

Abstract
Four heterozygous in-frame tandem duplications of different lengths in TNFRSF11A, the gene that encodes receptor activator of nuclear factor κB (RANK), constitutively activate RANK and lead to high turnover skeletal disease. Each duplication elongates the signal peptide of RANK. The 18-base pair (bp) duplication at position 84 (84dup18) causes familial expansile osteolysis (FEO), the 15-bp duplication at position 84 (84dup15) causes expansile skeletal hyperphosphatasia (ESH), the 12-bp duplication at position 90 (90dup12) causes panostotic expansile bone disease (PEBD), and the 27-bp duplication causes early-onset Paget’s disease of bone (PDB2). The severity of the associated skeletal disease seems inversely related to the duplication’s length. Additional 15- and 18-bp duplications of TNFRSF11A fit this pattern. Herein, we delineate the skeletal disease of a middle-aged man of Mexican descent who we found to harbor a novel 27-bp tandem duplication at position 77 (77dup27) of TNFRSF11A. His disorder shares features, particularly hand involvement, with the single Japanese (75dup27) and Chinese (78dup27) kindreds with PDB2 (PDB2Jpn and PDB2Chn, respectively). However, his distinct hearing loss developed later in adulthood compared to the other 27-bp families. He reported no morbidities during childhood, but in his late 20s developed unexplained tooth loss, low-trauma fractures, post-operative hypercalcemia, and painless enlargement of his hands. Biochemical studies showed elevated serum alkaline phosphatase (ALP), bone-specific ALP, C-telopeptide, and osteocalcin consistent with rapid bone remodeling. Radiologic imaging revealed remarkably lucent bones with vertebral compression fractures, calvarial lucencies, and thinned long bone cortices. DXA showed extremely low bone mineral density. His disorder genetically and phenotypically fits best with PDB2 and can be called PDB2Mex.
Keywords: Bone turnover, Early Paget’s disease of bone, TNFRSF11A, RANK, RANKL, Deafness, Hypercalcemia, Osteoclast, Tooth loss, Fracturing, Hyperphosphatasemia, NF-κB, HIV infection, Bone markers
Graphical abstract

1. Introduction
The receptor activator of nuclear factor κB (RANK) ligand (RANKL)/osteoprotegerin (OPG)/RANK/NF-κB signaling pathway importantly regulates skeletal remodeling by controlling osteoclast (OC) formation and activity [1,2]. In mouse models [2,3], this involves binding of RANKL to RANK, which then promotes osteoclastogenesis and OC-induced skeletal turnover [4]. If RANKL binds instead to its “decoy receptor” OPG, skeletal remodeling is attenuated [2,5].
In humans, monogenic disruption of RANKL/OPG/RANK/NF-κB signaling leads to several extremely rare skeletal disorders that illustrate the importance of this regulatory mechanism [6–11]. Among them, four autosomal dominant seemingly distinctive disorders result from heterozygous in-frame tandem duplications (12-, 15-, 18-, or 27-bp) within exon 1 of TNFRSF11A that encodes the signal peptide of RANK. These disorders reflect constitutive activation of RANK and comprise: 1) 18-bp duplication: familial expansile osteolysis (FEO) [12–20]; 2) 27-bp duplication: early-onset Paget’s disease of bone (PDB2) [21–23]; 3) 15-bp duplication: expansile skeletal hyperphosphatasia (ESH) [7,24–26]; and 4) 12-bp duplication: panostotic expansile bone disease (PEBD) [11]. Of interest, the severity of these disorders seems inversely related to the duplication length. Remarkably, as reviewed below, even the 27-base pair (bp) duplication disorder PDB2 seems conditioned by the specific 27-bp duplication.
We report a middle-aged man of Mexican descent and two family members who harbor a novel 27-bp tandem duplication in RANK (77dup27) and contrast their disease features (PDB2Mex) to the other two but different 27-bp tandem duplications causing PDB2 (i.e., Japanese [PDB2Jpn] and Chinese [PDB2Chn]) as well as FEO, ESH, and PEBD caused by tandem duplications in TNFRSF11A of progressively shorter lengths, respectively.
2. Materials and Methods
2.1. Propositus and Family
The propositus was referred in 2015 to us at the University of Colorado Hospital Endocrinology Metabolic Bone Clinic in Aurora, Colorado, for evaluation of multiple atraumatic fractures starting in adulthood and severely low bone mineral density (BMD). After obtaining his family’s medical history, his affected sister was also studied. Their father lived in Mexico but was also studied given his history of similar symptoms. Other family members were unavailable, but seemingly unaffected according to the propositus and his sister. The propositus’ son was 10 years old, lived in Mexico, and reportedly had had normal childhood development without any bone, dental or hearing problems. This study was in keeping with Institutional Review Board approval from the Washington University School of Medicine, St. Louis, MO. Informed written consent to participate was given by the patient, his sister, and their father prior to obtaining blood samples for mutation analysis of candidate genes.
2.2. Laboratory Testing
Routine biochemical testing of the propositus’ blood occurred during clinic visits at University of Colorado Hospital and included a comprehensive metabolic panel (including calcium, phosphorus, chloride, creatinine, and alkaline phosphatase [ALP]), bone-specific ALP, 25-hydroxyvitamin D, parathyroid hormone, C-telopeptide (CTX), and osteocalcin.
2.3. Radiologic Imaging
All radiological imaging occurred at the University of Colorado Hospital and included radiographs, whole-body bone scintigraphy, and dual-energy x-ray absorptiometry (DXA) (Lunar Prodigy Advance, GE Healthcare, Madison, WI, USA).
2.4. Mutation Analyses
To determine the etiology of our patient’s high-turnover bone disorder, peripheral blood DNA underwent mutation analyses of candidate genes in our research laboratory (Washington University School of Medicine): 1) Ion Torrent Next Generation Sequencing (NGS) of genes involved in bone turnover or elevated bone mass, specifically TNFRSF11A (RANK), TNFRSF11B (OPG), TNFSF11 (RANKL), VCP, SQSTM1, TGFB1, IFITM5, MAFB, CSF1, CSFR1, TRAF6, RELA, RELB, REL, NFKB1, NFKB2, TFEB, CA2, CLCN7, CTSK (CATHEPSINK), OSTM1, PLEKHM1, TCIRG1, SOST, SLC29A3, LRP4, LRP5, LRP6, SNX10, FAM206, FAM123B (AMER1), TYROBP, LEMD3, DLX3, and PTDSS1; 2) whole genome copy number microarray to identify large insertions or deletions (performed at the Washington University Genome Technology Access Center St. Louis, MO, USA); and 3) Sanger sequencing using primers and conditions we developed for exon 1 of TNFRSF11A encoding RANK, to look for duplications [11].
3. Results
The family pedigree is illustrated in Figure 1. The propositus (Figure 1; III.1) was a 48-year-old man from Mexico living in Colorado with well-controlled human immunodeficiency virus (HIV) infection treated with abacavir-dolutegravir-lamivudine combination therapy, and vitamin D deficiency treated with a single high-dose course of ergocalciferol followed by maintenance daily dosing with cholecalciferol.
Figure 1:

77dup27 Family Pedigree
He reported no fractures or deformities as a child, good sunlight exposure and nutrition, and maximum height of 5′6″ (167 cm) during adolescence. At age 28 years, a first episode of unexplained tooth loss occurred. In his early 30s, his tibias bowed anteriorly, and he fractured his right femur falling while running, which was repaired with intramedullary rod fixation. At age 36 years, his left femur fractured from tripping and was also repaired with intramedullary rod fixation. After only a few days of bed rest following surgical repair, he developed post-operative hypercalcemia which persisted for about one and one-half months. Kyphosis and thoracic compression fractures were noted on radiographs at age 37 years. By age 38 years, he became aware of difficulty hearing. An audiogram revealed bilateral mixed hearing loss and computed tomography (CT) of his temporal bones showed bilateral extensively demineralized middle ear ossicles.
Physical examination revealed height 5′2″ (157.5 cm), white sclerae, edentia, subjectively decreased hearing (right greater than left), thoracic kyphosis, enlarged fingers (Figure 2), and anteriorly bowed tibias.
Figure 2:

Propositus’ Enlarged Fingers
Laboratory testing revealed elevated serum ALP 330 U/L (n: 39-117 U/L) and bone-specific ALP 87.6 ug/L (n: 6.5-20.1 ug/L) whereas calcium, phosphorus, parathyroid hormone, 25-hydroxyvitamin D, chloride, and creatinine levels were normal. Two additional bone turnover markers were markedly elevated: CTX 2477 pg/mL (n: 60-700 pg/mL) and osteocalcin 281 ng/mL (n: 11-50 ng/mL).
Radiographic skeletal survey showed remarkably lucent bones with calvarial lucencies, vertebral compression fractures, thoracic kyphosis, thin long bone cortices, and hyperostosis with cortical lucencies in the pelvis (Fig. 3A–F). DXA demonstrated lumbar spine areal BMD 0.488 g/cm2 (Z-score−5.5). Whole body bone scintigraphy revealed symmetric significantly increased uptake of Tc99 tracer in his calvarium (including the vertex, maxilla, and mandible), pelvis, and appendicular skeleton including the large and small joints of his hands, wrists, and ankles with the greatest uptake diffusely in his anterior tibias (Figure 4).
Figure 3: Radiographs of Propositus.
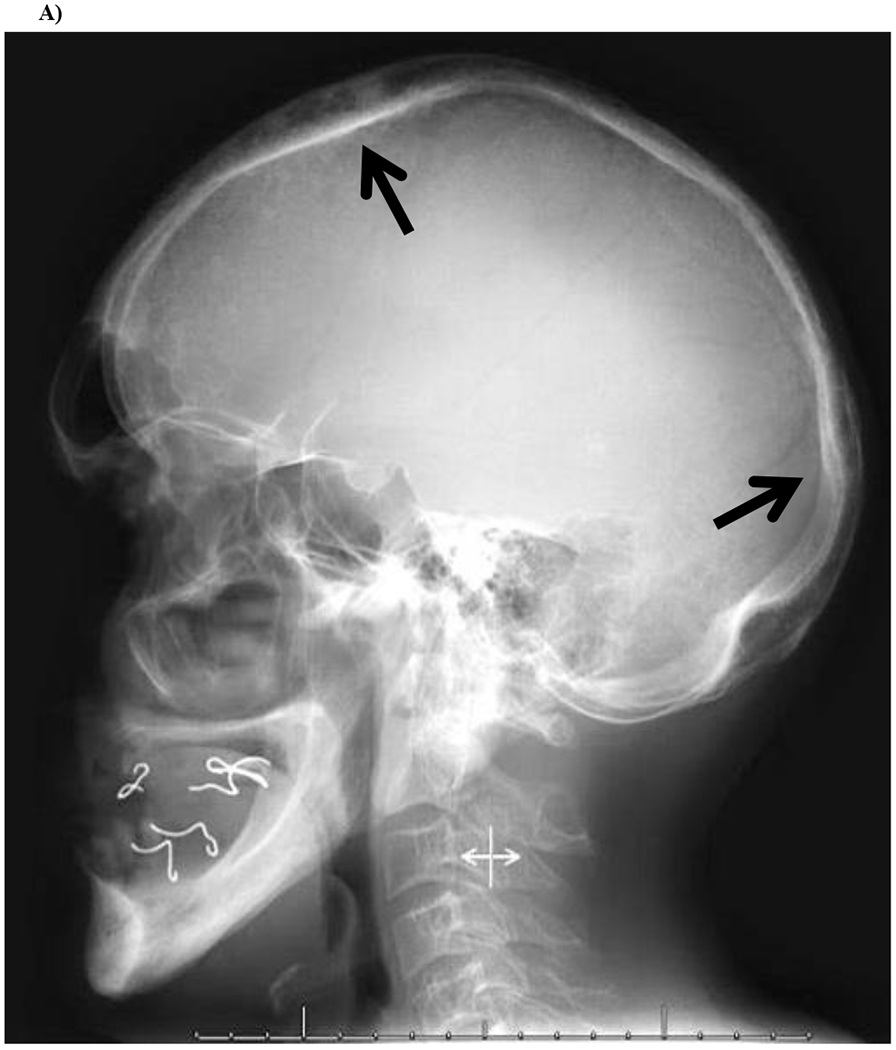





A) Skull
Lateral skull radiograph demonstrates slight calvarial expansion and rounded lucencies (arrows) in the frontal and occipital bones. There is also evidence that the propositus’ mouth was essentially edentulous.
B) Spine
Lateral radiograph of the thoracic spine is notable for multiple upper thoracic compression fractures with severe kyphosis.
C) Right Tibia and Fibula
AP radiograph of the right leg demonstrates enlargement and hyperostosis of the entire tibia and fibula, with cortical thickening and coarsening of the trabecular pattern. Similar features are noted in the distal femur and talus.
D) Left Tibia and Fibula
Lateral radiograph of the left leg demonstrates marked anterior bowing of the tibia (saber shin deformity) with cortical thickening, as well as central lucency of the medullary cavity and coarsened trabecular pattern.
E) Pelvis and Femurs
AP pelvic radiograph demonstrates healed bilateral femoral shaft fractures fixated with intramedullary nails. Note the thickened iliopectineal (arrow) and ilioischial (arrowhead) lines with lucent bones.
F) Right Humerus
AP radiograph of the right arm demonstrates a thickened cortex with coarse trabecular bone pattern, involving not only the humerus but also the proximal radius and ulna, similar to the findings in the propositus’ other long bones.
Figure 4: Whole Body Bone Scintigraphy.

Anterior and posterior planar views from Tc-99m MDP bone scintigraphy demonstrate mild varus bowing of the lower extremity long bones, with abnormally increased tracer in the appendicular long bones, particularly the tibias.
3.1. Mutation Analyses:
No causative mutation was identified by Ion Torrent NGS for genes involved in bone turnover and high/low bone density. Furthermore, whole genome-copy number microarray did not uncover any pathologic copy number variant. Sanger sequencing, however, identified a 27-bp duplication (77dup27) predicted to elongate the signal peptide of RANK by nine amino acid residues (Figure 5).
Figure 5:

Electropherograms showing RANK exon 1 duplication 77dup27 in the family members vs. control. The horizontal black line at the top of the control panel represents the duplicated 27 bp and the black arrows indicate the beginning of the duplication.
The patient’s 55-year old sister, who had multiple atraumatic maxillary tooth losses and kyphosis (but no history of deafness or fractures) as well as the father, aged 76 years, who reported multiple atraumatic fractures as an adult and hearing impairment of unknown duration (but without tooth loss), carried the same duplication in TNFRSF11A (Figure 5). Sanger sequencing of the father’s TNFRSF11A (from saliva) was not perfectly clear as the peaks for the candidate allele were smaller than the normal allele, suggesting he perhaps was mosaic for the RANK mutation.
4. Discussion
This family harbors a novel 27-bp tandem duplication (77dup27) in TNFRSF11A that predicts elongation of the signal peptide of RANK by nine amino acid residues. Similar RANK duplications explain the associated constitutively active RANK signaling, and our patient’s phenotype consistent with PDB2 [21,22] and having clinical features less closely resembling FEO, ESH, and PEBD (Table 1). We also note that while heritable disorders of the RANKL/OPG/RANK/NF-κB signaling pathway share many phenotypic features in common, only a limited number of families or kindreds, sometimes only one, have been studied [27].
Table 1:
Clinical Features of Increasing Tandem Duplication Length Within Exon 1 of TNFRSF11A Encoding RANK
| Clinical Syndrome | PEBD | ESH | FEO | PDB2Jpn | PDB2Chn | PDB2Mex |
|---|---|---|---|---|---|---|
| Signal Peptide Elongation | 12 | 15 | 18 | 27 | 27 | 27 |
| Mutation | 90dup12 | 84dup15 | 84dup18 | 75dup27 | 78dup27 | 77dup27 |
| Number of known kindreds/cases | 1/1 | 1/2 | 6/79 | 1/6 | 1/6 | 1/2 |
| Deafness | Yes, congenital | Yes, childhood | Yes, variable timing | Mild in childhood to early 20s | Mild in teens to early 20s | Mild in adulthood |
| Tooth loss in childhood | Yes | Yes | Yes | No | No | No |
| Hand involvement | Yes | Yes | No | Yes | Yes | Yes |
| Skull involvement | Yes | Yes | No | Yes | Yes | Yes |
| Mandible/maxilla involvement | Yes | No | No | Yes | Yes | Yes |
| Pelvic involvement | Yes | Yes | No | Yes | Yes | Yes |
| Hypercalcemia | No | Yes | No | Yes* | Rare/Mild | Yes* |
during immobilization
PEBD: panostotic expansile bone disease; ESH: expansile skeletal hyperphosphatasia; FEO: familial expansile osteolysis; PDB2: early-onset Paget’s disease of bone
Furthermore, as reviewed below, the tandem duplication length within exon 1 of TNFRSF11A that encodes the signal peptide of RANK (TNFRSF11A: OMIM 603499) [28] seems to correlate inversely with the severity of the associated skeletal disease (Figure 6). In 2000, Hughes et al. [6] reported that autosomal dominant FEO and PDB2 in Japan (PDB2Jpn) were caused by such heterozygous 18-bp and 27-bp tandem duplications, respectively. The two phenotypes are briefly outlined in Table 1. In 2002, Whyte and Hughes [7] reported another similar autosomal dominant disease ESH, caused by a 15-bp duplication in exon 1 of TNFRSF11A. That same year, Whyte et al. [8] reported autosomal recessive homozygous deletion of the different gene TNFRSF11B encoding OPG caused juvenile Paget’s disease (JPD). In 2014, Shafer et al. [11] reported a 32-year-old man from Mexico with extraordinarily severe generalized skeletal disease from a 12-bp duplication in TNFRSF11A. He had congenital deafness and developed severe limb deformity in childhood and then a large mandibular mass together comprising a disorder they called PEBD.
Figure 6: Duplications in RANK Causing FEO and Related Skeletal Diseases.

RANK exon 1 tandem duplications causing high-turnover skeletal disease. RANK protein is shown at the top, with important domains indicated, and normal DNA sequence just below. PEBD: panostotic expansile bone disease; ESH: expansile skeletal hyperphosphatasia; FEO: familial expansile osteolysis; PDB2: early-onset Paget’s disease of bone. Red C: One base difference between the Chinese PDB2 mutation compared to the Original Japanese and Mexican mutations; however, this base difference did not change the amino acid sequence. Note: Mutation numbering (e.g. 84dup18) is based on Hughes et al. (2000) using cDNA GenBank: AF018253.1, with the first base of exon 1 as the number one position instead of the current convention of using the A of the ATG start codon as number one [6]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Our patient’s presentation and clinical course seemed milder than in any of the aforementioned publications because his deafness did not present until adulthood and there was no juvenile tooth loss. His hypercalcemia was precipitated by immobilization as reported in PDB2Jpn [22]. Furthermore, the two siblings herein, despite carrying the same TNFRSF11A 77dup27 genotype, seemed to present differently, with the sister manifesting a milder form of the disease with tooth loss later in life. However, despite no formal complaints, she was kyphotic, had decreased hearing to finger rub, and showed some mild bowing of the tibia and thickened fingers. Unfortunately, we could not study her further.
After ensuring vitamin D repletion of our patient, we treated him with alendronate sodium, 70 mg orally once weekly. We chose an oral bisphosphonate rather than an intravenous bisphosphonate or denosumab, the monoclonal antibody that potently inhibits RANKL, concerned that hypocalcemia could occur due to his underlying rapid bone remodeling, as previously reported in a patient with JPD given zoledronic acid intravenously [29]. Family members with PDB2Jpn had incomplete and short-lived biochemical improvement with oral etidronate but later responded better to intravenous pamidronate or incadronate [23]. Our patient seemed to respond well to oral alendronate with no further fractures. Therefore, we are closely following his bone turnover markers (Table 2).
Table 2:
Propositus’ Laboratory Response to Alendronate Treatment
| Month/Year | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Test (units) | Normal Range | 8/15 | 10/16* | 1/17 | 4/17 | 7/17 | 12/17 | 5/18 | 9/18 | 7/19 |
| Calcium** (mg/dL) | 8.6-10.3 | 9.3 | 9.1 | 8.9 | 9.0 | 9.1 | 9.1 | 9.1 | n/a**** | 9.4 |
| 25(OH)vitamin D*** (ng/mL) | 13-62 | 68 | 64 | 34 | 36 | 37 | 33 | 37 | 20 | 28 |
| Alkaline phosphatase (U/L) | 39-117 | 251 | 364 | 339 | 243 | 236 | 180 | 206 | 190 | 167 |
| C-telopeptide (pg/mL) | 60-700 | 2477 | 2188 | 2201 | 1896 | 2003 | 1366 | 1169 | 1838 | 1736 |
| Osteocalcin (ng/mL) | 11-50 | 281 | 209 | 204 | 183 | 153 | 143 | 119 | 150 | 177 |
Started alendronate (70 mg po weekly).
Serum albumin ranged 3.9-4.3 g/dL.
Received ergocalciferol 50,000 units weekly until 1/17, then switched to cholecalciferol 2,000 units daily.
n/a: not available.
HIV infection and its treatment can be deleterious for bone health [29]. Patients with untreated HIV often have low bone mass which is multifactorial in etiology. Chronic inflammation, direct effects of HIV proteins, and high prevalence of traditional osteoporosis risk factors (e.g., tobacco and ethanol use, calcium and vitamin D deficiency, low body mass index) may all play a role [30]. Our patient became HIV infected in 2005, likely after he reached peak bone mass, but had experienced fractures and tooth and hearing loss earlier on. Thus, although his HIV infection and anti-retroviral therapy (ART) may have contributed to his low BMD, we suspected a genetic etiology for his primary skeletal disorder. His ART since 2015 has been abacavir-dolutegravir-lamivudine, although he had received other regimens including abacavir-lamivudine and efavirenz. The ART most likely to adversely impact bone health is tenofovir disoproxil fumarate (TDF) and, to a lesser extent, the protease inhibitors [31]. TDF can damage the proximal kidney tubule with subsequent hypophosphatemia and PTH elevation. Decreased glomerular filtration could also decrease 1-alpha-hydroxylase activity, key for the production of active 1,25 hydroxyvitamin D [30]. Regimens for pre-exposure prophylaxis (PrEP) containing TDF have been shown to decrease BMD, but that reverses after drug discontinuation [32]. While there are also concerns that protease inhibitors may play a role in ART-related bone loss, the mechanisms are less clear, and the degree of BMD changes has been smaller with more variability within the class among the medications [33].
5. Conclusions
Our patient’s high-turnover bone disease originated in a novel, heterozygous, 27-base pair tandem duplication in TNFRSF11A predicted to elongate the encoded signal peptide of RANK. Similar 27-base pair duplications have been reported in single Japanese (75dup27) and Chinese (78dup27) kindreds, predicting the identical 9-amino acid residue extension [21,22]. Our patient and perhaps his other affected family members share with the two Asian kindreds with PDB2 presentation in the late 20s, painless enlarged fingers, transient immobilization hypercalcemia, and absence of childhood tooth or hearing loss. Our experience supports mutation analysis in the diagnosis and understanding of high-turnover bone diseases, and consideration, if present, of the length of RANK signal peptide extension to anticipate distinctive skeletal effects and phenotypes.
Highlights.
77dup27 is a novel tandem duplication in TNFRSF11A, the gene that encodes RANK.
The associated phenotype resembles early-onset Paget’s disease of bone (PDB2).
PDB2Mex has later onset of hearing loss than PDB2Jpn and PDB2Chn (OMIM #602080).
6. Acknowledgments
The authors thank the patient and his family members for participating in our study. SJI also acknowledges support from an Advanced Fellowship in Geriatrics from the Geriatric Research, Education, and Clinical Center, Rocky Mountain Regional VA Medical Center, VA Eastern Colorado Health Care System, Office of Academic Affiliations, U.S. Department of Veterans Affairs.
Funding Sources
This work was supported in part by Shriners Hospitals for Children and The Clark and Mildred Cox Inherited Metabolic Bone Disease Research Fund and the Hypophosphatasia Research Fund at The Barnes-Jewish Hospital Foundation, Washington University School of Medicine; St. Louis, MO, USA. SJI also had support from NIH T32 DK007446.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Presented in Part at: Endocrine Society Annual Meeting. Orlando, FL. April 2017 (Endocrine Reviews, Volume 38, Issue 3, June 2017), and American Society of Bone and Mineral Research Annual Meeting. Denver, CO. September 2017 (J Bone Miner Res 32 (Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=ed3bbc8-c512-4de0-a8c8-f96388ab22f7.
Declarations of interest: None.
8. References
- [1].Martin TJ. Paracrine regulation of osteoclast formation and activity: milestones in discovery. J Musculoskelet Neuronal Interact. September 2004;4(3):243–53. [PubMed] [Google Scholar]
- [2].Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Archives of biochemistry and biophysics. May 15 2008;473(2): 139–46. 10.1016/j.abb.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Albagha OME, Rojas J, Van’t Hof R, Dorin J, Ralston SH. A mouse model of early onset Paget’s disease of bone caused by an insertion mutation affecting the rank signal peptide. Calcified Tissue Int. 2007;80:S34–S. [Google Scholar]
- [4].Park JH, Lee NK, Lee SY. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol Cells. October 2017;40(10):706–13. 10.14348/molcells.2017.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martin TJ, Sims NA. RANKL/OPG; Critical role in bone physiology. Rev Endocr Metab Disord. June 2015; 16(2): 131–9. 10.1007/s11154-014-9308-6. [DOI] [PubMed] [Google Scholar]
- [6].Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nature genetics. January 2000;24(1):45–8. 10.1038/71667. [DOI] [PubMed] [Google Scholar]
- [7].Whyte MP, Hughes AE. Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RANK and is allelic to familial expansile osteolysis. J Bone Miner Res. January 2002;17(1):26–9. 10.1359/jbmr.2002.17.1.26. [DOI] [PubMed] [Google Scholar]
- [8].Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. July 18 2002;347(3): 175–84. 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- [9].Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nature genetics. August 2007;39(8):960–2. 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- [10].Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. American journal of human genetics. July 2008;83(1):64–76. 10.1016/j.ajhg.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schafer AL, Mumm S, El-Sayed I, McAlister WH, Horvai AE, Tom AM, et al. Panostotic expansile bone disease with massive jaw tumor formation and a novel mutation in the signal peptide of RANK. J Bone Miner Res. April 2014;29(4):911–21. 10.1002/jbmr.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crone MD, Wallace RG. The radiographic features of familial expansile osteolysis. Skeletal Radiol. 1990;19(4):245–50. 10.1007/bf00191665. [DOI] [PubMed] [Google Scholar]
- [13].Dickson GR, Shirodria PV, Kanis JA, Beneton MN, Carr KE, Mollan RA. Familial expansile osteolysis: a morphological, histomorphometric and serological study. Bone. 1991; 12(5):331–8. 10.1016/8756-3282(91)90019-f. [DOI] [PubMed] [Google Scholar]
- [14].Elahi E, Shafaghati Y, Asadi S, Absalan F, Goodarzi H, Gharaii N, et al. Intragenic SNP haplotypes associated with 84dup18 mutation in TNFRSF11A in four FEO pedigrees suggest three independent origins for this mutation. J Bone Miner Metab. 2007;25(3):159–64. 10.1007/s00774-007-0748-x. [DOI] [PubMed] [Google Scholar]
- [15].Hughes AE, Shearman AM, Weber JL, Barr RJ, Wallace RG, Osterberg PH, et al. Genetic linkage of familial expansile osteolysis to chromosome 18q. Hum Mol Genet. February 1994;3(2):359–61. 10.1093/hmg/3.2.359. [DOI] [PubMed] [Google Scholar]
- [16].Johnson-Pais TL, Singer FR, Bone HG, McMurray CT, Hansen MF, Leach RJ. Identification of a novel tandem duplication in exon 1 of the TNFRSF11A gene in two unrelated patients with familial expansile osteolysis. J Bone Miner Res. February 2003;18(2):376–80. 10.1359/jbmr.2003.18.2.376. [DOI] [PubMed] [Google Scholar]
- [17].Osterberg PH, Wallace RG, Adams DA, Crone RS, Dickson GR, Kanis JA, et al. Familial expansile osteolysis. A new dysplasia. J Bone Joint Surg Br. March 1988;70(2):255–60. [DOI] [PubMed] [Google Scholar]
- [18].Palenzuela L, Vives-Bauza C, Femandez-Cadenas I, Meseguer A, Font N, Sarret E, et al. Familial expansile osteolysis in a large Spanish kindred resulting from an insertion mutation in the TNFRSF11A gene. J Med Genet. October 2002;39(10):E67 10.1136/jmg.39.10.e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wallace RG, Barr RJ, Osterberg PH, Mollan RA. Familial expansile osteolysis. Clin Orthop Relat Res. November 1989(248):265–77. [PubMed] [Google Scholar]
- [20].Whyte MP, Reinus WR, Podgornik MN, Mills BG. Familial expansile osteolysis (excessive RANK effect) in a 5-generation American kindred. Medicine (Baltimore). March 2002;81(2):101–21. 10.1097/00005792-200203000-00002. [DOI] [PubMed] [Google Scholar]
- [21].Ke YH, Yue H, He JW, Liu YJ, Zhang ZL. Early onset Paget’s disease of bone caused by a novel mutation (78dup27) of the TNFRSF11A gene in a Chinese family. Acta pharmacologica Sinica. August 2009;30(8): 1204–10. 10.1038/aps.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nakatsuka K, Nishizawa Y, Ralston SH. Phenotypic characterization of early onset Paget’s disease of bone caused by a 27-bp duplication in the TNFRSF11A gene. J Bone Miner Res. August 2003; 18(8):1381–5. 10.1359/jbmr.2003.18.8.1381. [DOI] [PubMed] [Google Scholar]
- [23].Riches PL, Imanishi Y, Nakatsuka K, Ralston SH. Clinical and biochemical response of TNFRSF11 A-mediated early-onset familial Paget disease to bisphosphonate therapy. Calcif Tissue Int. October 2008;83(4):272–5. 10.1007/s00223-008-9177-7. [DOI] [PubMed] [Google Scholar]
- [24].Chosich N, Long F, Wong R, Topliss DJ, Stockigt JR. Post-partum hypercalcemia in hereditary hyperphosphatasia (juvenile Paget’s disease). J Endocrinol Invest. Jul-Aug 1991; 14(7):591–7. 10.1007/BF03346877. [DOI] [PubMed] [Google Scholar]
- [25].Olsen CB, Tangchaitrong K, Chippendale I, Graham HK, Dahl HM, Stockigt JR. Tooth root resorption associated with a familial bone dysplasia affecting mother and daughter. Pediatr Dent. Sep-Oct 1999;21(6):363–7. [PubMed] [Google Scholar]
- [26].Whyte MP, Mills BG, Reinus WR, Podgornik MN, Roodman GD, Gannon FH, et al. Expansile skeletal hyperphosphatasia: a new familial metabolic bone disease. J Bone Miner Res. December 2000;15(12):2330–44. 10.1359/jbmr.2000.15.12.2330. [DOI] [PubMed] [Google Scholar]
- [27].Whyte MP. Medelian disorders of RANKL/OPG/RANK/NF-κB signaling, in Genetics of bone biology and skeletal disease, Thakker RV, et al. , Editors. 2018, Academic Press: San Diego, CA: p. 453–468. [Google Scholar]
- [28].Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD: MIM Number: 603499: 9/5/2018. World Wide Web URL: https://omim.org/entry/603499 (accessed 20 September 2019). [Google Scholar]
- [29].Polyzos SA, Anastasilakis AD, Litsas I, Efstathiadou Z, Kita M, Arsos G, et al. Profound hypocalcemia following effective response to zoledronic acid treatment in a patient with juvenile Paget’s disease. J Bone Miner Metab. November 2010;28(6):706–12. 10.1007/s00774-010-0198-8. [DOI] [PubMed] [Google Scholar]
- [30].Rothman MS, Bessesen MT. HIV infection and osteoporosis: pathophysiology, diagnosis, and treatment options. Curr Osteoporos Rep. December 2012;10(4):270–7. 10.1007/s11914-012-0125-0. [DOI] [PubMed] [Google Scholar]
- [31].Yin MT, Brown TT. HIV and Bone Complications: Understudied Populations and New Management Strategies. Curr HIV/AIDS Rep. December 2016; 13(6):349–58. 10.1007/sll904-016-0341-9. [DOI] [PubMed] [Google Scholar]
- [32].Glidden DV, Mulligan K, McMahan V, Anderson PL, Guanira J, Chariyalertsak S, et al. Brief Report: Recovery of Bone Mineral Density After Discontinuation of Tenofovir-Based HIV Pre-exposure Prophylaxis. J Acquir Immune Defic Syndr. October 1 2017;76(2):177–82. 10.1097/QAI.0000000000001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moran CA, Weitzmann MN, Ofotokun I. Bone Loss in HIV Infection. Curr Treat Options Infect Dis. March 2017;9(1):52–67. 10.1007/s40506-017-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]