

Abstract
In vitro approaches for predicting drug-drug interactions (DDIs) caused by alterations in transporter protein regulation are not well established. However, reports of transporter regulation via nuclear receptor (NR) modulation by drugs are increasing. This study examined alterations in transporter protein levels in sandwich-cultured human hepatocytes (SCHH; n = 3 donors) measured by liquid chromatography–tandem mass spectrometry–based proteomic analysis after treatment with N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]-N-(2,2,2-trifluoroethyl)benzenesulfonamide (T0901317), the first described synthetic liver X receptor agonist. T0901317 treatment (10 μM, 48 hours) decreased the levels of organic cation transporter (OCT) 1 (0.22-, 0.43-, and 0.71-fold of control) and organic anion transporter (OAT) 2 (0.38-, 0.38-, and 0.53-fold of control) and increased multidrug resistance protein (MDR) 1 (1.37-, 1.48-, and 1.59-fold of control). The induction of NR downstream gene expression supports the hypothesis that T0901317 off-target effects on farnesoid X receptor and pregnane X receptor activation are responsible for the unexpected changes in OCT1, OAT2, and MDR1. Uptake of the OCT1 substrate metformin in SCHH was decreased by T0901317 treatment. Effects of decreased OCT1 levels on metformin were simulated using a physiologically-based pharmacokinetic (PBPK) model. Simulations showed a clear decrease in metformin hepatic exposure resulting in a decreased pharmacodynamic effect. This DDI would not be predicted by the modest changes in simulated metformin plasma concentrations. Altogether, the current study demonstrated that an approach combining SCHH, proteomic analysis, and PBPK modeling is useful for revealing tissue concentration–based DDIs caused by unexpected regulation of hepatic transporters by NR modulators.
SIGNIFICANCE STATEMENT
This study utilized an approach combining sandwich-cultured human hepatocytes, proteomic analysis, and physiologically based pharmacokinetic modeling to evaluate alterations in pharmacokinetics (PK) and pharmacodynamics (PD) caused by transporter regulation by nuclear receptor modulators. The importance of this approach from a mechanistic and clinically relevant perspective is that it can reveal drug-drug interactions (DDIs) caused by unexpected regulation of hepatic transporters and enable prediction of altered PK and PD changes, especially for tissue concentration–based DDIs.
Introduction
Nuclear receptors (NRs) comprise a superfamily of transcription factors activated by ligands that can both activate and repress gene expression (Amacher, 2016). Drugs that target NRs are widely used and commercially successful. Indeed, a comprehensive review of US Food and Drug Administration (FDA)–approved small molecule drugs reported that 16% of these target NRs (Santos et al., 2017). NRs remain attractive targets for drug therapies in various diseases. For example, liver X receptors (LXRs), LXRα and LXRβ, are involved in cholesterol regulation and lipid metabolism and are potential therapeutic targets for atherosclerosis or dyslipidemia (Terasaka et al., 2003). Another NR, farnesoid X receptor (FXR), is involved in regulating bile acid, lipid, and glucose metabolism, and has been a promising therapeutic target for liver diseases such as primary biliary cirrhosis and nonalcoholic steatohepatitis (Han, 2018). However, the success of drugs targeting LXR and FXR has been limited because of off-target effects (Carotti et al., 2014; Ma et al., 2017; Han, 2018). NRs serve as master regulators and regulate multiple genes; therefore, unexpected effects could occur not only on pharmacological targets but also on drug metabolizing enzymes and transporters (Amacher, 2016). These off-target effects could alter drug pharmacokinetics (PK) and/or pharmacodynamics (PD) through drug-drug interactions (DDIs) and cause unwanted toxicity or decreased efficacy.
An increasing number of publications report NR-mediated regulation of proteins involved in drug absorption, distribution, metabolism, and excretion (ADME) (Amacher, 2016). Clinical DDI risks caused by cytochrome P450 (P450) induction are widely recognized, and in vitro approaches to evaluate P450 induction are well established (Kenny et al., 2018; Bernasconi et al., 2019). Many transporters involved in drug disposition and excretion are regulated by the same NRs as P450s, and, therefore, NR regulation might alter drug transport (Amacher, 2016). For example, multidrug resistance protein (MDR) 1 induction via pregnane X receptor (PXR) or constitutive androstane receptor is a well known transporter interaction involving NRs (Staudinger et al., 2013).
To decrease the risk of unexpected DDIs during clinical use, current DDI guidelines from the FDA and European Medicines Agency (EMA) recommend that sponsors conduct in vitro P450 induction studies during drug development (European Medicines Agency, 2012, Center for Drug Evaluation and Research, US Food and Drug Administration, 2020). Both the FDA and EMA note DDI risks caused by transporter induction via NRs. Although both DDI guidance documents especially mention MDR1 induction, NR-related DDIs also could be mediated by increases or decreases in the abundance of other transporters. Indeed, an induction study using human hepatocytes examined the effect of a wide variety of compounds on the mRNA expression of six hepatic transporters and found that all studied transporters were induced by several compounds and that NRs appeared to be involved (Badolo et al., 2015). However, in vitro methods to evaluate DDIs caused by changes in transporter abundance are not well established at this time and are not currently recommended by the FDA (Center for Drug Evaluation and Research, US Food and Drug Administration, 2020).
In vitro strategies need to be developed to predict the effects of alterations in transporters on drug disposition thereby avoiding unnecessary and costly in vivo studies. The most relevant system to assess changes in hepatic proteins is primary human hepatocytes. Sandwich-cultured human hepatocytes (SCHH) are considered to be the most physiologically relevant model that maintains transporter function, morphology, and regulatory machinery (LeCluyse et al., 2000; Brouwer et al., 2013). For regulation studies, mRNA quantification is popular because it is a simple and well established experimental procedure. However, transporter protein abundance is often poorly correlated with mRNA expression (Ohtsuki et al., 2012). Nonsynonymous single-nucleotide polymorphisms and posttranscriptional variability can cause differences in protein stability, leading to poor transcript and protein correlation (Prasad et al., 2019). Therefore, protein quantification is a better surrogate of in vivo activity (Prasad et al., 2017). Recently, there is an increasing number of reports applying liquid chromatography–tandem mass spectrometry (LC-MS/MS)–based proteomic analysis to quantify proteins involved in drug ADME (Bhatt and Prasad, 2018). Over the last decade, physiologically-based pharmacokinetic (PBPK) modeling has developed rapidly. Among a wide range of applications of PBPK modeling, predicting in vivo DDI risks is most popular (Bhatt and Prasad, 2018).
In this study, NR modulation of transporter protein levels in SCHH was examined. N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]-N-(2,2,2-trifluoroethyl) benzenesulfonamide (T0901317), the first described synthetic LXR agonist (Schultz et al., 2000), was chosen as a model NR agonist because of published data regarding its off-target effects on other NRs potentially involved in transporter regulation (Mitro et al., 2007). An approach combining SCHH, proteomic analysis, and PBPK modeling to simulate alterations in drug exposure was developed to predict effects of transporter alterations mediated by NR modulators.
Materials and Methods
Materials.
T0901317, metformin hydrochloride, and verapamil hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). Tritium-labeled metformin (27.0 Ci/mmol, radiochemical purity >98.9%) was purchased from Moravek (Brea, CA). The stable isotope–labeled peptides were generated by a peptide synthesis platform (PEPscreen, Custom Peptide Libraries; Sigma-Genosys). Transporter certified cryopreserved human hepatocytes HU8246, HU8192, and HH1107 purchased from Thermo Fisher Scientific (Waltham, MA) and In Vitro ADMET Laboratories (Columbia, MD) were obtained from two Caucasian female donors and one Caucasian/Hispanic female donor, respectively (age range, 37–55 years; body mass index range, 22–29). All Applied Biosystems Taqman assays were purchased from Thermo Fisher Scientific. QualGro Seeding medium, QualGro Hepatocyte Culture medium, and QualGro Hepatocyte Culture Induction medium were obtained from BioIVT (Durham, NC). Hanks’ balanced salt solution (HBSS) was purchased from Thermo Fisher Scientific. Collagen (type I)–coated Corning BioCoat 24-well cell culture plates and Matrigel were obtained from Thermo Fisher Scientific.
Hepatocyte Culture and T0901317 Treatment.
On day 0, hepatocytes were seeded at a density of 0.45 × 106 cells per well in BioCoat 24-well plates using QualGro Seeding Medium. Hepatocytes were overlaid the next day (day 1) with Matrigel diluted in QualGro Hepatocyte Culture Medium (0.25 mg/ml). On day 2, the culture medium was replaced with QualGro Hepatocyte Culture Induction Medium. SCHH were treated with T0901317 (10 μM) or vehicle (0.1% DMSO) in the induction medium for 48 hours starting from day 3. The T0901317 concentration of 10 μM was chosen because it is the industry standard for initial DDI risk screening in vitro for P450 inhibition (Wunberg et al., 2006; Lin et al., 2007). The medium was changed every 24 hours. On day 5, SCHH were processed for membrane protein extraction or RNA extraction or used for the uptake study.
Membrane Protein Extraction from SCHH.
The extraction of membrane proteins from SCHH was conducted by using the ProteoExtract Native Membrane Protein Extraction Kit (Calbiochem; EMD Biosciences Inc., Darmstadt, Germany) according to the manufacturer’s protocol, with minor modifications: homogenization was conducted after adding the extraction buffers, and the incubation time with extraction buffer I was 10–30 minutes and with extraction buffer II it was 30–60 minutes. A total of 12–16 wells of hepatocytes were pooled for each treatment group. The protein concentration was determined by using the Pierce BCA Protein assay kit (Thermo Fisher Scientific), and the samples were stored at −80°C until further use.
Preparation of Samples for LC-MS/MS–Based Proteomic Analysis.
The membrane protein samples were processed by protease digestion as described previously (Malinen et al., 2019) with minor modifications. Briefly, 18–37 μg of each membrane protein sample was mixed in ammonium bicarbonate buffer (25 mM ammonium bicarbonate) with 5% deoxycholate and 10 mM dithiothreitol, and incubated at 56°C for 30 minutes. Then, after the sample was mixed with iodoacetamide (final concentration of 15 mM) and incubated in the dark for 30 minutes at room temperature, the sample was diluted 5-fold with ammonium bicarbonate buffer. Lys-C protease (Thermo Fisher Scientific) was added to the sample to achieve a protein:protease ratio of 20:1 and incubated at 37°C for 4 hours. Then, trypsin was added to the sample at a protein:trypsin ratio of 20:1 followed by overnight incubation at 37°C. Formic acid was added to a final concentration of 2% (v/v), after which the mixture of stable isotope–labeled peptides (Supplemental Table 1) was added as internal standards. The sample was subjected to solid phase extraction using Oasis HLB (Waters Co., Milford, MA). Briefly, the cartridge was rinsed with methanol and equilibrated with 0.1% (v/v) formic acid in water, and the sample was passed through the cartridge. After rinsing the cartridge with 0.1% (v/v) formic acid in water, the peptides were eluted by acetonitrile with 0.1% (v/v) formic acid. After evaporation in a centrifugal evaporator, the eluate was reconstituted in 0.1% formic acid and 2% acetonitrile in water (v/v/v) and centrifuged at 21,000g for 1 minute, and the subsequent supernatant was analyzed by LC-MS/MS.
LC-MS/MS–based proteomic analysis was conducted as described previously (Malinen et al., 2019) with some modifications. The signature peptides for each transporter (Supplemental Table 1) were analyzed by LC-MS/MS [Thermo Scientific TSQ Quantum Ultra Triple Quadrupole mass spectrometer (Thermo Fisher Scientific) with nanoAcquity UPLC (Waters Co.)] in the selected reaction monitoring mode. Four sets of transitions were monitored for both signature and internal standard peptides. Samples for LC-MS/MS analysis were injected onto the Waters ACQUITY UPLC M-Class Symmetry C18 Trap Column (5 μm, 180 μm × 20 mm; Waters Co.) at a flow rate of 5 µl/min in 98% mobile phase A (0.1% formic acid in water) and 2% B (0.1% formic acid in acetonitrile) for 3 minutes. After trapping, the peptides were separated on an analytical column (Waters ACQUITY UPLC HSS T3 nanoACQUITY Column, 1.8 μm, 100 μm × 100 mm; Waters Co.) at a flow rate of 0.6 μl/min with linear gradient conditions detailed in Supplemental Table 2. Transporters were considered to be expressed when analyte peaks were detected in at least three sets of transitions. The peak area ratios of the analyte peptide to its internal standard were calculated for each set of transitions. The relative protein levels between T0901317- and DMSO-treated hepatocytes were calculated by dividing the peak area ratio in T0901317-treated hepatocytes by that in DMSO-treated hepatocytes for each lot.
Metformin Uptake in SCHH.
On day 5 of culture, a metformin uptake study in SCHH was conducted. SCHH were washed twice and preincubated for 10 minutes at 37°C with HBSS buffer. After preincubation, the uptake phase was initiated by incubating SCHH with 2 μM metformin (mixture of unlabeled and [3H]-labeled metformin, 6.75 µCi/ml) in HBSS buffer at 37°C for 10 minutes in the presence or absence of the organic cation transporter (OCT) 1 inhibitor verapamil (50 μM). This verapamil concentration was chosen to obtain complete inhibition of OCT1 based on the reported IC50 value (0.62 µM) of verapamil for OCT1 (Ahlin et al., 2011). At the end of the uptake phase, SCHH were washed with ice-cold HBSS buffer and then lysed with lysis solution containing 1× PBS with 0.5% Triton X-100. The total protein concentration of the cell lysates was determined using the Pierce BCA Protein Assay Kit. The radioactivity in the cell lysates was measured by liquid scintillation counting (Tri-Carb 3100TR; PerkinElmer), and the amount of metformin accumulated was normalized to the protein amount in the wells.
Gene Expression Studies.
On day 5 of culture, hepatocytes were harvested, and total RNA was extracted using the TRI Reagent according to the manufacturer’s protocol (Sigma-Aldrich). A NanoDrop spectrophotometer (Thermo Fisher Scientific) was used to measure the concentration and purity of the isolated RNA. Reverse transcription of the RNA (1 µg total RNA) to cDNA was performed using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) to study the gene expression of phospholipid-transporting ATPase [ATP-binding cassette transporter 1 (ABCA1)], CYP3A4, fibroblast growth factor 19 (FGF19), and sterol regulatory element-binding protein 1 (SREBF1). Real-time quantitative polymerase chain reaction (qPCR) was performed for each sample in duplicate with the QuantStudio 6 Flex System (Thermo Fisher Scientific) using 40 cycles. The analyzed genes and the gene-specific TaqMan assays (Thermo Fisher Scientific) used for real-time qPCR are listed in Table 1. The expression of small heterodimer partner (SHP) was analyzed as described previously (Jackson et al., 2016). Gene expression was calculated using the ∆∆Ct method, where glyceraldehyde-3-phosphate dehydrogenase was used as the reference gene and samples treated with 10 µM T0901317 were normalized to the control, 0.1% DMSO-treated, samples.
TABLE 1.
The target genes and TaqMan assays used in real-time qPCR experiments
| Target gene | Assay number | Purpose |
|---|---|---|
| GAPDH | Hs02758991_g1 | Reference gene |
| ABCA1 | Hs01059115_m1 | Marker of LXRα activation |
| SREBF1 | Hs00231674_m1 | Marker of LXRα activation |
| FGF19 | Hs00192780_m1 | Marker of FXR activation |
| SHP | Hs00222677_m1 | Marker of FXR activation |
| CYP3A4 | Hs00604506_m1 | Marker of PXR activation |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
PBPK Modeling to Simulate Effects of Transporter Alterations Caused by T0901317.
The impact of changes in transporter levels observed in T0901317-treated SCHH was examined using PBPK modeling. It was assumed that the alterations in transporter protein levels were directly translated to changes in transport activity. Two drugs, metformin, which is an OCT1 substrate, and digoxin, which is an MDR1 substrate, were used as model compounds for PBPK modeling. The modeling was performed using Simcyp software version 18 (Certara Ltd, Sheffield, UK) using the digoxin and metformin compound files. In these compound files, both digoxin and metformin are described by a full PBPK model with permeability-limited distribution to the liver (Neuhoff et al., 2013; Burt et al., 2016).
The OCT1-mediated hepatic uptake clearance of metformin was altered from control according to the extent of OCT1 protein down-regulation that was observed in each of the three lots of SCHH treated with T0901317. For the digoxin simulations, the degree of MDR1 induction in the liver was set as 1.5-fold of control, which is approximately the average level of induction in the three lots of SCHH treated with T0901317. The default values were used for other parameters in the compound files. The population representative of the Simcyp healthy volunteer population (“Sim-Healthy Volunteers”) was used for the simulations. The plasma and hepatic concentration–time profiles were simulated with the following dosing regimens: a single oral administration of 390 mg metformin (corresponding to a standard dose of 500 mg metformin hydrochloride) and a single 0.25 mg intravenous bolus of digoxin administered over 5 minutes. All simulated plasma concentrations refer to the total plasma concentration, and all hepatic concentrations refer to the unbound, intracellular concentration in hepatocytes.
To simulate changes in the PD effects of metformin, a PD model was linked to the simulated PK data. An indirect response model for the plasma glucose–lowering effect of metformin was presented previously by Lee and Kwon (2004). However, the model was constructed based on plasma concentrations of metformin, whereas the pharmacological target organ of metformin is the liver. To conduct the hepatic concentration–based simulation, the PD parameters (IC50, Kin, and Kout) of this indirect response model were converted to the corresponding hepatic concentration–based parameters as follows: Using the simulated plasma concentration–time profile of metformin in the “Sim-Healthy Volunteers” population representative, the PD response (plasma glucose concentration–time profile) was simulated. Then, the liver concentration–based PD parameters (IC50, Kin, Kout) were estimated by linking the simulated PD response to the intracellular unbound liver concentration–time profile of metformin using the indirect response model. The IC50, Kin, and Kout values derived from the estimation were 5.4 mg/l, 64, and 0.53 hour−1, respectively. By using these liver concentration–based PD parameters, the PD effects of metformin were simulated in cases where hepatic OCT1 levels were altered. All PD simulations were performed using Phoenix WinNonlin (version 8.1) with the PK data from Simcyp simulations.
Statistical Analysis.
Differences in protein abundance were analyzed by Student’s t test between T0901317 treatment and control (0.1% DMSO). In all cases, the criterion for statistical significance was P < 0.05.
Results
Effect of T0901317 Treatment on Transporter Protein Levels in SCHH.
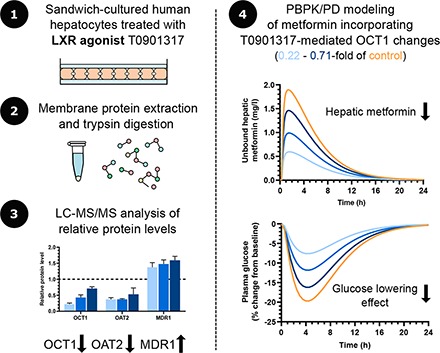
Ten transporters including MDR1, the bile salt export pump (BSEP), MDR3, multidrug resistance-associated protein (MRP) 2, OCT1, organic anion transporter (OAT) 2, Na+-taurocholate cotransporting polypeptide (NTCP), organic anion transporting polypeptide (OATP) 1B1, OATP1B3, and OATP2B1 were detected by LC-MS/MS–based proteomic analysis in all three lots of SCHH (Fig. 1). Among these transporters, T0901317 treatment significantly decreased OCT1 (0.22-, 0.43-, and 0.71-fold of control) and OAT2 (0.38-, 0.38-, and 0.53-fold of control), whereas MDR1 was increased (1.37-, 1.48-, and 1.59-fold of control). Breast cancer resistance protein (BCRP), multidrug and toxin extrusion protein (MATE) 1, MRP3, MRP4, OCT3, organic solute transporter (OST) α, and OSTβ were not detected.
Fig. 1.

Changes in protein levels of transporters in SCHH treated with T0901317 compared with vehicle control. Three lots of SCHH were treated with T0901317 (10 μM) or vehicle (0.1% DMSO) for 48 hours and then processed by LC-MS/MS–based proteomic analysis. Data are expressed as a relative value of protein abundance in T0901317-treated SCHH compared with the control and presented as mean ± S.D. (n = 3 to 4 transitions from one analysis). *P < 0.05 by Student’s t test between T0901315 treatment and DMSO treatment. BSEP, bile salt export pump; NTCP, Na+-taurocholate cotransporting polypeptide.
Effect of T0901317 Treatment on Metformin Uptake in SCHH.
To support the assumption that changes in protein levels correspond to functional changes, an uptake study with the OCT1 substrate metformin was performed using a representative hepatocyte lot. Hepatocyte Lot 1 exhibited the largest extent of OCT1 down-regulation. Therefore, metformin uptake was measured in this lot of SCHH exposed to T0901317 or vehicle (control). Uptake in the control SCHH was 13.0 ± 0.8 pmol/mg protein, and it was reduced to 8.0 ± 0.7 pmol/mg protein when the OCT1 inhibitor verapamil (50 µM) was included during the uptake. Uptake in T0901317-treated SCHH was 8.8 ± 1.1 pmol/mg protein. Verapamil at 50 µM, a concentration that is 80-fold higher than the reported IC50 (0.62 µM) (Ahlin et al., 2011), was considered to inhibit OCT1 completely; therefore, OCT1-mediated uptake was calculated by subtracting the measured uptake in the presence of verapamil from the uptake in the absence of verapamil assuming that the baseline, OCT1-independent metformin uptake was similar in control and T0901317-treated SCHH. As shown in Fig. 2, the OCT1-mediated uptake of metformin in the T0901317-treated SCHHs was reduced to 0.16-fold of control.
Fig. 2.

Effect of T0901317 treatment on the OCT1-mediated uptake of [3H]-metformin in SCHH. SCHH (Lot 1) were treated with T0901317 (10 μM) or vehicle (0.1% DMSO) for 48 hours, after which a 10-minute uptake study with [3H]-metformin (2 μM) was conducted. OCT1-mediated uptake was calculated by subtracting measured uptake in the presence of the OCT1 inhibitor verapamil (50 μM) from the uptake in the absence of verapamil. Data represent mean ± S.D. (n = 3 wells).
Effect of T0901317 Treatment on mRNA Expression of NR Downstream Genes in SCHH.
To verify the activation of NRs by T0901317 treatment in SCHH, several downstream genes of NRs were measured by real-time qPCR (Table 1). All of the studied genes were up-regulated in T0901317-treated SCHHs (Fig. 3). The mRNA expression of ABCA1 and SREBF1 was increased to more than 1.7- and 2.4-fold of control, respectively, suggesting LXR activation. As for the downstream genes of FXR, the mRNA expression of FGF19 and SHP was increased to more than 45-fold and 1.5-fold of control, respectively. Finally, CYP3A4, a marker of PXR activation, was induced to 4.3-fold or more of control (Fig. 3).
Fig. 3.

Effect of T0901317 treatment on mRNA expression levels of LXR downstream genes (ABCA1 and SREBF1), FXR downstream genes (FGF19 and SHP), and the PXR downstream gene CYP3A4 in three lots of SCHH. SCHH were treated with T0901317 (10 μM) or vehicle (0.1% DMSO) for 48 hours. Data are presented as relative gene expression using the ∆∆Ct method with the expression in T0901317-treated compared with control SCHH as mean ± S.D. (n = 3 wells).
PBPK Model Simulation of Potential Impact of T0901317-Mediated Changes in OCT1 Observed in SCHH.
The impact of T0901317-mediated changes in hepatic OCT1 and MDR1 protein levels on the PK of OCT1 substrate metformin and the MDR1 substrate digoxin was simulated. Hepatic OCT1 down-regulation slightly increased the plasma exposure of metformin to 1.08–1.22-fold of control for Cmax, plasma and 1.09–1.28-fold of control for area under the curve (AUC)plasma (Fig. 4A; Table 2). In contrast, the hepatic intracellular AUC of unbound metformin was decreased to 0.34–0.79-fold of that in the control subject (Fig. 4B; Table 2), depending on the extent of OCT1 down-regulation (0.22-, 0.43-, and 0.71-fold of control) that was observed in each of the T0901317-treated SCHH lots. The impact of OCT1 down-regulation on the PD of metformin was simulated based on unbound metformin concentrations in the hepatocyte intracellular water. Simulations showed up to a 61% decrease in the maximal plasma glucose–lowering effect of metformin with T0901317-mediated OCT1 down-regulation (Fig. 4C; Table 2). In contrast to metformin, both the plasma and hepatic AUC and Cmax of digoxin after intravenous administration were decreased to no less than 0.89-fold of control when hepatic MDR1 induction was included in the model (Fig. 5; Table 3).
Fig. 4.

Simulated plasma (A) and liver (B) concentration–time profiles of metformin and the plasma glucose–lowering effect of metformin (C) using a physiologically-based pharmacokinetic model incorporating the down-regulation of OCT1 protein levels by T0901317 treatment (10 µM, 48 hours) observed in three separate lots of sandwich-cultured human hepatocytes. Changes in OCT1 levels are shown as fold of control in the legend. A 390 mg oral dose of metformin was simulated using the “Sim-Healthy volunteers” population representative in Simcyp version 18. The pharmacodynamic simulations were based on the unbound intracellular hepatic concentrations (B) using an indirect response model modified from Lee and Kwon (2004).
TABLE 2.
Plasma and hepatic exposure of metformin simulated using a physiologically-based pharmacokinetic model incorporating T0901317-mediated changes in OCT1 protein levels observed in three lots of sandwich-cultured human hepatocytes
A 390 mg oral dose of metformin was simulated using the “Sim-Healthy volunteers” population representative in Simcyp version 18. Control simulations represent the conditions without any changes to OCT1.
| Hepatic OCT1 down-regulation | ||||
|---|---|---|---|---|
| Control | 0.22-fold of control (Lot 1) | 0.43-fold of control (Lot 2) | 0.71-fold of control (Lot 3) | |
| Plasma exposure | ||||
| Cmax, plasma (mg/l) | 0.65 | 0.79 | 0.75 | 0.70 |
| AUCplasma,0–24 h (mg/l・h) | 4.92 | 6.30 | 5.86 | 5.36 |
| Hepatic exposurea | ||||
| Cmax, liver (mg/l) | 1.89 | 0.60 | 0.99 | 1.46 |
| AUCliver,0–24 h (mg/l・h) | 12.4 | 4.22 | 6.84 | 9.81 |
| Effect | ||||
| Maximal change in plasma glucose (% of baseline) | −19.7 | −7.60 | −11.8 | −16.2 |
Liver concentrations are expressed as unbound concentration in liver intracellular water.
Fig. 5.

Simulated plasma (A) and liver (B) concentrations of digoxin using a physiologically-based pharmacokinetic model incorporating the 1.5-fold induction of hepatic MDR1 expression by T0901317 treatment (10 µM, 48 hours) observed in sandwich-cultured human hepatocytes. A 0.25 mg intravenous bolus dose of digoxin administered over 5 minutes was simulated using the “Sim-Healthy volunteers” population representative in Simcyp version 18.
TABLE 3.
Plasma and hepatic exposure of digoxin simulated using a physiologically-based pharmacokinetic model incorporating the average induction of hepatic MDR1 observed in T0901317-treated sandwich-cultured human hepatocytes
A 0.25 mg intravenous bolus dose of digoxin administered over 5 min was simulated using the “Sim-Healthy volunteers” population representative in Simcyp version 18.
| Hepatic MDR1 induction | Plasma exposure | Liver exposurea | ||
|---|---|---|---|---|
| Cmax, plasma (µg/l) | AUCplasma,0–24 h (µg/l・h) | Cmax, liver (µg/l) | AUCliver,0–24 h (µg/l・h) | |
| Control | 10.1 | 7.80 | 5.06 | 4.98 |
| 1.5-fold of control | 9.92 | 7.23 | 4.56 | 4.43 |
Liver concentrations expressed as unbound concentration in liver intracellular water.
Discussion
In vitro approaches for predicting DDIs caused by alterations in transporter protein regulation are immature even though the number of reports on transporter regulation via NR activation/suppression by drugs is increasing. In this study, proteomic analysis in SCHH revealed that the NR agonist T0901317 altered the protein levels of several important drug transporters. Using this information, PBPK modeling was applied to examine the potential impact of these changes on drug disposition. Importantly, a decrease in simulated metformin liver concentrations and a subsequent decrease in the simulated pharmacodynamic endpoint was revealed by T0901317-mediated OCT1 down-regulation that would not have been predicted based on slight changes in simulated metformin plasma concentrations.
T0901317 was used as a model compound because of its known effects on several NRs. Even though T0901317 was originally developed as an LXR agonist, later studies showed that T0901317 has affinity for other NRs, including FXR (EC50: 0.5 μM) and PXR (EC50: 0.023 μM) (Mitro et al., 2007). The measurement of downstream genes of FXR and PXR (FGF19 and SHP for FXR; CYP3A4 for PXR) revealed that all of the evaluated downstream genes were up-regulated by T0901317 treatment in SCHH (Fig. 3). This study is the first to demonstrate the modulation of OCT1, OAT2, and MDR1 protein levels in SCHH by T0901317 (Fig. 1). To the best of our knowledge, LXR has not been reported to be involved in regulation of these transporters (Amacher, 2016). However, the activation of FXR and PXR could explain the observed changes in transporter levels. Suppression of OCT1 and OAT2 protein levels via FXR activation has been reported previously (Popowski et al., 2005; Saborowski et al., 2006). It also is known that PXR activation induces MDR1 and decreases OCT1 levels (Harmsen et al., 2010; Hyrsova et al., 2016).
The regulation of transporter proteins by NRs is very complex because a single NR may be involved in the regulation of multiple transporters, and one transporter can be regulated by multiple NRs (Staudinger et al., 2013; Amacher, 2016). For instance, OATP1B1 induction is regulated by multiple NRs, including PXR, FXR, and constitutive androstane receptor, in addition to LXR, and there are reports suggesting that FXR regulates multiple transporters, including OATP1B1, OATP1B3, OAT2, MRP2, MRP3, and BSEP (Amacher, 2016). Because of the lack of direct one-to-one relationships between NR regulation and transporters, an in vitro system that maintains relevant in vivo–like NR regulation and transporter levels, localization, and function (e.g., SCHH) is essential to evaluate the complexities of transporter regulation by drugs. Choosing the appropriate in vitro system is critical since results may vary depending on the test system used.
Synthetic LXR agonists have been proposed to have potential utility in the treatment of metabolic diseases such as dyslipidemia, atherosclerosis, and diabetes (Mohan and Heyman, 2003). DDIs are a major concern in the treatment of these diseases because concomitant medications are often needed. Metformin, which is a first-line drug in the treatment of type 2 diabetes, is primarily taken up by OCT1 into hepatocytes, where its major pharmacological target resides (Gong et al., 2012). Simulations incorporating the observed OCT1 down-regulation in T0901317-treated SCHH revealed that the hepatic exposure of metformin (AUC) and its maximal plasma glucose–lowering effect decreased to 0.34-fold and 0.39-fold of control, respectively, with the highest level of OCT1 down-regulation (Fig. 4; Table 2). This is consistent with clinical studies reporting a decrease in metformin liver exposure and glucose-lowering effects in subjects with reduced function OCT1 genotypes (Shu et al., 2008; Sundelin et al., 2017). The predicted minor effects on the systemic exposure of metformin can be explained by predominant renal clearance of metformin (Gong et al., 2012). This interaction would, therefore, be missed by only monitoring plasma concentrations. Similarly, using plasma concentrations as the driving force for PD effects would have led to erroneous conclusions because plasma concentrations of metformin were actually slightly increased. Thus, this study demonstrated the ability of PBPK modeling incorporating proteomics data to simulate tissue-concentration–driven DDIs in both PK and PD, which are caused by transporter regulation by NR modulators.
MDR1 induction is the transporter regulation interaction highlighted by both the FDA and EMA DDI guidance ((European Medicines Agency, 2012, Center for Drug Evaluation and Research, US Food and Drug Administration, 2020)). Simulations using the MDR1 substrate digoxin, a cardiac medication with a narrow therapeutic range, were conducted. Although T0901317 increased MDR1 protein levels by 1.5-fold of control in SCHH, the PBPK model incorporating hepatic MDR1 induction revealed that this change did not significantly impact hepatic or systemic concentrations of digoxin (Fig. 5; Table 3).
The PBPK simulations conducted in this study assumed that alterations in protein levels quantitatively represented changes in activity. Consistent with this assumption, the decrease in OCT1-mediated metformin uptake in T0901317-treated SCHH was similar to the decrease in OCT1 protein levels (Figs. 1 and 2). A recent proteomic study reported that transporter protein levels quantitatively represent observed changes in in vitro activity for OATP1B1 and BCRP (Kumar et al., 2015). Using proteomic data and SCHH could facilitate investigation of functional changes in hepatic transporters. LC-MS/MS–based proteomic analysis can quantify multiple proteins simultaneously in one analysis with high specificity over a wide quantification range without the use of antibodies (Aebersold et al., 2013). These data can be further used in PBPK modeling to evaluate the combined effects of NR-mediated transporter regulation. This approach is supported by the increasing number of publications describing PBPK modeling using transporter level changes obtained by proteomic analysis. These models have been able to describe pharmacokinetic alterations caused by changes related to genotype, disease, and ontogeny (Prasad et al., 2014; Wang et al., 2016; Emoto et al., 2018).
This study was designed to demonstrate the utility of the outlined approach for identifying drug-mediated alterations in transporter regulation and predicting subsequent DDI risks, especially for initial DDI risk assessment in drug development. The results reveal that NR-mediated alterations are not limited to induction but also can manifest as suppression of protein expression leading to decreased protein levels. The use of in vitro techniques to detect the mechanism of NR-mediated DDIs is important because changes observed in clinical data could be falsely attributed to interindividual variability. In addition, the results from this study serve as a reminder that NR effects can impact the levels of not only MDR1 but other transporters, in this case OCT1 and OAT2. Recently, PBPK modeling was used to suggest induction of OATP1B by rifampicin during repeated dosing (Asaumi et al., 2019). These examples demonstrate that the true role of induction and suppression of protein expression and/or levels in DDIs will only be uncovered when effects are studied for a range of transporters instead of focusing on single proteins.
In conclusion, an approach combining SCHH, proteomic analysis, and PBPK modeling was used to predict alterations in PK and PD caused by transporter regulation by the NR agonist T0901317. This approach is useful because it can reveal DDIs caused by off-target effects of NR modulators and subsequent unexpected regulation of hepatic transporters. The changes in transporter levels observed in vitro can be incorporated into PBPK models and used to simulate potential alterations in PK and PD, which is important especially for tissue concentration–based DDIs.
Acknowledgments
The authors gratefully acknowledge Certara for providing Phoenix software to the University of North Carolina Eshelman School of Pharmacy, Division of Pharmacotherapy and Experimental Therapeutics, as part of the Pharsight Academic Center of Excellence Program. Certara UK (Simcyp Division) granted free access to the Simcyp Simulators through an academic license (subject to conditions). We gratefully acknowledge Dr. Paavo Honkakoski for his assistance with study design and helpful discussions regarding nuclear receptor modulation and related marker genes and Eric R. Hall and Nina Searcy for the analysis of SHP expression.
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- ADME
absorption, distribution, metabolism, and excretion
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- BSEP
bile salt export pump
- DDI
drug-drug interaction
- EMA
European Medicines Agency
- FDA
US Food and Drug Administration
- FGF19
fibroblast growth factor 19
- FXR
farnesoid X receptor
- HBSS
Hanks’ balanced salt solution
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LXR
liver X receptor
- MATE
multidrug and toxin extrusion
- MDR
multidrug resistance protein
- MRP
multidrug resistance-associated protein
- NR
nuclear receptor
- NTCP
Na+-taurocholate cotransporting polypeptide
- OAT
organic anion transporter
- OATP
organic anion transporting polypeptide
- OCT
organic cation transporter
- OST
organic solute transporter
- P450
cytochrome P450
- PBPK
physiologically-based pharmacokinetic
- PD
phamacodynamic
- PK
pharmacokinetic
- PXR
pregnane X receptor
- qPCR
quantitative polymerase chain reaction
- SCHH
sandwich-cultured human hepatocytes
- SHP
small heterodimer partner
- SREBF1
sterol regulatory element-binding protein 1
- T0901317
N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]-N-(2,2,2-trifluoroethyl) benzenesulfonamide
Authorship Contributions
Participated in research design: Ito, Sjöstedt, Malinen, Brouwer.
Conducted experiments: Ito, Sjöstedt, Malinen, Guo.
Performed data analysis: Ito, Sjöstedt, Malinen, Guo, Brouwer.
Wrote or contributed to the writing of the manuscript: Sjöstedt, Ito, Malinen, Guo, Brouwer.
Footnotes
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) [Award Number R35 GM122576 (K.L.R.B.)] and by Teijin Pharma Limited. N.S. was supported by the Sigrid Jusélius Foundation.
K.L.R.B. is a co-inventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, recently acquired by BioIVT. B-CLEAR is covered by US patent 6,780,580 and other US and international patents both issued and pending.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Aebersold R, Burlingame AL, Bradshaw RA. (2013) Western blots versus selected reaction monitoring assays: time to turn the tables? Mol Cell Proteomics 12:2381–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlin G, Chen L, Lazarova L, Chen Y, Ianculescu AG, Davis RL, Giacomini KM, Artursson P. (2011) Genotype-dependent effects of inhibitors of the organic cation transporter, OCT1: predictions of metformin interactions. Pharmacogenomics J 11:400–411. [DOI] [PubMed] [Google Scholar]
- Amacher DE. (2016) The regulation of human hepatic drug transporter expression by activation of xenobiotic-sensing nuclear receptors. Expert Opin Drug Metab Toxicol 12:1463–1477. [DOI] [PubMed] [Google Scholar]
- Asaumi R, Menzel K, Lee W, Nunoya K, Imawaka H, Kusuhara H, Sugiyama Y. (2019) Expanded physiologically‐based pharmacokinetic model of rifampicin for predicting interactions with drugs and an endogenous biomarker via complex mechanisms including organic anion transporting polypeptide 1B induction. CPT Pharmacometrics Syst Pharmacol 8:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badolo L, Jensen B, Säll C, Norinder U, Kallunki P, Montanari D. (2015) Evaluation of 309 molecules as inducers of CYP3A4, CYP2B6, CYP1A2, OATP1B1, OCT1, MDR1, MRP2, MRP3 and BCRP in cryopreserved human hepatocytes in sandwich culture. Xenobiotica 45:177–187. [DOI] [PubMed] [Google Scholar]
- Bernasconi C, Pelkonen O, Andersson TB, Strickland J, Wilk-Zasadna I, Asturiol D, Cole T, Liska R, Worth A, Müller-Vieira U, et al. (2019) Validation of in vitro methods for human cytochrome P450 enzyme induction: outcome of a multi-laboratory study. Toxicol In Vitro 60:212–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Prasad B. (2018) Critical issues and optimized practices in quantification of protein abundance level to determine interindividual variability in DMET proteins by LC-MS/MS proteomics. Clin Pharmacol Ther 103:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer KLR, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, Palm JE, Stieger B, Evers R, International Transporter Consortium (2013) In vitro methods to support transporter evaluation in drug discovery and development [published correction appears in Clin Pharmacol Ther (2013) 94:412]. Clin Pharmacol Ther 94:95–112. [DOI] [PubMed] [Google Scholar]
- Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, Rostami-Hodjegan A, Tucker GT, Rowland-Yeo K. (2016) Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci 88:70–82. [DOI] [PubMed] [Google Scholar]
- Carotti A, Marinozzi M, Custodi C, Cerra B, Pellicciari R, Gioiello A, Macchiarulo A. (2014) Beyond bile acids: targeting Farnesoid X Receptor (FXR) with natural and synthetic ligands. Curr Top Med Chem 14:2129–2142. [DOI] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research, US Food and Drug Administration (2020) In Vitro Drug Interaction Studies: Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Guidance for Industry, US Food and Drug Administration, Silver Spring, MD. [Google Scholar]
- Emoto C, Johnson TN, Neuhoff S, Hahn D, Vinks AA, Fukuda T. (2018) PBPK model of morphine incorporating developmental changes in hepatic OCT1 and UGT2B7 proteins to explain the variability in clearances in neonates and small infants. CPT Pharmacometrics Syst Pharmacol 7:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency (2012) Guideline on the Investigation of Drug Interactions, European Medicines Agency, London, UK
- Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. (2012) Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 22:820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CY. (2018) Update on FXR biology: promising therapeutic target? Int J Mol Sci 19:E2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JH. (2010) PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother Pharmacol 66:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrsova L, Smutny T, Carazo A, Moravcik S, Mandikova J, Trejtnar F, Gerbal-Chaloin S, Pavek P. (2016) The pregnane X receptor down-regulates organic cation transporter 1 (SLC22A1) in human hepatocytes by competing for (“squelching”) SRC-1 coactivator. Br J Pharmacol 173:1703–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JP, Freeman KM, Friley WW, St. Claire RLI, Black C, Brouwer KR. (2016) Basolateral efflux transporters: a potentially important pathway for the prevention of cholestatic hepatotoxicity. Appl In Vitro Toxicol 2:207–216. [Google Scholar]
- Kenny JR, Ramsden D, Buckley DB, Dallas S, Fung C, Mohutsky M, Einolf HJ, Chen L, Dekeyser JG, Fitzgerald M, et al. (2018) Considerations from the innovation and quality induction working group in response to drug-drug interaction guidances from regulatory agencies: focus on CYP3A4 mRNA in vitro response thresholds, variability, and clinical relevance. Drug Metab Dispos 46:1285–1303. [DOI] [PubMed] [Google Scholar]
- Kumar V, Prasad B, Patilea G, Gupta A, Salphati L, Evers R, Hop CE, Unadkat JD. (2015) Quantitative transporter proteomics by liquid chromatography with tandem mass spectrometry: addressing methodologic issues of plasma membrane isolation and expression-activity relationship. Drug Metab Dispos 43:284–288. [DOI] [PubMed] [Google Scholar]
- LeCluyse EL, Fix JA, Audus KL, Hochman JH. (2000) Regeneration and maintenance of bile canalicular networks in collagen-sandwiched hepatocytes. Toxicol In Vitro 14:117–132. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kwon KI. (2004) Pharmacokinetic-pharmacodynamic modeling for the relationship between glucose-lowering effect and plasma concentration of metformin in volunteers. Arch Pharm Res 27:806–810. [DOI] [PubMed] [Google Scholar]
- Lin T, Pan K, Mordenti J, Pan L. (2007) In vitro assessment of cytochrome P450 inhibition: strategies for increasing LC/MS-based assay throughput using a one-point IC(50) method and multiplexing high-performance liquid chromatography. J Pharm Sci 96:2485–2493. [DOI] [PubMed] [Google Scholar]
- Ma Z, Deng C, Hu W, Zhou J, Fan C, Di S, Liu D, Yang Y, Wang D. (2017) Liver X receptors and their agonists: targeting for cholesterol homeostasis and cardiovascular diseases. Curr Issues Mol Biol 22:41–64. [DOI] [PubMed] [Google Scholar]
- Malinen MM, Ito K, Kang HE, Honkakoski P, Brouwer KLR. (2019) Protein expression and function of organic anion transporters in short-term and long-term cultures of Huh7 human hepatoma cells. Eur J Pharm Sci 130:186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitro N, Vargas L, Romeo R, Koder A, Saez E. (2007) T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett 581:1721–1726. [DOI] [PubMed] [Google Scholar]
- Mohan R, Heyman RA. (2003) Orphan nuclear receptor modulators. Curr Top Med Chem 3:1637–1647. [DOI] [PubMed] [Google Scholar]
- Neuhoff S, Yeo KR, Barter Z, Jamei M, Turner DB, Rostami-Hodjegan A. (2013) Application of permeability-limited physiologically-based pharmacokinetic models: part I-digoxin pharmacokinetics incorporating P-glycoprotein-mediated efflux. J Pharm Sci 102:3145–3160. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Schaefer O, Kawakami H, Inoue T, Liehner S, Saito A, Ishiguro N, Kishimoto W, Ludwig-Schwellinger E, Ebner T, et al. (2012) Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos 40:83–92. [DOI] [PubMed] [Google Scholar]
- Popowski K, Eloranta JJ, Saborowski M, Fried M, Meier PJ, Kullak-Ublick GA. (2005) The human organic anion transporter 2 gene is transactivated by hepatocyte nuclear factor-4 alpha and suppressed by bile acids. Mol Pharmacol 67:1629–1638. [DOI] [PubMed] [Google Scholar]
- Prasad B, Achour B, Artursson P, Hop CECA, Lai Y, Smith PC, Barber J, Wisniewski JR, Spellman D, Uchida Y, et al. (2019) Toward a consensus on applying quantitative liquid chromatography-tandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin Pharmacol Ther 106:525–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Evers R, Gupta A, Hop CE, Salphati L, Shukla S, Ambudkar SV, Unadkat JD. (2014) Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos 42:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Vrana M, Mehrotra A, Johnson K, Bhatt DK. (2017) The promises of quantitative proteomics in precision medicine. J Pharm Sci 106:738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saborowski M, Kullak-Ublick GA, Eloranta JJ. (2006) The human organic cation transporter-1 gene is transactivated by hepatocyte nuclear factor-4alpha. J Pharmacol Exp Ther 317:778–785. [DOI] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al. (2000) Role of LXRs in control of lipogenesis. Genes Dev 14:2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, et al. (2008) Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther 83:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Woody S, Sun M, Cui W. (2013) Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver. Drug Metab Rev 45:48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundelin E, Gormsen LC, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, Christensen M, Brøsen K, Frøkiaer J, Jessen N. (2017) Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin Pharmacol Ther 102:841–848. [DOI] [PubMed] [Google Scholar]
- Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D, Inaba T. (2003) T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett 536:6–11. [DOI] [PubMed] [Google Scholar]
- Wang L, Collins C, Kelly EJ, Chu X, Ray AS, Salphati L, Xiao G, Lee C, Lai Y, Liao M, et al. (2016) Transporter expression in liver tissue from subjects with alcoholic or hepatitis C cirrhosis quantified by targeted quantitative proteomics. Drug Metab Dispos 44:1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunberg T, Hendrix M, Hillisch A, Lobell M, Meier H, Schmeck C, Wild H, Hinzen B. (2006) Improving the hit-to-lead process: data-driven assessment of drug-like and lead-like screening hits. Drug Discov Today 11:175–180. [DOI] [PubMed] [Google Scholar]