

Abstract
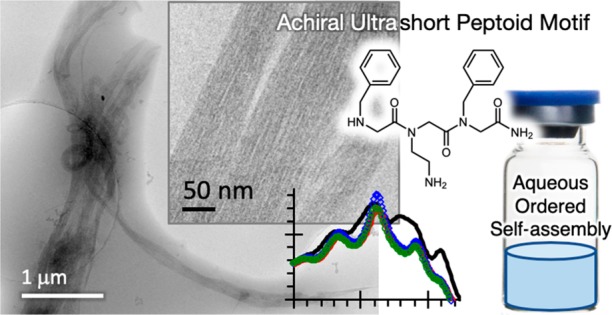
Peptoids are biofunctional N-substituted glycine peptidomimics. Their self-assembly is of fundamental interest because they demonstrate alternatives to conventional peptide structures based on backbone chirality and beta-sheet hydrogen bonding. The search for self-assembling, water-soluble “minimal” sequences, be they peptide or peptidomimic, is a further challenge. Such sequences are highly desired for their compatibility with biomacromolecules and convenient synthesis for broader application. We report the self-assembly of a set of trimeric, water-soluble α-peptoids that exhibit a relatively low critical aggregation concentration (CAC ∼ 0.3 wt %). Cryo-EM and angle-resolved DLS show different sequence-dependent morphologies, namely uniform ca. 6 nm wide nanofibers, sheets, and clusters of globular assemblies. Absorbance and fluorescence spectroscopies indicate unique phenyl environments for π-interactions in the highly ordered nanofibers. Assembly of our peptoids takes place when the sequences are fully ionized, representing a departure from superficially similar amyloid-type hydrogen-bonded peptide nanostructures and expanding the horizons of assembly for sequence-specific bio- and biomimetic macromolecules.
Control of self-assembly using sequence-specific polymers such as peptides and their mimics is a powerful approach to generating functional nanomaterials. Very short self-assembling peptides and their mimics (e.g., ≤ 5 residues) are of special interest since they are more easily scalable and they lead to insight into assembly requirements.1 Their discovery is nontrivial since assembly propensity (e.g., hydrophobic interactions and hydrogen bonding) and favorable solvent (water) interactions must be balanced among just a few residues.1−3 The realization that diphenylalanine (FF) is a key aggregating domain of amyloid peptides including amyloid β4−6 has had an immense impact since assembly from such a simple dipeptide was not anticipated. However, FF is not directly soluble in water, and its assembly requires dilution from an organic solution. Assembly of specific tripeptide derivatives of FF has also been reported, and a number of applications for both FF and its derivatives have been proposed.1,7,8
Peptoids are N-substituted glycine structural isomers of peptides, in which the functional side chains are attached to backbone amide nitrogen atoms instead of the α-carbons.9,10 This preserves the side chain spacing of peptides but removes backbone chirality. Like peptides, specific peptoid sequences may also give rise to bioactivity and secondary structures.11−14 Unlike peptides, the non-natural backbone structure confers great resistance to proteolysis,9,15 which greatly improves pharmacokinetics for therapeutic applications such as antimicrobial peptoids9,16−18 and benefits other long-term biomedical uses.10,19 The side chain shift also eliminates backbone hydrogen-bond (H-bond) donors, and hence intra- and interbackbone H-bonding. This also means that the peptoid backbone may be freely hydrated20,21 and, together with lack of backbone chirality, may exhibit great conformational flexibility.10,22
Peptoid self-assembly is an emerging area.9,10 To overcome flexible conformations and restricted H-bonding, it might appear that a relatively high number of residues would be required to confer sufficient attractive side chain interactions for assembly to occur. An initial report of self-assembled “peptoid gels” actually comprised of hybrids with peptides that could provide H-bonding.23 A strategy to recover H-bonding or constrain conformations is through Ugi-multicomponent synthesis to obtain N-substituted polyamides with amino acid side chains,24,25 and a derivative has been shown to form gels from organic–water mixtures.26 Otherwise, reported micellar assemblies have been driven by the relatively large hydrophobic blocks of long-chain polypeptoids or the hydrophobic alkyl “tails” of sequence-specific lipo-peptoids.27−31 Similarly, long amphiphilic sequences (16–36 residues) are required for the assembly of nanosheet or nanotube structures.32−35
We report the first examples of aqueous self-assembly from very short, water-soluble linear α-peptoid sequences without the directing influence of other components such as peptides36 or lipid tails.31 Inspired by FF and derivatives, we prepared a set of four achiral peptoid trimers comprised of analogs of the aromatic amino acid phenylalanine and the cationic lysine. In particular, we studied the effect of varying the sequence order as well as the side chain length of the lysine analog. In FF peptides and derivatives, various design rules have been reported to account for the importance of π–π stacking of the phenyl groups, interbackbone beta-sheet packing, and chirality.1,2,37 Chirality and inter- and intra-backbone H-bonding are however absent in peptoids. Our designs therefore test the minimal requirements for nanostructure assembly of peptoids/peptides that are directly soluble in water.
Our sequences are comprised of two residues of Nphe (N-benzylglycine), the analog of phenylalanine (Phe), and a residue of either Nlys (N-(4-aminobutyl)glycine) or Nae (N-(2-aminoethyl)glycine). Nlys is a direct lysine (Lys) analog, while Nae is a mimic with a shorter two methylene connection to the amine (Figure 1). The sequences are named with reference to the single letter codes of their amino acid counterparts as N(KFF), N(kFF), N(FKF), and N(FkF), with small “k” denoting Nae. The tripeptoids were synthesized by regular submonomer solid-phase synthesis and purified by preparative HPLC (see the SI). The purity and identity of the sequences were characterized by analytical HPLC and mass spectrometry (Figures S1–S3), the standard for solid-phase synthesized peptoids (and peptides).
Figure 1.

A) Chemical structures of tripeptoids studied. B) Schematic of sample preparation and assembly process.
In this first study, all samples were prepared by directly dissolving the sequences in deionized water (DIW). An acidic pH (e.g., pH ∼3 at 20 mg/mL) was measured for our assembly solutions due to small amounts of trifluoroacetic acid (TFA) typically retained from HPLC purification. The pH is far below the pKa of N-terminal and side chain amines (ca. 9–10). Thus, the amines on the Nlys and Nae side chains and the N-termini are expected to be ionized. This amine protonation increases solubility in water and electrostatic repulsion between the peptoids and would actually be expected to decrease assembly propensity.
Figure 2 shows that all four tripeptoids exhibited a similar critical aggregation concentration (CAC) in a fluorescence assay (0.3 ± 0.03 wt %, i.e., 3 ± 0.3 mg/mL), indicating the self-assembly of hydrophobic cores above the CAC that could sequester the hydrophobic dye. The measured CAC is actually comparable to those of a previously reported set of lipo-peptoids (ca. 0.1 wt %), which however required a long palmitoyl hydrophobic tail to drive assembly.31
Figure 2.

Examples of CAC determination from peptoid concentration dependence of ANS fluorescence: A) N(FkF), B) N(FKF), C) N(KFF), and D) N(kFF). I/I0 is the intensity ratio with and without peptoids. DIW was added directly to preweighed lyophilized peptoids to obtain the highest concentrations shown, and the samples were diluted further with DIW for measurements at lower concentrations. See Figure S4 for original spectra, SI 1.4 for sample preparation details, and example CAC data with pH control.
Analytical gradient RP-HPLC measurements (Figure S3) further characterized more sensitively that N(FkF) and N(FKF) partitioned more readily in water than N(kFF) and N(KFF) (i.e., eluted at a slightly higher 76% vs 72% water content in a water–acetonitrile (ACN) gradient). The overall similar CAC is presumably governed by the fixed number of hydrophobic Nphe residues and +2 charges from Nlys/Nae and the free N-terminus. The slightly higher hydrophobicity of N(kFF) and N(KFF) could be related to the proximity of the side chain primary and N-terminal secondary amines hindering double protonation.
In comparison, we recently showed that an Nphe dipeptoid and FF analogue are insoluble in water. For the dipeptoid, X-ray crystallography showed that π–π stacking of Nphe side chains induced crystallization of microneedles during evaporation from a DMSO–water mixture.38 Alternatively, lamellar nanostructures were observed when the dipeptoid was precipitated in water from an ACN solution.
For the present water-soluble tripeptoids also incorporating Nphe residues, we were able to confirm nanostructure formation by cryo-TEM (Figure 3). Remarkably, N(FkF) formed long, uniform ca. 6 nm wide nanofibers that extended many microns, which further collected into relatively straight bundles (Figure 3A–C and S5A–D). We note that the nanofibers were formed even at the acidic pH 3 of our 2 wt % samples, when we expect N(FkF) to be fully ionized. This behavior stands in contrast to peptide self-assembly, which is often triggered by adjusting the pH to deionize charged groups and enhance H-bonding.1,39
Figure 3.

Cryo-TEM images from 2 wt % (20 mg/mL) solutions of N(FkF) (A–C), N(FKF) (D–F), N(KFF) (G–I), and N(kFF) (J–L). The left column shows zoomed in areas indicated in the center column. The right column shows additional typical images. Further areas are shown in Figure S5. The insets in G show that the ∼50 nm N(KFF) features are clusters of the finest structures. Peptoid solutions were prepared in the same way as for CAC measurements (see the caption of Figure 2). See the SI for cryo-EM sample vitrification procedures.
Cryo-TEM further showed that tripeptoid assembly was highly sensitive to both the side chain length and the residue sequence order. N(FKF), which has Nlys with the longer side chain in the same central residue position as N(FkF), formed networks (Figure 3D–F) spanning a few hundred nanometers that are composed of globular assemblies ca. 15–20 nm wide (Figure 3D). However, nanofibers were occasionally observed to coexist (Figure S5H), indicating that the propensity for ordered assembly of this N(FxF) sequence is attenuated by the longer Nlys vs Nae side chain. It is not immediately clear why the seemingly small difference in side chain length between N(FkF) and N(FKF) has caused such a large shift in assembled morphology. However, the shift is corroborated by additional light scattering and spectroscopic evidence (see below). Moreover, it is well-known from peptide dimers and trimers that small changes in side chains and/or sequences can give rise to diverse assembly behavior.1−3 It is however possible that the longer side chain of Nlys is simply mismatched to or provides excessive conformational flexibility for potential ordered assembly.
N(kFF) and N(KFF), which have the cationic Nae/Nlys placed at the N-terminus, also formed interconnected assemblies (Figure 3G–L). Upon closer inspection, N(KFF) actually assembled into fine 5–10 nm features (insets in Figures 3G and S5K) that cluster into a second set of larger ca. 50 nm spherical assemblies. N(kFF), which has the shorter Nae side chain, also formed 5–10 nm fine features (Figure 3J). However, this sequence appeared to exhibit stronger interactions, since the fine features instead coalesced into globules ca. 50 nm in diameter (Figures 3J,K and S5O,P) as well as into nanosheets that spanned >100 nm (Figures 3L and S5O).
Dynamic light scattering (DLS) measurements corroborated the size and morphology of the nanoassemblies. N(FkF) shows a complex scattering behavior that could be fitted with subpopulations with hydrodynamic radii (RH) centered around 0.5 nm and 60 nm and another population > 1000 nm with a large dependence on a scattering angle (2θ) (Figure 4A). Since angular differences are characteristic of anisotropic particles, the micron-sized dimension should be related to the length of the nanofibers. The nonvarying sub-1 nm fraction was assigned to monomers, while the ca. 60 nm length scale could represent the effective averaged widths of the nanofiber bundles.
Figure 4.

Variation in hydrodynamic radii (RH) with a DLS scattering angle (2θ = 90°) for A) N(FkF), B) N(FKF), C) N(KFF), and D) N(kFF). Two wt % (20 mg/mL) solutions were used. The different symbols in each panel refer simply to the different size populations measured in each sample, as indicated by the labels of hydrodynamic radii (RH). They are unrelated between panels. Peptoid solutions were prepared in the same way as for CAC measurements (see SI 1.4 for sample preparation details).
Peptoid N(FKF) shows assemblies with RH centered around 108 nm (Figure 4B), which could indicate the loose networks of finer assemblies (Figures 2D–F). N(kFF) and N(KFF) show mainly the presence of structures with RH centered around 0.5 nm and 44–49 nm (Figures 3C,D), corresponding to respectively monomers and the clusters observed.
The high degree of molecular ordering implied by the uniformity of the N(FkF) nanofibers is reminiscent of some FF tripeptide derivatives assembling also into nanofibers.1,37 However, our peptoids assembled directly in acidified water. Solubility was likely promoted by the cationic Nae/Nlys side chains. Assembly however cannot be related to beta-sheet structures because there is no interbackbone H-bonding in peptoids. We speculate that, similar to the Nphe dipeptoid crystals we reported recently,38 nanofiber assembly was facilitated by Nphe π–π stacking as well as by flexible peptoid backbone twists that enable favorable positioning of interacting groups.38,40
We further characterized π–π stacking spectroscopically (Figure 5). First, N(FkF) showed a set of absorption fine structures in the 245–270 nm phenyl band distinct from other sequences as well as an additional absorption around 288 nm (Figure 5A), indicating a unique phenyl environment. This phenyl signature was retained at concentrations below the CAC (Figure S6A), indicating that they originate from the monomer state. On the other hand, while other tripeptoids produced fluorescence emissions at 282 and 288 nm, N(FkF) displayed a pair of especially well-separated emissions centered at 280 and 312 nm (Figure 5B). These peaks, separated by 32 nm (3663 cm–1), are assigned as monomer and strongly red-shifted excimer emission, respectively. An excimer was assigned based on its increasing emission with increasing concentration above the CAC (Figure S6). As the nanofibers assembled, the phenyl groups must be sufficiently close in space to form the excimer efficiently during the excited-state lifetime of the phenyl chromophore. This is analogous to the fluorescence behavior of FF peptide nanofibers37 and certain dimerized peptoids that are mediated by π-interactions.40,41
Figure 5.

A) Absorbance spectra of tripeptoids in water (2 wt %, 20 mg/mL; intensities normalized to local minimum at 243 nm). B) Fluorescence spectra of the same solutions (265 nm excitation; normalized to emission maxima). Peptoid solutions were prepared in the same way as for CAC measurements (see the caption of Figure 2 and SI 1.4 for details).
The excimer fluorescence features were already present 30 min after initially dissolving the peptoid in water, the time sample preparation and measurements took. They also held constant after long-term storage (tested up to 4 months at 4 °C; see Figure S7). Consistent with the CAC measured for N(FkF) (∼3 mg/mL; Figure 2), the 312 nm excimer peak grew more apparent as the peptoid concentration increased above 1 mg/mL (Figure S6).
In the context of sequence-specific assembly facilitated by Nphe π–π stacking, it is noteworthy that the less hydrophobic N(FxF) compared to N(xFF) (earlier elution in HPLC—Figure S1), which ordinarily would suggest lower assembly propensity, was actually able to form the more ordered nanofibers. Indeed, as discussed above, the sequences are expected to be fully charged to promote solubility. It is possible that stronger nonspecific attractive interactions due to higher hydrophobicities could be hindering the repositioning required during assembly to obtain ordered nanostructures. Interestingly, the 312 nm excimer emission remained when we raised the pH of the N(FkF) solution from pH 3 to pH 11 (Figure S8), indicating that the N(FkF) structural interactions were not controlled by charge or H-bonding. In comparison, the small separation of the 282 nm/288 nm peaks for N(FKF) is also unchanged from acidic to basic pH (Figure S9), although there is some slight overall shift of these peaks and some additional low intensity features appeared by pH 9. This lower pH stability could be due to the fact that the unordered N(FKF) structure may possibly be more susceptible to changes in environmental conditions.
In summary, we have identified a set of minimally short, water-soluble tripeptoids that assemble into uniform 6 nm wide nanofibers and other nanoassemblies. This is the first demonstration of self-assembly for such short linear α-peptoids. The assembled morphology depended on the sequence and was further controlled by the cationic side chain length. Spectroscopic results are consistent with unique π–π interactions that differentiate highly ordered nanofibers from other structures.
Our tripeptoids illustrate that ordered aqueous assembly of even very short peptidic chains can still be engineered without chirality, backbone–backbone H-bonding (e.g., beta-sheet structures2,37), and charge group deionization. Even if our tripeptoids may superficially resemble FF-peptides, peptoids and peptides appear to follow different assembly rules to strike a balance between solvation and intermolecular attraction. The flexibility of the peptoid backbone might even aid assembly, by accommodating favorable conformations for side chain H-bonding or π–π stacking, as previously implicated.38,40 Nae, the Lys-mimic included in our nanofiber- and sheet-forming tripeptoids, is also found in longer peptoid sequences that assemble into highly ordered bilayer nanosheets,32,34 suggesting a potential structural role. Future studies comprising additional nano characterization, molecular simulations, and a larger set of sequences may elucidate the molecular structures observed and clarify how the sequence and the Nae side chain may control the assembly. Overall, our tri-peptoid sequences display unique and novel self-assembly behavior distinct from di- and tripeptides and open the door to convenient studies of peptoid assembly directly in water. The proteolytically stable peptidic structures should be of fundamental interest as well as of value to applications such as stable biomaterials and other sequence-tunable nanostructures.
Acknowledgments
V.C. was supported by EPSRC Platform grant EP/L020599/1 to I.W.H. We acknowledge access to the Chemical Analysis Facility Laboratory (University of Reading). K.H.A.L. and K.M.T. thank the Human Frontier Science Program (RGY074) for financial support. A.H. thanks the Commonwealth Scholarship Commission for a Split Site award (INCN-2017-50). We also thank Aquib Jawed (Strathclyde/IIT-G) and Aman Bhardwaj (IIT-G) for supplementary measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmacrolett.9b01010.
Experimental details and supplementary data (PDF)
Author Present Address
⊥ School of Pharmacy, Department of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham NG7 2RD, UK.
Author Contributions
The manuscript was written through contributions of all authors. V.C. performed DLS and CAC measurements and, with I.W.H., drafted the manuscript. J.S. performed TEM, supervised by J.R. K.M.T. and A.H. synthesized, purified, and characterized the peptoids. L.M.P. co-supervised A.H. K.H.A.L. performed and together with R.M.E. supervised spectroscopic studies. I.W.H. and K.H.A.L. supervised the overall study and finalized the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lampel A.; Ulijn R. V.; Tuttle T. Guiding principles for peptide nanotechnology through directed discovery. Chem. Soc. Rev. 2018, 47, 3737–3758. 10.1039/C8CS00177D. [DOI] [PubMed] [Google Scholar]
- Frederix P. W. J. M.; Scott G. G.; Abul-Haija Y. M.; Kalafatovic D.; Pappas C. G.; Javid N.; Hunt N. T.; Ulijn R. V.; Tuttle T. Exploring the sequence space for (tri-) peptide self-assembly to design and discover. Nat. Chem. 2015, 7, 30–37. 10.1038/nchem.2122. [DOI] [PubMed] [Google Scholar]
- Wei G.; Su Z. Q.; Reynolds N. P.; Arosio P.; Hamley I. W.; Gazit E.; Mezzenga R. Self-assembling peptide and protein amyloids: from structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. 10.1039/C6CS00542J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamley I. W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- Gazit E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- Gazit E.Reductionist Approach in Peptide-Based Nanotechnology. In Annual Review of Biochemistry, Vol 87; Kornberg R. D., Ed.; Annual Reviews: Palo Alto, 2018; Vol. 87, pp 533–553, 10.1146/annurev-biochem-062917-012541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Gazit E. The physical properties of supramolecular peptide assemblies: from building block association to technological applications. Chem. Soc. Rev. 2014, 43, 6881–6893. 10.1039/C4CS00164H. [DOI] [PubMed] [Google Scholar]
- Tao K.; Makam P.; Aizen R.; Gazit E. Self-assembling peptide semiconductors. Science 2017, 358, eaam9756. 10.1126/science.aam9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight A. S.; Zhou E. Y.; Francis M. B.; Zuckermann R. N. Sequence Programmable Peptoid Polymers for Diverse Materials Applications. Adv. Mater. 2015, 27, 5665–5691. 10.1002/adma.201500275. [DOI] [PubMed] [Google Scholar]
- Lau K. H. A. Peptoids for biomaterials science. Biomater. Sci. 2014, 2, 627–633. 10.1039/C3BM60269A. [DOI] [PubMed] [Google Scholar]
- Reddy M. M.; Kodadek T. Protein ″fingerprinting″ in complex mixtures with peptoid microarrays. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12672–12677. 10.1073/pnas.0501208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveendra B. L.; Wu H.; Baccala R.; Reddy M. M.; Schilke J.; Bennett J. L.; Theofilopoulos A. N.; Kodadek T. Discovery of Peptoid Ligands for Anti-Aquaporin 4 Antibodies. Chem. Biol. 2013, 20, 351–359. 10.1016/j.chembiol.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma R. C.; Mulder G. E.; Kruijtzer J. A. W.; Posthuma G.; Rijkers D. T. S.; Liskamp R. M. J. Transformation of the amyloidogenic peptide amylin(20–29) into its corresponding peptoid and retropeptoid: Access to both an amyloid inhibitor and template for self-assembled supramolecular tapes. Bioorg. Med. Chem. Lett. 2007, 17, 1837–1842. 10.1016/j.bmcl.2007.01.042. [DOI] [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K.; Spellmeyer D. C.; Tan R. Y.; Frankel A. D.; Santi D. V.; Cohen F. E.; Bartlett P. A. Peptoids - a Modular Approach to Drug Discovery. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371. 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. Comparison of the Proteolytic Susceptibilities of Homologous L-Amino-Acid, D-Amino-Acid, and N-Substituted Glycine Peptide and Peptoid Oligomers. Drug Dev. Res. 1995, 35, 20–32. 10.1002/ddr.430350105. [DOI] [Google Scholar]
- Chongsiriwatana N. P.; Patch J. A.; Czyzewski A. M.; Dohm M. T.; Ivankin A.; Gidalevitz D.; Zuckermann R. N.; Barron A. E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2794–2799. 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzewski A. M.; Jenssen H.; Fjell C. D.; Waldbrook M.; Chongsiriwatana N. P.; Yuen E.; Hancock R. E. W.; Barron A. E. In Vivo, In Vitro, and In Silico Characterization of Peptoids as Antimicrobial Agents. PLoS One 2016, 11, e0135961. 10.1371/journal.pone.0135961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongsiriwatana N. P.; Lin J. S.; Kapoor R.; Wetzler M.; Rea J. A. C.; Didwania M. K.; Contag C. H.; Barron A. E. Intracellular biomass flocculation as a key mechanism of rapid bacterial killing by cationic, amphipathic antimicrobial peptides and peptoids. Sci. Rep. 2017, 7, 16718. 10.1038/s41598-017-16180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K. H. A.; Sileika T. S.; Park S. H.; Sousa A. M. L.; Burch P.; Szleifer I.; Messersmith P. B. Molecular Design of Antifouling Polymer Brushes Using Sequence-Specific Peptoids. Adv. Mater. Interfaces 2015, 2, 1400225. 10.1002/admi.201400225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung D. L.; Lau K. H. A. Atomistic Study of Zwitterionic Peptoid Antifouling Brushes. Langmuir 2019, 35, 1483–1494. 10.1021/acs.langmuir.8b01939. [DOI] [PubMed] [Google Scholar]
- Lau K. H. A.; Ren C.; Sileika T. S.; Park S. H.; Szleifer I.; Messersmith P. B. Surface-Grafted Polysarcosine as a Peptoid Antifouling Polymer Brush. Langmuir 2012, 28, 16099–16107. 10.1021/la302131n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murnen H. K.; Rosales A. M.; Dobrynin A. V.; Zuckermann R. N.; Segalman R. A. Persistence length of polyelectrolytes with precisely located charges. Soft Matter 2013, 9, 90–98. 10.1039/C2SM26849C. [DOI] [Google Scholar]
- Wu Z. D.; Tan M.; Chen X. M.; Yang Z. M.; Wang L. Molecular hydrogelators of peptoid-peptide conjugates with superior stability against enzyme digestion. Nanoscale 2012, 4, 3644–3646. 10.1039/c2nr30408b. [DOI] [PubMed] [Google Scholar]
- Tao Y.; Wang Z.; Tao Y. Polypeptoids synthesis based on Ugi reaction: Advances and perspectives. Biopolymers 2019, 110, e23288 10.1002/bip.23288. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wang S.; Liu J.; Xie Z.; Luan S.; Xiao C.; Tao Y.; Wang X. Ugi Reaction of Natural Amino Acids: A General Route toward Facile Synthesis of Polypeptoids for Bioapplications. ACS Macro Lett. 2016, 5, 1049–1054. 10.1021/acsmacrolett.6b00530. [DOI] [PubMed] [Google Scholar]
- Mangunuru H. P. R.; Yang H.; Wang G. J. Synthesis of peptoid based small molecular gelators by a multiple component reaction. Chem. Commun. 2013, 49, 4489–4491. 10.1039/c3cc41043a. [DOI] [PubMed] [Google Scholar]
- Sternhagen G. L.; Gupta S.; Zhang Y.; John V.; Schneider G. J.; Zhang D. Solution Self-Assemblies of Sequence-Defined Ionic Peptoid Block Copolymers. J. Am. Chem. Soc. 2018, 140, 4100–4109. 10.1021/jacs.8b00461. [DOI] [PubMed] [Google Scholar]
- Fetsch C.; Gaitzsch J.; Messager L.; Battaglia G.; Luxenhofer R. Self-Assembly of Amphiphilic Block Copolypeptoids – Micelles, Worms and Polymersomes. Sci. Rep. 2016, 6, 33491. 10.1038/srep33491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff N.; Ulbricht J.; Lorson T.; Schlaad H.; Luxenhofer R. Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering. Chem. Rev. 2016, 116, 1753–1802. 10.1021/acs.chemrev.5b00201. [DOI] [PubMed] [Google Scholar]
- Davidson E. C.; Rosales A. M.; Patterson A. L.; Russ B.; Yu B.; Zuckermann R. N.; Segalman R. A. Impact of Helical Chain Shape in Sequence-Defined Polymers on Polypeptoid Block Copolymer Self-Assembly. Macromolecules 2018, 51, 2089–2098. 10.1021/acs.macromol.8b00055. [DOI] [Google Scholar]
- Lau K. H. A.; Castelletto V.; Kendall T.; Sefcik J.; Hamley I. W.; Reza M.; Ruokolainen J. Self-assembly of ultra-small micelles from amphiphilic lipopeptoids. Chem. Commun. 2017, 53, 2178–2181. 10.1039/C6CC09888F. [DOI] [PubMed] [Google Scholar]
- Robertson E. J.; Battigelli A.; Proulx C.; Mannige R. V.; Haxton T. K.; Yun L.; Whitelam S.; Zuckermann R. N. Design, Synthesis, Assembly, and Engineering of Peptoid Nanosheets. Acc. Chem. Res. 2016, 49, 379–389. 10.1021/acs.accounts.5b00439. [DOI] [PubMed] [Google Scholar]
- Murnen H. K.; Rosales A. M.; Jaworski J. N.; Segalman R. A.; Zuckermann R. N. Hierarchical Self-Assembly of a Biomimetic Diblock Copolypeptoid into Homochiral Superhelices. J. Am. Chem. Soc. 2010, 132, 16112–16119. 10.1021/ja106340f. [DOI] [PubMed] [Google Scholar]
- Merrill N. A.; Yan F.; Jin H.; Mu P.; Chen C.-L.; Knecht M. R. Tunable assembly of biomimetic peptoids as templates to control nanostructure catalytic activity. Nanoscale 2018, 10, 12445–12452. 10.1039/C8NR03852J. [DOI] [PubMed] [Google Scholar]
- Sun J.; Jiang X.; Lund R.; Downing K. H.; Balsara N. P.; Zuckermann R. N. Self-assembly of crystalline nanotubes from monodisperse amphiphilic diblock copolypeptoid tiles. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3954–3959. 10.1073/pnas.1517169113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxton T. K.; Mannige R. V.; Zuckermann R. N.; Whitelam S. Modeling Sequence-Specific Polymers Using Anisotropic Coarse-Grained Sites Allows Quantitative Comparison with Experiment. J. Chem. Theory Comput. 2015, 11, 303–315. 10.1021/ct5010559. [DOI] [PubMed] [Google Scholar]
- Shlomo Z.; Vinod T. P.; Jelinek R.; Rapaport H. Stacking interactions by two Phe side chains stabilize and orient assemblies of even the minimal amphiphilic β-sheet motif. Chem. Commun. 2015, 51, 3154–3157. 10.1039/C4CC09673H. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Chippindale A. M.; Hamley I. W.; Barnett S.; Hasan A.; Lau K. H. A. Crystallization and lamellar nanosheet formation of an aromatic dipeptoid. Chem. Commun. 2019, 55, 5867–5869. 10.1039/C9CC02335F. [DOI] [PubMed] [Google Scholar]
- Raeburn J.; Cardoso A. Z.; Adams D. J. The importance of the self-assembly process to control mechanical properties of low molecular weight hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. 10.1039/c3cs60030k. [DOI] [PubMed] [Google Scholar]
- Jimenez C. J.; Tan J.; Dowell K. M.; Gadbois G. E.; Read C. A.; Burgess N.; Cervantes J. E.; Chan S.; Jandaur A.; Karanik T.; Lee J. J.; Ley M. C.; McGeehan M.; McMonigal A.; Palazzo K. L.; Parker S. A.; Payman A.; Soria M.; Verheyden L.; Vo V. T.; Yin J.; Calkins A. L.; Fuller A. A.; Stokes G. Y. Peptoids advance multidisciplinary research and undergraduate education in parallel: Sequence effects on conformation and lipid interactions. Biopolymers 2019, 110, e23256 10.1002/bip.23256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller A. A.; Huber J.; Jimenez C. J.; Dowell K. M.; Hough S.; Ortega A.; McComas K. N.; Kunkel J.; Asuri P. Solution effects on the self-association of a water-soluble peptoid. Biopolymers 2019, 110, e23248 10.1002/bip.23248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.