

Abstract
OBJECTIVE:
In view of our previous observations on differential expression of LIM and cysteine-rich domains 1 (LMCD1) in human versus rodents, we asked the question whether LMCD1 plays a species-specific role in the development of vascular lesions.
APPROACH AND REULTS:
A combination of genetic, molecular, cellular and disease models were used to test species-specific role of LMCD1 in the pathogenesis of vascular lesions. Here, we report species-specific regulation of LMCD1 expression in mediating vascular smooth muscle cell (VSMC) proliferation and migration during vascular wall remodeling in human versus mice. Thrombin induced LMCD1 expression in HASMCs but not MASMCs via activation of Par1-Gαq/11-PLCβ3-NFATc1 signaling. Furthermore, although LMCD1 mediates thrombin-induced proliferation and migration of both HASMCs and MASMCs via influencing E2F1-mediated CDC6 expression and NFATc1-mediated IL-33 expression, respectively, in humans it acts as an activator and in mice it acts as a repressor of these transcriptional factors. Interestingly, LMCD1 repressor activity was nullified by N-myristoyltransferase 2-mediated myristoylation in mouse. Besides, we found increased expression of LMCD1 in human stenotic arteries as compared to non-stenotic arteries. On the other hand, LMCD1 expression was decreased in neointimal lesions of mouse injured arteries as compared to non-injured arteries.
CONCLUSIONS:
Together, these observations reveal that LMCD1 acts as an activator and repressor of E2F1 and NFATc1 in human and mice, respectively, in the induction of CDC6 and IL-33 expression during development of vascular lesions. Based on these findings, LMCD could be a potential target for drug development against restenosis and atherosclerosis in humans.
Keywords: Activator, CDC6, cell migration and proliferation, E2F1, IL-33, LMCD1, NFATc1, repressor, smooth muscle cells
Graphical Abstract
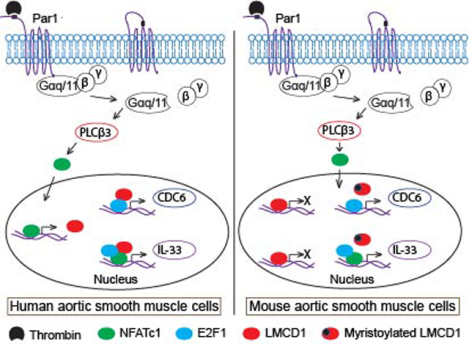
INTRODUCTION
LIM domain proteins are critical regulators of diverse cellular functions such as gene expression, cell growth, cell adhesion, cell differentiation and cytoskeleton remodeling [1]. LIM and cysteine-rich domains 1 (LMCD1)/Dyxin is a member of the LIM protein family, consists of a novel N-terminal cysteine-rich domain, a central PET (Prickle, Espinas, and Testin) domain, and two C-terminal LIM domains [2]. LIM and PET domains are typically involved in various protein–protein interactions [1]. Although many of the LIM proteins were well characterized, cellular functions of LMCD1 are poorly studied. Genetic studies have shown association of LMCD1 with nonsyndromic mitral valve prolapse in mouse [3] and low high-density lipoprotein cholesterol (HDL-C) levels in humans [4]. In addition, LMCD1 acts as a putative metastatic oncogene in human hepatocellular carcinoma [5], a transcriptional factor in epithelial to mesenchymal transition [6] and a potential biomarker for prostate cancer [7, 8] and alopecia [9]. Accumulating evidence suggests that LMCD1 plays a critical role in the development of cardiac hypertrophy via activation of calcineurin/nuclear factor of activated T cells (NFAT) signaling pathway in mice [10]. Overexpression of LMCD1 had led to cardiomyocyte hypertrophy and induction of the hypertrophic gene program in neonatal rat cardiomyocytes [11]. Moreover, LMCD1 also acts as a transcriptional repressor for GATA6, a zinc-finger transcription factor, in lung and cardiac tissues [12]. Our recent findings show that thrombin induces LMCD1 expression in human aortic smooth muscle cells (HASMCs), which in turn, acts as a transcriptional coactivator for E2F1 and NFATc1 in thrombin-induced CDC6 and IL-33 expression mediating HASMC proliferation and migration, respectively [13, 14]. In contrast, in rat aortic smooth muscle cells (RASMCs) and mouse aortic smooth muscle cells (MASMCs), the steady state levels of LMCD1 were not affected by thrombin and its downregulation did not affect thrombin-induced proliferation of these cells [13]. Based on these findings, we asked the question whether LMCD1 exhibits any species-specific expression in the regulation of thrombin-induced vascular smooth muscle cell (VSMC) proliferation and migration. In the present study, we show that LMCD1 acts as an activator of E2F1 and NFATc1 in the induction of CDC6 and IL-33 mediating VSMC proliferation and migration in the development of vascular lesions in humans. On the contrary, it acts as a repressor of these transcriptional factors and in response to thrombin its repressor activity was abolished by myristoylation in the induction of CDC6 and IL-33 expression mediating VSMC proliferation and migration, respectively, during development of vascular lesions in mice. Together these findings show for the first time that LMCD1 plays a differential role in the transactivation activity of E2F1 and NFATc1 in the regulation of their target genes influencing vascular lesions in human versus mice.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author on reasonable request.
Reagents:
Anti-β-Actin (SC-47778), anti-E2F1 (SC-251) and anti-PLCβ3 (SC-403) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Gαq (ab210004), anti- Gα11 (ab153951) and anti-LMCD1 (ab179454) antibodies were bought from Abcam (Cambridge, MA). Thrombin (T8885), anti-LMCD1 (HPA024059) and monoclonal anti-mouse SMCα-actin antibodies (A2547) were obtained from Sigma-Aldrich (St. Louis, MO). SCH79797 was purchased from TOCRIS Biosciences (Bristol, UK). pGL3 basic vector and Luciferase assay system (E4530) and PCR Master Mix (M750B) were obtained from Promega (Madison, WI). Human Gαq siRNA (ON-TARGETplus SMARTpool J-008562), human Gα11 siRNA (ON-TARGETplus SMARTpool J-010860), and control nontargeting siRNA (D-001810–10) were purchased from Dharmacon RNAi Technologies (Chicago, IL). Anti-IL33 (MBLPM033), anti-NFATc1 (MA3–024), goat anti-rabbit HRP (31460) and goat anti-mouse HRP (31437) antibodies, human LMCD1 siRNA (s26868), mouse LMCD1 siRNA (s78248), human NFATc1 siRNA (s9470), anti-NMT1 (PA5–54648), anti-NMT2 (MA5–25841) antibodies, mouse NMT1 siRNA (s70655) and mouse NMT2 siRNA (s70658), Quickchange Lightning Site-Directed Mutagenesis kit (210518–5), Biotin 3ʹ-end DNA labeling kit (89818), light-shift chemiluminescent EMSA kit (20148), Ibidi USA CULTURE-INSERTS (50305762), One ShotTM TOP10 Chemically Competent E. coli (C404003), Lipofectamine 3000 transfection reagent (L3000–015), Medium 231 (M231–500), smooth muscle growth supplements (S-007–25), gentamicin/amphotericin solution (R-015–10) and high sensitivity NeutrAvidin-HRP (Cat #31030) were obtained from Thermo Fisher Scientific (Waltham, MA). ECL Western blotting detection reagents (RPN2106) were purchased from GE Healthcare (Pittsburg, PA). Quick Ligation Kit (M2200S) was bought from New England Biolabs (Ipswich, MA). Neutralizing human IL-33 antibody (AF3625) and neutralizing mouse IL-33 antibody (AF3626) were obtained from R&D Systems (Minneapolis, MN). Phosphine-biotin (Item No. 13581) and 2-hydroxy myristic acid (Item No. 90390) were purchased from Cayman Chemical (Ann Arbor, MI). Azido myristic acid (Cat # 1345–25) was obtained from Click Chemistry Tools (Scottsdale, AZ). pRL-CMV vector (Cat # E2261) was bought from Promega (Madison, WI). Mouse LMCD1 (NM_144799) expression plasmid (Cat # MR205613) was purchased from Origene (Rockville, MD). All the primers used for RT-PCR, cloning, site-directed mutagenesis and EMSA were synthesized by IDT (Coralville, IA) and listed in Table 1.
Table 1.
List of primers used in the study
| Forward primer 5′ → 3′ | Reverse primer 5′ → 3′ | |
|---|---|---|
| Primers used for RT-PCR | ||
| Human LMCD1 Mouse LMCD1 Human β-Actin |
ACAGAGGGTGCCTTTTACCG GAGAAGCCAGGTGCTGTTTC AGCCATGTACGTTGCTAT |
GCTCGCAGACGTATTCCACT CGTCCAGGTCAGAGCTTAGG GATGTCCACGTCACACTTCA |
| Primers used for human LMCD1 promoter cloning into pGL3 basic vector | ||
| hLMCD1p(1.881kb) hLMCD1p(1.697kb) hLMCD1p(1.587kb) hLMCD1p(1.367kb) hLMCD1p(0.927kb) hLMCD1p(0.542kb) |
GGTTACGCGTGTTCAAGGTGATGGAAATCCTAAACACCCTGA GGTACCGAGCTCTTACGCGTTGGGAGCAAAC GGTACCGAGCTCTTACGCGTTAATAAAAAGTCACAATTCAG GTACCGAGCTCTTACGCGTCGGGACCCTG ACCGAGCTCTTACGCGTCTCTCAGGTTAAAGCC ACCGAGCTCTTACGCGTTGGGAAGGGGAG |
GGTTAGATCTCCTTAGCCACCTTTGCCATCC GGTTAGATCTCCTTAGCCACCTTTGCCATCC GGTTAGATCTCCTTAGCCACCTTTGCCATCC GGTTAGATCTCCTTAGCCACCTTTGCCATCC GGTTAGATCTCCTTAGCCACCTTTGCCATCC GGTTAGATCTCCTTAGCCACCTTTGCCATCC |
| Primers used for site-directed mutagenesis | ||
| hLMCD1p-NFAT-Mut mLMCD1p-Myr1-Mut |
CCAGGTTTGTTACACGTTAGCCAGCTCCTGCA CAAAAGATGTCCTTGCCCCAGCAGCAGTCCGCAA |
TGCAGGAGCTGGCTAACGTGTAACAAACCTGG TTGCGGACTGCTGCTGGGGCAAGGACATCTTTTG |
| Primers used for EMSA | ||
| LMCD1p-NFAT-WT LMCD1p-NFAT-Mut IL33p-NFAT-WT IL33p-NFAT-Mut CDC6p-E2F-WT CDC6p-E2F-Mut |
CCAGGTTTGGGAAACGTTAGCCAGCTCCTGCA CCAGGTTTGTTACACGTTAGCCAGCTCCTGCA CAAACTTTACTGAAATAATTATTTCCTCTATATGGTTATG CAAACTTTACTGAAATAATTATGTAATCTATATGGTTATG AATCGAGGCCGGGCTTTGGCGGGAGGTGGGAA AATCGAGGCCGGGCAATTCCGGGAGGTGGGAA |
TGCAGGAGCTGGCTAACGTTTCCCAAACCTGG TGCAGGAGCTGGCTAACGTGTAACAAACCTGG CATAACCATATAGAGGAAATAATTATTTCAGTAAAGTTTG CATAACCATATAGATTACATAATTATTTCAGTAAAGTTTG TTCCCACCTCCCGCCAAAGCCCGGCCTCGATT TTCCCACCTCCCGGAATTGCCCGGCCTCGATT |
| Primers used for ChIP assay | ||
| hCDC6-E2F mCDC6-E2F hIL33-NFAT mIL33-NFAT |
AAAAGAGGCGGTGCCCAAGG GAGTGACAACTAATCAGAAGCCT CCTCATTATGCAGTGCAGAGTACCATATC CTGCTTCCTACTTCGTGTTATCTT |
CACCACCACAAGCCCCTGAAC CGTAAAAACTCTCCCGCCACAAA GCTCACTACCAAATTGGTAACATAAC GGTTCCTCAAGAATAAATGGAGTAA |
Cell culture:
HASMCs (Cell systems; Cat # ACBRI 716) were isolated from normal human descending aorta and MASMCs (Cell Biologics; Cat # C57–6080) were isolated from aorta of pathogen-free C57BL/6 mice. Both HASMCs and MASMCs were subcultured in medium 231 containing smooth muscle cell growth supplements and DMEM with fetal bovine serum, respectively. Cultures were maintained in a humidified 95% air and 5% CO2 atmosphere at 37°C. Both HASMCs and MASMCs of different batches and passages between 4–10 were growth-arrested overnight in medium without any growth supplements or serum and used for the experiments unless otherwise indicated.
Animals:
C57BL/6J mice (both male and female) were obtained from Jackson Laboratory (Bar Harbor, ME) and maintained at the University of Tennessee Health Science Center’s animal facilities. All the experiments involving animals were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (Memphis, TN). Since we did not see gross differences between male and female in neointimal responses, the data was analyzed together.
Cell migration:
Cell migration was measured by wound closure assay as described previously [15]. Briefly, 80 μl of medium containing 2 × 105 cells was added in each chamber of ibidi culture inserts and grown to confluence. Then cells were growth-arrested and treated with and without thrombin (0.5 U/ml) in the presence of 5 mM hydroxyurea for 24 hrs. In the case of transfections, cells were transfected with either scrambled or specific siRNAs, and then plated in ibidi chambers before growing to confluence and growth-arrest. Cells were observed under Nikon Eclipse TS100 microscope before and after the experimental period with 10X/0.25 magnification and images were captured with CCD color camera (KP-D20AU) by using Apple iMovie 7.1.4 software. Cell migration was expressed as percentage of wound closure (total wound area at 0 hr – wound area at 24 hrs/total area at 0 hr × 100).
DNA synthesis:
HASMCs and MASMCs DNA synthesis was measured by [3H]-thymidine incorporation as described previously and expressed as counts/min/dish [13].
RT-PCR:
Total cellular RNA was isolated from HASMCs and MASMCs using TRIzol reagent as per the manufacturer’s instructions. Complementary DNA (cDNA) was prepared using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). LMCD1 and β-actin mRNAs were amplified with their specific primers (Table 1) using cDNA as a template. The amplified PCR products were separated on 1.5% agarose gels, stained with ethidium bromide, and the pictures were captured using a Kodak In-Vivo Imaging System (New Haven, CT).
Western blotting:
Cell or tissue extracts containing equal amounts of protein from control and treatments were resolved by electrophoresis on 0.1% (w/v) SDS and 10% (w/v) polyacrylamide gels. The proteins were transferred electrophoretically onto a nitrocellulose membrane. After blocking in 5% (w/v) non-fat dry milk, the membrane was incubated with the appropriate primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The antigen-antibody complexes were detected with the chemiluminescence detection reagent kit and the band intensities were quantified by densitometry using NIH ImageJ software.
Coimmunoprecipitation:
Cell extracts were prepared by cell lysis in 400 μL of lysis buffer (PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/mL PMSF, 100 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 mmol/L sodium orthovanadate) for 20 minutes on ice. The cell extracts were cleared by centrifugation at 12000 rpm for 15 minutes at 4°C. The cleared cell extracts containing an equal amount of protein from control and the indicated treatments were incubated with the indicated antibodies overnight at 4°C, followed by incubation with protein A/G-Sepharose CL4B beads for 3 hours with gentle rocking. The beads were collected by centrifugation at 1000 rpm for 2 minutes at 4°C and washed 4× with lysis buffer and once with PBS. The immunocomplexes were released by heating the beads in 40 μL of Laemmli sample buffer and analyzed by Western blotting for the indicated molecules using their specific antibodies.
Myristoylation:
HASMCs or MASMCs were growth-arrested and labeled with 40 μM 12-azidododecanoate (azidomyristate) for 3 hrs at 37°C as per the method described previously [16]. After labeling, cells were treated with and without thrombin (0.5 U/ml) for the indicated time periods, washed with cold PBS and cell extracts were prepared. Cell extracts containing equal amounts of protein from control and each treatment were incubated the indicated antibodies at 4°C overnight at which time protein A/G-Sepharose CL4B beads (50% slurry by w/v) were added and incubation continued for 3 hrs at 4°C with gentle rocking. The beads were collected by centrifugation and the immunocomplexes were eluted by heating in 40 μl of phosphine reaction buffer (0.1 M NaH2PO4, pH 7.2 and 1% SDS). Then the immunocomplexes were conjugated by incubation for 10 hrs at RT with 10 μl of 1 mM phosphine-biotin and separated by SDS-PAGE, transferred to PVDF membrane and blocked in blocking buffer. The membrane was incubated with neutravidin-HRP (1:10,000) and developed by ECL reagents. For inhibition of N-myristoylation, cells were pretreated with 2-hydroxymyristic acid (1 mM) for 1 hr at 37°C before labeling with azidomyristate.
Transfections:
HASMCs and MASMCs were transfected with nontargeted or ontargeted siRNA at a final concentration of 100 nM using Lipofectamine 3000 transfection reagent as per the manufacturer’s instructions. Wherever plasmid vectors were used, cells were transfected with plasmid DNAs at a final concentration of 5 μg/100 mm dish. After 6 hrs of transfection, cells were recovered in complete medium for 24 hrs and then growth-arrested overnight in serum-free medium and used as required.
Cloning and site-directed mutagenesis:
The human LMCD1 promoter encompassing −1748 to +133 nt relative to the transcription start site was amplified from human genomic DNA using forward primer hLMCD1p-(1.881 kb)F incorporating a Mlu1 restriction enzyme site at the 5′ end and a reverse primer hLMCD1p-(1.881 kb)R incorporating a BglII restriction enzyme site at the 5′ end. The PCR product was digested with Mlu1 and BglII, and the released fragment was cloned into Mlu1 and BglII sites of the pGL3 basic vector (Promega) to yield pGL3-hLMCD1p-(1.881 kb)-Luc. Various truncations of human LMCD1 promoter were cloned into pGL3 basic vector using the primers listed in Table 1. Mutation in the NFATc1 at −485 nt was introduced by the QuikChange Lightning site-directed mutagenesis kit. Plasmid DNAs were purified using the EndoFree plasmid maxi kit (Catalog No. 12362; Qiagen) and used in the transfection experiments.
Luciferase assay:
HASMCs and MASMCs were transfected with pCMV basic vector or pCMV-LMCD1 expression vector and/or the indicated IL-33 or CDC6 promoter-luciferase constructs using Lipofectamine 3000 transfection reagent for 6 hrs. Cells after 24 hrs of recovery in complete medium were growth-arrested in serum-free medium overnight. Cells were then treated with and without thrombin (0.5 U/ml) for 8 hrs, washed with cold PBS, and lysed in 200 μl of lysis buffer. The cell extracts were cleared by centrifugation at 12,000 rpm for 2 min at 4°C. The supernatants were assayed for luciferase activity using luciferase assay system (Promega) and a single tube luminometer (TD20/20; Turner Designs, Sunnyvale, CA). The values were normalized against pRL-CMV internal control vector and expressed as relative luciferase units.
EMSA:
HASMC and MASMC nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction kit (Catalog # 78833, Thermo Fisher Scientific) according to the manufacturer’s protocol. The protein content of the nuclear extracts was determined using a micro-BCA method (Pierce Biotechnology). Double stranded oligonucleotides encompassing NFAT binding element located at −485 nt of human LMCD1 promoter, E2F binding element located at −43 nt of human CDC6 promoter and NFAT binding element located at −100 nt of human IL-33 promoter were used as biotin-labeled probes (Table 1) to measure protein-DNA binding activity. Briefly, 5 μg of nuclear extract was incubated in a binding buffer (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM DTT, 2.5% glycerol) with 5 nM of biotin-labeled probe and 1 μg of poly(dI-dC) for 30 min at room temperature in a total volume of 20 μl on ice, and the protein-DNA complexes were resolved by electrophoresis on a 6% polyacrylamide gel using Tris borate-EDTA buffer (44.5 mM Tris-HCl, 44.5 mM borate, and 20 mM EDTA, pH 8.0). After separation, the protein-DNA complexes were transferred to nylon membrane using Tris borate-EDTA buffer, UV cross-linked, and visualized by chemiluminescence. To perform supershift EMSA, the complete reaction mix was incubated with 2 μg of the indicated antibody for 1 hr on ice before separating it by electrophoresis. Normal serum was used as negative control.
ChIP assay:
MASMCs with and without the indicated treatments were fixed with 1% formaldehyde for 10 min at 37°C. After washing, cells were scraped in PBS and centrifuged at 2000 rpm for 4 min at 4°C. The cell pellet was resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) containing protease inhibitors, sonicated and subjected to centrifugation at 13,000 rpm for 10 min at 4°C. The supernatant was collected, diluted with ChIP dilution buffer (16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, and 1.1% Triton X-100) and immunoprecipitated with the indicated antibodies. Mouse or rabbit pre-immune serum was used as a negative control. The immunoprecipitated DNA was un-cross-linked, subjected to proteinase K digestion, and purified using QIAquick columns (Cat # 28104, Qiagen). The ChIP and input samples were analyzed by PCR using the indicated primers (Table 1).
Guide wire injury (GI):
Mice were anesthetized using ketamine/xylazine and subjected to GI as described previously [17]. The left femoral artery was exposed by blunted dissection. Both the vein and artery were looped together proximally and distally with 4–0 silk sutures for blood flow control during the procedure. A small muscular branch of the femoral artery was isolated and a small incision was made on this exposed muscular branch and a straight spring wire (0.38 mm in diameter, No. C-SF-15–15, COOK, Bloomington, IN) was inserted into the lumen of the femoral artery. The wire was moved back and forth two times to denude the artery. After removal of the spring wire, the muscular branch of the artery was ligated. The blood flow in the injured femoral artery was restored by releasing the sutures that were placed in the proximal and distal femoral portions. At the indicated time periods of post GI, the animals were sacrificed with over dose of ketamine/xylazine, and femoral arteries were carefully harvested and either tissue extracts were prepared or fixed overnight in 4% PFA in PBS at 4°C. After fixing in PFA, the arteries were incubated overnight in 30% sucrose at 4°C, then embedded in OCT compound and 5–7 μm thick sections were cut in the middle of the artery.
Immunofluorescence:
Mouse femoral artery cryosections were permeabilized with 0.5% Triton X-100 for 15 min, and after blocking in 5% normal goat serum for 1 hr, the sections were probed with mouse anti-mouse SMCα-actin and rabbit anti-LMCD1 antibodies (1:100 dilution) overnight followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse or Alexa Fluor 568-conjugated goat anti-rabbit secondary antibodies (1:300 dilution). The sections were observed under a Zeiss Inverted Microscope (Zeiss AxioObserver Z1; Magnification at 10X/0.25 NA or 40X/0.6 NA), and the fluorescence images were captured with a Zeiss AxioCam MRm camera using the microscope operating software and Image Analysis Software AxioVision 4.7.2 (Carl Zeiss Imaging Solutions).
Human artery sections:
Human coronary artery specimens with stable moderate to unstable advanced atherosclerotic lesions or no lesion control sections from males with age group ranging from 34 to 66 years were obtained from CVPath Institute (Gaithersburg, MD) Sudden Death Registry. The use of human atherosclerotic and non-atherosclerotic artery sections for research purpose was approved by UTHSC Institutional Review Board. These samples were considered to be deemed non-human subjects, because they were postmortem samples. Briefly, the artery segments were fixed in formalin and 2 to 3 millimeter segments were embedded in paraffin. Cross-sections of five microns thick were cut from each of the segments, and mounted on slides. Slides were incubated with LMCD1 antibody (Sigma HPA024059, 1:1000 dilution), and SMα-actin antibody (DAKO, 1:200 dilution) overnight. Secondary anti-rabbit Alexa Fluor 555 and anti-mouse Alexa Fluor 488 antibodies (Thermo Fisher Scientific, 1:100 dilution) were used for fluorescence staining. DAPI was used as nuclei counter staining.
Statistics:
All the experiments were repeated 3 times, and the data are presented as Mean ± SD. Normality of the data (using Anderson-Darling Normality Test) and the equality of group variance (using F-test) were performed on all data using Minitab 18 software. The normally distributed data with similar variance were analyzed by 1-Way ANOVA followed by Tukey post hoc test. Two group comparisons with non-normal distributions were analyzed using nonparametric Mann-Whitney test. The p < 0.05 were considered statistically significant. In the case of EMSA, Western blotting and immunofluorescence, one set of the representative data is presented.
RESULTS
Thrombin induces LMCD1 expression in mediating HASMC but not MASMC proliferation and migration
Vascular injury elicits generation of thrombin, a potent mitogen and chemotactic factor, which in turn, modulates the expression of several genes in various cell types including VSMCs [18, 19]. As our recent findings showed a potential role of LMCD1 in thrombin-induced HASMC proliferation and migration and lack of a role in mouse aortic smooth muscle cell proliferation and migration [13], we asked the question whether LMCD1 exhibits any species-specific regulation of expression. To find this, first we studied a time course effect of thrombin on LMCD1 expression in HASMCs and MASMCs. Thrombin (0.5 U/mL) induced LMCD1 expression in a sustained manner at both mRNA and protein levels in HASMCs but not MASMCs (Figure 1A–D). In fact, LMCD1 expression seems to be down-regulated by thrombin in MASMCs (Figure 1C & D). To explore the functional significance of thrombin-induced LMCD1 expression in HASMCs, we tested its role in thrombin-induced proliferation and migration of HASMCs and MASMCs. LMCD1 knockdown using its siRNA attenuated thrombin-induced proliferation and migration of HASMCs but not MASMCs (Figure 1E–H). These results indicate species-specific role of LMCD1 in thrombin-induced VSMC growth and migration.
Figure 1. Species-specific role of LMCD1 in thrombin-induced VSMCs proliferation and migration.
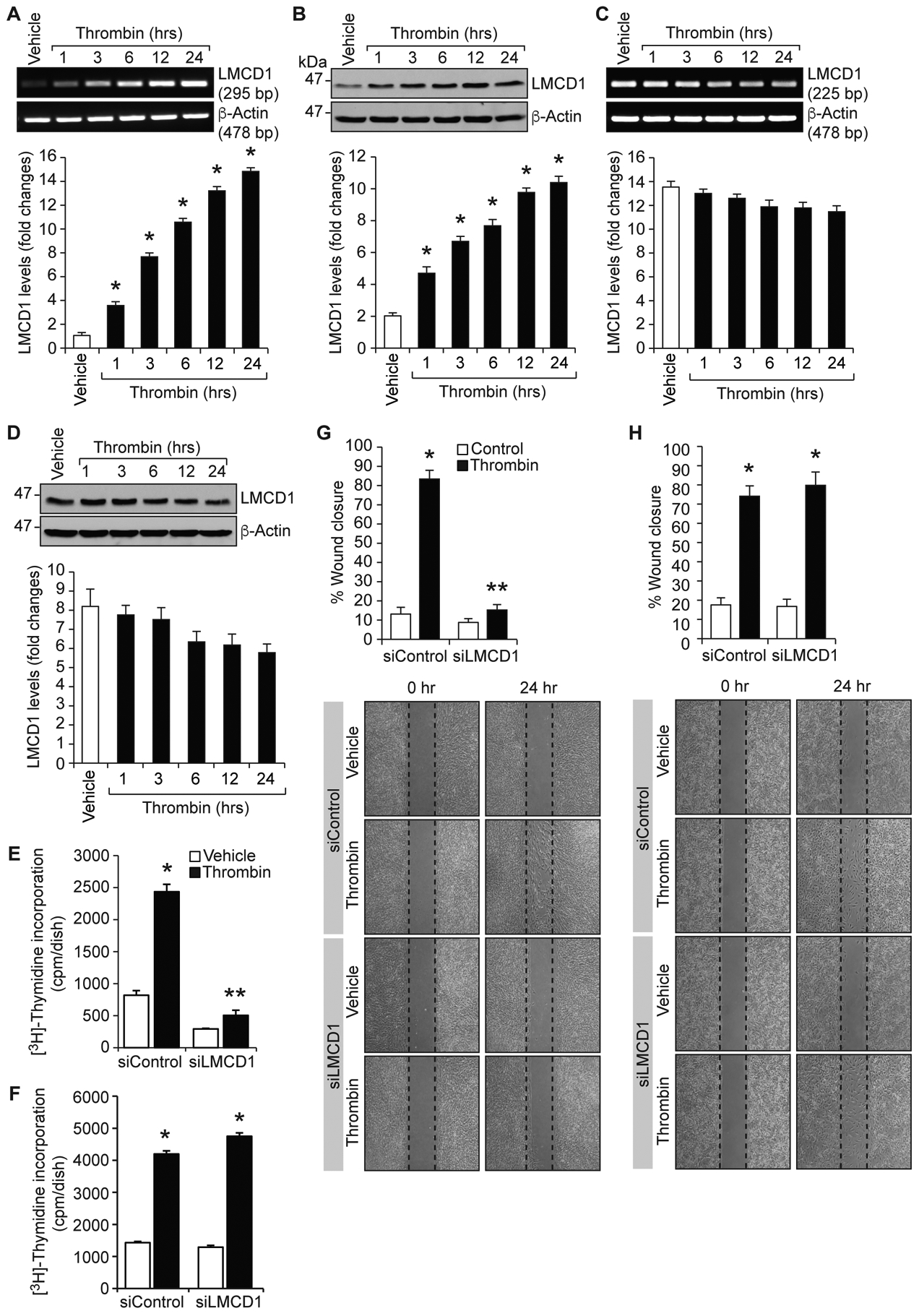
A-D. Quiescent HASMCs (A & B) or MASMCs (C & D) were treated with and without thrombin (0.5 U/ml) for the indicated time periods, and either total RNA was isolated and analyzed by RT-PCR for LMCD1 and β-actin mRNA levels (A & C) using their specific primers or cell extracts were prepared and analyzed by Western blotting for LMCD1 and β-actin protein levels (B & D) using their specific antibodies. HASMCs (E & G) or MASMCs (F & H) that were transfected with siControl or siLMCD1 (100 nM) were quiesced and tested for thrombin (0.5 U/ml)-induced DNA synthesis (E & F) and cell migration (G & H). The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl + vehicle; **, p < 0.05 versus siControl + Thrombin.
Thrombin-induced LMCD1 expression requires NFATc1 activation
To understand the molecular mechanisms involved in thrombin-induced LMCD1 expression in HASMCs and to explore the molecular basis for this species-specific expression divergence, we first analyzed human and mouse LMCD1 promoter sequences for potential transcription factor binding sites using TRANSFAC software [20]. We identified six binding sites for NFAT (at −261 nt; −485 nt; −1105 nt; −1365 nt; −1493 nt and −1736 nt); three binding sites for GATA (at −695 nt; −1398 nt and −1488); and one binding site for AP1 (at −185 nt) in human LMCD1 promoter, whereas in mouse LMCD1 promoter, there are three binding sites for NFAT (at −1174 nt; −1483 nt and −1505 nt); one binding site for GATA (at −517 nt); and one binding site for AP1 (at −180 nt) (Figure 2A & B). To find whether thrombin-induced LMCD1 expression in HASMCs exhibit any transcriptional regulation, we cloned a ~1.8 kb human LMCD1 promoter into pGL3 basic vector (pGL3-hLMCD1p-[1.8 kb]-Luc) and studied its activity. We observed a 5-fold increase in LMCD1 promoter activity in response to thrombin in HASMCs as compared to vehicle control (Figure 2C). These findings clearly indicate transcriptional regulation of thrombin-induced LMCD1 expression in humans. To identify the minimal LMCD1 promoter region required for thrombin-induced LMCD1 promoter activity in HASMCs, we performed serial promoter deletion analysis by subcloning the truncated regions −1564 to +133 nt (1.697 kb), −1454 to +133 nt (1.587 kb), −1234 to +133 nt (1.367 kb), −794 to +133 nt (0.927 kb), and −409 to +133 nt (0.542 kb) of human LMCD1 promoter into pGL3 basic vector. The constructs were named as pGL3-hLMCD1p-(1.697 kb)-Luc, pGL3-hLMCD1p-(1.587 kb)-Luc, pGL3-hLMCD1p-(1.367 kb)-Luc, pGL3-hLMCD1p-(0.927 kb)-Luc, and pGL3-hLMCD1p-(0.542 kb)-Luc, respectively. HASMCs were transfected with these constructs and their responsiveness to thrombin was measured. We observed a significant increase in LMCD1 promoter activity with all the constructs except with pGL3-hLMCD1p-(0.542 kb)-Luc construct indicating the presence of thrombin-responsive element(s) between −794 nt to −409 nt of human LMCD1 promoter from the transcriptional start site (Figure 2C). Human LMCD1 promoter region from −794 nt to −409 nt contains one NFAT-binding site at −485 nt and one GATA site at −695 nt. When we mutated NFAT-binding site at −485 nt by site-directed mutagenesis, thrombin-induced LMCD1 promoter activity was significantly blunted (Figure 2D). These results indicate the importance of the NFAT binding site at −485 nt in thrombin-induced LMCD1 expression in humans.
Figure 2. NFATc1 mediates LMCD1 promoter activity in HASMCs.
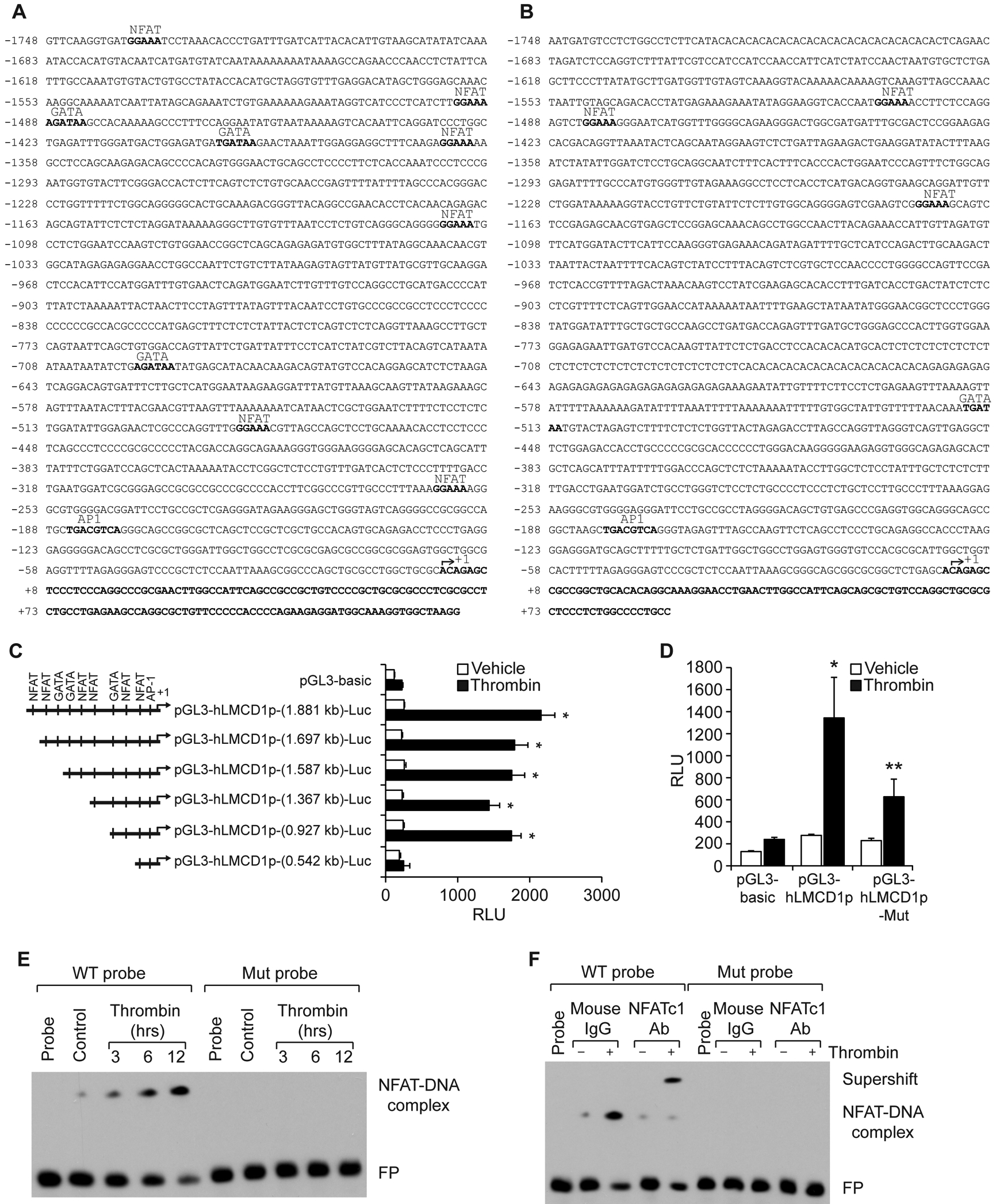
A & B. TRANSFAC analysis of human (A) and mouse (B) LMCD1 promoter sequence for the identification of potential transcription factor binding sites. C. HASMCs that were transfected with pGL3-basic vector or pGL3-hLMCD1 promoter-reporter gene constructs with serial 5′-deletions were quiesced and treated with and without thrombin (0.5 U/ml) for 8 hrs and analyzed for luciferase activity. D. HASMCs were transfected with pGL3-basic vector or pGL3-hLMCD1 promoter (0.927 kb) with and without the mutated NFAT-binding site (at −485 nt), quiesced, and treated with and without thrombin (0.5 U/ml) for 8 hrs, and luciferase activity was measured. E. Nuclear extracts of control and various time periods of thrombin (0.5 U/ml)-treated HASMCs were analyzed by EMSA using the NFAT-binding element at −485 nt as a biotin-labeled DNA probe. F. Nuclear extracts of control and 8 hrs of thrombin (0.5 U/ml)-treated HASMCs were analyzed for the presence of NFTAc1 in the protein-DNA complexes by supershift EMSA. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus vehicle control; **, p < 0.05 versus Thrombin. RLU, relative luciferase units; FP, free probe.
To gain further evidence for the role of the NFAT-binding site at −485 nt in thrombin-induced LMCD1 promoter activity in humans, we next studied a time course effect of thrombin on NFAT-DNA-binding activity by electrophoretic mobility shift assay (EMSA) using the NFAT-binding element at −485 nt as a biotin-labeled double-stranded oligonucleotide probe. Thrombin induced NFAT-DNA-binding activity in a time dependent manner with maximum effect at 12 hrs (Figure 2E). To confirm specificity of thrombin-induced NFAT-DNA-binding activity, we mutated NFAT-binding element at −485 nt and used it as a probe for EMSA. We found no protein-DNA binding activity with nuclear extracts of either control or thrombin-treated HASMCs using the mutant probe (Figure 2E). Furthermore, supershift EMSA using anti-NFATc1 antibodies showed the binding of NFATc1 to this site (Figure 2F). Because NFATc1 binds to NFAT-binding site at −485 nt of human LMCD1 promoter, we next tested the role of NFATc1 in thrombin-induced LMCD1 expression in HASMCs. Depletion of NFATc1 using its siRNA attenuated thrombin-induced LMCD1 expression and LMCD1 promoter activity (Figure 3A & E). These results suggest that NFATc1 binds to LMCD1 promoter and influences LMCD1 expression in response to thrombin.
Figure 3. Activation of PAR1-Gαq/11-PLCβ3 signaling is required for thrombin-induced NFATc1-mediated LMCD1 expression in HASMCs.

A-D. Quiescent HASMCs that were pretreated with Par1 antagonist (SCH79797) or HASMCs that were transfected with the indicated siRNA (100 nM) and quiesced were treated with and without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and analyzed by Western blotting for LMCD1 levels or for the indicated proteins for normalization or to show the efficacy of the siNRA on its target molecule using their specific antibodies. E-H. HASMCs were transfected with pGL3-basic vector or pGL3-hLMCD1 promoter (1.881 kb)-reporter construct in combination with the indicated siRNA were growth-arrested, treated with or without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and assayed for luciferase activity. In the case of Par1 antagonist, after transfection with basic vector or LMCD1 promoter-reporter construct and quiescence the cells were pretreated with and without the antagonist for 30 min before treatment with and without thrombin. The bar graphs represent the Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl + vehicle; **, p < 0.05 versus siControl + Thrombin. RLU, relative luciferase units; siCtrl, siControl.
PAR1-Gαq/11-PLCβ3 signaling is required for thrombin-induced NFATc1-mediated LMCD1 expression in HASMCs
Previously, we have shown that Par1, Gαq/11 and PLCβ3 mediate NFATc1 activation in the regulation of IL-33 expression by thrombin [14]. Based on these observations, we tested the role of Par1 and its downstream effectors Gαq/11 and PLCβ3 in thrombin-induced LMCD1 expression in HASMCs. Blocking of Par1 by its antagonist SCH79797 or downregulation of Gαq/11 or PLCβ3 levels using their respective siRNA suppressed thrombin-induced LMCD1 expression as well as its promoter activity (Figure 3B–D & F-H). These observations imply that Par1-Gαq/11-PLCβ3 act upstream to NFATc1 in thrombin-induced LMCD1 expression in HASMCs.
Thrombin-induced proliferation and migration of HASMCs and MASMCs require CDC6 and IL-33 expression, respectively, but are differentially regulated by LMCD1
To further explore the species-specific role of LMCD1 in VSMC proliferation and migration, we investigated role of CDC6 and IL-33 because our recent findings showed the involvement of CDC6 and IL-33 in thrombin-induced HASMC proliferation and migration, respectively [13, 14]. Towards this end, we first studied a time course effect of thrombin on CDC6 and IL-33 expression in HASMCs and MASMCs. In line with our previous findings [13, 14], thrombin induced both CDC6 and IL-33 expression in a time dependent manner in HASMCs (Figure 4A & C). Similarly, thrombin induced both CDC6 and IL-33 expression in a time dependent manner in MASMCs as well (Figure 4B & D). Furthermore, while depletion of CDC6 blunted thrombin-induced proliferation of HASMCs and MASMCs (Figure 4E & F), neutralization of IL-33 using its neutralizing antibody (IL-33 nAb) inhibited thrombin-induced migration of both HASMCs and MASMCs (Figure 4G & H). The role of CDC6 and IL-33 in thrombin-induced HASMC proliferation and migration, respectively, were similar to our previous observations [13, 14]. Together, these results suggest that thrombin-induced proliferation and migration of both HASMCs and MASMCs require CDC6 and IL-33 expression, respectively. Then, to explore the mechanisms by which LMCD1 differentially mediates thrombin-induced HASMC and MASMC proliferation and migration, we studied its role in thrombin-induced CDC6 and IL-33 expression. Interestingly, siRNA-mediated depletion of LMCD1 while diminishing thrombin-induced CDC6 and IL-33 expression in HASMCs, enhanced their expression in MASMCs (Figure 4I–L). These results indicate species-specific role of LMCD1 in the expression of CDC6 and IL-33.
Figure 4. While CDC6 and IL-33 expression are required for both HASMC and MASMC proliferation and migration, respectively, LMCD1 exhibits differential effects on their expression between human and mice.
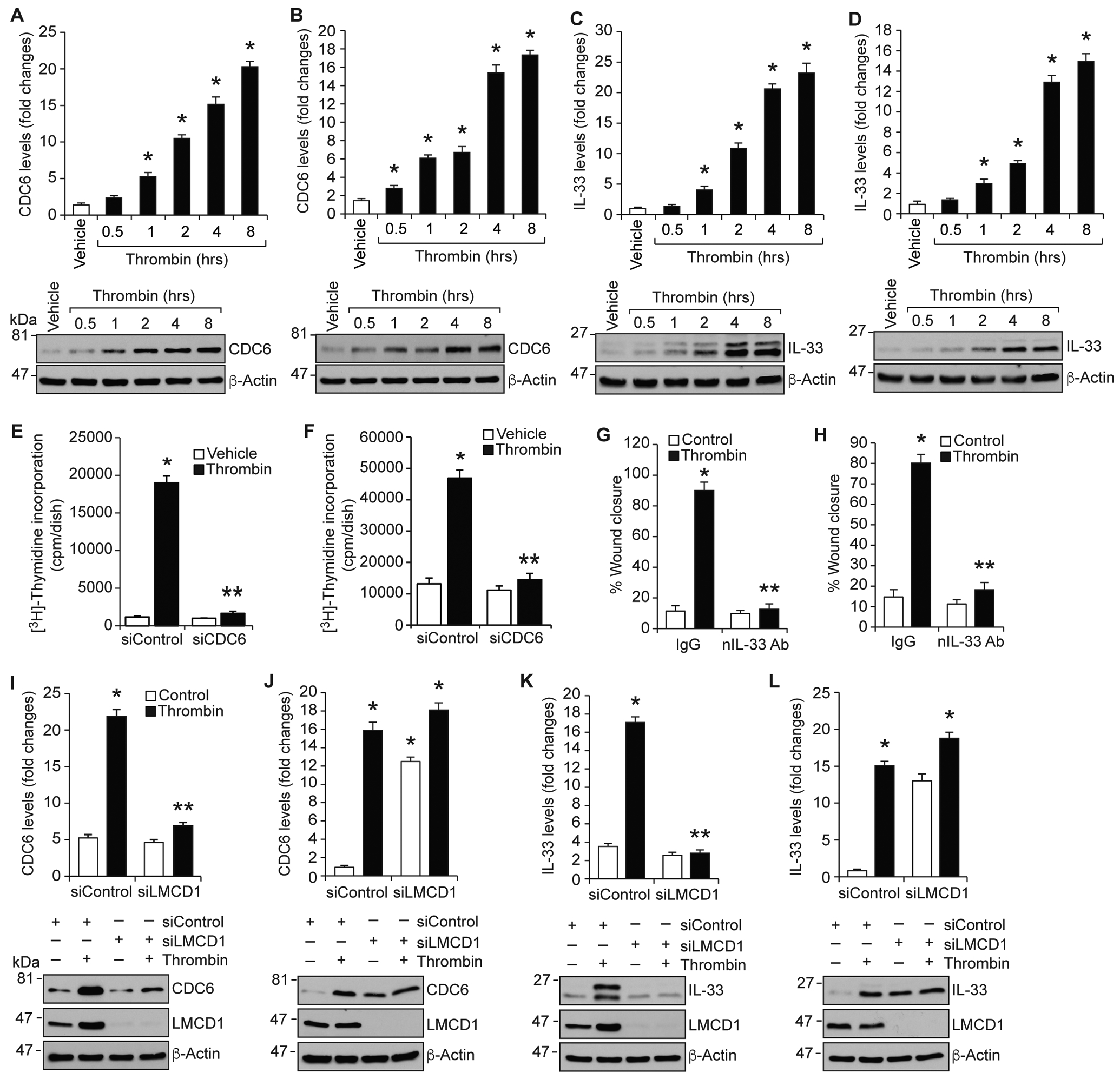
A-D. Quiescent HASMCs (A & C) or MASMCs (B & D) were treated with and without thrombin (0.5 u/ml) for the indicated time periods, cell extracts were prepared and analyzed by Western blotting for CDC6, IL-33 and β-actin levels using their specific antibodies. E-H. HASMCs (E & G) or MASMCs (F & H) that were transfected with siControl or siCDC6 (100 nM) or preincubated with IgG or neutralizing IL-33 antibody (100 ng/ml) were quiesced and tested for thrombin (0.5 U/ml)-induced DNA synthesis (E & F) or migration (G & H). HASMCs (I & K) or MASMCs (J & L) that were transfected with siControl or siLMCD1 (100 nM) and quiesced were treated with and without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and analyzed by Western blotting for CDC6, IL-33 and β-actin levels using their specific antibodies. The same membranes were also reprobed for LMCD1 levels to show its siRNA efficacy. The bar graphs represent the Mean ± S.D. values of three independent experiments. *, p < 0.05 versus siControl + vehicle; **, p < 0.05 versus siControl + Thrombin.
LMCD1 acts as an activator in human and a repressor in mouse of CDC6 and IL-33 promoters
To further elucidate species-specific role of LMCD1 in the expression of CDC6 and IL-33 in humans versus mice, we overexpressed human LMCD1 along with human CDC6 or IL-33 promoters in HASMCs and MASMCs and examined the promoter activities. Overexpression of LMCD1 while enhancing basal as well as thrombin-induced CDC6 and IL-33 promoter activities in HASMCs, completely inhibited their promoter activities in MASMCs (Figure 5A & B). These findings clearly indicate species-specific functional discrepancy of LMCD1 between human and mice in the expression of CDC6 and IL-33. As LMCD1 plays a role in thrombin-induced CDC6 and IL-33 promoter activities by modulating their DNA binding activity with trans-acting elements i.e., E2F1 in the case of CDC6 promoter and E2F1 and NFATc1 in the case of IL-33 promoter in HASMCs [13, 14], we asked whether LMCD1 has any role in thrombin-induced E2F1 or NFATc1-binding activities in MASMCs. To address this point, we performed super-shift EMSA using E2F (−43 nt) and NFAT (−100 nt) binding elements of human CDC6 and IL-33 promoters, respectively, as biotin-labeled double-stranded oligonucleotide probes and anti-LMCD1, anti-E2F1 and anti-NFATc1 antibodies both in HASMCs and MASMCs. Our results showed that in response to thrombin while the binding of both LMCD1 and E2F1 to E2F-binding site of CDC6 promoter increased in HASMCs, only E2F1 but not LMCD1 binding to E2F-binding site of CDC6 promoter was increased in MASMCs (Figure 5C & D). In fact, LMCD1 binding to E2F binding site of CDC6 promoter was decreased in MASCMs in response to thrombin as compared to control. Similarly, in response to thrombin, while the binding of LMCD1, E2F1 and NFATc1 to NFAT-binding site of IL-33 promoter was increased in HASMCs, only E2F1 and NFATc1 binding to NFAT-binding site of IL-33 promoter was increased in MASMCs (Figure 5E–G). LMCD1 binding to NFAT-binding site of IL-33 promoter was decreased in MASMCs in response to thrombin as compared to control. These findings were further supported by ChIP assay (Figure I in the online-only data supplement). These observations infer that in response to thrombin LMCD1 while associates with E2F1 or E2F1 and NFATc1 in binding to CDC6 and IL-33 promoters, respectively, in HASMCs, it dissociates from these transcriptional factors in binding to CDC6 and IL-33 promoters in MASMCs. Besides, our co-IP results also showed that while thrombin induces the association of LMCD1 with both E2F1 and NFATc1 in HASMCs, it dissociates from E2F1 and NFATc1 in MASMCs (Figure 5H & I). Based on these observations, it may be suggested that LMCD1 mediates thrombin-induced CDC6 and IL-33 expression by associating with E2F1 and NFATc1, respectively, in HASMCs. In contrast, LMCD1 mediates thrombin-induced CDC6 and IL-33 expression by its dissociation from E2F1 and NFATc1, respectively, in MASMCs.
Figure 5. LMCD1 acts as an activator in HASMCs and repressor in MASMCs of CDC6 and IL-33 promoter activities.
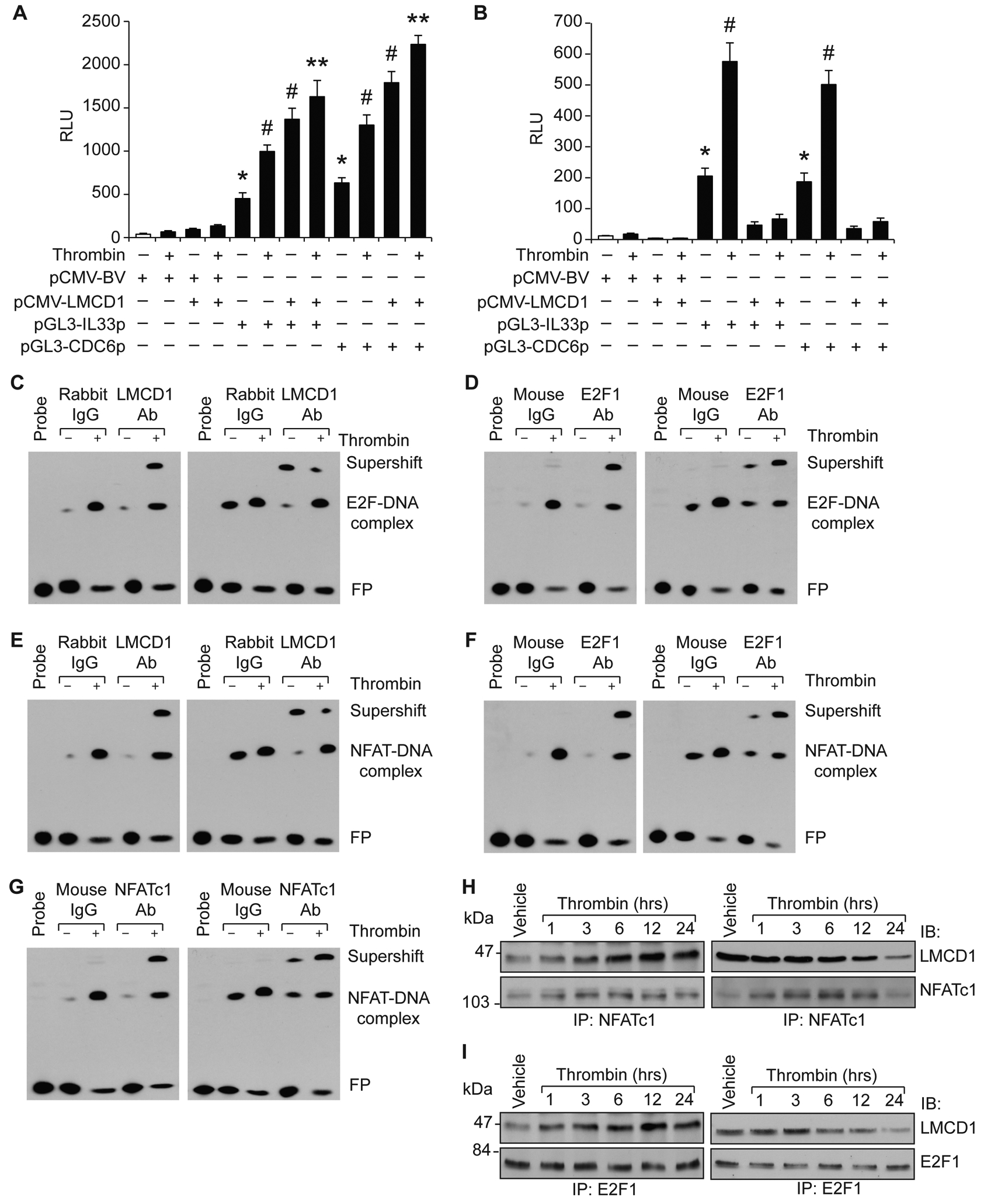
A & B. HASMCs (A) or MASMCs (B) were transfected with pCMV-basic or pCMV-LMCD1 expression vector alone or in combination with pGL3-hIL33p or pGL3-hCDC6p constructs and 36 hrs later cells were treated with and without thrombin (0.5 U/ml) for 8 hrs and luciferase activity was measured. C-G. Nuclear extract of control and 8 hrs of thrombin (0.5 U/ml)-treated HASMCs (left) or MASMCs (right) were analyzed by supershift EMSA for the presence of the indicated proteins in the protein-DNA complexes of CDC6 (C & D) or IL-33 (E-G) promoters using biotin-labeled E2F and NFAT-binding site DNA probes. H & I. Quiescent HASMCs (left) or MASMCs (right) were treated with and without thrombin (0.5 U/ml) for the indicated time periods, cell extracts were prepared and immunoprecipitated with anti-NFATc1 or E2F1 antibodies and the immunocomplexes were analyzed by Western blotting for LMCD1 using its specific antibody and the blots were reprobed for NFATc1 or E2F1. The bar graphs represent the Mean ± S.D. values of three independent experiments. *, p < 0.05 versus pCMV-basic vector; #, p < 0.05 versus pGL3-hIL-33p or pGL3-hCDC6p; **, p < 0.05 versus pGL3-hIL-33p + thrombin or pGL3-hCDC6p + thrombin.
Myristoylation of LMCD1 leads to its derepression of E2F1 and NFATc1 in the regulation of CDC6 and IL-33 expression in MASMCs
To explore the mechanisms involved in the repression of CDC6 and IL-33 promoter activities in MASMCs by LMCD1, we first analyzed human and mouse LMCD1 protein sequences for potential posttranslational modification sites. Both human and mouse LMCD1 contain 8 protein kinase C phosphorylation sites, 7 casein kinase II phosphorylation sites, 6 N-myristoylation sites, and one each for tyrosine phosphorylation, tyrosine sulfation and N-glycosylation sites, although with considerable sequence variations (Figure 6A). Based on this bioinformatic analysis, we have studied the role of its posttranslational modifications in its repressor activity on E2F1 and NFATc1 in CDC6 and IL-33 expression, respectively, in MASMCs. LMCD1 was found to be a phosphoprotein, but however, there were no apparent changes in its phosphorylation state at both Ser/Thr or Tyr residues between control and thrombin-treated cells (Figure 6B). Interestingly, we found that LMCD1 was myristylated in response to thrombin only in MASMCs but not HAMSCs (Figure 6C & D). In addition, inhibition of myristoylation prevented thrombin-induced LMCD1 myristoylation and its dissociation from both E2F1 and NFATc1 (Figure 6E–G). Inhibition of myristoylation also suppressed thrombin-induced dissociation of LMCD1 from both E2F-DNA and NFAT-DNA complexes of CDC6 and IL-33 promoters, respectively (Figure 6H & I and Figure II in the online-only data supplement). Consistent with these observations, inhibition of myristoylation also blocked thrombin-induced CDC6 and IL-33 promoter activity and their expression in MASMCs (Figure 6J–L).
Figure 6. N-Myristoylation of LMCD1 leads to its species-specific derepression of E2F1 and NFATc1 in the regulation of CDC6 and IL-33 expression in MASMCs.
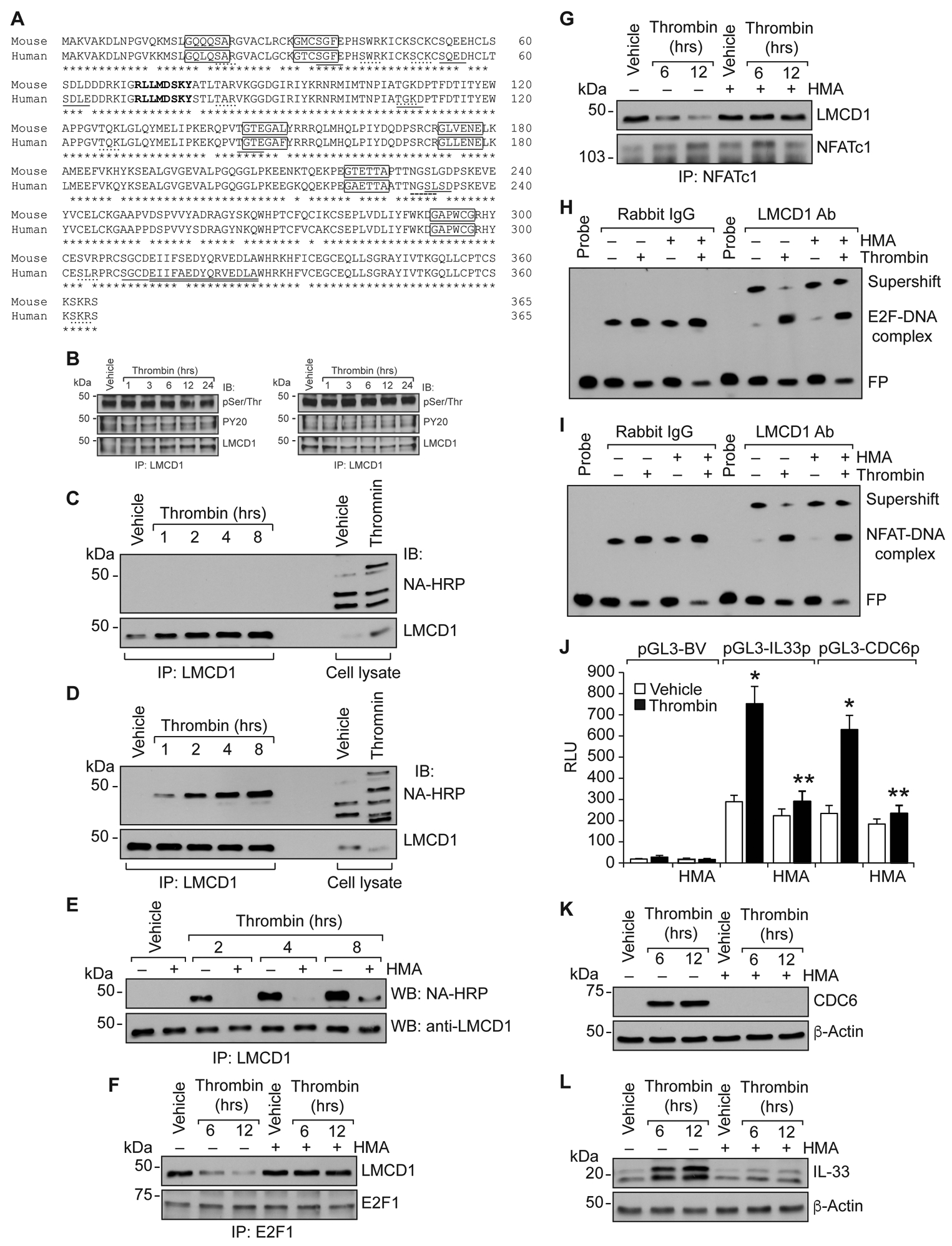
A. Alignment of mouse and human LMCD1 protein sequence with known functional sites: Boxed text, N-myristoylation sites; dotted underlined text, protein kinase c phosphorylation sites; underlined text, casein kinase II phosphorylation sites; bold text, tyrosine kinase phosphorylation site; double underlined text, tyrosine sulfation site; dashed underlined text, N-glycosylation site. B. Cell extracts of control and the indicated treatments of HASMCs (left panel) and MASMCs (right panel) were immunoprecipitated with anti-LMCD1 antibodies and the immunocomplexes were analyzed by Western blotting using pSer/Thr, PY20 or LMCD1 antibodies. C & D. Quiescent HASMCs (C) or MASMCs (D) were metabolically labeled with azidomyristate (40 μM) and treated with and without thrombin (0.5 U/mL) for the indicated time periods. Cell extracts were prepared, immunoprecipitated with anti-LMCD1 antibody, immunocomplexes were eluted with phosphine reaction buffer, conjugated to phosphine-biotine (250 μM), analyzed by Western blotting using neutravidin-HRP and normalized to LMCD1 levels. E-G. All the conditions were same as in D except that MASMCs were pre-treated with and without 2-hydroxy myristic acid before thrombin treatment. H & I. Nuclear extract of MASMCs with indicated treatments were analyzed by supershift EMSA for the presence of LMCD1 in protein-DNA complexes of CDC6 (H) or IL-33 (I) promoters using specific biotin-labeled probes. J. MASMCs were transfected with pGL3-basic or pGL3-hIL33p or pGL3-hCDC6p vectors and 36 h later cells were quiesced, treated with and without 2-hydroxy myristic acid for 1 h then with and without thrombin (0.5 U/mL) for 8 h, and the luciferase activity was measured. K & L. Quiescent MASMCs were treated with and without 2-hydroxy myristic acid for 1 h then with and without thrombin (0.5 U/mL) for indicated time periods. Cell extracts were prepared and analyzed by Western blotting using specific antibodies and normalized to β-actin levels. The bar graphs represent the mean ±S.D. values of three independent experiments. *, p < 0.05 versus pGL3-basic vector; **, p < 0.05 versus pGL3-hIL33p or pGL3-hCDC6p. HMA, hydroxy myristic acid; NA-HRP, neutravidin-HRP.
N-Myristoyl transferase 2 mediates thrombin-induced myristoylation of LMCD1 in MASMCs
To identify thrombin responsive myristoylation site(s) in mouse LMCD1, we first analyzed all the six myristoylation sites of mouse LMCD1 using myristoylator [21] and found high confidence score only for the proximal N-terminus myristoylation site (GQQQSA). Based on this bioinformatics analysis, we mutated glycine (GGC) of this myristoylation site to proline (CCC) by site-directed mutagenesis to generate mutant LMCD1 (PQQQSA, LMCD1-mt) expression vector (Figure III in the online-only data supplement) and tested its effect on thrombin-induced myristoylation of LMCD1. Expression of mutant LMCD1 negated thrombin-induced myristoylation of LMCD1 in MSAMCs as compared to wild type LMCD1 (Figure 7A). Based on these observations, we next studied its effect on LMCD1 binding to CDC6 and IL-33 promoters using supershift EMSA and ChIP assays. The EMSA and ChIP results show that forced expression of LMCD1-mt also blunted thrombin-induced dissociation of LMCD1 from both E2F-DNA and NFAT-DNA complexes of CDC6 and IL-33 promoters, respectively (Figure 7B–E). Consistent with these observations, over expression of LMCD1-mt inhibited thrombin-induced CDC6 and IL-33 promoter activities as well as their expression in MASMCs (Figure 7F–H). In vertebrates, N-myristoylation is catalyzed by two isoenzymes, namely, N-myristoyltransferase 1 (NMT1) and N-myristoyltransferase 2 (NMT2) [22]. Therefore, to explore the mechanisms of LMCD1 myristoylation, NMT1 and NMT2 levels were depleted by their respective siRNAs and tested their effects on thrombin-induced LMCD1 myristoylation. Depletion of NMT2 but not NMT1 substantially blunted thrombin-induced LMCD1 myristoylation in MASMCs (Figure 7I–L). Furthermore, downregulation of NMT2 also suppressed thrombin-induced CDC6 and IL-33 expression in MASMCs (Figure 7M & N).
Figure 7. N-Myristoyl transferase 2 mediates thrombin-induced myristoylation and derepression of LMCD1 in the regulation of E2F1-mediated CDC6 and NFATc1-mediated IL-33 expression in MASMCs.
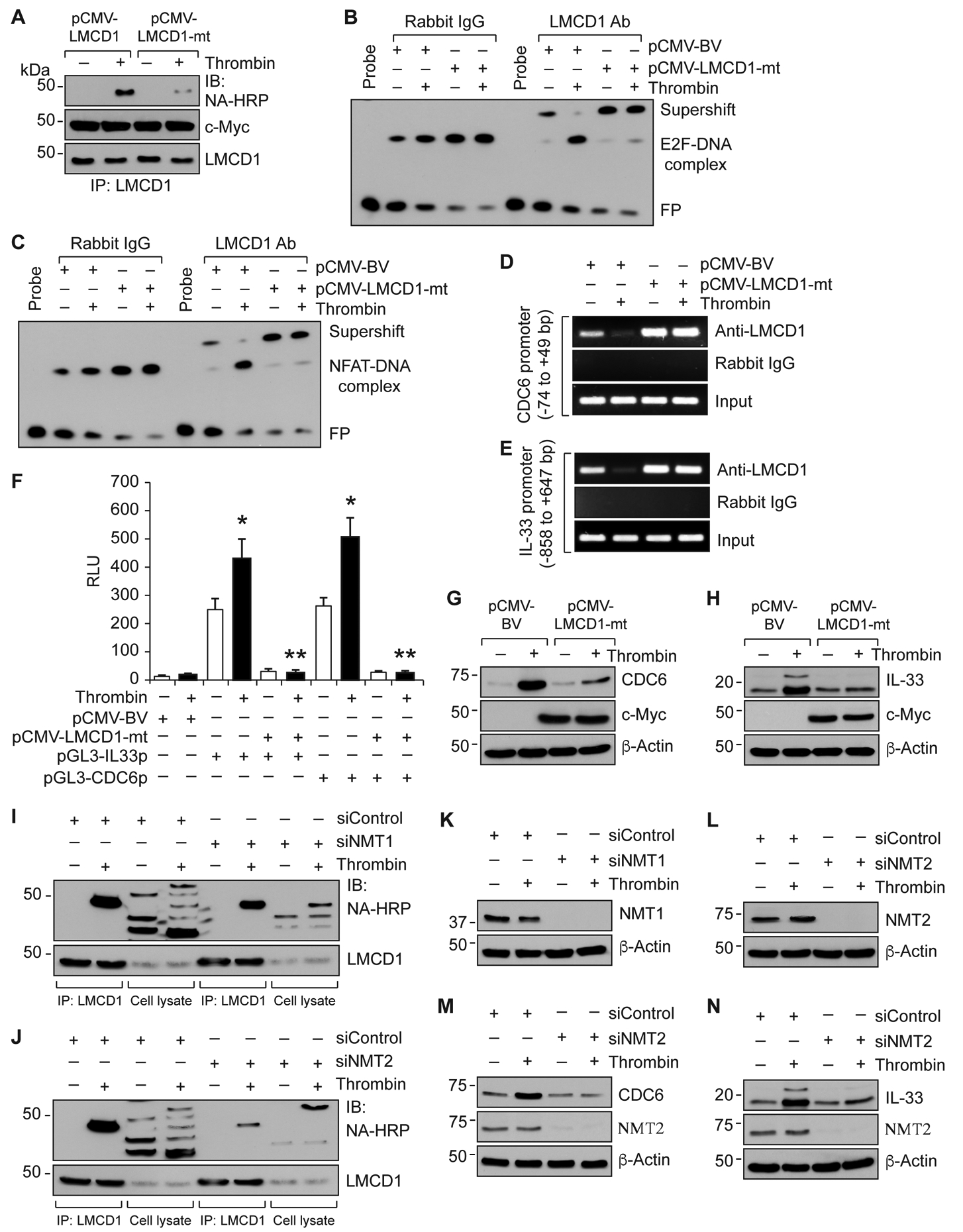
A. MASMCs were transfected with pCMV-LMCD1 or pCMV-LMCD1-mt vectors and 36 hrs later cells were quiesced, metabolically labeled with azidomyristate (40 μM) for 3 hrs and treated with and without thrombin (0.5 U/ml) for 8 hrs. Cell extracts were prepared, immunoprecipitated with anti-LMCD1 antibody, immunocomplexes were eluted with phosphine reaction buffer, conjugated to phosphine-biotine (250 μM) and analyzed by Western blotting using neutravidin-HRP and normalized to LMCD1 levels. B & C. MASMCs that were transfected with the indicated vectors and treated with and without thrombin (0.5 U/ml) for 8 hrs were analyzed by supershift EMSA for the presence of LMCD1 in the protein-DNA complexes of CDC6 (B) or IL-33 (C) promoters using their specific biotin-labeled probes. D & E. All the conditions were the same as in panels B & C except that cells were subjected to ChIP assay for CDC6 promoter region encompassing E2F site (D) or IL-33 promoter region encompassing NFAT site (E) using anti-LMCD1 antibodies. F. MASMCs were transfected with pCMV-basic or pCMV-LMCD1-mt vector in combination with pGL3-hIL33p or pGL3-hCDC6p and 36 hrs later cells were quiesced, treated with and without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and analyzed for luciferase activity. G & H. MASMCs that were transfected with pCMV-basic or pCMV-LMCD1-mt vectors were quiesced, treated with or without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and analyzed by Western blotting for CDC6 and IL-33 levels using their specific antibodies. The same membranes were reprobed for β-Actin for normalization. I-L. MASMCs were transfected with the indicated siRNA (100 nM) and 36 hrs later cells were quiesced, metabolically labeled with azidomyristate (40 μM) for 3 hrs and treated with and without thrombin (0.5 U/ml) for 8 hrs. Cell extracts were prepared, immunoprecipitated with anti-LMCD1 antibody, immunocomplexes were eluted with phosphine reaction buffer, conjugated to phosphine-biotine (250 μM) and analyzed by Western blotting using neutravidin-HRP and normalized to LMCD1 levels (I & J). Cell extracts were also analyzed for siRNA efficacy (K & L). M & N. MASMCs that were transfected with siNMT2 (100 nM) were quiesced, treated with or without thrombin (0.5 U/ml) for 8 hrs, cell extracts were prepared and analyzed by Western blotting for CDC6 and IL-33 levels. The membranes were reprobed for NMT2 and β-Actin. The bar graphs represent Mean ± S.D. values of three independent experiments. *, p < 0.05 versus pGL3-basic vector; **, p < 0.05 versus pGL3-hCDC6p or pGL3-hIL33p. NA-HRP, neutravidin-HRP; NMT1, N-Myristoyltransferase 1; NMT2, N-Myristoyltransferase 2.
Differential role of LMCD1 in vascular wall remodeling in human versus mice
VSMC proliferation and migration is a common phenomenon in restenosis and early development of atherosclerosis [23–25]. As our previous reports showed increased expression of LMCD1 in human atherosclerotic coronary artery along with increased CDC6 and IL-33 levels [13, 14], and because the present in vitro data shows species-specific regulation of LMCD1 in mediating VSMC proliferation and migration, we asked whether LMCD1 has any species-specific functional discrepancy in vascular wall remodeling between human and mice. To test this possibility, we examined the expression of LMCD1 in human stenotic and mouse neointimal lesions. It is indeed exciting to find that LMCD1 expression is increased in SMC-rich plaques in human stenotic arteries as compared to non-stenotic arteries (Figure 8A). On the other hand, LMCD1 expression was decreased in neointimal lesions of injured mouse femoral arteries as compared to uninjured arteries (Figure 8B). Furthermore, Western blot analysis showed that while CDC6 and IL-33 expressions were increased, LMCD1 expression was decreased in injured mouse femoral arteries as compared to uninjured arteries (Figure 8C). These results clearly demonstrate species-specific role of LMCD1 in the development of vascular lesions in human versus mice.
Figure 8. Differential expression of LMCD1 in human and mouse neointimal hyperplasia.
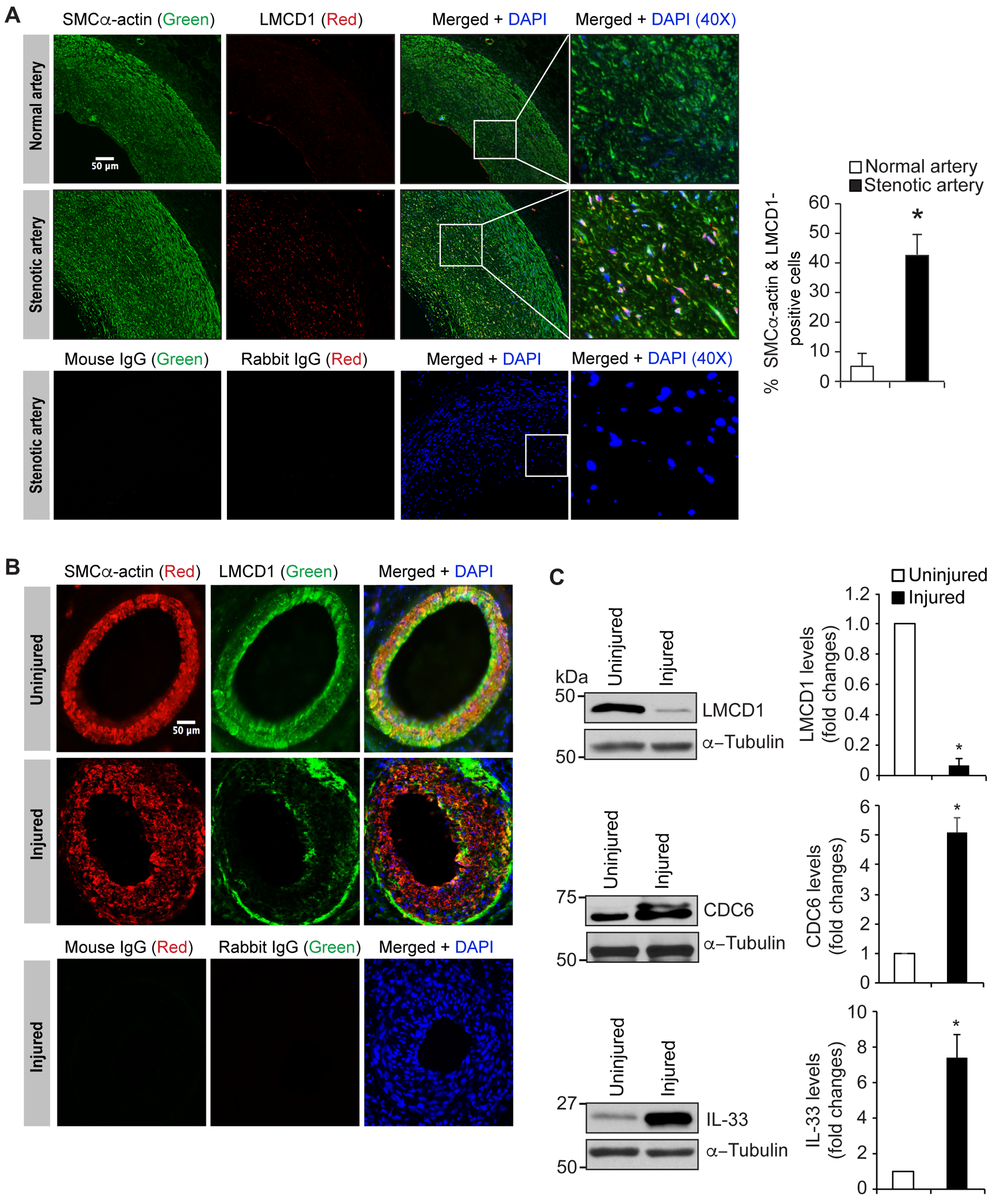
A. Human stenotic and non-stenotic artery sections were stained for SMCα-actin (green) and LMCD1 (red). The sections were also stained with normal mouse or rabbit IgG as negative controls. B. Uninjured and 3 weeks of post guidewire-injured mouse femoral artery cross-sections were stained for SMCα-actin (red) and LMCD1 (green). C. Tissue extracts prepared from uninjured and guide wire-injured mouse femoral arteries were analyzed by Western blotting for LMCD1, CDC6 and IL-33 levels using their specific antibodies and normalized to α-tubulin levels. The bar graphs represent Mean ± S.D. values of six animals (two pooled arteries for each Western blotting analysis). *, p < 0.05 versus uninjured. Scale bar is 50 μm.
DISCUSSION
Increased VSMC migration and proliferation play a key role in the pathogenesis of restenosis and atherosclerosis [23–25]. Thrombin, a procoagulant and a proinflamatory serine protease, is one of the most potent mitogen and chemotactic factors that stimulate the migration and proliferation of VSMCs by altering the expression of a vast number of genes [26]. Recently, we have demonstrated that LMCD1 plays a role in HASMC growth and migration with increased expression in SMC of human atherosclerotic lesions [13, 14]. In addition CDC6 and IL-33 that were regulated by LMCD1 were also involved in injury-induced neointimal development in a mouse model. However, while LMCD1 expression was required for HASMC multiplication it was not essential for MASMC proliferation [13]. Human LMCD1 was mapped to 3p26-p24 whereas mouse LMCD1 was mapped to the central region of chromosome 6 [2]. Furthermore, LMCD1 gene has high degree of evolutionary conservation across species [27]. This information may suggest that a potential mechanism behind its species-specific expression by thrombin in human versus mice could be related to its rapid promoter evolution. Accumulating evidence suggests that the divergent expression of conserved genes could be due to consequence of the evolution of the cis-acting regulatory elements of their promoters rather than specific features of cell culture systems [28–30]. In line with this view, by using TRANSFAC software [20], we observed significant differences in the number and location of the transcription factor binding sites between human and mouse LMCD1 promoters. Mainly, human LMCD1 promoter contains a potential thrombin-responsive NFAT element at −485 nt, which is completely absent in mouse LMCD1 promoter. Site-directed mutagenesis of this site blunted thrombin-induced LMCD1 promoter activity in HASMCs. Consistent with this observation, we found that thrombin induces LMCD1 expression in HASMCs but not MASMCs. More interestingly, our results revealed a significant role of LMCD1 in thrombin-induced proliferation and migration of HASMCs but not MASMCs. As our recent studies have revealed a role for CDC6 and IL-33 in thrombin-induced HASMC proliferation and migration, respectively [13, 14], we tested the role of LMCD1 in thrombin induced CDC6 and IL-33 expression in HASMCs and MASMCs. Depletion of LMCD1 levels blunted thrombin-induced CDC6 and IL-33 expression in HASMCs, but surprisingly enhanced their expression in MASMCs. In addition, LMCD1 over-expression significantly induced CDC6 and IL-33 promoter activity in HASMCs, while it diminished them in MASMCs. All these results infer species-specific functional divergence of LMCD1 between human and mice. Moreover, our super-shift EMSA, ChIP and co-IP results showed an elegant mechanism linking LMCD1 species-specificity between human and mice. In response to thrombin, LMCD1 while formed a complex with E2F1 and NFATc1 in HASMCs, it was found to be dissociated from these transcriptional factors in MASMCs. E2F1 and/or NFATc1 are essential modulators of thrombin-induced CDC6 and IL-33 expression mediating VSMC proliferation and migration, respectively [13, 14]. These results infer that LMCD1 acts as an activator in human and as a repressor in mouse of CDC6 and IL-33 promoters in mediating thrombin-induced VSMC proliferation and migration. It is interesting to note that LMCD1 is myristoylated by thrombin in the regulation of E2F1 and NFATc1 in the modulation of CDC6 and IL-33 expression mediating VSMC growth and migration in mice. Although many studies have demonstrated a role for myristoylation in protein trafficking to plasma membrane and cellular signaling [31–33], its role in the regulation of transcriptional factor coactivator/repressor activity is not known. In this aspect, the present findings for the first time show that LMCD1, by myristoylation dissociates from E2F1 and NFATc1 and thus alleviates its repression of these transcriptional factors in the regulation of CDC6 and IL-33 expression in mediating VSMC proliferation and migration in mouse. Our findings also identify that myristoylation of LMCD1 at a proximal N-terminus region, GQQQSA (amino acids 18–23) is required for its dissociation from CDC6 and IL-33 promoters in enhancing their expression in response to thrombin. In understanding the mechanisms, our findings further show that, of the two NMTs expressed in vertebrates, NMT2 mediates thrombin-induced myristoylation of LMCD1 in the regulation of both CDC6 and IL-33 expressions in mediating MASMC proliferation and migration.
In view of the present in vitro data showing species-specific regulation of LMCD1 in VSMC proliferation and migration, and our previous findings that showed increased expression of LMCD1 in human atherosclerotic coronary artery along with increased CDC6 and IL-33 expressions [13, 14], we have tested the role of LMCD1 in the development of neointimal lesions. Our results showed that while LMCD1 expression was increased in SMC-rich plaques in stenotic human arteries as compared to non-stenotic arteries, its expression was decreased in injured mouse femoral arteries as compared to uninjured arteries. Although LMCD1 levels were decreased, CDC6 and IL-33 levels were however increased in injured mouse femoral arteries as compared to uninjured arteries. These observations are consistent with our previous reports showing decreased levels of LMCD1 in rat common carotid artery in response to ligation and mouse carotid artery in response to balloon injury [13]. The differential expression of LMCD1 in human stenotic artery as compared to neointimal lesions of injured mouse femoral artery (present study) clearly demonstrate the species-specific regulation of LMCD1 in human versus rodents during vascular wall remodeling. However, although its expression levels were differentially regulated between human and mice, LMCD1 by acting as an activator in human and as a repressor in mice in the modulation of E2F1 and NFATc1 led to increased expression of CDC6 and IL-33 in influencing VSMC proliferation and migration, respectively, during the development of vascular lesions in both the species. Since NMT2 is mediating LMCD1 myristoylation in the regulation E2F1 and NFATc1 and thereby their target genes, CDC6 and IL-33, respectively, it is likely NMT2 may also play a role in mouse vascular wall remodeling. It is noteworthy that although thrombin does not affect the steady state levels of LMCD1 in RASMCs or MASMCs in vitro significantly, its levels were decreased substantially in both rat and mouse carotid arteries or mouse femoral artery in response to injury. These findings may suggest that besides thrombin, other players might also be involved in the downregulation of LMCD1 in mediating vascular wall remodeling in rat and mouse in vivo.
Supplementary Material
HIGHLIGHTS.
LMCD1 by acting as an activator of E2F1 and NFATc1 mediates thrombin-induced CDC6 and IL-33 expression leading to increased proliferation and migration of HASMCs.
On the other hand, LMCD1 acts as a repressor of these transcriptional factors in mouse, that is abolished by its NMT2-mediated myristoylation in thrombin-induced CDC6 and IL-33 expression promoting proliferation and migration of MASMCs.
In line with its specific-specific activator and repressor role in human versus mouse in E2F1-mediated CDC6 expression and NFATc1-mediated IL-33 expression, LMCD1 expression was increased in human stenotic arterial lesions and decreased in mouse neointimal lesions.
Due to its activator role of E2F1 and NFTAc1 in the regulation of CDC6 and IL-33 in humans, LMCD1 could be a potential therapeutic target for human vascular diseases.
ACKNOWLEDGMENTS
SG performed RT-PCR, Western blotting, pulldown assays, myristoylation, EMSA, ChIP, promoter luciferase activities, cell migration, DNA synthesis, cloning, site-directed mutagenesis and wrote the article; PP performed guidewire injury, immunostaining and Western blotting; JJ performed cloning and site-directed mutagenesis; LG performed immunofluorescence staining; RV provided critical reagents and edited manuscript; GNR provided overall experimental design, interpreted data and edited the article. All authors have reviewed and approved the manuscript.
SOURCES OF FUNDING
This work was supported by a grant HL069908 from NIH to GNR.
Nonstandard Abbreviations and Acronyms
- CDC6
Cell division cycle 6
- E2F1
E2F transcription factor 1
- EMSA
Electrophoretic mobility shift assay
- Gαq/11
Gα protein q/11
- HASMC
Human aortic smooth muscle cell
- HMA
Hydroxy myristic acid
- IL-33
Interleukin-33
- LMCD1
LIM and cysteine-rich domains 1
- MASMC
Mouse aortic smooth muscle cell
- NA-HRP
Neutravidin-HRP
- NFATc1
Nuclear factor of activated T cells 1
- NMT
N-Myristoyltransferase
- Par1
Protease-activated receptor 1
- PLCβ3
Phospholipase Cβ3
- VSMC
Vascular smooth muscle cells
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931. [DOI] [PubMed] [Google Scholar]
- 2.Bespalova IN, Burmeister M. Identification of a novel LIM domain gene, LMCD1, and chromosomal localization in human and mouse. Genomics. 2000;63(1):69–74. [DOI] [PubMed] [Google Scholar]
- 3.Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wakil SM, Ram R, Muiya NP, Andres E, Mazhar N, Hagos S, Alshahid M, Meyer BF, Morahan G, Dzimiri N. A common variant association study reveals novel susceptibility loci for low HDL-cholesterol levels in ethnic Arabs. Clin Genet. 2016;90:518–525. [DOI] [PubMed] [Google Scholar]
- 5.Chang CY, Lin SC, Su WH, Ho CM, Jou YS. Somatic LMCD1 mutations promoted cell migration and tumor metastasis in hepatocellular carcinoma. Oncogene. 2012;31:2640–2652. [DOI] [PubMed] [Google Scholar]
- 6.Du L, Yamamoto S, Burnette BL, Huang D, Gao K, Jamshidi N, Kuo MD. Transcriptome profiling reveals novel gene expression signatures and regulating transcription factors of TGFβ-induced epithelial-to-mesenchymal transition. Cancer Med. 2016;5:1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dmitriev AA, Rosenberg EE, Krasnov GS, Gerashchenko GV, Gordiyuk VV, Pavlova TV, et al. Identification of Novel Epigenetic Markers of Prostate Cancer by NotI-Microarray Analysis. Dis Markers. 2015;2015:241301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vozianov SO, Kashuba VI, Grygorenko VM, Gordiyuk VV, Danylets RO, Bondarenko YM, Vikarchuk MV. Identification of a new diagnostic markers of prostatic cancer, using noti-microchips. Klin Khir. 2016;4:54–57. [PubMed] [Google Scholar]
- 9.Muhammad SA, Fatima N, Paracha RZ, Ali A, Chen JY. A systematic simulation-based meta-analytical framework for prediction of physiological biomarkers in alopecia. J Biol Res (Thessalon). 2019;26:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bian ZY, Huang H, Jiang H, Shen DF, Yan L, Zhu LH, Wang L, Cao F, Liu C, Tang QZ, Li H. LIM and cysteine-rich domains 1 regulates cardiac hypertrophy by targeting calcineurin/nuclear factor of activated T cells signaling. Hypertension 2010;55:257–263. [DOI] [PubMed] [Google Scholar]
- 11.Frank D, Frauen R, Hanselmann C, Kuhn C, Will R, Gantenberg J, Füzesi L, Katus HA, Frey N. Lmcd1/Dyxin, a novel Z-disc associated LIM protein, mediates cardiac hypertrophy in vitro and in vivo. J Mol Cell Cardiol. 2010;49:673–682. [DOI] [PubMed] [Google Scholar]
- 12.Rath N, Wang Z, Lu MM, Morrisey EE. LMCD1/Dyxinis a novel transcriptional cofactor that restricts GATA6 function by inhibiting DNA binding. Mol Cell Biol. 2005;25:8864–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janjanam J, Zhang B, Mani AM, Singh NK, Traylor JG Jr, Orr AW, Rao GN. LIM and cysteine-rich domains 1 is required for thrombin induced smooth muscle cell proliferation and promotes atherogenesis. J Biol Chem. 2018;293:3088–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Govatati S, Pichavaram P, Janjanam J, Zhang B, Singh NK, Mani AM, Traylor JG Jr, Orr AW, Rao GN. NFATc1-E2F1-LMCD1-Mediated IL-33 Expression by Thrombin Is Required for Injury-Induced Neointima Formation. Arterioscler Thromb Vasc Biol. 2019;39:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kundumani-Sridharan V, Van Quyen D, Subramani J, Singh NK, Chin YE, Rao GN. Novel interactions between NFATc1 (Nuclear Factor of Activated T cells c1) and STAT-3 (Signal Transducer and Activator of Transcription-3) mediate G protein-coupled receptor agonist, thrombin-induced biphasic expression of cyclin D1, with first phase influencing cell migration and second phase directing cell proliferation. J Biol Chem. 2012;287:22463–22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin DD, Vilas GL, Prescher JA, Rajaiah G, Falck JR, Bertozzi CR, Berthiaume LG. Rapid detection, discovery, and identification of post-translationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. FASEB J. 2008;22:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kundumani-Sridharan V, Singh NK, Kumar S, Gadepalli R, Rao GN. Nuclear factor of activated T cells c1 mediates p21-activated kinase 1 activation in the modulation of chemokine-induced human aortic smooth muscle cell F-actin stress fiber formation, migration, and proliferation and injury-induced vascular wall remodeling. J Biol Chem. 2013;288:22150–22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bar-Shavit R, Kahn A, Fenton JW 2nd, Wilner GD. Chemotactic response of monocytes to thrombin. J Cell Biol. 1983;96:282–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McNamara CA, Sarembock IJ, Gimple LW, Fenton JW 2nd, Coughlin SR, Owens GK. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J Clin Invest. 1993;91:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases ontranscriptional regulation. TRANSFAC, TRRD, and COMPEL. Nucleic Acids Res. 1998;26:362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bologna G, Yvon C, Duvaud S, Veuthey AL. N-terminal Myristoylation Predictions by Ensembles of Neural Networks. Proteomics. 2004;4:1626–1632. [DOI] [PubMed] [Google Scholar]
- 22.Thinon E, Serwa RA, Broncel M, Brannigan JA, Brassat U, Wright MH, Heal WP, Wilkinson AJ, Mann DJ, Tate EW. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat Commun. 2014;5:4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doran AC, Meller N, McNamara CA. Role of Smooth Muscle Cells in the Initiation and Early Progression of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.llahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML.Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaberi N, Soleimani A, Pashirzad M, Abdeahad H, Mohammadi F, Khoshakhlagh M, Khazaei M, Ferns GA, Avan A, Hassanian SM. Role of thrombin in the pathogenesis of atherosclerosis. J Cell Biochem. 2019;120:4757–4765. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Deng CY, Xiong YZ, Zuo B, Xing L, Li FE, Lei MG, Zheng R, Jiang SW. cDNA cloning, sequence analysis of the porcine LIM and cysteine-rich domain 1 gene. Acta Biochim Biophys Sin (Shanghai). 2005;37:843–850. [DOI] [PubMed] [Google Scholar]
- 28.Young R, Bush SJ, Lefevre L, McCulloch MEB, Lisowski ZM, Muriuki C, Waddell LA, Sauter KA, Pridans C, Clark EL, Hume DA. Species-Specific Transcriptional Regulation of Genes Involved in Nitric Oxide Production and Arginine Metabolism in Macrophages. Immunohorizons. 2018;2:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Müller M, Krause SW, Rehli M. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem. 2003;278:21502–21509. [DOI] [PubMed] [Google Scholar]
- 30.Tjärnlund-Wolf A, Hultman K, Blomstrand F, Nilsson M, Medcalf RL, Jern C. Species-Specific Regulation of t-PA and PAI-1 Gene Expression in Human and Rat Astrocytes. Gene Regul Syst Bio. 2014;8:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Udenwobele DI, Su RC, Good SV, Ball TB, Varma Shrivastav S, Shrivastav A. Myristoylation: An Important Protein Modification in the Immune Response. Front Immunol. 2017;8:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverman L, Resh MD. Lysine residues form an integral component of a novel NH2-terminal membrane targeting motif for myristylated pp60v-src. J Cell Biol. 1992;119:415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bastidas AC, Deal MS, Steichen JM, Keshwani MM, Guo Y, Taylor SS. Role of N-terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J Mol Biol. 2012;422:215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.