

Abstract
Fragment antigen-binding domains (Fabs) from anti-Frizzled and anti-LRP6 monoclonal antibodies were conjugated using SpyTag-SpyCatcher chemistry via a one-pot reaction. The resulting synthetic heterodimeric agonist outperformed the natural ligand, Wnt-3a, in activating canonical Wnt signaling in mammalian cells. This approach should be broadly applicable to activate receptor-mediated cellular signaling.
Graphical Abstract
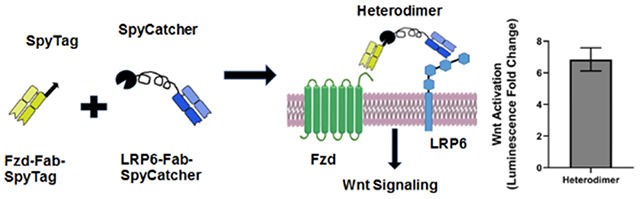
Heterodimers synthesized by conjugating anti-Frizzled and anti-LRP6 Fabs using SpyTag-SpyCatcher chemistry activate the canonical Wnt signaling pathway.
Wnt/β-catenin signaling is essential for proper embryonic development as it plays a crucial role in stem cell proliferation, self-renewal and differentiation1. Mutations in this pathway have been implicated in several diseases, which include different forms of cancer, Alzheimer’s disease, and type II diabetes2. In the absence of Wnt signaling, cytosolic (active) β-catenin is efficiently degraded by “destruction complexes”3 and maintained at a low concentration. The binding of the canonical Wnt ligand to its co-receptors Frizzled (Fzd) and LRP6 leads to the translocation of a fraction of the destruction complexes to the plasma membrane and a partial disassembly of these complexes4, 5. The resulting decrease in the rate of degradation of intracellular β-catenin results in an increase in its concentration and an upregulation of the transcription of downstream target genes6, 7.
Efficient purification of Wnt ligands represents a major impediment in this field of research. Wnt ligands undergo myriad post-translational modifications which are critical for retention of their activity8. For example, the canonical Wnt-3a ligand contains two N-linked glycosylation sites, which are important for its secretion and folding9, 10. Furthermore, the addition of a palmitoleic acid moiety which is essential for binding to Fzd makes the Wnt ligand hydrophobic and water insoluble, which necessitates the use of detergents in workflows designed to purify these ligands. These added difficulties and expenses have traditionally hindered the use of Wnt proteins as therapeutic agents11, 12.
To circumvent these issues, several alternate approaches of activating the Wnt signaling pathway have been developed in the past which do not require the natural Wnt ligands. Satisfactorily mimicking the endogenous dynamics of canonical Wnt signalling, however, remains challenging13. One such alternate approach involved treating the cells with Li+. The Li+ inhibits the degradation of β-catenin by disrupting the activity of glycogen synthase kinase 3 (GSK3)14. However, Li+ targets several other cellular components such as G-protein coupled receptors, the transcription factor AP-1 and a variety of phosphatases, and is thus not a specific activator of canonical Wnt signaling15. An alternative is the small molecule CHIR99021, which similarly inhibits GSK3 but with much greater specificity14. However, it activates Wnt signaling through an alternate mechanism of action and not by the fractional inactivation of the destruction complexes5. Furthermore, the GSK3 enzyme plays a role in transducing other cellular signaling pathways beyond the destruction complex, e.g., insulin signaling, and so inhibition of GSK3 by CHIR99021 may lead to undesired interference with other pathways16.
Cong et al. previously showed that heterodimerization of Frizzled and LRP6 receptors may be sufficient to trigger the downstream canonical Wnt signaling cascade17. In a recent elegant study, Janda et al. reported single-chain protein-based canonical Wnt agonists18. These agonists consisted of either an engineered bacterial protein or a single-chain variable fragment specific to Frizzled (Fzd) fused to the C-terminal domain of the natural Wnt antagonist, Dickkopf-1 (DKK1), which binds LRP618. These surrogate agonists simultaneously bound to Fzd and LRP6 and activated canonical Wnt signaling. While this report represents a key milestone in developing surrogate agonists, the approach was based on re-engineering a natural ligand of the Wnt co-receptor. We reasoned that it would be advantageous to design a strategy that is broadly applicable to designing synthetic agonists for a variety of signaling pathways. Natural ligands for a target receptor may not be readily available or easy to express, purify, or re-engineer. Moreover it would be advantageous to develop a modular strategy that allowed experimentalists to easily engineer the agonist by varying properties including but not limited to: specificity (e.g., to change the target receptor or to target a subset of receptors in a family); affinity for one or more target receptors; the binding epitope on the target receptor; and the avidity of the agonist. In contrast, the binding epitope for a natural ligand, its affinity for a target receptor, and the avidity of the resulting agonist, are not readily tuneable. To address these limitations, we have developed an alternate approach for designing synthetic heterodivalent agonists that activate canonical Wnt signaling. Specifically, we used SpyTag-SpyCatcher chemistry to covalently link two fragment antigen-binding domains (Fabs) that bound to Frizzled and LRP6 respectively (Fig. 1).
Figure 1. Schematic of a heterodimer agonist binding to the membrane proteins Frizzled and LRP6.
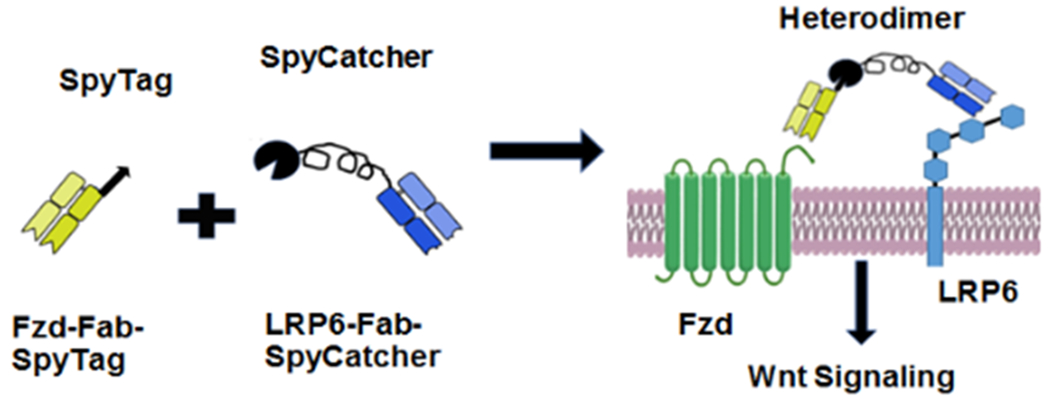
Fusing Frizzled (Fzd) and LRP6 Fabs via SpyCatcher-SpyTag interaction forms a heterodimer that binds to the canonical Wnt pathway receptors Fzd and LRP6.
First, to express wild-type Fabs, we obtained sequences of full-length anti-Frizzled and anti-LRP6 antibodies from Gurney et al. and Jenkins et al. respectively19, 20. The sequences of the variable regions within the heavy and light chains of the antibodies were inserted into two separate vectors, TGEX-FH and TGEX-LC respectively (Fig. S1). The anti-Fzd and anti-LRP6 Fabs were expressed by co-transfecting HEK (human embryonic kidney) 293F cells with the plasmids encoding the corresponding heavy and light chains. The Fabs were subsequently purified using immobilized metal affinity chromatography (IMAC) and size-exclusion chromatography in succession. We characterized the purified Fabs using SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE; Fig. 2a) and quantified the binding of the anti-Fzd and anti-LRP6 Fabs to Frizzled and LRP6 respectively by performing an ELISA (Fig. 2b). We observed significant binding of both purified Fabs to their corresponding antigens suggesting that the anti-Fzd and anti-LRP6 Fabs were capable of specifically binding to Frizzled and LRP6 respectively (Fig. 2b). To further quantify the activity of the anti-Fzd and anti-LRP6 Fabs, we transfected HEK 293T cells with the Wnt-responsive 7x TCF-luciferase reporter and treated the cells simultaneously with Wnt-3a and either the anti-Fzd or the anti-LRP6 Fab21. We observed that both anti-Fzd and anti-LRP6 Fabs inhibited canonical Wnt signalling (Fig. 2c), confirming that both purified Fabs were capable of binding to their respective receptors on mammalian cells and competing with Wnt-3a binding.
Figure 2. Characterization of expression and binding of anti-Fzd and anti-LRP6 Fabs.
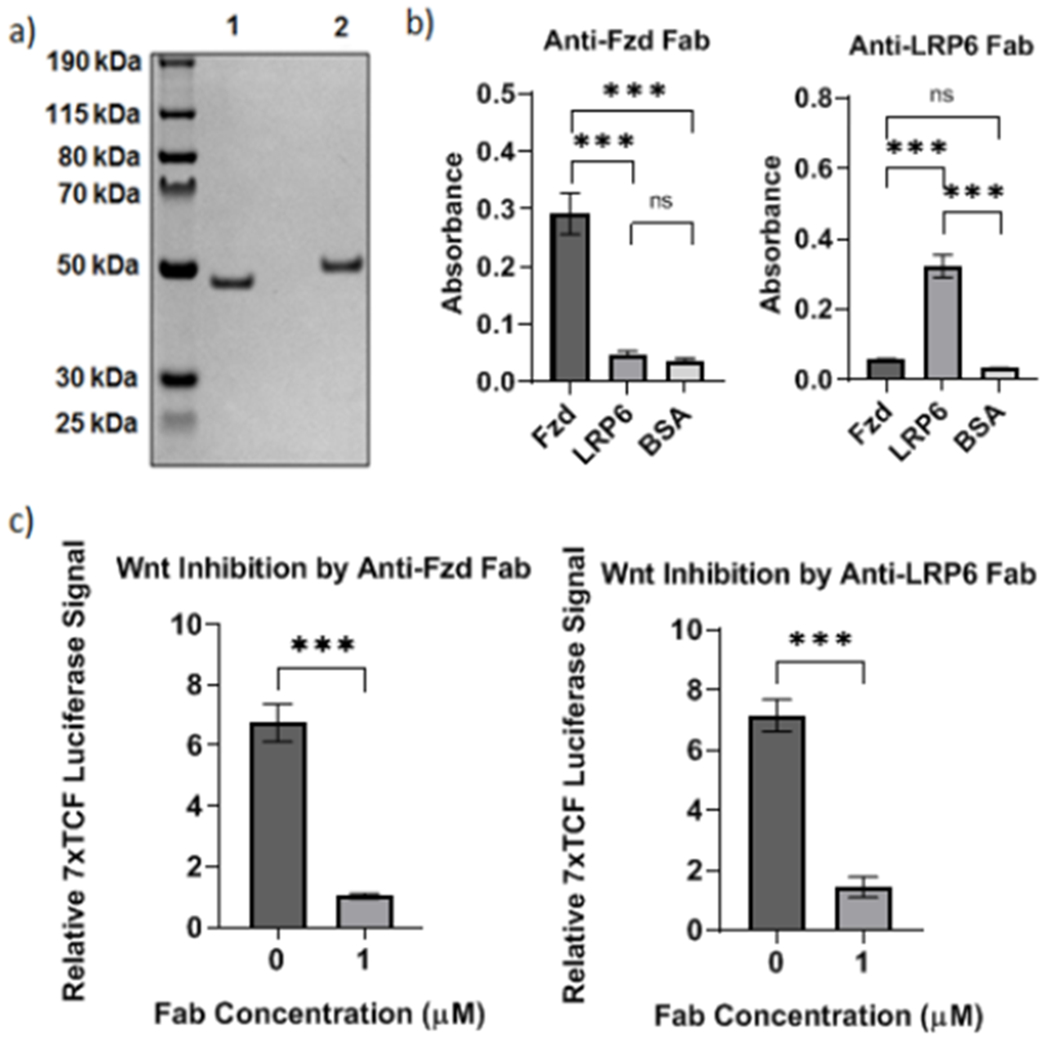
a) Characterization of purified recombinant anti-Fzd Fab (Lane 1) and anti-LRP6 Fab (Lane 2) by SDS-PAGE. b) ELISAs showing specific binding of anti-Fzd and anti-LRP6 Fabs to the recombinant proteins Fzd2 and LRP6 fused to the human Fc region (Fzd2-Fc and LRP6-Fc respectively). BSA served as the negative control. ***p < 0.0001 by ANOVA and Tukey post hoc test; ns: not significant. c) Luciferase assays showing level of canonical Wnt signaling activation in HEK 293T cells when treated with 15 nM of exogenous Wnt-3a in the absence or presence of 1 μM anti-Fzd and anti-LRP6 Fabs respectively. ***p < 0.0001 by Student’s t-test. Plots show mean ± 1 standard deviation (s.d.).
Next, to covalently link the two Fabs, we used the SpyTag-SpyCatcher system, which was developed by engineering a bacterial adhesin protein22. It consists of a short peptide, SpyTag, which spontaneously forms a covalent isopeptide bond in the presence of its protein partner, SpyCatcher (Fig. 3a). The SpyTag was fused to the C-terminus of the anti-Fzd Fab’s heavy chain while the SpyCatcher was fused to the C-terminus of the anti-LRP6 Fab’s light chain using flexible (Glycine)4Serine-based linkers. The linkers ensured proper folding of the fusion proteins and allowed for greater accessibility of the SpyTag and SpyCatcher during reaction. We expressed these modified fusion proteins, termed Fzd-Fab-SpyTag and LRP6-Fab-SpyCatcher respectively, in HEK 293F cells and confirmed that the engineered Fabs retained their binding characteristics by performing an ELISA against immobilized Frizzled and LRP6 proteins (Fig. 3b).
Figure 3. Synthesis and characterization of the purified Fab Heterodimer and its components.
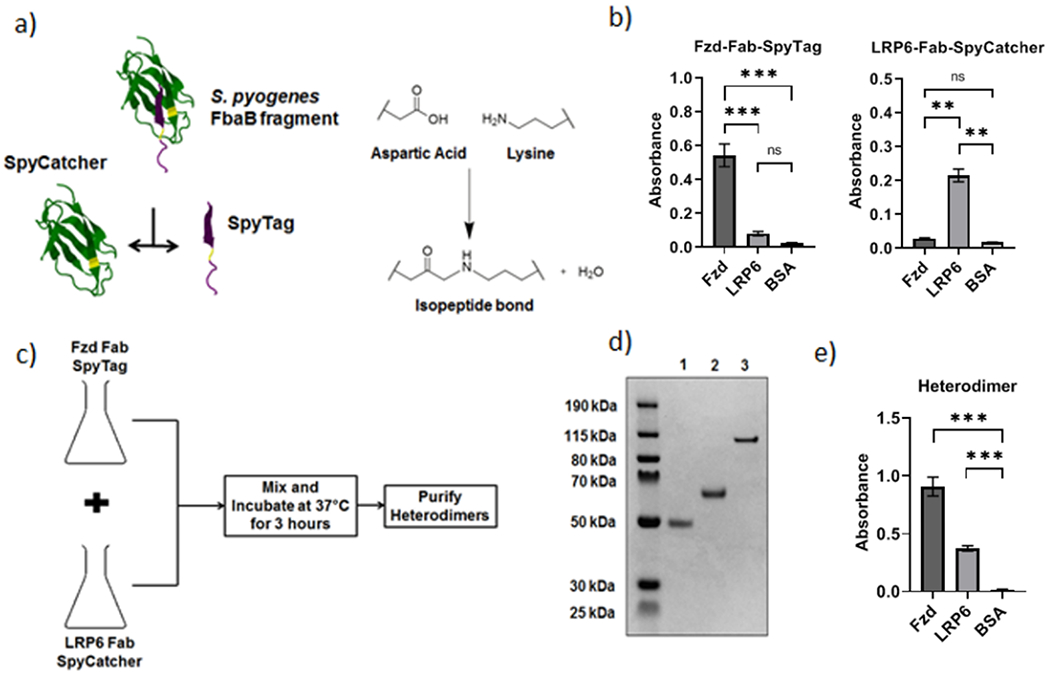
a) Schematic showing SpyTag and SpyCatcher partner proteins isolated from S. pyogenes FbaB protein22 and reacting covalently via the formation of an isopeptide bond between them. b) Characterization of the binding of Fzd-Fab-SpyTag and LRP6-Fab-SpyCatcher fusion proteins to Fzd2-Fc, LRP6-Fc, and BSA by ELISA. ***p < 0.0001, **p < 0.01 by ANOVA and Tukey post hoc test; ns: not significant. c) Schematic showing the one-pot synthesis of Fzd-LRP6-Fab heterodimer from individual fusion protein components. d) SDS-PAGE gel confirming expression and purification of Fzd-Fab-SpyTag (Lane 1), LRP6-Fab-SpyCatcher (Lane 2) and the Fab heterodimer product (Lane 3) formed from the one-pot reaction. e) Characterization of the binding of the Fab heterodimer to Fzd2-Fc, LRP6-Fc, and BSA by ELISA. ***p < 0.0001 by ANOVA and Tukey post hoc test. Plots show mean ± 1 s.d.
We proceeded to generate the Fzd-LRP6 heterodimers via a single-step “one-pot” reaction (Fig. 3c), instead of purifying the respective fusion proteins individually. We reacted equimolar amounts of Fzd-Fab-SpyTag and LRP6-Fab-SpyCatcher by mixing the requisite volumes of crude HEK 293F cell culture media containing the respective expressed fusion proteins. The resulting mixture contained the fully reacted and folded anti-Fzd-LRP6-heterodimer and the unreacted Fzd-Fab-SpyTag and LRP6-Fab-SpyCatcher “monomers”. The one-pot strategy significantly reduces protein loss and degradation because the reaction takes place in HEK 293F cell media, which has been optimized to maximize recombinant protein yield. This strategy also eliminated the need for two protein purification steps, which would otherwise be needed to obtain the individual reactants.
We purified the Fzd-LRP6-heterodimer using size-exclusion chromatography (SEC) which separated the dimer from the fusion protein “monomers” and characterized the product by SDS Polyacrylamide Gel Electrophoresis (Fig. 3d). We further characterized the purified heterodimer by dynamic light scattering to confirm that the SpyTag-SpyCatcher reaction did not cause aggregation (Fig. S2), liquid chromatography mass spectrometry (Fig. S3), and SEC (Fig. S4). We also performed an ELISA to confirm that the heterodimers bound to both Frizzled and LRP6, but not to the control protein BSA (Fig. 3e).
Next, we proceeded to check whether the Fzd-LRP6-heterodimer could activate canonical Wnt signaling. As mentioned before, we transfected HEK 293T cells with the Wnt-responsive 7x TCF-luciferase reporter plasmid and treated the cells with the heterodimer and compared the level of activation to that in cell populations treated with wild-type Wnt-3a. The synthetic heterodimer showed clear activation of canonical Wnt signaling at levels higher than those seen upon stimulation with wild-type Wnt-3a (Fig. 4a). In contrast, cells treated with the individual Fab monomers, i.e., Fzd-Fab-SpyTag and LRP6-Fab-SpyCatcher, or with an orthogonal heterodimeric fusion protein composed of a pair of Fabs which bind to epitopes completely orthogonal to Frizzled and LRP6, showed no significant activation of Wnt signalling (Fig. 4a). These results established that the synthetic Fzd-LRP6-heterodimer is capable of activating the canonical Wnt signaling pathway.
Figure 4. Characterizing Fab Heterodimer-Induced canonical Wnt signaling activation.
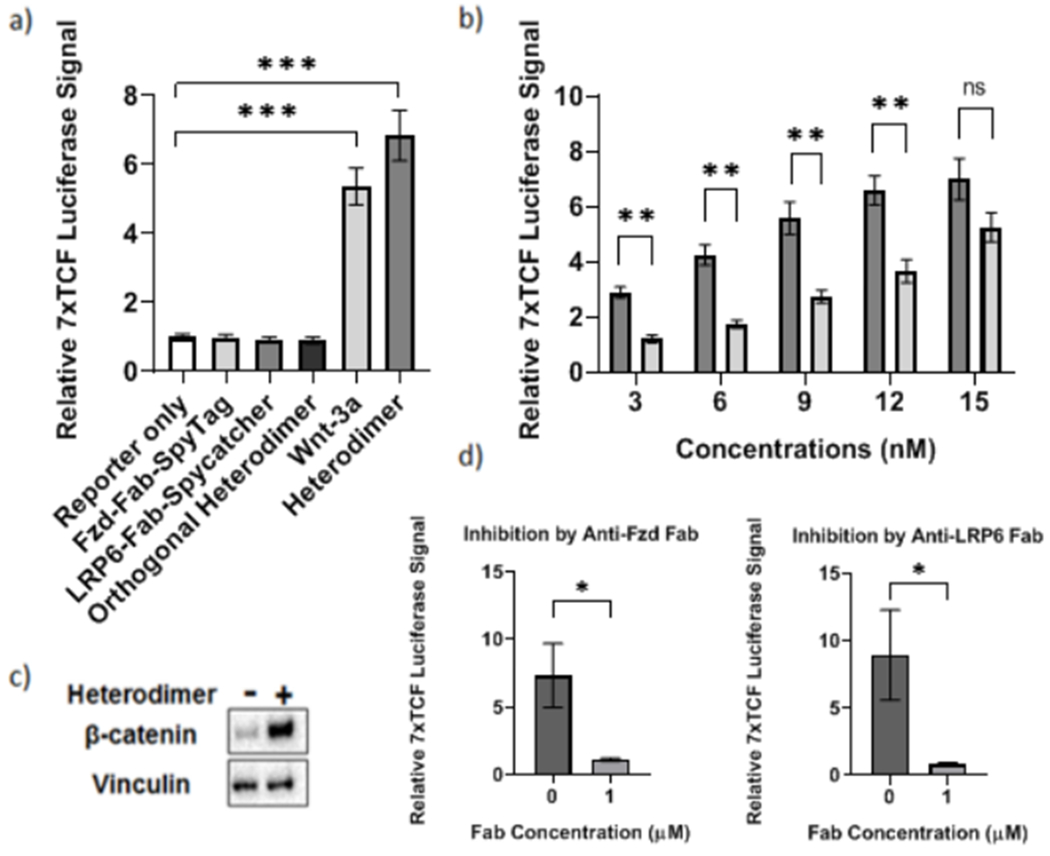
a) Luciferase assays showing level of canonical Wnt signaling activation in HEK 293T cells when treated with 15 nM of Fab heterodimer, the component Fab fusions, Wnt-3a, and an orthogonal Fab heterodimer. ***p < 0.0001 by ANOVA. b) Bar graph comparing Wnt-responsive luminescence signals at concentrations of Fab heterodimer (dark bar) and Wnt-3a (light bar) ranging from 0 to 15 nM. **p < 0.01 by multiple Bonferroni corrected student’s t-tests; ns: not significant. c) Western blot showing levels of cytosolic (active) β-catenin in HEK 293T cell lysates in the absence (−) and presence (+) of 15 nM Fab heterodimer overnight. d) Luciferase assays showing level of canonical Wnt signaling activation in HEK 293T cells when treated with 15 nM of Fab heterodimer in the absence or presence of 1 μM Fzd and LRP6 Fabs respectively. *p < 0.05 by Student’s t-test. Plots show mean ± 1 s.d.
To further characterize the heterodimer, we compared the extent of Wnt signaling activation caused by Wnt-3a and the synthetic heterodimer at concentrations ranging from 0 to 15 nM (Fig. 4B). At all concentrations tested, the synthetic heterodimer exhibited higher fold changes in Wnt-responsive luciferase signals than Wnt-3a, indicating improved signaling potency. The difference in fold change of activation between the heterodimer and Wnt-3a was the highest at the lowest concentration, i.e., 3 nM (Fig. 4b).
To further confirm that the heterodimer activates canonical Wnt signalling, we characterized the effect of the heterodimer on intracellular β-catenin levels, which increase upon activation of the Wnt signaling pathway. We treated HEK 293T cells with 15 nM heterodimer for 18 hours and performed a western blot on the cytosolic fractions isolated after lysis (Fig. 4c). Lysates of cells treated with the heterodimer had significantly increased levels of β-catenin compared to untreated cells, confirming the activation of the Wnt signaling pathway.
Additionally, we tested the ability of the anti-LRP6 and anti-Fzd Fab to inhibit signaling induced by the heterodimer. We transfected HEK 293T cells with the 7x TCF-luciferase reporter and treated with the heterodimer in the absence or presence of either anti-Fzd Fab or anti-LRP6 Fab (Fig. 4d). The extent of signaling was greatly reduced by both the anti-Fzd Fab and the anti-LRP6 Fab confirming that they competed with the heterodimer for binding to Wnt receptors on the cell surface. Linking the Fabs by SpyTag-SpyCatcher chemistry thus fundamentally changes their properties – by converting the two Wnt antagonists to a Wnt agonist – while simultaneously enhancing the avidity (Fig. S5).
To conclude, we have generated water-soluble synthetic heterodimers that potently bind to both Frizzled and LRP6 in the plasma membrane. The one-pot protocol described minimizes protein loss and degradation and circumvents the need for two additional protein purification steps, thus decreasing overall expense. The resulting Fzd-LRP6-heterodimers activate the canonical Wnt signaling pathway. The activation of Wnt signaling for the cells treated with the heterodimer is significantly greater than for those treated with wild-type Wnt-3a at lower ligand concentrations. In addition to canonical Wnt signaling, this approach can be readily extended to generate synthetic agonists of a variety of other signalling pathways23–26 with tunable properties. The approach described could thus have far-reaching implications for future research and therapeutic applications.
Supplementary Material
Acknowledgements
RSK and DVS acknowledge support from NIH grants R01 NS087253 and R01 NS083856. RSK acknowledges support from the National Science Foundation under Grant No. EEC-1648035. This work was also supported by Georgia Institute of Technology’s Parker H. Petit Institute for Bioengineering and Bioscience including the Systems Mass Spectrometry Core Facility.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Clevers H, Cell, 2006, 127, 469–480. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H and Nusse R, Cell, 2012, 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- 3.Stamos JL and Weis WI, Cold Spring Harbor Perspectives in Biology, 2013, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M and Niehrs C, Science, 2007, 316, 1619–1622. [DOI] [PubMed] [Google Scholar]
- 5.Mukherjee A, Dhar N, Stathos M, Schaffer DV and Kane RS, Iscience, 2018, 6, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez AR, Klein AM and Kirschner MW, Science, 2012, 338, 1337–1340. [DOI] [PubMed] [Google Scholar]
- 7.Li VSW, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJR, Maurice MM, Mahmoudi T and Clevers H, Cell, 2012, 149, 1245–1256. [DOI] [PubMed] [Google Scholar]
- 8.Willert K and Nusse R, Cold Spring Harbor Perspectives in Biology, 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komekado H, Yamamoto H, Chiba T and Kikuchi A, Genes to Cells, 2007, 12, 521–534. [DOI] [PubMed] [Google Scholar]
- 10.Kurayoshi M, Yamamoto H, Izumi S and Kikuchi A, Biochemical Journal, 2007, 402, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janda CY, Waghray D, Levin AM, Thomas C and Garcia KC, Science, 2012, 337, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T and Takada S, Developmental Cell, 2006, 11, 791–801. [DOI] [PubMed] [Google Scholar]
- 13.Beurel E, Grieco SF and Jope RS, Pharmacology & Therapeutics, 2015, 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM, Lobbardi RM, Brockmann J, Keller C, Wu X and Langenau DM, Proceedings of the National Academy of Sciences, 2014, 111, 5349–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roux M and Dosseto A, Metallomics, 2017, 9, 1326–1351. [DOI] [PubMed] [Google Scholar]
- 16.Maurer U, Preiss F, Brauns-Schubert P, Schlicher L and Charvet C, Journal of Cell Science, 2014, 127, 1369–1378. [DOI] [PubMed] [Google Scholar]
- 17.Cong F, Schweizer L and Varmus H, Development, 2004, 131, 5103–5115. [DOI] [PubMed] [Google Scholar]
- 18.Janda CY, Dang LT, You CJ, Chang JL, de Lau W, Zhong ZDA, Yan KS, Marecic O, Siepe D, Li XN, Moody JD, Williams BO, Clevers H, Piehler J, Baker D, Kuo CJ and Garcia KC, Nature, 2017, 545, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurney A, US Patent 9,499,630 B2, 2016.
- 20.Jenkins D, Lei M, Loew A and Zhou L, US Patent 8,883,735 B2, 2014.
- 21.Fuerer C and Nusse R, Plos One, 2010, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT and Howarth M, Proceedings of the National Academy of Sciences of the United States of America, 2012, 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devi LA, Trends in Pharmacological Sciences, 2001, 22, 532–537. [DOI] [PubMed] [Google Scholar]
- 24.Saito Y, Haendeler J, Hojo Y, Yamamoto K and Berk BC, Molecular and Cellular Biology, 2001, 21, 6387–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, Viardot A, Marks R, Diedrich H, Faul C, Reichle A, Horst H-A, Brüggemann M, Wessiepe D, Holland C, Alekar S, Mergen N, Einsele H, Hoelzer D and Bargou RC, Journal of Clinical Oncology, 2014, 32, 4134–4140. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Fu J, Zhang M and Liu D, Journal of Hematology & Oncology, 2015, 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.