

Abstract
Background
Pneumonia is a common acute lower respiratory infection in children and elders. Circular RNAs (circRNAs) have recently been uncovered to play important roles in pneumonia. However, the function and mechanism of circ_0038467 in pneumonia remain elusive.
Methods
Cell viability and apoptosis were determined using the Cell Counting Kit‐8 (CCK‐8) assay and flow cytometry, respectively. The levels of interleukin 6 (IL‐6), IL‐8 and IL‐1β were detected by enzyme‐linked immunosorbent assay (ELISA). Western blot analysis was performed to assess the expression of related proteins. Circ_0038467 was characterized by Ribonuclease R (RNase) digestion and subcellular localization assays. The levels of circ_0038467 and miR‐338‐3p were evaluated by quantitative real‐time polymerase chain reaction (qRT‐PCR). The direct interaction between circ_0038467 and miR‐338‐3p was validated by the dual‐luciferase reporter and RNA immunoprecipitation (RIP) assays.
Results
Our data indicated that lipopolysaccharide (LPS) induced an inflammatory injury in 16HBE cells by repressing cell viability and enhancing cell apoptosis and proinflammatory cytokines production. Circ_0038467 was upregulated and miR‐338‐3p was downregulated in LPS‐treated 16HBE cells. Circ_0038467 knockdown or miR‐338‐3p overexpression attenuated LPS‐induced 16HBE cell inflammatory injury. Moreover, circ_0038467 acted as a sponge of miR‐338‐3p in 16HBE cells. MiR‐338‐3p mediated the alleviated effect of circ_0038467 knockdown on LPS‐induced 16HBE cell inflammatory injury. Additionally, the Janus kinase/ signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathway was involved in the circ_0038467/miR‐338‐3p axis‐mediated regulation in LPS‐induced 16HBE cell inflammatory injury.
Conclusions
The current work had led to the identification of circ_0038467 knockdown that alleviated LPS‐induced inflammatory injury in 16HBE cells at least partly through sponging miR‐338‐3p and regulating JAK/STAT3 pathway, highlighting novel molecular targets for the treatment of pneumonia.
Keywords: 16HBE cells, circ_0038467, inflammatory injury, miR‐338‐3p, pneumonia
Introduction
Pneumonia is a common acute lower respiratory infection with high mortality and morbidity in children and elders.1 Despite great advances in preventive, diagnostic, and therapeutic agents, pneumonia remains a major cause of infection‐related death worldwide.2 In patients with severe pneumonia, treatment failure is relevant to excessive inflammation and worse outcomes.3 Hence, the elucidation of the regulatory mechanism of the inflammatory process is very important to identify more effective targets for pneumonia treatment.
Circular RNAs (circRNAs) are endogenous non‐coding RNAs, which are characterized by their covalently closed loop structures without 5′ caps or 3′ polyadenylated tails.4 Recent studies have demonstrated that circRNAs function as crucial regulators in the pathogenesis of human inflammatory diseases, including pneumonia.5, 6 For instance, Yang et al. reported that circRNA vacuolar ATPase assembly factor (circVMA21) protected WI‐38 cell from lipopolysaccharide (LPS)‐triggered inflammatory injury possibly by regulating microRNA (miRNA)‐142‐3p and nuclear factor‐κB (NF‐κB) signaling.7 Guo and colleagues revealed that the knockdown of circRNA ankyrin repeat domain 36 (circANKRD36) relieved LPS‐evoked cytotoxicity in MRC‐5 cells via sponging miRNA‐31 and regulating myeloid differentiation factor 88 (MyD88).8 As for circ_0038467, derived from the back‐splicing of ubiquinol‐cytochrome‐c reductase core protein 2 (UQCRC2), it was found to be significantly upregulated in neodymium oxide‐induced 16HBE cell line using the microarray circRNA analysis.9 In the current work, the function and mechanism of circ_0038467 in inflammatory injury were explored by using LPS‐induced 16HBE cells.
MiRNAs are small regulatory RNA molecules that serve as key players of inflammatory response in pneumonia.10, 11 Until now, many miRNAs, such as miR‐1247 and miR‐3941 have been reported to regulate LPS‐induced inflammatory damage in acute pneumonia.12, 13 Previous evidence described that miR‐338‐3p was involved in the inflammatory damage in the rat spinal cord.14 Additionally, miR‐338‐3p was reported to weaken TNF‐α‐induced lipogenesis in sebocytes.15 However, the role of miR‐338‐3p in pneumonia is still elusive. CircRNAs are proposed to regulate the abundance of available miRNAs through acting as molecular sponges, highlighting the importance of the regulatory networks in human diseases.16 A putative target sequence between circ_0038467 and miR‐338‐3p was predicted using the online database Circinteractome, which prompted us to examine miR‐338‐3p as a potential mediator of circ_0038467 in LPS‐induced inflammatory injury.
In the present study, we firstly established pneumonia in vitro model by LPS stimulation. Subsequently, we investigated the effect and mechanism of circ_0038467 in LPS‐induced inflammatory injury in 16HBE cells.
Methods
Cell culture and LPS treatment
16HBE cell line, an immortalized human bronchial epithelial cell line, was purchased from BeNa Culture Collection (Beijing, China) and grown in high glucose Dulbecco's Modified Eagle Medium (DMEM‐H, Gibco, Zug, Switzerland) plus 10% (FBS, Gibco) and 1% antibiotics (Solarbio, Beijing, China) at 37°C with a 5% CO2 atmosphere. When it reached approximately 70%–80% confluence, the cells were treated with various concentrations (0, 1, 5 and 10 μg/mL) or 5 μg/mL of LPS (Sigma‐Aldrich, Castle Hill, Australia) for 24 hours. Additionally, to observe the effect of the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway on LPS‐induced inflammatory injury, the cells were exposed to 10 μM of ruxolitinib (Sigma‐Aldrich), a JAK1/JAK2 inhibitor, for 24 hours.
Generation of knockdown cells and overexpression cells
For circ_0038467 knockdown studies, 16HBE cells were transiently transfected with small interfering RNA (siRNA) against circ_0038467 (30 nM, si‐circ#1, si‐circ#2 and si‐circ#3, GenePharma, Shanghai, China), and a scrambled oligonucleotide sequence (30 nM, si‐NC, GenePharma) were used as the negative control. For miR‐338‐3p overexpression studies, 16HBE cells were introduced with commercial miR‐338‐3p mimic (30 nM, GenePharma) or a nontarget negative control (30 nM, miR‐NC mimic, GenePharma). MiR‐338‐3p knockdown cells were produced using a commercial inhibitor of miR‐338‐3p (30 nM, anti‐miR‐338‐3p, GenePharma), with a matched scrambled sequence (30 nM, anti‐NC, GenePharma) as the negative control. The HiPerFect transfection reagent (Qiagen, Surrey, UK) was used for each transfection, referring to the protocols of producers.
Detection of cell viability and apoptosis
The cell viability test was conducted by the Cell Counting Kit‐8 (CCK‐8, Dojindo, Kumamoto, Japan) based on the quantity of a formazan dye, referring to the suggestion of producers. The ChroMate 4300 microplate reader (Awareness Technology, Palm City, FL, USA) was used to evaluate the absorbance set at OD 450 nm. The flow cytometry for cell apoptosis was carried out using fluorescein isothiocyanate labeled Annexin V (Annexin V‐FITC, BD Biosciences, Cowley, UK) and propidium iodide (PI, BD Biosciences) staining as described previously.17 A FACScan flow cytometer (BD Biosciences) was employed to detect and quantify apoptotic cells.
Enzyme‐linked immunosorbent assay (ELISA)
The production of interleukin‐6 (IL‐6), IL‐8 and IL‐1β were quantified by a corresponding human ELISA Kit (Invitrogen, Basel, Switzerland) based on the producer's recommendations. The absorbance of each plate was read by the ChroMate 4300 microplate reader using 450 nm as the primary wavelength (620 nm as optional reference wavelength).
Western blot
Cell extracts were prepared using the Complete Lysis‐M reagent (Roche Molecular Systems, CA, USA), and western blot was performed as previously described.12 Protein samples (50 μg) were resolved on 8%–10% sodium dodecyl sulfate polyacrylamide gel (SDS‐PAGE), electrophoretically blotted onto a Hybond P polyvinylidene fluoride membrane (PVDF, Millipore, Vienna, Austria), and then probed with primary antibodies: anti‐IL‐6 (ab6672), anti‐IL‐8 (ab7747), anti‐IL‐1β (ab9722), antiphosphorylated JAK (anti‐p‐JAK, ab138005), anti‐JAK (ab133666), anti‐STAT3 (ab119352), anti‐p‐STAT3 (ab76315) and anti‐glyceraldehyde 3‐phosphate dehydrogenase (anti‐GAPDH, ab181602, Abcam, Cambridge, UK).
RNA extraction and Ribonuclease R (RNase) digestion
Total RNA from 16HBE cells after various treatments or transfections was isolated by homogenization in TRIzol reagent (Invitrogen) following the manufacturer's guidelines. The quantity and quality of the RNA were evaluated by the NanoDrop ND‐1000 spectrophotometer (Thermo Fisher Scientific, Courtaboeuf, France). For the analysis of RNase R digestion, the RNAs were incubated in 20 μL of reactions containing 0.5 μL of RNase R (10 U, TaKaRa, Dalian, China) at 37°C for 15 minutes, and reactions were purified by the RNeasy MinElute cleaning Kit (Qiagen) based on the manufacturer's protocols.
Quantitative real‐time polymerase chain reaction (qRT‐PCR)
For the quantification of circ_0038467, reverse‐transcription (RT) reaction was performed using the Sensiscript II reverse transcriptase (Qiagen), and qRT‐PCR was conducted by the SYBR Green PCR master mix (Qiagen). GAPDH was used as a housekeeping gene for normalization. Using the Roche LightCycle 96 qRT‐PCR System, the following PCR primers were used: circ_0038467: 5'‐TCCCAGCTGACCTAAAGTCAAT‐3′ (forward) and 5'‐TGGTGACATTGAGCAGGAAC‐3′ (reverse), GAPDH: 5'‐GTCAGCCGCATCTTCTTTTG‐3′ (forward) and 5'‐GCGCCCAATACGACCAAATC‐3′ (reverse). Human miR‐338‐3p and U6 (as the endogenous control) were determined using the Qiagen miScript RT Kit and miScript SYBR Green Kit, with following primers: miR‐338‐3p: 5'‐GCGTCCAGCATCAGTGATT‐3′ (forward) and 5'‐GTGCAGGGTCCGAGGT‐3′ (reverse), U6: 5'‐CTCGCTTCGGCAGCACA‐3′ (forward) and 5'‐AACGCTTCACGAATTTGCGT‐3′ (reverse). The relative levels of circ_0038467 and miR‐338‐3p were calculated using the 2−ΔΔCt formula.
Subcellular fractionation
Total RNA from both cytoplasmic and nuclear fractions was extracted by the Cytoplasmic & Nuclear RNA Purification Kit (Norgen Biotek, Thorold, ON, Canada) based on the suggestion of producers. GAPDH and U6 were used as internal controls.
Dual‐luciferase reporter and RNA immunoprecipitation (RIP) assays
Bioinformatic analysis for the directly targeted miRNAs of circ_0038467 was carried out using the online database Circinteractome (https://circinteractome.nia.nih.gov/miRNA_Target_Sites/mirna_target_sites.html). The spartial sequences of circ_0038467 harboring wild‐type miR‐338‐3p‐binding sites (UGCUGG) and the site‐directed mutant (ACGACC) in the target region were cloned in the pmirGLO vector (Promega, Southampton, UK) downstream from the luciferase reporter gene, respectively. Each reporter construct (20 ng) was transfected into 16HBE cells together with miR‐338‐3p mimic (30 nM) or miR‐NC mimic (30 nM) using the HiPerFect transfection reagent. Cell extracts were prepared 48 hours post‐transfection, and the luciferase activity was determined by the Promega Dual‐luciferase Reporter Assay System.
For RIP studies, cell extracts were incubated with antibodies against Argonaute2 (ab156870, anti‐Ago2, Abcam) or a negative IgG (ab205718, anti‐IgG, Abcam) at 4°C for two hours, followed by the incubated with protein A/G beads (Sigma‐Aldrich) for two hours. Total RNA was obtained from the beads, and circ_0038467 enrichment was assessed by qRT‐PCR.
Statistical analysis
Results are presented as mean ± standard error (SEM). A two‐tailed Student's t‐test or one‐way analysis of variance (ANOVA) was used for the statistical analysis of all tests. Significance values were indicated as *P < 0.05.
Results
Inflammatory injury of LPS on 16HBE cells
First, we assessed the influence of LPS on 16HBE cells by measuring cell viability, apoptosis and proinflammatory cytokines production. CCK‐8 assays showed that cell viability was dose‐dependently weakened by LPS compared with the negative control (Fig 1a). Flow cytometry analyses revealed that LPS prominently accelerated cell apoptosis in a dose‐dependent manner (Fig 1b). Moreover, the data of ELISA and western blot assays confirmed that LPS treatment resulted in increased expression of proinflammatory cytokines (IL‐6, IL‐8 and IL‐1β) in a dose‐dependent manner in 16HBE cells (Fig 1c–f). These results together indicated that LPS induced the inflammatory injury in 16HBE cells.
Figure 1.
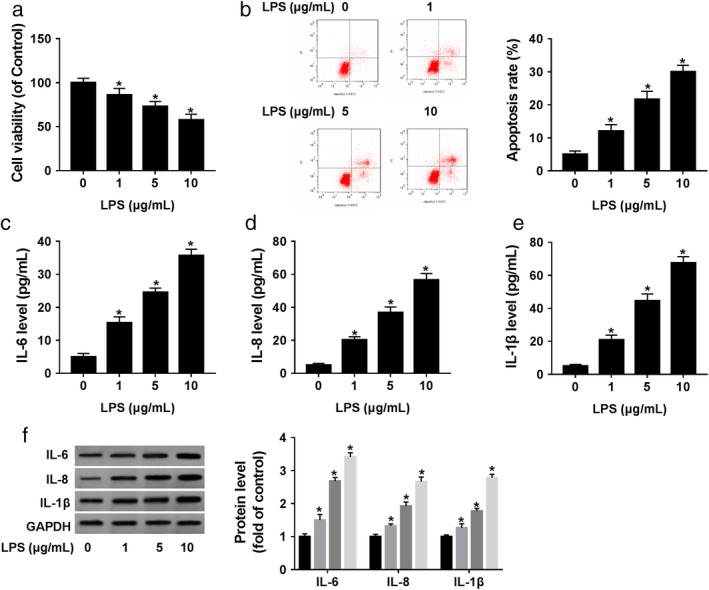
LPS induced 16HBE cell inflammatory injury. 16HBE cells were treated with various concentrations (0, 1, 5 and 10 μg/mL) of LPS for 24 hours. (a) Cell viability was detected by CCK‐8 assay. (b) Cell apoptosis was assessed by flow cytometry. (c–f) The levels of IL‐6, IL‐8 and IL‐1β were determined by ELISA and western blot. *P < 0.05.
Upregulation of circ_0038467 in LPS‐treated 16HBE cells
For preliminary observation of the involvement of circ_0038467 in pneumonia, we determined its expression pattern in 16HBE cells after LPS treatment. The data of qRT‐PCR assays revealed that LPS treatment triggered a significant increase in circ_0038467 expression compared with the negative group (Fig 2a). We then validated whether circ_0038467 was indeed a circular transcript using RNase R assays. In comparison to the negative control, GAPDH level was remarkably reduced by RNase R, and circ_0038467 was resistant to digestion with RNase R (Fig 2b). Additionally, subcellular localization assays demonstrated that circ_0038467 was strikingly enriched in the cytoplasm fraction of 16HBE cells after LPS treatment (Fig 2c). These data together pointed a notion that circ_0038467 was upregulated in LPS‐treated 16HBE cells.
Figure 2.
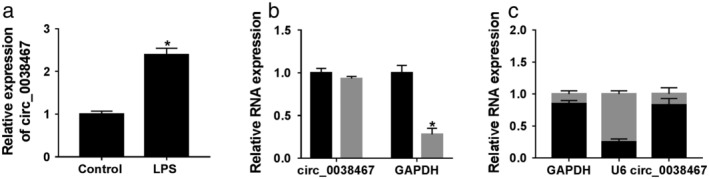
Circ_0038467 was highly expressed in LPS‐induced 16HBE cells. 16HBE cells were treated with 5 μg/mL of LPS for 24 hours. (a) Circ_0038467 expression was assessed by qRT‐PCR. (b) Total RNA was digested by RNase R, and then circ_0038467 level was detected, with GAPDH as the loading control.  RNase R−,
RNase R−,  RNase R−. (c) Circ_0038467 expression was evaluated by qRT‐PCR in the nuclear and cytoplasm fractions of treated 16HBE cells. *P < 0.05.
RNase R−. (c) Circ_0038467 expression was evaluated by qRT‐PCR in the nuclear and cytoplasm fractions of treated 16HBE cells. *P < 0.05.
Circ_0038467 deficiency alleviated LPS‐induced inflammatory injury in 16HBE cells
To determine the function of circ_0038467 in LPS‐induced 16HBE cell inflammatory injury, we carried out “phenocopy” silencing using siRNA against circ_0038467 (si‐circ#1, si‐circ#2 and si‐circ#3). Transient transfection of si‐circ, but not a scrambled sequence, prominently downregulated the expression of circ_0038467 in 16HBE cells (Fig 3a). Remarkably, si‐circ#2 introduction led to a more distinct downregulation in circ_0038467 level (Fig 3a). Hence, we selected si‐circ#2 for further experiments. The results of CCK‐8 and flow cytometry analyses revealed that in contrast to the negative group, LPS‐induced antiviability (Fig 3b) and proapoptosis (Fig 3c) effects were significantly reversed by circ_0038467 depletion. Moreover, the promotional impact of LPS on IL‐6, IL‐8 and IL‐1β levels was strongly abolished by circ_0038467 knockdown (Fig 3d–g). All these data implied that circ_0038467 silencing protected 16HBE cell from LPS‐induced inflammatory injury.
Figure 3.
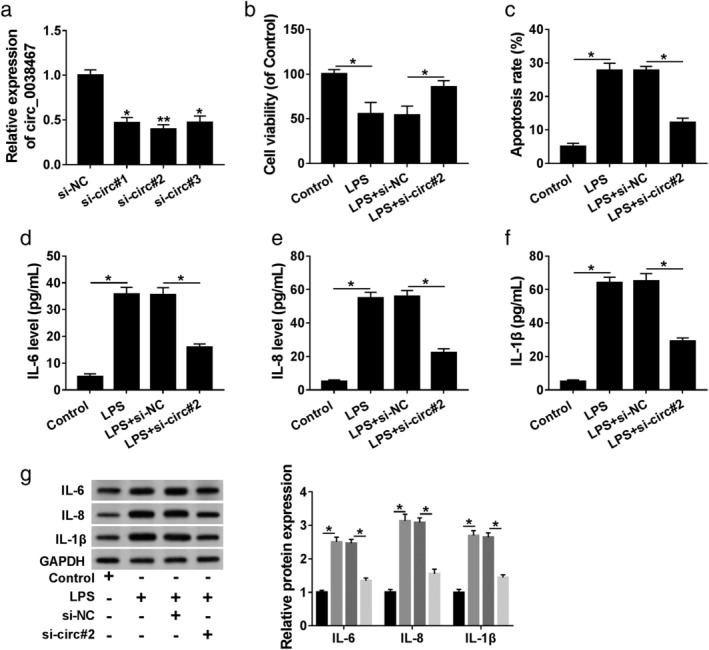
LPS induced 16HBE cell inflammatory injury via upregulating circ_0038467. (a) Circ_0038467 expression was detected by qRT‐PCR in 16HBE cells after transfection with si‐NC, si‐circ#1, si‐circ#2 and si‐circ#3. 16HBE cells were transfected with or without si‐NC or si‐circ#2, and then treated with 5 μg/mL of LPS for 24 hours, followed by (b) the measurement of cell viability by CCK‐8 assay; (c) cell apoptosis by flow cytometry; (d–f) the levels of IL‐6, IL‐8 and IL‐1β by ELISA, and (g) western blot (g). si‐circ#1: si‐circ_0038467#1, si‐circ#2: si‐circ_0038467#2, si‐circ#3: si‐circ_0038467#3. *P < 0.05.
Circ_0038467 acted as a sponge of miR‐338‐3p
To understand the mechanism by which circ_0038467 relieved LPS‐induced 16HBE cell inflammatory injury, we performed a detailed analysis for the potential miRNAs that bind to circ_0038467. Using Circinteractome online database, a putative target sequence (UGCUGG) for miR‐338‐3p was discovered within circ_0038467 (Fig 4a). Luciferase reporter plasmids containing the wild‐type miR‐338‐3p‐binding sites, or a site‐directed mutant in the target sequence were transfected into 16HBE cells together with miR‐338‐3p mimic or miR‐NC mimic. With wild‐type reporter construct and miR‐338‐3p mimic caused a prominent reduction in luciferase activity (Fig 4b). Upon transfection of the mutant construct, miR‐338‐3p mimic‐mediated activity reduction in luciferase was highly abrogated (Fig 4b), hinting the validity of these binding sites for interaction. The data of qRT‐PCR showed that miR‐338‐3p expression was inhibited by LPS treatment in 16HBE cells (Fig 4c). Moreover, circ_0038467 silencing resulted in increased miR‐338‐3p expression as compared to the negative group (Fig 4d). MiRNAs silence gene expression in the RNA‐induced silencing complex (RISC), which also contains Ago2.18 Thus, RIP experiments were conducted using anti‐Ago2 antibody. As a result, in contrast to the negative control, circ_0038467 and miR‐338‐3p were synchronously enriched by anti‐Ago2 antibody (Fig 4e), suggesting the endogenous relationship between circ_0038467 and miR‐338‐3p. Together, these results strongly established that circ_0038467 sequestered miR‐338‐3p via acting as a miR‐338‐3p sponge.
Figure 4.
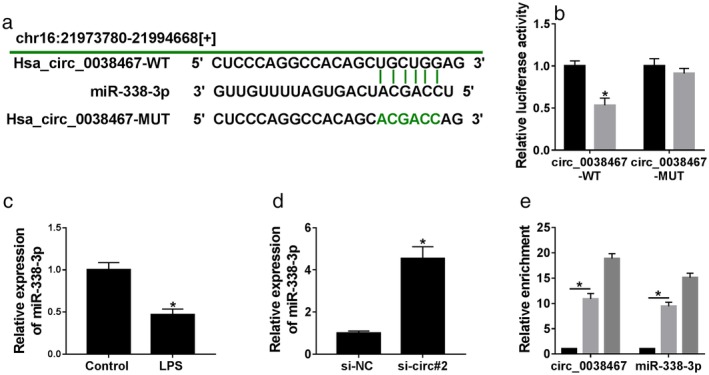
Circ_0038467 acted as a sponge of miR‐338‐3p in 16HBE cells. (a) Schematic model of the miR‐338‐3p‐binding sites within circ_0038467 identified by the online database Circinteractome and the mutant in the target sequence. (b) Luciferase activity was monitored in 16HBE cells cotransfected with circ_0038467 wild‐type reporter construct (circ_0038467‐WT) or circ_0038467 mutant‐type construct (circ_0038467‐MUT) and miR‐338‐3p mimic or miR‐NC mimic.  miR‐NC,
miR‐NC,  miR‐338‐3p. (c) MiR‐338‐3p expression was detected by qRT‐PCR in 16HBE cells after LPS treatment. (d) MiR‐338‐3p expression was determined in 16HBE cells transfected with si‐NC or si‐circ#2. si‐circ#2: si‐circ_0038467#2. (e) Cell lysates were incubated with anti‐Ago2 or anti‐IgG antibody, and enrichment of circ_0038467 and miR‐338‐3p were evaluated by qRT‐PCR. *P < 0.05.
miR‐338‐3p. (c) MiR‐338‐3p expression was detected by qRT‐PCR in 16HBE cells after LPS treatment. (d) MiR‐338‐3p expression was determined in 16HBE cells transfected with si‐NC or si‐circ#2. si‐circ#2: si‐circ_0038467#2. (e) Cell lysates were incubated with anti‐Ago2 or anti‐IgG antibody, and enrichment of circ_0038467 and miR‐338‐3p were evaluated by qRT‐PCR. *P < 0.05.
MiR‐338‐3p overexpression attenuated LPS‐induced 16HBE cell inflammatory injury
Then, we observed the effect of miR‐338‐3p in LPS‐induced inflammatory injury. In comparison to the negative group, the downregulation of LPS on miR‐338‐3p expression was prominently reversed by the transfection of miR‐338‐3p mimic (Fig 5a). CCK‐8 and flow cytometry assays revealed that compared with the negative group, LPS‐mediated viability diminishment (Fig 5b) and apoptosis enhancement (Fig 5c) in 16HBE cells were remarkably abolished by miR‐338‐3p overexpression. Moreover, the results of ELISA and western blot presented that miR‐338‐3p upregulation significantly weakened the levels of IL‐6, IL‐8 and IL‐1β, which were elevated by LPS treatment (Fig 5d–g). All these data strongly suggested that miR‐338‐3p overexpression alleviated LPS‐induced inflammatory damage in 16HBE cells.
Figure 5.
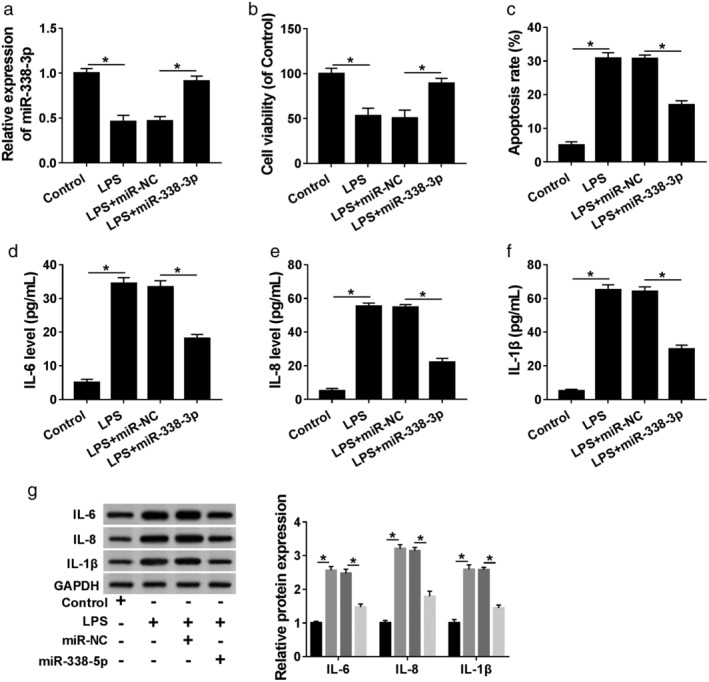
LPS induced 16HBE cell inflammatory injury by downregulating miR‐338‐3p. MiR‐338‐3p expression by qRT‐PCR: (a) cell viability by CCK‐8 assay; (b) cell apoptosis by flow cytometry; (c) the levels of IL‐6, IL‐8 and IL‐1β by ELISA; (d–f) western blot, and (g) in 16HBE cells transfected with or without miR‐338‐3p mimic or miR‐NC mimic before LPS treatment. *P < 0.05.
MiR‐338‐3p mediated alleviated effect of circ_0038467 knockdown on LPS‐induced inflammatory injury in 16HBE cells
Given our data that circ_0038467 acted as a sponge of miR‐338‐3p, we further validated whether circ_0038467 depletion exerted a protective role in LPS‐induced 16HBE cells by miR‐338‐3p. qRT‐PCR data revealed that in contrast to the anti‐NC group, miR‐338‐3p expression was prominently reduced by anti‐miR‐338‐3p introduction (Fig 6a). Subsequent experiment results revealed that compared to the negative group, circ_0038467 knockdown‐mediated viability promotion (Fig 6b) and apoptosis suppression (Fig 6c) were significantly abrogated by anti‐miR‐338‐3p introduction. Furthermore, the reduced effect of circ_0038467 knockdown on IL‐6, IL‐8 and IL‐1β production was strikingly reversed by the transfection of anti‐miR‐338‐3p (Fig 6d–g). Taken together, these data hinted that circ_0038467 deficiency relieved LPS‐induced inflammatory injury by miR‐338‐3p in 16HBE cells.
Figure 6.
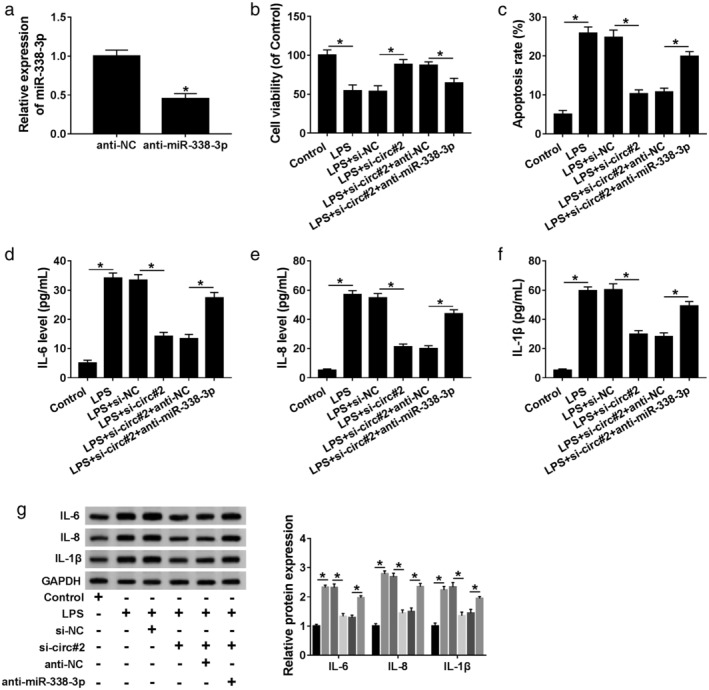
MiR‐338‐3p mediated the alleviated effect of circ_0038467 knockdown on LPS‐induced 16HBE cell inflammatory injury. (a) MiR‐338‐3p expression was evaluated by qRT‐PCR in 16HBE cells transfected with anti‐NC or anti‐miR‐338‐3p. 16HBE cells were transfected with si‐NC, si‐circ#2, si‐circ#2+anti‐NC or si‐circ#2+anti‐miR‐338‐3p before LPS treatment, followed by the determination of cell viability by CCK‐8 assay; (b) cell apoptosis by flow cytometry; (c) the levels of IL‐6, IL‐8 and IL‐1β by ELISA; (d–f) western blot, and (g) si‐circ#2: si‐circ_0038467#2. *P < 0.05.
JAK/STAT3 signaling pathway was involved in circ_0038467/miR‐338‐3p axis‐mediated regulation in LPS‐induced inflammatory injury in 16HBE cells
JAK/STAT3 signaling pathway, an important mediator in inflammation, has been accepted to be associated with pneumonia pathogenesis.19, 20 Herein, we further determined whether the pathway was correlated with the regulation of the circ_0038467/miR‐338‐3p axis in LPS‐induced inflammatory injury in 16HBE cells. In comparison to the negative control, LPS treatment resulted in increased expression of p‐JAK and p‐STAT3 (Fig 7a,b), indicating that LPS activated the pathway. Moreover, these data showed that the levels of p‐JAK and p‐STAT3 were prominently reduced by circ_0038467 depletion, and this effect was strongly reversed by anti‐miR‐338‐3p introduction (Fig 7a,b). All these data suggested that circ_0038467 knockdown repressed the activation of JAK/STAT3 signaling pathway by miR‐338‐3p in LPS‐induced 16HBE cells. Additionally, the activation of JAK/STAT3 pathway was significantly weakened by ruxolitinib in LPS‐induced 16HBE cells (Fig 8a). Furthermore, the inhibition of JAK/STAT3 pathway led to a distinct enhancement in cell viability and a clear suppression in cell apoptosis, as well as a prominent reduction in the production of IL‐6, IL‐8 and IL‐1β (Fig 8b–g), suggesting that this pathway inhibition alleviated LPS‐induced inflammatory injury in 16HBE cells. These results together established a notion that the JAK/STAT3 pathway was involved in the circ_0038467/miR‐338‐3p axis‐mediated regulation in LPS‐induced 16HBE cell inflammatory injury.
Figure 7.
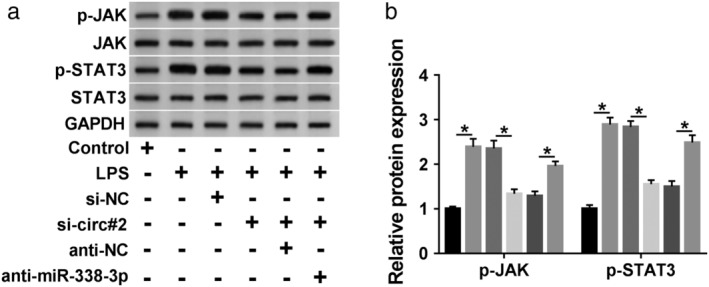
JAK/STAT3 signaling pathway was involved in the regulation of the circ_0038467/miR‐338‐3p axis in LPS‐induced 16HBE cells. (a,b) 16HBE cells were transfected with si‐NC, si‐circ#2, si‐circ#2 + anti‐NC or si‐circ#2 + anti‐miR‐338‐3p, and then were treated with LPS for 24 hours, followed by the detection of JAK, p‐JAK, STAT3, p‐STAT3 levels by western blot. si‐circ#2: si‐circ_0038467#2. *P < 0.05.
Figure 8.
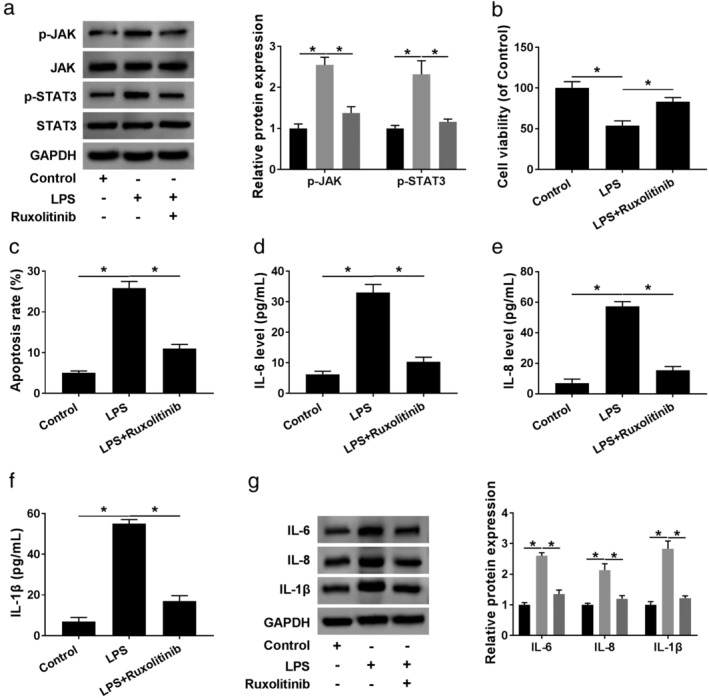
The inhibition of JAK/STAT3 signaling pathway weakened LPS‐induced inflammatory injury in 16HBE cells. 16HBE cells were synchronously treated with LPS and ruxolitinib for 24 hours, followed by the measurement of JAK, p‐JAK, STAT3, p‐STAT3 levels by: (a) western blot; control, LPS, LPS + ruxolitinib; (b) cell viability by CCK‐8 assay; (c) cell apoptosis by flow cytometry, (d–f) levels of IL‐6, IL‐8 and IL‐1β by ELISA, and (g) western blot. *P < 0.05.
Discussion
Pneumonia is a worldwide public health concern with high morbidity rates and elevated economic costs.21 Excessive inflammatory response contributes to the development and progression of pneumonia.22 LPS, the outer membrane of Gram‐negative, can activate the inflammatory response, and it has been widely used to construct the pneumonia in vitro model.23, 24 In the present work, our data validated that LPS induced inflammatory injury in 16HBE cells, as evidenced by a repression in cell viability and an enhancement in cell apoptosis, as well as the augment in proinflammatory cytokines (IL‐6, IL‐8 and IL‐1β) levels, in agreement with recent work.25 The current study had led to the identification of circ_0038467 silencing that alleviated LPS‐induced inflammatory injury in 16HBE cells through regulating miR‐338‐3p/JAK/STAT3 pathway.
In recent years, circRNAs are attracting attention owing to their potential contributes to the pathogenesis of human diseases, including pneumonia.6, 26 Using the microarray analysis of circRNAs, circ_0038467 was discovered to be highly expressed in inflammatory 16HBE cells, eliciting its potential involvement in inflammatory diseases.9 Therefore, our research started with the hypothesis that circ_0038467 might implicate in the progression of pneumonia. To validate this, we determined the expression of circ_0038467 in LPS‐treated 16HBE cells, and our data demonstrated a striking upregulation of circ_0038467. Due to the closed loop structures with neither 5′ caps nor 3′ polyadenylated tails, circRNAs are resistant to RNase R.27 Using RNase R assays, we uncovered that circ_0038467 was indeed a circular transcript. Moreover, we were first to highlight that the silencing of circ_0038467 alleviated LPS‐induced 16HBE cell inflammatory injury. Additionally, subcellular localization assays indicated that circ_0038467 was enriched in the cytoplasm fraction of LPS‐treated 16HBE cells, which provided the possibility for the direct interaction between circ_0038467 and miRNAs.
Then, we predicted the potential miRNAs that bind to circ_0038467 using the online database Circinteractome and validated that circ_0038467 acted as a sponge of miR‐338‐3p in 16HBE cells. MiR‐338‐3p has been reported to repress tumorigenesis and progression of multiple human tumors, such as ovarian cancer, gastric cancer and NSCLC.28, 29, 30 MiR‐338‐3p was also demonstrated to be downregulated in acute kidney injury.31 Moreover, in acute cerebral infarction patients, plasma miR‐338‐3p level was associated with the expression of C‐reactive protein, which was a crucial inflammatory factor in atherosclerosis.32 Additionally, miR‐338‐3p was abnormally expressed in the serum of patients with asthma and chronic obstructive pulmonary diseases.33 In the present research, for the first time, we validated that the upregulation of miR‐338‐3p alleviated LPS‐induced inflammatory injury in 16HBE cells. More interestingly, our data substantiated that miR‐338‐3p mediated the protective effect of circ_0038467 knockdown on LPS‐induced 16HBE cell inflammatory injury.
JAK/STAT3 signaling is an important oncogenic pathway that is involved in the carcinogenesis of many types of human cancers, including colorectal cancer, hepatocellular carcinoma and pancreatic cancer.34, 35, 36 Emerging studies have indicated that the JAK/STAT3 signaling plays an important role in the inflammatory response.37, 38 Moreover, JAK/STAT3 signaling was reported to be implicated in the lung during the acute inflammatory injury, illuminating a potential target for anti‐inflammatory treatment.20 In this study, for the first time, we demonstrated that JAK/STAT3 signaling was involved in the regulatory network of the circ_0038467/miR‐338‐3p axis in LPS‐induced inflammatory injury in 16HBE cells. The current work was limited in vitro researches, and more in vivo researches about the novel mechanism in pneumonia using animal model will be implemented in further work.
In conclusion, the current work suggested that circ_0038467 deficiency protected 16HBE cells from LPS‐induced inflammatory injury possibly through sponging miR‐338‐3p and blocking the JAK/STAT3 signaling pathway. Circ_0038467 and miR‐338‐3p were thus identified as promising molecular targets for the treatment of pneumonia.
Disclosure
The authors declare that they have no financial conflicts of interest.
Acknowledgment
This work was supported by Science and Technology Project of Xinjiang Uygur Autonomous Region (No. 201517103).
References
- 1. Quinton LJ, Walkey AJ, Mizgerd JP. Integrative physiology of pneumonia. Physiol Rev 2018; 98: 1417–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sungurlu S, Balk RA. The role of biomarkers in the diagnosis and management of pneumonia. Clin Chest Med 2018; 39: 691–701. [DOI] [PubMed] [Google Scholar]
- 3. Yang Y, Tang H. Aberrant coagulation causes a hyper‐inflammatory response in severe influenza pneumonia. Cell Mol Immunol 2016; 13: 432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qu S, Yang X, Li X et al Circular RNA: A new star of noncoding RNAs. Cancer Lett 2015; 365: 141–8. [DOI] [PubMed] [Google Scholar]
- 5. Dimmeler S, Nicolas J, Boeckel N et al Circular RNA for the diagnosis of cardiovascular and inflammatory diseases. Google patents 2018.
- 6. Wan Q‐Q, Wu D, Ye Q‐F. The expression profiles of circRNAs in lung tissues from rats with lipopolysaccharide‐induced acute respiratory distress syndrome: A microarray study. Biochem Biophys Res Commun 2017; 493: 684–9. [DOI] [PubMed] [Google Scholar]
- 7. Yang P, Gao R, Zhou W et al Protective impacts of circular RNA VMA21 on lipopolysaccharide‐engendered WI‐38 cells injury via mediating microRNA‐142‐3p. BioFactors 2019. 10.1002/biof.593. [DOI] [PubMed] [Google Scholar]
- 8. Guo R, Zhang L, Meng J. Circular RNA ANKRD36 attends to lipopolysaccharide‐aroused MRC‐5 cell injury via regulating microRNA‐31‐3p. BioFactors 2019. 10.1002/biof.592. [DOI] [PubMed] [Google Scholar]
- 9. Hua Q, Chen Y, Liu Y et al Circular RNA 0039411 is involved in neodymium oxide‐induced inflammation and antiproliferation in a human bronchial epithelial cell line via sponging miR‐93‐5p. Toxicol Sci 2019; 170: 69–81. [DOI] [PubMed] [Google Scholar]
- 10. Abd‐El‐Fattah AA, Sadik NAH, Shaker OG et al Differential microRNAs expression in serum of patients with lung cancer, pulmonary tuberculosis, and pneumonia. Cell Biochem Biophys 2013; 67: 875–84. [DOI] [PubMed] [Google Scholar]
- 11. Griss K, Han M, Nell C et al miRNAs as biomarkers in pneumonia and COPD‐exacerbation. Pneumologie 2016; 70: P29. [Google Scholar]
- 12. Guo J, Cheng Y. MicroRNA‐1247 inhibits lipopolysaccharides‐induced acute pneumonia in A549 cells via targeting CC chemokine ligand 16. Biomed Pharmacother 2018; 104: 60–8. [DOI] [PubMed] [Google Scholar]
- 13. Fei S, Cao L, Pan L. microRNA‐3941 targets IGF2 to control LPS‐induced acute pneumonia in A549 cells. Mol Med Rep 2018; 17: 4019–26. [DOI] [PubMed] [Google Scholar]
- 14. Liu CC, Cheng JT, Li TY, Tan PH. Integrated analysis of microRNA and mRNA expression profiles in the rat spinal cord under inflammatory pain conditions. Eur J Neurosci 2017; 46: 2713–28. [DOI] [PubMed] [Google Scholar]
- 15. Liu J, Cao L, Feng Y, Li Y, Li T. MiR‐338‐3p inhibits TNF‐α‐induced lipogenesis in human sebocytes. Biotechnol Lett 2017; 39: 1343–9. [DOI] [PubMed] [Google Scholar]
- 16. Kartha RV, Subramanian S. Competing endogenous RNAs (ceRNAs): New entrants to the intricacies of gene regulation. Front Genet 2014; 5: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb Protoc 2016; 2016: pdb.prot087288 10.1101/pdb.prot087288. [DOI] [PubMed] [Google Scholar]
- 18. Iwakawa H‐O, Tomari Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol 2015; 25: 651–65. [DOI] [PubMed] [Google Scholar]
- 19. Hodge DR, Hurt EM, Farrar WL. The role of IL‐6 and STAT3 in inflammation and cancer. Eur J Cancer 2005; 41: 2502–12. [DOI] [PubMed] [Google Scholar]
- 20. Gao H, Ward PA. STAT3 and suppressor of cytokine signaling 3: Potential targets in lung inflammatory responses. Expert Opin Ther Targets 2007; 11: 869–80. [DOI] [PubMed] [Google Scholar]
- 21. Komiya K, Ishii H, Kadota J‐I. Healthcare‐associated pneumonia and aspiration pneumonia. Aging Dis 2014; 6: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bordon JM, Fernandez‐Botran R, Wiemken TL et al Bacteremic pneumococcal pneumonia: Clinical outcomes and preliminary results of inflammatory response. Infection 2015; 43: 729–38. [DOI] [PubMed] [Google Scholar]
- 23. Liu X, Meng J. Luteolin alleviates LPS‐induced bronchopneumonia injury in vitro and in vivo by down‐regulating microRNA‐132 expression. Biomed Pharmacother 2018; 106: 1641–9. [DOI] [PubMed] [Google Scholar]
- 24. Yu C, Xiang Q, Zhang H. Xianyu decoction attenuates the inflammatory response of human lung bronchial epithelial cell. Biomed Pharmacother 2018; 102: 1092–8. [DOI] [PubMed] [Google Scholar]
- 25. Li Y, Song D, Bo F et al Diazepam inhibited lipopolysaccharide (LPS)‐induced pyroptotic cell death and alleviated pulmonary fibrosis in mice by specifically activating GABA(A) receptor α4‐subunit. Biomed Pharmacother 2019; 118: 109239. [DOI] [PubMed] [Google Scholar]
- 26. Haque S, Harries LW. Circular RNAs (circRNAs) in health and disease. Genes 2017; 8: 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Su M, Xiao Y, Ma J et al Circular RNAs in cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol Cancer 2019; 18: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang R, Shi H, Ren F et al MicroRNA‐338‐3p suppresses ovarian cancer cells growth and metastasis: Implication of Wnt/catenin beta and MEK/ERK signaling pathways. J Exp Clin Cancer Res 2019; 38: 494. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Sun F, Yu M, Yu J et al miR‐338‐3p functions as a tumor suppressor in gastric cancer by targeting PTP1B. Cell Death Dis 2018; 9: 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang P, Shao G, Lin X, Liu Y, Yang Z. MiR‐338‐3p inhibits the growth and invasion of non‐small cell lung cancer cells by targeting IRS2. Am J Cancer Res 2017; 7: 53–63. [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang L, Liu X‐Q, Ma Q et al hsa‐miR‐500a‐3P alleviates kidney injury by targeting MLKL‐mediated necroptosis in renal epithelial cells. FASEB J 2019; 33: 3523–35. [DOI] [PubMed] [Google Scholar]
- 32. Teng L, Meng R. Long non‐coding RNA MALAT1 promotes acute cerebral infarction through miRNAs‐mediated hs‐CRP regulation. J Mol Neurosci 2019; 69: 494–504. [DOI] [PubMed] [Google Scholar]
- 33. Deshpande DA, Dileepan M, Walseth TF, Subramanian S, Kannan MS. MicroRNA regulation of airway inflammation and airway smooth muscle function: Relevance to asthma. Drug Dev Res 2015; 76: 286–95. [DOI] [PubMed] [Google Scholar]
- 34. Xue X, Ramakrishnan SK, Weisz K et al Iron uptake via DMT1 integrates cell cycle with JAK‐STAT3 signaling to promote colorectal tumorigenesis. Cell Metab 2016; 24: 447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamaguchi Y, Mori A, Fujimoto Y et al Longer warm ischemia can accelerate tumor growth through the induction of HIF‐1α and the IL‐6‐JAK‐STAT3 signaling pathway in a rat hepatocellular carcinoma model. J Hepatobiliary Pancreat Sci 2016; 23: 771–9. [DOI] [PubMed] [Google Scholar]
- 36. Hu H, Zhang Q, Chen W et al MicoRNA‐301a promotes pancreatic cancer invasion and metastasis through the JAK/STAT3 signaling pathway by targeting SOCS5. Carcinogenesis 2019; bgz121. [DOI] [PubMed] [Google Scholar]
- 37. Fang J, Chu L, Li C et al JAK2 inhibitor blocks the inflammation and growth of esophageal squamous cell carcinoma in vitro through the JAK/STAT3 pathway. Oncol Rep 2015; 33: 494–502. [DOI] [PubMed] [Google Scholar]
- 38. Yu Q, Zeng K, Ma X et al Resokaempferol‐mediated anti‐inflammatory effects on activated macrophages via the inhibition of JAK2/STAT3, NF‐κB and JNK/p38 MAPK signaling pathways. Int Immunopharmacol 2016; 38: 104–14. [DOI] [PubMed] [Google Scholar]