

Summary
Establishing causal links between non-coding variants and human phenotypes is an increasing challenge. Here we introduce a high-throughput mouse reporter assay for assessing the pathogenic potential of human enhancer variants in vivo and examine nearly a thousand variants in an enhancer repeatedly linked to polydactyly. We show that 71% of all rare non-coding variants previously proposed as causal led to reporter gene expression in a pattern consistent with their pathogenic role. Variants observed to alter enhancer activity were further confirmed to cause polydactyly in knock-in mice. We also used combinatorial and single-nucleotide mutagenesis to evaluate the in vivo impact of mutations affecting all positions of the enhancer and identified additional functional substitutions, including potentially pathogenic variants hitherto not observed in humans. Our results uncover the functional consequences of hundreds of mutations in a phenotype-associated enhancer and establish a widely applicable strategy for systematic in vivo evaluation of human enhancer variants.
Keywords: Cis-regulatory element, enhancer, rare non-coding variant, mutation, limb development, Polydactyly, CRISPR/Cas9, ZRS, Sonic hedgehog (Shh), genome editing
Graphical Abstract
In Brief
Development of a scalable in vivo mouse enhancer-reporter assay and its use to systematically interrogate the ZRS canonical enhancer, which drives Sonic hedgehog expression in mammalian limb development, broadly illuminates enhancer variant pathogenicity.
Introduction
Genome-wide association and whole-genome sequencing studies generate rapidly expanding lists of non-coding variants potentially causing or contributing to human diseases. However, there is a major gap in establishing conclusive links between individual variants and specific phenotypes observed in human patients. Many non-coding variants are hypothesized to affect distant-acting transcriptional enhancers, a predominant class of DNA regulatory sequences that activate the expression of target genes in a tissue-specific manner (Furlong and Levine, 2018; Long et al., 2016; Shlyueva et al., 2014; Visel et al., 2009b). Computational and in vitro approaches can provide ) useful initial prioritization strategies (Kircher et al., 2019; Smith et al., 2013; Tewhey et al., 2016; van Arensbergen et al., 2019), but a conclusive demonstration of the pathogenic potential of variants and a mechanistic understanding of their effects generally require the functional assessment of enhancer variants through in vivo experiments. Mouse transgenesis is a powerful system for this purpose, but traditional transgenic approaches have limited throughput and high cost, and so-called ‘position effects’ can complicate the interpretation of results (Inoue and Ahituv, 2015; Kvon, 2015).
In the present study, we developed a highly scalable mouse enhancer-reporter assay named enSERT (enhancer inSERTion) that relies on site-specific integration of a transgene into the mouse genome and avoids position effects (Figure 1A). To demonstrate the utility of enSERT, we systematically interrogated all nucleotides in one of the best-studied human enhancers, the Zone of Polarizing Activity [ZPA] Regulatory Sequence (ZRS, also known as MFCS1) (Lettice et al., 2003). The ZRS is a limb-specific enhancer of the Sonic hedgehog (SHH) gene and is located at the extreme distance of nearly one million base pairs from its target promoter. The enhancer is active in the posterior margins of developing fore- and hindlimb buds (Figure 1B) and is critically required for normal limb development in mice (Sagai et al., 2005). Changes in the ZRS enhancer have been implicated in the evolution of vertebrate appendages (Kvon et al., 2016; Leal and Cohn, 2016; Letelier et al., 2018). In humans, as well as in several other tetrapods, single nucleotide mutations within the ZRS enhancer cause limb malformations, most commonly preaxial polydactyly (Table S1) (Hill and Lettice, 2013; Lettice et al., 2003; VanderMeer and Ahituv, 2011). ZRS mutations implicated in polydactyly cause ectopic activation of Shh expression in the anterior margin of limb bud mesenchyme in addition to the normal activity domain in the posterior margin of the limb bud (Lettice et al., 2014; Masuya et al., 2007). Misexpression of Shh in the anterior domain results in erroneous digit outgrowth and polydactyly. To date, 21 different human mutations in this enhancer have been identified in patients with polydactyly and reported in the literature (Table S1). Each mutation is individually rare, with most described in a single case or family, and none are present in current public genetic variant databases (gnomAD r.2.0.1 or dbSNP Build 150). Despite the extensive body of work on this enhancer, the majority (86%) of these rare variants have never been validated in vivo for their effect on gene expression, and only two have been experimentally demonstrated to cause abnormal limb development when introduced into the mouse genome (Lettice et al., 2014; Xu et al., 2019). Given this remarkable concentration of mutations implicated in both human limb malformations and vertebrate evolution, the ZRS enhancer provides a rich testbed for the systematic assessment of the effects of sequence variation on enhancer activity.
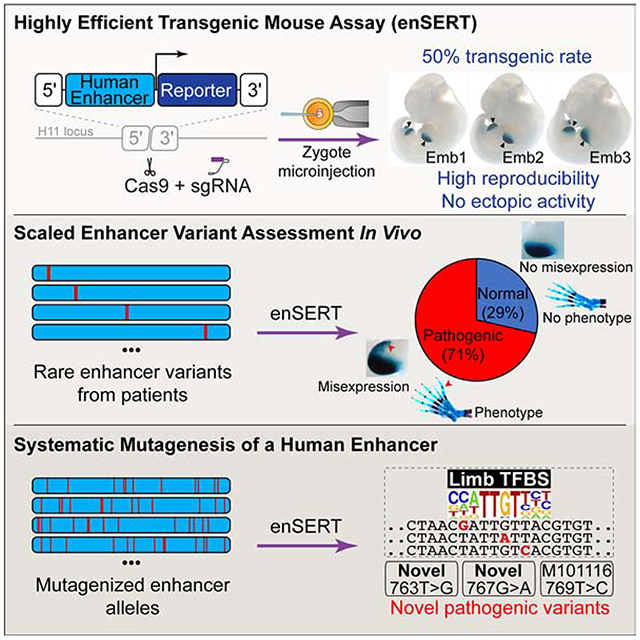
Figure 1: EnSERT allows large-scale and robust enhancer variant assessment.
(A) Overview of the method. The conventional enhancer-reporter assay relies on random integration of the transgene into the mouse genome upon zygote microinjection (top), which results in a low transgenic rate and limited reproducibility due to randomectopic effects. EnSERT (bottom) uses CRISPR/Cas9 to direct transgene integration to a specific genomic location, which results in higher reproducibility, no ectopic effects, and higher efficiency. Shown on the right are independently injected LacZ-stained embryos that resulted from random integration (top) or enSERT (bottom) of a transgene containing a human forebrain enhancer (hs200) or human limb enhancer (ZRS). Arrows denote reproducible enhancer activity at E11.5.
(B) The human ZRS limb enhancer of SHH is located approximately 1 Mb from its target promoter. When tested in a transgenic mouse reporter assay, ZRS activates reporter expression in the posterior margins of both fore- and hindlimb buds (ZPA) of E11.5 mouse embryos (left). Sequence variants in ZRS cause Shh misexpression in the anterior limb bud (middle), which results in polydactyly in multiple vertebrate species (right; shown here: polydactylous cat; photo by Jonna Austin).
(C) EnSERT reproducibly detects the anterior lacZ misexpression (red arrowheads) caused by the ‘Cuban’ allele of the human ZRS enhancer (see Figure S3A for details).Numbers of embryos with LacZ staining in the anterior limb bud (red) over the total number of transgenic embryos screened (black) are indicated. Only transgenic embryos that carried at least two copies of the reporter transgene at the H11 locus were considered in the analysis (see STAR methods for details). TFBS, transcription factor binding site.
In this study, we used enSERT to systematically mutagenize all nucleotides of the human ZRS enhancer (789 bp) either individually (80 variants) or in combination with other positions (67 compound constructs containing 16-40 mutations each). We first describe the functional assessment of all reported human ZRS enhancer mutations implicated in polydactyly, showing that 71% (15/21) drive abnormal reporter gene expression in the limb, which supports their pathogenicity. In contrast, 29% (6/21) of the published variants had no apparent impact on gene expression, raising the possibility that these are rare but potentially benign variants coincidentally present in polydactyly patients. We confirm these findings using knock-in mice carrying point mutations corresponding to the human variants, in which only mutations altering reporter gene expression as determined by enSERT result in abnormal limb development. We next explored this approach as a prospective functional screening tool after resequencing the ZRS in 61 cases of polydactyly of unknown genetic etiology. Of eight newly discovered rare variants in these individuals, we could functionally implicate three as likely pathogenic for polydactyly. Finally, we perform systematic mutagenesis of the ZRS enhancer and demonstrate that many nucleotides, spread across the entire length of the enhancer, are critical for its normal function. Notably, random mutagenesis uncovered novel gain-of-function mutations that have not yet been reported in human patients with polydactyly. We propose that preemptive in vivo systematic mutagenesis screens of disease-associated enhancers to establish supportive functional data will facilitate the interpretation of future human genetics findings.
Results
Scaled site-directed enhancer-reporter assay
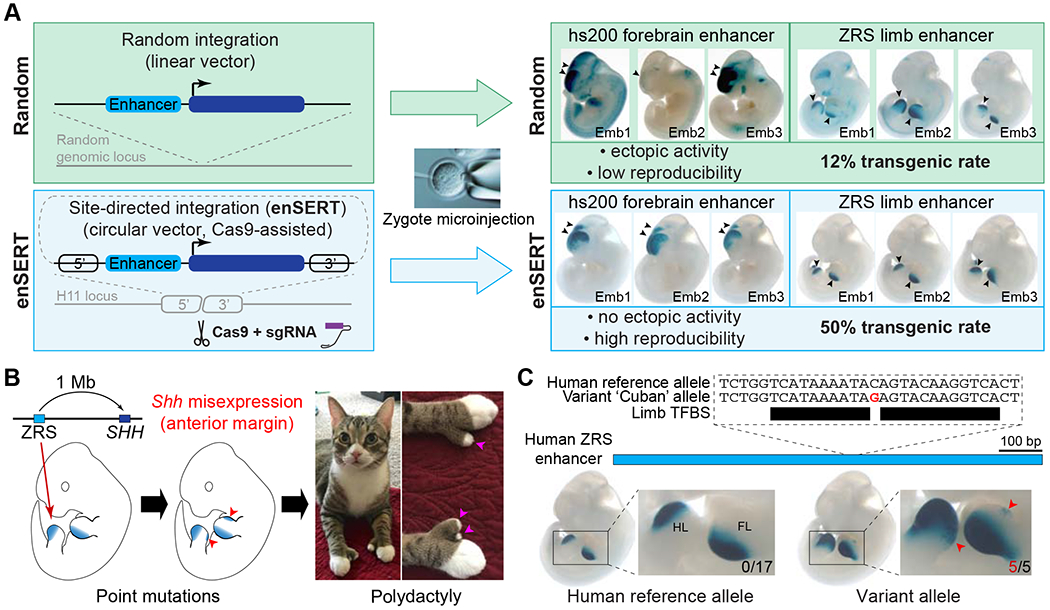
Conventional mouse enhancer reporter assays (Kothary et al., 1989; Pennacchio et al., 2006; Visel et al., 2007; Zákány, 1988) are based on random integration of a transgene into the mouse genome. While this method has been a gold standard for assessing the effects of human sequence variants on in vivo enhancer activity [e.g., (Fakhouri et al., 2014; Lettice et al., 2008; Turner et al., 2017)], it suffers from low efficiency of transgene integration and position effects that complicate the interpretation of experimental data. Thus, it requires generating a large number of transgenic animals and makes large-scale variant assessment prohibitively expensive. To overcome these limitations, we developed enSERT (enhancer inSERTion), a transgenic approach based on Cas9-mediated integration (Figure 1A). We identified a safe harbor integration site in the mouse genome that resulted in high CRISPR/Cas9-mediated homologous recombination (the H11 locus) (Tasic et al., 2011), as well as a minimal transgene promoter (from the Shh gene) with no background expression in this genomic location (Figures 1A and S1; STAR Methods). With enSERT, we achieved an average transgenic rate of 50% for transgenes as large as 11 kb (based on >1200 transgenic mice resulting from the injection of >150 independent transgenic constructs), compared to a 12% transgenic rate observed in conventional random transgenesis (based on >3000 independent transgenic constructs; Figures 1A and S1; STAR Methods) (Kothary et al., 1989; Pennacchio et al., 2006; Visel et al., 2007; Zákány, 1988). Moreover, due to the site-specific integration, enSERT results in more reproducible enhancer activity detection and is compatible with expression in all major mouse embryonic tissues (Figures 1A and S2A). In comparison with previous site-directed methods (Tasic et al., 2011), enSERT does not require maintaining a mouse strain with a ‘landing pad’ and, therefore, can be applied to any mouse strain of interest. The enSERT method overcomes the limitations of conventional enhancer-reporter transgenesis and enables a more than four-fold increase in throughput of enhancer assessment in vivo.
Robust in vivo assessment of human enhancer variants with enSERT
We first demonstrated the ability of enSERT to robustly detect the effects of single variants on enhancer activity by testing a previously characterized pathogenic human ZRS allele (Figure 1C) [referred to as the ‘Cuban’ variant in Ref. (Lettice et al., 2003)]. Towards this goal, we developed an enhancer variant scoring system that classifies the limb enhancer activity patterns from LacZ staining into five different categories: 1) complete loss of activity, 2) reduced activity, 3) normal activity, 4) a gain of activity in the anterior limb bud, and 5) strong gain of activity in the anterior limb bud. Scoring was done by multiple annotators blinded to genotype (see STAR Methods). As a control, we tested the reference human ZRS enhancer allele (789 bp) in parallel. For this reference allele, all examined embryos with site-specific integration of the transgene displayed normal activity at the region of the posterior margins of fore- and hindlimb buds (ZPA) where the Shh target gene is normally expressed, and none (0/17) displayed staining in the anterior margins of the limb buds (Figures 1C and S3A) (Lettice et al., 2003). For comparison, a conventional transgenic approach that relies on random transgene integration resulted in ZPA-specific activity in only 1/5 embryos, likely due to position effects (Figure S3B). We next introduced the Cuban variant into the human ZRS enhancer allele, and in all embryos examined by enSERT (5/5) we detected strong gain of enhancer activity in the anterior limb bud, consistent with a previously shown pathogenic role for this variant (Figures 1C and S3A) (Lettice et al., 2008). These results indicate that enSERT robustly and reproducibly assesses the impact of non-coding variants on enhancer activity in vivo.
Systematic assessment of rare non-coding variants in a human limb enhancer
To functionally assess and classify rare non-coding, putatively pathogenic variation within the ZRS enhancer, we compiled all published ZRS variants that have been implicated in polydactyly in humans and other species (Figure S2B and Table S1). In total, this represents a panel of 29 variants (21 from human and nine from other species, with one in common between human and mouse; Figure S2B and Table S1). We introduced each of these variants into the human ZRS enhancer and individually assessed their impact on limb enhancer activity using enSERT in mice. The majority of these enhancer variants (15/21, 71% of human variants; 5/9, 56% of orthologous variants from other species) showed reproducible gain of enhancer activity in the anterior limb bud in both hind- and forelimbs (Figure 2 and Table S2), consistent with a pathogenic role in polydactyly. In contrast, 29% (6/21) of the human variants previously proposed to be causal displayed normal enhancer activity that was indistinguishable from the reference ZRS sequence, suggesting that they may be benign (Figure 2, Table S2 and STAR Methods for more details on orthologous variants from other species).
Figure 2: Systematic assessment of all variants in the human ZRS enhancer.
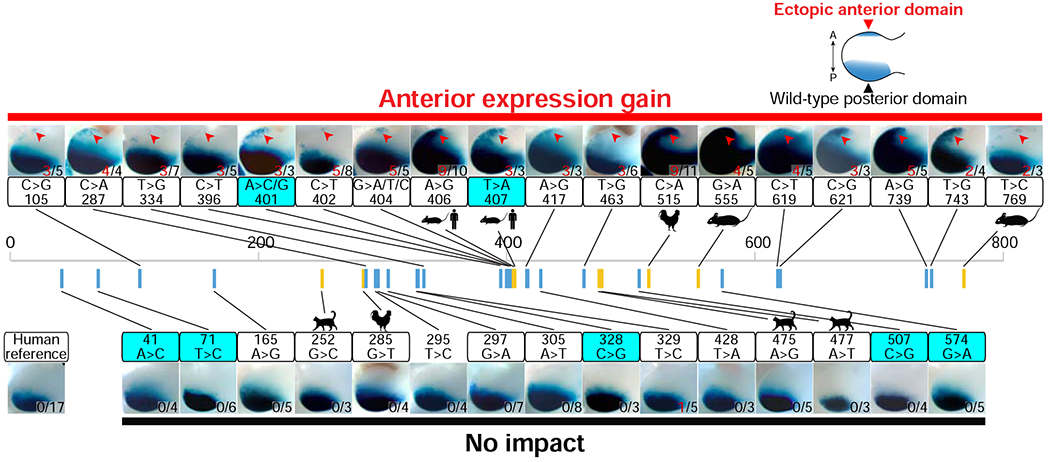
Enhancer activities for each of the 36 ZRS variant alleles implicated in preaxial polydactyly, including 21 human variants reported in the literature, nine ZRS mutations from other vertebrate species, and eight additional human variants identified in this study (cyan boxes). Shown are representative forelimb buds of transgenic E11.5 mouse embryos. Human ZRS enhancer (789 bp; chr7:156,791,087-156,791,875; hg38) variants are shown as blue bars, whereas variants discovered at orthologous positions in the ZRS enhancer of other species are shown as yellow bars. Red arrows indicate ectopic anterior LacZ staining. For positions with multiple reported variants, results for only one variant are shown (401A>C and 404G>A), but the respective other variants at the same position also show anterior expression gain (Table S2). Numbers of embryos with LacZ staining in the anterior limb bud (red) over the total number of transgenic embryos screened are indicated. See Table S2 for details.
Classifying newly discovered human ZRS variants linked to polydactyly
To assess the utility of this functional screen for interpreting clinically obtained genetic data, we resequenced the ZRS in patients with preaxial polydactyly, including a total of 61 unrelated individual probands or families. We identified 10 rare variants in the ZRS enhancer in these cases, eight of which were not previously reported (Figure 2 and Table S1). To assess these novel variants, we individually introduced each of them into the reference human ZRS sequence. Using enSERT, we observed that three of the eight variants (38%; 401A>C, 401A>G, and 407T>A) caused a gain of enhancer activity in the anterior limb bud that is consistent with a causal role in preaxial polydactyly. In contrast, the remaining five rare variants produced normal enhancer activity, suggesting that they are potentially benign (Figure 2). Upon further genetic analysis, we observed that 3 of these 5 putatively benign variants are found in public variant databases (dbSNP), albeit at rare frequencies (MAF≤0.02%), providing additional evidence against their role in human polydactyly. These data support the use of functional interrogation of rare human enhancer variants in mice to reveal their impact on gene expression, and the translational application of this data to aid in the interpretation of clinically generated genetic information.
Human variant knock-ins validate pathogenic and potentially benign variants.
To assess the extent to which the observed changes in enhancer activity affect limb morphology in vivo, we used CRISPR/Cas9 to generate a series of 11 knock-in mice where we introduced a human variant into the endogenous mouse ZRS enhancer (Figure 3A). We first individually introduced four of the variants (295T>C, 305A>T, 329A>C, and 297G>A) that did not cause misexpression of the reporter gene in the enSERT assay, and we observed that each resulted in normal limbs, further supporting that these variants may be benign (Figure 3B). In contrast, following the introduction of seven variants that caused misexpression of the reporter gene in the anterior portion of the limb bud (396C>T, 401A>G [newly reported in this study], 404G>A, 417A>G, 463C>G, 621C>G, and 739A>G), we observed five (396C>T, 401A>G, 404G>A, 417A>G, and 739A>G) that resulted in the formation of extra digits, reproducing phenotypes observed in humans with polydactyly and confirming that these variants are indeed pathogenic (Figure 3 and S4). In the two cases that did not result in polydactyly, the variants were in an area of increased sequence divergence between the human and mouse ZRS enhancers (621C>G; Figures S2B and S4) or displayed weaker and more variable reporter gene misexpression (463C>G; Figure S4 and Table S2). Taken together, these data support the power of enSERT for the scalable in vivo assessment of enhancer variants observed in patients.
Figure 3: Variant knock-in mice accurately reproduce human phenotypes.
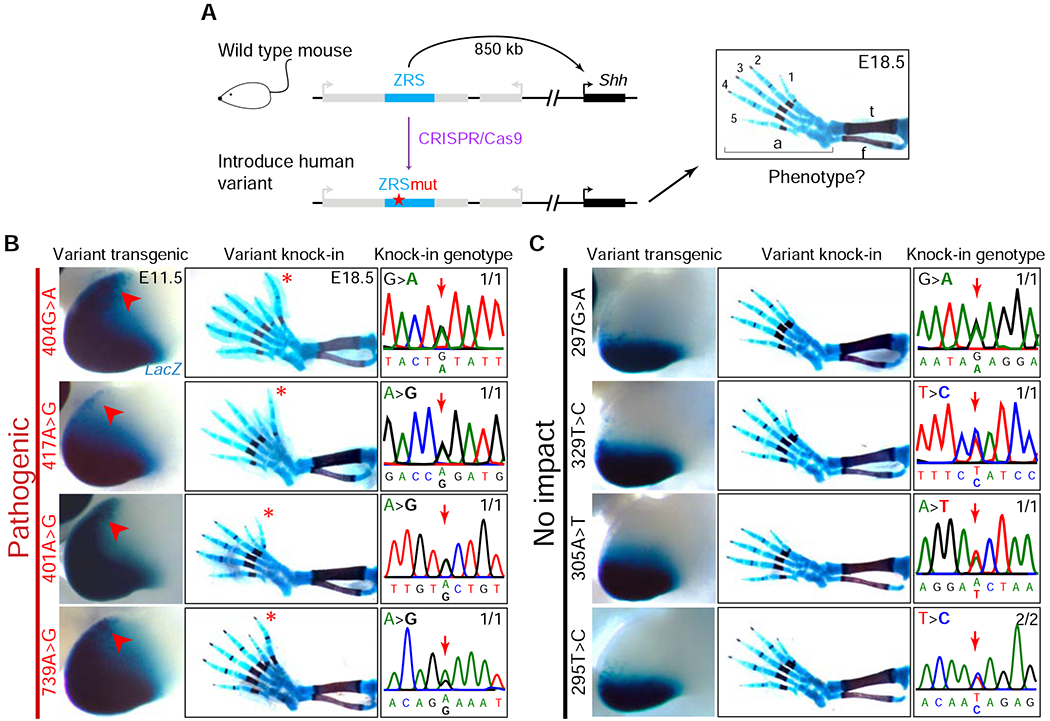
(A) CRISPR/Cas9-mediated human variant knock-in into the mouse ZRS enhancer. Schematic of the mouse Shh locus is shown (left, not to scale). The ZRS is located in intron five of the Lmbr1 gene (intron-exon structure not shown), 850 kb away from the promoter of Shh. A CRISPR/Cas9-modified mouse Shh locus with a human ZRS variant is shown below. A representative image of a wild-type E18.5 mouse hindlimb skeletal preparation, stained for bone (red) and cartilage (blue), is shown on the right; f, fibula; t, tibia; a, autopod; 1-5, digit numbers.
(B-C) LacZ staining in the hindlimbs of transgenic E11.5 mouse embryos containing human ZRS enhancer alleles (first column). Red arrows indicate ectopic LacZ staining in the anterior limb bud. Hindlimb skeletal preparations from E18.5 mice (second column), with genotyping sequence traces confirming the variant knock-ins at the endogenous mouse ZRS enhancer locus (third column) shown. Numbers indicate how many embryos exhibited the representative limb phenotype (B: polydactyly, C: wild-type) over the total number of embryos with the targeted genotype. Red asterisk: extra digit. See Figure S4 and STAR Methods for all knock-in mice and details.
Systematic mutagenesis of the human ZRS enhancer
To identify novel gain-of-function mutations in the human ZRS enhancer and to explore its functional robustness to sequence perturbation, we used enSERT to assess the general consequences of mutagenesis on ZRS activity in vivo. We systematically introduced point mutations in batches, where we changed either ~5% or ~2% of bases within the enhancer. We first designed 17 non-overlapping 5% mutation constructs (40 base pair substitutions per construct) that in combination cover all nucleotides of the ZRS enhancer, except for base pairs that overlapped or are immediately adjacent to variants implicated in polydactyly, which we tested separately (see below). In 82% of constructs (14/17), 5% mutagenesis completely abolished enhancer activity (Figure 4). To reduce the mutational load, we next designed 44 non-overlapping 2% mutation constructs (16 base pair substitutions per construct) covering the same set of base pairs. In 68% of cases, 2% mutagenesis either completely abolished enhancer activity (26%, 11/44) or weakened the enhancer activity (44%, 19/44), further supporting the fragile nature of this enhancer. In contrast, in 23% of cases (10/44), 2% mutagenesis did not cause any observable changes in ZRS enhancer activity, suggesting that these nucleotides are not essential for enhancer function. Finally, in 7% of mutant constructs (3/44), we observed a gain of lacZ reporter expression in the anterior limb buds, suggesting the presence of additional, yet to be identified, mutations that could cause polydactyly in humans (Figure 4).
Figure 4: Systematic mutagenesis of the ZRS enhancer.
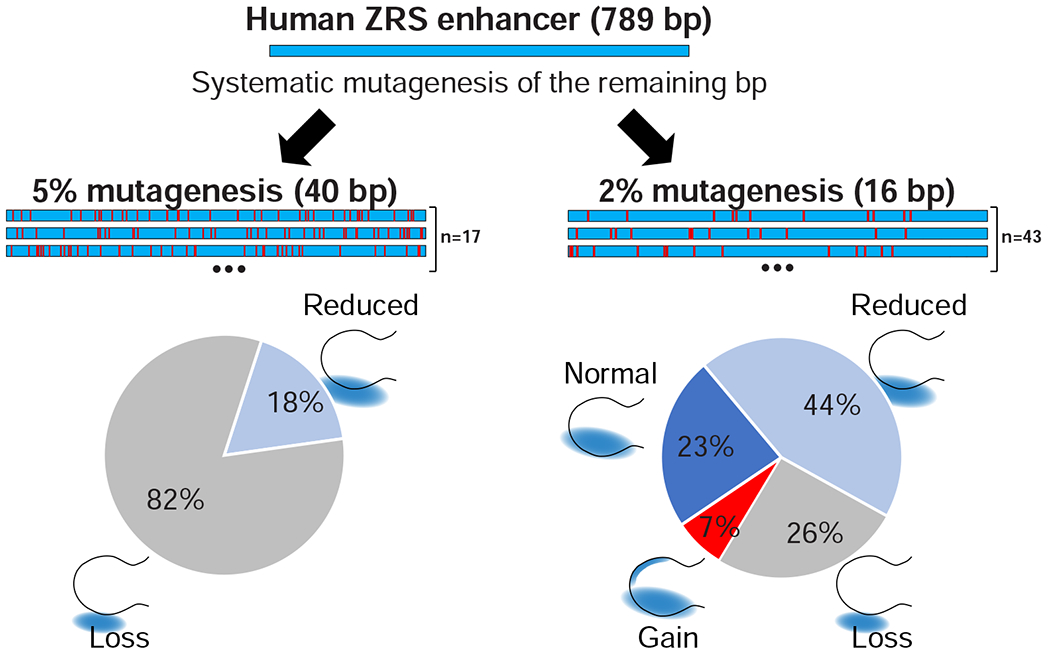
Enhancer mutagenesis strategy and results. Shown is the human ZRS enhancer in which we introduced either 40 base pair substitutions (left, 5% mutagenesis) or 16 base pair substitutions (right, 2% mutagenesis). The known variants from Figure 2 and nucleotides immediately neighboring them were excluded (Table S1). Pie charts below summarize results for all mutagenesis alleles tested in each of the groups. Schematic illustration of limb buds with corresponding LacZ staining is shown.
Identification of novel pathogenic variants that cause gain of enhancer function
To identify the exact mutation(s) causing the gain of function observed within the 2% mutagenized constructs, we selected one construct (hs2496.69) for further dissection. We individually introduced each of 16 variants from this allele into the human ZRS enhancer and tested them using enSERT (Figure 5A). We observed that 14/16 individually tested variants did not result in gain of enhancer activity (constructs hs2496.111-123; Figure 5A and Table S2). However, we found that each of the two remaining variants (765T>G [construct Hs2496.124] and 771T>C [Hs2496.125]; Figure 5A and Table S2) was individually sufficient to cause anterior misexpression of the ZRS, consistent with the pattern seen for the composite 2% mutagenesis (hs2496.69) construct (Figure 5A). Notably, both variants are located within seven base pairs of each other and reside near a previously characterized polydactyly mutation (769T>C, M101116). All three mutations appear to affect the same predicted SOX transcription factor binding site, which matches best to SOX5/SOX6/SOX9, all of which are crucial for limb development (Figure 5A) (Akiyama et al., 2002). This observed clustering of gain-of-function mutations raised the question of whether this is a general phenomenon. Indeed, when we generated compound mutant constructs in which 30 base pairs immediately adjacent to gain-of-function variants were mutated, we observed a strong gain of ZRS activity in the anterior limb (Figure S5B and Table S2). These data support that additional pathogenic variants are likely to be uncovered in polydactyly cases as resequencing is applied to increasing numbers of patients.
Figure 5: Identification of novel pathogenic variants.
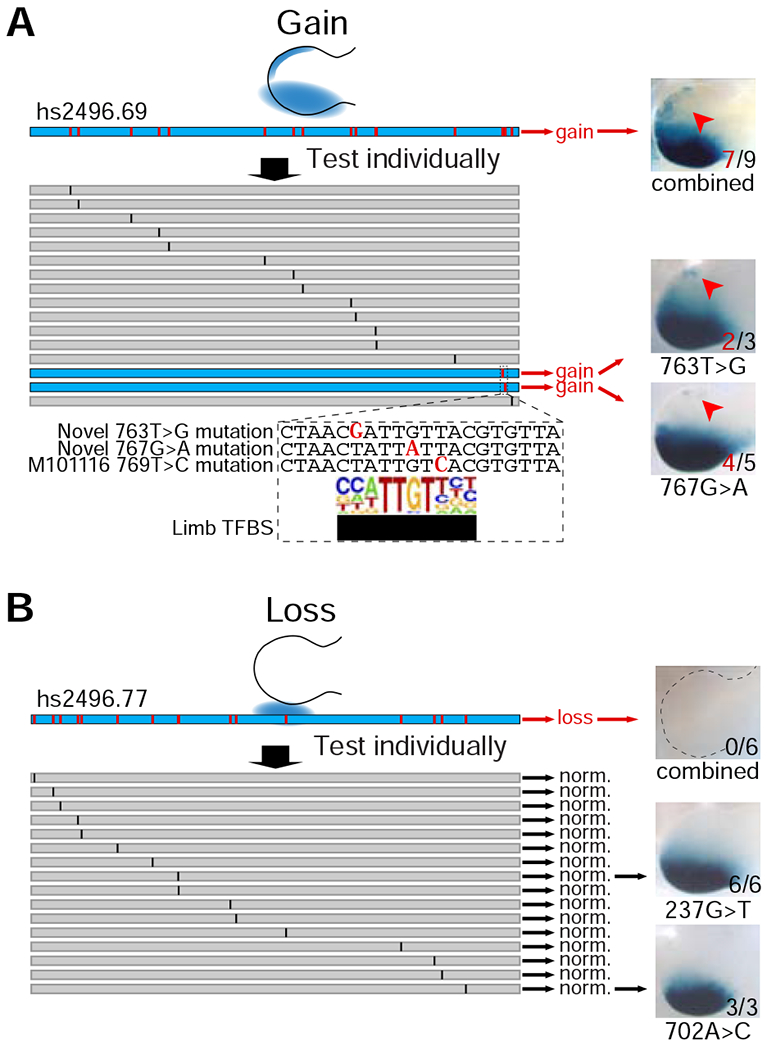
(A) Dissection of gain-of-function compound mutant. Representative LacZ-stained forelimbs of transgenic E11.5 mouse embryos (right) containing mutagenized human enhancer alleles (left) are shown. Red arrows indicate ectopic LacZ staining in the anterior portion of the limb bud. Numbers of embryos with LacZ staining in the anterior limb bud over the total number of transgenic embryos screened are indicated.
(B) Dissection of loss-of-function compound mutant. All individual variants resulted in normal enhancer activity. Two of 16 constructs with normal activity are shown as examples. Numbers of embryos with LacZ staining in the posterior margins of limb buds over the total number of transgenic embryos screened are indicated. See STAR Methods for details.
Discussion
Recent human genomics studies have revealed that most disease- and phenotype-associated variants discovered from genome-wide association studies do not affect protein-coding sequences but rather lie in non-coding genome. Additionally, whole genome sequencing of individuals is identifying a growing list of non-coding variants within the human population that could have a potential role in human biology (Albert and Kruglyak, 2015; Consortium et al., 2011). Therefore, reliably differentiating non-coding variants with and without phenotypic consequences represents a growing challenge. In the present study, we developed enSERT, with a focus on the robust and efficient in vivo assessment of human variants in distant-acting transcriptional enhancers. We used enSERT to systematically interrogate all published, as well as newly discovered unpublished, rare variants in the human ZRS enhancer found in patients with preaxial polydactyly. Our functional data show that 71% of published rare variants that were previously assumed to be causal result in ectopic in vivo enhancer activity. A majority of these gain-of-function variants that were knocked into the endogenous mouse enhancer result in polydactyly, thus further supporting their pathogenicity. In contrast, 29% of published rare variants fail to cause reporter misexpression in transgenic mice, or limb phenotypes in variant knock-in mice, suggesting that these variants may be benign incidental observations in cases whose preaxial polydactyly is caused by other environmental and/or genetic factors. Indeed, for the majority of novel cases of preaxial polydactyly that we examined in this study (80%, 51/61 families) no rare base pair variants in the ZRS enhancer were identified.
The human population exhibits an excess of extremely rare variants, leading to the recommendation that low variant frequency alone must be taken with caution as evidence for pathogenicity (Consortium et al., 2011; Keinan and Clark, 2012; Li et al., 2017). Our finding that nearly a third of reported ZRS variants are possibly benign reinforces this precaution and is comparable to the proportion of putative pathogenic coding variants identified in a large, genetically diverse, panel of sequenced exomes (Lek et al., 2016; Wright et al., 2019). Notably, variants demonstrated to be pathogenic in our functional assay, displayed higher levels of nucleotide conservation than apparently benign variants, suggesting that pathogenic nucleotides are subject to purifying selection (p-value <0.01 by Mann-Whitney test, Figure S5C). However, there is a substantial overlap in the ranges of conservation scores between the two variant classes, highlighting the difficulty of using conservation alone to distinguish between pathogenic and potentially benign variants (Figure S5C). Additionally, neither transcription factor motif analysis (Figure S5D) nor reported human genetics data (Table S1 and Figure S5E) could explain the observed difference between those alleles experimentally classified as pathogenic or potentially benign in this study. These data emphasize the importance of functional assays to aid in distinguishing rare non-coding variants that are likely pathogenic from those potentially benign.
The extensive number of variants reported for the ZRS enhancer suggests that it is a hotspot for gain-of-function pathogenic mutations and that it could potentially harbor many more polydactyly-causing variants that have not yet been identified in human patients (Hill and Lettice, 2013; Lettice et al., 2008; Sagai et al., 2004). Our random mutagenesis data indicates that there are, indeed, a number of novel gain-of-function mutations yet to be discovered through patient sequencing. Based on our results, we expect that there are approximately 38 total base pairs that cause an ectopic gain of enhancer activity in the entire human ZRS enhancer (see STAR Methods). However, these gain-of-function mutations account only for a small fraction (7%) of all ZRS base pairs that are deeply conserved between human and fish (Figure S2B) suggesting that gain-of-function mutations in enhancers may be rare, and that, in isolation, most point mutations in enhancers will likely have a subtle effect on enhancer function (Figure 5B).
In summary, our work illustrates the power of large-scale transgenesis to comprehensively interrogate how non-coding variants affect human biology and raises the intriguing possibility of performing preemptive in vivo saturation mutagenesis screens for disease-associated enhancers to interpret new human genetics findings.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact Len Pennacchio (LAPennacchio@lbl.gov). The PCR4-Shh::lacZ-H11 and PCR4-Hsp68::lacZ-H11 plasmids have been deposited to Addgene and are available at www.addgene.org (Addgene plasmids #139098 and #139098 respectively). All other vectors described in this study are freely available from the authors upon request. In addition, archived surplus transgenic embryos for many constructs can be made available upon request for complementary studies.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Model
All animal work was reviewed and approved by the Lawrence Berkeley National Laboratory Animal Welfare and Research Committee. All mice used in this study were housed at the Animal Care Facility (ACF) of LBNL. Mice were monitored daily for food and water intake, and animals were inspected weekly by the Chair of the Animal Welfare and Research Committee and the head of the animal facility in consultation with the veterinary staff. The LBNL ACF is accredited by the American Association for the Accreditation of Laboratory Animal Care International (AAALAC). Transgenic mouse assays and enhancer knock-ins were performed in Mus musculus FVB strain mice. The following developmental ages were used in this study: embryonic day E11.5, E12.5, E13.5, E14.5 and E18.5 mice. Animals of both sexes were used in the analysis. See method details for sample size selection and randomization strategies.
Patients
For detailed clinical phenotypes of unrelated cases with ZRS variants see Table S1. The human subjects committee at Washington University in St Louis approved this study (for data on human subjects that was collected in the USA). The analyses have been performed in a diagnostic basis (not research), in accordance with the bioethics rules of French law (for data on human subjects that was collected in France). All patients or, in the case of minors, their parents gave written consent for the study.
METHOD DETAILS
Embryo Microinjection
All transgenic and variant knock-in mice in this study were generated using a CRISPR/Cas9 microinjection protocol, as previously described (Kvon et al., 2016). Briefly, a mix of Cas9 protein (final concentration of 20 ng/ul; IDT Cat. No. 1074181), sgRNA (50 ng/ul) and donor plasmid (25 ng/ul) in injection buffer (10 mM Tris, pH 7.5; 0.1 mM EDTA) was injected into the pronucleus of FVB embryos. Female mice (CD-1 strain) were used as surrogate mothers. Super-ovulated female FVB mice (7-8 weeks old) were mated to FVB stud males, and fertilized embryos were collected from oviducts. The injected zygotes were cultured in M16 with amino acids at 37°C under 5% CO2 for approximately 2 hours. After that, zygotes were transferred into the uterus of pseudopregnant CD-1 females. F0 embryos were collected at E11.5 (for LacZ staining) or E18.5 (for skeletal preparations). LacZ staining and skeletal preparations were performed as previously described (Kvon et al., 2016). The procedures for generating transgenic and engineered mice were reviewed and approved by the Lawrence Berkeley National Laboratory (LBNL) Animal Welfare and Research Committee.
High-Throughput in vivo Enhancer Reporter Assay (enhancer inSERTion, enSERT)
Generation of enSERT mice using CRISPR/Cas9.
Transgenic mice carrying site-specific integration of the enhancer-reporter transgene were created using a modified CRISPR/Cas9 protocol. gRNAs targeting each of the integration sites (5qB1: 5′-gaaaagcatttagcag-3’; 14qE1: 5′-agacagccagcacgcttgtg-3′; H11: 5′-gctgatggaacaggtaacaa-3′) were designed using CHOPCHOP (Montague et al., 2014) and synthesized as previously described (Kvon et al., 2016) or ordered from IDT. To create a targeting vector containing the enhancer-reporter transgene, we first ordered chemically synthesized enhancer sequences flanked by homologous arms for Gibson cloning (for all ZRS enhancer variants; IDT) or PCR amplified enhancer sequences using primers with homology arm overhangs (for all other enhancers). All enhancer sequences and primers are available on the VISTA Enhancer Browser (https://enhancer.lbl.gov/). We then cloned each individual enhancer into the Hsp68::lacZ (hs200 enhancer only) (Pennacchio et al., 2006) or Shh::lacZ reporter vector (all other enhancers) flanked by homology arms to each of the three integration regions and incorporated it into the pCR4-TOPO (Thermo Fisher Scientific) backbone using Gibson (New England Biolabs [NEB]) cloning (Gibson et al., 2009) (Figure S1A). The map and the sequence of the PCR4-Shh::lacZ-H11 vector (with Shh promoter) is available at Addgene (Addgene plasmid #139098). After pronuclear microinjections (see above), the F0 embryos were harvested at embryonic days E11.5, E12.5, E13.5 or E14.5 and processed for LacZ staining. The embryos were genotyped by PCR and Sanger sequencing using primers 5′F and 5′R (for 5′ homology arm) and 3′F and 3′R (for 3′ homology arm).
Screening for a landing site with a high frequency of integration.
To develop enSERT, we first selected three landing sites in the mouse genome that contained previously integrated transgenes with no ectopic expression in mouse embryos (5qB1, 14qE1, and H11) (Ruf et al., 2011; Tasic et al., 2011). We designed sgRNAs targeting each of the three loci (Figure S1A). We first tested a sensor targeting vector containing the human hs200 forebrain enhancer (Pennacchio et al., 2006), along with the minimal Hsp68 promoter and lacZ reporter gene, at each of these integration sites. We chose the H11 locus for downstream studies because it had a high (65%) knock-in efficiency with 93% (28/30) of knock-in embryos showing LacZ staining in the forebrain (Figure S1, B and D).
Shh promoter is an optimal minimal promoter for enSERT.
In an initial test using a known forebrain enhancer (hs200) linked to the Hsp68 minimal promoter and lacZ gene, we observed the expected forebrain staining. However, we also observed unexpected but consistent background expression outside of the forebrain, including in the neural tube, heart, dorsal root ganglion, trigeminal and midbrain (Figure S1E). 25 out of 28 embryos (89%) that harbored hs200-Hsp68::lacZ inserted at the H11 locus displayed this unexpected ectopic activity. Because previous transgene insertions at the H11 locus did not display similar activity in E11.5 embryos (Tasic et al., 2011) we hypothesized that it was more likely to be caused by the hs200 enhancer or the Hsp68 promoter than gene regulatory elements in the vicinity of the H11 integration site. To determine if this staining was due to the enhancer or the Hsp68 promoter, we generated transgenic embryos harboring only the Hsp68::lacZ inserted at the H11 locus (same vector as before except with no enhancer). The ectopic expression persisted in these embryos, indicating that the background expression was not caused by the enhancer but by the promoter (Figure S1F). Small amounts of background expression (in neural tube) were already known for the Hsp68 promoter when it was used for random transgenesis, and this background activity was more pronounced and widespread in the H11 system (Figure S1F), necessitating the search for a new promoter. We, therefore, replaced the Hsp68 promoter with a minimal promoter of the Shh gene (mm10: chr5:28,466,764-28,467,284). Shh is a developmental gene tightly regulated by multiple tissue-specific enhancers that are active in various embryonic tissues and is, therefore in principle, expected to be suitable for enhancer analysis. Indeed, all embryos (seven out of seven) that contained Shh::lacZ (with no enhancer) inserted at H11 locus did not display any lacZ activity in E11.5 embryos (Figure S1G and S1F), while insertion of hs200-Shh::lacZ (with hs200 enhancer) at the H11 locus resulted in only the expected forebrain-specific expression (Figure S1G).
enSERT captures enhancer activities in all major embryonic tissues.
To test if a combination of a minimal Shh promoter and H11 integration site will support specific expression in various mouse tissues and for a majority of enhancers, we used enSERT to test a panel of human and mouse enhancers collectively active across all major embryonic tissues of the mid-gestation mouse embryo. This set included enhancers active in the heart (hs1760), dorsal root ganglion (hs215), neural tube (hs1043), tail 5 (hs1472), face (mm1917), branchial arch (hs2580), limb (ZRS and hs1473), trigeminal (hs215), forebrain (hs200), midbrain (hs2594), hindbrain (hs2597), and eye (hs1473) (see VISTA Enhancer Browser for details, https://enhancer.lbl.gov). All enhancers were active in the respective tissues, confirming that enSERT is compatible with expression analysis across different mouse tissues (Figure S2A).
Tandem integration at H11.
In ~50% of transgenic embryos, we observed integration of multiple copies of the enhancer-lacZ transgene at the H11 locus, which resulted in more robust lacZ reporter expression than the single-copy integrations. Multicopy integration did not result in an increase in the background activity (Figure S1F) and most likely resulted from the tandem insertion of the entire donor plasmid at the H11 locus, with both 5′ and 3′ homology arms integrated correctly (Figure S1C). To detect tandem integration at the H11 locus, we used the 3′F and 3′R H11 primers, along with the Tandem-F and Tandem-R primers that amplified part of the donor plasmid backbone (PCR4-TOPO). Embryos that were positive by PCR-based genotyping for the correctly targeted plasmid integration into H11 but negative for the donor plasmid backbone were presumed to harbor a single-copy transgene insertion at H11 locus (Figure S1A), while those positive for both the correctly targeted insertion AND the backbone sequence were assumed to be tandem integrations. For most enhancers that we tested at the H11 locus, single copy transgene integration was sufficient to drive robust lacZ activity in E11.5 mouse embryos (Figure S2A). However, a single copy of a transgene containing the ZRS enhancer drove weak lacZ activity that did not allow comparisons between different ZRS enhancer mutants. We therefore only compared ZRS enhancer activity between transgenic embryos with multiple copies of the transgene.
ZRS enhancer variant assessment in vivo using enSERT
The specificity of enSERT in detecting ZRS enhancer misexpression.
To assess the specificity of enSERT in detecting limb enhancer misexpression, we individually introduced 12 point mutations into the human ZRS enhancer that were outside of the known variants. All 12 mutations produced normal enhancer activity patterns that were indistinguishable from a reference allele, and none of the 12 mutations caused a gain of enhancer activity (constructs Hs2496.14, Hs2496.19, Hs2496.20, Hs2496.23-27, Hs2496.30, Hs2496.35, Hs2496.36, Hs2496.43; Figure S5A and Table S2). When we mutated base pairs that were immediately adjacent to potentially benign variants (from Figure 2), this resulted in normal enhancer activity, as well (constructs Hs2496.146 and Hs2496.147; Figure S5A and Table S2). These data support the specificity of enSERT in detecting ZRS enhancer misexpression in the anterior portion of the limb buds.
Introduction of ZRS mutations from other species into the human ZRS enhancer.
When we assessed human ZRS variants using enSERT in mice (Figure 2), we used the 789 bp human ZRS allele with the introduced variant to avoid potential effects caused by enhancer sequence divergence between human and mouse. To assess ZRS variants from other species and to test how ZRS sequence divergence affects variant assessment, we first introduced four previously characterized mouse mutations of spontaneous (Hemimelic extra-toes [Hx] mouse) (Knudsen and Kochhar, 1981) or mutagen-induced (M101116, M100081, and DZ mouse strains) (Masuya et al., 2007; Sagai et al., 2004; Zhao et al., 2009) origin; Figure S2B and Table S1) into the human ZRS enhancer sequence. All four orthologous mutations resulted in anterior lacZ reporter misexpression (Figure 2) when introduced into a human enhancer background, suggesting that humans harboring the orthologous mutations will likely have polydactyly. Indeed, patients who have heterozygous DZ (407T>A) and M100081 (406A>G) variants display severe polydactyly (Table S1) (Norbnop et al., 2014). We then introduced putative polydactyly mutations from cats (UK1, UK2, and Hemingway) and chicken (Silkiel and Silkie2) into the human ZRS enhancer (Figure S2B and Table S1). Of these five alleles, only the Silkie2 mutation caused anterior lacZ reporter misexpression, while variant human ZRS enhancers with orthologous Silkie1 or any of the orthologous cat mutations did not cause anterior lacZ reporter misexpression and displayed enhancer activity patterns that were indistinguishable from the reference allele (Figure 2). All three cat mutations are located in regions of significant sequence divergence between human, mouse, and cat, which could potentially explain why the cat variants are likely non-pathogenic when embedded into the sequence context of the human enhancer.
Estimating the total number of nucleotides that cause a gain of ZRS enhancer activity.
To estimate a total number of pathogenic gain-of-function nucleotide positions in the 789 bp ZRS enhancer, we first started with 18 nucleotide positions that overlapped pathogenic variants from our initial screen (Figure 2). We next added nucleotides that were immediately adjacent to pathogenic nucleotides. Since we tested them combined in two different batches, the number of additional pathogenic nucleotides from these mutation-adjacent positions is somewhere between two (lower estimate) and 30 (higher estimate) (Figure S5B). Finally, we estimated 14 more gain-of-function positions in the remaining 741 nucleotides based on the 7% frequency of gain-of-function 2% mutants in our systematic mutagenesis and considering positions that were potentially masked by loss-of-function 2% mutants (Figure 5A). This resulted in a total estimate of approximately 38 pathogenic nucleotides.
Identification of variants that cause loss of enhancer function.
To determine the sequence basis of the loss-of-function mutations, we selected a 2% mutant (hs2496.77) that resulted in complete abolishment of enhancer activity and individually introduced each of the variants into the human ZRS enhancer (Figure 4). We could not identify critical base pairs because all 16/16 mutants with individual variants displayed normal enhancer activity (constructs Hs2496.126-142; Figure 5B and Table S2). These results suggest potential redundancy between ZRS base pairs or a cumulative effect of small undetectable changes, with individual point mutations having little to no effect on enhancer activity, but multiple combined mutations having deleterious effects. This intra-enhancer robustness is similar to the redundancy observed between multiple individual highly conserved enhancers (Osterwalder et al., 2018).
Generation of variant knock-in mice using CRISPR/Cas9
Mouse strains carrying a human variant knocked into the ZRS enhancer were created using a CRISPR/Cas9 protocol (see Figure S4A for details). The sgRNA targeting the ZRS enhancer region was designed using CHOPCHOP (Montague et al., 2014) to position the guide target sequence outside the conserved ZRS core near its 5′ border (sgRNA recognition sequence was 5′- gaatgcatgcaggaactcagGGG -3′, where GGG is the PAM). To create a donor plasmid, a mouse ZRS enhancer with the corresponding human single nucleotide variant and mutagenized sgRNA recognition site (Figure S4A) was chemically synthesized (IDT), flanked by homology arms and incorporated into the pCR4-TOPO (Thermo Fisher Scientific) backbone using Gibson cloning as previously described (Kvon et al., 2016). After pronuclear microinjections, F0 mice were collected at embryonic day E18.5 and genotyped by PCR and Sanger sequencing using primers LF, LR, RF, and RR (see Figure S4A and Key Resources Table).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gibson Assembly® Master Mix | NEB | Cat# E2611S/L |
| Alcian blue 8GX | Sigma | A-3157 |
| Alizarin red S | Sigma | A-5533 |
| Alt-R® S.p. Cas9 Nuclease | IDT | 1081058 |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB | Charles River | http://www.criver.com/ |
| Mouse: FVB/Shh-ZRSem7Axvi (396C>T variant knock-in) | This paper | N/A |
| Sequence-Based Reagents | ||
| For primer sequences, please see STAR Methods | IDT | N/A |
| For chemically synthetized DNA of mutagenized ZRS enhancer alleles, please see Table S2 | IDT | N/A |
| For sgRNA sequences, please see STAR Methods | IDT | N/A |
| Recombinant DNA | ||
| PCR4-Shh::lacZ-H11 vector | This paper | Addgene #139098 |
| PCR4-Hsp68::lacZ-H11 vector | This paper | Addgene #139099 |
| PCR4-hs200-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs200-Hsp68::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs200-Hsp68::lacZ-5qB1 vector | This paper | N/A; available from the authors |
| PCR4-hs200-Hsp68::lacZ-14qE1 vector | This paper | N/A; available from the authors |
| PCR4-ZRS-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-ZRSmut-Shh::lacZ-H11 vectors. For a complete list of mutagenized ZRS sequences, please see Table S2 | This paper | N/A; available from the authors |
| PCR4-hs2594-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs2597-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs1043-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs1760-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs2580-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-mm1917-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs1473-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs1472-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| PCR4-hs215-Shh::lacZ-H11 vector | This paper | N/A; available from the authors |
| Primers | ||
| 5qB1 5’F primer (outside HA-5’): gccacaaagcaagagtgtcgaa | IDT | N/A |
| 5’R primer (inside hs200): gtggtgaagctttgtgtccgag | IDT | N/A |
| 3’F primer (inside SV40 poly(A)): cctccccctgaacctgaaacat | IDT | N/A |
| 5qB1 3’R primer (outside HA-3’): actggactgctgctatttccgt | IDT | N/A |
| 14qE1 5’F primer (outside HA-5’): agagacctcaggctaaaagttggt | IDT | N/A |
| 14qE1 3’R primer (outside HA-3’): ctgccgccatgtcgtcttttag | IDT | N/A |
| H11 5’F primer (outside HA-5’): acactaaggaaccctggctgtg | IDT | N/A |
| H11 3’R primer (outside HA-3’): ctacactcctcccacccagttg | IDT | N/A |
| Tandem-F primer (inside PCR4-TOPO backbone): tctgacgctcagtggaacgaaa | IDT | N/A |
| Tandem-R primer (inside PCR4-TOPO backbone): agactgggcggttttatggaca | IDT | N/A |
| LF primer (outside HA-L): ggtagaggccaggaagtcg | IDT | N/A |
| LR primer (inside HA-R)*: gaCAtgtCaGtagtcGctcaGa | IDT | N/A |
| RF primer (inside HA-L)*: atcagatGtTCtGtgtaCgtGacc | IDT | N/A |
| RR primer (outside HA-R): gtcatttcaactttcttatttcagtata | IDT | N/A |
| Software and Algorithms | ||
| CHOPCHOP | (Montague et al., 2014) | https://chopchop.rc.fas.harvard.edu/ |
| MAFFT | (Katoh and Standley, 2013) | http://mafft.cbrc.jp/alignment/software/ |
Uppercase letters highlight nucleotides that were changed in the donor plasmid to allow for distinguishing between the knock-in and unmodified versions of the mouse ZRS locus.
Skeletal preparations
Skeletal preparations were performed as previously described (Kvon et al., 2016) according to a standard Alcian blue/Alizarin red protocol (Ovchinnikov, 2009). The stained embryos were dissected in 80% glycerol and limbs were imaged at 1x using a Leica MZ16 microscope and a Leica DFC420 digital camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
ZRS enhancer activity pattern scoring
The stained transgenic embryos were imaged from both sides using a Leica MZ16 microscope and Leica DFC420 digital camera. We only considered transgenic embryos that contained tandem integration of the transgene at H11 locus, as single integration of ZRS-lacZ transgene did not produce sufficiently robust staining in the limb buds (see above for details). A total of 1243 embryo images for all tested alleles of the human ZRS enhancer were shuffled and their labels removed for scoring. Annotation was performed by five independent reviewers blinded to the ZRS allele genotype. The reviewers classified each image to one of the following enhancer activity patterns in the limb buds: 1) lost, 2) reduced, 3) normal (i.e., indistinguishable from the reference allele), 4) gain (i.e., staining was present in the most anterior portion of the limb buds), and 5) strong gain (i.e., strong staining was present in the most anterior portion of the limb buds) (Figure S3C). Both sides of the embryo were annotated for most embryos. Final annotations for each transgenic embryo were determined by the staining type with the most reviewer votes and by vote consistency between the left and the right limb buds. An allele was classified as altering ZRS activity if it resulted in a statistically significant increase in transgenic embryos with activity patterns that deviated from normal (i.e., activity loss or gain) compared to the reference human ZRS enhancer allele (Fisher’s exact test p-value < 0.05, Figure S3C and S3D and Table S2).
Sample Selection and Blinding
Sample sizes for transgenic assays were selected empirically based on our previous experience of performing transgenic mouse assays for >4,000 total putative enhancers (Attanasio et al., 2013; Blow et al., 2010; May et al., 2012; Pennacchio et al., 2006; Visel et al., 2007; 2009a). Mouse embryos were excluded from further analysis if they did not carry the reporter transgene at the H11 locus, contained only a single copy of the reporter or contained ectopic staining outside the limb (suggesting random integration). All transgenic mice were treated with identical experimental conditions. Randomization and experimenter blinding were performed during the scoring of embryo images (see above).
Supplementary Material
Figure S1. Highly-efficient site-specific mouse transgenesis using enhancer inSERTion (enSERT), Related to Figure 1
(A) Schematic overview of the strategy. The donor targeting vector contained two homology arms (grey, indicated as HA-5′ and HA-3′) and a corresponding enhancer-reporter transgene with an enhancer (light blue), minimal promoter (brown), lacZ reporter (dark blue), and SV40 poly(A) sequence. The sgRNA recognition site is indicated in purple. PCR primers used for genotyping are shown as arrows (5′F, 3′R-outside of the homology arms; 5′R, 3′F - transgene specific). See STAR Methods for more details.
(B) Table showing integration efficiencies, enhancer expression, and coordinates of three different landing sites that we targeted with a forebrain enhancer-reporter transgene using the strategy shown in (A).
(C) PCR genotyping analysis of F0 transgenic E11.5 mice using primer pairs 5′F/5′R and 3′F/3′R to confirm the correct integration of the 5′ (HA-5′) and the 3′ (HA-3′) homology arms at H11 locus, respectively. Numbers indicate independent mice. Mice with lacZ reporter staining in forebrain are indicated in blue.
(D) 48-well plate with LacZ-stained E11.5 embryos in which the forebrain enhancer lacZ reporter was targeted to the H11 locus.
(E) LacZ-stained E11.5 embryos with the forebrain enhancer and Hsp68 minimal promoter driving lacZ reporter expression from the H11 locus. Red arrows point to ectopic activity caused by the Hsp68 promoter.
(F) Comparison of background activities between the Hsp68 and Shh promoters at the H11 locus. The Hsp68 promoter on its own (without enhancer) drives lacZ reporter expression around the neural tube, heart, trigeminal nerve, and the head of the E11.5 embryo. The Shh promoter (without enhancer) does not drive detectable lacZ reporter expression in E11.5, E12.5, E13.5, or E14.5 embryos.
(G) LacZ-stained E11.5 embryos with the forebrain enhancer and Shh promoter driving lacZ reporter gene expression from the H11 locus. Specific enhancer-driven staining in the forebrain, with no background LacZ staining, is observed.
Figure S2, Related to Figure 1
(A) EnSERT captures in vivo enhancer activities in all major mouse embryonic tissues. Shown are representative LacZ-stained embryos with the enhancer reporter construct integrated at the H11 locus. In vivo enhancer activities at E11.5 correspond to 10 different tissue-specific enhancers (nine human and one mouse enhancers; see also VISTA Enhancer Browser: https://enhancer.lbl.gov/).
(B) Position and evolutionary conservation of published and newly reported ZRS variants. Shown is the human ZRS enhancer (789 bp) aligned with the orthologous sequences from five different vertebrate species, including cartilaginous and bony fishes (elephant shark and coelacanth, respectively), chicken, cat, and mouse. Human mutations are shown in blue boxes; mouse mutations are shown in pink boxes; cat and chicken mutations are shown in yellow boxes. * Novel ZRS variants and families reported in this study.
Figure S3. Highly reproducible single nucleotide enhancer variant assessment using enSERT, Related to Figure 2
(A) EnSERT is able to reproducibly detect anterior lacZ misexpression upon the introduction of the ‘Cuban’ variant into the human ZRS enhancer. Shown are independently injected LacZ-stained mouse embryos with transgene integration at the H11 locus showing the activity of the human ZRS reference allele (left) or ‘Cuban’ allele. Red arrowheads indicate anterior LacZ staining caused by the ‘Cuban’ variant.
(B) Random transgenesis results in ectopic staining and low reproducibility (data from (Kvon et al., 2016)). Shown are independently injected LacZ-stained mouse embryos with random transgene integration showing the activity of the ZRS human reference allele. Red arrowheads indicate ectopic anterior LacZ staining.
(C) ZRS enhancer activity pattern scoring. ZRS limb enhancer activity patterns from LacZ staining were classified into five different categories: 1) complete loss of activity, 2) reduced activity, 3) normal activity, 4) a gain of activity in the anterior limb bud, and 5) strong gain of activity in the anterior limb bud. Scoring was done independently at least five annotators blinded to genotype. See STAR Methods for more details.
(D) Bar chart showing the scoring summary for each of the alleles (x-axis). The y-axis shows the percentage of analyzed transgenic embryos for each allele that were annotated in each category (bottom). Alleles on x-axis were sorted by their final categorization (top). See Table S2 and STAR Methods for more details.
Figure S4. Limb phenotypes of knock-in mice with human variants, Related to Figure 3
(A) Schematic overview of the human variant knock-in strategy. A 4.5 kb mouse genomic region containing the ZRS enhancer (light blue) is shown together with the vertebrate phyloP conservation (dark blue). The donor vector contained two homology arms (gray, labeled HA-L and HA-R) with vector-specific sequences for genotyping (green) and a corresponding replaced region (blue) containing a human variant (red) and mutagenised sgRNA recognition site (5′-agtaccatgcgtgtgtTtTagCC-3′) but otherwise identical to the mouse reference ZRS sequence. The sgRNA recognition site is indicated in purple. PCR primers used for genotyping are shown as arrows (LF, RR - mouse-specific, outside of the homology arms; RF, LR - donor vector-specific). See STAR Methods for more details.
(B) Forelimb and hindlimb skeletal preparations from E18.5 mice, with genotyping sequence traces confirming the variant knock-ins at the endogenous mouse ZRS enhancer (third column) shown. Numbers indicate how many embryos exhibited the representative limb phenotype. Skeletal preparations for 396C>T mice were prepared from heterozygous E18.5 mice from an established breeding line. All other skeletal preparations were prepared from F0 E18.5 mice. One variant (621C>G) that displayed strong reporter gene misexpression but resulted in normal limbs upon variant introduction into the endogenous mouse enhancer was located within a subregion of increased sequence divergence between humans and mice, which may explain the inconsistency between the human enhancer enSERT results and the mouse enhancer knock-in phenotype (Figure S2B). Another variant (463C>G) that displayed gain of enhancer activity but resulted in normal limbs upon introduction in the mouse genome caused weaker and more variable reporter gene misexpression, which may explain the absence of polydactyly in the F0 knock-in mice (table S2).
(C) Forelimbs and hindlimbs from E18.5 mice homozygous for the 396C>T variant knock-in (ZRS396T/ZRS396T), along with genotyping results (third column). Numbers indicate how many mice exhibited the representative limb phenotype out of the total number of ZRS396T/ZRS396T E18.5 mice screened.
*Extra digit.
Figure S5. Variant assessment in the ZRS enhancer, Related to Figure 4
(A) Enhancer activities for human ZRS alleles that contain point mutations (black bars) in highly conserved TF binding sites outside of the known variants in the forelimb buds of transgenic e11.5 mouse embryos. Numbers of embryos with lacZ activity in the anterior limb bud (red) over the total number of transgenic embryos screened (black) are indicated.
(B) Mutagenesis of all pathogenic variants (all gain combined), nucleotides immediately adjacent to pathogenic variants (all +1 positions combined [Hs2496.144] and all −1 positions combined [Hs2496.145]) and nucleotides that are immediately adjacent to human variants that were classified as potentially benign by this study (all +1 positions combined [Hs2496.146] and all −1 positions combined [Hs2496.147]). Shown are enhancer activities for each of the constructs and the reference human allele in the forelimb buds of transgenic E11.5 mouse embryos. Numbers of embryos with LacZ staining in the anterior limb bud (red) over the total number of transgenic embryos screened (black) are indicated. Red arrowheads indicate ectopic anterior LacZ staining.
(C) Comparison of evolutionary sequence conservation (based on PhyloP score for 46 vertebrates) for potentially benign, gain-of-function (GoF), and common variants within the human ZRS enhancer. * p-value (by Mann-Whitney test) <0.01. ** p-value <0.001. n.s. - not significant. See Table S1 for PhyloP scores for each of the variants.
(D) P-value (TF motif match) change for potentially benign, gain-of-function (GoF), and common variants within the human ZRS enhancer that overlap predicted TF binding sites. See Table S1 for P-value scores for each of the variants.
(E) Comparison of human genetic and clinical data for pathogenic and potentially benign ZRS variants. Shown are all human ZRS variants tested in this study (columns) and the supporting human genetics data and activity in a transgenic reporter assay (rows). Variants that caused a gain of expression in anterior limb bud are highlighted in red. Novel variants reported in this study are highlighted in blue boxes. See Table S1 for details. TFBS, transcription factor binding site; cntrl, control.
Table S1, Related to Figure 2 [see separate Excel file]
All ZRS variants tested in this study, including previously published human variants (Al-Qattan et al., 2012; Albuisson et al., 2011; Baas et al., 2017; Cai et al., 2019; Cho et al., 2013; Farooq et al., 2010; Furniss et al., 2008; Girisha et al., 2014; Gurnett et al., 2007; Heutink et al., 1994; Lettice et al., 2003; 2008; Lodder, 2009; Norbnop et al., 2014; Semerci et al., 2009; VanderMeer et al., 2012; 2014; Vanlerberghe et al., 2015; Wieczorek et al., 2010; Wu et al., 2016; Zguricas et al., 1999; Zhang et al., 2019), variants from animals (Dorshorst et al., 2010; Dunn et al., 2011; Knudsen and Kochhar, 1981; Lettice et al., 2008; Masuya et al., 2007; Zhao et al., 2009) and novel variants reported in this study.
Table S1 contains the following columns: variant coordinate in GRCh38 genome assembly (1st column), relative position within the ZRS enhancer (2nd column), reference and variant alleles (3rd column), variant name (4th column), organism of origin (5th column), VISTA ID of the tested construct containing the human ZRS enhancer with the variant (6th column), variant classification based on the enSERT result (7th column), reference (8-9th columns), clinical phenotypes and human genetics data for variants from human patients (10-24th columns), PhyloP scores (25th column), predicted TF binding sites (26th column),and corresponding P-values (TF motif match scores) for reference and variant alleles (27th and 28th columns). Note that variants in the ZRS enhancer from other species are shown at orthologous positions in the human ZRS enhancer.
Table S2, Related to Figure 4 [see separate Excel file]
All ZRS enhancer mutants tested in this study together with the summary of the mouse transgenic reporter results.
Table S2 contains the following columns: VISTA ID of the tested transgenic construct containing mutagenized ZRS enhancer (1st column), type of introduced mutation(s) (2nd column), relative position within the ZRS enhancer (for point mutations and variants only; 3nd column), reference and variant alleles (for point mutations and variants only; 4rd column), sequence of mutagenized ZRS enhancer (5rd column), numbers of embryos per transgenic construct that displayed complete loss of enhancer activity in the limb buds (6th column), reduced activity (7th column), normal activity (8th column), gain of enhancer activity in the anterior margin of limb buds (9th column), strong gain of enhancer activity (10thcolumn), p-values by Fisher’s exact test (for loss [11th column] and 20 gain [12th column]), and final annotation based on results in all embryos (13th column).
Highlights.
enSERT– a highly efficient CRISPR/Cas9-mediated site-specific transgenic mouse assay
In vivo assessment of all rare variants linked to polydactyly in a human enhancer
In vivo testing showed normal enhancer activity for 30% of presumed pathogenic variants
Systematic mutagenesis of this human enhancer identifies novel pathogenic Variants
Acknowledgments
The authors would like to acknowledge Muriel Holder-Espinasse (Guy’s Hospital, London), Ghislaine Plessis (CHU Caen), Alain Verloes (CHU Paris), and Carine Abel (Hopital de la Croix Rousse, Lyon), the French reference centers for developmental anomalies and ERN-ITHACA for patient recruitment. The authors also thank Valentina Snetkova and Marco Osterwalder for help with enhancer pattern annotation and J. Omar Yanez-Cuna for help with designing mutant enhancer alleles. The authors would also like to thank Jonna Austin for photo permissions.
Funding: This work was supported by National Institutes of Health grants R01HG003988 (to L.A.P.), 5K99HG009682 (to E.Z.K.), and R01AR067715 (to C.A.G and M.B.D). E.Z.K. was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation funded by the Howard Hughes Medical Institute. The research was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231, University of California. IO and SH were supported by the Intramural Research Program of the National Library of Medicine at the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
Images of whole-mount-stained embryos and are available online (http://enhancer-staging.lbl.gov:2002/index.php/s/JMBTXbJ6MKgt4f5).
References
- Akiyama H, Chaboissier MC, Martin JF, Schedl A, and De Crombrugghe B (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes & Development 16, 2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qattan MM, Abdulkareem, Al I, Haidan, Al Y, and Balwi, Al M. (2012). A novel mutation in the SHHlong-range regulator (ZRS) is associated with preaxial polydactyly, triphalangeal thumb, and severe radial ray deficiency. Am. J. Med. Genet. A 158A, 2610–2615. [DOI] [PubMed] [Google Scholar]
- Albert FW, and Kruglyak L (2015). The role of regulatory variation in complex traits and disease. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- Albuisson J, Isidor B, Giraud M, Pichon O, Marsaud T, David A, Le Caignec C, and Bezieau S (2011). Identification of two novel mutations in Shh long-range regulator associated with familial preaxial polydactyly. Clin. Genet. 79, 371–377. [DOI] [PubMed] [Google Scholar]
- Attanasio C, Nord AS, Zhu Y, Blow MJ, Li Z, Liberton DK, Morrison H, Plajzer-Frick I, Holt A, Hosseini R, et al. (2013). Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas M, Potuijt JWP, Hovius SER, Hoogeboom AJM, Galjaard R-JH, and van Nieuwenhoven CA (2017). Intrafamilial variability of the triphalangeal thumb phenotype in a Dutch population: Evidence for phenotypic progression over generations? Am. J. Med. Genet. A 173, 2898–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Mcculley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2010). ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Ma J, Pan R, Wang C, Li W, Cai C, Lin S, and Shu J (2019). Genetic analysis of one family with congenital limb malformations. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36, 890–892. [DOI] [PubMed] [Google Scholar]
- Cho T-J, Baek GH, Lee H-R, Moon HJ, Yoo WJ, and Choi IH (2013). Tibial hemimelia-polydactyly-five-fingered hand syndrome associated with a 404 G>A mutation in a distant sonic hedgehog cis-regulator (ZRS). Journal of Pediatric Orthopaedics B 22, 219–221. [DOI] [PubMed] [Google Scholar]
- Consortium, T.1.G.P., author, C., committee, S., Medicine, P.G.B.C.O., Max Planck Institute for Molecular Genetics, Science, R.A., Technologies, A.G.A., Medicine, B.C.O., BGI-Shenzhen, College, B., et al. (2011). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorshorst B, Okimoto R, and Ashwell C (2010). Genomic Regions Associated with Dermal Hyperpigmentation, Polydactyly and Other Morphological Traits in the Silkie Chicken. Journal of Heredity 101, 339–350. [DOI] [PubMed] [Google Scholar]
- Dunn IC, Paton IR, Clelland AK, Sebastian S, Johnson EJ, McTeir L, Windsor D, Sherman A, Sang H, Burt DW, et al. (2011). The chicken polydactyly (Po) locus causes allelic imbalance and ectopic expression of Shh during limb development. Dev. Dyn. 240, 1163–1172. [DOI] [PubMed] [Google Scholar]
- Fakhouri WD, Rahimov F, Attanasio C, Kouwenhoven EN, Ferreira De Lima RL, Felix TM, Nitschke L, Huver D, Barrons J, Kousa YA, et al. (2014). An etiologic regulatory mutation in IRF6 with loss- and gain-of-function effects. Hum Mol Genet 23, 2711–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq M, Troelsen JT, Boyd M, Eiberg H, Hansen L, Hussain MS, Rehman SU, Azhar A, Ali A, Bakhtiar SM, et al. (2010). Preaxial polydactyly/triphalangeal thumb is associated with changed transcription factor-binding affinity in a family with a novel point mutation in the long-range cis-regulatory element ZRS. Eur. J. Hum. Genet. 18, 733–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EEM, and Levine MS (2018). Developmental enhancers and chromosome topology. Science 361, 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furniss D, Lettice LA, Taylor IB, Critchley PS, Giele H, Hill RE, and Wilkie AOM (2008). A variant in the sonic hedgehog regulatory sequence (ZRS) is associated with triphalangeal thumb and deregulates expression in the developing limb. Hum Mol Genet 17, 2417–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Girisha KM, Bidchol AM, Kamath PS, Shah KH, Mortier GR, Mundlos S, and Shah H (2014). A novel mutation (g.106737G>T) in zone of polarizing activity regulatory sequence (ZRS) causes variable limb phenotypes in Werner mesomelia. Am. J. Med. Genet. A 164, 898–906. [DOI] [PubMed] [Google Scholar]
- Gurnett CA, Bowcock AM, Dietz FR, Morcuende JA, Murray JC, and Dobbs MB (2007). Two novel point mutations in the long-range SHH enhancer in three families with triphalangeal thumb and preaxial polydactyly. Am. J. Med. Genet. A 143A, 27–32. [DOI] [PubMed] [Google Scholar]
- Heutink P, Zguricas J, Vanoosterhout L, Breedveld GJ, Testers L, Sandkuijl LA, Snijders P, Weissenbach J, Lindhout D, Hovius S, et al. (1994). The Gene for Triphalangeal Thumb Maps to the Subtelomeric Region of Chromosome 7q. Nat Genet 6, 287–292. [DOI] [PubMed] [Google Scholar]
- Hill RE, and Lettice LA (2013). Alterations to the remote control of Shh gene expression cause congenital abnormalities. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 368, 20120357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue F, and Ahituv N (2015). Decoding enhancers using massively parallel reporter assays. Genomics 106, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, and Clark AG (2012). Recent Explosive Human Population Growth Has Resulted in an Excess of Rare Genetic Variants. Science 336, 740–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, and Ahituv N (2019). Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat Commun 10, 1478–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen TB, and Kochhar DM (1981). The role of morphogenetic cell death during abnormal limb-bud outgrowth in mice heterozygous for the dominant mutation Hemimelia-extra toe (Hmx). J Embryol Exp Morphol 65 Suppl, 289–307. [PubMed] [Google Scholar]
- Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, and Rossant J (1989). Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707–714. [DOI] [PubMed] [Google Scholar]
- Kvon EZ (2015). Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics 106, 185–192. [DOI] [PubMed] [Google Scholar]
- Kvon EZ, Kamneva OK, Melo US, Barozzi I, Osterwalder M, Mannion BJ, Tissieres V, Pickle CS, Plajzer-Frick I, Lee EA, et al. (2016). Progressive Loss of Function in a Limb Enhancer during Snake Evolution. Cell 167, 633–642. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal F, and Cohn MJ (2016). Loss and Re-emergence of Legs in Snakes by Modular Evolution of Sonic hedgehog and HOXD Enhancers. Curr Biol. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letelier J, la Calle-Mustienes de, E., Pieretti J, Naranjo S, Maeso I, Nakamura T, Pascual-Anaya J, Shubin NH, Schneider I, Martinez-Morales JR, et al. (2018). A conserved Shh cis-regulatory module highlights a common developmental origin of unpaired and paired fins. Nat Genet 50, 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJH, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, and de Graaff E (2003). A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12, 1725–1735. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Hill AE, Devenney PS, and Hill RE (2008). Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum Mol Genet 17, 978–985. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Williamson I, Devenney PS, Kilanowski F, Dorin J, and Hill RE (2014). Development of five digits is controlled by a bipartite long-range cis-regulator. Development 141, 1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, Hess GT, Zappala Z, Strober BJ, Scott AJ, et al. (2017). The impact of rare variation on gene expression across tissues. Nature 550, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder E (2009). Keeping Sonic Hedgehog Under the Thumb: Genetic Regulation of Limb Development. [Google Scholar]
- Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuya H, Sezutsu H, Sakuraba Y, Sagai T, Hosoya M, Kaneda H, Miura I, Kobayashi K, Sumiyama K, Shimizu A, et al. (2007). A series of ENU-induced single-base substitutions in a long-range cis-element altering Sonic hedgehog expression in the developing mouse limb bud. Genomics 89, 207–214. [DOI] [PubMed] [Google Scholar]
- May D, Blow MJ, Kaplan T, Mcculley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. (2012). Large-scale discovery of enhancers from human heart tissue. Nat Genet 44, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, and Valen E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research 42, W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbnop P, Srichomthong C, Suphapeetiporn K, and Shotelersuk V (2014). ZRS 406A>G mutation in patients with tibial hypoplasia, polydactyly and triphalangeal first fingers. J Hum Genet 59, 467–470. [DOI] [PubMed] [Google Scholar]
- Osterwalder M, Barozzi I, Tissieres V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 489, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov D (2009). Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb Protoc 2009, pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. (2006). In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502. [DOI] [PubMed] [Google Scholar]
- Ruf S, Symmons O, Uslu VV, Dolle D, Hot C, Ettwiller L, and Spitz F (2011). Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat Genet 43, 379–386. [DOI] [PubMed] [Google Scholar]
- Sagai T, Hosoya M, Mizushina Y, Tamura M, and Shiroishi T (2005). Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 132, 797–803. [DOI] [PubMed] [Google Scholar]
- Sagai T, Masuya H, Tamura M, Shimizu K, Yada Y, Wakana S, Gondo Y, Noda T, and Shiroishi T (2004). Phylogenetic conservation of a limb-specific, cis-acting regulator of Sonic hedgehog ( Shh). Mamm. Genome 15, 23–34. [DOI] [PubMed] [Google Scholar]
- Semerci CN, Demirkan F, Ozdemir M, Biskin E, Akin B, Bagci H, and Akarsu NA (2009). Homozygous feature of isolated triphalangeal thumb-preaxial polydactyly linked to 7q36: no phenotypic difference between homozygotes and heterozygotes. Clin. Genet. 76, 85–90. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, and Stark A (2014). Transcriptional enhancers: from properties to genome wide predictions. Nat Rev Genet 15, 272–286. [DOI] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, and Ahituv N (2013). Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, and Luo L (2011). Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci USA 108, 7902–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. (2016). Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, Kronenberg ZN, Hormozdiari F, Raja A, Pennacchio LA, et al. (2017). Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722. e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, van der Weide RH, Teunissen H, Võsa U, Franke L, et al. (2019). High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet 52, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderMeer JE, and Ahituv N (2011). cis-regulatory mutations are a genetic cause of human limb malformations. Dev. Dyn. 240, 920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderMeer JE, Afzal M, Alyas S, Haque S, Ahituv N, and Malik S (2012). A novel ZRS mutation in a Balochi tribal family with triphalangeal thumb, pre-axial polydactyly, post-axial polydactyly, and syndactyly. Am. J. Med. Genet. A 158A, 2031–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderMeer JE, Lozano R, Sun M, Xue Y, Daentl D, Jabs EW, Wilcox WR, and Ahituv N (2014). A novel ZRS mutation leads to preaxial polydactyly type 2 in a heterozygous form and Werner mesomelic syndrome in a homozygous form. Hum. Mutat. 35, 945–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlerberghe C, Faivre L, Petit F, Fruchart O, Jourdain AS, Clavier F, Gay S, Manouvrier-Hanu S, and Escande F (2015). Intrafamilial variability of ZRS-associated syndrome: characterization of a mosaic ZRS mutation by pyrosequencing. Clin. Genet. 88, 479–483. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2009a). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Research 35, D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, and Pennacchio LA (2009b). Genomic views of distant-acting enhancers. Nature 461, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek D, Pawlik B, Li Y, Akarsu NA, Caliebe A, May KJW, Schweiger B, Vargas FR, Balci S, Gillessen-Kaesbach G, et al. (2010). A specific mutation in the distant sonic hedgehog ( SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum. Mutat. 31, 81–89. [DOI] [PubMed] [Google Scholar]
- Wright CF, West B, Tuke M, Jones SE, Patel K, Laver TW, Beaumont RN, Tyrrell J, Wood AR, Frayling TM, et al. (2019). Assessing the Pathogenicity, Penetrance, and Expressivity of Putative Disease-Causing Variants in a Population Setting. The American Journal of Human Genetics 104, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-F, Guo S, Fan X-F, Fan L-L, Jin J-Y, Tang J-Y, and Xiang R (2016). A Novel ZRS Mutation in a Chinese Patient with Preaxial Polydactyly and Triphalangeal Thumb. Cytogenet Genome Res 149, 171–175. [DOI] [PubMed] [Google Scholar]
- Xu C, Yang X, Zhou H, Li Y, Xing C, Zhou T, Zhong D, Lian C, Yan M, Chen T, et al. (2019). A novel ZRS variant causes preaxial polydactyly type I by increased sonic hedgehog expression in the developing limb bud. Genet. Med. 149, 171–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zákány J (1988). Spatial regulation of homeobox gene fusions in the embryonic central nervous system of transgenic mice. Neuron 1, 679–691. [DOI] [PubMed] [Google Scholar]
- Zguricas J, Heus H, Morales-Peralta E, Breedveld G, Kuyt B, Mumcu EF, Bakker W, Akarsu N, Kay SP, Hovius SE, et al. (1999). Clinical and genetic studies on 12 preaxial polydactyly families and refinement of the localisation of the gene responsible to a 1.9 cM region on chromosome 7q36. Journal of Medical Genetics 36, 32–40. [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lyu Y, Li-Ling J, and Liu C (2019). [Mutation analysis in a large Chinese pedigree affected with preaxial polydactyly II]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36, 610–612. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ding J, Li Y, Ren K, Sha J, Zhu M, and Gao X (2009). HnRNP U mediates the long-range regulation of Shh expression during limb development. Hum Mol Genet 18, 3090–3097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Highly-efficient site-specific mouse transgenesis using enhancer inSERTion (enSERT), Related to Figure 1
(A) Schematic overview of the strategy. The donor targeting vector contained two homology arms (grey, indicated as HA-5′ and HA-3′) and a corresponding enhancer-reporter transgene with an enhancer (light blue), minimal promoter (brown), lacZ reporter (dark blue), and SV40 poly(A) sequence. The sgRNA recognition site is indicated in purple. PCR primers used for genotyping are shown as arrows (5′F, 3′R-outside of the homology arms; 5′R, 3′F - transgene specific). See STAR Methods for more details.
(B) Table showing integration efficiencies, enhancer expression, and coordinates of three different landing sites that we targeted with a forebrain enhancer-reporter transgene using the strategy shown in (A).
(C) PCR genotyping analysis of F0 transgenic E11.5 mice using primer pairs 5′F/5′R and 3′F/3′R to confirm the correct integration of the 5′ (HA-5′) and the 3′ (HA-3′) homology arms at H11 locus, respectively. Numbers indicate independent mice. Mice with lacZ reporter staining in forebrain are indicated in blue.
(D) 48-well plate with LacZ-stained E11.5 embryos in which the forebrain enhancer lacZ reporter was targeted to the H11 locus.
(E) LacZ-stained E11.5 embryos with the forebrain enhancer and Hsp68 minimal promoter driving lacZ reporter expression from the H11 locus. Red arrows point to ectopic activity caused by the Hsp68 promoter.
(F) Comparison of background activities between the Hsp68 and Shh promoters at the H11 locus. The Hsp68 promoter on its own (without enhancer) drives lacZ reporter expression around the neural tube, heart, trigeminal nerve, and the head of the E11.5 embryo. The Shh promoter (without enhancer) does not drive detectable lacZ reporter expression in E11.5, E12.5, E13.5, or E14.5 embryos.
(G) LacZ-stained E11.5 embryos with the forebrain enhancer and Shh promoter driving lacZ reporter gene expression from the H11 locus. Specific enhancer-driven staining in the forebrain, with no background LacZ staining, is observed.
Figure S2, Related to Figure 1
(A) EnSERT captures in vivo enhancer activities in all major mouse embryonic tissues. Shown are representative LacZ-stained embryos with the enhancer reporter construct integrated at the H11 locus. In vivo enhancer activities at E11.5 correspond to 10 different tissue-specific enhancers (nine human and one mouse enhancers; see also VISTA Enhancer Browser: https://enhancer.lbl.gov/).
(B) Position and evolutionary conservation of published and newly reported ZRS variants. Shown is the human ZRS enhancer (789 bp) aligned with the orthologous sequences from five different vertebrate species, including cartilaginous and bony fishes (elephant shark and coelacanth, respectively), chicken, cat, and mouse. Human mutations are shown in blue boxes; mouse mutations are shown in pink boxes; cat and chicken mutations are shown in yellow boxes. * Novel ZRS variants and families reported in this study.
Figure S3. Highly reproducible single nucleotide enhancer variant assessment using enSERT, Related to Figure 2
(A) EnSERT is able to reproducibly detect anterior lacZ misexpression upon the introduction of the ‘Cuban’ variant into the human ZRS enhancer. Shown are independently injected LacZ-stained mouse embryos with transgene integration at the H11 locus showing the activity of the human ZRS reference allele (left) or ‘Cuban’ allele. Red arrowheads indicate anterior LacZ staining caused by the ‘Cuban’ variant.
(B) Random transgenesis results in ectopic staining and low reproducibility (data from (Kvon et al., 2016)). Shown are independently injected LacZ-stained mouse embryos with random transgene integration showing the activity of the ZRS human reference allele. Red arrowheads indicate ectopic anterior LacZ staining.
(C) ZRS enhancer activity pattern scoring. ZRS limb enhancer activity patterns from LacZ staining were classified into five different categories: 1) complete loss of activity, 2) reduced activity, 3) normal activity, 4) a gain of activity in the anterior limb bud, and 5) strong gain of activity in the anterior limb bud. Scoring was done independently at least five annotators blinded to genotype. See STAR Methods for more details.
(D) Bar chart showing the scoring summary for each of the alleles (x-axis). The y-axis shows the percentage of analyzed transgenic embryos for each allele that were annotated in each category (bottom). Alleles on x-axis were sorted by their final categorization (top). See Table S2 and STAR Methods for more details.
Figure S4. Limb phenotypes of knock-in mice with human variants, Related to Figure 3
(A) Schematic overview of the human variant knock-in strategy. A 4.5 kb mouse genomic region containing the ZRS enhancer (light blue) is shown together with the vertebrate phyloP conservation (dark blue). The donor vector contained two homology arms (gray, labeled HA-L and HA-R) with vector-specific sequences for genotyping (green) and a corresponding replaced region (blue) containing a human variant (red) and mutagenised sgRNA recognition site (5′-agtaccatgcgtgtgtTtTagCC-3′) but otherwise identical to the mouse reference ZRS sequence. The sgRNA recognition site is indicated in purple. PCR primers used for genotyping are shown as arrows (LF, RR - mouse-specific, outside of the homology arms; RF, LR - donor vector-specific). See STAR Methods for more details.
(B) Forelimb and hindlimb skeletal preparations from E18.5 mice, with genotyping sequence traces confirming the variant knock-ins at the endogenous mouse ZRS enhancer (third column) shown. Numbers indicate how many embryos exhibited the representative limb phenotype. Skeletal preparations for 396C>T mice were prepared from heterozygous E18.5 mice from an established breeding line. All other skeletal preparations were prepared from F0 E18.5 mice. One variant (621C>G) that displayed strong reporter gene misexpression but resulted in normal limbs upon variant introduction into the endogenous mouse enhancer was located within a subregion of increased sequence divergence between humans and mice, which may explain the inconsistency between the human enhancer enSERT results and the mouse enhancer knock-in phenotype (Figure S2B). Another variant (463C>G) that displayed gain of enhancer activity but resulted in normal limbs upon introduction in the mouse genome caused weaker and more variable reporter gene misexpression, which may explain the absence of polydactyly in the F0 knock-in mice (table S2).
(C) Forelimbs and hindlimbs from E18.5 mice homozygous for the 396C>T variant knock-in (ZRS396T/ZRS396T), along with genotyping results (third column). Numbers indicate how many mice exhibited the representative limb phenotype out of the total number of ZRS396T/ZRS396T E18.5 mice screened.
*Extra digit.
Figure S5. Variant assessment in the ZRS enhancer, Related to Figure 4
(A) Enhancer activities for human ZRS alleles that contain point mutations (black bars) in highly conserved TF binding sites outside of the known variants in the forelimb buds of transgenic e11.5 mouse embryos. Numbers of embryos with lacZ activity in the anterior limb bud (red) over the total number of transgenic embryos screened (black) are indicated.
(B) Mutagenesis of all pathogenic variants (all gain combined), nucleotides immediately adjacent to pathogenic variants (all +1 positions combined [Hs2496.144] and all −1 positions combined [Hs2496.145]) and nucleotides that are immediately adjacent to human variants that were classified as potentially benign by this study (all +1 positions combined [Hs2496.146] and all −1 positions combined [Hs2496.147]). Shown are enhancer activities for each of the constructs and the reference human allele in the forelimb buds of transgenic E11.5 mouse embryos. Numbers of embryos with LacZ staining in the anterior limb bud (red) over the total number of transgenic embryos screened (black) are indicated. Red arrowheads indicate ectopic anterior LacZ staining.
(C) Comparison of evolutionary sequence conservation (based on PhyloP score for 46 vertebrates) for potentially benign, gain-of-function (GoF), and common variants within the human ZRS enhancer. * p-value (by Mann-Whitney test) <0.01. ** p-value <0.001. n.s. - not significant. See Table S1 for PhyloP scores for each of the variants.
(D) P-value (TF motif match) change for potentially benign, gain-of-function (GoF), and common variants within the human ZRS enhancer that overlap predicted TF binding sites. See Table S1 for P-value scores for each of the variants.
(E) Comparison of human genetic and clinical data for pathogenic and potentially benign ZRS variants. Shown are all human ZRS variants tested in this study (columns) and the supporting human genetics data and activity in a transgenic reporter assay (rows). Variants that caused a gain of expression in anterior limb bud are highlighted in red. Novel variants reported in this study are highlighted in blue boxes. See Table S1 for details. TFBS, transcription factor binding site; cntrl, control.
Table S1, Related to Figure 2 [see separate Excel file]
All ZRS variants tested in this study, including previously published human variants (Al-Qattan et al., 2012; Albuisson et al., 2011; Baas et al., 2017; Cai et al., 2019; Cho et al., 2013; Farooq et al., 2010; Furniss et al., 2008; Girisha et al., 2014; Gurnett et al., 2007; Heutink et al., 1994; Lettice et al., 2003; 2008; Lodder, 2009; Norbnop et al., 2014; Semerci et al., 2009; VanderMeer et al., 2012; 2014; Vanlerberghe et al., 2015; Wieczorek et al., 2010; Wu et al., 2016; Zguricas et al., 1999; Zhang et al., 2019), variants from animals (Dorshorst et al., 2010; Dunn et al., 2011; Knudsen and Kochhar, 1981; Lettice et al., 2008; Masuya et al., 2007; Zhao et al., 2009) and novel variants reported in this study.
Table S1 contains the following columns: variant coordinate in GRCh38 genome assembly (1st column), relative position within the ZRS enhancer (2nd column), reference and variant alleles (3rd column), variant name (4th column), organism of origin (5th column), VISTA ID of the tested construct containing the human ZRS enhancer with the variant (6th column), variant classification based on the enSERT result (7th column), reference (8-9th columns), clinical phenotypes and human genetics data for variants from human patients (10-24th columns), PhyloP scores (25th column), predicted TF binding sites (26th column),and corresponding P-values (TF motif match scores) for reference and variant alleles (27th and 28th columns). Note that variants in the ZRS enhancer from other species are shown at orthologous positions in the human ZRS enhancer.
Table S2, Related to Figure 4 [see separate Excel file]
All ZRS enhancer mutants tested in this study together with the summary of the mouse transgenic reporter results.
Table S2 contains the following columns: VISTA ID of the tested transgenic construct containing mutagenized ZRS enhancer (1st column), type of introduced mutation(s) (2nd column), relative position within the ZRS enhancer (for point mutations and variants only; 3nd column), reference and variant alleles (for point mutations and variants only; 4rd column), sequence of mutagenized ZRS enhancer (5rd column), numbers of embryos per transgenic construct that displayed complete loss of enhancer activity in the limb buds (6th column), reduced activity (7th column), normal activity (8th column), gain of enhancer activity in the anterior margin of limb buds (9th column), strong gain of enhancer activity (10thcolumn), p-values by Fisher’s exact test (for loss [11th column] and 20 gain [12th column]), and final annotation based on results in all embryos (13th column).