 20 min at 37°C. Reactions were quenched by the addition of Laemmli SDS-PAGE loading buffer and subjected to 8% SDS-PAGE prior to imaging on an Azure c600 instrument. After fluorescence images were acquired, gels were stained with Coomassie (GelCode Blue).
Supplementary Material
ACKNOWLEDGMENTS
We thank Eugenia Wulff for the provision of plasmid pEAW9 used in this study, Clive Wells for electron microscopy, and Stephanie Kellogg for critical review of the manuscript.
This study was supported by grants AI134660, AI128219, and AI132927 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, Participating National Healthcare Safety Network Facilities . 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 2.Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM. 2016. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol 37:1288–1301. doi: 10.1017/ice.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller WR, Murray BE, Rice LB, Arias CA. 2016. Vancomycin-resistant enterococci: therapeutic challenges in the 21st century. Infect Dis Clin North Am 30:415–439. doi: 10.1016/j.idc.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Kristich CJ, Rice LB, Arias CA. 2014. Enterococcal infection-treatment and antibiotic resistance In Gilmore MS, Clewell DB, Ike Y, Shankar N. (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 5.Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in Enterococcus. Virulence 3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller WR, Munita JM, Arias CA. 2014. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther 12:1221–1236. doi: 10.1586/14787210.2014.956092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray BE. 1990. The life and times of the Enterococcus. Clin Microbiol Rev 3:46–65. doi: 10.1128/cmr.3.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmeli Y, Eliopoulos GM, Samore MH. 2002. Antecedent treatment with different antibiotic agents as a risk factor for vancomycin-resistant Enterococcus. Emerg Infect Dis 8:802–807. doi: 10.3201/eid0808.010418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepard BD, Gilmore MS. 2002. Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect 4:215–224. doi: 10.1016/S1286-4579(01)01530-1. [DOI] [PubMed] [Google Scholar]
- 10.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, Pamer EG. 2008. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. 2000. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343:1925–1932. doi: 10.1056/NEJM200012283432604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 14.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 15.Arbeloa A, Segal H, Hugonnet JE, Josseaume N, Dubost L, Brouard JP, Gutmann L, Mengin-Lecreulx D, Arthur M. 2004. Role of class A penicillin-binding proteins in PBP5-mediated beta-lactam resistance in Enterococcus faecalis. J Bacteriol 186:1221–1228. doi: 10.1128/jb.186.5.1221-1228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rice LB, Carias LL, Rudin S, Hutton R, Marshall S, Hassan M, Josseaume N, Dubost L, Marie A, Arthur M. 2009. Role of class A penicillin-binding proteins in the expression of beta-lactam resistance in Enterococcus faecium. J Bacteriol 191:3649–3656. doi: 10.1128/JB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inglis B, Matthews PR, Stewart PR. 1988. The expression in Staphylococcus aureus of cloned DNA encoding methicillin resistance. J Gen Microbiol 134:1465–1469. doi: 10.1099/00221287-134-6-1465. [DOI] [PubMed] [Google Scholar]
- 18.Tesch W, Strässle A, Berger-Bächi B, O’Hara D, Reynolds P, Kayser FH. 1988. Cloning and expression of methicillin resistance from Staphylococcus epidermidis in Staphylococcus carnosus. Antimicrob Agents Chemother 32:1494–1499. doi: 10.1128/aac.32.10.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Signoretto C, Boaretti M, Canepari P. 1994. Cloning, sequencing and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis. FEMS Microbiol Lett 123:99–106. doi: 10.1111/j.1574-6968.1994.tb07207.x. [DOI] [PubMed] [Google Scholar]
- 20.Kristich CJ, Little JL. 2012. Mutations in the beta subunit of RNA polymerase alter intrinsic cephalosporin resistance in enterococci. Antimicrob Agents Chemother 56:2022–2027. doi: 10.1128/AAC.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vesic D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob Agents Chemother 56:2443–2451. doi: 10.1128/AAC.05984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mesnage S, Chau F, Dubost L, Arthur M. 2008. Role of N-acetylglucosaminidase and N-acetylmuramidase activities in Enterococcus faecalis peptidoglycan metabolism. J Biol Chem 283:19845–19853. doi: 10.1074/jbc.M802323200. [DOI] [PubMed] [Google Scholar]
- 23.Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H, Devine KM. 2007. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol Microbiol 65:180–200. doi: 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- 24.Gee KR, Kang HC, Meier TI, Zhao G, Blaszcak LC. 2001. Fluorescent Bocillins: synthesis and application in the detection of penicillin-binding proteins. Electrophoresis 22:960–965. doi:. [DOI] [PubMed] [Google Scholar]
- 25.Moon TM, D’Andréa ÉD, Lee CW, Soares A, Jakoncic J, Desbonnet C, Garcia-Solache M, Rice LB, Page R, Peti W. 2018. The structures of penicillin-binding protein 4 (PBP4) and PBP5 from enterococci provide structural insights into beta-lactam resistance. J Biol Chem 293:18574–18584. doi: 10.1074/jbc.RA118.006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice LB, Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Lonks J, Moon TM, D’Andréa ÉD, Page R, Peti W. 2018. Structural and regulatory changes in PBP4 trigger decreased beta-lactam susceptibility in Enterococcus faecalis. mBio 9:e00361-18. doi: 10.1128/mBio.00361-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duez C, Hallut S, Rhazi N, Hubert S, Amoroso A, Bouillenne F, Piette A, Coyette J. 2004. The ponA gene of Enterococcus faecalis JH2-2 codes for a low-affinity class A penicillin-binding protein. J Bacteriol 186:4412–4416. doi: 10.1128/JB.186.13.4412-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hujer AM, Kania M, Gerken T, Anderson VE, Buynak JD, Ge X, Caspers P, Page MG, Rice LB, Bonomo RA. 2005. Structure-activity relationships of different beta-lactam antibiotics against a soluble form of Enterococcus faecium PBP5, a type II bacterial transpeptidase. Antimicrob Agents Chemother 49:612–618. doi: 10.1128/AAC.49.2.612-618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez AJ, Cesbron Y, Shaw SL, Bazan Villicana J, Tsui HT, Boersma MJ, Ye ZA, Tovpeko Y, Dekker C, Holden S, Winkler ME. 2019. Movement dynamics of divisome proteins and PBP2x:FtsW in cells of Streptococcus pneumoniae. Proc Natl Acad Sci U S A 116:3211–3220. doi: 10.1073/pnas.1816018116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morlot C, Zapun A, Dideberg O, Vernet T. 2003. Growth and division of Streptococcus pneumoniae: localization of the high molecular weight penicillin-binding proteins during the cell cycle. Mol Microbiol 50:845–855. doi: 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- 31.Cho H, Wivagg CN, Kapoor M, Barry Z, Rohs PDA, Suh H, Marto JA, Garner EC, Bernhardt TG. 2016. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat Microbiol 1:16172. doi: 10.1038/nmicrobiol.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. 2016. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537:634–638. doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taguchi A, Welsh MA, Marmont LS, Lee W, Sjodt M, Kruse AC, Kahne D, Bernhardt TG, Walker S. 2019. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat Microbiol 4:587–594. doi: 10.1038/s41564-018-0345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emami K, Guyet A, Kawai Y, Devi J, Wu LJ, Allenby N, Daniel RA, Errington J. 2017. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat Microbiol 2:16253. doi: 10.1038/nmicrobiol.2016.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kellogg SL, Little JL, Hoff JS, Kristich CJ. 2017. Requirement of the CroRS two-component system for resistance to cell-wall-targeting antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61 doi: 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. [DOI] [PubMed] [Google Scholar]
- 37.Vesic D, Kristich CJ. 2013. A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J Bacteriol 195:1815–1824. doi: 10.1128/JB.02135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji Y, Marra A, Rosenberg M, Woodnutt G. 1999. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J Bacteriol 181:6585–6590. doi: 10.1128/JB.181.21.6585-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji Y, Woodnutt G, Rosenberg M, Burnham MK. 2002. Identification of essential genes in Staphylococcus aureus using inducible antisense RNA. Methods Enzymol 358:123–128. doi: 10.1016/s0076-6879(02)58084-8. [DOI] [PubMed] [Google Scholar]
- 40.Ji Y, Van Zhang B, Horn SF, Warren P, Woodnutt G, Burnham MK, Rosenberg M. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293:2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- 41.Papp-Wallace KM, Senkfor B, Gatta J, Chai W, Taracila MA, Shanmugasundaram V, Han S, Zaniewski RP, Lacey BM, Tomaras AP, Skalweit MJ, Harris ME, Rice LB, Buynak JD, Bonomo RA. 2012. Early insights into the interactions of different beta-lactam antibiotics and beta-lactamase inhibitors against soluble forms of Acinetobacter baumannii PBP1a and Acinetobacter sp. PBP3. Antimicrob Agents Chemother 56:5687–5692. doi: 10.1128/AAC.01027-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.