

Abstract
As a member of the Cullin-RING ligase family, Cullin-RING ligase 4 (CRL4) has drawn much attention due to its broad regulatory roles under physiological and pathological conditions, especially in neoplastic events. Based on evidence from knockout and transgenic mouse models, human clinical data, and biochemical interactions, we summarize the distinct roles of the CRL4 E3 ligase complexes in tumorigenesis, which appears to be tissue- and context-dependent. Notably, targeting CRL4 has recently emerged as a noval anti-cancer strategy, including thalidomide and its derivatives that bind to the substrate recognition receptor cereblon (CRBN), and anticancer sulfonamides that target DCAF15 to suppress the neoplastic proliferation of multiple myeloma and colorectal cancers, respectively. To this end, PROTACs have been developed as a group of engineered bi-functional chemical glues that induce the ubiquitination-mediated degradation of substrates via recruiting E3 ligases, such as CRL4 (CRBN) and CRL2 (pVHL). We summarize the recent major advances in the CRL4 research field towards understanding its involvement in tumorigenesis and further discuss its clinical implications. The anti-tumor effects using the PROTAC approach to target the degradation of undruggable targets are also highlighted.
Keywords: CRL4, Cullin 4, E3 ligases, PROTACs, Tumorigenesis, Targeted therapy
Introduction
The ubiquitin-proteasome system (UPS)
The ubiquitin-proteasome system (UPS) is an evolutionarily conserved apparatus that serves as a major regulator of proteostasis in eukaryotic cells1. The UPS generally consists of ubiquitin, ubiquitination enzymes and the 26S proteasome, which synergistically form an enzymatic cascade to transfer ubiquitin in a substrate-specific manner to promote subsequent proteolysis and degradation of the target protein2. The machinery of the ubiquitin-proteasome cascade has been reviewed extensively3,4. Briefly, the ATP-dependent activation of ubiquitin by the E1 activating enzyme is indispensable for the initiation of the enzymatic cascade5. As a result, activated ubiquitin is then transferred to an E2 conjugating enzyme, which then assist in the recruitment of an E3 ligase into a complex with the ubiquitin moiety. Subsequently, E3 ligase determines the substrate specificity and facilitates the formation of a covalent isopeptide bond between the ubiquitin and lysine residues of target proteins, leading to substrate ubiquitination. Additionally, E3 ligases also mediate the attachment of the ubiquitin moiety to existing ubiquitin chains on a substrate protein, resulting in poly-ubiquitination and diverse consequences to the target protein, such as degradation2,6, altered activity, or subcellular localization of substrates7.
Physiological significance and pathological roles of the UPS
Consistent with a broad central role that the UPS plays in proteostatic control, nearly every aspect of cellular biology is regulated in some manner by the UPS pathway, especially the cell cycle, cell growth, immune homeostasis, and metabolic stability6. For instance, proteasomal degradation of cell cycle regulators, such as p21 and p27, is a critical to control cell cycle progression6. In addition, proteolytic activities provide necessary raw materials for intracellular recycling and rebuilding, such as generation of amino acids6,8. Meanwhile, via proteasomal cleavage of endogenous proteins, the UPS governs the production of MHC (major histocompatibility complex) Class I antigens, which are critical for the functions of the immune response9,10. Dysregulation of the UPS is involved in the pathogenesis of multiple disorders, especially neurodegeneration, neoplastic transformation and autoimmune diseases11. Pathologically, neurodegenerative disorders often feature the accumulation of misfolded proteins, such as tau aggregates and Aβ plaques in Alzheimer’s disease12,13. Autoimmune diseases are often induced by the mis-recognition of endogenous proteins as exogenous antigens14, which was tightly controlled by the UPS. For instance, increased generation of MHC Class I antigen HLA (human leukocyte antigen)-B27 closely correlates to Ankylosing Spondylitis15,16.
Cancer relevant roles of the UPS
Dysregulation of the UPS is associated with tumorigenic events17,18. E3 ligases are involved in the final enzymatic activity leading to ubiquitination and dictates the specificity of substrate selection. Hence, depending on the tumorigenic properties of specific substrates, E3 ligases play a context-dependent role by degrading tumor suppressors or oncoproteins19,20. It is noteworthy that E3 ligase may also cause altered activity or subcellular re-localization of target proteins. For example, MDM2 (mouse double minute 2 homolog), serving as an E3 ligase to destabilize tumor suppressor p53, has been regarded as one of the most frequently mutated oncoproteins in lung and breast carcinoma21–23. On the other hand, SPOP (speckle-type POZ protein) is frequently mutated adaptor protein in prostate cancer serving as a tumor suppressor by targeting TRIM24 (tripartite motif-containing 24) and the androgen receptor24,25. Due to their critically important role in substrate selection and ubiquitin transfer during the UPS cascade, E3 ligases are broadly acknowledged as a key target for anti-cancer therapeutics, thus the use of small molecules targeting UPS for cancer treatment has been developed for diverse malignancies, especially multiple myeloma and lymphoma26,27,28, which will be discussed in the following sections.
DUBs: the antagonizing force in counter-balancing the UPS
Similar to other post-translational modifications, there are also mechanisms in place to reverse and therefore antagonize the role of ubiquitination and thus maintain protein stability, which is largely mediated by a cluster of proteins termed deubiquitinating enzymes (DUBs)29,30. Currently, nearly 100 DUBs have been identified in humans31, which are divided into 7 subgroups by a specific functional domain, including ubiquitin C-terminal hydrolase (UCH) DUBs, ubiquitin specific protease (USP) DUBs, ovarian tumor (OTU) DUBs, Josephin DUBs, JAB1/MPN+/MOV34 (JAMM) DUBs32 and another two non-canonical DUBs, MINDY DUBs33 and ZUFSP DUBs34,35. Consistent with the roles for the UPS participating in critically important cellular biological processes, DUBs are likewise involved in nearly every aspect of cellular biology17,36. For instance, USP9X (ubiquitin specific peptidase 9) is found to be negatively correlated with carcinogenesis, which deubiquitinates and functionally stabilizes its substrate LATS1 (large tumor suppressor kinase 1), leading to the inactivation of the Hippo pathway37. USP2 (ubiquitin specific peptidase 2) triggers malignant progression in prostate cancer cells via deubiquitination and stabilization of fatty acid synthase (FAS), which positively correlates with aggressiveness and negatively predicts survival38.
CRL4 E3 ligases in tumorigenesis
E3 ligases and the Cullin-RING family
There are hundreds of E3 ligases identified in mammalian ubiquitination cascades39,40. Based on structural and functional differences, E3 ligases are subdivided into two groups, namely the HECT (homologous to the E6AP carboxyl terminus) family and the RING (really interesting new gene) family41. Specifically, RING E3 ligases bind to ubiquitin-E2 heterodimer and protein substrates in a simultaneous manner, facilitating an efficient transfer of ubiquitin onto selected substrates. Whereas, ubiquitination mediated by HECT E3 ligases is separated into two steps, formation of a thioester bond with ubiquitin followed by substrate recognition and ubiquitin transfer40,42. In addition, the RING ligases have been further categorized into several subfamilies, including BARD1 (BRCA1-associated RING domain 1), c-Cbl (Casitas B-lineage lymphoma), anaphase-promoting complex (APC), heterodimer of BRCA1 (breast cancer 1) and Cullin-RING ligases (CRLs), each harboring a RING catalytic domain40.
Characterized by being composed of multi-subunit complexes, the Cullin-RING ligases are the most extensively studied subfamily of RING ligases. Structurally, a Cullin-RING ligase complex is composed of four essential subunits, including a Cullin scaffold, RING-finger protein, adaptor protein, and a substrate recognition protein43. As a central coordinator of complex formation, the Cullin scaffold provides the platform for both the RING-finger protein and adaptor protein. The RING-finger protein serves as a docking site for both the ubiquitin-E2 complex and the substrate recognition module to facilitate the transfer of ubiquitin to the recruited substrate. The substrate recognition module that associates within the complex through its interaction with the adaptor protein and dictate substrate specificity43,44. To date, eight Cullin scaffolds have been characterized in mammalian cells (Cullin 1, Cullin 2, Cullin 3, Cullin 4A, Cullin 4B, Cullin 5, Cullin 7, and Cullin 9) as well as two RING-finger proteins (RBX1 and RBX2), four adaptor proteins (SKP1, ElonginB, ElonginC, and DDB1) and more than 400 substrate recognition receptor proteins45. These subunits are assembled into hundreds of unique Cullin-RING ligase complexes that are responsible for nearly 20% of ubiquitination events mediated by the UPS46,47.
Introduction of Cullin 4 E3 ligases
The Cullin 4-RING ligases (CRL4s) contain two homogenous scaffolds, defined as Cullin 4A (CUL4A) and Cullin 4B (CUL4B), respectively. Commonly, both scaffolds bind to damaged DNA binding protein 1 (DDB1) and the catalytic subunit RING-finger protein RBX1 through their N- and C-terminus respectively to support the structure of entire complex48. However, despite of 82% identity with regard to their genomic sequences, each scaffold targets a unique set of substrates. Compared to CUL4A, CUL4B features a longer N-terminus, which contains an extra nuclear localization signal (NLS) that governs its targeting to the nucleus, in addition to interaction with DDB1. On the other hand, the majority of CUL4A resides in the cytoplasm to regulate substrate ubiquitination49, although a small fraction is found in the nucleus to target nuclear proteins50 (Table 1). CUL4 scaffolds are also neddylated by the ubiquitin-like protein NEDD8 (neural precursor cell expressed, developmentally down-regulated 8), which is essential to stimulate the activity of CRL4s complexes51. Moreover, RBX1 acts as a regulatory subunit of the CRL4 complex through recruitment of ubiquitin E2 complex and transferring the ubiquitin moiety. The adaptor DDB1 serves as the bridging factor between the Cullin scaffold and substrate recognition subunit, DDB1-CUL4-associated factor (DCAF) (Figure 1)51,52.
Table 1.
Pathological involvements of mutated components of CRL4
| Gene | Component category | Alteration | Consequence |
|---|---|---|---|
| Cul4b | Scaffold | Mutation | X-linked mental retardation77,78; Cerebral malformations80 |
| Dcaf8 | Substrate recognition protein | Mutation | Axonal hereditary motor and sensory neuropathy (HMSN2)81 |
| Dcaf14 | Substrate recognition protein | Mutation | Developmental retardation, intellectual defects, obesity and dysmorphic features82 |
| Dcaf17 | Substrate recognition protein | Mutation | Woodhouse Sakati syndrome83 |
| Ddb2 | Substrate recognition protein | Mutation | Xeroderma pigmentosum84 |
| Gnb3 | Substrate recognition protein | Mutation | Hypertension85 |
| Wdr62 | Substrate recognition protein | Mutation | Cerebral malformations80 |
Figure 1. Overview of neoplastic roles of CRL4 components.

Physiological roles of Cullin 4 E3 ligases -evidence from knockout mouse models
According to the phenotypic studies derived from knockout mouse models on CRL4 components, CRL4s participate in embryonic development as well as maturation and homeostasis of multiple tissues and organs. Unlike the essential roles of most Cullins, germline knockouts of Cul4a or Cul4b are both viable and shows no overt growth abnormalities48,53–55, which is likely due to redundancy between CUL4A and CUL4B. Abrogation of both CUL4A and CUL4B in mouse embryonic fibroblasts and tumor cell lines led to growth arrest and loss of viability55. Moreover, germline knockout of the Ddb1 adaptor is embryonic lethal56,57, highlighting the essential role of the CRL4 ubiquitin ligase in maintaining growth and survival of mammals. Although a previous study suggested an embryonic requirement of Cul4a58, other studies have shown that systemic ablation of Cul4a does not lead to embryonic lethality55,59, and the embryonic phenotype seen in the earlier study may be attributed to an unintended deletion of the adjacent Pcid2 gene on the complementary strand of DNA around the Cul4a locus55.
The hematopoietic lineage is another system where CRL4 activity regulates protein homeostasis to control biological outcome. Earlier proteomic/yeast two-hybrid assays identified the HOX Homeodomain transcription factors to be targeted by the CRL4 ubiquitin ligase for ubiquitination and degradation49,60. As the hematopoietic stem cells (HSCs) and progenitors undergo differentiation, HOX genes are transcriptionally downregulated61–63 and the HOX proteins are targeted for degradation via the CRL4 ubiquitin ligase to ensure proper differentiation49. Importantly, a conserved LXCXE motif was identified in the Helix I region of HOX HD which serves as the CRL4 degron motif that is conserved among all 39 HOX family members60. Failure of HOXA9 degradation by CRL4 blocks granulocytic differentiation49, while transduction of an engineered degradation-resistant HOXB4 into adult HSCs could effectively promote ex vivo expansion of HSCs and multipotent progenitors, and enhance bone marrow engraftment of transduced human adult CD34+ HSCs60.
The potential significance of CRL4 in the reproductive system was further revealed in the Cul4a and Cul4b knockout animals: Cul4a-null mice exhibit male infertility, yet had no major effect on reproduction of females was observed, revealing the essential roles of CRL4A in male reproduction64–66. Cul4b−/Y males are also infertile, while no Cul4b−/− females can be derived as CUL4B is x-linked. While the redundancy of the two CUL4s accounts for the viability and normal development of Cul4a−/− or Cul4b−/Y mice, CUL4A and CUL4B genes are differentially expressed at distinct stages of male meiosis64–66. As such, Cul4a−/− spermatocytes are arrested at pachytene to diplotene transitions of Meiosis I, while Cul4b−/Y sperms are defective at the later stage of spermiogenesis64–66. Accumulation of CRL4 target CDT1, as well as p53 was observed, consistent with the increased apoptosis among germ cells64–66. While female reproduction remains normal in individual Cul4a−/− mice, abrogation of the entire CRL4 ubiquitin ligase led to infertility in females67. Interestingly, conditional deletion of Ddb1 or CRL4 substrate receptors VprBP/DCAF1 or DCAF2 cause female infertility possibly via diverse mechanisms including, massive DNA damage and disruption of the cell cycle, TET (Ten-eleven translocation methylcytosine dioxygenase) inactivation-mediated ovulation defects, oocyte loss, or repression on PI3K/Akt pathway68–70.
Another core system that CRL4 regulates is the central nervous system. Conditional deletion of Ddb1 in the murine brains leads to neonatal lethality by increased accumulation of p53, although p53 is not considered a direct substrate of CRL457. Meanwhile, specific ablation of Ddb1 in the hippocampus and cerebral cortex generates an epileptic phenotype in experimental mice, mechanistically via changes in restriction on the activity of the BK (Ca2+ and voltage-activated K+) channel, which further indicates the potentially pathological role of CRL4 in neuroelectrophysiological disorders71.
CRL4s are also found to function in other organ systems. For instance, hepatocyte-specific deletion of Ddb1 disables liver gluconeogenesis in part due to increased accumulation of the CRL4 substrate CRY1 (cryptochrome 1), which in turn leads to downregulation of the FOXO1 (forkhead box protein O1)-driven gluconeogenic responses72. However, CRL4s seem to play contradictory roles towards hepatic cell expansion under various circumstances. Deletion of Ddb1 accelerates liver regeneration and trigger spontaneous onset of hepatic carcinogenesis, suggesting a role in controlling cellular proliferation73. This phenotypic disparity may be explained by DDB1 engaging in other regulatory networks besides CRL4, which could cloud our understanding of the effect of DDB1 loss due to reduction in CRL4 activity. Therefore, the phenotypic information from Cul4a knockout mouse models could be more informative. Moreover, CRL4s may have a role in regulating metabolism, since depletion of Cul4b in adipocytes and pancreatic δ cells produces opposing outcomes, namely enhanced insulin sensitivity by stabilizing PPARγ (peroxisome proliferator-activated receptor γ), or glucose intolerance by blocking PRC2 (polycomb repressive complex 2)-induced somatostatin secretion, respectively74,75. Meanwhile, CRL4s also demonstrate neoplastic involvement in skin carcinogenesis through restricting the capacity of nucleotide excision repair55,76, which will be further discussed in subsequent sections (Table 2).
Table 2.
Summarized phenotypes of knockout mouse models on CRL4 components
| Gene | Component category | Knockout mode | Phenotype |
|---|---|---|---|
| Cul4a−/− | Scaffold | Germline | Normal development/lifespan. No gross abnormalities55. Male infertility64,66; mild Cardiac hypertrophy and hypertension in male but not female mice85,249 |
| Conditional (skin) | Resistant to UV-induced skin carcinogenesis55 | ||
| Conditional (liver) | Reduced hepatocyte proliferation after exposure to liver toxins59 | ||
| Cul4b−/Y | Scaffold | Germline (epiblast) | Normal development/lifespan; No obvious gross abnormalities54 ; Infertility65 |
| Conditional (pancreatic δ cell) | Decreased glucose intolerance and insulin secretion75 | ||
| Conditional (adipocyte) | Enhanced expansion of adipose tissue; Increased glucose tolerance and insulin sensitivity74 | ||
| Conditional (germ cell) | Male infertility65 | ||
| Conditional (hematopoietic cell) | Increased accumulation and activity of MDSCs post LPS250 | ||
| Conditional (nervous system) | Elevated amount of astrocytes251 | ||
| Conditional (myeloid system) | Enhanced peritonitis induced by LPS administration252 | ||
| Systematic | Embryonic lethality resulting from defective extra-embryonic development54; Growth retardation of embryos253 | ||
| Germline | Embryonic lethality57,254 | ||
| Ddbr−/− | Adaptor | Conditional (brain and lens) Conditional (skin) |
Neonatal lethality57 Postnatal lethality56 |
| Conditional (hematopoietic cell) | Bone marrow failure254 | ||
| Conditional (oocyte) | Ovulation defect69,70 | ||
| Conditional (liver) | Impaired hepatic gluconeogenesis72; Liver regeneration and spontaneous development of hepatocellular carcinoma73 | ||
| Conditional (hippocampus and cerebral cortex) | Epilepsy71 | ||
| Germline | Embryonic lethality255 | ||
| Dcaf1−/− | Substrate recognition protein | Conditional (B cell) | Impaired B cell maturity and development256 |
| Conditional (T cell) | Insufficiency of T cells257 | ||
| Conditional (oocyte) | Oocyte death and infertility69,70 | ||
| Conditional (oocyte) | Female infertility68 | ||
| Dcaf2−/− | Substrate recognition protein | Germline | Susceptible to UV-induced skin carcinogenesis76 |
Abbreviations: MDSCs: myeloid-derived suppressor cells; LPS: lipopolysaccharide; UV: ultraviolet;
Pathological involvement of Cullin 4 E3 ligases-evidence from clinical studies
Consistent with their diverse physiological roles in mouse models, mutations of CRL4 components have also been linked to multiple human pathological conditions, especially in the nervous system.
X-linked mental retardation (XLMR), also known as Cabezas Syndrome, is genetically characterized by specific mutations in the X chromosome, which causes short stature, hypogonadism, learning disability, obesity, aggressive outbursts, intentional tremor, pes cavus and seizures among adolescents77,78. Current studies have confirmed that mutations in X-linked Cul4b gene account for the onset of XLMR, which may be partially attributed to the stability of its substrate WDR5 (WD repeat-containing protein 5) that facilitates the epigenetic silencing of neuronal genes and thus inhibits neurite outgrowth77–79, although WDR5 accumulation was not observed in Cul4b−/− MEFs54. Genetic rescue of Cul4b-mutant mice significantly decreases the occurrence of XLMR, suggesting a potential of Cul4b-targeted therapy53. In addition, mutation of Cul4b also leads to cerebral malformations, possibly through dysregulation of the function of WDR62 (WD repeat-containing protein 62), a substrate recognition receptor (DCAF) of CRL4 and also a protein whose mutation is frequently detected in patients with microcephaly80. Additionally, a specific mutation in Dcaf8, leading to the amino acid substitution R317C, disrupts its binding to the DDB1 adaptor and the formation of a functional CRL4 complex, leading to axonal hereditary motor and sensory neuropathy (HMSN2) and giant axons81. Moreover, patients bearing a mutant of Dcaf14 feature developmental retardation, intellectual defects, obesity and dysmorphic characteristics82. Dcaf17 is the causative gene of Woodhouse Sakati Syndrome (WSS), which is characterized by progressive extrapydamidal symptoms, together with hearing loss, hypogonadism, diabetes and learning disability83. This experimental evidence verifies the close correlation between CRL4 mal-function and pathogenesis in the central nervous system.
Meanwhile, mutated Gnb3, encoding a substrate recognition receptor GNB3 (G protein subunit beta 3), pathologically associates with hereditary hypertension while mutation on Ddb2 results in xeroderma pigmentosum, which features impaired DNA repair upon UV exposure and higher susceptibility to skin carcinogenesis84,85. Considering the diverse physiological roles of CRL4, we may predict that more pathological correlations between CRL4 dysregulation and human disorders may be identified in the near future (Table 3).
Table 3.
Major mammalian substrates of CRL4 and their biological impacts
| Substrate recognition protein | Scaffold | Substrate | Modification | Biological impact |
|---|---|---|---|---|
| DCAF1 | CUL4A | LATS1 | Degradation | Oncogenic contribution139,140 |
| CUL4A | LATS2 | Altered activity | Oncogenic contribution139 | |
| CUL4A | HLTF | Degradation | Triggering HIV replication110 | |
| CUL4A | Dicer1 | Degradation | Enhanced HIV infection258; Colon cancer progression109 | |
| CUL4A/B | FOXM1 | Degradation | Cell cycle arrest136 | |
| CUL4A/B | PP2A | Degradation | Crucial for oocyte meiosis and female fertility67 | |
| CUL4A/B | MyoD | Degradation | Halted differentiation of skeletal muscle137 | |
| CUL4A | MCM10 | Degradation | Cell cycle arrest259,260 | |
| CUL4B | TR4 | Degradation | Resistant to high fat caused fatty liver261 | |
| CUL4A/B | SAMHD1 | Degradation | Facilitating HIV replication262 | |
| CUL4A/B | TET1/2/3 | Altered activity | Essential for ovary maturity69; Tumor263 suppression |
|
| CUL4A/B | RORα | Degradation | Transcriptional repression138 | |
| CUL4A | UNG2 | Degradation | Cell cycle arrest264,265 | |
| CUL4A | SMUG1 | Degradation | Cell cycle arrest264 | |
| CUL4A | NF2 | Degradation | Activation of oncogenic pathways141 | |
| DCAF2 | CUL4A | CRY1 | Degradation | Promotion of liver gluconeogenesis; Homeostasis of molecular circadian behavior72,147 |
| CUL4A/B | CDT1 | Degradation | Stimulating the proliferation of melanoma150,152 | |
| CUL4A/B | p21 | Degradation | Stimulating the proliferation of melanoma150; Regulation of replication licensing98 | |
| CUL4A | p53 | Degradation | Cell cycle control266 | |
| CUL4A/B | SET7 | Degradation | Maintaining genomic integrity143–145 | |
| CUL4A/B | SET8 | Degradation | Stimulating the proliferation of melanoma150; DNA damage control145,267 | |
| CUL4A/B | PCNA | Altered activity | Enhanced translesion DNA synthesis268 | |
| CUL4A/B | p12 | Degradation | DNA repair control269 | |
| CUL4A | CHK1 | Degradation | Cell cycle progression146 | |
| CUL4A/B | MMSET | Degradation | Cell cycle arrest270 | |
| CUL4A/B | XPG | Degradation | DNA repair271 | |
| CUL4A/B | TDG | Degradation | Homeostasis of DNA replication272 | |
| CUL4A | SDE2 | Degradation | Protection of genomic stability against replication stresses273 | |
| CUL4A/B | CDC6 | Degradation | Cell cycle regulation274 | |
| CUL4A | Tob | Degradation | Apoptotic response to DNA damage275 | |
| CUL4A | GCN5 | Degradation | Transcriptional suppression148 | |
| DCAF4L2 | CUL4A | PPM1B | Degradation | Increased invasion of colorectal cancer276 |
| DCAF7 | CUL4A/B | LigI | Degradation | NS277 |
| DCAF8 | CUL4A | H3 | Altered activity | Postnatal liver maturation278 |
| CUL4B | CDC25A | Degradation | NS170 | |
| DCAF9 | CUL4A/B | H2A | Altered activity | Suppressed adipogenesis279 |
| DCAF11 | CUL4A | NRF2 | Degradation | More sensitive to chemotherapies171 |
| CUL4A | SLBP | Degradation | Cell cycle homeostasis280,281 | |
| CUL4B | p21 | Degradation | Inhibiting cell cycle progression172 | |
| DCAF15 | CUL4A/B | CAPERα | Degradation | Anti-growth effect in cancer cells173 |
| AhR | CUL4B | ERα | Degradation | NS282 |
| COP1 | CUL4A | ETV5 | Degradation | Lung homeostasis and tumor suppression166 |
| CUL4A | c-Jun | Degradation | Inhibition on oncogenic transcriptions167 | |
| COPS8 | CUL4A | CENP-A | Subcellular relocalization | Homeostasis of mitosis283 |
| CRBN | CUL4A/B | IKZF1/3 | Degradation | Anti-tumor impacts against multiple myeloma cells162 |
| CUL4A | ZFP91 | Degradation | NS284 | |
| CUL4A | GSPT1 | Degradation | Anti-tumor impacts against leukemic cells163 | |
| CUL4A/B | APP | Degradation | Alleviation of neurodegeneration159 | |
| CUL4A | GS | Degradation | NS185 | |
| CUL4A/B | CK1α | Degradation | Inhibition on myelodysplastic syndrome164 | |
| CUL4A/B | CLC-1 | Degradation | Homeostasis of membrane excitability160 | |
| CUL4A | SLO1 | Altered activity | Preventing epileptogenesis71 | |
| DDB2 | CUL4A | p27 | Degradation | Cell cycle progression156 |
| CUL4A | AR | Degradation | Suppression of prostate cancer proliferation154,285 | |
| CUL4A/B | H2A | Altered activity | DNA repair84 | |
| CUL4A/B | HBO1 | Degradation | Suppressed cell proliferation158 | |
| CUL4A | DDB2 | Degradation | Regulation of DNA damage286,287 | |
| FBXO44 | CUL4B | RGS2 | Degradation | NS288 |
| FBXW5 | CUL4A | DLC1 | Degradation | Growth of lung cancer cells289 |
| CUL4A/B | TSC2 | Degradation | Homeostasis of cell growth290 | |
| GNB2 | CUL4A | GRK2 | Degradation | Cardiovascular protection249 |
| GNB3 | CUL4A | GRK2 | Degradation | Cardiovascular homeostasis85 |
| HBx | CUL4A/B | SMC5/6 | Degradation | Increased replication of HBV128 |
| HOXB4 | CUL4A | Geminin | Degradation | Elevated proliferation of hematopoietic stem and progenitor cells291 |
| RBBP7 | CUL4A/B | CENP-A | Subcellular relocalization | Homeostasis of mitosis292 |
| STRAP | CUL4A | PNKP | Degradation | Susceptible to oxidative DNA damage293 |
| WDR70 | CUL4A/B | H2B | Altered activity | Stability of cell division294 |
| Other substrates* | CUL4A/B | γ-tubulin | Degradation | Stability of centrosome295 |
| CUL4B | HUWE1 | Degradation | Reduced apoptosis after DNA damage296 | |
| CUL4A/B | AMBRA1 | Degradation | Autophagy termination297 | |
| CUL4B | p53 | Degradation | Impeding stress-induced cellular senescence298 | |
| CUL4A | PEX7P | Degradation | Control of peroxisome biogenesis299 | |
| CUL4A | HOXB4 | Degradation | Decreased proliferation of hematopoietic stem cells60 | |
| CUL4B | CSN5 | Degradation | Regulation of bone morphogenetic signaling300 | |
| CUL4A | p73 | Altered activity | Transcriptional repression301 | |
| CUL4A | GRK5 | Degradation | NS302 | |
| CUL4A | ORCA | Degradation | Cell cycle regulation303 | |
| CUL4B | WDR5 | Degradation | Promoting neurite outgrowth79 | |
| CUL4B | Peroxiredoxin III | Degradation | Increased production of cellular reactive oxygen species304 | |
| CUL4A | RASSF1A | Degradation | Cell cycle progression305 | |
| CUL4B | cyclin E | Degradation | Cell cycle progression306 | |
| CUL4A | CHK1 | Degradation | Genomic instability307 | |
| CUL4B | H3/H4 | Altered activity | Facilitating cellular response to DNA damage308 | |
| CUL4A | p27 | Degradation | Cell proliferation50 | |
| CUL4A | HOXA9 | Degradation | Myelocytic maturation49 | |
| CUL4A | REDD1 | Degradation | Activation of mTOR signaling309 |
Those proteins are assumed to be the substrates of CRL4 without detailed description of binding motif and interactions with substrate recognition proteins.
Abbreviations:COPS8: COP9 signalosome subunit 8; CENP-A: centromere protein A; DCAF2: alias CDT2; CRY1: cryptochrome 1; TR4: nuclear receptor subfamily 2 group C member 2 (alias NR2C2); CRBN: cereblon; ZFP91: zinc finger protein 91; NS: not specified; DCAF11: alias WDR23; CAPERα: coactivator of activating protein-1 and estrogen receptor α; NRF2: nuclear factor erythroid 2 like 2; LATS1: large tumor suppressor 1; FOXM1: forkhead box M1; ETV5: ETS variant 5; AR: androgen receptor; H3: histone H3 protein; ERα: estrogen receptor α; SDE2: SDE2 telomere maintenance homolog; HBx: hepatitis B virus regulatory protein X; SMC5: structural maintenance of chromosome 5; LigI: DNA ligase I; GNB3: G protein subunit beta 3; GRK2: G protein-coupled receptor kinase 2; GSPT1: G1 to S phase transition 1; CDT1: chromatin licensing and DNA replication factor 1; SET8: lysine methyltransferase 5A (alias KMT5A); APP: amyloid precursor protein; SLBP: stem-loop binding protein; PPM1B: protein phosphatase, Mg2+/Mn2+ dependent 1B; WDR70: WD repeat domain 70; GS: glutamine synthetase; HLTF: helicase-like transcription factor; MMSET: nuclear receptor binding SET domain protein 2 (alias NSD2); HBO1: lysine acetyltransferase 7 (alias KAT7); PP2A: phosphatase 2A; CK1α: casein kinase 1A1; MCM10: minichromosome maintenance 10 replication initiation factor; CLC-1: chloride voltage-gated channel 1; FBXO44: F-box protein 44; RGS2: regulator of G protein signaling 2; TET: tet methylcytosine dioxygenase; XPG: Xeroderma pigmentosum group G protein; TDG: thymine DNA glycosylase; SLO1: potassium calcium-activated channel subfamily M alpha 1 (alias KCNMA1); CDC6: cell division cycle 6; FBXW5: F-box and WD repeat domain containing 5; DLC1: DLC1 Rho GTPase activating protein; CHK1: checkpoint kinase 1; RORα: RAR related orphan receptor A; STRAP: serine-threonine kinase receptor associated protein; PNKP: polynucleotide kinase 3’-phosphatase; UNG2: uracil DNA glycosylase 2; SMUG1: single-strand-selective monofunctional uracil-DNA glycosylase 1; GCN5: lysine acetyltransferase 2A (alias KAT2A); PCNA: proliferating cell nuclear antigen; β-TRCP: beta-transducin repeat containing E3 ubiquitin protein ligase; REDD1: regulated in development and DNA damage responses 1; TSC2: TSC complex subunit 2; NF2: neurofibromin 2; AhR: aryl hydrocarbon receptor; ERα: estrogen receptor α; COP1: ring finger and WD repeat domain 2 (RFWD2); CDC25A: cell division cycle 25A; HUWE1: HECT, UBA and WWE domain containing 1; AMBRA1: autophagy and beclin 1 regulator 1; PEX7P: peroxisomal biogenesis factor 7p; HOXB4: homeobox B4; CSN5: COP9 signalosome subunit 5; ORCA: origin recognition complex subunit 1; RASSF1A: RAS association domain family 1 isoform A; CHK1: checkpoint kinase 1; SKP2: S-phase kinase associated protein 2; HOXA9: homeobox A9; SAMHD1: SAM and HD domain containing deoxynucleoside triphosphate triphosphohydrolase 1.
Cullin 4 E3 ligases in tumorigenesis
According to the canonical definition of cancer, there are six hallmarks of cancer pathology, including sustaining proliferative signaling, resisting cell death, evading growth suppression, activating invasion and metastasis, inducing angiogenesis, and enabling replicative immortality86. Any regulators that effects these hallmarks are believed to be involved in tumorigenesis, either in a positive or negative manner87. Due to the participation of CRL4 in multiple cellular processes, including cell cycle progression and regulation of apoptotic death, its potential role in tumorigenesis has drawn much attention. Current studies have verified a close, but complicated, and context-dependent correlation between CRL4 activity and malignancies, on basis of the evidence from genetic mouse models, clinical pathological specimens and cellular and molecular experiments, which will be discussed in detail below (also see a recent review on pathological role of the CRL4 ubiquitin ligase88).
Cullin 4 scaffold protein in tumorigenesis
As described above, there are two homologous CRL4scaffold proteins, namely CUL4A and CUL4B, which have shared substrates but possess non-overlapping activities. Current investigations implicate that both scaffold proteins exert oncogenic functions in a variety of malignancies, suggesting that the entire CRL4 system may play a role in tumorigenesis despite having distinct interactions towards cellular substrates, since the scaffold protein exclusively corresponds to the activity of the whole complex89–92,51,93.
Mounting evidence suggests critical roles that CUL4A plays in cellular response to DNA damage. Following genotoxic stress, CUL4A targets the CDT1 DNA replication licensing factor, the PR-Set7/Set8 histone methyltransferase, and the p21 cyclin dependent kinase inhibitor for ubiquitin-proteasomal degradation to ensure that damaged DNA is not replicated94–100. However, CUL4A also restricts DNA repair capacity post UV. Conditional knockout of Cul4a in skin rendered the mice hyper-resistant to UV-induced dermatological carcinogenesis55. In response to UV irradiation, cells activate the nucleotide excision repair (NER) pathway for removing UV-induced cyclobutane pyrimidine dimers and 6, 4-photoproducts, and the G1/S DNA damage checkpoint pathway to stop the cell cycle until DNA lesions are repaired by NER. Liu et al showed that CUL4A is a potent inhibitor of DNA damage response by ubiquitin-dependent degradation of DDB2 and XPC, two rate-limiting NER factors responsible for recognition of DNA damage, as well as the p21 effector of the G1/S DNA damage checkpoint pathway. As such, Cul4a−/− cells displayed dramatically enhanced DNA repair and DNA damage checkpoint activities, and Cul4a knockout mice are hyper-resistant to UV-induced skin carcinogenesis55. Therefore, blocking CUL4A activity represents an attractive new strategy for cancer prevention.
Genetic and pathological evidence suggests that CUL4A is frequently dysregulated in neoplastic events. Gene amplification and transcriptional upregulation are both shown to account for CUL4A overexpression in tumors (e.g. CUL4A and CUL4B are both targets of LEF/TCF transcription factors of the canonical Wnt signaling)101 Overexpression of CUL4A leads to initiation and progression of lung cancer in mice89–91. Consistently, numerous pathological studies have detected upregulation of CUL4A in various human cancer specimens, such as gastric cancer102–104, breast cancer104–106, colorectal cancer104,107–109 and lung cancer93,110,111, which is also negatively correlated with prognostic survival93,110,111,112–116. As the core component of the CRL4 complex, the tumorigenic effects of CUL4A overexpression mainly depend on its interactions with downstream targets (details will be discussed in subsequent sections), which trigger cell cycle progression and/or epithelial mesenchymal transition, leading to tumor proliferation, invasion or drug resistance106,107,117.
CUL4B has also been recognized as a tumorigenic protein in many malignancies. Transgenic upregulation of Cul4b induces spontaneous hepatic carcinogenesis92. Analyses from clinical samples show that aberrant expression of CUL4B has been observed in a wide spectrum of malignant tumors, especially lung cancer93,118, colorectal cancer115,116 and pancreatic cancer119, serving as a negative indicator of patient survival as well116,120. Analogous to CUL4A, abnormal interactions between CRL4B and substrates directly link to activation of proliferative pathways (such as the Wnt/β-catenin pathway) and epigenetic silencing, participating in nearly every aspect of tumor progression121,122. On the other side, downregulation of certain miRNAs such as miR-194 and miR-300 may explain, in part, the mechanisms inducing the overexpression of CUL4B overexpression in several cancers, although its upstream mechanisms remain largely unknown118,119 (Table 4).
Table 4.
Neoplastic roles of CRL4 components
| Component | Neoplastic role | Physiological evidence (knockout or transgenic mouse models) | Pathological evidence (cancer relevant human specimens) |
|---|---|---|---|
| CUL4A | Oncogenic | Lung cancer initiation and progression (transgenic overexpression of Cul4a)89–91;Resistant to UV-induced skin carcinogenesis (skin-specific knockout of Cul4a)55 | Overexpressed in multiple myeloma310, cholangiocarcinoma113,114, gastric cancer102–104, colorectal cancer104,107–109, ovarian cancer104,112,311, lung cancer93,110,111, breast cancer104–106, osteosarcoma312,313, hepatocellular cancer117,314, malignant pleural mesothelioma315,316, pituitary adenoma317, prostate cancer318 |
| CUL4B | Oncogenic | Spontaneous development of liver cancer (transgenic overexpression of Cul4b)92 | Overexpressed in cholangiocarcinoma120, pancreatic carcinoma119, lung cancer93·118, colorectal cancer115,116, glioma319, liver cancer122, cervical cancer121, esophageal cancer320, ovarian cancer112 |
| RBX1 | Oncogenic | N/A | Overexpressed in ovarian cancer123, lung cancer124, gastric cancer125, bladder cancer126 |
| DDB1 | Context-dependent | Spontaneous development of liver cancer (hepatocyte-specific knockout of Ddbl)73 | Overexpressed in ovarian cancer123 |
The RING-finger protein RBX1 and adaptor DDB1 in tumorigenesis
Similar to the Cul4A and Cul4B scaffold proteins, the catalytic component, RING-finger RBX1 is also regarded as an oncoprotein in ovarian cancer123, lung cancer124, gastric cancer125, and bladder cancer126. Nevertheless, both genetically engineered mouse models and mechanistic studies are still limited.
The adaptor DDB1, it displays a context-dependent role in tumorigenesis as well. Results from knockout mouse models show that hepatocyte-specific ablation of Ddb1 facilitates spontaneous development of liver cancer73, whereas overexpression of DDB1 is detected in ovarian cancer, consistent with the oncogenic role of other CRL4 complex components123. This context-dependent role of DDB1 may reflect its diversity on neoplastic contributions of the CLR4 complexes, or is possibly a result of other non-CLR4 related activities, since DDB1 is not an exclusive protein for CRL4 and could involve other regulatory networks in cancer cells. In this regard, DDB1 was initially identified as a component of the UV-DDB complex that surveys the chromosomes for UV-induced DNA lesions and recruits the nucleotide excision repair apparatus to sites of DNA damage (Table 4)127.
Cullin 4 substrate proteins in tumorigenesis
Oncogenic members
Similar to the oncogenic functions of the CUL4A/B scaffolds, the majority of the CRL4 substrate recognition receptors play tumorigenic roles. HBx (hepatitis B virus regulatory protein X) is a recombinant protein encoded by the HBV genome (hepatitis B virus) and expressed following infection in human hepatocytes. This protein can function as a substrate recognition receptor of CRL4 leading to the destabilization of SMC5/6 (structural maintenance of chromosome 5/6) to increase the replication of HBV128. Transgenic overexpression of Hbx induces liver carcinogenesis in rodent models129,130, which is also found to be upregulated in human liver cancer specimens131. Increased HBx expression is also observed in intrahepatic cholangiocarcinoma132 and adenoid cystic carcinoma133, suggesting a general involvement of HBx in promoting tumorigenesis. Mechanistically, apart from its involvement in HBV replication, SMC5/6 also has a vital role in regulating cell division and proliferation while HBx could interact with apoptotic machinery to stimulate downstream oncogenes134,135, which together explain the oncogenic impact of HBx in human cancers (Table 3 and Table 5).
Table 5.
Major oncogenic substrate recognition proteins
| Substrate recognition protein | Physiological evidence (knockout or transgenic mouse models) | Pathological evidence (cancer relevant human specimens) | Biochemical evidence (cancer relevant substrates) |
|---|---|---|---|
| DCAF1 | NA | Overexpressed in ovarian cancer136, colon cancer109 | LATS1/2139,140, Dicer1109, NF2141 |
| DCAF2 | NA | Overexpressed in head and neck squamous cell carcinoma149, melanoma150, ovarian cancer123, Ewing sarcoma151 | CDT1150,152, p21150, SET8150 |
| DCAF4L2 | NA | Overexpressed in colorectal cancer276 | PPM1B276 |
| DCAF6 | NA | Overexpressed in prostate cancer154 | NA |
| DCAF13 | NA | Overexpressed in hepatocellular carcinoma155 | NA |
| AhR | NA | Overexpressed in liver cancer321, thyroid cancer322 | NA |
| COPS8 | NA | NA | CENP-A323 |
| FBXO44 | NA | Overexpressed in breast cancer324 | NA |
| FBXW5 | NA | NA | DLC1289, TSC2290,325 |
| HBx | Hepatocarcinogenesis in transgenic Hbx mice129,130 | Overexpressed in liver cancer131, intrahepatic cholangiocarcinoma132, adenoid cystic carcinoma133 | NA |
| RBBP7 | NA | NA | CENP-A292,323 |
| STRAP | NA | Overexpressed in colon cancer326, lung cancer326 | NA |
| HOXB4 | NA | Overexpressed in acute myeloid leukemia327, nephroblastoma328 | Geminin329 |
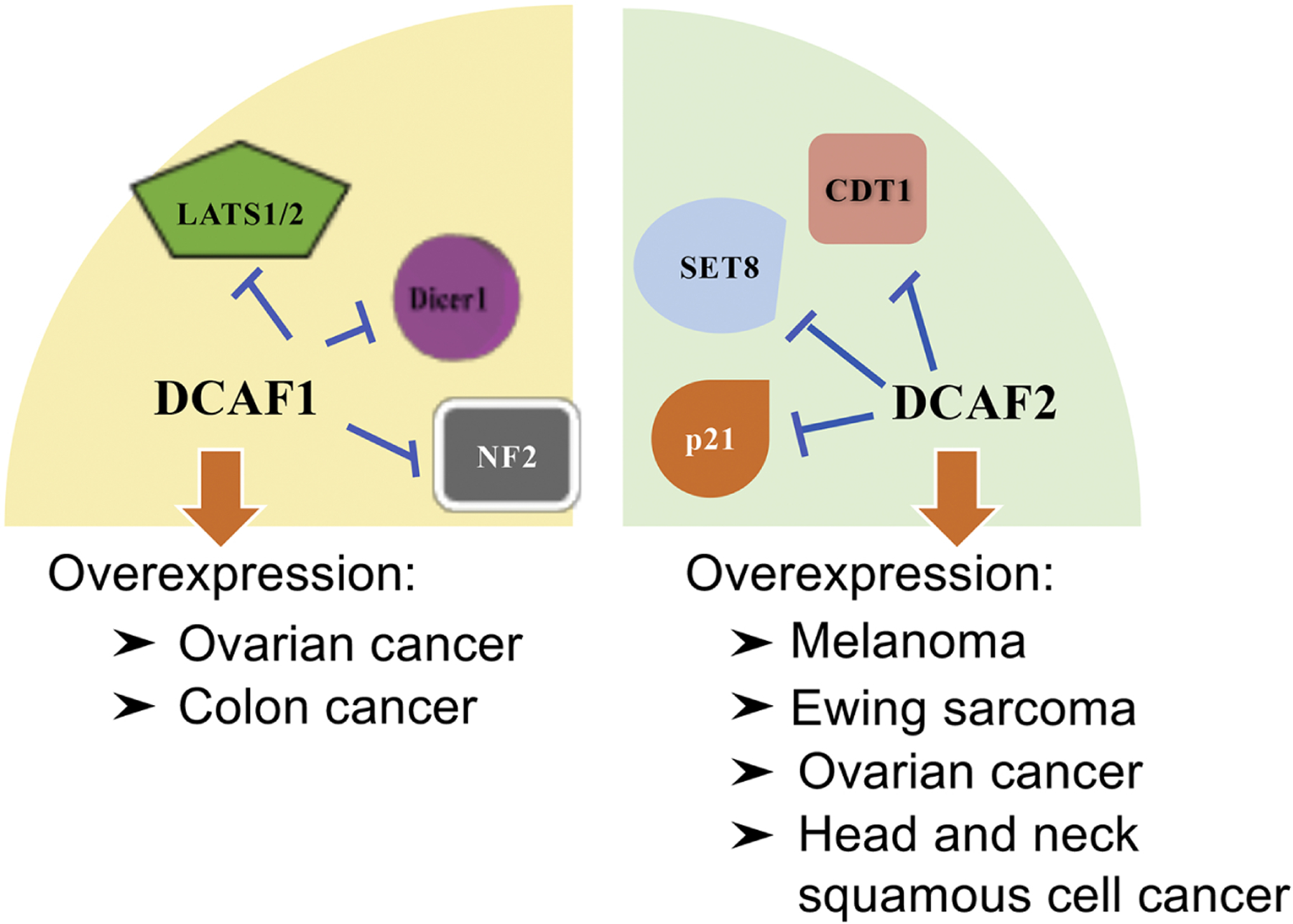
DCAF1 is one of the primary and well-defined substrate recognition receptors of CLR4 complexes, where more than 15 proteins have been identified as substrates of DCAF1. By interacting with target substrates, resulting in either degradation or non-degradation outcomes, DCAF1 participates in multiple physiological and pathological processes, including cell cycle arrest136, germ cell meiosis67, virus replication110, skeletal muscle differentiation137 and transcriptional repression138. As for its neoplastic roles, DCAF1 has been regarded as an important oncogenic component of the CRL4 complex, where it is found to be upregulated in both ovarian cancer136 and colon cancer specimens109. This oncogenic role may be mechanistically attributed to its destabilizing effects on substrates such as LATS1 (large tumor suppressor 1)139,140, Dicer1109, and NF2 (neurofibromin 2)141, which normally act as tumor suppressors to inhibit the activity of multiple downstream oncogenic pathways, including Hippo/YAP139,140 and JAK/STAT3109. Interestingly, the tumor suppressor Merlin (NF2) is a direct inhibitor of CRL4-DCAF1/VPRBP, and tumor-associated mutations of Merlin either fail to bind DCAF1 or are unable to move into the nucleus to block DCAF1 ubiquitin ligase activity142. However, genetically engineered mouse models are still necessary to fully appreciate the oncogenic roles of DCAF1 (Table 3 and Table 5).
Besides DCAF1, DCAF2 is also a well-investigated substrate receptor of CRL4, with more than 15 defined downstream ubiquitin substrates. By destabilizing or activating specific substrates, it is involved in the regulation of various biological events, including genomic stability143–145, cell cycle progression146, gluconeogenesis72,147 and transcriptional suppression148. As for a role in tumorigenesis, DCAF2 has been recognized as a major oncogenic receptor of CRL4. Elevated levels of DCAF2 are frequently detected in head and neck squamous cell carcinoma149, melanoma150, ovarian cancer123 and Ewing sarcoma151. Mechanistic studies have confirmed that degradation of CDT1, p21, and SET8 (set-domain histone methyltransferase-8) by CRL4 (DCAF2) regulates histone methylation and stimulates proliferation of melanoma, serving as a potential target of anti-melanoma therapeutics150,152. However, biochemical evidence of a role for DCAF2 in other malignancies remains limited, let alone in rodent models (Table 3 and Table 5). Recently, WDR4 (WD repeat 4-containing cullin-RING ubiquitin ligase 4) has been reported as an oncogenic receptor of CRL4 by promoting the degradation of the tumor suppressor PML (Promyelocytic leukemia) via ubiquitination to re-modulate an immunosuppressive tumor microenvironment in lung cancer setting153.
In addition to the aforementioned receptors, there are CRL4 substrate recognition receptors responsible for triggering the onset and progression of malignancies, including overexpression of DCAF6 in prostate cancer154, DCAF13 in hepatocellular carcinoma155. The majority of substrate-mediated mechanisms are centered on cell cycle progression, genomic instability, and regulation of apoptosis, suggesting therapeutic potential in targeting CRL4 ubiquitin ligase (Figure 2, Table 3 and Table 5).
Figure 2. Major oncogenic substrates of CRL4.
Tumor suppressive members
Despite mounting evidence of an oncogenic role of CRL4, many substrate receptors of CRL4 have demonstrated tumor suppressive effects, adding diversity and complexity into the regulatory landscape of CRL4 in tumorigenesis.
DDB2 (damaged DNA binding 2)
DDB2 is recognized as one of the most consequential tumor suppressors in human cancer. Physiologically, the majority of CRL4 (DDB2)-substrate interactions lead to alterations of cell cycle regulation and DNA repair, hinting at an underlying role in neoplastic control84,156. Genetic ablation of Ddb2 sensitizes rodent models to UV-induced skin tumorigenesis76 while downregulated expression of DDB2 is commonly observed in cases of human prostate cancer154, skin cancer84 and colorectal cancer157, which support the tumor suppressive role of DDB2. Moreover, studies have shown that destabilization of oncogenic substrates, including HBO1 (histone acetyltransferase bound to ORC 1)158, may contribute to the cancer inhibitory effects of DDB2 (Table 3 and Table 6).
Table 6.
Major tumor suppressive substrate recognition proteins
| Substrate recognition protein | Physiological evidence (knockout or transgenic mouse models) | Pathological evidence (cancer relevant human specimens) | Biochemical evidence (cancer relevant substrates) |
|---|---|---|---|
| DCAF8 | NA | NA | CDC25A170 |
| DCAF11 | NA | NA | NRF2171, p21172 |
| DCAF15 | NA | NA | CAPERα173 |
| COP1 | NA | Downregulated in renal cell carcinoma165 | ETV5166, c-Jun167 |
| CRBN | NA | Downregulated in multiple myeloma161 | IKZF1/3162, GSPT1163, CK1α164 |
| DDB2 | Susceptible to UV-induced skin carcinogenesis (systemic Ddb2 knockout)76 | Downregulated in prostate cancer154, skin cancer84, colorectal cancer157 | AR154,285, HBO1158 |
| WDR70 | NA | Loss-of-function mutation in ovarian cancer169 | H2B294,330 |
Cereblon (CRBN)
Besides DDB2, cereblon (CRBN) is another vital substrate receptor of CRL4 found negatively correlated with tumorigenic behaviors. Owing to the variety of downstream substrates, CRL4 (CRBN) has multiple impacts on cellular biology, especially in neurodegeneration159, anti-epileptogenesis71 and homeostasis of membrane excitability160. The anti-tumor role of CRBN is usually centered on hematological malignancies, whose expression level is dramatically decreased in multiple myeloma cells161. Although IKZF1/3 (Ikaros family zinc finger protein 1/3), GSPT1 (G1-to-S phase transition 1) and CK1 (casein kinase 1α) may not be traditional substrates of CRBN, the induced turnover of IKZF1/3162, GSPT1163 and CK1α164 by CRL4 (CRBN) accounts for the inhibitory effects of thalidomide-based molecules (thalidomide, lenalinomide, and pomalidomide) against multiple myeloma, leukemia, and myelodysplastic syndrome, respectively, which may mechanistically correlate to the inhibition on cell cycle progression (Table 3 and Table 6). The role of thalidomide-based molecules inducing CRBN-dependent degradation of various key tumorigenic factors will be discussed further below.
COP1 (constitutive photomorphogenesis 1)
Expression of COP1 is suppressed in renal cell carcinoma165. Moreover, biochemical studies suggest that by destabilizing its substrate ETV5 (ETS variant 5) and c-Jun, CRL4 (COP1) suppresses multiple oncogenic transcription factors in lung tumorigenesis, suggesting a tumor suppressive function166,167. These results have indicated a role for COP1 in cancer regulation. Cop1 knockout mice are embryonic lethal, whereas Cop1+/− mice are viable and fertile168. Through the generation of the Cop1 hypomorphic alleles, in which the Cop1 protein level was reduced by 90%, shows a 15–20% reduction in body weight compared to wild-type mice, coupled with decreased organ sizes168 (Table 3 and Table 6).
WDR70 (WD repeat domain 70)
Loss-of-function mutations of WDR70 have been identified in ovarian cancer169. WDR70 mediates mono-ubiquitination of histone H2B, thereby ensuring H2B stability during cell division and preventing malignant transformation169. DCAF8, DCAF11 and DCAF15 have been identified as potential tumor suppressors due to their effects on regulating the stability of CDC25A (cell division cycle 25A)170, NRF2 (nuclear factor erythroid 2-related factor 2)171, p21172 and CAPERα (coactivator of activating protein-1 and estrogen receptor α)173, respectively (Figure 3, Table 3 and 6).
Figure 3. Major tumor suppressive substrates of CRL4.

Context-dependent members
Unlike those members with specific neoplastic contributions, context-dependent substrate recognition receptors are rarely identified. Both GNB2 (G protein subunit beta 2) and GNB3 are substrate receptors for CRL4, which degrade GRK2 (G protein-coupled receptor kinase 2) resulting in cardiovascular protective effects. However, due to the inconsistent roles of GRK2 in tumorigenesis, it is thought to be an oncoprotein in breast cancer174 and tumor suppressor in hepatocellular carcinoma175, these two receptors are accordingly considered as context-dependent members, despite a lack of clinical or mouse models studies175–177 (Table 3 and Table 7).
Table 7.
Major context-dependent substrate recognition proteins
Degrons recognized by Cullin 4 substrate receptors
Degrons, specific short amino acid within a target protein, is critical for the interaction of target protein with their E3 ligase receptors178. Degrons have been identified within many E3 ligase receptor substrates, such as the ETGE degron motif for KEAP1179; and the D-box, the KEN box and the ABBA motif for the APC/C E3 ligase complex180. More importantly, regulation of degron recognition and/or availability including through post-translational modifications such as phosphorylation, methylation, acetylation and hydroxylation, has been reported to promote or inhibit substrate recognition by E3 ligases in response to environmental stimuli. For instance, the substrate recognition receptor β-TRCP recognizes a phosphorylated DSG degron181, and pVHL utilizes the prolyl hydroxylated degrons182. Conceivably, inherited/somatic mutations have been identified in degrons contributing to the accumulation of oncoproteins through escaping E3 ligase-mediated degradation, leading to various diseases, including cancer178. Thus, defining the degrons within substrates of E3 receptors is not only crucial to validate the direct interaction of a target protein with its E3 ligase receptor, but also highlight the potential to design therapeutics to target the oncoproteins for degradation.
CRL4 substrate receptors also display recognition of their substrates in a degron dependent manner. For instance, COP1 has been characterized to bind degrons within substrates with the consensus amino acid sequence EExxxVP[D/E] (Figure 4A)183, and Cdt2, also termed DCAF2, recognized its substrates containing a PCNA-interacting peptide (PIP) degron (Figure 4B)184. Interestingly, DCAF1, harboring a putative chromo domain, has been demonstrated to read a “monomethyl degron” (Figure 4C)138, whereas, Cereblon (CRBN), a direct thalidomide teratogenicity target protein, recognizes an “acetylated degron” in the presence of high glutamine (Figure 4D)185. However, due to limited number of substrates identified for CRL4 receptors, more effort is necessary in identifying CRL4 substrates, and degrons within those substrates, to clarify the exact degron requirements. These will benefit biomedical studies and further improve development of cancer treatments, especially in utilizing the PROTAC technology186, which has been considered a novel mechanism targeting what have been long thought of as “undruggable” targets.
Figure 4. A schematic illustration of degrons that are recognized by CRL4 receptors.

Harnessing the ubiquitin ligase for targeted degradation of cellular proteins
During the past two decades, targeted therapy has become the most promising strategy in modern medicine through the identification and development of small therapeutic molecules targeting mutated or modified factors driving cancer. In addition, biologics such as antibody-based therapeutics, RNA interference, and CRISPR-mediated genetic modifications, have gained much interest recently in drug discovery.
The primary mechanism behind antibody-based biologics is based on antigen-antibody interactions that may specifically and potently block the activity of pathogenic antigens to achieve therapeutic goals, which has been successful in cases such as bevacizumab for VEGFR (vascular endothelial growth factor receptor) and cetuximab for EGFR (epidermal growth factor receptor)187. However, since their ionic properties are naturally rejected by the non-ionic cell membrane, these antibody-based biologic drugs are only effective to extracellular or membrane proteins and also costly in nature, which greatly limit their clinical applications186. In addition, the RNAi (RNA interference) technology has emerged as a vital addition to antibody-based regimens, which focuses on degrading intracellular mRNA to restrict the translation and expression of specific proteins via pre-designed siRNA. Nevertheless, because of the instability of siRNA (small interfering RNA) under physiological circumstances, RNAi treatment displays unfavorably oral bioavailability and often poor tissue distribution, which has largely limited its usage thus far to liver diseases188. In consideration of these challenges, a better small molecule with intracellular permeability, increased bioavailability, and widespread distribution may therefore provide wider bioavailability and efficacy.
Protein Knockout
Most intracellular proteins are directed to the ubiquitin machinery for proteolytic destruction. Owing to the outstanding selectivity and degradative proficiency of the ubiquitin machinery, nature has chosen ubiquitin ligases as a prime apparatus for eliminating key target proteins, thereby altering a desired cellular process or pathway (Figure 5). In 1993, Scheffner and colleagues described the first case of viral hijacking of the ubiquitin ligase to promote tumorigenesis: the high-risk human papillomavirus (HPV) types 16 and 18 utilize their oncogenic E6 oncoprotein to recruit p53 tumor suppressor to the cellular E6AP ubiquitin ligase for ubiquitination and proteasomal degradation189. Here E6 functions as a bridging peptide that brings E6AP and p53 in close proximity to facilitate ubiquitin transfer onto p53.
Figure 5. Timeline illustrating crucial discoveries of the protein knockout and PROTAC techniques.
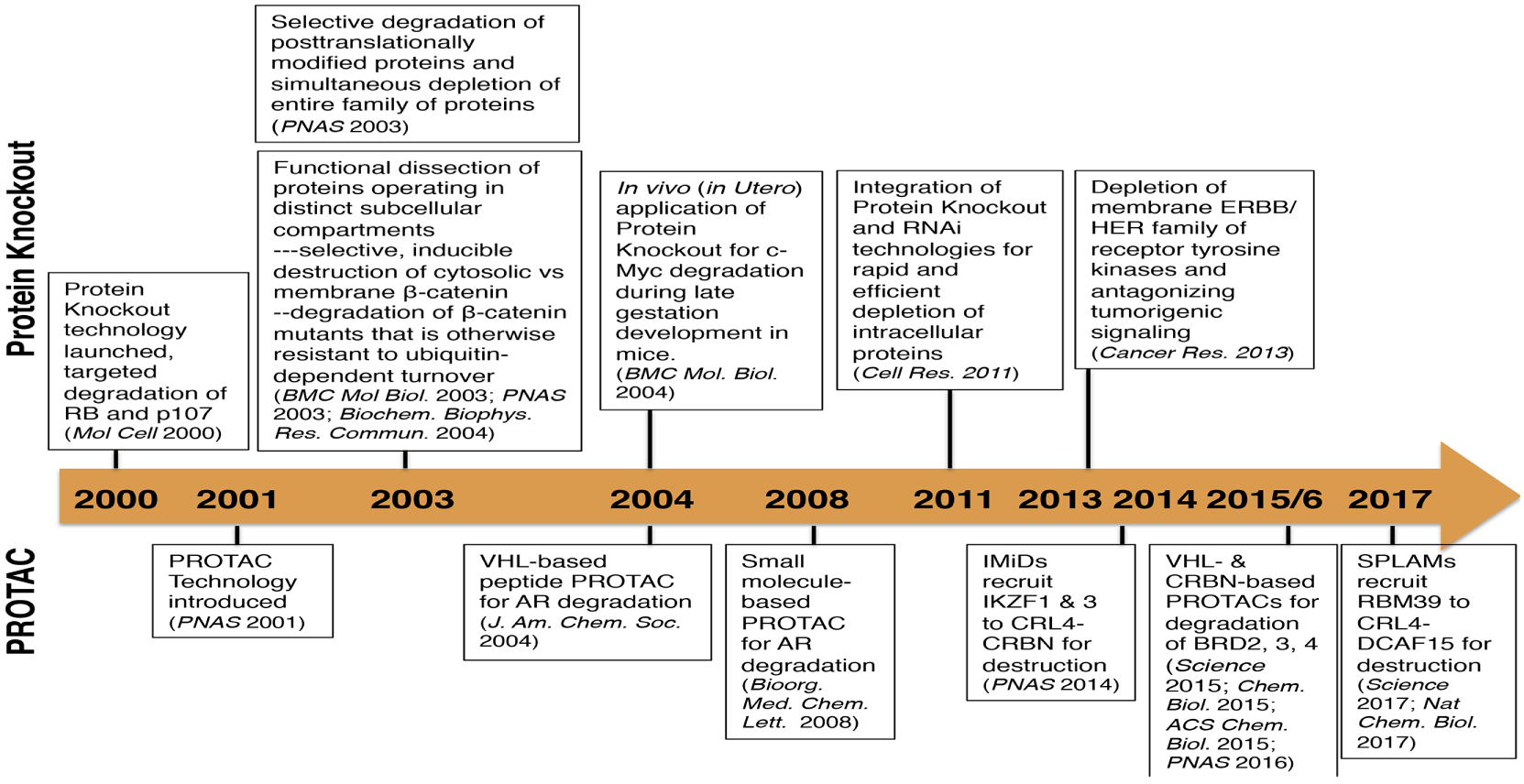
Given that ubiquitin ligases are modular proteins and substrate specificity is dictated by protein-protein interactions, and inspired by the hijacking mechanism of the HPVs, Zhou et al. set out to engineer SCFβ-TRCP ubiquitin ligase to target cellular proteins which are otherwise not substrates of β-TRCP or escaped recognition by β-TRCP due to oncogenic mutations of the DSG degron190,191. This is achieved by covalent attachment of specific binding peptide of the desired target to β-TRCP or truncated β-TRCP deleted of the substrate-binding WD40 domain, thereby enabling recruitment of desired target protein to the SCF machinery for ubiquitination and proteasomal degradation. This engineered ubiquitination machinery, designated protein knockout (PKO), was employed to successfully deplete a variety of intracellular or membrane proteins of interest, including pRB, p107, p130, β-catenin, c-Myc, EGFR, ErbB2/HER2, ErbB3/HER3, as well as viral oncoproteins (HIV16 E1)191–197. Moreover, the use of PKO demonstrated the remarkable versatility in selective targeting of post-translationally modified subpopulation of desired cellular proteins, a unique function that are not attainable by CRIPSPR-Cas9, RNAi and antisense technologies191–197. Furthermore, PKO can be integrated with RNAi to block protein synthesis and speed up protein destruction simultaneously, thereby achieving more rapid and effective depletion of stable proteins198. Taken together, these studies set the stage for harnessing ubiquitin ligases in degrading cellular proteins of interest, and opened up new avenues to design and develop therapeutic strategies in drug discovery.
PROTAC (PROteolysis TArgeting Chimera)
PROTAC technique has been established as an alternative to current small molecule therapies. PROTACs feature a bimodal molecule that simultaneously connects an E3 ligase and the target protein, leading to ubiquitin-dependent degradation on specific substrates199. This method creates an opportunity to degrade specific proteins that currently could not be pharmaceutically targeted by small molecules or without characterized E3 ligases, and could theoretically lead to the proteolysis of any substrate via only a single E3 ligase. Therefore, identifying appropriate binding ligands for E3 ligases and substrates is the most critical and challenging step for PROTAC design (Figure 5).
The first successful model of PROTAC was reported in 2001 by Sakamoto and colleagues, who creatively designed a chimeric molecule with a small ligand for MetAP2 (methionine aminopeptidase 2) and a 10-amino-acid phosphopeptide for β-TRCP, a substrate recognition receptor for CRL1200. Later, both the Ivan and Jaakkola groups described a VHL (von Hippel-Lindau)-based PROTAC design which promoted the degradation of FKBP12 (the 12-kDa FK506-binding protein) and AR via the VHL E3 ligase202. The HIFα (hypoxia inducible factor α) moiety, which was a natural substrate for CRL2 (VHL), was therefore incorporated into the PROTAC molecule as a ligand for VHL, a substrate receptor of CRL2. The hydroxylation of Proline-564 on HIFα was pre-requisite and necessary for recruiting VHL182,201. Therefore, this hydroxyproline-containing chimeric molecule became a classical representative of peptide-based PROTACs. Nonetheless, due to the peptidic nature of HIFα moiety in this PROTAC, its inefficient intracellular delivery limits in vivo applications. The hydroxyproline core is actually a non-ionic subunit of the HIFα moiety, and a re-designed VHL ligand featuring a sole hydroxyproline core greatly reduces the peptidic nature, while still retains binding affinity, which is therefore defined as small molecule-based PROTAC and more amenable for delivery to intracellular targets203 (Details will be discussed in subsequent sections) (Figure 6 and Table 9).
Figure 6. Major CRL4 targeted drugs and pharmacological mechanisms.
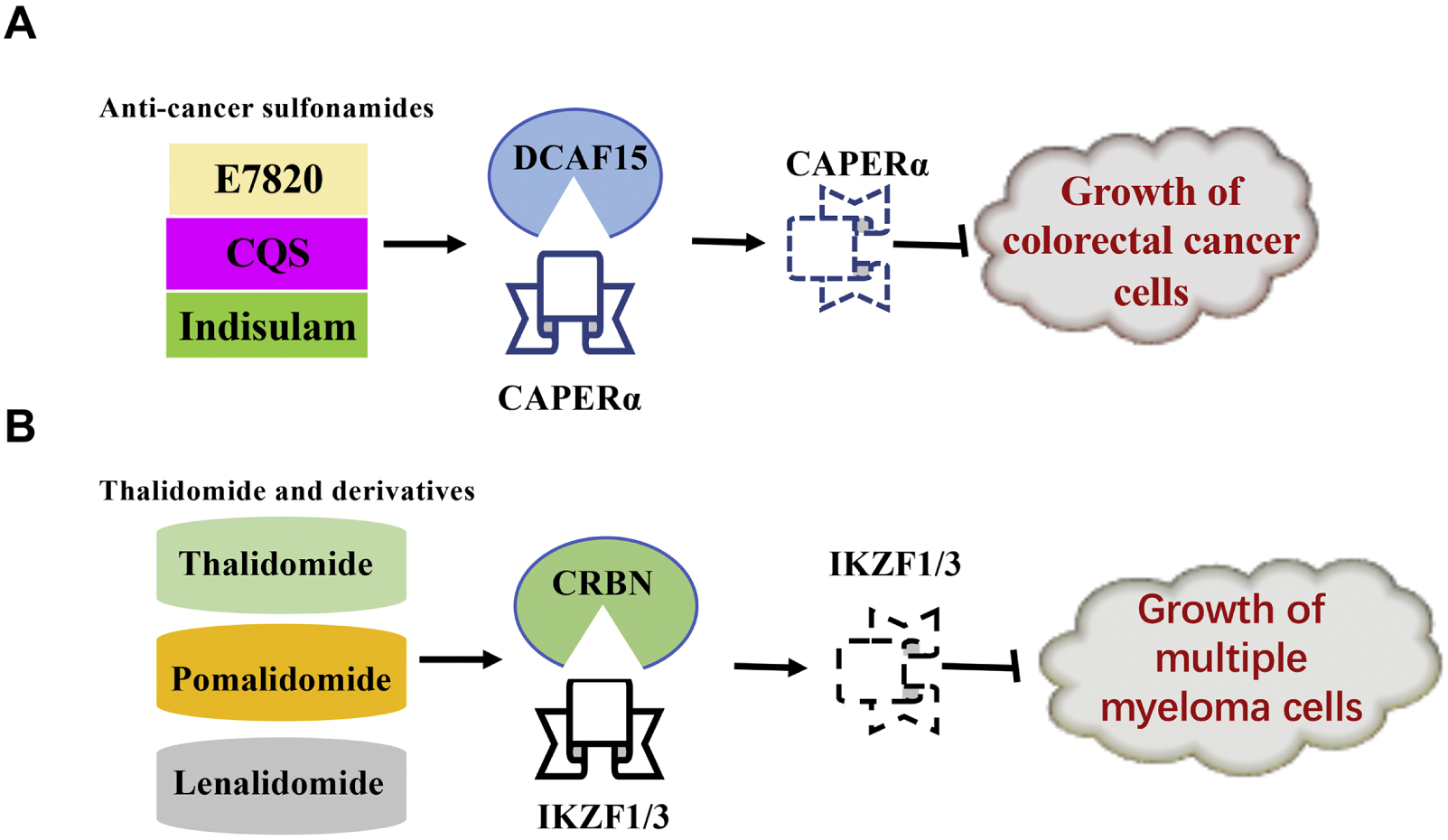
Table 9.
Major peptide PROTACs and their biological impacts
| Name | Target | Target ligand | E3 ligase | E3 ligase ligand | Biological impacts |
|---|---|---|---|---|---|
| Api-PROTAC | AHR | Apigenin | CRL2 (VHL) | A pentapeptide sequence derived from HIF1α | Potential roles against AHR positive disorders332,333 |
| Tri_a-PROTAC | AKT | PCC | CRL2 (VHL) | A heptapeptide derived from HIF1α | Potential roles against AKT positive disorders227 |
| PROTAC-3 | AR | DHT | CRL1 (β-TRCP) | A phosphopeptide derived from IκBα | Potential roles against AR positive disorders228 |
| PROTAC-5 | AR | DHT | CRL2 (VHL) | A heptapeptide derived from HIF1α | Potential roles against AR positive disorders202 |
| PROTAC-2 | ER | Estradiol | CRL1 (β-TRCP) | A phosphopeptide derived from IκBα | Potential roles against ER positive disorders228 |
| E2-SMPI | ER | Estradiol | CRL2 (VHL) | An octapeptide or pentapeptide derived from HIF1α | Inhibition of endothelial cell differentiation220–222 |
| PROTAC-4 | FKBP12 | AP21998 | CRL2 (VHL) | A heptapeptide derived from HIF1α | Potential roles against FKBP12 positive disorders202 |
| phosphoPROTAC-1 | FRS2α | Tyrosine phosphorylation sequences of TrkA | CRL2 (VHL) | A heptapeptide derived from HIF1α | Inhibition of neuronal differentiation223 |
| HBx-PROTAC | HBx | The oligomerization domain of HBx | CRL2 (VHL) | ODD domain of HIF1α | Potential roles against HBV infection and development of liver cancer225 |
| Fu-SMPI | MetAP-2 | Fumagillol | CRL2 (VHL) | An octapeptide derived from HIF1α | Potential roles against MetAP-2 positive disorders220 |
| phosphoPROTAC-2 | PI3K | Tyrosine phosphorylation sequences of ErbB3 | CRL2 (VHL) | A heptapeptide derived from HIF1α | Loss of viability of breast cancer cells223 |
| Smad3-PROTAC | Smad3 | SMC | CRL2 (VHL) | A pentapeptide derived from HIF1α | Potential roles against the development of renal fibrosis226 |
| TH006 | Tau | Sequences derived from β-tubulin | CRL2 (VHL) | A heptapeptide derived from HIF1α | Reduced proteotoxic effects among AD mice224 |
Abbreviations: NS: not specific; AHR: aryl hydrocarbon receptor; PCC: protein-catalyzed capture agent; DHT: dihydroxytestosterone; FRS2α: fibroblast growth factor receptor substrate 2 α; TrkA: tropomyosin receptor kinase A; PI3K: phosphatidylinositol-3-kinase; ErbB3: erythroblastosis oncogene B3; MetAP-2: methionine aminopeptidase-2; SMC: small molecule compound; ERK: extracellular regulated protein kinase; ODD: oxygen-dependent degradation; E2-SMPI: Estradiol-Small molecule proteolysis inducer; SNIPER: specific and nongenetic IAPs-dependent protein erasers; AR: androgen receptor; ER: estrogen receptor; AD: Alzheimer’s Disease.
Small molecule-based PROTACs
CRL2 (VHL)-based PROTACs
The peptidic features of PROTACs limited its clinical application, which led to the design of less ionic and therefore a more bioavailable type of PROTAC, later defined as small molecule PROTACs186. The first report of a CRL2 (VHL)-based small molecule PROTAC emerged in 2012, which featured a hydroxyproline core inside the ligand for VHL with the unnecessary residues of HIFα moiety trimmed to minimize the molecular weight and peptide nature, allowing greater penetration of the cell plasma membrane204–206. In 2015, the degrading effects of this small molecule CRL2 (VHL)-based PROTAC was demonstrated in preclinical cancer models where this PROTAC displayed marked degradation of specific proteins of more than 90%, including RIPK2 (receptor interacting serine/threonine kinase 2), ERRα (estrogen-related receptor α) and HaloTag fusion proteins203,207. Therefore, these results demonstrate potential for PROTACs as a novel therapeutic option against numerous diseases, especially cancers. Currently, more preclinical data is necessary before assessing therapeutic efficacy in clinical trials (Table 10).
Table 10.
Major small molecule PROTACs and their biological impacts
| Name | Target | Target ligand | E3 ligase | E3 ligase ligand | Biological impacts |
|---|---|---|---|---|---|
| SNIPER-1 | AR | DHT | cIAP1 | BE04 | Potential roles against AR positive disorders216 |
| SARM-nutlin PROTAC | AR | SARM | MDM2 | Nutlin | Potential roles against AR positive disorders214 |
| TKI-PROTAC-1 | BCR-ABL | TKI moiety (bosutinib and dasatinib) | CRL4 (CRB) | Pomalidomide | Reduced viability of chronic myelogenous leukemic cells238 |
| Compound MZ1 | BRD4 | JQ1 | CRL2 (VHL) | VHL binders | Suppression on oncogenic transcriptions associated with BRD4235 |
| ARV-771 | BRD4 | OTX015 | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Tumor suppression in CRPC mouse models233; Induced apoptosis in MCL cells334 |
| Compound 23 | BRD4 | Azacarbazole | CRL4 (CRB) | Thalidomide | Growth suppression against xenograft leukemic tumors234 |
| dBET1 | BRD4 | JQ1 | CRL4 (CRB) | Phthalimide | Delayed leukemia progression in mice211 |
| CLIPTAC-1 | BRD4 | TCO-tagged JQ1 | CRL4 (CRB) | Thalidomide | Potential roles against BRD4 positive disorders237 |
| ARV-825 | BRD4 | OTX015 | CRL4 (CRB) | Pomalidomide | Suppressed proliferation and enhanced apoptosis in Burkitt’s lymphoma210; Induced apoptosis in MCL cells334 |
| TKI-PROTAC-2 | c-ABL | TKI moiety (dasatinib) | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Potential roles against c-ABL positive disorders238 |
| TKI-PROTAC-3 | c-ABL | TKI moiety (bosutinib and dasatinib) | CRL4 (CRB) | Pomalidomide | Potential roles against c-ABL positive disorders238 |
| CDK9-PROTAC | CDK9 | Aminopyrazole analog | CRL4 (CRB) | Thalidomide | Potential roles against CDK9 positive disorders213 |
| Compound 4 | CRABP I/II | ATRA | cIAP1 | MeBS | Inhibited cell migration of neuroblastoma cells218,219 |
| brequinar-PROTAC | DHODH | Brequinar | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Cytotoxicity against cancer cells239 |
| SNIPER-2 | ER | Estrone | cIAP1 | BE04 | Potential roles against ER positive disorders216,217 |
| CLIPTAC-2 | ERK1/2 | Probe 1 | CRL4 (CRB) | Thalidomide | Potential roles against ERK1/2 positive disorders237 |
| PROTAC_ ERRα | ERRα | Thiazolidinedione-based ligand | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Potential roles against ERRα positive disorders203 |
| HT7-PROTAC-1 | FKBP12 | NS | ß-TRCP-HT7 | Chloroalkane | Potential roles against FKBP12 positive disorders230 |
| dFKBP | FKBP12 | Steel factor | CRL4 (CRB) | Phthalimide | against FKBP12 positive disorders211 |
| HT7-PROTAC-2 | FKBP12 | NS | Parkin-HT7 | Chloroalkane | Potential roles against FKBP12 positive disorders230 |
| SNIPER-3 | RAR | Ch55 | cIAP1 | BE04 | Potential roles against RAR positive disorders216 |
| PROTAC_ RIPK2 | RIPK2 | Vandetanib | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Potential roles against RIPK2 positive disorders203 |
| SirReal-PROTAC | Sirtuin 2 | SirReal | CRL4 (CRB) | Thalidomide | Enhanced acetylation of the microtubule network212 |
| SNIPER (TACC3) | TACC3 | KHS101 | APC/C (CDH1) | Bestatin | Cytotoxic effects against cancer cells229 |
| PROTAC 3i | TBK1 | Aminopyrimidine chemotype | CRL2 (VHL) | HIF1α-derived and hydroxyproline-contained ligand | Potential roles against TBK1 positive disorders335 |
| Homo-PROTAC | VHL | VH032 | CRL2 (VHL) | VH298 | Self-degradation of E3 ligases231 |
Abbreviations: NS: not specific; ODD: oxygen-dependent degradation; E2-SMPI: Estradiol-Small molecule proteolysis inducer; SNIPER: specific and nongenetic IAPs-dependent protein erasers; AHR: aryl hydrocarbon receptor; AR: androgen receptor; ER: estrogen receptor; FKBP12: the 12-kDa FK506-binding protein; RAR: retinoic acid receptor; cIAP1: cellular inhibitor of apoptosis protein 1; SARM: non-steroidal androgen receptor ligand; MDM2: mouse double minute 2 homolog; BCR-ABL: TKI: tyrosine kinase inhibitor; BCR-ABL: breakpoint cluster region-abelson murine leukemia viral oncogene; BRD4: JQ1: a BET bromodomain inhibitor; BE04: a bestatin derivative that has affinity to cIAP1; CRPC: castration-resistant prostate cancer; CRABP: cellular retinoic acid-binding protein; ATRA: all-trans retinoic acid; MeBS: methyl bestatin; ERRα: estrogen-related receptor α; RIPK2: receptor interacting serine/threonine kinase 2; TACC3: transforming acidic coiled coil-containing protein 3; DHODH: dihydroorotate dehydrogenase; TBK1: TANK-binding kinase 1; MCL: mantle cell lymphoma; CDK9: cyclin-dependent kinase 9; SirReal: sirtuin rearranging ligand; SMC: small molecule compound; ERK: extracellular regulated protein kinase.
Thalidomide derivatives and CRL4 (CRBN)-based PROTAC
As mentioned above, thalidomide and its derivatives are able to directly target the substrate receptor CRBN of CRL4, promoting the degradation of multiple substrates for anti-tumor effects especially in multiple myeloma162,208,209. Since these drugs feature non-ionic structures and are therefore suitable as small molecule ligands, many CRL4 (CRBN)-based PROTACs have been constructed via utilizing the moiety of thalidomide derivatives as the ligand for CRBN in conjugation with specific inhibitors as the substrate ligands.
In 2015, Winter and colleagues reported that by assembling phthalimide moiety and BET antagonist, a heterobifunctional PROTAC was formed which bridged CRBN and BRD4 (bromodomain-containing protein 4), leading to the highly selective degradation of BRD4 and consequently demonstrating anti-proliferative and tumor inhibitory effects in mouse models210,211. Additionally, with a FKBP12 inhibitor as the substrate ligand, the phthalimide-equipped PROTAC could also connect CRBN with FKBP12 to facilitate its ubiquitin-dependent degradation211. Apart from the phthalimide-characterized PROTACs, thalidomide-equipped PROTACs have also emerged, which lead to the degradation of Sirtuin 2 (SIRT2)212 and CDK9 (cyclin-dependent kinase 9)213, implicating their possible applications in regulating cell cycle and epigenetic stability (Table 10).
Other major E3 ligases for PROTAC technology
The MDM2-based PROTAC was the first reported example of small molecule PROTACs214. It was constructed by a nonsteroidal AR ligand for AR recognition, while the MDM2 ligand was a polyethylene glycol (PEG)-contained nutlin, a pharmaceutical inhibitor of MDM2. Schneekloth and colleagues discovered that the expression of AR was effectively inhibited in HeLa cells after administration of MDM2-based PROTAC which could be reversed by the proteasome inhibitor epoxomicin, displaying therapeutic potential against AR-positive human malignancies214,215.
Additionally, the cIAP1-based PROTAC has also been designed, featuring a bestatin-incorporated ligand for E3 ligase cIAP1215. According to different target ligands for substrates, this set of PROTACs could successfully lead to the degradation of ERα216,217, RAR (retinoic acid receptor)216 and CRABP I/II (cellular retinoic acid binding protein I/II)218,219, involved in multiple biological activities such as metastasis of neuroblastoma cells (Table 10).
PROTAC: Major biological impacts Peptide PROTAC
As the first-generation of PROTAC technology, peptide PROTACs had limited tissue distributions and membrane penetration. However, these PROTACs still exhibited various biological effects via degrading specific substrates.
CRL2 (VHL) is the major E3 ligase that is utilized for experimental design of peptide PROTACs. Targeted turnover of ER (estrogen receptor), FRS2α (fibroblast growth factor receptor substrate 2 α) and tau, VHL-based PROTACs lead to inhibition of endothelial cell differentiation220–222, neuronal differentiation223 and reduced proteotoxic effects in AD (Alzheimer’s Disease) mouse models224, respectively, demonstrating vital physiological participation and therapeutic potential against pathological conditions. Additionally, the HBx-targeted PROTAC also displayed protective roles against HBV (hepatitis B virus) infection and liver carcinogenesis225 while the Smad3-targeted PROTAC prevented the progression of renal fibrosis226. Meanwhile, despite a lack of in vivo evidence, the remaining PROTACs are believed to have crucial roles in multiple biological events, especially for those targeting AKT227, AR202 and MetAP-2220 for degradation.
Besides CRL2 (VHL), there are also two PROTACs, namely PROTAC-3 and PROTAC-2, that utilize CRL1 (β-TRCP) as E3 ligases to destabilize AR and ER, respectively228. Nonetheless, although the technology works well to degrade target proteins in vitro, more in vivo evidence are necessary to evaluate their potential efficacy.
Currently, although peptide PROTACs are no longer a primary focus in the development of PROTAC technology due to difficulties described above, there may still be instances where there is therapeutic value of peptide PROTACs under certain circumstances, which maybe act as alternative options to small molecule PROTACs in the future (Table 9).
Small molecule PROTAC
Due to its limited molecule weight and non-ionic features, small molecule PROTAC is thought to provide greater bioavailability and efficacy. CRL2 (VHL) and CRL4 (CRBN) are two key E3 ligases for small molecule PROTACs. Currently, owing to the incurability of malignant diseases, all evidence regarding VHL-based PROTACs is tumor associated, which serve as potential anti-cancer interventions (Table 10). Similarly, the pathological role of CRBN-based PROTACs is also centered on neoplastic relevant behaviors (Table 10). Besides this direct evidence, the biological functions of the remaining small molecule PROTACs still require in vivo verification.
In addition, other PROTAC members including cIAP1-based, APC/C (CDH1)-based and MDM2-based PROTACs also emerge as protein degraders for different targets, displaying potential therapeutic effects against RAR (retinoic acid receptor)-positive216, TACC3 (transforming acidic coiled coil-containing protein 3)-positive229 and AR-positive214 disorders respectively. Moreover, apart from these traditional small molecule PROTACs, two types of novel PROTACs have been reported. The first type, HaloTag7-fused PROTACs (HT7-PROTACs), where unlike other PROTACs the E3 ligases (β-TRCP) is engineered and fused to a HT7 tag. And then a chloroalkane ligand in the PROTAC is applied to attach the E3 ligase and leads to substrate degradation230. This suggests that if it is difficult to design a small molecule ligand for certain E3 ligases, engineered modifications of E3 ligases may be an alternative approach for cell and molecular based studies. Secondly, a homo-PROTAC has also been designed, which is a bivalent small-molecule dimerizer of VHL E3 ligase that induces its self-degradation231. This likewise provides therapeutic implications against those disorders with an elevated expression of undruggable E3 ligases (Table 10).
PROTAC: anti-tumor functionality by degrading oncogenic proteins
Most of the PROTACS have thus far been designed with the purpose of treating cancers via degradation of specific oncogenic proteins, such as BRD4. Currently, investigations have confirmed that BRD4 acts as an oncoprotein in multiple malignancies such as prostate and hematological cancers, primarily by interacting with histones in order to trigger transcription of oncogenes232. Nevertheless, due to toxicities and progressive inefficacies of BRD4 inhibitors including BET, more specific and tolerable targeted medications are required, such as BRD4-targeted PROTACs. So far, there are six PROTACs reported to effectively target and destabilize BRD4, either VHL-based or CRBN-based. Among them, four PROTACs have shown anti-tumor efficacy, including ARV-771, compound 23, dBET1 and ARV-825, which display great inhibitory effects against the malignant progression of castration-resistant prostate cancer (CRPC)233, xenograft leukemic models234, leukemic cells211 and Burkitt’s lymphoma210 respectively. All these PROTACs, especially ARV-771 and ARV-825, are waiting for verification in animal studies before being assessed in the clinic. Furthermore, despite a lack of direct evidence, the other two PROTACs including compound MZ1235,236 and CLIPTAC-1236,237 also target BRD4, which are believed to have potential contributions against BRD4-positive neoplasms.
In addition to BRD4, other PROTACs have also been regarded as anti-tumor PROTACs. As the only peptide PROTAC, phosphoPROTAC-2 directly degrades PI3K via recruiting CRL2 (VHL), culminating in reduced viability of breast cancer cells223. BCR-ABL, is an oncogenic fusion protein for chronic myelogenous leukemia, can be targeted by TKI-PROTAC-1 which has shown in vitro effects on survival of chronic myelogenous leukemic cells238. CRABP I/II are cellular binding proteins of retinoic acid and mediate the biologic impact of retinoic acid under diverse circumstances. The oncogenic role of CRABP I/II in the pathogenesis of neuroblastoma has been widely shown, and correspondingly, compound 4, a CRABP I/II-targeted PROTAC, has also been shown to have a tumor suppressive role against the migration of neuroblastoma cells218,219. Moreover, both brequinar-PROTAC and SNIPER (TACC3) are characterized by their tumor inhibitory effect among colon and breast cancer cells, respectively, via targeting DHODH (dihydroorotate dehydrogenase)239 and TACC3 (transforming acidic coiled-coil-containing protein 3), respectively229. These results have shown the therapeutic potential of PROTACs as a novel class of anti-tumor therapeutics.
Apart from those with direct evidence, the majority of current PROTACs are assumed to potentially play anti-neoplastic roles, which have not been shown yet in vivo despite their pro-degrading effects on certain oncogenic substrates in cells. Among those, AR and ER are the most popular targets for PROTACs. At present, there have been four PROTACs, either peptide or small molecule, believed to successfully and specifically destabilize AR in vitro and in vivo, including PROTAC-3228,240, PROTAC-5202,240, SNIPER-1216,240 and SARM-nutlin PROTAC214,240. The AR-targeted PROTACs are considered as potential therapeutic agents given the extensive oncogenic role of AR in a variety of cancers, especially prostate cancer. Moreover, ER, a notorious oncoprotein in gynecological malignancies241, has been targeted by three PROTACs, namely PROTAC-2228,242, E2-SMPI220–222,242 and SNIPER-2216,217,243, all demonstrating degrading effects of the ER protein despite limited studies showing suppression of tumorigenesis.
Taken together, although currently no PROTAC has entered the clinic, the anti-tumor PROTACs are still expected to become important supplements to current targeted therapies. Compared to the direct evidence regarding the anti-tumor effects of small molecule PROTACs, only one peptide PROTAC has reported a direct effect on cancer cell viability, which highlights the advantage and therapeutic potential of small molecules PROTACs. Nevertheless, there are issues with the current status of PROTACs that should be addressed in future investigations. First, PROTAC related studies have lacked comparisons with standard of care agents, which may lower their credibility as an alternative to currently approved options. Second, although there has been direct evidence towards the anti-tumor impact of certain PROTACs, in vivo efficacy in animals has been limited, let alone clinical assessment, therefore more studies are necessary to demonstrate the clinical benefits of this promising technology (Figure 7 and Table 11).
Figure 7. Anti-tumor PROTACs and their working mechanisms.
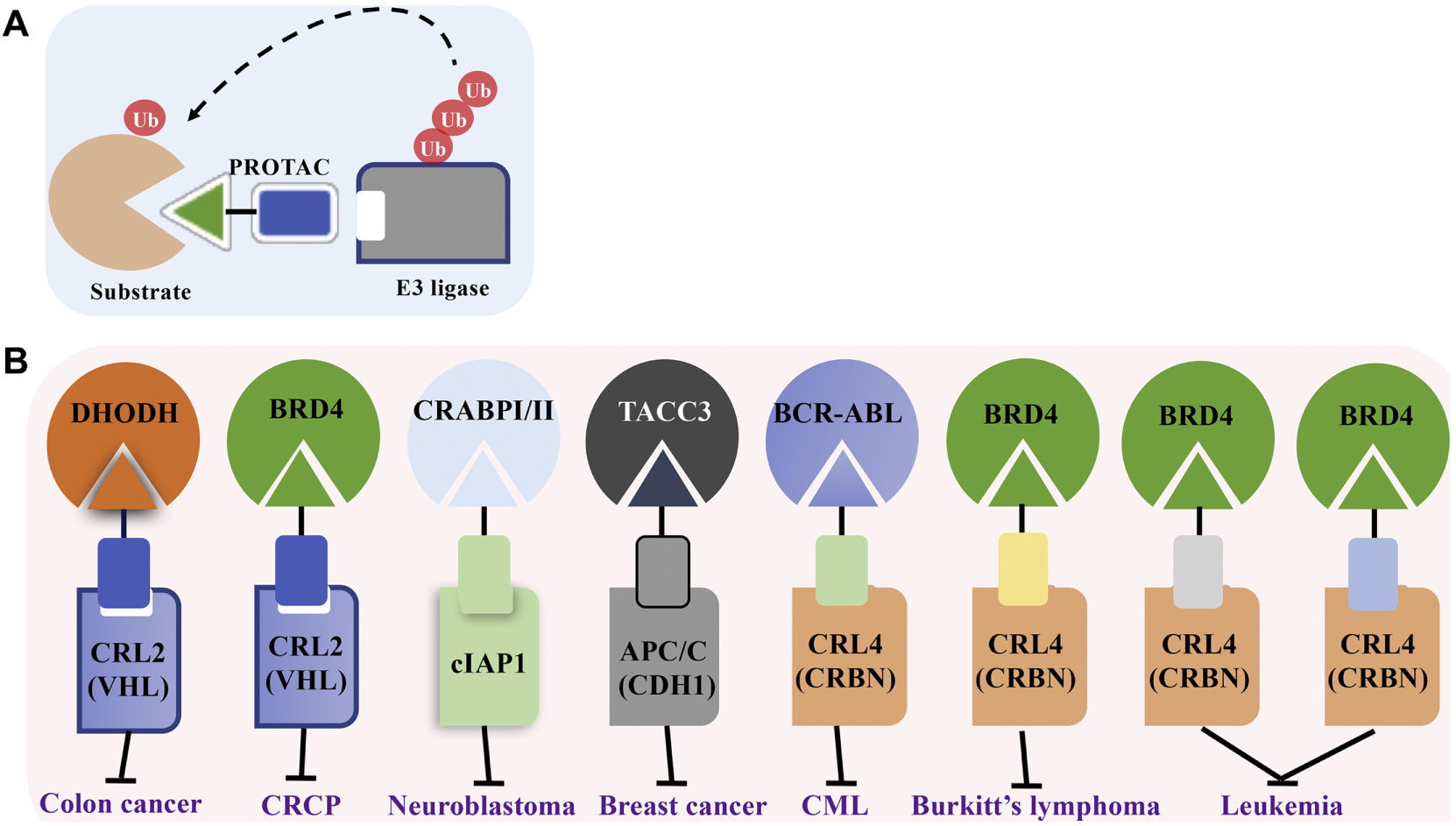
Table 11.
Major anti-tumor PROTACs and their contributions
| Name | Category | Target | E3 ligase | Evidence* |
|---|---|---|---|---|
| phosphoPROTAC-2 | Peptide | PI3K | CRL2 (VHL) | Direct roles against viability of breast cancer cells223 |
| TKI-PROTAC-1 | Small molecule | BCR-ABL | CRL4 (CRB) | Direct roles against viability of chronic myelogenous leukemic cells238 |
| ARV-771 | Small molecule | BRD4 | CRL2 (VHL) | Direct roles against the tumor growth in CRPC mouse models233 and MCL cells334 |
| Compound 23 | Small molecule | BRD4 | CRL4 (CRB) | Direct roles against the growth of xenograft leukemic tumors234 |
| dBET1 | Small molecule | BRD4 | CRL4 (CRB) | Direct roles against the progression of leukemia211 |
| ARV-825 | Small molecule | BRD4 | CRL4 (CRB) | Direct roles against the proliferation of Burkitt’s lymphoma210 and MCL cells334 |
| Compound 4 | Small molecule | CRABP I/II | cIAP1 | Direct roles against the migration of neuroblastoma cells218,219 |
| brequinar-PROTAC | Small molecule | DHODH | CRL2 (VHL) | Direct roles against the viability of colon cancer cells239 |
| SNIPER (TACC3) | Small molecule | TACC3 | APC/C (CDH1) | Direct roles against the viability of breast cancer cells229 |
| Api-PROTAC | Peptide | AHR | CRL2 (VHL) | Potential roles against multiple AHR positive cancers332,333,336 |
| Tri_a-PROTAC | Peptide | Akt | CRL2 (VHL) | Potential roles against multiple Akt positive cancers227,337 |
| PROTAC-3 | Peptide | AR | CRL1 (β-TRCP) | Potential roles against multiple AR positive cancers228,240 |
| PROTAC-5 | Peptide | AR | CRL2 (VHL) | Potential roles against multiple AR positive cancers202,240 |
| PROTAC-2 | Peptide | ER | CRL1 (β-TRCP) | Potential roles against multiple ER positive cancers228,242 |
| E2-SMPI | Peptide | ER | CRL2 (VHL) | Potential roles against multiple ER positive cancers220–222,242 |
| phosphoPROTAC-1 | Peptide | FRS2α | CRL2 (VHL) | Potential roles against multiple FRS2α positive cancers223,338 |
| HBx-PROTAC | Peptide | HBx | CRL2 (VHL) | Potential roles against development of liver cancer225 |
| Fu-SMPI | Peptide | MetAP-2 | CRL2 (VHL) | Potential roles against multiple MetAP-2 positive cancers220,339 |
| SNIPER-1 | Small molecule | AR | cIAP1 | Potential roles against multiple AR positive cancers216,240 |
| SARM–nutlin PROTAC | Small molecule | AR | MDM2 | Potential roles against multiple AR positive cancers214,240 |
| Compound MZ1 | Small molecule | BRD4 | CRL2 (VHL) | Potential roles against multiple BRD4 positive cancers235,236 |
| CLIPTAC-1 | Small molecule | BRD4 | CRL4 (CRB) | Potential roles against multiple BRD4 positive cancers236,237 |
| TKI-PROTAC-2 | Small molecule | c-ABL | CRL2 (VHL) | Potential roles against multiple c-ABL positive cancers238,340 |
| TKI-PROTAC-3 | Small molecule | c-ABL | CRL4 (CRB) | Potential roles against multiple c-ABL positive cancers238,340 |
| CDK9-PROTAC | Small molecule | CDK9 | CRL4 (CRB) | Potential roles against multiple CDK9 positive cancers213,341 |
| SNIPER-2 | Small molecule | ER | cIAP1 | Potential roles against multiple ER positive cancers216,217,243 |
| CLIPTAC-2 | Small molecule | ERK1/2 | CRL4 (CRB) | Potential roles against multiple ERK1/2 positive cancers237,342 |
| PROTAC_ ERRα | Small molecule | ERRα | CRL2 (VHL) | Potential roles against multiple ERRα positive cancers203,343 |
| SNIPER-3 | Small molecule | RAR | cIAP1 | Potential roles against multiple RAR positive cancers216,344 |
| PROTAC_ RIPK2 | Small molecule | RIPK2 | CRL2 (VHL) | Potential roles against multiple RIPK2 positive cancers203,345 |
| SirReal-PROTAC | Small molecule | Sirtuin 2 | CRL4 (CRB) | Potential roles against multiple Sirtuin 2 positive cancers212,346 |
| PROTAC 3i | Small molecule | TBK1 | CRL2 (VHL) | Potential roles against multiple TBK1 positive cancers335,347 |
If there are direct descriptions or results demonstrating the anti-tumor efficacy of specific PROTAC, then these kinds of evidences are defined as “Direct”, while if there are no direct descriptions, the anti-tumor effects can only be predicted from the role of substrates, then these kinds of evidences are defined as “Potential”.
Perspective and therapeutic implications
As we have described, many substrate receptors demonstrate tumor-inhibitory effects by degrading oncogenic substrates, serving as potential therapeutic targets for cancer treatment. Currently, there are two major types of medications that specially aim at the interactions between substrates and recognition receptors in order to exert anti-cancer impacts, namely sulfonamides and thalidomide derivatives, each characterized by specific mechanisms. Three members of anticancer sulfonamides E7820, CQS (chloroquinoxaline sulfonamide) and indisulam have displayed efficacy against various cancers, including colorectal cancer and melanoma244–246. However, their mechanisms of action remain poorly defined. Investigations have found that their anti-cancer contributions in colorectal cancer cells may rely on the interaction between CRL4 (DCAF15) and the substrate CAPERα, promoting its degradation173,247. Knockout of DCAF15 or overexpression of CAPERα confers resistance to the effects by sulfonamides, further proving DCAF15 as the mechanistic target of sulfonamides247. So far, all these drugs have undergone clinical trials and are waiting for approval by the FDA for clinical application against colorectal cancer.
Thalidomide and its derivatives, known as immunomodulatory drugs (IMiDs), have drawn much attention owing to their great anti-tumor effectiveness against hematological malignancies, especially multiple myeloma248. Current investigations have confirmed that IMiDs facilitates interaction between CRBN (cereblon) and its substrates. By binding to CRBN, IMiDs, including thalidomide, pomalidomide and lenalidomide, can trigger the degradation of substrates such as IKZF1/3248 and CK1α164, thus halting tumor expansion of multiple myeloma and myelodysplastic syndrome. All these IMiDs have gained FDA approval against multiple myeloma. Besides IMiDs, there is another type of CRBN modulator CC-885 which promotes the interaction between CRBN and GSPT1 to trigger the degradation of GSPT1, showing tumor suppressive effects against leukemic cells163. In addition, DCAF2 seems to function as a therapeutic target since its interaction with substrates SET8 and p21 could be specifically inhibited by Pevonedistat150 (Table 8).
Table 8.
Major CRL4s targeted therapies
| Drug | Mechanism | Function | Preclinical evidence | Clinical trial | FDA approval |
|---|---|---|---|---|---|
| E7820 | Promoting DCAF15-CAPERα degradation | Anti-growth in colorectal cancer cells247 | Yes | Completed | Not yet |
| CQS | Promoting DCAF15-CAPERα degradation | Anti-growth in colorectal cancer cells247 | Yes | Completed | Not yet |
| Indisulam | Promoting DCAF15-CAPERα degradation | Anti-growth in colorectal cancer cells173,247 | Yes | Completed | Not yet |
| CC-220 | Promoting CRBN- IKZF1 and IKZF3 degradation | Anti-tumor effect against multiple myeloma cells331 | Yes | Ongoing | Not yet |
| Lenalidomide | Promoting CRBN- CK1α degradation | Anti-tumor effect against myelodysplastic syndrome164 | Yes | Completed | Approved |
| Promoting CRBN- IKZF1 and IKZF3 degradation | Anti-tumor effect against multiple myeloma cells162,208,209 | Yes | Completed | Approved | |
| Thalidomide | Promoting CRBN- IKZF1 and IKZF3 degradation | Anti-tumor effect against multiple myeloma cells248 | Yes | Completed | Approved |
| Pomalidomide | Promoting CRBN- IKZF1 and IKZF3 degradation | Anti-tumor effect against multiple myeloma cells248 | Yes | Completed | Approved |
| CC-885 | Promoting CRBN- GSPT1 degradation | Anti-tumor effect against leukemic cells163 | Yes | Not yet | Not yet |
| Pevonedistat | Inactivation of DCAF2-SET8/p21 degradation | Anti-tumor effect against melanoma cells150 | Yes | Completed | Not yet |
Abbreviations: CQS: chloroquinoxaline sulfonamide; CC-220: alias Compound 6.
As a lesser understood member of the Cullin-RING ligase family, CRL4 has drawn much attention due to its extensive involvement in physiological and pathological conditions, especially in tumorigenesis. Based on the evidence described above from knockout and transgenic mouse models, human clinical data, and biochemical interactions, the scaffold protein CUL4A/B and RING finger protein RBX1 are believed to serve as oncoproteins in a variety of malignancies. Nevertheless, the adaptor DDB1 and substrate recognition receptors seem to play context dependent roles in tumorigenesis, adding diversity to the actual role of CRL4 in tumorigenesis.
Moreover, owing to its critical role in cancer regulation, many CRL4-targeted medications have been identified, including thalidomide and its derivatives that target CRBN and the sulfonamides that target DCAF15, which significantly suppress proliferation of multiple myeloma and colorectal cancer, respectively. PROTACs are a group of artificially assembled protein degraders that induce the ubiquitination-mediated degradation of substrates via recruiting endogenous E3 ligases, such as CRL4 (CRBN) and CRL2 (VHL). Due to their ability to increase the number of druggable targets and potential for high specificity, more than 30 PROTACs have been designed and display anti-tumor effects, especially BRD4-targeted PROTACs. Current and future studies in animal tumor models and clinical trials would lead to mechanistic and functional understanding of PROTACs as drug candidates, and set the stage for their clinical usage in cancer patients.
Acknowledgements
The authors sincerely apologize to all those colleagues whose important work was not cited in this paper owing to space limitations. They thank the members of Wei laboratory for critical reading and discussion of the manuscript. W.W. is a Leukemia & Lymphoma Society (LLS) research scholar. This work was supported in part by Scientific Research Training Program for Young Talents (Union Hospital, Tongji Medical College, Huazhong University of Science and Technology) to J.C., by National Natural Science Foundation of China (81572413) to K.T., by the V Foundation for Cancer Research to P.Z., and by US National Institutes of Health (NIH) grants to P. Z. (CA159925 and CA213992) and W.W. (GM094777 and CA177910).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing interests. Correspondence and requests for materials should be addressed to W.W. (wwei2@bidmc.harvard.edu) or P.Z. (pez2001@med.cornell.edu).
Reference
- 1.Leithe E Regulation of connexins by the ubiquitin system: Implications for intercellular communication and cancer. Biochim Biophys Acta 1865, 133–146, doi: 10.1016/j.bbcan.2016.02.001 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Varshavsky A The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu Rev Biochem 86, 123–128, doi: 10.1146/annurev-biochem-061516-044859 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Cheng J et al. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochim Biophys Acta 1869, 11–28, doi: 10.1016/j.bbcan.2017.11.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo J, Cheng J, North BJ & Wei W Functional analyses of major cancer-related signaling pathways in Alzheimer’s disease etiology. Biochim Biophys Acta 1868, 341–358, doi: 10.1016/j.bbcan.2017.07.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciechanover A The unravelling of the ubiquitin system. Nat Rev Mol Cell Biol 16, 322–324, doi: 10.1038/nrm3982 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Dikic I Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem 86, 193–224, doi: 10.1146/annurev-biochem-061516-044908 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Gilberto S & Peter M Dynamic ubiquitin signaling in cell cycle regulation. J Cell Biol 216, 2259–2271, doi: 10.1083/jcb.201703170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VerPlank JJS & Goldberg AL Regulating protein breakdown through proteasome phosphorylation. Biochem J 474, 3355–3371, doi: 10.1042/BCJ20160809 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vigneron N, Abi Habib J & Van den Eynde BJ Learning from the Proteasome: How To Fine-Tune Cancer Immunotherapy. Trends Cancer 3, 726–741, doi: 10.1016/j.trecan.2017.07.007 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Lazaro S, Gamarra D & Del Val M Proteolytic enzymes involved in MHC class I antigen processing: A guerrilla army that partners with the proteasome. Mol Immunol 68, 72–76, doi: 10.1016/j.molimm.2015.04.014 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Fierabracci A Proteasome inhibitors: a new perspective for treating autoimmune diseases. Curr Drug Targets 13, 1665–1675 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Deger JM, Gerson JE & Kayed R The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration. Aging Cell 14, 715–724, doi: 10.1111/acel.12359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Q et al. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front Aging Neurosci 8, 303, doi: 10.3389/fnagi.2016.00303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coutant F & Miossec P Altered dendritic cell functions in autoimmune diseases: distinct and overlapping profiles. Nat Rev Rheumatol 12, 703–715, doi: 10.1038/nrrheum.2016.147 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Magnacca A et al. Characterization of a proteasome and TAP-independent presentation of intracellular epitopes by HLA-B27 molecules. J Biol Chem 287, 30358–30367, doi: 10.1074/jbc.M112.384339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaur G & Batra S Emerging role of immunoproteasomes in pathophysiology. Immunol Cell Biol 94, 812–820, doi: 10.1038/icb.2016.50 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Kwon SK, Saindane M & Baek KH p53 stability is regulated by diverse deubiquitinating enzymes. Biochim Biophys Acta 1868, 404–411, doi: 10.1016/j.bbcan.2017.08.001 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Micel LN, Tentler JJ, Smith PG & Eckhardt GS Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J Clin Oncol 31, 1231–1238, doi: 10.1200/JCO.2012.44.0958 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama KI & Nakayama K Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6, 369–381, doi: 10.1038/nrc1881 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Zheng N, Zhou Q, Wang Z & Wei W Recent advances in SCF ubiquitin ligase complex: Clinical implications. Biochim Biophys Acta 1866, 12–22, doi: 10.1016/j.bbcan.2016.05.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wade M et al. Functional analysis and consequences of Mdm2 E3 ligase inhibition in human tumor cells. Oncogene 31, 4789–4797, doi: 10.1038/onc.2011.625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honda R, Tanaka H & Yasuda H Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420, 25–27 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Ding F, Xiao H, Wang M, Xie X & Hu F The role of the ubiquitin-proteasome pathway in cancer development and treatment. Front Biosci (Landmark Ed) 19, 886–895 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Groner AC et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 29, 846–858, doi: 10.1016/j.ccell.2016.04.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geng C et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 74, 5631–5643, doi: 10.1158/0008-5472.CAN-14-0476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dou QP & Zonder JA Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets 14, 517–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuo Y et al. Proteasome inhibitor MG132 inhibits angiogenesis in pancreatic cancer by blocking NF-kappaB activity. Dig Dis Sci 55, 1167–1176, doi: 10.1007/s10620-009-0814-4 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Kravtsova-Ivantsiv Y et al. KPC1-mediated ubiquitination and proteasomal processing of NF-kappaB1 p105 to p50 restricts tumor growth. Cell 161, 333–347, doi: 10.1016/j.cell.2015.03.001 (2015). [DOI] [PubMed] [Google Scholar]
- 29.de Poot SAH, Tian G & Finley D Meddling with fate: The proteasomal deubiquitinating enzymes. J Mol Biol, doi: 10.1016/j.jmb.2017.09.015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mevissen TET & Komander D Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem 86, 159–192, doi: 10.1146/annurev-biochem-061516-044916 (2017). [DOI] [PubMed] [Google Scholar]
- 31.He M et al. The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics. Cell Biosci 6, 62, doi: 10.1186/s13578-016-0127-1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eletr ZM & Wilkinson KD Regulation of proteolysis by human deubiquitinating enzymes. Biochim Biophys Acta 1843, 114–128, doi: 10.1016/j.bbamcr.2013.06.027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdul Rehman SA et al. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol Cell 63, 146–155, doi: 10.1016/j.molcel.2016.05.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwasna D et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol Cell 70, 150–164 e156, doi: 10.1016/j.molcel.2018.02.023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haahr P et al. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol Cell 70, 165–174 e166, doi: 10.1016/j.molcel.2018.02.024 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Kumari N et al. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta 1868, 456–483, doi: 10.1016/j.bbcan.2017.09.002 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Toloczko A et al. Deubiquitinating Enzyme USP9X Suppresses Tumor Growth via LATS Kinase and Core Components of the Hippo Pathway. Cancer Res 77, 4921–4933, doi: 10.1158/0008-5472.CAN-16-3413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graner E et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 5, 253–261 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Fajner V, Maspero E & Polo S Targeting HECT-type E3 ligases - insights from catalysis, regulation and inhibitors. FEBS Lett 591, 2636–2647, doi: 10.1002/1873-3468.12775 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Zheng N & Shabek N Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 86, 129–157, doi: 10.1146/annurev-biochem-060815-014922 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Wong CC, Gong B & Yu J Functional significance and therapeutic implication of ring-type E3 ligases in colorectal cancer. Oncogene, doi: 10.1038/onc.2017.313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Natarajan C & Takeda K Regulation of various DNA repair pathways by E3 ubiquitin ligases. J Cancer Res Ther 13, 157–169, doi: 10.4103/0973-1482.204879 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Bulatov E & Ciulli A Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J 467, 365–386, doi: 10.1042/BJ20141450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lydeard JR, Schulman BA & Harper JW Building and remodelling Cullin -RING E3 ubiquitin ligases. EMBO Rep 14, 1050–1061, doi: 10.1038/embor.2013.173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui D, Xiong X & Zhao Y Cullin-RING ligases in regulation of autophagy. Cell Div 11, 8, doi: 10.1186/s13008-016-0022-5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, Sui J, Zhang F & Zhang C Cullin family proteins and tumorigenesis: genetic association and molecular mechanisms. J Cancer 6, 233–242, doi: 10.7150/jca.11076 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y & Sun Y Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des 19, 3215–3225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson S & Xiong Y CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci 34, 562–570, doi: 10.1016/j.tibs.2009.07.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y et al. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. EMBO J 22, 6057–6067, doi: 10.1093/emboj/cdg577 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li B, Jia N, Kapur R & Chun KT Cul4A targets p27 for degradation and regulates proliferation, cell cycle exit, and differentiation during erythropoiesis. Blood 107, 4291–4299, doi: 10.1182/blood-2005-08-3349 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hannah J & Zhou P Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene 573, 33–45, doi: 10.1016/j.gene.2015.08.064 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sang Y, Yan F & Ren X The role and mechanism of CRL4 E3 ubiquitin ligase in cancer and its potential therapy implications. Oncotarget 6, 42590–42602, doi: 10.18632/oncotarget.6052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CY et al. Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X-linked mental retardation. Hum Mol Genet 21, 4270–4285, doi: 10.1093/hmg/dds261 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Liu L et al. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res 22, 1258–1269, doi: 10.1038/cr.2012.48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu L et al. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol Cell 34, 451–460, doi: 10.1016/j.molcel.2009.04.020 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cang Y et al. DDB1 is essential for genomic stability in developing epidermis. Proc Natl Acad Sci U S A 104, 2733–2737, doi: 10.1073/pnas.0611311104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cang Y et al. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 127, 929–940, doi: 10.1016/j.cell.2006.09.045 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Li B, Ruiz JC & Chun KT CUL-4A is critical for early embryonic development. Mol Cell Biol 22, 4997–5005 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kopanja D et al. Proliferation defects and genome instability in cells lacking Cul4A. Oncogene 28, 2456–2465, doi: 10.1038/onc.2009.86 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee J et al. Improved ex vivo expansion of adult hematopoietic stem cells by overcoming CUL4-mediated degradation of HOXB4. Blood 121, 4082–4089, doi: 10.1182/blood-2012-09-455204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sauvageau G et al. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci U S A 91, 12223–12227 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giampaolo A et al. HOXB gene expression and function in differentiating purified hematopoietic progenitors. Stem Cells 13 Suppl 1, 90–105 (1995). [PubMed] [Google Scholar]
- 63.Pineault N, Helgason CD, Lawrence HJ & Humphries RK Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp Hematol 30, 49–57 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Yin Y et al. The E3 ubiquitin ligase Cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev Biol 356, 51–62, doi: 10.1016/j.ydbio.2011.05.661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yin Y et al. Cell Autonomous and Nonautonomous Function of CUL4B in Mouse Spermatogenesis. J Biol Chem 291, 6923–6935, doi: 10.1074/jbc.M115.699660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kopanja D et al. Cul4A is essential for spermatogenesis and male fertility. Dev Biol 352, 278–287, doi: 10.1016/j.ydbio.2011.01.028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu C, Ji SY, Sha QQ, Sun QY & Fan HY CRL4-DCAF1 ubiquitin E3 ligase directs protein phosphatase 2A degradation to control oocyte meiotic maturation. Nat Commun 6, 8017, doi: 10.1038/ncomms9017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu YW et al. Maternal DCAF2 is crucial for maintenance of genome stability during the first cell cycle in mice. J Cell Sci 130, 3297–3307, doi: 10.1242/jcs.206664 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Yu C et al. CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science 342, 1518–1521, doi: 10.1126/science.1244587 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Yu C, Xu YW, Sha QQ & Fan HY CRL4DCAF1 is required in activated oocytes for follicle maintenance and ovulation. Mol Hum Reprod 21, 195–205, doi: 10.1093/molehr/gau103 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Liu J et al. CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat Commun 5, 3924, doi: 10.1038/ncomms4924 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Tong X et al. DDB1-Mediated CRY1 Degradation Promotes FOXO1-Driven Gluconeogenesis in Liver. Diabetes 66, 2571–2582, doi: 10.2337/db16-1600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamaji S et al. Hepatocyte-specific deletion of DDB1 induces liver regeneration and tumorigenesis. Proc Natl Acad Sci U S A 107, 22237–22242, doi: 10.1073/pnas.1015793108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li P et al. Lack of CUL4B in Adipocytes Promotes PPARgamma-Mediated Adipose Tissue Expansion and Insulin Sensitivity. Diabetes 66, 300–313, doi: 10.2337/db16-0743 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Li Q et al. A cullin 4B-RING E3 ligase complex fine-tunes pancreatic delta cell paracrine interactions. J Clin Invest 127, 2631–2646, doi: 10.1172/JCI91348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoon T et al. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene 24, 469–478, doi: 10.1038/sj.onc.1208211 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zou Y et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet 80, 561–566, doi: 10.1086/512489 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tarpey PS et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 80, 345–352, doi: 10.1086/511134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakagawa T & Xiong Y X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol Cell 43, 381–391, doi: 10.1016/j.molcel.2011.05.033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vulto-van Silfhout AT et al. Variants in CUL4B are associated with cerebral malformations. Hum Mutat 36, 106–117, doi: 10.1002/humu.22718 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klein CJ et al. Ubiquitin ligase defect by DCAF8 mutation causes HMSN2 with giant axons. Neurology 82, 873–878, doi: 10.1212/WNL.0000000000000206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Webster E et al. De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb Mol Case Stud 2, a001172, doi: 10.1101/mcs.a001172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ali RH et al. Exome sequencing revealed a novel biallelic deletion in the DCAF17 gene underlying Woodhouse Sakati syndrome. Clin Genet 90, 263–269, doi: 10.1111/cge.12700 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Kapetanaki MG et al. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A 103, 2588–2593, doi: 10.1073/pnas.0511160103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zha Z et al. Hypertension-associated C825T polymorphism impairs the function of Gbeta3 to target GRK2 ubiquitination. Cell Discov 2, 16005, doi: 10.1038/celldisc.2016.5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 88.Lee J & Zhou P Pathogenic Role of the CRL4 Ubiquitin Ligase in Human Disease. Front Oncol 2, 21, doi: 10.3389/fonc.2012.00021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang YL et al. Lung tumourigenesis in a conditional Cul4A transgenic mouse model. J Pathol 233, 113–123, doi: 10.1002/path.4352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Y et al. PR-Set7 is Degraded in a Conditional Cul4A Transgenic Mouse Model of Lung Cancer. Zhongguo Fei Ai Za Zhi 18, 345–350, doi: 10.3779/j.issn.1009-3419.2015.06.15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y et al. Analysis of lung tumor initiation and progression in transgenic mice for Cre-inducible overexpression of Cul4A gene. Thorac Cancer 6, 480–487, doi: 10.1111/1759-7714.12257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yuan J et al. Accelerated hepatocellular carcinoma development in CUL4B transgenic mice. Oncotarget 6, 15209–15221, doi: 10.18632/oncotarget.3829 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jia L et al. Dysregulation of CUL4A and CUL4B Ubiquitin Ligases in Lung Cancer. J Biol Chem 292, 2966–2978, doi: 10.1074/jbc.M116.765230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jin J, Arias EE, Chen J, Harper JW & Walter JC A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 23, 709–721, doi: 10.1016/j.molcel.2006.08.010 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Higa LA, Mihaylov IS, Banks DP, Zheng J & Zhang H Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol 5, 1008–1015, doi: 10.1038/ncb1061 (2003). [DOI] [PubMed] [Google Scholar]
- 96.Hu J, McCall CM, Ohta T & Xiong Y Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 6, 1003–1009, doi: 10.1038/ncb1172 (2004). [DOI] [PubMed] [Google Scholar]
- 97.Zhong W, Feng H, Santiago FE & Kipreos ET CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature 423, 885–889, doi: 10.1038/nature01747 (2003). [DOI] [PubMed] [Google Scholar]
- 98.Kim Y, Starostina NG & Kipreos ET The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev 22, 2507–2519, doi: 10.1101/gad.1703708 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abbas T et al. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 22, 2496–2506, doi: 10.1101/gad.1676108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Centore RC et al. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell 40, 22–33, doi: 10.1016/j.molcel.2010.09.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miranda-Carboni GA et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev 22, 3121–3134, doi: 10.1101/gad.1692808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gong Y et al. CUL4A promotes cell invasion in gastric cancer by activating the NF-kappaB signaling pathway. Biologics 11, 45–53, doi: 10.2147/BTT.S127650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deng J et al. Cullin 4A (CUL4A), a direct target of miR-9 and miR-137, promotes gastric cancer proliferation and invasion by regulating the Hippo signaling pathway. Oncotarget 7, 10037–10050, doi: 10.18632/oncotarget.7048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y et al. CUL4A induces epithelial-mesenchymal transition and promotes cancer metastasis by regulating ZEB1 expression. Cancer Res 74, 520–531, doi: 10.1158/0008-5472.CAN-13-2182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen LC et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res 58, 3677–3683 (1998). [PubMed] [Google Scholar]
- 106.Wang Y et al. Involvement of CUL4A in regulation of multidrug resistance to P-gp substrate drugs in breast cancer cells. Molecules 19, 159–176, doi: 10.3390/molecules19010159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sui X et al. CUL4A promotes proliferation and metastasis of colorectal cancer cells by regulating H3K4 trimethylation in epithelial-mesenchymal transition. Onco Targets Ther 10, 735–743, doi: 10.2147/OTT.S118897 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ren W et al. Aberrant Expression of CUL4A Is Associated with IL-6/ STAT3 Activation in Colorectal Cancer Progression. Arch Med Res 47, 214–222, doi: 10.1016/j.arcmed.2016.07.001 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Ren W et al. Jak-STAT3 pathway triggers DICER1 for proteasomal degradation by ubiquitin ligase complex of CUL4A(DCAF1) to promote colon cancer development. Cancer Lett 375, 209–220, doi: 10.1016/j.canlet.2016.02.055 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Hrecka K et al. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc Natl Acad Sci U S A 113, E3921–3930, doi: 10.1073/pnas.1605023113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y et al. CUL4A overexpression enhances lung tumor growth and sensitizes lung cancer cells to erlotinib via transcriptional regulation of EGFR. Mol Cancer 13, 252, doi: 10.1186/1476-4598-13-252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Birner P et al. Human homologue for Caenorhabditis elegans CUL-4 protein overexpression is associated with malignant potential of epithelial ovarian tumours and poor outcome in carcinoma. J Clin Pathol 65, 507–511, doi: 10.1136/jclinpath-2011-200463 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Huang G et al. CUL4A overexpression as an independent adverse prognosticator in intrahepatic cholangiocarcinoma. BMC Cancer 17, 395, doi: 10.1186/s12885-017-3389-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang TJ et al. Cullin 4A is associated with epithelial to mesenchymal transition and poor prognosis in perihilar cholangiocarcinoma. World J Gastroenterol 23, 2318–2329, doi: 10.3748/wjg.v23.i13.2318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Song B, Zhan H, Bian Q & Li J Knockdown of CUL4B inhibits proliferation and promotes apoptosis of colorectal cancer cells through suppressing the Wnt/beta-catenin signaling pathway. Int J Clin Exp Pathol 8, 10394–10402 (2015). [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang T et al. Cullin 4B is a novel prognostic marker that correlates with colon cancer progression and pathogenesis. Med Oncol 30, 534, doi: 10.1007/s12032-013-0534-7 (2013). [DOI] [PubMed] [Google Scholar]
- 117.Pan Y et al. CUL4A facilitates hepatocarcinogenesis by promoting cell cycle progression and epithelial-mesenchymal transition. Sci Rep 5, 17006, doi: 10.1038/srep17006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mi J et al. Dysregulation of the miR-194-CUL4B negative feedback loop drives tumorigenesis in non-small-cell lung carcinoma. Mol Oncol 11, 305–319, doi: 10.1002/1878-0261.12038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang JQ et al. MicroRNA-300 promotes apoptosis and inhibits proliferation, migration, invasion and epithelial-mesenchymal transition via the Wnt/beta-catenin signaling pathway by targeting CUL4B in pancreatic cancer cells. J Cell Biochem, doi: 10.1002/jcb.26270 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Li P et al. Cul4B is a novel prognostic marker in cholangiocarcinoma. Oncol Lett 14, 1265–1274, doi: 10.3892/ol.2017.6297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang Y et al. CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene 34, 104–118, doi: 10.1038/onc.2013.522 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Yuan J et al. CUL4B activates Wnt/beta-catenin signalling in hepatocellular carcinoma by repressing Wnt antagonists. J Pathol 235, 784–795, doi: 10.1002/path.4492 (2015). [DOI] [PubMed] [Google Scholar]
- 123.Pan WW et al. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J Biol Chem 288, 29680–29691, doi: 10.1074/jbc.M113.495069 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xing R et al. RBX1 expression is an unfavorable prognostic factor in patients with non-small cell lung cancer. Surg Oncol 25, 147–151, doi: 10.1016/j.suronc.2016.05.006 (2016). [DOI] [PubMed] [Google Scholar]
- 125.Migita K et al. Prognostic impact of RING box protein-1 (RBX1) expression in gastric cancer. Gastric Cancer 17, 601–609, doi: 10.1007/s10120-013-0318-y (2014). [DOI] [PubMed] [Google Scholar]
- 126.Wang W et al. Overexpression of RING box protein-1 (RBX1) associated with poor prognosis of non-muscle-invasive bladder transitional cell carcinoma. J Surg Oncol 107, 758–761, doi: 10.1002/jso.23317 (2013). [DOI] [PubMed] [Google Scholar]
- 127.Dualan R et al. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics 29, 62–69, doi: 10.1006/geno.1995.1215 (1995). [DOI] [PubMed] [Google Scholar]
- 128.Murphy CM et al. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep 16, 2846–2854, doi: 10.1016/j.celrep.2016.08.026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang C et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology 55, 108–120, doi: 10.1002/hep.24675 (2012). [DOI] [PubMed] [Google Scholar]
- 130.Koike K et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 19, 810–819 (1994). [PubMed] [Google Scholar]
- 131.Ding R et al. Overexpressed Id-1 is associated with patient prognosis and HBx expression in hepatitis B virus-related hepatocellular carcinoma. Cancer Biol Ther 10, 299–307 (2010). [DOI] [PubMed] [Google Scholar]
- 132.Zhou YM, Cao L, Li B, Zhang XZ & Yin ZF Expression of HBx protein in hepatitis B virus-infected intrahepatic cholangiocarcinoma. Hepatobiliary Pancreat Dis Int 11, 532–535 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Xie L, Wang W, Xu B & Liu Y [Experimental study on hepatitis B-virus X gene expression in adenoid cystic carcinoma]. Hua Xi Kou Qiang Yi Xue Za Zhi 32, 328–330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hwang G et al. SMC5/6 is required for the formation of segregation-competent bivalent chromosomes during meiosis I in mouse oocytes. Development 144, 1648–1660, doi: 10.1242/dev.145607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jiang T, Liu M, Wu J & Shi Y Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc Natl Acad Sci U S A 113, 2074–2079, doi: 10.1073/pnas.1525616113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang X et al. VprBP/DCAF1 Regulates the Degradation and Nonproteolytic Activation of the Cell Cycle Transcription Factor FoxM1. Mol Cell Biol 37, doi: 10.1128/MCB.00609-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jung ES et al. Jmjd2C increases MyoD transcriptional activity through inhibiting G9a-dependent MyoD degradation. Biochim Biophys Acta 1849, 1081–1094, doi: 10.1016/j.bbagrm.2015.07.001 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Lee JM et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell 48, 572–586, doi: 10.1016/j.molcel.2012.09.004 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Li W et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 26, 48–60, doi: 10.1016/j.ccr.2014.05.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ni W et al. A novel lncRNA uc.134 represses hepatocellular carcinoma progression by inhibiting CUL4A-mediated ubiquitination of LATS1. J Hematol Oncol 10, 91, doi: 10.1186/s13045-017-0449-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huang J & Chen J VprBP targets Merlin to the Roc1-Cul4A-DDB1 E3 ligase complex for degradation. Oncogene 27, 4056–4064, doi: 10.1038/onc.2008.44 (2008). [DOI] [PubMed] [Google Scholar]
- 142.Li W et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 140, 477–490, doi: 10.1016/j.cell.2010.01.029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oda H et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 40, 364–376, doi: 10.1016/j.molcel.2010.10.011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tardat M et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12, 1086–1093, doi: 10.1038/ncb2113 (2010). [DOI] [PubMed] [Google Scholar]
- 145.Abbas T et al. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 40, 9–21, doi: 10.1016/j.molcel.2010.09.014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Huh J & Piwnica-Worms H CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol 33, 213–226, doi: 10.1128/MCB.00847-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tong X et al. CUL4-DDB1-CDT2 E3 Ligase Regulates the Molecular Clock Activity by Promoting Ubiquitination-Dependent Degradation of the Mammalian CRY1. PLoS One 10, e0139725, doi: 10.1371/journal.pone.0139725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li Y, Jaramillo-Lambert A, Hao J, Yang Y & Zhu W The stability of histone acetyltransferase general control non-derepressible (Gcn) 5 is regulated by Cullin4-RING E3 ubiquitin ligase. J Biol Chem 286, 41344–41352, doi: 10.1074/jbc.M111.290767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vanderdys V et al. The Neddylation Inhibitor Pevonedistat (MLN4924) Suppresses and Radiosensitizes Head and Neck Squamous Carcinoma Cells and Tumors. Mol Cancer Ther, doi: 10.1158/1535-7163.MCT-17-0083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Benamar M et al. Inactivation of the CRL4-CDT2-SET8/p21 ubiquitylation and degradation axis underlies the therapeutic efficacy of pevonedistat in melanoma. EBioMedicine 10, 85–100, doi: 10.1016/j.ebiom.2016.06.023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mackintosh C et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31, 1287–1298, doi: 10.1038/onc.2011.317 (2012). [DOI] [PubMed] [Google Scholar]
- 152.Higa LA et al. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol 8, 1277–1283, doi: 10.1038/ncb1490 (2006). [DOI] [PubMed] [Google Scholar]
- 153.Wang YT et al. Ubiquitination of tumor suppressor PML regulates prometastatic and immunosuppressive tumor microenvironment. J Clin Invest 127, 2982–2997, doi: 10.1172/JCI89957 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen HH et al. NRIP/DCAF6 stabilizes the androgen receptor protein by displacing DDB2 from the CUL4A-DDB1 E3 ligase complex in prostate cancer. Oncotarget 8, 21501–21515, doi: 10.18632/oncotarget.15308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cao J et al. The overexpression and prognostic role of DCAF13 in hepatocellular carcinoma. Tumour Biol 39, 1010428317705753, doi: 10.1177/1010428317705753 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Yan Y, Zhang X & Legerski RJ Artemis interacts with the Cul4A-DDB1DDB2 ubiquitin E3 ligase and regulates degradation of the CDK inhibitor p27. Cell Cycle 10, 4098–4109, doi: 10.4161/cc.10.23.18227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Roy N et al. DDB2 suppresses epithelial-to-mesenchymal transition in colon cancer. Cancer Res 73, 3771–3782, doi: 10.1158/0008-5472.CAN-12-4069 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Matsunuma R et al. UV Damage-Induced Phosphorylation of HBO1 Triggers CRL4DDB2-Mediated Degradation To Regulate Cell Proliferation. Mol Cell Biol 36, 394–406, doi: 10.1128/MCB.00809-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Del Prete D, Rice RC, Rajadhyaksha AM & D’Adamio L Amyloid Precursor Protein (APP) May Act as a Substrate and a Recognition Unit for CRL4CRBN and Stub1 E3 Ligases Facilitating Ubiquitination of Proteins Involved in Presynaptic Functions and Neurodegeneration. J Biol Chem 291, 17209–17227, doi: 10.1074/jbc.M116.733626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen YA et al. The Cullin 4A/B-DDB1-Cereblon E3 Ubiquitin Ligase Complex Mediates the Degradation of CLC-1 Chloride Channels. Sci Rep 5, 10667, doi: 10.1038/srep10667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Broyl A et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood 121, 624–627, doi: 10.1182/blood-2012-06-438101 (2013). [DOI] [PubMed] [Google Scholar]
- 162.Kronke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305, doi: 10.1126/science.1244851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Matyskiela ME et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257, doi: 10.1038/nature18611 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Kronke J et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 523, 183–188, doi: 10.1038/nature14610 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ta L, Xuan C, Xing N & Zhu X COP1 is downregulated in renal cell carcinoma (RCC) and inhibits the migration of RCC ACHN cells in vitro. Mol Med Rep 14, 1371–1378, doi: 10.3892/mmr.2016.5373 (2016). [DOI] [PubMed] [Google Scholar]
- 166.Zhang Z et al. Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc Natl Acad Sci U S A 114, 3903–3908, doi: 10.1073/pnas.1621177114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wertz IE et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science 303, 1371–1374, doi: 10.1126/science.1093549 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Migliorini D et al. Cop1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. J Clin Invest 121, 1329–1343, doi: 10.1172/JCI45784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Guo LD et al. [Functional Analysis of DNA Damage Repair Factor WDR70 and Its Mutation in Ovarian Cancer]. Sichuan Da Xue Xue Bao Yi Xue Ban 47, 501–506 (2016). [PubMed] [Google Scholar]
- 170.Wu Y et al. A genome-scale CRISPR-Cas9 screening method for protein stability reveals novel regulators of Cdc25A. Cell Discov 2, 16014, doi: 10.1038/celldisc.2016.14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lo JY, Spatola BN & Curran SP WDR23 regulates NRF2 independently of KEAP1. PLoS Genet 13, e1006762, doi: 10.1371/journal.pgen.1006762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chen Z et al. CRL4BDCAF11 E3 ligase targets p21 for degradation to control cell cycle progression in human osteosarcoma cells. Sci Rep 7, 1175, doi: 10.1038/s41598-017-01344-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, doi: 10.1126/science.aal3755 (2017). [DOI] [PubMed] [Google Scholar]
- 174.Nogues L et al. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. EBioMedicine 13, 132–145, doi: 10.1016/j.ebiom.2016.09.030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ma Y, Han CC, Huang Q, Sun WY & Wei W GRK2 overexpression inhibits IGF1-induced proliferation and migration of human hepatocellular carcinoma cells by downregulating EGR1. Oncol Rep 35, 3068–3074, doi: 10.3892/or.2016.4641 (2016). [DOI] [PubMed] [Google Scholar]
- 176.Rivas V et al. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest 123, 4714–4730, doi: 10.1172/JCI67333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhou L et al. G-protein-coupled receptor kinase 2 in pancreatic cancer: clinicopathologic and prognostic significance. Hum Pathol 56, 171–177, doi: 10.1016/j.humpath.2016.06.012 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Meszaros B, Kumar M, Gibson TJ, Uyar B & Dosztanyi Z Degrons in cancer. Sci Signal 10, doi: 10.1126/scisignal.aak9982 (2017). [DOI] [PubMed] [Google Scholar]
- 179.Lo SC, Li X, Henzl MT, Beamer LJ & Hannink M Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J 25, 3605–3617, doi: 10.1038/sj.emboj.7601243 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chang L & Barford D Insights into the anaphase-promoting complex: a molecular machine that regulates mitosis. Curr Opin Struct Biol 29, 1–9, doi: 10.1016/j.sbi.2014.08.003 (2014). [DOI] [PubMed] [Google Scholar]
- 181.Lau AW, Fukushima H & Wei W The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front Biosci (Landmark Ed) 17, 2197–2212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ivan M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468, doi: 10.1126/science.1059817 (2001). [DOI] [PubMed] [Google Scholar]
- 183.Uljon S et al. Structural Basis for Substrate Selectivity of the E3 Ligase COP1. Structure 24, 687–696, doi: 10.1016/j.str.2016.03.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Havens CG et al. Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J Biol Chem 287, 11410–11421, doi: 10.1074/jbc.M111.337683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nguyen TV et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell 61, 809–820, doi: 10.1016/j.molcel.2016.02.032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Neklesa TK, Winkler JD & Crews CM Targeted protein degradation by PROTACs. Pharmacol Ther 174, 138–144, doi: 10.1016/j.pharmthera.2017.02.027 (2017). [DOI] [PubMed] [Google Scholar]
- 187.Punt CJ & Tol J More is less -- combining targeted therapies in metastatic colorectal cancer. Nat Rev Clin Oncol 6, 731–733, doi: 10.1038/nrclinonc.2009.168 (2009). [DOI] [PubMed] [Google Scholar]
- 188.Bobbin ML & Rossi JJ RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol 56, 103–122, doi: 10.1146/annurev-pharmtox-010715-103633 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Scheffner M, Huibregtse JM, Vierstra RD & Howley PM The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 (1993). [DOI] [PubMed] [Google Scholar]
- 190.Zhou P, Bogacki R, McReynolds L & Howley PM Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol Cell 6, 751–756 (2000). [DOI] [PubMed] [Google Scholar]
- 191.Cong F, Zhang J, Pao W, Zhou P & Varmus H A protein knockdown strategy to study the function of beta-catenin in tumorigenesis. BMC Mol Biol 4, 10, doi: 10.1186/1471-2199-4-10 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang J, Zheng N & Zhou P Exploring the functional complexity of cellular proteins by protein knockout. Proc Natl Acad Sci U S A 100, 14127–14132, doi: 10.1073/pnas.2233012100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Su Y, Ishikawa S, Kojima M & Liu B Eradication of pathogenic beta-catenin by Skp1/Cullin/F box ubiquitination machinery. Proc Natl Acad Sci U S A 100, 12729–12734, doi: 10.1073/pnas.2133261100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liu J, Stevens J, Matsunami N & White RL Targeted degradation of beta-catenin by chimeric F-box fusion proteins. Biochem Biophys Res Commun 313, 1023–1029 (2004). [DOI] [PubMed] [Google Scholar]
- 195.Cohen JC et al. Transient in utero knockout (TIUKO) of C-MYC affects late lung and intestinal development in the mouse. BMC Dev Biol 4, 4, doi: 10.1186/1471-213X-4-4 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chen W, Lee J, Cho SY & Fine HA Proteasome-mediated destruction of the cyclin a/cyclin-dependent kinase 2 complex suppresses tumor cell growth in vitro and in vivo. Cancer Res 64, 3949–3957, doi: 10.1158/0008-5472.CAN-03-3906 (2004). [DOI] [PubMed] [Google Scholar]
- 197.Kong F et al. Engineering a single ubiquitin ligase for the selective degradation of all activated ErbB receptor tyrosine kinases. Oncogene 33, 986–995, doi: 10.1038/onc.2013.33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hannah J & Zhou P Maximizing target protein ablation by integration of RNAi and protein knockout. Cell Res 21, 1152–1154, doi: 10.1038/cr.2011.89 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sakamoto KM Protacs for treatment of cancer. Pediatr Res 67, 505–508, doi: 10.1203/PDR.0b013e3181d35017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sakamoto KM et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559, doi: 10.1073/pnas.141230798 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jaakkola P et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472, doi: 10.1126/science.1059796 (2001). [DOI] [PubMed] [Google Scholar]
- 202.Schneekloth JS Jr. et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc 126, 3748–3754, doi: 10.1021/ja039025z (2004). [DOI] [PubMed] [Google Scholar]
- 203.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611–617, doi: 10.1038/nchembio.1858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Buckley DL et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl 51, 11463–11467, doi: 10.1002/anie.201206231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Buckley DL et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134, 4465–4468, doi: 10.1021/ja209924v (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Van Molle I et al. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem Biol 19, 1300–1312, doi: 10.1016/j.chembiol.2012.08.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Buckley DL et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol 10, 1831–1837, doi: 10.1021/acschembio.5b00442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kronke J, Hurst SN & Ebert BL Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 3, e941742, doi: 10.4161/21624011.2014.941742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309, doi: 10.1126/science.1244917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lu J et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763, doi: 10.1016/j.chembiol.2015.05.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Winter GE et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381, doi: 10.1126/science.aab1433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schiedel M et al. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J Med Chem, doi: 10.1021/acs.jmedchem.6b01872 (2017). [DOI] [PubMed] [Google Scholar]
- 213.Robb CM et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 53, 7577–7580, doi: 10.1039/c7cc03879h (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Schneekloth AR, Pucheault M, Tae HS & Crews CM Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett 18, 5904–5908, doi: 10.1016/j.bmcl.2008.07.114 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ottis P & Crews CM Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol 12, 892–898, doi: 10.1021/acschembio.6b01068 (2017). [DOI] [PubMed] [Google Scholar]
- 216.Itoh Y, Kitaguchi R, Ishikawa M, Naito M & Hashimoto Y Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg Med Chem 19, 6768–6778, doi: 10.1016/j.bmc.2011.09.041 (2011). [DOI] [PubMed] [Google Scholar]
- 217.Demizu Y et al. Design and synthesis of estrogen receptor degradation inducer based on a protein knockdown strategy. Bioorg Med Chem Lett 22, 1793–1796, doi: 10.1016/j.bmcl.2011.11.086 (2012). [DOI] [PubMed] [Google Scholar]
- 218.Itoh Y, Ishikawa M, Naito M & Hashimoto Y Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc 132, 5820–5826, doi: 10.1021/ja100691p (2010). [DOI] [PubMed] [Google Scholar]
- 219.Okuhira K et al. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett 585, 1147–1152, doi: 10.1016/j.febslet.2011.03.019 (2011). [DOI] [PubMed] [Google Scholar]
- 220.Zhang D, Baek SH, Ho A & Kim K Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorg Med Chem Lett 14, 645–648 (2004). [DOI] [PubMed] [Google Scholar]
- 221.Bargagna-Mohan P, Baek SH, Lee H, Kim K & Mohan R Use of PROTACS as molecular probes of angiogenesis. Bioorg Med Chem Lett 15, 2724–2727, doi: 10.1016/j.bmcl.2005.04.008 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cyrus K et al. Jostling for position: optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem 5, 979–985, doi: 10.1002/cmdc.201000146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hines J, Gough JD, Corson TW & Crews CM Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A 110, 8942–8947, doi: 10.1073/pnas.1217206110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chu TT et al. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem Biol 23, 453–461, doi: 10.1016/j.chembiol.2016.02.016 (2016). [DOI] [PubMed] [Google Scholar]
- 225.Montrose K & Krissansen GW Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem Biophys Res Commun 453, 735–740, doi: 10.1016/j.bbrc.2014.10.006 (2014). [DOI] [PubMed] [Google Scholar]
- 226.Wang X et al. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem Pharmacol 116, 200–209, doi: 10.1016/j.bcp.2016.07.017 (2016). [DOI] [PubMed] [Google Scholar]
- 227.Henning RK et al. Degradation of Akt using protein-catalyzed capture agents. J Pept Sci 22, 196–200, doi: 10.1002/psc.2858 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sakamoto KM et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics 2, 1350–1358, doi: 10.1074/mcp.T300009-MCP200 (2003). [DOI] [PubMed] [Google Scholar]
- 229.Ohoka N et al. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis 5, e1513, doi: 10.1038/cddis.2014.471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ottis P et al. Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem Biol 12, 2570–2578, doi: 10.1021/acschembio.7b00485 (2017). [DOI] [PubMed] [Google Scholar]
- 231.Maniaci C et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun 8, 830, doi: 10.1038/s41467-017-00954-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.French CA Small-Molecule Targeting of BET Proteins in Cancer. Adv Cancer Res 131, 21–58, doi: 10.1016/bs.acr.2016.04.001 (2016). [DOI] [PubMed] [Google Scholar]
- 233.Raina K et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 113, 7124–7129, doi: 10.1073/pnas.1521738113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zhou B et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem, doi: 10.1021/acs.jmedchem.6b01816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zengerle M, Chan KH & Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 10, 1770–1777, doi: 10.1021/acschembio.5b00216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S & Haendler B Targeting BET bromodomains for cancer treatment. Epigenomics 7, 487–501, doi: 10.2217/epi.14.91 (2015). [DOI] [PubMed] [Google Scholar]
- 237.Lebraud H, Wright DJ, Johnson CN & Heightman TD Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent Sci 2, 927–934, doi: 10.1021/acscentsci.6b00280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lai AC et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl 55, 807–810, doi: 10.1002/anie.201507634 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Madak JT, Cuthbertson CR, Chen W, Showalter HD & Neamati N Design, Synthesis, and Characterization of Brequinar Conjugates as Probes to Study DHODH Inhibition. Chemistry 23, 13875–13878, doi: 10.1002/chem.201702999 (2017). [DOI] [PubMed] [Google Scholar]
- 240.Culig Z & Santer FR Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev 33, 413–427, doi: 10.1007/s10555-013-9474-0 (2014). [DOI] [PubMed] [Google Scholar]
- 241.Cristobal I et al. PP2A regulates signaling through hormonal receptors in breast cancer with important therapeutic implications. Biochim Biophys Acta 1868, 435–438, doi: 10.1016/j.bbcan.2017.08.005 (2017). [DOI] [PubMed] [Google Scholar]
- 242.Huang D, Yang F, Wang Y & Guan X Mechanisms of resistance to selective estrogen receptor down-regulator in metastatic breast cancer. Biochim Biophys Acta 1868, 148–156, doi: 10.1016/j.bbcan.2017.03.008 (2017). [DOI] [PubMed] [Google Scholar]
- 243.Wu D, Cheung A, Wang Y, Yu S & Chan FL The emerging roles of orphan nuclear receptors in prostate cancer. Biochim Biophys Acta 1866, 23–36, doi: 10.1016/j.bbcan.2016.06.001 (2016). [DOI] [PubMed] [Google Scholar]
- 244.Bekaii-Saab TS et al. A phase II study of chloroquinoxaline sulfonamide (CQS) in patients with metastatic colorectal carcinoma (MCRC). Invest New Drugs 24, 343–346, doi: 10.1007/510637-005-4827-3 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Baur M, Gneist M, Owa T & Dittrich C Clinical complete long-term remission of a patient with metastatic malignant melanoma under therapy with indisulam (E7070). Melanoma Res 17, 329–331, doi: 10.1097/CMR.0b013e3282ef4189 (2007). [DOI] [PubMed] [Google Scholar]
- 246.Mita M et al. Phase I study of E7820, an oral inhibitor of integrin alpha-2 expression with antiangiogenic properties, in patients with advanced malignancies. Clin Cancer Res 17, 193–200, doi: 10.1158/1078-0432.CCR-10-0010 (2011). [DOI] [PubMed] [Google Scholar]
- 247.Uehara T et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol 13, 675–680, doi: 10.1038/nchembio.2363 (2017). [DOI] [PubMed] [Google Scholar]
- 248.Fischer ES et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53, doi: 10.1038/nature13527 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zha Z et al. A Non-Canonical Function of Gbeta as a Subunit of E3 Ligase in Targeting GRK2 Ubiquitylation. Mol Cell 58, 794–803, doi: 10.1016/j.molcel.2015.04.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Qian Y et al. The CUL4B/AKT/beta-Catenin Axis Restricts the Accumulation of Myeloid-Derived Suppressor Cells to Prohibit the Establishment of a Tumor-Permissive Microenvironment. Cancer Res 75, 5070–5083, doi: 10.1158/0008-5472.CAN-15-0898 (2015). [DOI] [PubMed] [Google Scholar]
- 251.Zhao W et al. Lack of CUL4B leads to increased abundance of GFAP-positive cells that is mediated by PTGDS in mouse brain. Hum Mol Genet 24, 4686–4697, doi: 10.1093/hmg/ddv200 (2015). [DOI] [PubMed] [Google Scholar]
- 252.Hung MH, Jian YR, Tsao CC, Lin SW & Chuang YH Enhanced LPS-induced peritonitis in mice deficiency of cullin 4B in macrophages. Genes Immun 15, 404–412, doi: 10.1038/gene.2014.32 (2014). [DOI] [PubMed] [Google Scholar]
- 253.Jiang B et al. Lack of Cul4b, an E3 ubiquitin ligase component, leads to embryonic lethality and abnormal placental development. PLoS One 7, e37070, doi: 10.1371/journal.pone.0037070 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Gao J et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. Elife 4, doi: 10.7554/eLife.07539 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.McCall CM et al. Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol Cell Biol 28, 5621–5633, doi: 10.1128/MCB.00232-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kassmeier MD et al. VprBP binds full-length RAG1 and is required for B-cell development and V(D)J recombination fidelity. EMBO J 31, 945–958, doi: 10.1038/emboj.2011.455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Guo Z et al. DCAF1 controls T-cell function via p53-dependent and -independent mechanisms. Nat Commun 7, 10307, doi: 10.1038/ncomms10307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Casey Klockow L et al. The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology 444, 191–202, doi: 10.1016/j.virol.2013.06.010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Romani B, Shaykh Baygloo N, Aghasadeghi MR & Allahbakhshi E HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J Biol Chem 290, 17380–17389, doi: 10.1074/jbc.M115.641522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kaur M, Khan MM, Kar A, Sharma A & Saxena S CRL4-DDB1-VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res 40, 7332–7346, doi: 10.1093/nar/gks366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Karim MF, Yoshizawa T, Sobuz SU, Sato Y & Yamagata K Sirtuin 7-dependent deacetylation of DDB1 regulates the expression of nuclear receptor TR4. Biochem Biophys Res Commun 490, 423–428, doi: 10.1016/j.bbrc.2017.06.057 (2017). [DOI] [PubMed] [Google Scholar]
- 262.Sharifi HJ, Furuya AK, Jellinger RM, Nekorchuk MD & de Noronha CM Cullin4A and cullin4B are interchangeable for HIV Vpr and Vpx action through the CRL4 ubiquitin ligase complex. J Virol 88, 6944–6958, doi: 10.1128/JVI.00241-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Nakagawa T et al. CRL4(VprBP) E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol Cell 57, 247–260, doi: 10.1016/j.molcel.2014.12.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wen X, Casey Klockow L, Nekorchuk M, Sharifi HJ & de Noronha CM The HIV1 protein Vpr acts to enhance constitutive DCAF1-dependent UNG2 turnover. PLoS One 7, e30939, doi: 10.1371/journal.pone.0030939 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ahn J et al. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J Biol Chem 285, 37333–37341, doi: 10.1074/jbc.M110.133181 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Banks D et al. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle 5, 1719–1729, doi: 10.4161/cc.5.15.3150 (2006). [DOI] [PubMed] [Google Scholar]
- 267.Jorgensen S et al. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol 192, 43–54, doi: 10.1083/jcb.201009076 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Terai K, Abbas T, Jazaeri AA & Dutta A CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell 37, 143–149, doi: 10.1016/j.molcel.2009.12.018 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang S et al. A novel function of CRL4(Cdt2): regulation of the subunit structure of DNA polymerase delta in response to DNA damage and during the S phase. J Biol Chem 288, 29550–29561, doi: 10.1074/jbc.M113.490466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Evans DL et al. MMSET is dynamically regulated during cell-cycle progression and promotes normal DNA replication. Cell Cycle 15, 95–105, doi: 10.1080/15384101.2015.1121323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Han C et al. Cdt2-mediated XPG degradation promotes gap-filling DNA synthesis in nucleotide excision repair. Cell Cycle 14, 1103–1115, doi: 10.4161/15384101.2014.973740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Shibata E, Dar A & Dutta A CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J Biol Chem 289, 23056–23064, doi: 10.1074/jbc.M114.574210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Jo U et al. PCNA-Dependent Cleavage and Degradation of SDE2 Regulates Response to Replication Stress. PLoS Genet 12, e1006465, doi: 10.1371/journal.pgen.1006465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Clijsters L & Wolthuis R PIP-box-mediated degradation prohibits re-accumulation of Cdc6 during S phase. J Cell Sci 127, 1336–1345, doi: 10.1242/jcs.145862 (2014). [DOI] [PubMed] [Google Scholar]
- 275.Suzuki T et al. Inhibition of DNA damage-induced apoptosis through Cdc7-mediated stabilization of Tob. J Biol Chem 287, 40256–40265, doi: 10.1074/jbc.M112.353805 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang H et al. DCAF4L2 promotes colorectal cancer invasion and metastasis via mediating degradation of NFkappab negative regulator PPM1B. Am J Transl Res 8, 405–418 (2016). [PMC free article] [PubMed] [Google Scholar]
- 277.Peng Z, Liao Z, Matsumoto Y, Yang A & Tomkinson AE Human DNA Ligase I Interacts with and Is Targeted for Degradation by the DCAF7 Specificity Factor of the Cul4-DDB1 Ubiquitin Ligase Complex. J Biol Chem 291, 21893–21902, doi: 10.1074/jbc.M116.746198 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Li G et al. CRL4DCAF8 Ubiquitin Ligase Targets Histone H3K79 and Promotes H3K9 Methylation in the Liver. Cell Rep 18, 1499–1511, doi: 10.1016/j.celrep.2017.01.039 (2017). [DOI] [PubMed] [Google Scholar]
- 279.Groh BS et al. The antiobesity factor WDTC1 suppresses adipogenesis via the CRL4WDTC1 E3 ligase. EMBO Rep 17, 638–647, doi: 10.15252/embr.201540500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Djakbarova U, Marzluff WF & Koseoglu MM DDB1 and CUL4 associated factor 11 (DCAF11) mediates degradation of Stem-loop binding protein at the end of S phase. Cell Cycle 15, 1986–1996, doi: 10.1080/15384101.2016.1191708 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Brodersen MM et al. CRL4(WDR23)-Mediated SLBP Ubiquitylation Ensures Histone Supply during DNA Replication. Mol Cell 62, 627–635, doi: 10.1016/j.molcel.2016.04.017 (2016). [DOI] [PubMed] [Google Scholar]
- 282.Ohtake F et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 446, 562–566, doi: 10.1038/nature05683 (2007). [DOI] [PubMed] [Google Scholar]
- 283.Niikura Y et al. CENP-A K124 Ubiquitylation Is Required for CENP-A Deposition at the Centromere. Dev Cell 32, 589–603, doi: 10.1016/j.devcel.2015.01.024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.An J et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase. Nat Commun 8, 15398, doi: 10.1038/ncomms15398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Chang SW et al. DDB2 is a novel AR interacting protein and mediates AR ubiquitination/degradation. Int J Biochem Cell Biol 44, 1952–1961, doi: 10.1016/j.biocel.2012.07.023 (2012). [DOI] [PubMed] [Google Scholar]
- 286.El-Mahdy MA et al. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem 281, 13404–13411, doi: 10.1074/jbc.M511834200 (2006). [DOI] [PubMed] [Google Scholar]
- 287.Nag A, Bondar T, Shiv S & Raychaudhuri P The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol 21, 6738–6747, doi: 10.1128/MCB.21.20.6738-6747.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Sjogren B, Swaney S & Neubig RR FBXO44-Mediated Degradation of RGS2 Protein Uniquely Depends on a Cullin 4B/DDB1 Complex. PLoS One 10, e0123581, doi: 10.1371/journal.pone.0123581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kim TY et al. CRL4A-FBXW5-mediated degradation of DLC1 Rho GTPase-activating protein tumor suppressor promotes non-small cell lung cancer cell growth. Proc Natl Acad Sci U S A 110, 16868–16873, doi: 10.1073/pnas.1306358110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hu J et al. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev 22, 866–871, doi: 10.1101/gad.1624008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ohno Y et al. Hoxb4 transduction down-regulates Geminin protein, providing hematopoietic stem and progenitor cells with proliferation potential. Proc Natl Acad Sci U S A 107, 21529–21534, doi: 10.1073/pnas.1011054107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Mouysset J et al. CRL4(RBBP7) is required for efficient CENP-A deposition at centromeres. J Cell Sci 128, 1732–1745, doi: 10.1242/jcs.162305 (2015). [DOI] [PubMed] [Google Scholar]
- 293.Parsons JL et al. Phosphorylation of PNKP by ATM prevents its proteasomal degradation and enhances resistance to oxidative stress. Nucleic Acids Res 40, 11404–11415, doi: 10.1093/nar/gks909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zeng M et al. CRL4(Wdr70) regulates H2B monoubiquitination and facilitates Exo1-dependent resection. Nat Commun 7, 11364, doi: 10.1038/ncomms11364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Thirunavukarasou A et al. Cullin 4A and 4B ubiquitin ligases interact with gamma-tubulin and induce its polyubiquitination. Mol Cell Biochem 401, 219–228, doi: 10.1007/s11010-014-2309-7 (2015). [DOI] [PubMed] [Google Scholar]
- 296.Yi J et al. DNA damage-induced activation of CUL4B targets HUWE1 for proteasomal degradation. Nucleic Acids Res 43, 4579–4590, doi: 10.1093/nar/gkv325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Antonioli M et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev Cell 31, 734–746, doi: 10.1016/j.devcel.2014.11.013 (2014). [DOI] [PubMed] [Google Scholar]
- 298.Wei Z et al. CUL4B impedes stress-induced cellular senescence by dampening a p53-reactive oxygen species positive feedback loop. Free Radic Biol Med 79, 1–13, doi: 10.1016/j.freeradbiomed.2014.11.010 (2015). [DOI] [PubMed] [Google Scholar]
- 299.Miyauchi-Nanri Y, Mukai S, Kuroda K & Fujiki Y CUL4A-DDB1-Rbx1 E3 ligase controls the quality of the PTS2 receptor Pex7p. Biochem J 463, 65–74, doi: 10.1042/BJ20130861 (2014). [DOI] [PubMed] [Google Scholar]
- 300.He F et al. X-linked intellectual disability gene CUL4B targets Jab1/CSN5 for degradation and regulates bone morphogenetic protein signaling. Biochim Biophys Acta 1832, 595–605, doi: 10.1016/j.bbadis.2013.01.015 (2013). [DOI] [PubMed] [Google Scholar]
- 301.Malatesta M et al. The Cul4A-DDB1 E3 ubiquitin ligase complex represses p73 transcriptional activity. Oncogene 32, 4721–4726, doi: 10.1038/onc.2012.463 (2013). [DOI] [PubMed] [Google Scholar]
- 302.Wu Z et al. Targeted ubiquitination and degradation of G-protein-coupled receptor kinase 5 by the DDB1-CUL4 ubiquitin ligase complex. PLoS One 7, e43997, doi: 10.1371/journal.pone.0043997 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Shen Z & Prasanth SG Orc2 protects ORCA from ubiquitin-mediated degradation. Cell Cycle 11, 3578–3589, doi: 10.4161/cc.21870 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Li X et al. Cullin 4B protein ubiquitin ligase targets peroxiredoxin III for degradation. J Biol Chem 286, 32344–32354, doi: 10.1074/jbc.M111.249003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Jiang L, Rong R, Sheikh MS & Huang Y Cullin-4A.DNA damage-binding protein 1 E3 ligase complex targets tumor suppressor RASSF1A for degradation during mitosis. J Biol Chem 286, 6971–6978, doi: 10.1074/jbc.M110.186494 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Zou Y et al. Characterization of nuclear localization signal in the N terminus of CUL4B and its essential role in cyclin E degradation and cell cycle progression. J Biol Chem 284, 33320–33332, doi: 10.1074/jbc.M109.050427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Leung-Pineda V, Huh J & Piwnica-Worms H DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res 69, 2630–2637, doi: 10.1158/0008-5472.CAN-08-3382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wang H et al. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell 22, 383–394, doi: 10.1016/j.molcel.2006.03.035 (2006). [DOI] [PubMed] [Google Scholar]
- 309.Katiyar S et al. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep 10, 866–872, doi: 10.1038/embor.2009.93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Yang Y, Wang S, Li J, Qi S & Zhang D CUL4A as a marker and potential therapeutic target in multiple myeloma. Tumour Biol 39, 1010428317703923, doi: 10.1177/1010428317703923 (2017). [DOI] [PubMed] [Google Scholar]
- 311.Han X et al. CUL4A functions as an oncogene in ovarian cancer and is directly regulated by miR-494. Biochem Biophys Res Commun 480, 675–681, doi: 10.1016/j.bbrc.2016.10.114 (2016). [DOI] [PubMed] [Google Scholar]
- 312.Li X, Xu R, Liu H & Fang K CUL4A expression in pediatric osteosarcoma tissues and its effect on cell growth in osteosarcoma cells. Tumour Biol 37, 8139–8144, doi: 10.1007/s13277-015-4715-1 (2016). [DOI] [PubMed] [Google Scholar]
- 313.Song J, Zhang J & Shao J Knockdown of CUL4A inhibits invasion and induces apoptosis in osteosarcoma cells. Int J Immunopathol Pharmacol 28, 263–269, doi: 10.1177/0394632015586656 (2015). [DOI] [PubMed] [Google Scholar]
- 314.Yasui K et al. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology 35, 1476–1484, doi: 10.1053/jhep.2002.33683 (2002). [DOI] [PubMed] [Google Scholar]
- 315.Yang YL et al. Cul4A overexpression associated with Gli1 expression in malignant pleural mesothelioma. J Cell Mol Med 19, 2385–2396, doi: 10.1111/jcmm.12620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Hung MS et al. Cul4A is an oncogene in malignant pleural mesothelioma. J Cell Mol Med 15, 350–358, doi: 10.1111/j.1582-4934.2009.00971.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Xu Y, Wang Y, Ma G, Wang Q & Wei G CUL4A is overexpressed in human pituitary adenomas and regulates pituitary tumor cell proliferation. J Neurooncol 116, 625–632, doi: 10.1007/s11060-013-1349-2 (2014). [DOI] [PubMed] [Google Scholar]
- 318.Ren S et al. Oncogenic CUL4A determines the response to thalidomide treatment in prostate cancer. J Mol Med (Berl) 90, 1121–1132, doi: 10.1007/s00109-012-0885-0 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Dong J, Wang XQ, Yao JJ, Li G & Li XG Decreased CUL4B expression inhibits malignant proliferation of glioma in vitro and in vivo. Eur Rev Med Pharmacol Sci 19, 1013–1021 (2015). [PubMed] [Google Scholar]
- 320.Hu H et al. CRL4B catalyzes H2AK119 monoubiquitination and coordinates with PRC2 to promote tumorigenesis. Cancer Cell 22, 781–795, doi: 10.1016/j.ccr.2012.10.024 (2012). [DOI] [PubMed] [Google Scholar]
- 321.Liu Z et al. AhR expression is increased in hepatocellular carcinoma. J Mol Histol 44, 455–461, doi: 10.1007/s10735-013-9495-6 (2013). [DOI] [PubMed] [Google Scholar]
- 322.Mian C et al. AHR over-expression in papillary thyroid carcinoma: clinical and molecular assessments in a series of Italian acromegalic patients with a long-term follow-up. PLoS One 9, e101560, doi: 10.1371/journal.pone.0101560 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Gu XM et al. Expression and prognostic relevance of centromere protein A in primary osteosarcoma. Pathol Res Pract 210, 228–233, doi: 10.1016/j.prp.2013.12.007 (2014). [DOI] [PubMed] [Google Scholar]
- 324.Lu Y et al. The F-box protein FBXO44 mediates BRCA1 ubiquitination and degradation. J Biol Chem 287, 41014–41022, doi: 10.1074/jbc.M112.407106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kang YJ, Lu MK & Guan KL The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ 18, 133–144, doi: 10.1038/cdd.2010.82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Halder SK et al. Oncogenic function of a novel WD-domain protein, STRAP, in human carcinogenesis. Cancer Res 66, 6156–6166, doi: 10.1158/0008-5472.CAN-05-3261 (2006). [DOI] [PubMed] [Google Scholar]
- 327.Umeda S et al. Prognostic significance of HOXB4 in de novo acute myeloid leukemia. Hematology 17, 125–131, doi: 10.1179/102453312X13376952196250 (2012). [DOI] [PubMed] [Google Scholar]
- 328.Koller K et al. Identification of the transcription factor HOXB4 as a novel target of miR-23a. Genes Chromosomes Cancer 52, 709–715, doi: 10.1002/gcc.22066 (2013). [DOI] [PubMed] [Google Scholar]
- 329.Blanchard Z et al. Geminin overexpression promotes imatinib sensitive breast cancer: a novel treatment approach for aggressive breast cancers, including a subset of triple negative. PLoS One 9, e95663, doi: 10.1371/journal.pone.0095663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Zhang K et al. Loss of H2B monoubiquitination is associated with poor-differentiation and enhanced malignancy of lung adenocarcinoma. Int J Cancer 141, 766–777, doi: 10.1002/ijc.30769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Matyskiela ME et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem, doi: 10.1021/acs.jmedchem.6b01921 (2017). [DOI] [PubMed] [Google Scholar]
- 332.Lee H, Puppala D, Choi EY, Swanson H & Kim KB Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem 8, 2058–2062, doi: 10.1002/cbic.200700438 (2007). [DOI] [PubMed] [Google Scholar]
- 333.Puppala D, Lee H, Kim KB & Swanson HI Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol 73, 1064–1071, doi: 10.1124/mol.107.040840 (2008). [DOI] [PubMed] [Google Scholar]
- 334.Sun B et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia, doi: 10.1038/leu.2017.207 (2017). [DOI] [PubMed] [Google Scholar]
- 335.Crew AP et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem, doi: 10.1021/acs.jmedchem.7b00635 (2017). [DOI] [PubMed] [Google Scholar]
- 336.Feng S, Cao Z & Wang X Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta 1836, 197–210, doi: 10.1016/j.bbcan.2013.05.001 (2013). [DOI] [PubMed] [Google Scholar]
- 337.Spangle JM, Roberts TM & Zhao JJ The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim Biophys Acta 1868, 123–131, doi: 10.1016/j.bbcan.2017.03.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Sato T & Gotoh N The FRS2 family of docking/scaffolding adaptor proteins as therapeutic targets of cancer treatment. Expert Opin Ther Targets 13, 689–700, doi: 10.1517/14728220902942330 (2009). [DOI] [PubMed] [Google Scholar]
- 339.Yin SQ, Wang JJ, Zhang CM & Liu ZP The development of MetAP-2 inhibitors in cancer treatment. Curr Med Chem 19, 1021–1035 (2012). [DOI] [PubMed] [Google Scholar]
- 340.Hantschel O & Superti-Furga G Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol 5, 33–44, doi: 10.1038/nrm1280 (2004). [DOI] [PubMed] [Google Scholar]
- 341.Morales F & Giordano A Overview of CDK9 as a target in cancer research. Cell Cycle 15, 519–527, doi: 10.1080/15384101.2016.1138186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Samatar AA & Poulikakos PI Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13, 928–942, doi: 10.1038/nrd4281 (2014). [DOI] [PubMed] [Google Scholar]
- 343.Ranhotra HS Estrogen-related receptor alpha and cancer: axis of evil. J Recept Signal Transduct Res 35, 505–508, doi: 10.3109/10799893.2015.1049362 (2015). [DOI] [PubMed] [Google Scholar]
- 344.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR & Gronemeyer H RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 6, 793–810, doi: 10.1038/nrd2397 (2007). [DOI] [PubMed] [Google Scholar]
- 345.Wu S, Kanda T, Nakamoto S, Imazeki F & Yokosuka O Knockdown of receptor-interacting serine/threonine protein kinase-2 (RIPK2) affects EMT-associated gene expression in human hepatoma cells. Anticancer Res 32, 3775–3783 (2012). [PubMed] [Google Scholar]
- 346.Bruzzone S et al. Rejuvenating sirtuins: the rise of a new family of cancer drug targets. Curr Pharm Des 19, 614–623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Chen W et al. TBK1 Promote Bladder Cancer Cell Proliferation and Migration via Akt Signaling. J Cancer 8, 1892–1899, doi: 10.7150/jca.17638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]