

Abstract
Next-generation sequencing has enabled genetic and genomic characterization of melanoma to an unprecedent depth. However, the high mutational background plus the limited deep-coverage whole-genome sequencing performed on cutaneous melanoma samples, make difficult the identification of novel driver mutations. We sought to explore the somatic mutation portfolio in exonic and gene regulatory regions in human melanoma samples, for which we performed targeted sequencing of tumors and matched germline DNA samples from 89 melanoma patients, identifying known and novel recurrent mutations. Two recurrent mutations found in the RPS27 promoter associated with decreased RPS27 mRNA levels in vitro. Data mining and IHC analyses revealed a bimodal pattern of RPS27 expression in melanoma, with RPS27-low patients displaying worse prognosis. In vitro characterization of RPS27-high and -low melanoma cell lines, as well as loss-of-function experiments, demonstrated that high RPS27 status provides increased proliferative and invasive capacities, while low RPS27 confers survival advantage in low-attachment and resistance to therapy. Additionally, we demonstrate that 10 other cancer types harbor bimodal RPS27 expression and in those, similarly to melanoma, RPS27-low expression associates with worse clinical outcomes. RPS27 promoter mutation could thus represent a mechanism of gene expression modulation in melanoma patients, which may have prognostic and predictive implications.
Keywords: High-throughput sequencing, RPS27, non-coding, biomarker, promoter analysis, drug resistance
Introduction
Next-generation sequencing studies (Cancer Genome Atlas, 2015; Hayward et al., 2017; M. Krauthammer et al., 2012) have confirmed previously known driver mutations of melanoma, such as BRAF V600E and NRAS Q61 mutations, while revealing novel drivers, such as recurrent RAC P29S. These efforts have also identified genetic interactions that inform melanoma sub-classification and can more effectively guide precision medicine. However, the ability to detect novel driver mutations at lower frequency varies between studies and is hampered by the high mutational burden of melanoma (the high background noise from non-driver mutations at low frequency), resulting in a critical need for mutational analysis of large sample cohorts. In addition, limited deep-coverage, whole-genome sequencing has been performed on melanoma samples, thus sequencing data for non-exonic regions, such as enhancers and promoters, remains limited. Targeted sequencing approaches can provide a feasible alternative for a more in-depth analysis of melanoma genomic landscape by identifying novel, clinically relevant mutations, including those in the non-coding genome.
In this study, we performed targeted, hybrid-capture-based sequencing of 89 melanoma tumors and patient-matched germline DNA samples. Our study confirms previously known mutations and identifies new recurrent point mutations, which may contribute to melanoma pathogenesis. Among these, we report recurrent and non-recurrent mutations in the promoter of the ribosomal protein S27, RPS27 (also known as metallopanstimulin 1 or MPS-1), a core ribosomal gene that encodes a component of the 40S subunit, confirming previous findings (Dutton-Regester et al., 2014; Hayward et al., 2017). We also report additional novel mutations at this genomic region and test the functional impact of RPS27 promoter mutations, as this has not been previously assessed. Our studies reveal that RPS27 promoter mutations associate with reduced expression, and expose a bimodal expression of RPS27 in melanoma that correlates with patient prognosis and drug sensitivity.
Methods
Study population
The study population consisted of 89 patients of self-reported non-Hispanic Caucasian descent diagnosed at NYU Langone Health (NYULH). The clinical and demographic characteristics of the patient cohort are summarized in Supplementary Table 1.
Targeted sequencing, somatic mutation calling and filtering
Targeted genomic sequencing (TGS) was performed on 89 matched tumor-normal samples at a mean coverage of 250x. The selection of the specific regions targeted for sequencing was based on evidence from prior sequencing data in TCGA, literature search and genetic loci of interest based on prior biological rationale generated by our group or other IMCG investigators. Specifically, the selected loci included 140 protein-coding genes (targeting coding exons as well as UTRs and proximal promoters(~2kb)), 20 lncRNA, 10 miRNA, 1 circRNA, 16 melanoma enhancers/super-enhancers (Fontanals-Cirera et al., 2017), all mitochondrial genes and selected genes with highest mutation burden reported in metastatic melanoma (Snyder et al., 2014) (Fig. 1B, Supplementary Table 2). The rationale for such design is two-fold: 1) the sequencing of exonic regions allowed for an evaluation of previously identified exonic mutations, and 2) the expansion of the sequencing targets by ~2kb of proximal promoter sites and other non-coding regions enabled the identification of mutations that may regulate expression of putative drivers, representing alternative genetic mechanisms disrupting these genes/pathways. The total genomic target of the sequencing was 3.19 Mb. The sample processing and pipeline used for the TGS of melanoma samples is depicted in Figure 1C.
Figure 1.

Description of the study population, gene targets, and sequencing workflow and data.
(A) Patient population by tumor stage, clinical classification and tumor type (primary/metastatic). (B) Genomic target selection for the sequencing of 89 melanomas. The proportion distribution reflects the genomic size of each target type by the total number of bases sequenced. (C) Sequencing and analysis pipeline. (D) Number of somatic mutations per patient found in the NYU TGS cohort. (E) Distribution of mutations by type per patient from the NYU cohort. (F) Oncoprint of non-synomynous and splice-site SNVs and coding and splice-site InDels predicted to impact protein coding mRNA. Selected known and previously nominated melanoma-associated genes, in addition to novel genes, are included.
Evaluating novelty of recurrent mutations with TCGA data
Somatic mutation data from 403 matched tumor-normal samples from The Cancer Genome Atlas Skin Cutaneous Melanoma Whole Exome Sequencing dataset (TCGA SKCM WES) was retrieved in Mutation Annotation Format (MAF) using the Genomic Data Commons (GDC) portal (Grossman et al., 2016). The frequencies of mutations called in the TCGA WES population were calculated in R and compared with the frequencies in the NYU TGS cohort. Because the sequencing target in TGS did not represent only exonic regions covered by TCGA WES, mutations that were recurrent in the NYU TGS cohort but not identified in the TCGA WES cohort were further evaluated. For this purpose, the sequencing coverage at the genomic positions of these mutations was calculated across the TCGA WES samples using samtools (H. Li et al., 2009), in order to determine if the mutation was targeted by the sequencing protocol. TGS-recurrent mutations not targeted and such, not detected in TCGA WES, were further interrogated.
Cell culture
All cells were grown in a humidified incubator at 37°C and 5% CO2. STR profiling was performed and authenticated 451Lu, A375, SK-MEL-5 and WM278. Cells were maintained in culture for no more than 20 passages, excluding passaging prior to receipt in the Hernando lab. All cells were routinely tested for Mycoplasma contamination. Short-term cultures (STCs) were established at NYU Langone Medical Center (Iman Osman lab) from surgically resected patient tissues (de Miera, Friedman, Greenwald, Perle, & Osman, 2012). Informed consent was obtained from all participating patients.
PCR of genomic DNA
RPS27 promoter from human samples and cell lines was PCR-amplified using a high-fidelity polymerase (Phusion Hot Start II DNA Polymerase, ThermoFisher). Primers were designed to cover the whole proximal promoter region and 5’UTR of the RPS27 gene (384 bp):
Forward primer: GCTCCAACTGTCAAGGTATGGC
Reverse primer: AATCAGCGTGAGGGGAGAGTTC
Reporter assay
RPS27 promoter sequence cloned into a pLightSwitch vector was purchased from Switchgear Genomics (#S702164). The vector contains 911 nt from RPS27 human promoter (including the 5’UTR) followed by the RenSP luciferase gene. Reporter assay was performed in HEK293T and SK-Mel-147 cell lines, seeding 15,000 cells in 96-well plates. The day after, cells were transfected with 100 ng of reporter plasmid (Renilla luciferase) and 20 ng of TK-Cypridina co-reporter. Cells were transfected using 0.5 μL of Lipo3000 per well, seeding triplicates per condition. 48h after transfection, luciferase activity was determined with the LighSwitch Dual Assay System (DA010), measuring in a FlexStation 3 microplate reader. Renilla luciferase units of each well were divided by its internal Cypridina control value. Average of wt RPS27 construct was considered as 1 and all values were referred to that control value.
RNA silencing
shRNA
pLKO-shRNA vectors were purchased from Millipore-Sigma (SHCLNG-NM_001030, clones TRCN0000117592 and TRCN0000117593). TRIPZ inducible shRNA for RPS27 was purchased from Dharmacon (RHS4740-EG6232).
siRNA
SMARTpool ON-Target plus siRNA were purchased from Dharmacon, both siRPS27 (L-050195–00-0005) and Non-targeting control (D-001810–10-05). Transfection of siRNA was performed using RNAiMax (ThermoScientific #13778).
In vitro invasion
Cell invasion was measured using 24-well Fluoroblok transwell inserts (Becton Dickinson, 8 μm pore), coating them with Matrigel (Becton Dickinson/Corning) diluted 1:30 in coating buffer (0.01M pH8 Tris-HCl, 0.7% NaCl), as previously described (Agrawal et al., 2017).
Protein isolation and Western blot analysis
Whole cell protein extractions and Western Blot were performed as previously described (Fontanals-Cirera et al., 2017). Primary antibodies used were: RPS27 (Abcam, ab4385, 1:5,000), α-tubulin (Sigma, T9026, 1:10,000), β-actin-peroxidase (Sigma, A3854, 1:50,000), GFP (Abcam, ab6556, 1:1,000) and Renilla luciferase (Abcam, ab184925, 1:20,000). Uncropped blots can be found in Figure S12.
IHC
Unconjugated murine clone 4A12 anti-human Metallopanstiumlin-1 (MPS-1), raised against a purified recombinant fragment of MPS-1 (Thermo Fischer Scientific Cat# MA5–15522 Lot# SE2382905), was used for immunohistochemistry. For RPS27 expression in melanoma stages, IHC was performed in a tissue microarray (Biomax, ME208), containing 30 cases of primary melanoma, 30 cases of metastatic melanoma and 9 normal skin tissue, with triplicate cores per case. Melanoma cases from the NYU cohort that were stained for RPS27 correspond to 29 wt and 9 mutant samples, paraffin-embedded 4-micron human melanoma slides. RPS27 expression categories were identified and assigned to each sample in a blind way by an expert pathologist.
Drug dose-response curves
Drugs were purchased from Selleckchem (Olaparib, S1060; Cisplatin, S1166; Palbociclib, S1116; Vemurafenib, S1267; Navitoclax, S1001; AR-42, S2244; Belinostat, S1085; YM155, S1130).
Additional and expanded methods can be found in Supporting Information.
Results
Targeted genomic sequencing of melanoma patients reveals novel recurrent mutations.
Eighty-nine melanoma tissue specimens, including 24 primary and 65 metastatic tumors and matched whole-blood germline DNA samples, collected by the NYU Interdisciplinary Melanoma Cooperative Group (IMCG) between years 2000 and 2014, were selected for the study (Fig. 1A). The clinical characteristics are described in Supplementary Table 1. We developed a sequencing panel of specific genomic regions of interest through literature search and internal studies, including most reported and putative driver mutations (Cancer Genome Atlas, 2015; Dutton-Regester et al., 2014; Hodis et al., 2012; Michael Krauthammer et al., 2015; Vinagre et al., 2013) (Fig. 1B, Supplementary Table 2). The sample processing and pipeline used for the targeted genomic sequencing (TGS) of melanoma samples is depicted in Figure 1C. The average sequencing depth of targeted regions in both tumor and normal was above 250x (257±88x for normal and 266±138x for tumor samples). Additionally, around 80% of the sequenced bases had a coverage higher than 50x (85±11% for germline and 78±18% for tumor samples). Overall, we detected 9,680 mutations in 89 melanomas, comprising 9,551 SNVs and 129 indels (Supplementary Table 3, Fig 1D). As expected, a high presence of C>T transitions was found in most patients (Fig. 1E). Figure 1F shows by patient the non-synonymous exonic mutations predicted to impact protein coding mRNA of selected genes, including melanoma drivers (e.g. BRAF, NRAS, NF1, PTEN) as well as novel mutations in plausible driver genes that have not been functionally characterized in melanoma (e.g. CMYA5, MERTK, STK11).
We assessed the most recurrent mutations (n=43) in coding or non-coding genomic regions that were present in tumors from at least 3 individuals (Table 1). To validate the observed frequencies and to assess the novelty of these mutations, we compared the most recurrent 43 mutations with TCGA SKCM WES data (Table 1). Of these mutations, 20 were not targeted by WES, and therefore could not be compared with our TGS frequencies. To interrogate these, we analyzed 75 melanoma patients from the TCGA SKCM dataset with WGS data available and assessed the observed frequencies for these mutations. However, the low average sequencing depth (~5x) of the WGS data provided only a limited sensitivity for the detection of non-exonic mutations identified by TGS.
Table 1.
Most frequent recurrent somatic mutations in coding and non-coding genomic loci from 89 melanomas tested in this study.
Recurrent mutations observed in our targeted genome sequencing (TGS) are listed by gene location and sorted by percentage of mutated samples. For comparison, the percentage of mutated samples observed in TCGA WES is listed. The mutations are further annotated by the presence and identified tumor site in COSMIC database. NA denotes “not assessed” by TCGA WES.
| Gene | Mutation | Location | TGS NYU (%) | TCGA WES(%) | Exonic Variant Function | AA Change | Cosmic |
|---|---|---|---|---|---|---|---|
| BRAF | chr7:140453136:A:T | exonic | 31.5 | 45.2 | non-synonymous SNV | V600E | Skin, others (COSM476) |
| RPS27 | chr1:153963239:C:T | UTR5 | 10.1 | 8.9 | . | . | |
| NRAS | chr1:115256529:T:C | exonic | 9.0 | 12.2 | non-synonymous SNV | Q61R | Skin, others (COSM584) |
| BRAF | chr7:140453137:C:T | exonic | 7.9 | 9.2 | non-synonymous SNV | V600M | Skin, others (COSM1130) |
| SOX17,RP1 | chr8:55526923:A:G | intergenic | 6.7 | NA | . | . | |
| SOX17,RP1 | chr8:55527362:A:G | intergenic | 6.7 | NA | . | . | |
| MGAM | chr7:141759821:G:A | intronic | 6.7 | NA | . | . | |
| NRAS | chr1:115256530:G:T | exonic | 5.6 | 7.9 | non-synonymous SNV | Q61K | Skin, others (COSM580) |
| IDH1 | chr2:209113113:G:A | exonic | 5.6 | 3.0 | non-synonymous SNV | R132C | Skin, others (COSM28747) |
| ANK3 | chr10:62334377:G:A | intronic | 5.6 | 0.2 | . | . | |
| THSD7B | chr2:138414689:G:A | exonic | 4.5 | 2.0 | non-synonymous SNV | R1445Q | Urinary tract (COSM1305663) |
| RPS27 | chr1:153963227:C:T | upstream | 4.5 | 4.5 | . | . | |
| RP1 | chr8:55541960:G:A | exonic | 4.5 | 0.5 | non-synonymous SNV | E1840K | |
| TLR4 | chr9:120477803:C:T | UTR3 | 3.4 | NA | . | . | |
| STK19 | chr6:31940123:G:A | exonic | 3.4 | 1.2 | non-synonymous SNV | D89N | Skin (COSM226579) |
| SOX17,RP1 | chr8:55526940:T:C | intergenic | 3.4 | NA | . | . | |
| PWRN1,NPAP1 | chr15:24918764:G:A | intergenic | 3.4 | NA | . | . | |
| PTENP1-AS | chr9:33677430:G:A | ncRNA_exonic | 3.4 | NA | . | . | |
| PTCH1 | chr9:98238105:G:A | intronic | 3.4 | NA | . | . | |
| PCLO | chr7:82385834:C:T | UTR3 | 3.4 | NA | . | . | |
| PCLO | chr7:82450880:G:A | UTR3 | 3.4 | NA | . | . | |
| PADI1 | chr1:17552678:C:T | intronic | 3.4 | 1.0 | . | . | |
| MAP2K1 | chr15:66679112:C:T | upstream | 3.4 | NA | . | . | |
| MAP2K1 | chr15:66774131:G:A | exonic | 3.4 | 0.5 | non-synonymous SNV | E203K | Skin, others (COSM232755) |
| LRP1B | chr2:141777573:C:T | exonic | 3.4 | 0.7 | non-synonymous SNV | E630K | |
| LOC389705,TTC39B | chr9:15083360:T:A | intergenic | 3.4 | NA | . | . | |
| HYDIN | chr16:70889118:G:A | exonic | 3.4 | 0.7 | non-synonymous SNV | S4119F | Skin (COSM137067) |
| GRM3 | chr7:86394514:G:A | exonic | 3.4 | 0.7 | non-synonymous SNV | G18E | Skin (COSM141835) |
| GRM3 | chr7:86468214:G:A | exonic | 3.4 | 0.5 | non-synonymous SNV | G462R | |
| GNA11 | chr19:3119404:-:G | intronic | 3.4 | NA | . | . | |
| DNAH8 | chr6:38705576:C:T | intronic | 3.4 | 0.7 | . | . | |
| DNAH6 | chr2:85014280:C:T | intronic | 3.4 | NA | . | . | |
| DNAH5 | chr5:13692194:G:A | exonic | 3.4 | 0.7 | stopgain | R4592X | Skin, other (COSM267977) |
| DNAH5 | chr5:13753599:G:A | exonic | 3.4 | 1.2 | non-synonymous SNV | R3539C | Skin (COSM1695413) |
| DCC | chr18:51058443:G:A | UTR3 | 3.4 | NA | . | . | |
| DCC | chr18:51060254:C:T | UTR3 | 3.4 | NA | . | . | |
| CSMD3 | chr8:114185963:G:A | exonic | 3.4 | 0.2 | non-synonymous SNV | P193S | |
| CSMD1 | chr8:2795273:C:T | UTR3 | 3.4 | NA | . | . | |
| CSMD1 | chr8:3265481:G:A | exonic | 3.4 | 0.2 | non-synonymous SNV | R671C | |
| ANK3 | chr10:61835777:G:A | exonic | 3.4 | 0.2 | non-synonymous SNV | S1621F | |
| ALK | chr2:29524646:C:T | intronic | 3.4 | NA | . | . | |
| ALK | chr2:29911572:C:G | intronic | 3.4 | NA | . | . | |
| ALK | chr2:30141580:A:G | intronic | 3.4 | NA | . | . |
As shown in Table 1, the known established non-synonymous drivers were among the most recurrent mutations identified in TGS (e.g. BRAF V600E/V600M, NRAS Q61R/Q61K, IDH1 R132C). We also identified novel plausible driver mutations that have not been functionally characterized in melanoma (e.g. MAP2K1, TLR4). One of the genes most frequently associated with mutations in our cohort was the ribosomal gene RPS27. We observed ~24% of patients with at least one mutation in close proximity to the TSS of RPS27 (Fig. 2A, S1A), with two recurrent mutations in the 5’UTR and proximal promoter, observed in 10% and 4.5% of the cases, respectively. We confirmed these mutations by PCR and Sanger sequencing in a subset of patients (Fig. S1B). Pairwise analyses of co-occurrence and mutual exclusivity of RPS27 mutations vs. established melanoma driver mutations showed no significant association after FDR adjustment (Fig. S2A–F). RPS27 mutations have been previously described with similar percentages in other melanoma cohorts (Dutton-Regester et al., 2014; Hayward et al., 2017) (Fig. S1A) and, even though a potential effect on translation for the 5’UTR mutation was suggested (Dutton-Regester et al., 2014), RPS27 promoter mutations have not yet been functionally characterized.
Figure 2.

Description of the mutations found in RPS27 and analysis of their effect on gene expression.
(A) Location (x-axis) and number of mutations (y-axis) detected in the RPS27 locus in the NYU TGS cohort. 1.3kb of the proximal promoter and the entire RPS27 gene are displayed. (B) Luciferase reporter assay of the RPS27 promoter (∼900 bp), using the WT and mutated constructs (at −8 and +5 bases from TSS). Error bars represent standard deviation of 5 independent experiments. (C) Quantitative PCR of Renilla luciferase mRNA expressed in HEK293T cells, using the same vectors as in (B). Renilla luciferase mRNA levels were relativized to GFP mRNA levels, control of transfection. Error bars represent SD. Data shows one of 3 independent experiments. (D) Renilla luciferase protein levels of HEK293T cells transfected with the reporter vectors (see B,C) analyzed by Western blot. For control of transfection, a vector expressing GFP was used. ∝-tubulin was used as loading control. Numbers represent the average quantification of Renilla luciferase from 3 independent experiments. (E) Percentage of WT (n=29) and MUT (n=9) melanoma samples from the TGS NYU cohort that present different levels of RPS27 protein expression, determined by IHC. Numbers 1 and 2 represent low and high RPS27 intensity, respectively. Fisher’s exact test was applied. (F) Representative 40x images of RPS27 IHC performed in metastatic melanoma patients from NYU TGS cohort. The percentage of positive cells is described by letters “d” (diffuse, >50%) and “f” (focal, <50%). ∗p < 0.05, ∗∗∗p < 0.001 by unpaired t-test.
Characterization of RPS27 recurrent mutations in the promoter region.
The region of the RPS27 locus sequenced comprised 1.3 kb of the proximal promoter, all the exonic and some intronic sequences. No mutations were identified in coding exon regions of RPS27, suggesting that such mutations may be deleterious for the cellular function, and hence undergo negative selection pressure. This is supported by a pan-cancer analysis using data from the cBioPortal cohort of 39,802 sequenced patients (Cerami et al., 2012), which reported mutations in RPS27 coding region in only 17 cases. Since the majority of the RPS27-associated mutations identified are near the TSS (Fig. 2A), we hypothesized that they could impact gene transcription. To test this hypothesis, we performed a dual luciferase reporter assay in which the RPS27 promoter was cloned upstream of the Renilla luciferase gene, using Firefly luciferase for normalization. The regulatory region tested corresponds to 911 nt encompassing the proximal promoter and the 5’UTR of RPS27. We mutated this construct to generate variants harboring the two most frequent mutations, at the 5’UTR (+5 of TSS) and the promoter (−8 of TSS). In HEK293T and the melanoma cell line SK-MEL-147, both tested RPS27 promoter mutations cause a significant decrease in luciferase activity (Fig. 2B). These data suggest that RPS27 promoter mutations may result in decreased RPS27 gene expression. This effect could be caused by reduced transcript levels or translation. We confirmed that the 5’UTR of the cloned reporter construct was efficiently transcribed in our assays, as shown by RT-PCR amplification (Fig. S3A). Quantitative RT-PCR showed decreased levels of luciferase mRNA when either mutation was present (Fig. 2C). Accordingly, Renilla luciferase protein levels determined by Western blot were also decreased in cells transfected with RPS27 mutant constructs (Fig. 2D).
Next, we assessed the presence of RPS27 mutations in human melanoma cell lines, including 25 established cell lines and 13 short-term cultures, which were analyzed by PCR and Sanger sequencing. Surprisingly, only 1 of the established melanoma cell lines (4%) contained mutations in the promoter of RPS27 at −8 and −63 from the TSS (Fig. S3B, C), the latter not found in patient samples. In the sequenced short-term cultures, 1 sample (8%) harbored a mutation at position −1 from the TSS (Fig. S3D, E). This relatively low frequency of RPS27 mutations suggests a negative selection during adaptation to adherent culture.
To determine if mutations in RPS27 could impact protein expression in human melanoma, we performed RPS27 immunohistochemistry in 37 patient samples from our melanoma cohort, 28 WT and 9 with RPS27 mutations (MUT; 7 in the 5’UTR and 2 in the promoter). Tumor staining was scored based on signal intensity and was generally observed to be bimodal (1 = low, 2 = high). Moreover, we observed a higher percentage of tumors with high RPS27 expression in the WT group compared with MUT tumors (Fig. 2E). Representative images of RPS27 staining in both WT and MUT groups are shown (Fig. 2F). These data support that mutations adjacent to the TSS of RPS27, which cumulatively are present in 24% of our melanoma cohort (21/89 cases), tend to associate with a decrease in expression levels. To further validate these results, we examined the association between RPS27 promoter mutations and RPS27 expression in 403 patients with WES data from the TCGA SKCM dataset (Cancer Genome Atlas, 2015), which captured a part of the proximal promoter. Also, 70 of these patients had WGS data information available. By interrogating these TCGA datasets, we found that 75 of the 403 patients (19% of samples) harbored RPS27 promoter mutations (Fig. S4A). In these samples, average RPS27 expression was not significantly lower in MUT vs. WT patients (Fig. S4B). However, similar to our IHC analyses, we observed that RPS27 displays a bimodal expression pattern, with most tumors expressing high RPS27 levels and a subset with substantially reduced expression. Importantly, RPS27 expression did not correlate with tumor purity in metastatic melanoma samples, as determined by three independent methods (Aran, Sirota, & Butte, 2015; B. Li & Li, 2014; Yoshihara et al., 2013) (Fig. S4C–E). This finding indicates that contamination by stroma is not the likely source of the starkly different RPS27 expression observed and supports the existence of two distinct RPS27 expression states in human melanoma.
RPS27 is overexpressed in human melanoma and displays a bimodal distribution.
The finding of recurrent mutations in the promoter region of RPS27, their surprising effects on RPS27 expression, and the variable expression of this ribosomal gene across human melanoma samples led us to further investigate the functional and clinical significance of RPS27 expression in melanoma. First, we analyzed RPS27 protein levels in a collection of human cultured melanocytes (n=4), and primary (n=7) and metastatic (n=11) melanoma cell lines. We observed undetected/low expression of RPS27 in melanocytes derived from both perinatal and adult skin, while all melanoma cell lines presented higher RPS27 levels (Fig. 3A). Interestingly, cell lines of metastatic origin generally expressed more RPS27 than primary melanoma-derived cell lines (Fig. 3B). To further assess RPS27 expression in relation to melanoma progression, we performed IHC of a commercial TMA containing primary melanoma tissues (n=30), lymph node metastases (LN; n=30) and control normal skin (n=9). For this analysis, we followed the same protocol and scoring method used for IHC of the NYU patient cohort (0, 1 and 2, represent undetected, low and high RPS27 intensity, respectively). All melanocytes present in the 9 skin samples showed undetectable RPS27 expression (Fig. 3C, D). In this collection of patient tissues, we again observed bimodal expression of RPS27, which was independent of tumor stage. Approximately 50% of samples had low/undetected RPS27 expression (value 0) and 50% high RPS27 (Fig. 3C, D). In contrast to melanoma cell lines, we found no difference in RPS27 expression between primary and metastatic melanoma, though all metastatic samples were of lymph node origin in this collection. Furthermore, in the NYU TGS melanoma cohort analyzed by IHC (Fig. 2E, F), lymph node metastases expressed lower RPS27 when compared with distant and in transit metastases (Fig. S5). We also analyzed the full TCGA melanoma cohort, containing 450+ cases of primary and metastatic melanoma with RNA-seq data (primary n=103, metastatic n=369). RPS27 expression was again observed to be bimodal in both primary and metastatic melanoma (Fig. 3E). Moreover, we also observed that RPS27 mRNA was significantly more abundant in metastatic compared with primary melanomas (Fig. 3E). Surprisingly, despite the general increase in RPS27 expression during melanoma progression, stratifying patients into bimodal expression groups revealed that low RPS27 expression is associated with worse prognosis (Fig. 3F), with a similar but non-significant trend when restricting to the subset of primary cases (Fig. 3G). Meanwhile, patient stratification based on RPS27 mutational status (rather than expression) did not associate with differential prognosis (Fig. S6).
Figure 3.
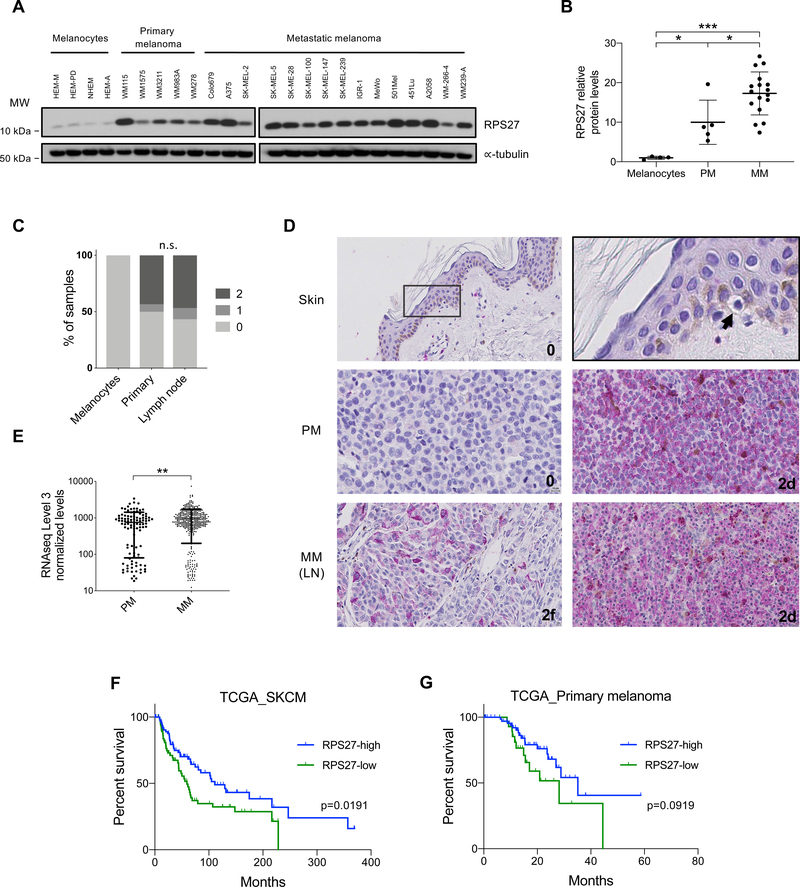
Bimodal RPS27 expression in human melanoma.
(A) RPS27 protein levels in cultured human epidermal melanocytes (neonatal, HEM-M and HEM-D; and adult, NHEM and HEM-A), primary and metastatic human melanoma cell lines, detected by Western blot. ∝-tubulin was used as loading control. (B) Quantification of RPS27 protein levels in (A), using Fiji software and normalized to ∝-tubulin. Data shows one representative experiment. ∗p < 0.05, ∗∗∗p < 0.001 by unpaired t-test. (C) Percentage of skin melanocytes, primary and lymph node melanoma samples from a tissue microarray (TMA) by level of RPS27 expression, determined by IHC. Numbers 0, 1 and 2 represent undetected, low and high RPS27 intensity, respectively. Fisher’s exact test was applied. (D) Representative 40x images of RPS27 IHC performed in the human melanoma TMA. Skin was zoomed 4x to show a melanocyte with undetected RPS27 expression (arrowhead). (E) RPS27 normalized mRNA levels in melanoma patients from TCGA (n=474), comparing primary and metastatic samples. ∗∗p < 0.01 by Mann-Whitney test. (F) Kaplan-Meier curves for overall survival of all PM and MM combined melanoma patients, stratified by expression of RPS27 (Top 20%, n=92; Low 20%, n=92). P-value was calculated using Log-rank test. (G) Kaplan-Meier curves for overall survival of primary melanoma patients, stratified by bimodal expression of RPS27 (Top, n=66; Low, n=34). P-value was calculated using Log-rank test.
Modulating RPS27 levels impacts proliferation, invasion and survival in low attachment
To test the biological functions of RPS27 in melanoma, we modulated its expression in human metastatic melanoma cell lines. We attempted to mimic bimodal RPS27-low melanoma by downregulating RPS27 in RPS27-high melanoma cell lines. Depletion of RPS27 mRNA with constitutive shRNA had a severe anti-proliferative effect in melanoma cells (Fig. S7A–C, left panels). In contrast, doxycycline-inducible shRNAs only led to a minor decrease of RPS27 levels without any phenotypic effect on cell proliferation assays (Fig. S7A–C, right panels). This result suggests that the expression of RPS27 is tightly regulated, and that a minimum threshold is required for cell growth. Transient transfection of siRNA allowed us to partially downregulate RPS27 expression to intermediate levels, as determined by qRT-PCR and Western blot (Fig. 4A, B). RPS27 suppression in 501Mel and A375 melanoma cell lines caused a modest but significant reduction in proliferation (Fig. 4C). Additionally, the invasive capacity of these melanoma cells through a Matrigel membrane was partially impaired upon RPS27 silencing (Fig. 4D, E). Interestingly, RPS27 silencing in these same cells resulted in enhanced growth capacity in ultra-low attachment conditions (Fig. 4F), with clear differences in growth patterns in suspension: RPS27-high cells formed more intercellular contacts and RPS27-low cells grew more disperse and established less contacts (Fig. 4G). In addition, the adhesion capacity of RPS27-low cells to gelatin (Fig. 4H, I), fibronectin (Fig. 4J, K) and collagen I (Fig. 4L, M) was reduced when compared to RPS27-high cells. Overall, these data suggest the existence of two different states in melanoma cell lines: RPS27-high, characterized by higher proliferation/invasion, and RPS27-low, characterized by lower adherence and higher anchorage-independent survival.
Figure 4.

RPS27 modulation impacts proliferation, invasion, adhesion and survival in low attachment conditions.
(A) RPS27 mRNA levels of control (NTC) and siRNA transfected 501Mel and A375 cell lines, determined by quantitative PCR. Bars represent SD from one representative experiment. (B) Western blot detection of RPS27 protein levels in the control and silenced melanoma cell lines. ∝-tubulin was used as loading control. (C) Proliferation curves of cell lines characterized in (A,B). A representative experiment out of 3 is shown. (D) Invasive capacity of melanoma cells silenced for RPS27. Data shows the average of 4 independent experiments. Bars represent SD. (E) Representative 20x fluorescent images of invading 501Mel and A375 cells with RPS27 modulation. (F) Relative growth in low attachment of melanoma cell lines silenced for RPS27. Data shows the average of independent experiments (501Mel, n=6; A375, n=4). Bars represent SD. (G) Representative 4x images of the control and siRPS27 conditions growing in ultra-low attachment plates. (H,I,J,K,L,M) Relative cell adhesion to (H) gelatin, (J) fibronectin and (L) collagen I of melanoma cell lines silenced for RPS27. Representative fluorescent images of cells adhering to gelatin, fibronectin and collagen I are shown in (I), (K), and (M), respectively. Data shows the average of 3 independent experiments, with bars representing SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by unpaired t-test.
RPS27 levels correlate with drug sensitivity
The increased ability of RPS27-low cells to grow in low attachment conditions could reflect higher survival capacity in adverse conditions. Thus, we examined if RPS27 levels could associate to sensitivity to specific therapeutics. To test this question, we analyzed the Genomics of Drug Sensitivity in Cancer (GDSC) database (Yang et al., 2013) which, to date, has profiled 1,065 cancer cell lines with 266 compounds. RPS27 RNA-seq expression data was extracted for the available cell lines from the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012). We separated melanoma cell lines (n=31) for their RPS27 expression into RPS27-high vs. RPS27-low (top 25% vs. lower 25%) and compared their sensitivity to different drugs, generating the ratio of their median IC50. A ratio of 1 corresponds to equal sensitivity between RPS27-high vs. RPS27-low cell lines. Interestingly, for most of the molecules (72%) the ratio was lower than 1 (Fig. 5A), which supports the hypothesis that RPS27-low cells are generally more resistant to drugs. For some of the drug categories, RPS27 seemed to be better predictor of sensitivity, since all or most of the molecules of the category had an IC50 ratio lower than 1 (e.g. cytoskeleton, DNA replication, JNK and p38 signaling or protein stability). When examining the whole collection of melanoma cell lines, RPS27 expression was found inversely correlated with sensitivity to drugs impacting genome integrity (PARP inhibitors), DNA replication (TOP inhibitors) and cell cycle (CDK4–6, AURKA-B inhibitors) (Pearson’s r value < −0.5; Fig. 5B, C). Interestingly, even though many MAPK inhibitors showed an IC50 ratio less than 1 (Fig. 5A), none of them exhibited a strong correlation with RPS27 levels (Fig. 5B, C). To validate our in silico findings, we characterized 10 cell lines, selected by RPS27 expression (Fig. 5D, E), for their sensitivity to selected drugs. For these experiments, we chose a PARP inhibitor (olaparib), a CDK4–6 inhibitor (palbociclib) and a DNA crosslinker (cisplatin). Dose-response curves of the 10 cell lines showed differential sensitivity between the RPS27-high and RPS27-low to olaparib (Fig. 5F, J), palbociclib (Fig. 5G, K) and cisplatin (Fig. 5H, L), with RPS27-low cell lines being more resistant to these drugs. In addition, we tested a BRAF inhibitor (PLX-4032, vemurafenib), which is part of the standard of care for many metastatic melanoma patients harboring the BRAF V600E mutation. We tested it in 7 of the 10 cell lines, which were BRAF mutant. RPS27-low cell lines showed increased resistance to BRAF inhibition (Fig. 5I, M). Some other drugs that our in silico data had suggested as potentially more effective in RPS27-high cell lines, targeting histone deacetylases (AR-42 and belinostat) and survival pathways (navitoclax, a Bcl-2 inhibitor; YM155, a survivin inhibitor), showed no differential sensitivity between RPS27-high and RPS27-low cell lines (Fig. S8). To test if RPS27, in addition to being a marker of sensitivity to several drugs, could be a functional mediator of resistance, we silenced its expression in melanoma cell lines and treated them with olaparib, palbociclib, cisplatin and vemurafenib. Dose-response curves showed no significant difference in drug sensitivity of control and RPS27-silenced cells (Fig. S9), which supports RPS27 as a marker - not a mediator - of the resistance.
Figure 5.

RPS27 expression levels predict drug sensitivity in human melanoma cell lines.
(A) Drug sensitivity in human cell lines from the Cancer Cell Line Encyclopedia (CCLE). Y-axis represents the ratio of the average IC50 of melanoma cell lines with high RPS27 vs cell lines with low RPS27 (25% top vs 25% bottom). X-axis shows the drug categories analyzed in Genomics of Drug Sensitivity in Cancer (GDSC). (B) Pearson’s correlation between RPS27 expression and IC50 for the indicated drugs. Two-tailed p-value is also included. (C) Examples of drugs that show negative Pearson’s correlation between RPS27 expression and sensitivity to the drug. (D) RPS27 relative mRNA levels in the 10 melanoma cell lines with high and low expression of the gene, determined by qRT-PCR. ∗∗p < 0.01 by unpaired t-test. (E) Western blot detection of RPS27 protein levels in cell lines from (D). β-actin was used as loading control. (F,G,H,I) Dose-response curves of the indicated drugs (olaparib, palbociclib, cisplatin, PLX-4032, respectively). IC50 for each cell line is shown next to the name in the legend. (J,K,L,M) Comparison of the average IC50 of human melanoma cell lines with high vs. low RPS27 levels, for the drugs (J) olaparib, (K) palbociclib, (L) cisplatin and (M) PLX-4032. Data is the average of independent experiments (olaparib, palbociclib and cisplatin, n=4; PXL-4032, n=3). ∗p < 0.05, by unpaired t-test.
RPS27, a core ribosomal gene, is expected to be expressed in other human tumors. Thus, we examined RPS27 expression and its potential correlation with outcomes and response to therapy in other cancer types. In the TCGA repository we found a similar RPS27 bimodal expression in many other tumor types in addition to melanoma (Fig. S10A). We also tested if RPS27 expression would separate the patients by differential prognosis. Patients with RPS27-high levels generally have better prognosis than RPS27-low patients, being the difference statistically significant for low grade glioma (LGG), liver hepatocellular carcinoma (LIHC), and thyroid carcinoma (THCA) (Fig. S10B). In addition, a pan-cancer analysis of human cell lines comprising 29 tumor types (Fig. S10C) predicts that, similarly to melanoma, RPS27-low cell lines may be more resistant to various drugs (Fig. S10D).
Overall, these data suggest that RPS27 expression may serve as a marker of drug sensitivity for different therapeutics, in melanoma and possibly other cancer types. This is especially important for the response to currently approved targeted therapies (i.e., vemurafenib) and drugs that have recently gained more attention for the treatment of melanoma (i.e., palbociclib). The predictive potential of RPS27 could be linked to distinct functional ‘states’ associated to high vs low RPS27.
Discussion
Large scale NGS efforts have confirmed previously known melanoma drivers and identified and nominated novel mutations as possible melanoma players. Moreover, these efforts elucidated previously unknown aspects of melanoma biology, offering a promise for important clinical benefits through therapeutic targeting of novel driver mutations and associated pathways, similar to BRAF mutant inhibitors. In this study, we sequenced 89 melanoma samples at high depth for selected loci, inclusive of both exonic and non-exonic regions, such as promoters of genes previously proposed as possible drivers of melanoma. Overall, our data further confirmed previously reported mutations and identified several recurrent mutations in coding and non-coding loci that have not been reported to-date in melanoma.
Among the genes with novel recurrent mutations, TLR4 (Toll-like receptor 4) primarily functions as a pathogen-associated molecular pattern recognition receptor (i.e., bacterial LPS) in the innate immune system. Additionally, TLR4 is overexpressed in different cancer types, and several tumor ligands that stimulate TLR4 signaling through NF-κB have been described (Pandey, Chauhan, & Jain, 2018). Here, we identified a recurrent mutation in the 3’UTR of TLR4, which might affect the post-transcriptional modulation of TLR4 via miRNA. TLR4 regulation by miRNAs has been described in different cell types (Curtale et al., 2018; Wang et al., 2018; Yan et al., 2018), including melanoma (Jin et al., 2019). Another novel recurrent mutation identified in this study is in MAP2K1, which encodes MEK1. Although activating exonic mutations in the MAP2K1 gene have been reported (Emelyanova et al., 2017; Nikolaev et al., 2011), to our knowledge, there are no reports on MAP2K1 promoter mutations. Differential binding of transcription factors resulting from mutations in the promoter could affect MAP2K1 gene expression. Both TLR4 and MAP2K1 mutations as well as other SNVs in Supplementary Table 3 warrant future investigation.
Our data also revealed many non-recurrent mutations in the RPS27 locus and confirmed previously reported recurrent RPS27 promoter mutations (Dutton-Regester et al., 2014; Hayward et al., 2017), with 24% of patient samples harboring at least one mutation adjacent to the TSS of RPS27. Despite their prior identification, the phenotypic effect of RPS27 promoter mutations and general RPS27 function have not been investigated in melanoma. A previous study proposed that the recurrent mutation in the 5’UTR of RPS27 (chr1:153963239) would alter the transcriptional start to position −1 (from the annotated TSS), extending the 5’UTR by one nucleotide, which could promote the expression of this gene via mTOR signaling (Dutton-Regester et al., 2014). However, the presence of additional mutations upstream of the TSS, including additional recurrent mutations, suggest a possible role in transcriptional regulation of RPS27 instead. For the promoter mutation at the position −8 from the TSS, Hayward et al. did not predict any gained or lost putative transcription factor binding sites (Hayward et al., 2017). However, when we compared mutant with wild type promoter reporter constructs, the two most frequent mutations at the RPS27 promoter (−8 and +5 from TSS) reduced transcriptional activity, suggesting some level of differential transcriptional control. Consistent with reporter assay observations, IHC analysis showed that more melanoma tumors with mutations in the RPS27 promoter region harbor reduced RPS27 protein levels than WT samples, although the difference was not statistically significant.
Interestingly, compared to the 24% of melanoma patients harboring RPS27 mutations, we only found mutations in 2/38 cultured melanoma cell lines (5%). Despite the limited sample size, it is possible that RPS27 mutations are negatively selected during the process of adhesion and in vitro expansion, such that RPS27-mutant tumors fail to establish and/or are outcompeted by RPS27 WT cells. Our in vitro experiments support this hypothesis: cells with RPS27-low levels (with which RPS27 mutations associate) present lower adherent and proliferative capabilities, and could be lost during the establishment of cell lines.
The wide range of RPS27 levels among melanoma samples is surprising since, as a core ribosomal gene, RPS27 has been described as a candidate housekeeping gene for human gene expression studies (Caracausi et al., 2017). We confirmed a bimodal expression of RPS27 protein abundance by tissue microarray, and observed a similar pattern in other cancer types in TCGA. Compared to melanocytes, we found overexpression of RPS27 in melanoma, both in cell lines and human samples. Consistently, a previous study documented increased RPS27 staining by IHC in melanoma compared with nevi (Santa Cruz, Hamilton, Klos, & Fernandez-Pol, 1997). This supports a model in which RPS27 expression increases during melanomagenesis. In addition, we detected increased RPS27 mRNA in metastatic vs. primary melanoma samples from TCGA, consistent with the observed protein levels in a collection of metastatic and primary melanoma cell lines. We found no difference between 30 primary and 30 lymph node metastatic samples stained for RPS27. However, lymph node metastases have been reported to present lower RPS27 levels compared to other metastatic sites (Santa Cruz et al., 1997), an observation that we reproduced in our NYU cohort IHC study. Therefore, an increase in RPS27 expression during the metastatic process cannot be dismissed. Future studies should examine different metastatic sites and investigate the role of RPS27 in melanoma progression.
Functionally, RPS27 depletion in metastatic melanoma cells reduced their invasive capacity, in agreement with its higher levels in metastasis compared to primary melanoma. RPS27-silenced cells also showed slightly lower proliferation in adherent conditions. At first glance, these results seem to confront the clinical data, in which RPS27-low patients display worse prognosis. However, we have shown that low RPS27 confers a decrease in cell adherence, a growth advantage in low-attachment conditions and associates with multidrug resistance, suggesting a more aggressive behavior. We confirmed increased resistance of RPS27-low cell lines to olaparib, palbociclib and cisplatin, drugs related to genome integrity and cell cycle progression. However, RPS27 suppression was not sufficient to sensitize melanoma cells to these drugs, which supports RPS27 as a marker rather than a direct determinant of resistance. We have observed that RPS27-low cell lines, except for IGR-1, exhibit lower proliferation rates than RPS27-high cell lines (data not shown), consistent with our in vitro data of RPS27 silencing. This could partially contribute to their differential sensitivity to drugs targeting DNA replication and cell cycle. In addition, RPS27 levels correlated with response to BRAF inhibitors, which together with MEKi represent the first line of treatment for many BRAF V600E metastatic melanoma patients. Given the clinical relevance of this treatment, RPS27 association with BRAF inhibition should be further characterized in future studies.
The mechanism by which distinct RPS27 levels impact melanoma cell properties could be explained by the ribosomal function of the protein. Recent works support the idea that multiple ribosome subtypes have different functional characteristics, impacting translation globally or by selective mRNA translation (Guo, 2018). Modulation of ribosome function can occur via variations in ribosomal protein (RP) composition (Kondrashov et al., 2011) or in stoichiometry of ribosomal proteins (Slavov, Semrau, Airoldi, Budnik, & van Oudenaarden, 2015). Additionally, RPS27 could have extra-ribosomal functions, as has been reported for many RP genes (Xiong, Zhao, He, & Sun, 2011; Zhou, Liao, Liao, Liao, & Lu, 2015). Future studies should explore these possibilities.
Based on our data, we propose two compatible possible models to explain how distinct RPS27 levels may contribute to melanoma progression (Fig. S11). A) A dynamic model, in which RPS27 expression is highly plastic during melanoma progression. Thus, RPS27 may be increased during the invasive phase, downregulated during early stages of metastatic adaptation to support anchorage-independent survival, and restored after colonization to enhance proliferation. Mutations in the RPS27 promoter, such as the ones characterized here, represent one such mechanism that could decrease its expression and favor anchorage-independent melanoma cell survival. The effect of these mutations, occurring only in regulatory regions, could be overridden by additional mechanisms (e.g., transcriptional, post-transcriptional or translational) controlling mRNA and protein expression. Such a scenario could explain why not all cases with RPS27 mutation display low RPS27 levels. B) Static model, in which two subtypes of melanoma exist, with invariable RPS27 levels -either high or low- that manage to metastasize (the first by higher proliferation and invasive potential, the second by higher survival in anchorage-independent conditions), but those with low levels would have a more aggressive and drug resistant behavior.
In the era of melanoma targeted therapy and immunotherapy, there are still many non-responding patients, as well as patients who fail treatments due to emergence of drug resistance. Several new combinatorial treatments are being explored, aiming to prevent or counteract therapeutic resistance. In order to maximize the efficacy of these treatments, there is an urgent need for markers that predict drug sensitivity. Our data suggest RPS27 as a potential prognostic marker in cutaneous melanoma and a plausible predictive biomarker of multidrug sensitivity. Some of these compounds, such as CDK4/6 inhibitors, have recently been tested clinically for the treatment of melanoma and other solid tumors (Jerby-Arnon et al., 2018; Schettini et al., 2018). Interestingly, we found that the bimodal RPS27 expression extends to 10 other human cancer types in addition to melanoma. In fact, RPS27 expression also correlates with patient outcomes in low-grade glioma, liver hepatocellular carcinoma, thyroid carcinoma and adrenocortical carcinoma. Future studies will address the role of RPS27 as a prognostic and predictive biomarker in other cancers.
In sum, RPS27 displays a bimodal pattern of expression in melanoma, which may correspond to two functional states characterized by distinct proliferation, invasion and survival in adverse conditions, and potentially hold prognostic and predictive value.
Supplementary Material
Supplementary figure 1. RPS27 mutations in the NYU cohort vs. other datasets.
(A) Table comparing the percentage of melanoma patients with RPS27 mutations in the TGS NYU cohort vs the percentages in Hayward et al., Dutton-Regester et al. and TCGA WGS cohorts. The two recurrent mutations that we characterize in this study are indicated blue. N/A in the columns with Dutton-Regester et al. and Hayward et al. data mean “not publicly available”. (B) Sanger sequencing of melanoma samples from TGS NYU cohort. Two examples of RPS27 promoter (−8) and 5’UTR (5+) mutations are shown, with the corresponding whole blood control compared to the melanoma sample. Arrowheads point to the mutated base.
Supplementary figure 2. Pairwise associations of mutual exclusivity and co-occurrence of RPS27 mutations vs. main melanoma driver genes.
(A, B) Pairwise associations of (A) co-occurrence and (B) mutual exclusivity of mutations in selected genes among TGS NYU patients. FDR-adjusted p-values (q-values) are represented in the upper diagonal, and unadjusted p-values are represented in the lower diagonal. Significant associations are highlighted. (C) Oncoplot of selected altered genes across TGS NYU patients. (D, E) Pairwise associations of (D) co-occurrence and (E) mutual exclusivity of mutations in selected genes among SKCM TCGA (WES). FDR-adjusted p-values (q-values) are represented in the upper diagonal, and unadjusted p-values are represented in the lower diagonal. Significant associations are highlighted in green. (F) Oncoplot of selected altered genes across SKCM TCGA (WES) patients.
Supplementary figure 3. RPS27 mutations in human melanoma cell lines and short-term cultures.
(A) RT-PCR amplification of the fusion between the RPS27 5’-UTR and the luciferase gene, in HEK293T transfected cells (lanes 2 and 3). -RT: “no retrotranscriptase” control. pLightSwitch vector DNA was used as a positive control (lane 1) and H2O as negative control of the PCR (lane 4). (B) Table describing RPS27 mutational status of 25 established human melanoma cell lines. (C) Sanger sequencing of the RPS27 promoter in two melanoma cell lines. Arrowheads point to the mutated bases. (D) Table describing the mutational status of RPS27 in 13 human short-term cultures. (E) Sanger sequencing of the RPS27 promoter in 3 melanoma short-term cultures. Arrowheads point to the mutated base.
Supplementary figure 4. Bimodal expression of RPS27 in human melanoma from TCGA dataset.
(A) Needle plot depicting the location (x-axis) and number of mutations (y-axis) detected in the RPS27 locus in the TCGA SKCM WES cohort (n=403 patients). 200 bp of the proximal promoter and 200 bp of the RPS27 gene, containing the 5’UTR and intron 1 (blue arrowheads), are represented. (B) RPS27 mRNA expression levels in 403 melanoma patients from TCGA WES, separated by RPS27 mutational status. Mann-Whitney test was applied for the group comparison. (C,D,E) RPS27 expression in metastatic melanoma samples from TCGA SKCM dataset was compared to tumor purity estimated by three independent methods: (C) ESTIMATE, based on gene expression signatures; (D) LUMP, based on CpG methylation; and (E) CHAT, based on sCNAs and SNVs.
Supplementary figure 5. RPS27 expression at different stages of the metastatic process.
(A) Percentage of cases with different RPS27 levels in “in transit”, lymph node and distant metastatic samples from the NYU TGS cohort, determined by IHC. Numbers 1 and 2 represent low and high RPS27 intensity, respectively. Representative 40x images of RPS27 IHC of (B) in transit, (C) lymph node and (D) distant metastasis analyzed in (A). % of positive cells are described by letters “d” (diffuse, >50%) and “f” (focal, <50%).
Supplementary figure 6. Study of the survival data of melanoma patients from TCGA based on their RPS27 mutational status.
Kaplan-Meier curves for overall survival of SKCM TCGA patients. Comparison groups include the two most common mutations in the RPS27 gene, promoter (−8) and 5’UTR (+5), (A) separated or (B) combined, vs. RPS27 wt or other mutations in the gene; (C) any mutations in the RPS27 promoter or 5’UTR vs. RPS27 wt or other mutations in the gene; and (D) any mutations in the RPS27 gene vs. RPS27 wt.
Supplementary figure 7. RPS27 expression modulation in 501Mel human melanoma cell line.
RPS27 expression was modulated using two shRNA methods, constitutive (pLKO, marked RPS27 reduction, left panels) and doxycycline-inducible (TRIPZ, moderate RPS27 decrease, right panels). (A) RPS27 mRNA levels detected by qRT-PCR. One representative experiment in shown. Bars correspond to SD. (B) Western blot characterization of RPS27 protein levels. ∝-tubulin was used as loading control. (C) Growth curves of 501Mel cell line with RPS27 expression modulation. One representative experiment out of 2 is shown. Bars correspond to SD. ∗∗∗p < 0.001 by unpaired t-test of the AUC.
Supplementary figure 8. Drug sensitivity analysis of RPS27-high vs. RPS27-low cell lines.
Dose-response curves of the indicated drugs: (A,B) HDAC inhibitors, AR-42 and belinostat, respectively; (C) Bcl-2 inhibitor, navitoclax; and (D) Survivin inhibitor, YM155. IC50 for each cell line is shown next to the name in the legend. One out of 2 independent experiments is shown.
Supplementary figure 9. Drug sensitivity analysis of human melanoma cells with down-regulation of RPS27.
Dose-response curves of the indicated drugs: (A,B) olaparib, (C,D) palbociclib, (E,F) cisplatin, and (G,H) PLX-4032, in A375 and 501Mel, respectively for each drug. The IC50 for each cell line and condition is shown close to the name in the legend. Curves represent the average of independent experiments (n=5, except for PLX-4032, for which n=3).
Supplementary figure 10. RPS27 expression and prognostic value in human cancer.
(A) RPS27 expression levels in 11 human cancers from TCGA, including cutaneous melanoma. Genomic alterations are color-coded as indicated. Graph was generated using cBioPortal. (B) Kaplan-Meier curves for overall survival of patients from the 10 cancer types with bimodal RPS27 expression, stratified by expression of RPS27, separating the patients by the median. Plots were generated by Oncolnc.org, which includes the p-value calculated by the Log-rank test. For ACC and THCA, data was downloaded and graphs generated manually with GraphPad Prism. (C) RPS27 RNA-seq expression levels in human cell lines from The Cancer Cell Line Encyclopedia (CCLE), grouped by tumor type of origin (TCGA acronyms are used). Broken lines divide the 20% top and 20% bottom with high and low RPS27 expression, respectively. (D) Pan-cancer analysis of drug sensitivity in human cell lines from CCLE. Y-axis represents the ratio of the average IC50 of cell lines with high RPS27 vs. cell lines with low RPS27 (20% top vs 20% bottom, n=250 cell lines total). X-axis indicates the drug categories analyzed in Genomics of Drug Sensitivity in Cancer (cancerrxgene.org, Yang et al., 2013).
Supplementary figure 11. Potential models of RPS27 expression and its contribution to melanoma progression.
(A) Dynamic model. Tumors would be heterogeneous for RPS27 expression and RPS27 levels would be modulated in a plastic manner during melanoma progression. Cells with distinct RPS27 levels could drive different stages of tumor progression: RPS27 may be increased during the invasive phase, downregulated during early stages of metastatic adaptation to support anchorage-independent survival, and restored after colonization to enhance proliferation. Mutations in the RPS27 promoter represent one such mechanism that could decrease its expression and favor anchorage-independent melanoma cell survival. (B) Static model. Two subtypes of melanoma may exist, with invariable RPS27 levels -either high (top) or low (bottom)- that manage to metastasize (the first by higher proliferation and invasive potential, the second by higher survival in anchorage-independent conditions).
Supplementary figure 12. Uncropped blots from Western blot analyses.
Uncropped blots from figures (A) 2D, (B) 3A, (C) 4B, (D) 5E and (E) S7D.
Significance.
Functional characterization of recurrent exonic mutations, such as BRAFV600E, led to a deeper understanding of melanoma and development of novel treatments. Additionally, recent whole-genome sequencing efforts are revealing alterations affecting non-coding regions. However, the functional significance of those mutations remains elusive. Our targeted sequencing of melanoma-relevant loci identified new low-frequency mutations in non-coding regions, in particular outlining the mutational landscape of the RPS27 promoter. Moreover, we found a striking bimodal expression of RPS27 which correlates with distinct in vitro phenotypes and response to therapies, including vemurafenib and palbociclib. In sum, our data suggest RPS27 holds prognostic and predictive potential in melanoma.
Acknowledgments
E.H. is funded by NIH P01CA206980, R01CA2022027 and NYU Melanoma SPORE NCI P50 CA225450. A.F. is supported by Fundacion Ramon Areces (“Ampliacion Estudios en el Extranjero” fellowship) and D.H. and A.U-M. have been supported by the NIH/NCI 5 T32 CA009161-37 (Training Program in Molecular Oncology and Immunology, PI: Dr. David E. Levy).
We thank the NYU Experimental Pathology Research Laboratory (Drs. Cindy Loomis and Luis Chiriboga), the Center for Biospecimen Research & Development, and the Genomics Technology Center (Dr. Adriana Heguy), supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant NIH/NCI P30CA016087, and National Institute of Health S10 Grant S10 OD021747.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D, … Hernando E. (2017). A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer Cell, 31(6), 804–819 e807. doi: 10.1016/j.ccell.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Sirota M, & Butte AJ (2015). Systematic pan-cancer analysis of tumour purity. Nat Commun, 6, 8971. doi: 10.1038/ncomms9971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, … Garraway LA (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483(7391), 603–607. doi: 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. (2015). Genomic Classification of Cutaneous Melanoma. Cell, 161(7), 1681–1696. doi: 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caracausi M, Piovesan A, Antonaros F, Strippoli P, Vitale L, & Pelleri MC (2017). Systematic identification of human housekeeping genes possibly useful as references in gene expression studies. Mol Med Rep, 16(3), 2397–2410. doi: 10.3892/mmr.2017.6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, … Schultz N (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov, 2(5), 401–404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtale G, Renzi TA, Mirolo M, Drufuca L, Albanese M, De Luca M, … Locati M. (2018). Multi-Step Regulation of the TLR4 Pathway by the miR-125a~99b~let-7e Cluster. Front Immunol, 9, 2037. doi: 10.3389/fimmu.2018.02037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miera EV-S, Friedman EB, Greenwald HS, Perle MA, & Osman I. (2012). Development of five new melanoma low passage cell lines representing the clinical and genetic profile of their tumors of origin. Pigment Cell & Melanoma Research, 25(3), 395–397. doi: 10.1111/j.1755-148x.2012.00994.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton-Regester K, Gartner JJ, Emmanuel R, Qutob N, Davies MA, Gershenwald JE, … Samuels Y (2014). A highly recurrent RPS27 5’UTR mutation in melanoma. Oncotarget, 5(10), 2912–2917. doi: 10.18632/oncotarget.2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emelyanova M, Ghukasyan L, Abramov I, Ryabaya O, Stepanova E, Kudryavtseva A, … Nasedkina T. (2017). Detection of BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K½ mutations in Russian melanoma patients using LNA PCR clamp and biochip analysis. Oncotarget, 8(32), 52304–52320. doi: 10.18632/oncotarget.17014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanals-Cirera B, Hasson D, Vardabasso C, Di Micco R, Agrawal P, Chowdhury A, … Bernstein E (2017). Harnessing BET Inhibitor Sensitivity Reveals AMIGO2 as a Melanoma Survival Gene. Mol Cell, 68(4), 731–744 e739. doi: 10.1016/j.molcel.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, & Staudt LM (2016). Toward a Shared Vision for Cancer Genomic Data. N Engl J Med, 375(12), 1109–1112. doi: 10.1056/NEJMp1607591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H. (2018). Specialized ribosomes and the control of translation. Biochem Soc Trans, 46(4), 855–869. doi: 10.1042/BST20160426 [DOI] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, … Mann GJ (2017). Whole-genome landscapes of major melanoma subtypes. Nature, 545(7653), 175–180. doi: 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, … Chin L. (2012). A landscape of driver mutations in melanoma. Cell, 150(2), 251–263. doi: 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, … Regev A. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell, 175(4), 984–997 e924. doi: 10.1016/j.cell.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Wang A, Liu L, Wang G, Li G, & Han Z. (2019). miR-145–5p inhibits tumor occurrence and metastasis through the NF-kappaB signaling pathway by targeting TLR4 in malignant melanoma. J Cell Biochem. doi: 10.1002/jcb.28388 [DOI] [PubMed] [Google Scholar]
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Ishijima J, … Barna M (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell, 145(3), 383–397. doi: 10.1016/j.cell.2011.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, … Halaban R. (2015). Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nature Genetics, 47(9), 996–1002. doi: 10.1038/ng.3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, … Halaban R (2012). Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet, 44(9), 1006–1014. doi: 10.1038/ng.2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, & Li JZ (2014). A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data. Genome Biol, 15(9), 473. doi: 10.1186/s13059-014-0473-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, … Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, … Antonarakis SE (2011). Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet, 44(2), 133–139. doi: 10.1038/ng.1026 [DOI] [PubMed] [Google Scholar]
- Pandey N, Chauhan A, & Jain N. (2018). TLR4 Polymorphisms and Expression in Solid Cancers. Mol Diagn Ther, 22(6), 683–702. doi: 10.1007/s40291-018-0361-9 [DOI] [PubMed] [Google Scholar]
- Santa Cruz DJ, Hamilton PD, Klos DJ, & Fernandez-Pol JA (1997). Differential expression of metallopanstimulin/S27 ribosomal protein in melanocytic lesions of the skin. J Cutan Pathol, 24(9), 533–542. [DOI] [PubMed] [Google Scholar]
- Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, … Del Mastro L. (2018). CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front Oncol, 8, 608. doi: 10.3389/fonc.2018.00608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N, Semrau S, Airoldi E, Budnik B, & van Oudenaarden A. (2015). Differential Stoichiometry among Core Ribosomal Proteins. Cell Rep, 13(5), 865–873. doi: 10.1016/j.celrep.2015.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, … Chan TA (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med, 371(23), 2189–2199. doi: 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, … Soares P (2013). Frequency of TERT promoter mutations in human cancers. Nat Commun, 4, 2185. doi: 10.1038/ncomms3185 [DOI] [PubMed] [Google Scholar]
- Wang W, Zhao E, Yu Y, Geng B, Zhang W, & Li X. (2018). MiR-216a exerts tumor-suppressing functions in renal cell carcinoma by targeting TLR4. Am J Cancer Res, 8(3), 476–488. [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Zhao Y, He H, & Sun Y. (2011). Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene, 30(15), 1798–1811. doi: 10.1038/onc.2010.569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Liu G, Jin C, Wang Z, Duan Q, Xu J, & Xu D. (2018). MicroRNA-6869–5p acts as a tumor suppressor via targeting TLR4/NF-kappaB signaling pathway in colorectal cancer. J Cell Physiol, 233(9), 6660–6668. doi: 10.1002/jcp.26316 [DOI] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, … Garnett MJ. (2013). Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res, 41(Database issue), D955–961. doi: 10.1093/nar/gks1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, … Verhaak RG (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun, 4, 2612. doi: 10.1038/ncomms3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Liao WJ, Liao JM, Liao P, & Lu H. (2015). Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol, 7(2), 92–104. doi: 10.1093/jmcb/mjv014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. RPS27 mutations in the NYU cohort vs. other datasets.
(A) Table comparing the percentage of melanoma patients with RPS27 mutations in the TGS NYU cohort vs the percentages in Hayward et al., Dutton-Regester et al. and TCGA WGS cohorts. The two recurrent mutations that we characterize in this study are indicated blue. N/A in the columns with Dutton-Regester et al. and Hayward et al. data mean “not publicly available”. (B) Sanger sequencing of melanoma samples from TGS NYU cohort. Two examples of RPS27 promoter (−8) and 5’UTR (5+) mutations are shown, with the corresponding whole blood control compared to the melanoma sample. Arrowheads point to the mutated base.
Supplementary figure 2. Pairwise associations of mutual exclusivity and co-occurrence of RPS27 mutations vs. main melanoma driver genes.
(A, B) Pairwise associations of (A) co-occurrence and (B) mutual exclusivity of mutations in selected genes among TGS NYU patients. FDR-adjusted p-values (q-values) are represented in the upper diagonal, and unadjusted p-values are represented in the lower diagonal. Significant associations are highlighted. (C) Oncoplot of selected altered genes across TGS NYU patients. (D, E) Pairwise associations of (D) co-occurrence and (E) mutual exclusivity of mutations in selected genes among SKCM TCGA (WES). FDR-adjusted p-values (q-values) are represented in the upper diagonal, and unadjusted p-values are represented in the lower diagonal. Significant associations are highlighted in green. (F) Oncoplot of selected altered genes across SKCM TCGA (WES) patients.
Supplementary figure 3. RPS27 mutations in human melanoma cell lines and short-term cultures.
(A) RT-PCR amplification of the fusion between the RPS27 5’-UTR and the luciferase gene, in HEK293T transfected cells (lanes 2 and 3). -RT: “no retrotranscriptase” control. pLightSwitch vector DNA was used as a positive control (lane 1) and H2O as negative control of the PCR (lane 4). (B) Table describing RPS27 mutational status of 25 established human melanoma cell lines. (C) Sanger sequencing of the RPS27 promoter in two melanoma cell lines. Arrowheads point to the mutated bases. (D) Table describing the mutational status of RPS27 in 13 human short-term cultures. (E) Sanger sequencing of the RPS27 promoter in 3 melanoma short-term cultures. Arrowheads point to the mutated base.
Supplementary figure 4. Bimodal expression of RPS27 in human melanoma from TCGA dataset.
(A) Needle plot depicting the location (x-axis) and number of mutations (y-axis) detected in the RPS27 locus in the TCGA SKCM WES cohort (n=403 patients). 200 bp of the proximal promoter and 200 bp of the RPS27 gene, containing the 5’UTR and intron 1 (blue arrowheads), are represented. (B) RPS27 mRNA expression levels in 403 melanoma patients from TCGA WES, separated by RPS27 mutational status. Mann-Whitney test was applied for the group comparison. (C,D,E) RPS27 expression in metastatic melanoma samples from TCGA SKCM dataset was compared to tumor purity estimated by three independent methods: (C) ESTIMATE, based on gene expression signatures; (D) LUMP, based on CpG methylation; and (E) CHAT, based on sCNAs and SNVs.
Supplementary figure 5. RPS27 expression at different stages of the metastatic process.
(A) Percentage of cases with different RPS27 levels in “in transit”, lymph node and distant metastatic samples from the NYU TGS cohort, determined by IHC. Numbers 1 and 2 represent low and high RPS27 intensity, respectively. Representative 40x images of RPS27 IHC of (B) in transit, (C) lymph node and (D) distant metastasis analyzed in (A). % of positive cells are described by letters “d” (diffuse, >50%) and “f” (focal, <50%).
Supplementary figure 6. Study of the survival data of melanoma patients from TCGA based on their RPS27 mutational status.
Kaplan-Meier curves for overall survival of SKCM TCGA patients. Comparison groups include the two most common mutations in the RPS27 gene, promoter (−8) and 5’UTR (+5), (A) separated or (B) combined, vs. RPS27 wt or other mutations in the gene; (C) any mutations in the RPS27 promoter or 5’UTR vs. RPS27 wt or other mutations in the gene; and (D) any mutations in the RPS27 gene vs. RPS27 wt.
Supplementary figure 7. RPS27 expression modulation in 501Mel human melanoma cell line.
RPS27 expression was modulated using two shRNA methods, constitutive (pLKO, marked RPS27 reduction, left panels) and doxycycline-inducible (TRIPZ, moderate RPS27 decrease, right panels). (A) RPS27 mRNA levels detected by qRT-PCR. One representative experiment in shown. Bars correspond to SD. (B) Western blot characterization of RPS27 protein levels. ∝-tubulin was used as loading control. (C) Growth curves of 501Mel cell line with RPS27 expression modulation. One representative experiment out of 2 is shown. Bars correspond to SD. ∗∗∗p < 0.001 by unpaired t-test of the AUC.
Supplementary figure 8. Drug sensitivity analysis of RPS27-high vs. RPS27-low cell lines.
Dose-response curves of the indicated drugs: (A,B) HDAC inhibitors, AR-42 and belinostat, respectively; (C) Bcl-2 inhibitor, navitoclax; and (D) Survivin inhibitor, YM155. IC50 for each cell line is shown next to the name in the legend. One out of 2 independent experiments is shown.
Supplementary figure 9. Drug sensitivity analysis of human melanoma cells with down-regulation of RPS27.
Dose-response curves of the indicated drugs: (A,B) olaparib, (C,D) palbociclib, (E,F) cisplatin, and (G,H) PLX-4032, in A375 and 501Mel, respectively for each drug. The IC50 for each cell line and condition is shown close to the name in the legend. Curves represent the average of independent experiments (n=5, except for PLX-4032, for which n=3).
Supplementary figure 10. RPS27 expression and prognostic value in human cancer.
(A) RPS27 expression levels in 11 human cancers from TCGA, including cutaneous melanoma. Genomic alterations are color-coded as indicated. Graph was generated using cBioPortal. (B) Kaplan-Meier curves for overall survival of patients from the 10 cancer types with bimodal RPS27 expression, stratified by expression of RPS27, separating the patients by the median. Plots were generated by Oncolnc.org, which includes the p-value calculated by the Log-rank test. For ACC and THCA, data was downloaded and graphs generated manually with GraphPad Prism. (C) RPS27 RNA-seq expression levels in human cell lines from The Cancer Cell Line Encyclopedia (CCLE), grouped by tumor type of origin (TCGA acronyms are used). Broken lines divide the 20% top and 20% bottom with high and low RPS27 expression, respectively. (D) Pan-cancer analysis of drug sensitivity in human cell lines from CCLE. Y-axis represents the ratio of the average IC50 of cell lines with high RPS27 vs. cell lines with low RPS27 (20% top vs 20% bottom, n=250 cell lines total). X-axis indicates the drug categories analyzed in Genomics of Drug Sensitivity in Cancer (cancerrxgene.org, Yang et al., 2013).
Supplementary figure 11. Potential models of RPS27 expression and its contribution to melanoma progression.
(A) Dynamic model. Tumors would be heterogeneous for RPS27 expression and RPS27 levels would be modulated in a plastic manner during melanoma progression. Cells with distinct RPS27 levels could drive different stages of tumor progression: RPS27 may be increased during the invasive phase, downregulated during early stages of metastatic adaptation to support anchorage-independent survival, and restored after colonization to enhance proliferation. Mutations in the RPS27 promoter represent one such mechanism that could decrease its expression and favor anchorage-independent melanoma cell survival. (B) Static model. Two subtypes of melanoma may exist, with invariable RPS27 levels -either high (top) or low (bottom)- that manage to metastasize (the first by higher proliferation and invasive potential, the second by higher survival in anchorage-independent conditions).
Supplementary figure 12. Uncropped blots from Western blot analyses.
Uncropped blots from figures (A) 2D, (B) 3A, (C) 4B, (D) 5E and (E) S7D.