

Abstract
We report a new immunodeficiency disorder in mice caused by a viable hypomorphic mutation of Snrnp40, an essential gene encoding a subunit of the U5 small nuclear ribonucleoprotein (snRNP) complex of the spliceosome. Snrnp40 is ubiquitous but strongly expressed in lymphoid tissue. Homozygous mutant mice showed hypersusceptibility to infection by mouse cytomegalovirus (MCMV) and multiple defects of lymphoid development, stability, and function. Cell-intrinsic defects of hematopoietic stem cell differentiation also affected homozygous mutants. Snrnp40 deficiency in primary hematopoietic stem cells or T cells or the EL4 cell line increased the frequency of splicing errors, mostly intron retention, in several hundred mRNAs. Diminished expression of proteins associated with immune cell function was also observed in Snrnp40-mutant cells. The immunologic consequences of Snrnp40 deficiency presumably result from cumulative, moderate effects on processing of many different mRNA molecules and secondary reductions in the expression of critical immune proteins, yielding a syndromic immune disorder.
Introduction
All blood cells develop from hematopoietic stem cells (HSCs), which differentiate via phenotypically defined and progressively more committed steps into mature immune cells1. Differentiation of HSCs to multipotent progenitors (MPPs), or MPPs to specific lineages, involves global changes in transcription orchestrated by transcription factors, chromatin modifiers, and other molecules that control gene expression. We have pursued a forward genetic study to identify novel genes with non-redundant function in hematopoiesis, surveying by flow cytometry immune cell populations in peripheral blood from mice carrying N-ethyl-N-nitrosourea (ENU)-induced germline mutations. Here, we describe a mutation of Snrnp40 that impairs HSC differentiation to MPPs, and subsequent differentiation to common lymphoid progenitors (CLPs), T cells, B cells and natural killer (NK) cells.
Snrnp40 is a component of the U5 small nuclear ribonucleoprotein (snRNP) of the major (U2-dependent) and minor (U12-dependent) spliceosomes, but its precise function is not known2–4. The U5 snRNP consists of a single snRNA after which the complex is named, seven Sm proteins, and approximately ten particle specific proteins2, 4. An RNA-free salt-stable subcomplex containing Snrnp40, Prpf8, Snrnp200 (Brr2), and Eftud2 (Snu114) can be isolated from native HeLa cell U5 snRNPs4, suggesting that these proteins function together in the U5 snRNP. During splicing, the U5 snRNP is preassembled with the U4/U6 snRNP to form the U4/U6.U5 tri-snRNP. The tri-snRNP is recruited to the pre-spliceosome, a complex containing the pre-mRNA, U1 snRNP, and U2 snRNP. Within this pre-catalytic spliceosome complex, major RNA and protein rearrangements occur, resulting in release of U1 and U4 snRNPs and formation of the activated spliceosome. The catalytic steps of splicing depend on the U5 snRNP for 5′ splice site recognition and positioning (Prpf8)5–7, alignment of the 5′ and 3′ splice sites for ligation (U5 snRNA and Prpf8)2, 8–10, and regulation of U4/U6 and U2/U6 snRNA unwinding (Prpf8, Snrnp200, and Eftud2)11–14. Because Snrnp40 is tightly bound to Prpf8, Eftud2, and Snrnp200, it is likely that Snrnp40 also participates and facilitates these processes.
We detected numerous retained introns in RNA isolated from Snrnp40-mutant cells, as well as a preponderance of normally spliced exon-exon junctions. In addition, reduced expression of several hundred transcripts was detected in mutant cells. Thus, Snrnp40 is necessary for accurate implementation of the transcriptional program necessary for HSC differentiation, with particular importance for lymphoid differentiation and effector function.
Results
A viable hypomorphic allele of Snrnp40
In a forward genetic screen for mutations affecting immune cell development or function15, we identified the same single base substitution in 17 unrelated pedigrees derived from the C57BL/6J stock obtained from The Jackson Laboratory (JAX). The mutation (chr4: 130378042(+) G->C) was within Snrnp40, and was associated with a complex and strictly recessive immune phenotype, which we named skywarp (swp) (Supplementary Fig. 1). We demonstrated that the Snrnp40swp allele was a spontaneous mutation in the C57BL/6J stock (Supplementary Fig. 2a). Homozygous swp mice were born to heterozygous parents at normal Mendelian frequencies (Supplementary Table 1).
Human and mouse orthologues of Snrnp40 are 98% identical at the amino acid level4 and contain seven WD40 repeats accounting for most of the protein sequence (Supplementary Fig. 2b). The swp mutation is situated 3 bp proximal to the start of Snrnp40 exon 5, and was predicted to cause skipping of exon 5, leading to an in-frame deletion of 41 amino acids affecting the third and fourth WD40 repeats of the 358-residue protein (Fig. 1a,b). Exon skipping was confirmed by RT-PCR of splenic RNA, which showed a majority of truncated transcripts and a small amount of normal transcripts in Snrnp40swp/swp mice (Fig. 1c and Supplementary Table 2). Immunoblot analysis of tagged versions of wild-type and swp mutant proteins expressed in 293T cells, or of endogenous Snrnp40 in splenocytes, indicated that the truncated protein was highly unstable (Fig. 1d,e). However, a small amount of wild-type Snrnp40 was detected in swp/swp splenocytes, consistent with RT-PCR data (Fig. 1c,e and Supplementary Table 2). Snrnp40 mRNA and protein were widely expressed throughout the body, with particularly high levels in lymphoid organs (Supplementary Fig. 3a–c). Snrnp40 protein was detected in T cells, B cells, and NK cells (Supplementary Fig. 3d).
Figure 1. The skywarp mutation.
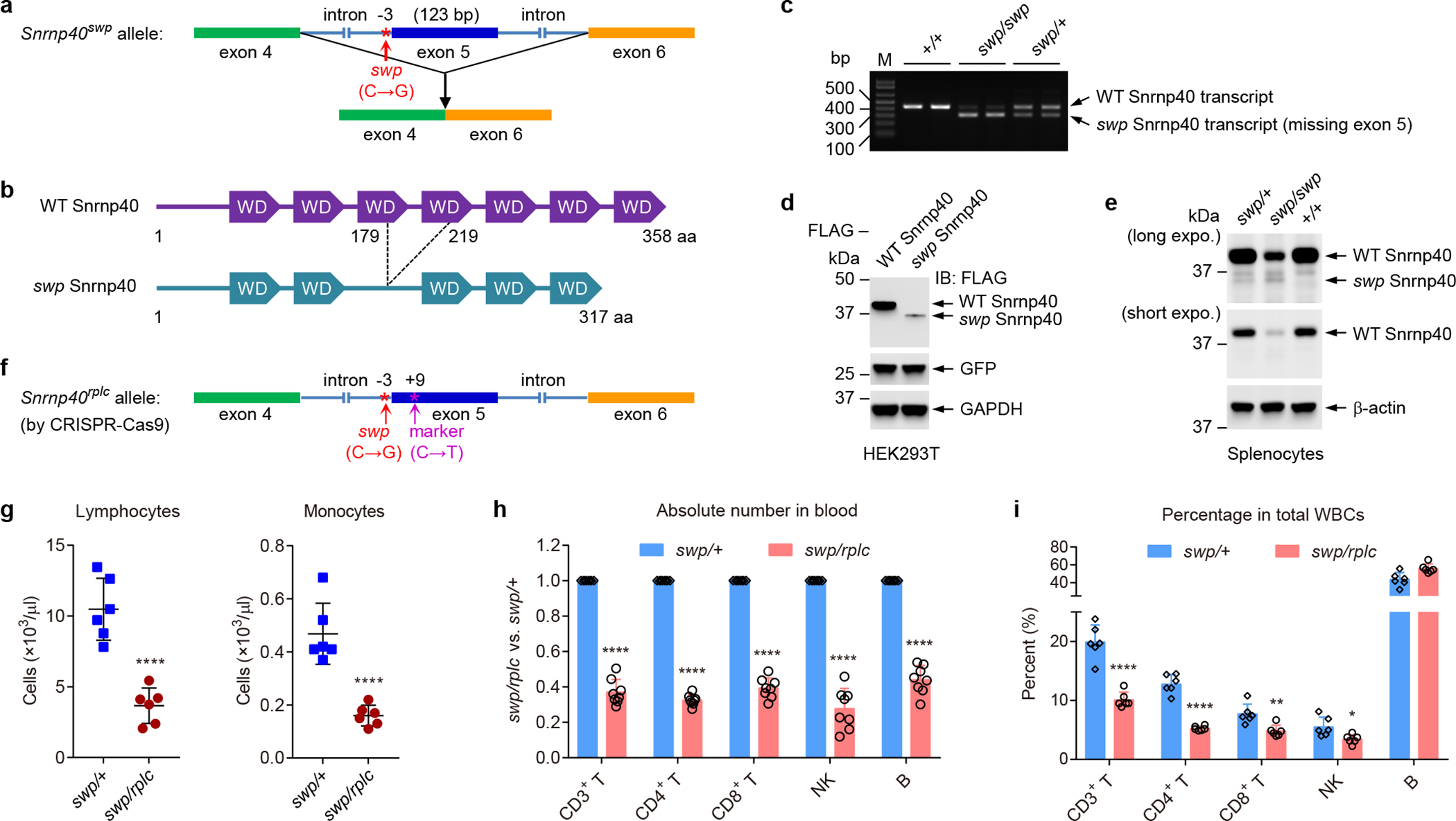
a, Schematic of the skywarp (swp) mutation, a C to G transversion 3 bp proximal to exon 5 predicted to damage the intron 4 3′ splice site leading to skipping of exon 5. b, Schematic of the Snrnp40swp mutant protein. c, RT-PCR amplification across exon 5 of Snrnp40 mRNA from splenocytes of the indicated genotypes. d, Immunoblot analysis of FLAG-tagged wild-type Snrnp40 and Snrnp40swp expression in HEK 293T cells. Co-transfected GFP was used as a transfection efficiency control and GAPDH was used as an internal control. e, Immunoblot analysis of endogenous Snrnp40 in lysates of splenocytes from mice of the indicated genotypes. f, Schematic of the rplc allele generated by CRISPR-Cas9 gene targeting. The original swp single base substitution was recreated in cis with a synonymous marker mutation (C to T) 11 bp downstream from the swp mutation. g, Lymphocyte and monocyte counts from hematological analysis of the blood (n=6 mice per genotype; ****P<0.0001, unpaired, two-tailed Student’s t-test). h, Ratio (Snrnp40swp/rplc to Snrnp40swp/+) of the number of each cell type in the blood (n=8 mice per genotype; ****P<0.0001, unpaired, two-tailed Student’s t-test). i, Frequency of each cell type among total white blood cells in the blood (n=6 mice per genotype; *P=0.018, **P=0.0041, ****P<0.0001, unpaired, two-tailed Student’s t-test). Data are representative of two (c,d), three (h), four (g), or six (e,i) independent experiments (mean ± s.d. in g-i). The gel (c) and blots (d,e) were cropped to show relevant bands and their original images are presented in the Source Data.
To validate that the Snrnp40swp mutation was causative for the immunological phenotypes observed in swp/swp mice, we used CRISPR-Cas9–mediated gene targeting to create a null allele (Snrnp40–) and a “replacement” allele (Snrnp40rplc) in which the original swp single base substitution was recreated in cis with a synonymous marker mutation 11 bp downstream from the swp mutation (Fig. 1f and Supplementary Fig. 3e). Interestingly, even though the marker mutation had no effect on the Snrnp40 amino acid sequence, the rplc allele caused a greater reduction of wild-type Snrnp40 transcript levels than the swp allele, suggesting that the marker mutation also impairs splicing (Supplementary Table 2). RT-PCR confirmed that the rplc allele caused exon 5 skipping and immunoblotting showed reduced wild-type Snrnp40 protein in swp/rplc splenocytes (Supplementary Fig. 3f,g). Wild-type, rplc, swp, and knockout (KO) alleles were variously combined to assess dosage effects on transcript abundance, viability, and immunological phenotypes (Supplementary Table 1 and 2). Immunodeficiency, reduced body weight (Supplementary Fig. 3h), and white-spotted ventral fur (Supplementary Fig. 3i) were observed in Snrnp40swp/rplc mice, in which the wild-type Snrnp40 transcript was expressed at 6.7% the amount present in Snrnp40+/+ mice (Supplementary Table 2). An immune phenotype and reduced body size (Supplementary Fig. 3h) without spotting or with minimal spotting was observed in Snrnp40swp/swp mice, in which the wild-type Snrnp40 transcript was expressed at 10.2% the amount present in Snrnp40+/+ mice (Supplementary Table 2). These findings are consistent with the more damaging effect of the rplc mutation compared to the swp mutation on Snrnp40 expression. No phenotype was detected with 50% expression (Snrnp40+/−). The genotypes Snrnp40–/–, Snrnp40rplc/–, Snrnp40rplc/rplc, and Snrnp40swp/– mice, resulting in wild-type Snrnp40 transcript abundance ranging from 0 to 5.1% of the Snrnp40+/+ level, were uniformly embryonic lethal (Supplementary Table 2). These data demonstrate that the Snrnp40 viability threshold lies somewhere between 5.1% and 6.7% of the wild-type expression level, while the threshold for normal immune development and function lies somewhere between 10.2% and 50% of the wild-type expression level in mice.
Skywarp immunological phenotypes
Snrnp40swp/rplc mice exhibited significantly reduced numbers of lymphocytes and monocytes (Fig. 1g), but normal numbers of neutrophils, eosinophils, and basophils (Supplementary Fig. 4a–c). Complete blood count (CBC) testing showed reduced numbers of red blood cells, correlating with a reduced blood concentration of hemoglobin (Supplementary Fig. 4d,e); hematocrit (HCT) was reduced whereas red blood cell distribution width (RDW) was increased in Snrnp40swp/rplc mice (Supplementary Fig. 4f–j). Blood platelet counts were elevated in Snrnp40swp/rplc mice, but mean platelet volume (MPV) was normal (Supplementary Fig. 4k,l). Further analyses of lymphocyte populations indicated deficiencies of T cells, B cells, and NK cells (Fig. 1h,i and Supplementary Fig. 4m). Macrophage and neutrophil numbers were normal (Supplementary Fig. 4n,o).
Snrnp40swp/rplc mice had both decreased percentages and absolute numbers of CD3+ T cells, CD4+ T cells, and CD8+ T cells in the blood (Fig. 1h,i). Greater frequencies of Snrnp40swp/swp CD8+ T cells in the blood expressed the activation marker CD44 (Fig. 2a), and both CD4+ and CD8+ T cells expressed CD44 at higher levels than did wild-type cells (Fig. 2b). Consistent with these findings, the frequency of naïve CD8+ T cells was reduced while those of central and effector memory CD8+ T cells were increased in Snrnp40swp/rplc spleen and lymph node compared to the corresponding Snrnp40swp/+ tissues (Fig. 2c–f). A similar effect was observed for naïve and effector memory CD4+ T cells in Snrnp40swp/rplc spleen and lymph node; Snrnp40swp/rplc central memory CD4+ T cells were found at near normal frequencies (Fig. 2c–f). In contrast, the CD3ε chain of the TCR was expressed at lower levels on Snrnp40swp/swp T cells than on wild-type T cells (Fig. 2g). Consistent with their hyperactivated state and reduced TCR signaling potential, Snrnp40swp/swp CD4+ and CD8+ T cells failed to proliferate and underwent apoptosis in response to homeostatic expansion signals after transfer into irradiated C57BL/6J wild-type recipient mice (Fig. 2h,i). Defective cell cycle progression was also observed in Snrnp40swp/swp T cells (Supplementary Fig. 5a,b). However, antigen-specific T cell proliferation, T-dependent and T-independent antibody responses, as well as cytotoxic T lymphocyte (CTL) activity, appeared to be normal in Snrnp40-mutant mice (Supplementary Fig. 5c–h).
Figure 2. T cell deficiency in Snrnp40swp/rplc mice.
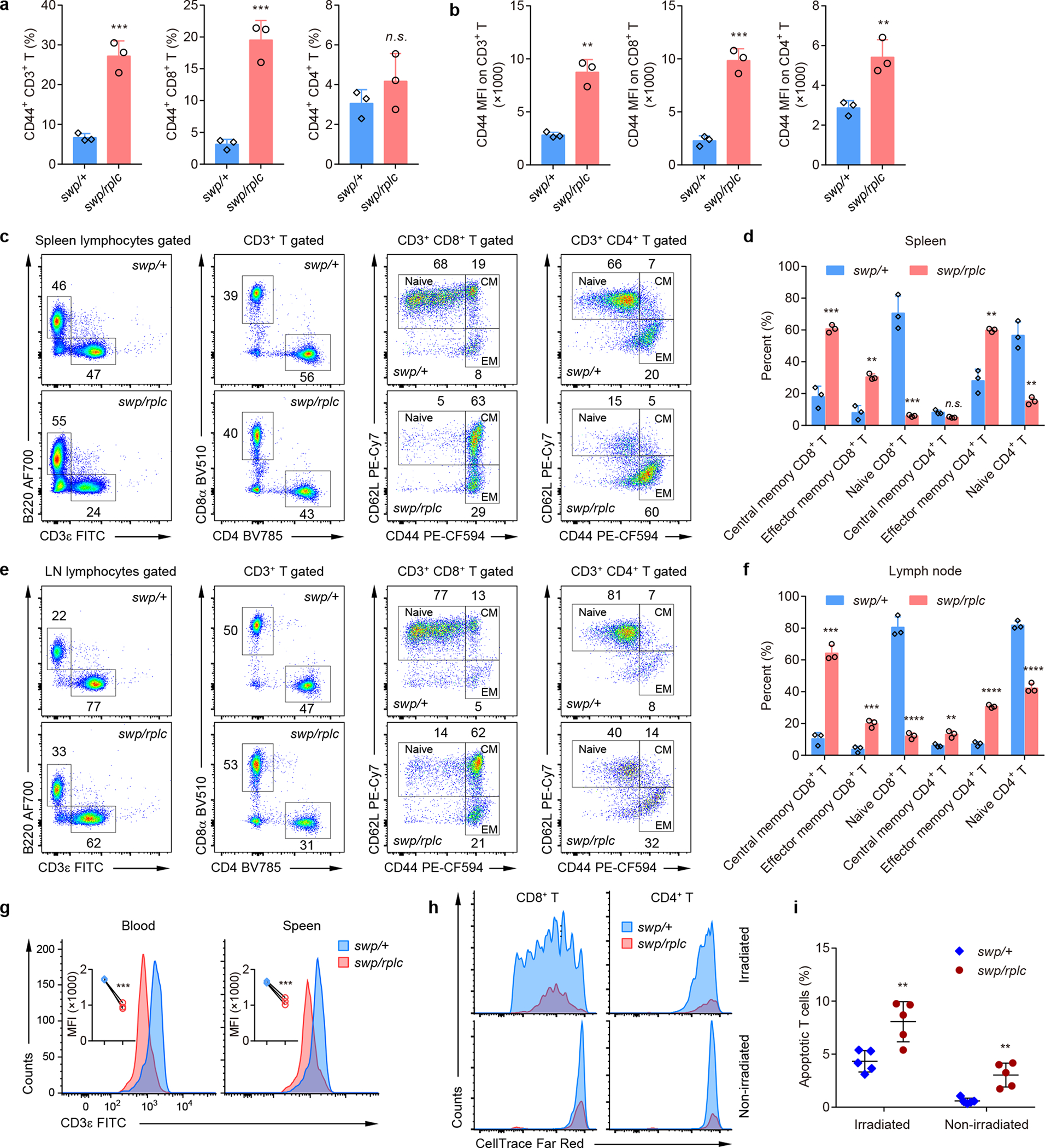
a, Percentage of CD44+ CD3+ T cells, CD44+ CD8+ T cells, and CD44+ CD4+ T cells in the blood of Snrnp40swp/rplc and Snrnp40swp/+ littermates (n=3 mice per genotype; ***P=0.0009, ***P=0.0008, n.s., not significant (P=0.2833), unpaired, two-tailed Student’s t-test). b, Mean fluorescence intensity (MFI) of CD44 staining on T cells in the blood of Snrnp40swp/rplc and Snrnp40swp/+ littermates (n=3 mice per genotype; **P=0.0011, ***P=0.0004, **P=0.0096, unpaired, two-tailed Student’s t-test). c-f, Flow cytometry of lymphocytes from spleen (c-d) and lymph node (e-f) of Snrnp40swp/rplc and Snrnp40swp/+ littermates (n=3 mice per genotype), assessing the expression of CD44 and CD62L on CD8+ and CD4+ T cells (c,e). Quantification of the percentage of central memory (CD44hiCD62L+), effector memory (CD44hiCD62L–) and naïve (CD44loCD62L+) T cells (d,f). ***P=0.00046, **P=0.0012, ***P=0.00049, **P=0.0016, **P=0.0013; n.s., not significant (P=0.055) in d, and ***P=0.00012, ***P=0.00081, ****P=0.000066, **P=0.006, ****P=0.00002, ****P=0.000061 in f (unpaired, two-tailed Student’s t-test). g, Flow cytometry of blood or spleen cells from Snrnp40swp/rplc and Snrnp40swp/+ mice, assessing the expression of CD3ε on T cells. Quantification of MFI of CD3ε staining on T cells is shown (inset) (n=3 mice per genotype; ***P=0.0001, ***P=0.0010, unpaired, two-tailed Student’s t-test). h,i, Flow cytometry assessing the proliferation (h) and apoptosis (i) of adoptively transferred CD8+ and CD4+ T cells in the spleens of recipient mice. An equal mixture of CellTrace™ Far Red-labeled pan T cells isolated from the spleens of Snrnp40swp/rplc and Snrnp40swp/+ mice was adoptively transferred into irradiated C57BL/6J wild-type hosts. Histograms showing CellTrace™ Far Red dilution in cells harvested from spleens of irradiated or non-irradiated hosts seven days after transfer (h). Percentage of annexin V-positive T cells four days after transfer (i) (n=5 recipient mice; **P=0.0046, **P=0.0015, unpaired, two-tailed Student’s t-test). Data are representative of two (h,i), three (c-f), five (a,b), or six (g) independent experiments (mean ± s.d. in a,b,d,f,i).
Thymi of Snrnp40swp/rplc mice displayed diminished size and cellularity compared to those of Snrnp40swp/+ mice (Fig. 3a,b). Thymocyte development was blocked at the CD4–CD8– double-negative (DN) stage, within which the DN1 and DN4 populations were reduced, and at the CD4+CD8+ double-positive (DP) stage, resulting overall in a shortage of single-positive CD4+ and CD8+ thymocytes (Fig. 3c–f). Together, these data indicate that hypomorphic mutation of Snrnp40 results in T cell deficiency due to impaired T cell development.
Figure 3. Defective T cell development in the thymus of Snrnp40swp/rplc mice.
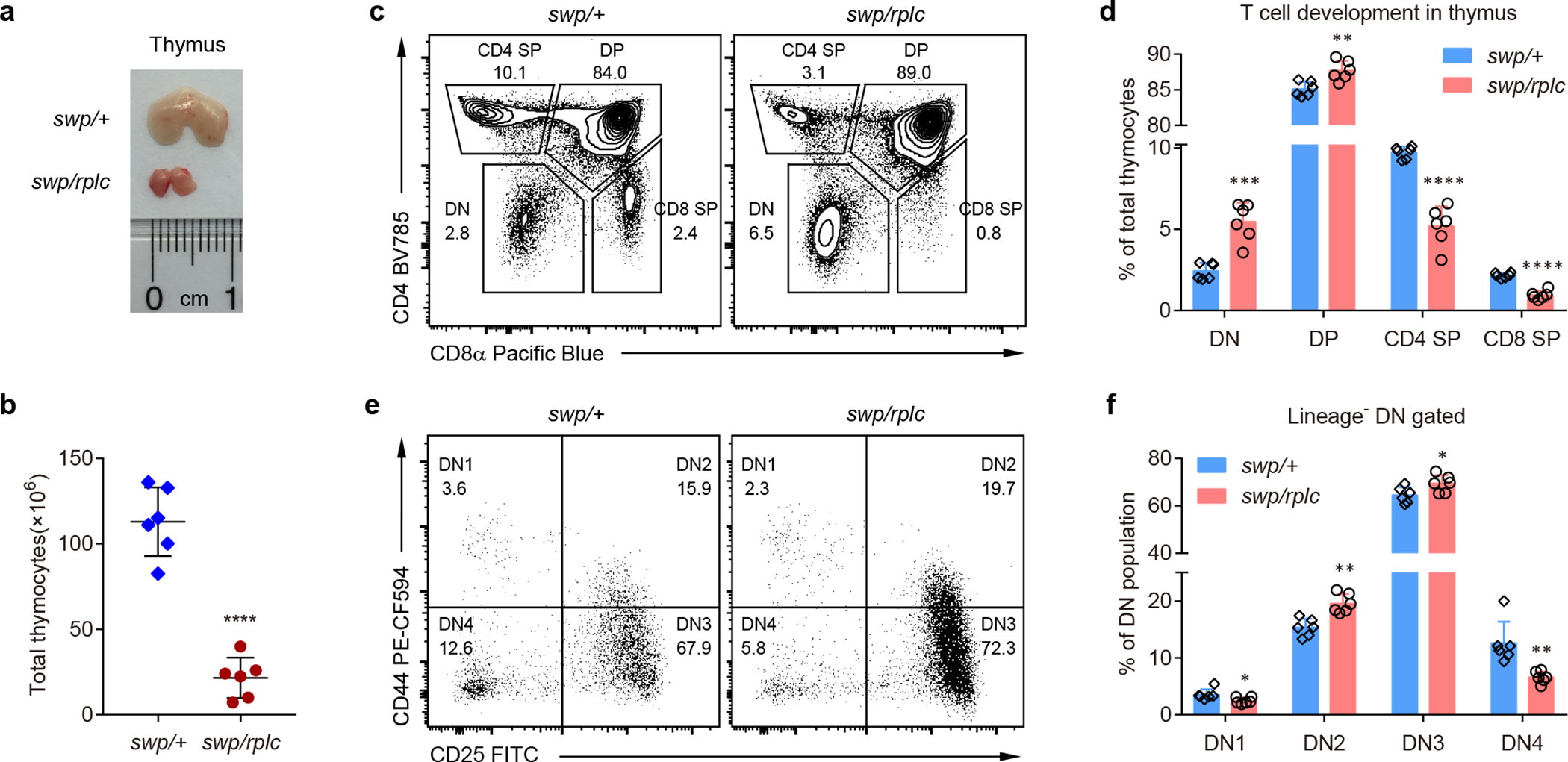
a, Representative image of the thymus from Snrnp40swp/rplc and Snrnp40swp/+ littermates. b, Total number of thymocytes in Snrnp40swp/rplc and Snrnp40swp/+ littermates (n=6 mice per genotype; ****P<0.0001, unpaired, two-tailed Student’s t-test). c,d, Flow cytometry of thymocytes from Snrnp40swp/rplc and Snrnp40swp/+ littermates, assessing the expression CD4 and CD8α (c). Numbers indicate percent cells in outlined areas. Quantification of the percentage of DN (CD4–CD8–), DP (CD4+CD8+), CD4 SP (CD4+CD8–) and CD8 SP (CD8+CD4–) T cells among total thymocytes (d) (n=6 mice per genotype; ***P=0.00016, **P=0.0044, ****P<0.00001, ****P<0.00001, unpaired, two-tailed Student’s t-test). e,f, Flow cytometry of Lineage– DN thymocytes from Snrnp40swp/rplc and Snrnp40swp/+ littermates, assessing the expression of CD25 and CD44 (e). Numbers indicate percent cells in each quadrant. Quantification of the percentage of DN1 (CD25–CD44+), DN2 (CD25+CD44+), DN3 (CD25+CD44–) and DN4 (CD25–CD44–) thymocytes among the Lineage– DN population (f) (n=6 mice per genotype; *P=0.036, **P=0.0011, *P=0.037, **P=0.0038, unpaired, two-tailed Student’s t-test). Data are representative of three (e,f), five (c,d), or seven (a,b) independent experiments (mean ± s.d. in b,d,f).
Snrnp40swp/swp and Snrnp40swp/rplc mice were highly susceptible to infections with normally non-lethal inocula of mouse cytomegalovirus (MCMV) or herpes simplex virus type 1 (HSV-1), accumulating elevated virus titers in the spleen and liver compared to wild-type mice (Fig. 4a,b). Whereas 100% of infected wild-type mice survived, 100% (8/8) or 75% (6/8) of Snrnp40swp/swp mice died by six days after infection with MCMV (Fig. 4c) or HSV-1 (Fig. 4d), respectively. Consistent with an inability to control the virus, elevated serum cytokine concentrations were detected in Snrnp40swp/swp mice 36 hours after inoculation with MCMV, with the notable exceptions of interferon-γ (IFN-γ) and interleukin-17A (IL-17A), which were downregulated (Fig. 4e–j). NK cells are critical for defense against MCMV16, and in Snrnp40swp/swp mice NK cells were impaired in their ability to kill target cells in an in vivo assay system (Fig. 4k,l) despite normal degranulation, granzyme B release, and tumor necrosis factor (TNF) and IFN-γ production in response to PMA/ionomycin stimulation in vitro (Supplementary Fig. 6a–d). The cytokine responses of Snrnp40swp/swp bone marrow-derived dendritic cells to several DNA and RNA viruses were largely normal; only TNF responses to MCMV were slightly diminished (Supplementary Fig. 6e–h). These data suggest that defective NK cell function and reduced NK cell numbers account for enhanced susceptibility of Snrnp40swp/swp mice to MCMV.
Figure 4. Viral susceptibility and impaired NK cell function in Snrnp40swp/swp and Snrnp40swp/rplc mice.
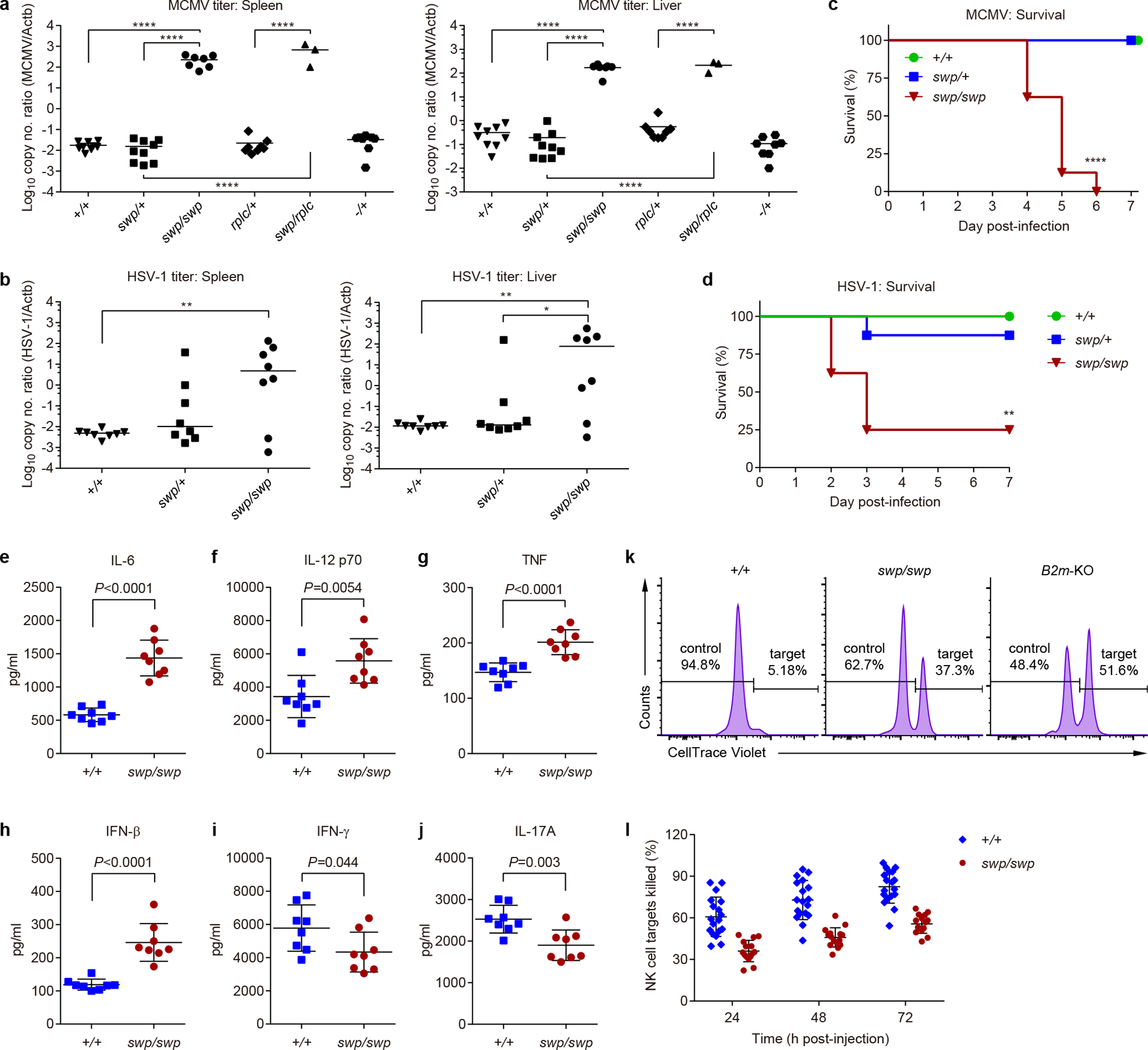
a-j, Mice were infected with MCMV (a,c,e-j) or HSV-1 (b,d). a, MCMV titers in the spleens (left) and livers (right) five days after MCMV infection (n=9 mice for +/+ and swp/+, n=8 mice for and rplc/+ and −/+, n=7 mice for swp/swp, n=3 mice for swp/rplc). Statistical significance was determined by one-way ANOVA with Sidak’s multiple comparisons using Log10 of viral titers. ****P<0.0001. b, HSV-1 titers in the spleens (left) and livers (right) seven days after HSV-1 infection (n=8 mice per genotype). Statistical significance was determined by one-way ANOVA with Sidak’s multiple comparisons using Log10 of viral titers. **P=0.0084, **P=0.0063, *P=0.048. c, Survival curves after MCMV infection (n=8 mice per genotype; ****P<0.0001, two-sided Log-rank (Mantel-Cox) test). The experiment was concluded after 7 days, but no additional death was observed for at least 10 additional days. d, Survival curves after HSV-1 infection (n=8 mice per genotype; **P=0.0011, two-sided Log-rank test). e-j, Serum concentrations of IL-6 (e), IL-12 p70 (f), TNF (g), IFN-β (h), IFN-γ (i), and IL-17A (j) 36 h after MCMV infection (n=8 mice per genotype; the indicated P values were determined by unpaired, two-tailed Student’s t-test). k,l, Equal numbers of CellTrace™ Violet-labeled C57BL/6J splenocytes (control cells) and B2m-KO splenocytes (NK target cells) were injected i.v. into recipient mice of the indicated genotypes. k, Flow cytometry of control and NK target cells remaining in the blood of recipient mice 3 days after injection. Numbers indicate percent of injected control and NK target cells. l, Quantification of the percentage of NK target cells killed in the recipient mice (n=18 mice for +/+, n=15 mice for swp/swp; ****P<0.0001, two-way ANOVA with Sidak’s multiple comparisons. Data are representative of two (b-d), three (a,e-j), or four (k,l) independent experiments (line indicates median in a,b; mean ± s.d. in e-j,l).
Reciprocal bone marrow transplantation using wild-type and Snrnp40swp/swp mice demonstrated that T cell, B cell, and NK cell deficiencies, and MCMV susceptibility of Snrnp40swp/swp mice were attributable to impaired Snrnp40 function in the hematopoietic compartment (Fig. 5a–f). Moreover, the hematopoietic defects were cell-intrinsic, as shown in mixed bone marrow chimeras generated by reconstituting lethally irradiated Rag2/Il2rg double-KO recipient mice with a 50:50 mixture of Snrnp40swp/rplc (CD45.2) and wild-type (CD45.1) bone marrow. After eight weeks, flow cytometry analysis demonstrated reconstitution of B cell, T cell, and NK cell compartments with 95–100% wild-type cells to the near complete exclusion of Snrnp40swp/rplc cells (Fig. 5g and Supplementary Fig. 7a–h). The development of Snrnp40swp/rplc macrophages and neutrophils was also slightly impaired compared to the wild-type cells in this system.
Figure 5. Hematopoietic cell-intrinsic defect in lymphoid development in Snrnp40swp/swp and Snrnp40swp/rplc mice.
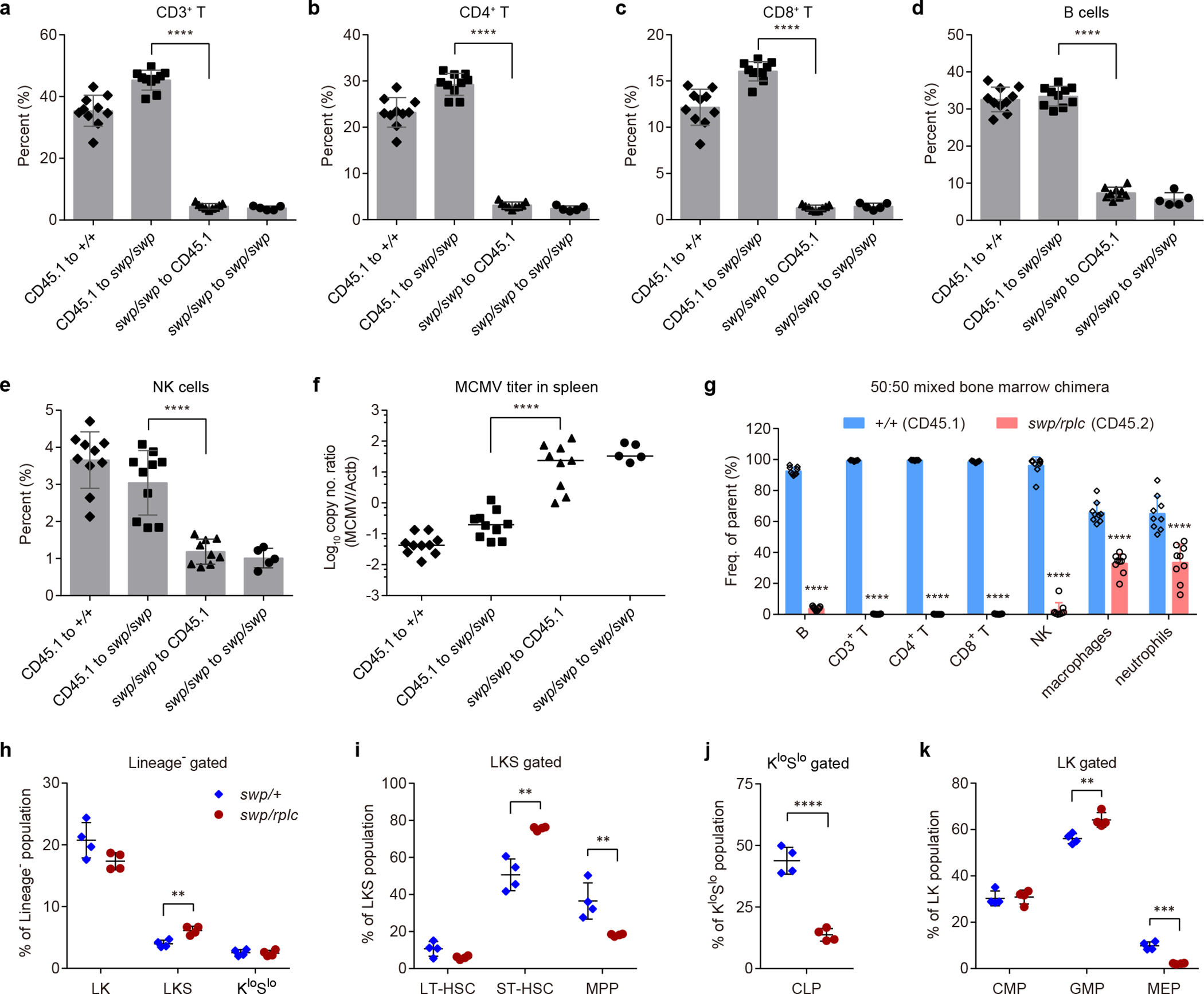
a-f, Reciprocal bone marrow transplantation. Quantification of the percentage of CD3+ T cells (a), CD4+ T cells (b), CD8+ T cells (c), B cells (d), and NK cells (e) in the blood of Snrnp40swp/swp or wild-type recipient mice (n=10 mice for CD45.1 to +/+ and CD45.1 to swp/swp, n=9 mice for swp/swp to CD45.1, n=5 mice for swp/swp to swp/swp; ****P<0.0001, one-way ANOVA with Sidak’s multiple comparisons). f, MCMV titers in the spleens of Snrnp40swp/swp or wild-type recipient mice five days after MCMV infection (n values same as in a-d, ****P<0.0001, one-way ANOVA with Sidak’s multiple comparisons using Log10 of viral titers). g, Mixed bone marrow chimeras. Quantification of the percentage of wild-type (CD45.1) and Snrnp40swp/rplc (CD45.2) immune cells in the blood of recipient mice (n=9 recipient mice per group; ****P<0.0001, unpaired, two-tailed Student’s t-test). Averaged data from two independent experiments are plotted. h-k, Analysis of hematopoietic stem and progenitor cell subpopulations in bone marrow of Snrnp40swp/+ or Snrnp40swp/rplc mice (n=4 mice per genotype). h, Quantification of the percentage of LKS, LK, and KloSlo cells in the Lineage– population (**P=0.0029, unpaired, two-tailed Student’s t-test). i, Quantification of the percentage of LT-HSC, ST-HSC, and MPPs in the LKS population (**P=0.0012, **P=0.0097, unpaired, two-tailed Student’s t-test). j, Quantification of the percentage of CLPs in the KloSlo population (****P<0.0001, unpaired, two-tailed Student’s t-test). k, Quantification of the percentage of CMPs, GMPs, and MEPs in the LK population (**P=0.0055, ***P=0.00012, unpaired, two-tailed Student’s t-test). Data are representative of two (a-g) or three (h-k) independent experiments (line indicates median in f; mean ± s.d. in a-e,g-k).
Analysis of hematopoietic stem and progenitor cells (HSPCs) in the bone marrow of Snrnp40swp/rplc mice by flow cytometry (Supplementary Fig. 7i,j) revealed that while long-term and short-term HSCs (LT-HSC and ST-HSC, respectively) were present at normal or elevated frequencies, MPPs were reduced ~45% (Fig. 5h,i). Importantly, the frequency of CLPs was reduced by ~70%, consistent with the decrease in mature lymphocytes observed in Snrnp40swp/rplc mice (Fig. 5j). Common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), and megakaryocyte–erythroid progenitors (MEPs) were mildly affected or not affected in Snrnp40swp/rplc mice (Fig. 5k). These findings indicate a hematopoietic cell-intrinsic requirement for Snrnp40 from the early stages of their development, during a time soon after commitment to the hematopoietic lineage. In addition, Snrnp40 is necessary for differentiation of MPPs specifically to the lymphoid lineage.
Impaired splicing and transcript expression in Snrnp40-deficient cells
We hypothesized that the immunological defects observed in Snrnp40-deficient mice originated from impaired splicing. Diminished expression of Snrnp200 but not Eftud2, components of the U5 snRNP subcomplex containing Snrnp404, was detected in Snrnp40swp/swp splenocytes (Supplementary Fig. 8a). Cd2bp2 (also known as U5–52K), another U5 snRNP component thought to aid in formation of the U4/U6.U5 tri-snRNP17, was expressed at normal levels in Snrnp40swp/swp splenocytes (Supplementary Fig. 8a). We identified by mass spectrometry dozens of splicing factors and regulators that coimmunoprecipitated with Snrnp40 from lysates of EL4 cells, a T cell line, or NK-92 cells, an NK cell line (Supplementary Fig. 8b–g, Supplementary Datasets 1 and 2). Therefore, we sought to identify transcripts dependent upon Snrnp40 for splicing in lymphoid cells. We analyzed the effect of Snrnp40 mutation on splicing in EL4 cells, T cells, and HSPCs. For EL4 cells, we used CRISPR-Cas9 targeting to knock out Snrnp40 (Fig. 6a) and then performed RNA-seq analysis under conditions of PMA/ionomycin stimulation or no stimulation. A total of 281 splicing errors were identified, of which 241 occurred more frequently in Snrnp40-KO cells vs. wild-type cells and 40 occurred more frequently in wild-type cells vs. Snrnp40-KO cells (Fig. 6b,c and Supplementary Dataset 3); all errors were detected in both mutant and wild-type cells. Three types of errors, intron retention, exon skipping, and cryptic splice site usage, were detected. Strikingly, intron retention accounted for 89% (n = 214) of the splicing errors that occurred more frequently in mutant cells, compared to only 30% (n = 12) of those more frequent in wild-type cells (Fig. 6b–e, Supplementary Table 3, and Supplementary Dataset 4). In contrast, exon skipping was the most common type of error among splicing errors that were more frequent in wild-type vs. Snrnp40-KO cells (Fig. 6b,c and Supplementary Table 3). Although the numbers and percentages of aberrant transcripts sufficient to cause phenotypic effects are not known and will vary by gene, imposing criteria of <10% aberrant of a particular transcript in wild-type cells and >30% aberrant of that transcript in mutant cells excludes all but eight distinct errors of the 281 total (unstimulated condition: Amdhd2, Pcnx3, Polm, Rsbn1l, Mapk8ip3; PMA/ionomycin condition: Polrmt, Pcnx3, Polm, Traf4, Ppp1r16a, Mapk8ip3; all with retained introns). That is, few errors were found at vastly different frequencies in Snrnp40-KO vs. wild-type cells (Fig. 6b,c). These data indicate that complete deficiency of Snrnp40 moderately increases the frequency of missplicing at hundreds of exon-intron-exon junctions, predominantly resulting in intron retention.
Figure 6. Splicing errors in Snrnp40-mutant cells.
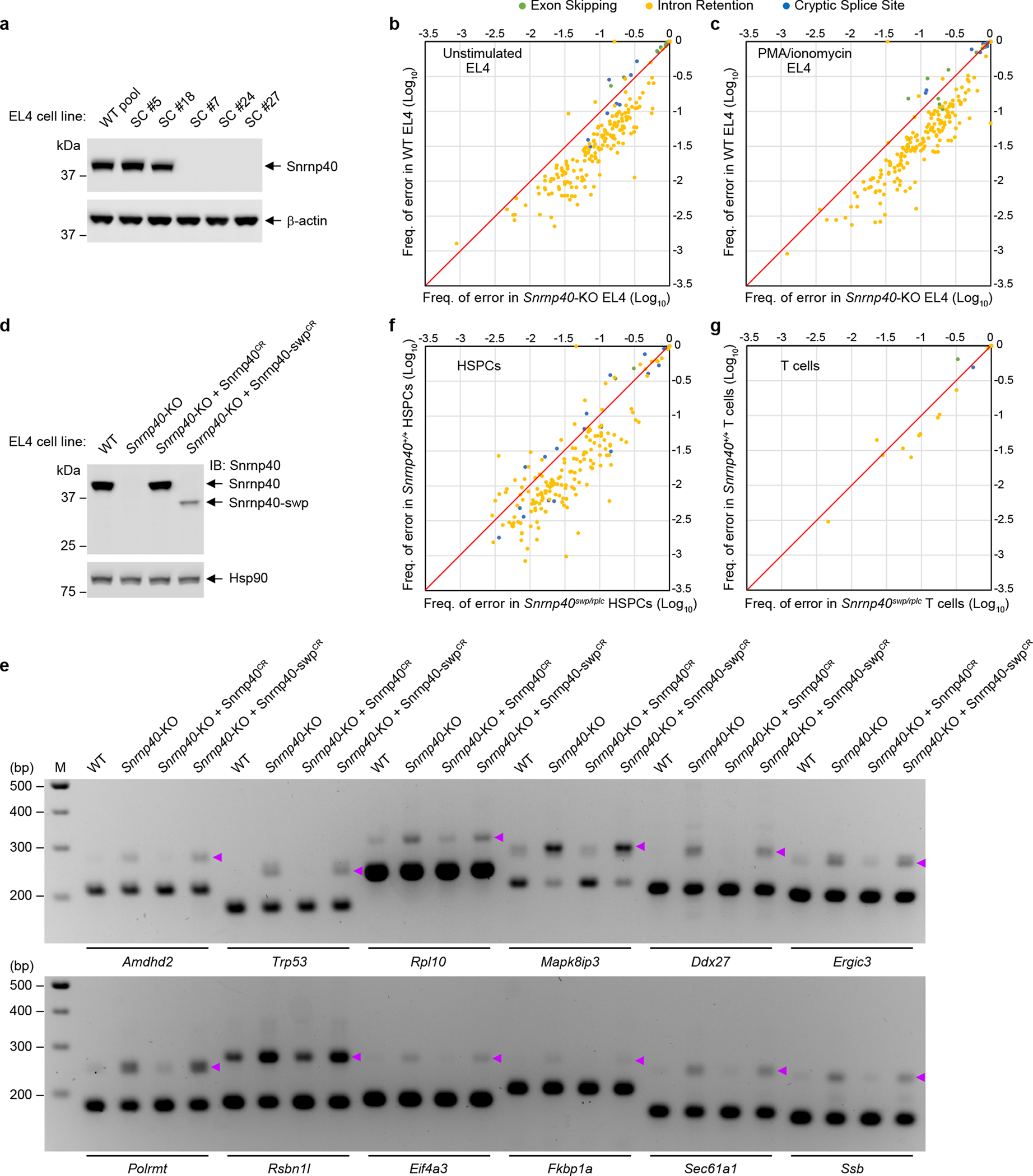
a, Immunoblot analysis of Snrnp40 expression in five clonal EL4 cell lines targeted by CRISPR-Cas9. DNA sequencing showed that three clones (#7, #24, #27) were compound heterozygous for different Snrnp40 null alleles, and no Snrnp40 expression was detected by immunoblot. #5 had the wild-type Snrnp40 sequence, while #18 was heterozygous for a null allele. b,c, Comparative analysis of the frequency of 281 splicing errors in Snrnp40-KO EL4 cells vs. wild-type EL4 cells not stimulated (b) or stimulated with PMA/ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin) (c). d, Immunoblot analysis of Snrnp40 expression in wild-type (WT), Snrnp40-KO, Snrnp40-KO reconstituted with CRISPR-resistant WT Snrnp40 (Snrnp40-KO + Snrnp40CR), and Snrnp40-KO reconstituted with CRISPR-resistant swp form of Snrnp40 (Snrnp40-KO + Snrnp40-swpCR) EL4 cells. Note that Snrnp40-swpCR expression level was much lower than that of Snrnp40CR, similar to Fig. 1d. e, RT-PCR analysis of splicing errors resulting in retained introns in EL4 cells described in d. Retained introns were detected in genes including Amdhd2, Trp53, Rpl10, Mapk8ip3, Ddx27, Ergic3, Polrmt, Rsbn1l, Eif4a3, Fkbp1a, Sec61a1, and Ssb. Purple arrowheads indicate longer PCR products containing retained introns. f,g, Comparative analysis of the frequency of 218 splicing errors in Snrnp40swp/rplc HSPCs vs. Snrnp40+/+ HSPCs (f) and the frequency of 25 splicing errors in Snrnp40swp/rplc T cells vs. Snrnp40+/+ T cells (g). Each symbol represents one splicing error in a particular transcript; the frequency of the error among total sequences containing that junction (PIR or PSI) in Snrnp40-mutant cells or in wild-type cells is plotted. Diagonal line represents equality in Snrnp40-mutant cells and wild-type cells. Experiment was performed two (d,e) or five (a) times with similar results. RNA-seq experiment was performed one time (b,c,f,g). The blots (a,d) and gels (e) were cropped to show relevant bands and their original images are presented in the Source Data.
Since null alleles of Snrnp40 are homozygous lethal in mice, we isolated HSPCs and T cells from Snrnp40swp/rplc mice for RNA-seq analysis. Among Snrnp40 viable allelic combinations examined, swp/rplc resulted in expression of wild-type Snrnp40 at 6.7% of levels in Snrnp40+/+ mice, and was phenotypically the most severe (Supplementary Table 2). A total of 218 and 25 splicing errors were identified, respectively, in HSPCs and T cells (Supplementary Datasets 4, 5 and 6) and as for EL4 cells the majority of the errors were more frequent in Snrnp40swp/rplc cells compared to wild-type cells (Fig. 6f,g). Moreover, intron retention again represented the most common type of splicing error that occurred more frequently in mutant HSPCs or T cells, representing 86.7% and 84% of those errors, respectively (Fig. 6f,g). Thus, in all three cell types examined the main effect of reduced Snrnp40 function was impaired splicing resulting in intron retention.
We also examined transcript abundance in Snrnp40-mutant EL4 cells, HSPCs, and T cells since we expected that differential expression of many genes may result from nonsense-mediated decay of misspliced mRNAs. In EL4 cells, we identified 341 and 511 differentially expressed genes (≥1.5-fold change), respectively, in unstimulated and PMA/ionomycin-stimulated Snrnp40-KO EL4 cells compared to wild-type cells under the same conditions (Fig. 7a,b and Supplementary Dataset 7). 197 of these genes were commonly affected in both unstimulated and stimulated conditions (Fig. 7b and Supplementary Dataset 7). Among all 655 affected genes, functional categorization by gene ontology (GO) annotations showed that 101 (15.4%) had immune system functions (65 downregulated and 36 upregulated in Snrnp40-KO cells) (Fig. 7a and Supplementary Dataset 8). We verified the downregulation of seven candidate genes (Themis, Ikzf3, Slamf6, Cd226, Tnfsf10, Nt5e and Arg1) in Snrnp40-KO EL4 cells (Fig. 7c) and Snrnp40swp/rplc splenocytes (Fig. 7d) by RT-qPCR. Downregulation of the hematopoietic-specific transcription factor Ikzf3 was verified in Snrnp40swp/rplc bone marrow cells (Fig. 7e,f). Downregulation at the protein level was further confirmed for Themis in thymocytes (Fig. 7g), and downregulation of IL-17A and IL-17F at the mRNA and protein levels was confirmed in Snrnp40-KO EL4 cells (Fig. 7h–k).
Figure 7. Diminished expression of mRNAs and proteins with immune system functions in Snrnp40-KO EL4 cells.
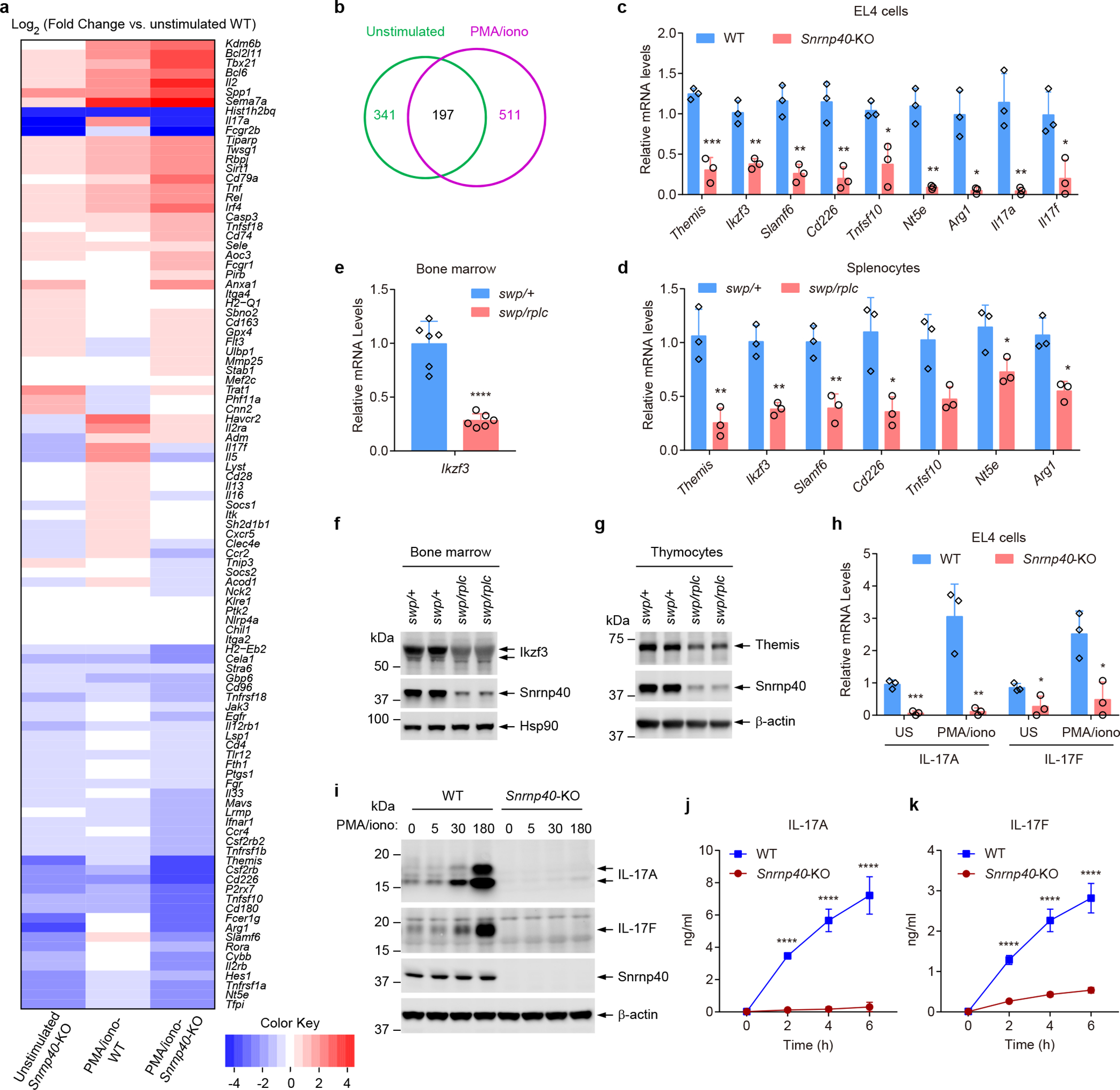
a, Heatmap for differentially expressed immune genes in unstimulated or PMA/ionomycin stimulated Snrnp40-KO and wild-type EL4 cells. Data represent Log2 of fold change vs. unstimulated WT. b, Venn diagram for 655 unique differentially expressed genes. 30.0% (197) were affected in both unstimulated and stimulated conditions. c, RT-qPCR analysis of the indicated mRNAs in Snrnp40-KO and wild-type EL4 cells (n=3 independent cell lines per group; ***P=0.00063, **P=0.0024, **P=0.0023, **P=0.0043, *P=0.014, **P=0.0014, *P=0.01, **P=0.0062, *P=0.044, unpaired, two-tailed Student’s t-test). d, RT-qPCR analysis of the indicated mRNAs in Snrnp40swp/+ and Snrnp40swp/rplc splenocytes (n=3 mice per genotype; **P=0.0080, **P=0.0025, **P=0.0058, *P=0.021, *P=0.022, *P=0.039, **P=0.0078, unpaired, two-tailed Student’s t-test). e, RT-qPCR analysis of Ikzf3 in Snrnp40swp/+ and Snrnp40swp/rplc bone marrow (n=6 mice per genotype; ****P<0.0001, unpaired, two-tailed Student’s t-test). f,g, Immunoblot analysis of Ikzf3 expression in bone marrow (f) and Themis expression in thymocytes (g) of Snrnp40swp/+ and Snrnp40swp/rplc mice. h, RT-qPCR analysis of Il17a and Il17f in unstimulated (US) and PMA/ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin) stimulated Snrnp40-KO and wild-type EL4 cells (n=3 independent cell lines per group; ***P=0.00045, **P=0.0073, *P=0.041, *P=0.016, unpaired, two-tailed Student’s t-test). i, Immunoblot analysis of IL-17A and IL-17F expression in Snrnp40-KO and wild-type EL4 cells at indicated time points after PMA/ionomycin stimulation. j,k, Concentration of IL-17A (j) and IL-17F (k) in the supernatants of Snrnp40-KO and wild-type EL4 cells (n=3 independent cell lines per group) at different time points after PMA/ionomycin stimulation. ****P<0.0001, two-way ANOVA with Sidak’s multiple comparisons test). RNA-seq experiment was performed one time (a,b). Data are representative of two (c-e,j,k) or three (f-i) independent experiments (mean ± s.d. in c-e,h,j,k). Quantities of mRNA are expressed relative to Gapdh transcript level (c-e,h). The blots (f,g,i) were cropped to show relevant bands and their original images are presented in the Source Data.
In HSPCs, we identified 1,867 genes that were differentially expressed between Snrnp40swp/rplc and wild-type cells (genes with fold-changes of any magnitude); 1,047 (56.1%) had lower expression in Snrnp40swp/rplc cells while 820 (43.9%) had lower expression in wild-type cells (Fig. 8a and Supplementary Dataset 9). Of the 1,867 affected genes, 452 (24.2%) were classified as having immune system functions by GO annotations (257 downregulated and 195 upregulated in Snrnp40swp/rplc cells) (Supplementary Dataset 10). For T cells, we found 1,745 differentially expressed genes between Snrnp40swp/rplc and wild-type cells (genes with fold-changes of any magnitude); 400 (22.9%) had lower expression in Snrnp40swp/rplc cells while 1,345 (77.1%) had lower expression in wild-type cells (Fig. 8a and Supplementary Dataset 11). Of the 1,745 affected genes, 473 (27.1%) had immune system functions according to GO annotations (71 downregulated and 402 upregulated in Snrnp40swp/rplc cells) (Supplementary Dataset 12). The favored candidate genes for aspects of the swp immunological phenotypes are listed in Supplementary Table 4. We validated the expression changes (Fig. 8b) and intron retention events (Fig. 8c) in several candidate genes using mRNA from HSPCs isolated from a separate group of mice. Comparing the differentially expressed genes from EL4 cells to those from T cells, we initially expected to find significant overlap in the affected genes and in their directions of change. However, we found that there was no discernable correlation between the effect of Snrnp40 mutation on gene expression in EL4 cells and the effect on gene expression in T cells (R2=0.0002; Fig. 8d). Taken together, these data suggest that the primary effect of Snrnp40 deficiency is aberrant splicing, particularly intron retention, which leads to cell type-specific expression of mutant proteins as well as diminished expression of proteins that collectively account for the immune deficiency in mice with deficits of Snrnp40 function.
Figure 8. Impaired splicing and transcript expression in primary Snrnp40swp/rplc HSPCs and T cells.
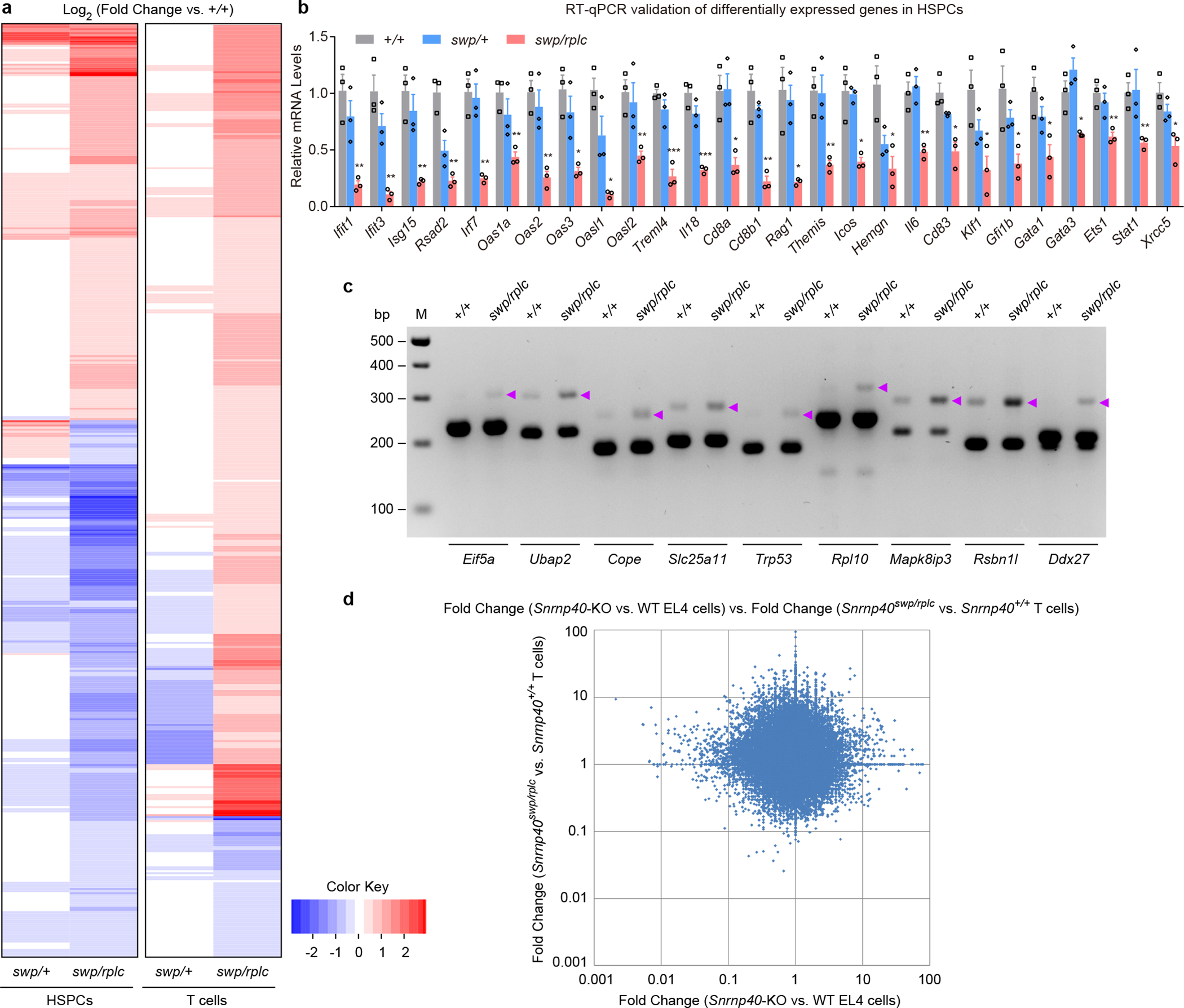
a, Heatmap for differentially expressed immune genes in HSPCs and T cells. b, RT-qPCR analysis of the indicated mRNAs in Snrnp40+/+, Snrnp40swp/+, and Snrnp40swp/rplc HSPCs (n=3 mice per genotype). Data are mean ± s.e.m.; P values (Snrnp40+/+ vs. Snrnp40swp/rplc HSPCs) were determined by unpaired, two-tailed Student’s t-test. **P=0.0051, 0.0033, 0.0049, 0.0013, 0.0027, 0.0040, 0.0029, *P=0.018, 0.013, **P=0.0082, ***P=0.00051, 0.00061, *P=0.013, **P=0.0063, *P=0.020, **P=0.0069, *P=0.013, 0.049, **P=0.0061, *P=0.011, 0.029, 0.038, 0.025, 0.021, **P=0.0081, 0.0088, *P=0.018. Quantities of mRNA are expressed relative to Gapdh transcript level. c, RT-PCR analysis of splicing errors resulting in retained introns in Snrnp40+/+ and Snrnp40swp/rplc HSPCs. Retained introns in genes Eif5a, Ubap2, Cope, Slc25a11, Trp53, Rpl10, Mapk8ip3, Rsbn1l, and Ddx27 are shown. Purple arrowheads indicate longer PCR products containing retained introns. d, Comparison between the effect of Snrnp40 mutation on gene expression in EL4 cells vs. the effect in T cells. Differential expression data from EL4 cell and T cell RNA-seq experiments were plotted. The graph shows the fold-changes of exons that were differentially expressed in Snrnp40-KO EL4 cells plotted vs. the fold-changes of the corresponding exons in Snrnp40swp/rplc T cells (R2=0.0002). Fold-changes for each cell type are relative to the corresponding wild-type cells. RNA-seq experiment was performed one time (a,d). Data are representative of two independent experiments (b,c). The gel (c) was cropped to show relevant bands and it original image is presented in the Source Data.
Discussion
In mice, approximately 1/3 of all genes are essential to permit survival from conception through weaning age18–21. Null alleles of these genes are not easily studied without conditional expression. However, viable hypomorphic alleles causing quantitative or qualitative effects on the encoded protein may permit investigation of post-developmental functions, and affect a particular cell or tissue type disproportionately. Here we describe one such example, in which a mutation that diminished the quantity of U5 snRNP component Snrnp40 caused prominent immune dysfunction. Snrnp40 is most highly expressed in lymphoid tissue in the adult mouse, and phenotypic effects limited to a few tissues appear to be a typical outcome of hypomorphic mutations of core spliceosomal proteins in humans or mice. For example, mutations of six core snRNP proteins are reported to cause retinitis pigmentosa in humans22 (PRPF3, PRPF4, and PRPF31 in U4/U6 snRNP; PRPF6, PRPF8, and SNRNP200 in U5 snRNP). Mutations in core proteins of the minor snRNPs also cause tissue-specific effects, most notably to the nervous system23. Our observations of hematopoietic and lymphoid defects in Snrnp40-deficient mice corroborate findings that mutations in Prpf8 and Eftud2, also components of the salt-stable U5 snRNP subcomplex, are associated with hematopoietic defects. PRPF8 mutations are associated with myelodysplastic syndrome in humans24 and heterozygous null mutations of Eftud2 result in decreased numbers of leukocytes, lymphocytes, and neutrophils in mice19.
Data obtained using yeast, fly, or mammalian systems indicate that only a small fraction of splicing events is affected by mutations of various core snRNP proteins25–28, consistent with our finding that only a few hundred splicing errors were amplified in Snrnp40-KO EL4 cells or primary Snrnp40swp/rplc HSPCs compared to wild-type cells. Interestingly, we detected far fewer splicing errors in Snrnp40swp/rplc primary T cells than in Snrnp40swp/rplc HSPCs or Snrnp40-KO EL4 cells, a phenomenon that we do not presently understand. In all three cell types examined, most splicing errors were intron retention, possibly because spliceosome assembly might proceed normally up to the stage of the pre-spliceosome association with an intron, after which U4/U6.U5 tri-snRNP recruitment and the catalytic steps of splicing fail due to U5 snRNP dysfunction. Our data support the proposal that pre-mRNA substrates differ in their requirement for core components of the spliceosome29. Surprisingly, few (in HSPCs and T cells) to none (in EL4 cells) of the unspliced or aberrantly spliced genes corresponded with those found to be differentially expressed in the mutant cells. Thus, we conclude that reduced levels of specific mRNA molecules are generally an indirect effect of Snrnp40 mutation.
No single gene deficiency appeared to be solely responsible for the skywarp immunological phenotypes, and both the translation of unspliced/misspliced transcripts and altered gene expression may contribute. We favor the hypothesis that reductions in the expression of several immune regulators in aggregate may account for the developmental and functional defects observed in skywarp homozygous mice. Indeed, we could identify no single gene deficiency that phenocopied Snrnp40 deficiency, likely due to the simultaneous reduction of the expression of many genes in Snrnp40swp/swp mice. We had anticipated that comparison of gene expression changes caused by Snrnp40 mutation in EL4 cells and primary T cells might reveal numerous genes similarly affected in both cell types as key molecules responsible for the skywarp T cell phenotype. However, complete knockout of Snrnp40 expression in EL4 cells results in quite different effects than the reduction of Snrnp40 expression in T cells.
Themis deficiency, observed in both Snrnp40-KO EL4 cells and in Snrnp40swp/rplc primary T cells, is an attractive candidate that may contribute to the T cell phenotypes in Snrnp40-mutant mice. Heterozygosity for a hypomorphic mutation of Themis results in reduced CD4+ and CD8+ T cells in blood30, supporting the hypothesis that reductions of Themis transcript (~70% reduction) and protein levels in Snrnp40-deficient thymi and spleens could be responsible for the T cell developmental phenotypes of Snrnp40-deficient mice; these phenotypes are highly similar to those of Themis-deficient mice31–33. In particular, both Snrnp40-deficient and Themis-deficient mice showed a block in thymocyte development from the DP to SP stage, and a decrease in CD4+ and CD8+ T cells. Themis regulates positive selection by controlling TCR signal strength31, 34, and its reduction in Snrnp40swp/rplc mice may contribute to the observed impairment in DP to SP thymocyte differentiation.
The elevated susceptibility of Snrnp40-deficient mice to MCMV infection is likely due to the combination of NK cell deficiency and impaired NK cell function. An intact IL-17 pathway is necessary for development of functional competence by NK cells, and mice deficient in IL-17RA, IL-17A, or IL-17F display impaired NK cell-mediated cytokine responses and increased susceptibility to MCMV35. We detected 96% and 60% reductions, respectively, in Il17a and Il17f mRNA expression in unstimulated Snrnp40-KO EL4 cells, which were confirmed at the protein level. Moreover, homozygous skywarp mice infected with MCMV had reduced serum IL-17A compared to wild-type mice. Thus, NK cell dysfunction in Snrnp40swp/swp mice may be due to reduced IL-17 signaling. IFN-γ, a cytokine produced abundantly by NK cells, was also diminished in the serum of MCMV infected Snrnp40swp/swp mice. Other antiviral protein-encoding genes including Oas1a, Irf7, and Ifit1 showed reduced expression in Snrnp40swp/rplc HSPCs and T cells. We predict that Snrnp40swp/swp mice may be susceptible to other types of viral infection.
In HSPCs, the expression of numerous transcriptional regulators was reduced as a consequence of Snrnp40 mutation and we hypothesize that deficiencies of key immune transcription factors may contribute to the impaired development of MPPs, CLPs, and subsequent lymphocyte lineages. For example, the transcriptional activator GATA-3 is required for thymocyte and early lineage T progenitor development36–39 and its expression was reduced approximately 40% in Snrnp40swp/rplc HSPCs. Ets1 expression was also reduced by about 40% in Snrnp40swp/rplc HSPCs; this transcription factor is necessary for NK cell and CD8+ T cell development40–42. Reductions of Klf143, 44 and Gfi1b45–47 may be partially responsible for the erythrocyte and platelet defects observed in Snrnp40swp/rplc mice. The broad effect of Snrnp40 mutation on transcript expression in HSPCs, particularly those encoding transcription factors, again suggests cumulative effects of these abnormalities causes the skywarp phenotype.
Mice with up to 93.3% reductions in wild-type Snrnp40 expression were viable despite a broad effect of Snrnp40 deficiency on mRNA expression levels. However, levels below 6.7% of wild-type expression were insufficient for survival, reflecting the fundamental importance of splicing for cellular functions. In contrast to the embryonic lethality of mice with < 6.7% Snrnp40 expression, T cells of the EL4 line completely lacking Snrnp40 expression were viable. We found that among 341 and 511 genes with expression level changes greater than 1.5-fold in unstimulated and PMA/ionomycin-stimulated Snrnp40-KO EL4 cells, respectively, compared to wild-type cells, a total of only 11 genes completely failed to be expressed in Snrnp40-KO cells (seven for each condition, three overlapping). We infer that sufficient quantities of all cell-essential genes are expressed and adequately processed and translated in Snrnp40-KO cells, permitting survival. Essential developmental processes appear to be more sensitive to Snrnp40 deletion than processes necessary for continued survival of cultured T cells. We note that while hematopoietic phenotypes were prominent in our analyses of Snrnp40-mutant mice, other phenotypes including a defect in melanoblast migration were also observed, but their mechanistic causes have yet to be investigated.
In conclusion, we have identified Snrnp40 as a key molecule necessary for the development of lymphocytes and monocytes, and for the antiviral innate immune response. The high level of conservation between human and mouse SNRNP40 protein sequences raises the possibility that viable hypomorphic mutations of SNRNP40 may in some cases also be responsible for immune dysfunction in humans.
Online Methods
Experimental Animals
C57BL/6J, Rag2 KO, and Rag2/Il2rg double-KO mice were purchased from The Jackson Laboratory. The swp homozygotes were obtained from intercrossing of C57BL/6J mice carrying the spontaneous swp mutation. Mice were maintained at the University of Texas Southwestern Medical Center, and all husbandry and experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee.
Generation of Snrnp40-mutant mice and cells
CRISPR-Cas9–mediated gene targeting system was used to generate Snrnp40 knockout (KO) and replacement (rplc) alleles.
For generation of the Snrnp40 KO allele, the sgRNA sequence was 5′-TCACCCCAATGGATCCACCC-3′. The KO allele contains a 4-bp deletion within the sequence 5′-CCCAATGGA[TCCA]CCCTGGCTTCTGCAGG-3′ (deletion in brackets) in exon 2 of Snrnp40, and is predicted to cause a frame shift beginning after amino acid 79 of the wild type Snrnp40 protein and terminating after inclusion of nine aberrant amino acids.
For generation of the Snrnp40rplc allele, the sgRNA sequence was 5′-CUUGCUUUCUGCUUCAGCUU-3′, and the DNA template used for the replacement was: 5′-GGAGGATGCGCTGTGACTGCTGTGGCTGCCCTGAGAACAGTGTTACTTACAGTGCTTGCTTTCTGCTTGAGCTTTGGGATATCCGGAAGAAAGCAGCTGTCCAGACATTTCAGAACACGTACCAGGTGTTGGCTGTGACCTTCAATGAC-3′. The rplc allele contains the original swp mutation in cis with a marker mutation consisting of a synonymous exonic nucleotide substitution 11 bp 3′ to the primary mutation site (sense strand): 5′-CAGTGCTTGCTTTCTGCTT(C→G)AGCTTTGGGA(C→T)ATCCGGAAGAAAGCAGCTGT-3′. The marker mutation affects the ninth nucleotide in exon 5.
For genotyping, Snrnp40 genomic DNA amplified across the mutation site was sequenced. For genotyping of Snrnp40 KO alleles, the PCR primers were: 5′-AGGAAGGTCTCTTGTGAAAGAC-3′ (fwd) and 5′-CGAAGCAACCTACTTTCCAATTAG-3′ (rev), and the sequencing primer was: 5′-ATGTTAAGATTCTGAAGCATAA-3′. For genotyping of rplc as well as swp alleles, the PCR primers were: 5′-AGGGAGTCAGATCCTTTGGAATTGG-3′ (fwd) and 5′-GGACAGAGCAAGAGAAGTGCATTTATTT-3′ (rev); and the sequencing primer was: 5′-AAGTGGGTGCAGGGAATCATACG-3′.
For generation of Snrnp40-KO EL4 T cells, cells were infected with lentivirus (lentiCRISPRv2) encoding sgRNA 5′- GAUAACUAUGCGACGUUGAA-3′ and Cas9. After puromycin selection, single cells were selected for subculture and immunoblotting was used to confirm knockout of Snrnp40 expression. For reconstitution of Snrnp40 or Snrnp40swp expression in the Snrnp40-KO cells, lentiviral vector pLV-EF1a-MCS-IRES-Hyg (BioSettia) harboring CRISPR-resistant sequence 5′- GAcAAtTAcGCcACcTTaAA-3′ (without changing amino acids) was used.
Antibodies
Primary antibodies used for immunoblotting were: anti-Snrnp40 (HPA026527) from Atlas; anti-FLAG M2 (F1804) from Sigma-Aldrich; anti-Themis (06–1328) and anti-GAPDH (MAB374) from Millipore; anti-Snrnp200 (A303–453A-T) from Bethyl; anti-Eftud2 (10208–1-AP) from Proteintech; anti-Cd2bp2 (PA5–18286) from Invitrogen; anti-Ikzf3 (Aiolos, 15103), anti-IL-17A (13838), anti-IL-17F (13186), anti-α-tubulin (3873), anti-β-actin (3700), anti-Hsp90 (4874) and anti-GFP (2956) from Cell Signaling. Antibodies used for flow cytometry were: anti-CD3ε (145–2C11), anti-CD4 (RM4–5), anti-CD8α (53–6.7), anti-B220 (RA3–6B2), anti-NK-1.1 (PK136), anti-CD25 (PC61.5), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD11c (HL3), anti-CD11b (M1/70), anti-F4/80 (BM8), anti-mouse Lineage Cocktail (CD3/Ly-6G/CD11b/B220/Ter-119), anti-c-Kit (CD117, 2B8), anti-Sca-1 (Ly-6A/E, D7), anti-IL-7Rα (CD127, SB/199), anti-CD16/32 (93), anti-Flk-2 (CD135, A2F10), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD107a (Lamp-1, 1D4B), anti-TNF (MP6-XT22) from BioLegend or BD Biosciences, and anti-CD34 (RAM34), anti-granzyme B (NGZB), anti-IFN-γ (XMG1.2) from eBioscience.
Hematological Analysis
Hematological analyses were performed on a HemaVet 950FS Hematology Analyzer (Drew Scientific).
Flow Cytometry
Flow cytometry was performed on an LSRFortessa cell analyzer (BD Biosciences) and analyzed with FlowJo software (Tree Star). Immune cells were analyzed using the following markers: CD3+ T cells (CD3ε+); CD4+ T cells (CD3ε+CD4+); CD8+ T cells (CD3ε+CD8+); B cells (B220+); NK cells (NK-1.1+CD3ε–); neutrophils (CD11b+F4/80–); macrophages (CD11b+F4/80+); DN (CD4–CD8α–); DP (CD4+CD8α+); CD4 SP (CD4+CD8α–); CD8 SP (CD8α+CD4–); DN1 (CD44+CD25–); DN2 (CD44+CD25+); DN3 (CD44–CD25+); DN4 (CD44–CD25–); central memory T cells (CD44hiCD62L+); effector memory T cells (CD44hiCD62L–); naïve T cells (CD44loCD62L+).
For flow cytometry analysis of HSCs and progenitor cells48, Lineage– cells from the single cells were further analyzed based on c-Kit and Sca-1 expression to define three populations: LKS (c-Kit+Sca-1+), LK (c-Kit+Sca-1lo), and KloSlo (c-KitloSca-1lo). CLPs were identified as IL-7Rα+ cells in the KloSlo population. LT-HSC, ST-HSC, and MPPs were respectively identified as CD34–Flk-2–, CD34+Flk-2–, and CD34+ Flk-2+ cells in the LKS population. MEPs, CMPs, and GMPs were respectively identified as CD34–CD16/32–, CD34+CD16/32–, and CD34+CD16/32+ cells in the LK population.
MCMV Susceptibility Screen and MCMV Quantification
The generation of MCMV (Smith strain) stock from the salivary glands of 4-week-old MCMV-infected BALB/c mice, as well as the protocol for in vivo MCMV susceptibility screen of ENU germline mutants, can be found at https://mutagenetix.utsouthwestern.edu/protocol/protocol_list.cfm. Briefly, MCMV was administered i.p. at 105 PFU per 20 g body weight. Five days later, ~30 mg of spleen or liver was harvested from each mouse. Total genomic DNA (including from mouse host and MCMV virus) was extracted by incubating the tissues in 200 μl of PBND buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2, 0.1 mg/ml Gelatin, 0.45% IGEPAL CA-630, and 0.45% Tween 20) supplemented with 100 μg/ml proteinase K at 56 °C overnight. For liver samples, pre-incubation in PBND buffer supplemented with 0.5% SDS and 10 mM EDTA at 75°C for 30 min was necessary to inactivate the high-content DNase. The second day, the supernatant DNA was incubated at 95°C for 15 min to inactivate proteinase K, and then diluted 100× and directly used for quantification of the copy number of both mouse genome (represented by mouse gene Actb) and MCMV genome (represented by MCMV immediate-early gene IE1) by real-time quantitative PCR (RT-qPCR). The viral titer was expressed by the copy number ratio of MCMV genome versus mouse genome (shown in Log10). Our experiments demonstrated a strong correlation between this qPCR assay and the standard plaque assay in NIH/3T3 cells. The RT-qPCR primers for MCMV-IE1 were: 5′-GCATCGAAAGACAACGCAAG-3′ (fwd) and 5′-ACGTAGCTCATCCAGACTCTC-3′ (rev). The RT-qPCR primers for mouse Actb were: 5′-AGCTCACCATTCACCATCTTG −3′ (fwd) and 5′-GACTCATCGTACTCCTGCTTG-3′ (rev). For the survival assay, mice were inoculated i.p. with 1.5 × 105 PFU MCMV per 20 g body weight and survival was monitored daily for 17 days.
HSV-1 Infection and Quantification
Age-matched mice were infected i.p. with HSV-1 (KOS strain, provided by the laboratory of Z.J. Chen, University of Texas Southwestern Medical Center, Dallas, TX) at 2 × 107 PFU per mouse. The viability of the infected mice was monitored daily for 7 days. On day 7 after infection or on the day they died, spleens and livers from live or dead mice were collected for HSV-1 quantification by RT-qPCR as described above for MCMV, but with the RT-qPCR primers for HSV-1 DNA polymerase gene: 5′-GAAAAGACGTTCACCAAGCTG-3′ (fwd) and 5′-CTGGAGGTGCGGTTGATAAA-3′ (rev).
Cytokine Detection
36 h after MCMV infection, mice were bled from the retro-orbital sinus, and the concentration of IL-6, IL-12 (p70), TNF, IFN-β, IFN-γ and IL-17A in the serum was assayed by ELISA using cytokine-specific kits according to the manufacturer’s instructions (IFN-β and IL-17A, BioLegend; IL-6, IL-12 (p70), TNF, and IFN-γ, eBioscience). EL4 cells were plated in 96-well plates and stimulated with PMA/ionomycin. Supernatants at different time points were collected for ELISA measurement of IL-17A and IL-17F (Thermo).
NK Cell Assays
For the in vivo NK cell cytotoxicity assay, splenocytes from C57BL/6J mice (control) and B2m-deficient mice (NK target) were, respectively, labeled with a high (5 μM) and low (1 μM) concentration of CellTrace™ Violet (Life Technologies) at 37°C for 10 min. The labeling was stopped by adding 10% cold FBS. Cells were washed twice, counted, and resuspended at 5 × 107 cells/ml. The two populations were mixed at a 1:1 ratio and injected i.v. into recipient mice. One, two, and three days after injection, blood from the recipients was analyzed by flow cytometry for the number of target cells and control cells. The disappearance of the NK target population relative to the control population reflects the in vivo cytotoxic ability of NK cells.
For the in vitro NK cell assays, NK cells were negatively purified from spleens using the NK Cell Isolation Kit II (Miltenyi Biotec). The cells were unstimulated or stimulated with PMA/ionomycin for 1 h, after which protein transport inhibitors (Brefeldin A and monensin, eBioscience) were added and incubated for additional 5 h. Cells were then collected and stained for NK cell markers (NK-1.1+CD3ε–) and surface CD107a (Lamp-1, a degranulation marker). After wash, the NK cells were fixed and permeabilized using Intracellular Fixation & Permeabilization Buffer Set (eBioscience), and stained for intracellular granzyme B, TNF, and IFN-γ. Samples were analyzed by flow cytometry.
Homeostatic T Cell Proliferation
Splenic T cells negatively purified using the Pan T Cell Isolation Kit II (Miltenyi Biotec) from either CD45.1 wild-type or CD45.2 swp mutant mice were labeled with CellTrace™ Far Red at a final concentration of 3 μM. Equal number of T cells from each population were combined and adoptively transferred into irradiated (750 rad) or non-irradiated C57BL/6J wild-type recipient mice. Four and seven days later, spleen cells in recipient mice were examined for the proliferation of CD4+ and CD8+ T cells by flow cytometry. Apoptotic T cells were determined using Annexin V Apoptosis Detection Kit (BD Biosciences).
In Vivo Antigen Specific T Cell Proliferation
Snrnp40swp/swp mice were crossed with OT-I transgenic or OT-II transgenic mice to produce Snrnp40swp/swp;OT-I Tg and Snrnp40swp/swp;OT-II Tg mice (CD45.2). CD4+ T cells were isolated from the spleens of Snrnp40swp/swp;OT-II Tg mice and Snrnp40+/+;OT-II Tg mice, and CD8+ T cells were isolated from the spleens of Snrnp40swp/swp;OT-I Tg mice and Snrnp40+/+;OT-I Tg mice, using CD4+ T and CD8+ T Cell Isolation Kits (Miltenyi Biotec). These cells were then labeled with 3 μM CellTrace™ Far Red. Labeled cells of a single genotype were injected into individual CD45.1 wild-type mice (2 × 106 cells/mouse). The following day, mice were i.p. injected with or without 250 μg of freshly prepared EndoFit™ ovalbumin (OVA, InvivoGen). Two or three days after OVA immunization, spleen cells in recipient mice were harvested for analysis of OVA-specific CD4+ T and CD8+ T cell proliferation by flow cytometry.
Cell Cycle Analysis of Stimulated Primary T Cells
Negatively purified splenic T cells were stimulated with Dynabeads™ Mouse T-Activator CD3/CD28 (Life Technologies) in round-bottom 96-wells for three days. 10 μM of EdU was then added to the cells and incubated for 6 h. Cell cycle was analyzed by assaying the incorporation of EdU into the newly synthesized DNA and 7-AAD staining by flow cytometry according to manufacturer’s instructions (Life Technologies).
Antibody Responses to Immunization
On day 0, each mouse was i.p. immunized with 2 × 106 infectious units (IU) of rSFV vector encoding β-galactosidase (rSFV-β-gal). On day 7, each mouse was further i.p. immunized with 40 μg of NP-AECM-Ficoll (Biosearch Technologies). On day 13, blood from the sub-mandibular vein of mice was collected, and the T-dependent antibody to rSFV-β-gal (β-galactosidase specific IgG) and the T-independent antibody to NP-Ficoll (NP specific IgM) in the serum were measured by ELISA.
In Vivo CTL Cytotoxicity
Mice were unimmunized or immunized i.p. with 2 × 106 infectious units (IU) of rSFV-β-gal. Eleven days later, the same mice were retro-orbitally injected with equal numbers (3 × 106) of two cell populations labeled with different intensities of CellTrace™ Violet: control C57BL/6J splenocytes (1 μM, low Violet) and antigen-specific CTL targets (syngeneic splenocytes externally loaded with β-galactosidase peptide 497–504 (ICPMYARV) (5 μM, high Violet). After 48 h, the blood was collected from the sub-mandibular vein of mice and analyzed by flow cytometry for the number of remaining control and target cells. The disappearance of the high Violet-containing CTL target population relative to the low Violet-containing control population reflects the in vivo cytotoxic ability of CTLs. Specific killing was defined as the ratio = target cells/control cells. The percentage of target cell lysis = (1 – ratio in immunized mice/ratio in unimmunized mice) × 100.
Bone Marrow-Derived Dendritic Cell (BMDC) Infection
Mouse bone marrow cells were isolated and cultured in 40 ng/ml of GM-CSF in petri dishes (3 × 106 cells/dish). Seven days later, the suspension cells and loosely attached cells were collected and purified for BMDCs using the Pan DC Isolation Kit (Miltenyi Biotec). The BMDCs (≥ 95% CD11c+) were seeded in 96-well round-bottom plates (1 × 105 cells per well). MCMV, HSV-1, rSFV, Sendai virus (SeV), or influenza virus (H1N1) were added at the indicated multiplicity of infection (MOI). 16 h later, the supernatants were harvested for measurement of TNF, IL-6, IFN-α, and IFN-β by ELISA according to the manufacturer’s instructions (IFN-α, InvivoGen).
Reciprocal Bone Marrow Transplantation and Mixed Bone Marrow Chimeras
For bone marrow chimeras, bone marrow cells from CD45.1 wild-type or CD45.2 Snrnp40swp/swp mutant mice were mixed at a 1:1 ratio (3 × 106 for each population) and retro-orbitally injected into irradiated (two doses of 5 Gy) Rag2/Il2rg double-KO mice. For reciprocal bone marrow transplantation, donor bone marrow cells were retro-orbitally injected into irradiated recipient mice. Eight weeks later, peripheral blood from the recipients was collected and analyzed by flow cytometry for the development of various immune cells.
Immunoprecipitation/Mass Spectrometry Analysis for Snrnp40-interacting proteins
Mouse T cell lymphoma EL4 cells or human NK-92 cells (ATCC) were infected with lentivirus encoding either N-terminal FLAG-tagged Snrnp40 (mouse Snrnp40 for EL4, human SNRNP40 for NK-92) or FLAG epitope solely. Stable cell lines were amplified to ten 150-mm dishes and harvested in 15 ml cell lysis buffer [25 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% IGEPAL CA-630, plus Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo)]. The cleared lysates were immunoprecipitated with 200 μl anti-FLAG M2 agarose beads for 4 h at 4°C. After extensive wash, the agarose bound proteins were eluted using 160 μl of 3x FLAG peptide. The eluted proteins were subjected to both silver staining and immunoblotting analysis. 100 μl of the final samples were loaded to SDS-PAGE. The bands on the gel were visualized by staining with GelCode™ Blue Safe Protein Stain (Thermo), and whole stained lanes were subjected to semiquantitative mass spectrometry analysis (LC-MS/MS) by the Proteomics Core facility at the University of Texas Southwestern Medical Center.
RNA Sequencing and Differential Expression and Splicing Analysis
Two Snrnp40-KO clonal EL4 cell lines, one wild-type clonal EL4 cell line, and one pool of polyclonal wild-type EL4 cells were unstimulated or stimulated with PMA/ionomycin (50 ng/ml PMA and 1 μg/ml ionomycin) for 4 h. HSPCs and T cells were negatively purified from the bone marrow and spleen of three each Snrnp40+/+, Snrnp40swp/+, and Snrnp40swp/rplc mice using EasySep™ Mouse Hematopoietic Progenitor Cell Isolation Kit (STEMCELL Technologies) and mouse Pan T Cell Isolation Kit II (Miltenyi Biotec), respectively. The purity was 92–94% for HSPCs (Lineage–) and 94–98% for T cells (CD3+) as determined by flow cytometry. Total RNAs from these cells were extracted using the RNeasy Plus kit with genomic DNA elimination (Qiagen). Libraries were generated using the KAPA Stranded RNA-Seq Kit with RiboErase (HMR) (Kapa Biosystems). Paired-end 2 × 100 bp sequencing was performed using an Illumina HiSeq 2500. For RNA-seq data analysis, reads were de-multiplexed using CASAVA (v1.8.2) and tophat (v2.0.10) was used to map the reads to the mouse transcriptome (build mm10). Cufflinks and cuffmerge (v2.1.1) were then used to calculate reads per kilo base per million and consolidate results across the samples (data were averaged for each genotype/treatment combination). Finally, cuffdiff (v2.1.1) was used to calculate log-fold changes and the associated statistics. Log10(FPKM + 1) values were plotted using R package heatmap.2, and the results scaled by row to obtain values for all data between −1 and 1. DAVID 6.849, 50 was used to classify differentially expressed genes into functionally related groups based on Gene Ontology (GO) annotations51, 52 and the number of genes with immune-related GO annotations was summed.
For splicing analysis, reads from mapped bam files for each sample were separated by chromosome, due to size. The chromosomal bam files were then fed to ASpli (v1.4.0)53, using the GRCm38 reference gene transfer file (.gtf) for gene annotation and read quantification. Counts were gathered of reads spanning junctions as well as those that read from exon into intron. These counts were then analyzed via the AsDiscover function of ASpli to annotate junctions. Percent Intron Retention (PIR) or Percent Sequence Inclusion (PSI) metrics were calculated for all alternate splicing events; data from the Snrnp40-KO lines and the wild-type lines were combined (summed) in these calculations. Once junctions were collected and analyzed for alternate splicing, the junctionDUReport function was used to generate lists of differentially expressed junctions, with their associated P values.
Immunoblotting
Cells or tissues (with homogenization) were lysed in RIPA buffer with protease inhibitors, and protein contents were determined by BCA assay. In some cases, cells were directly lysed in 1× SDS sample buffer (50 mM Tris-Cl, pH 6.8, 2% SDS, 5% β-mercaptoethanol, 0.1% bromophenol blue, and 10% glycerol). Protein extracts were separated by electrophoresis on 4–12% NuPAGE Bis-Tris Mini Gel (Invitrogen) and transferred to a nitrocellulose membrane. The immunoreactivity was detected by the ECL system.
RNA Preparation, Reverse Transcription, and RT-qPCR
Tissue or cell samples were lysed in TRIzol (Invitrogen) for RNA isolation following a standard protocol, and 1 μg of RNA was used for reverse transcription by iScript™ Reverse Transcription Supermix (Bio-Rad). RT-qPCR was performed using PowerUp SYBR Green Master Mix (Life Technologies). The 2–ΔΔCt method was used for relative quantification. The primers used were: Snrnp40 (for Supplementary Fig. 3a and b), 5′-AGGTTTGTGTACGTGTGGG-3′ (fwd), 5′-CTCTTGTCACTGGATGCTGAG-3′ (rev); and Gapdh, 5′-AACAGCAACTCCCACTCTTCC-3′ (fwd), 5′-GTGGTCCAGGGTTTCTTACTC-3′ (rev). The RT-PCR primers used in Fig. 1c and Supplementary Fig. 3f were spanning exon 5 of Snrnp40: 5′-GACAAAACTGTGGCGGTGTG-3′ (fwd), 5′-CTCTCTTTGGGAGCAAATGGC-3′ (rev). The RT-qPCR primers used for measuring wild-type Snrnp40 transcript in Snrnp40swp/swp and Snrnp40swp/rplc mice in Supplementary Table 2 were: 5′-CCCCAACTTGTCTGTACTGG-3′ (fwd, located within exon 5) and 5′-TCTGATCGCTCGTGTCATTG-3′ (rev). RT-PCR primers used for the validation of intron retention and RT-qPCR primers used for the validation of differential expression of genes identified by RNA-seq are listed in Supplementary Table 5.
Statistical Analysis
Data represent the mean ± s.d. or s.e.m. as indicated in graphs depicting error bars. GraphPad Prism 7 was used to determine the statistical significance. Comparisons between groups were made using unpaired, two-tailed Student’s t-test (two groups) and one-way or two-way analysis of variance (ANOVA) with Sidak’s multiple comparisons (multiple groups). Survival curve P values were calculated by Log-rank (Mantel-Cox) test. Differences with P < 0.05 were considered statistically significant.
Reporting Summary
Further information on research design is available in the Life Sciences Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grants AI100627, AI125581, and GM067759 (to B.B.), and by the Rheumatology Research Foundation (to E.N.-G.).
Footnotes
Data Availability
The main data that support the findings of this study are available in the article and its Supplementary Figures. Data are available from the corresponding author upon reasonable request.
Code Availability
The code that supports the findings of this study is available from the corresponding author upon request.
Competing Interests
The authors declare no competing interests.
References
- 1.Rieger MA & Schroeder T Hematopoiesis. Cold Spring Harb Perspect. Biol 4, 10.1101/cshperspect.a008250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner IA, Norman CM, Churcher MJ & Newman AJ Roles of the U5 snRNP in spliceosome dynamics and catalysis. Biochem. Soc. Trans 32, 928–931 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Will CL & Luhrmann R Spliceosome structure and function. Cold Spring Harb Perspect. Biol 3, 10.1101/cshperspect.a003707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achsel T, Ahrens K, Brahms H, Teigelkamp S & Luhrmann R The human U5–220kD protein (hPrp8) forms a stable RNA-free complex with several U5-specific proteins, including an RNA unwindase, a homologue of ribosomal elongation factor EF-2, and a novel WD-40 protein. Mol. Cell. Biol 18, 6756–6766 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reyes JL, Kois P, Konforti BB & Konarska MM The canonical GU dinucleotide at the 5′ splice site is recognized by p220 of the U5 snRNP within the spliceosome. RNA 2, 213–225 (1996). [PMC free article] [PubMed] [Google Scholar]
- 6.Teigelkamp S, Whittaker E & Beggs JD Interaction of the yeast splicing factor PRP8 with substrate RNA during both steps of splicing. Nucleic Acids Res 23, 320–326 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyatt JR, Sontheimer EJ & Steitz JA Site-specific cross-linking of mammalian U5 snRNP to the 5′ splice site before the first step of pre-mRNA splicing. Genes Dev 6, 2542–2553 (1992). [DOI] [PubMed] [Google Scholar]
- 8.Chiara MD, Palandjian L, Feld Kramer R & Reed R Evidence that U5 snRNP recognizes the 3’ splice site for catalytic step II in mammals. EMBO J 16, 4746–4759 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teigelkamp S, Newman AJ & Beggs JD Extensive interactions of PRP8 protein with the 5′ and 3’ splice sites during splicing suggest a role in stabilization of exon alignment by U5 snRNA. EMBO J 14, 2602–2612 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umen JG & Guthrie C A novel role for a U5 snRNP protein in 3’ splice site selection. Genes Dev 9, 855–868 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Bartels C, Klatt C, Luhrmann R & Fabrizio P The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO Rep 3, 875–880 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner TJ & Guthrie C Genetic analysis reveals a role for the C terminus of the Saccharomyces cerevisiae GTPase Snu114 during spliceosome activation. Genetics 170, 1063–1080 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeder C, Kutach AK & Guthrie C ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase requires the C terminus of Prp8. Nat. Struct. Mol. Biol 16, 42–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Small EC, Leggett SR, Winans AA & Staley JP The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol. Cell 23, 389–399 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang T et al. Real-time resolution of point mutations that cause phenovariance in mice. Proc. Natl. Acad. Sci. U. S. A 112, E440–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biron CA, Nguyen KB, Pien GC, Cousens LP & Salazar-Mather TP Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol 17, 189–220 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Laggerbauer B et al. The human U5 snRNP 52K protein (CD2BP2) interacts with U5–102K (hPrp6), a U4/U6.U5 tri-snRNP bridging protein, but dissociates upon tri-snRNP formation. RNA 11, 598–608 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang T et al. Probability of phenotypically detectable protein damage by ENU-induced mutations in the Mutagenetix database. Nat. Commun 9, 441-017-02806-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickinson ME et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayadi A et al. Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm. Genome 23, 600–610 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White JK et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154, 452–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruzickova S & Stanek D Mutations in spliceosomal proteins and retina degeneration. RNA Biol 14, 544–552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma B, Akinyi MV, Norppa AJ & Frilander MJ Minor spliceosome and disease. Semin. Cell Dev. Biol 79, 103–112 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Kurtovic-Kozaric A et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 29, 126–136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park JW, Parisky K, Celotto AM, Reenan RA & Graveley BR Identification of alternative splicing regulators by RNA interference in Drosophila. Proc. Natl. Acad. Sci. U. S. A 101, 15974–15979 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pleiss JA, Whitworth GB, Bergkessel M & Guthrie C Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 5, e90 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papasaikas P, Tejedor JR, Vigevani L & Valcarcel J Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 57, 7–22 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Inoue D, Bradley RK & Abdel-Wahab O Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 30, 989–1001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wahl MC, Will CL & Luhrmann R The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Kakugawa K et al. A novel gene essential for the development of single positive thymocytes. Mol. Cell. Biol 29, 5128–5135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu G et al. Themis controls thymocyte selection through regulation of T cell antigen receptor-mediated signaling. Nat. Immunol 10, 848–856 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson AL et al. Themis is a member of a new metazoan gene family and is required for the completion of thymocyte positive selection. Nat. Immunol 10, 831–839 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesourne R et al. Themis, a T cell-specific protein important for late thymocyte development. Nat. Immunol 10, 840–847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu G et al. Themis sets the signal threshold for positive and negative selection in T-cell development. Nature 504, 441–445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bar E, Whitney PG, Moor K, Reis e Sousa C & LeibundGut-Landmann S IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity 40, 117–127 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Hosoya T et al. GATA-3 is required for early T lineage progenitor development. J. Exp. Med 206, 2987–3000 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pai SY et al. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity 19, 863–875 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Ting CN, Olson MC, Barton KP & Leiden JM Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384, 474–478 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Wang L et al. Distinct functions for the transcription factors GATA-3 and ThPOK during intrathymic differentiation of CD4(+) T cells. Nat. Immunol 9, 1122–1130 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barton K et al. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity 9, 555–563 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Clements JL, John SA & Garrett-Sinha LA Impaired generation of CD8+ thymocytes in Ets-1-deficient mice. J. Immunol 177, 905–912 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Higuchi T et al. Thymomegaly, microsplenia, and defective homeostatic proliferation of peripheral lymphocytes in p51-Ets1 isoform-specific null mice. Mol. Cell. Biol 27, 3353–3366 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nuez B, Michalovich D, Bygrave A, Ploemacher R & Grosveld F Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature 375, 316–318 (1995). [DOI] [PubMed] [Google Scholar]
- 44.White RA et al. Hematologic characterization and chromosomal localization of the novel dominantly inherited mouse hemolytic anemia, neonatal anemia (Nan). Blood Cells Mol. Dis 43, 141–148 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Foudi A et al. Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J. Exp. Med 211, 909–927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vassen L et al. Growth factor independence 1b (gfi1b) is important for the maturation of erythroid cells and the regulation of embryonic globin expression. PLoS One 9, e96636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saleque S, Cameron S & Orkin SH The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev 16, 301–306 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayle A, Luo M, Jeong M & Goodell MA Flow cytometry analysis of murine hematopoietic stem cells. Cytometry A 83, 27–37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Huang da W, Sherman BT & Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45, D331–D338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huber W et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.