

Abstract
The ability of growth hormone (GH) to induce adipose tissue lipolysis has been known for over five decades; however, the molecular mechanisms that mediate this effect, as well as the ability of GH to inhibit insulin-stimulated glucose uptake, have been scarcely documented. In this same timeframe, our understanding of adipose tissue has evolved to reveal a complex structure with distinct types of adipocytes, depot-specific differences, a biologically significant extracellular matrix and important endocrine properties mediated by adipokines. All of these aforementioned features, in turn, can influence lipolysis. In this Review, we provide a historical and current overview of the lipolytic effect of GH in humans, mice and cultured cells. More globally, we explain lipolysis in terms of GH-induced intracellular signaling and its effect on obesity, insulin resistance and lipotoxicity. In this regard, findings that define molecular mechanisms by which GH induces lipolysis are described. Finally, data are presented for the differential effect of GH on specific adipose tissue depots and on distinct classes of metabolically active adipocytes. Together, these cellular, animal and human studies reveal novel cellular phenotypes and molecular pathways regulating the metabolic effects of GH on adipose tissue.
Introduction
Since the 1980s, the global prevalence of obesity has significantly increased in adults and children, which is associated with excess morbidity and mortality. Obesity is a complex trait that is influenced by diet, physical activity, age and genetics1,2. The storage and release of triglycerides from adipose tissue are important processes involved in the development of obesity and are controlled by afferent signals including a variety of hormones, for example, catecholamines, growth hormone (GH) and insulin.
GH is a peptide hormone that is secreted by somatotrophs in the anterior pituitary gland and has a variety of tissue-specific effects, including anabolic effects on muscle and bone and catabolic action on white adipose tissue3. Of note, the ability of GH to induce lipolysis in adipose tissue and affect fat volume and distribution is well accepted3; however, the molecular mechanisms that underpin these effects are still not firmly established. In addition, the effect of GH on inhibiting the action of insulin (that is, the diabetogenic effect of GH) has been known for more than half a century; yet, the molecular mechanisms supporting this effect in humans are only now being elucidated4,5. Clinical evidence demonstrates that GH regulates insulin sensitivity in humans as a direct result of its induction of lipid catabolism in adipose tissue, which liberates free fatty acids (FFA)6. Moreover, in patients with GH deficiency, insulin resistance caused by GH treatment is reversed by pharmacological blockade of lipolysis7,8.
In this Review, we summarize the lipolytic effects of GH in adipose tissue with emphasis on studies in humans. We will also discuss experimental data in mice and cultured cells, emerging data documenting the underlying molecular mechanisms and the growing understanding of how these factors might be influenced by the cellular composition and location of adipose tissue.
An overview of lipolysis
Lipolysis is a catabolic branch of the fatty acid cycle that provides fatty acids in times of metabolic need. Fatty acids are essential as energy substrates and for the synthesis of most lipids. However, despite their fundamental physiological importance, oversupply of FFAs cause lipotoxicity, which can disrupt the integrity of membranes, alter cellular acid–base homeostasis and elicit the generation of harmful bioactive lipids9. Furthermore, the high concentrations of circulating FFAs and triglycerides observed in obesity and lipodystrophy cause insulin resistance in skeletal muscle and decreased glucose tolerance10–13. Importantly, GH is now known to regulate the balance of fatty acid esterification and triglyceride lipolysis, thereby having a central role in regulating whole body metabolism and glucose homeostasis3,14.
Adipose tissue lipolysis of stored triglycerides to FFAs and glycerol is coordinated by a number of proteins, including several enzymes and lipid droplet-associated proteins (Figure 1). Lipolysis involves the action of three different lipases: adipose triglyceride lipase (ATGL); hormone-sensitive lipase (HSL); and monoacylglycerol lipase (MGL). ATGL, which is associated with lipid droplets and encoded by PNPLA2, is the rate-limiting enzyme for lipolysis and catalyzes the first step of hydrolysis of triglyceride to diacylglycerol15–19. Various other proteins are also involved in the lipolytic machinery. For example, in adipocytes, CGI-58 is an activator of ATGL. Under basal conditions, Perilipin A (PLIN1), a major lipid droplet coat protein in mature adipocytes, prevents the access of CGI-58 to ATGL, thereby decreasing lipolysis18,20–22. Upon β-adrenergic stimulation, protein kinase A (PKA) activation results in phosphorylation of PLIN1, causing the release of CGI-58 that then binds and stimulates ATGL on lipid droplets20,23. In addition, PKA phosphorylates HSL, which then translocates to lipid droplets and together with ATGL and MGL leads to acute activation of triglyceride hydrolysis.
Figure 1: GH-induced lipolysis and insulin resistance.
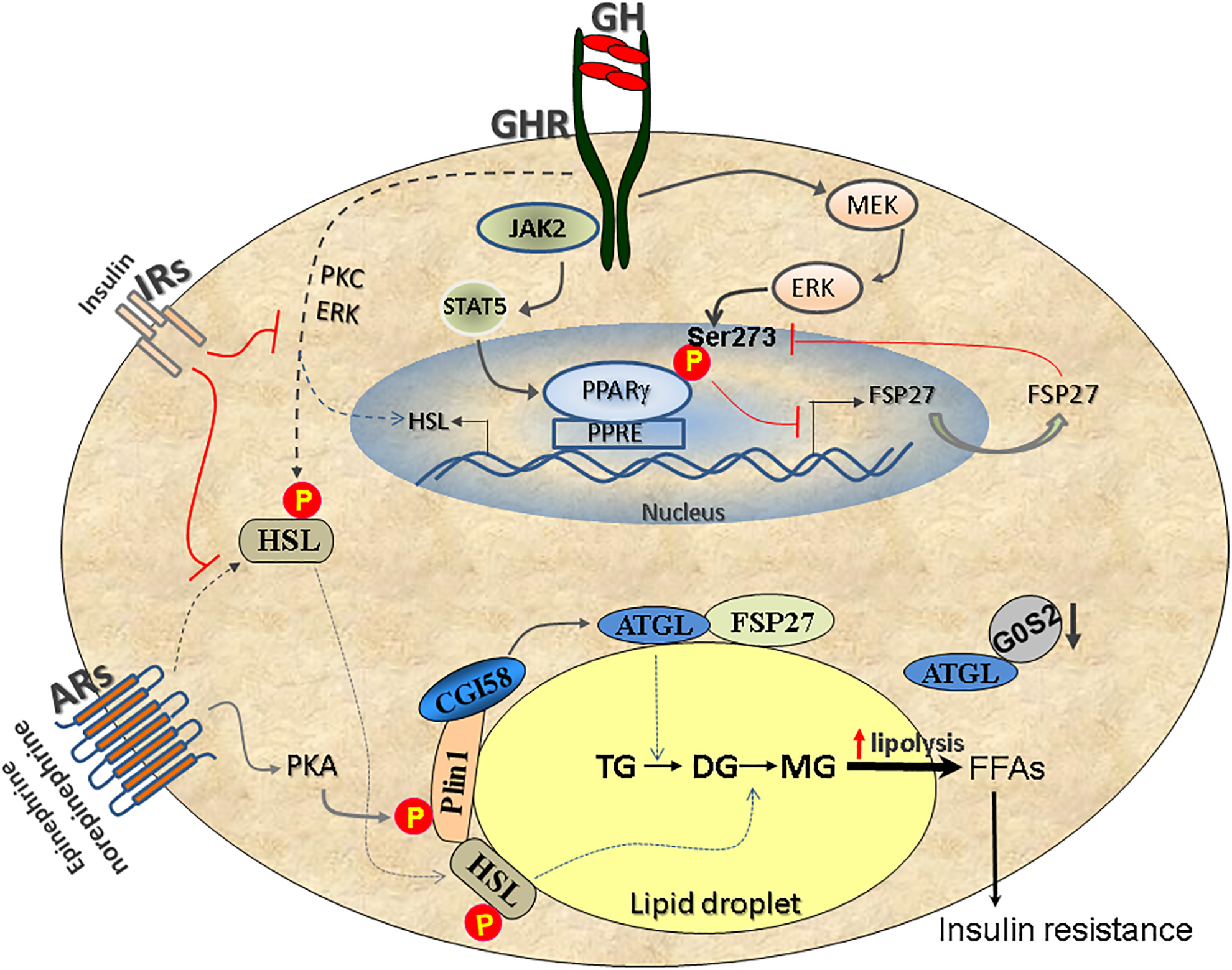
GH-induced signaling activates the MEK–ERK pathway, which causes phosphorylation of PPARγ at Ser273. This event results in PPARγ inactivation and FSP27 downregulation. The decrease in FSP27 expression leads to increased lipolysis and higher circulatory free fatty acids (FFAs), a hallmark of insulin resistance and type 2 diabetes mellitus. PPARγ-regulated FSP27 feeds-back to stabilize PPARγ in the nucleus. Parallel to the above pathway, GH binding to its receptors also activates STAT5, which is a positive regulator of PPARγ, but the MEK–ERK activation pathway predominates to inactivate PPARγ. In addition, GH induces lipolysis by activating HSL and by increasing the de novo expression of HSL mRNA via the activation of PKC and ERK. Adrenergic signaling via adrenergic receptors (ARs) has a parallel role in the stimulation of lipolysis via activating the lipolytic cascade involving the lipases ATGL, HSL and MGL. Finally, insulin signalling via insulin receptors (IRs) inhibits lipolysis by suppressing GH-induced PKC and ERK activity as well as the adrenergic pathway.
ATGLis also regulated by two inhibitory proteins: G0S2 and FSP27. For example, G0S2 binds directly to ATGL and attenuates ATGL-mediated lipolysis via inhibiting its triglyceride hydrolase activity24. Furthermore, FSP27 is a lipid droplet-associated protein that regulates lipid droplet dynamics25–29 and lipolysis, through direct interaction with ATGL to inhibit its catalytic capacity30. In addition, FSP27 also transcriptionally represses ATGL expression during insulin signalling (Figure 1)31.
The lipolytic effects of GH in humans
Historical perspectives.
Maurice Raben was among the first to purify and test pituitary-derived human GH in human s and he noted that the rise observed in serum FFA levels after treatment was “perhaps the most sensitive response to GH of any yet described”32. Interestingly, he studied treatment responses in adult volunteers, including elderly patients with panhypopituitarism, where a single physiological dose of GH could induce a lipolytic33. Raben also demonstrated that the lipolytic effect induced by GH treatment is suppressed by food intake and amplified by fasting33, which suggests a physiological role of GH to partition substrate metabolism between fat mobilization and protein synthesis that is dependent on ambient nutrient status. In addition, Raben hypothesized that the suppressive effect of food intake on GH-induced lipolysis provided a mechanism of control “not requiring a change in GH secretion” and that GH was present “at all times”33.
The advent of a GH radioimmunoassay disproved Raben’s GH secretion hypothesis and demonstrated that serum levels of GH are secreted in a pulsatile manner, especially during night time and after exercise34,35. Furthermore, serum GH levels were suppressed postprandially and elevated during fasting36, which is consistent with the aforementioned lipolytic effects. Experimental studies on the metabolic effects of GH and insulin in human volunteers6 confirmed the lipolytic action of GH and demonstrated that GH-induced lipolysis was accompanied by increased FFA uptake and oxidation in skeletal muscle in vivo. Moreover, GH treatment was shown to acutely and directly induce resistance to insulin-stimulated glucose uptake in human skeletal muscle37. As the circulating pattern of insulin inversely correlates with that of GH (that is, high insulin levels postprandially and low levels during fasting), the following hypotheses were proposed: first, in the immediate postprandial period, insulin acts alone to increase the storage of glucose and other nutrients; second, in the fasting state, GH acts alone to facilitate mobilization and oxidation of endogenous lipid stores; third, in between these two phases, GH and insulin might act in synergy to promote protein anabolism6. This model has largely stood the test of time, even though we now know that the anabolic effects of GH largely depend on the concerted actions of GH, insulin and insulin-like growth factor 1 (IGF1)3.
Further evidence of GH-induced lipolysis in humans.
Regarding the mechanisms whereby GH increases lipolysis in vivo, experimental studies in human volunteers demonstrate that this effect of GH is blocked by treatment with acipimox, an antilipolytic agent that suppresses cAMP formation and ultimately the lipolytic action of HSL by inhibition of the niacin receptor7,8. Furthermore, these human studies also show that suppression of lipolysis with acipimox abrogated the antagonistic effects of GH on insulin-stimulated muscle glucose uptake7,8. In addition to effects on lipolysis, GH treatment for 5 weeks in female individuals with obesity suppresses the activity of lipoprotein lipase, an enzyme that plays a part in lipid clearance from the blood stream by hydrolyzing triglycerides from circulating chylomicrons and VLDLs38. However, short-term GH treatment in young lean male individuals has no effect on the turnover rate of VLDL-associated triglycerides39. Taken together, these findings suggest that in humans, GH actively increases circulating FFA levels by increasing lipolysis and by inhibiting FFA uptake into adipose tissue. Theoretically, novel compounds could be developed that specifically target the lipolytic and anabolic effects of GH, respectively.
Patients with GH-deficiency and acromegaly.
The physiological and clinical significance of the lipolytic effects of GH have been tested in adult patients with GH-deficiency (GHD) during fasting40 as well as during a hypoglycaemic clamp41. Interestingly, these studies show that GH is critical for lipid mobilization and utilization when glucose availability is limited. Also well-documented is that long term GH replacement in patients with GHD induces sustained lipolysis and a gradual reduction in fat mass towards normal levels (Figure 2)42.
Figure 2: Body composition assessed by CT in adults with GH-deficiency.
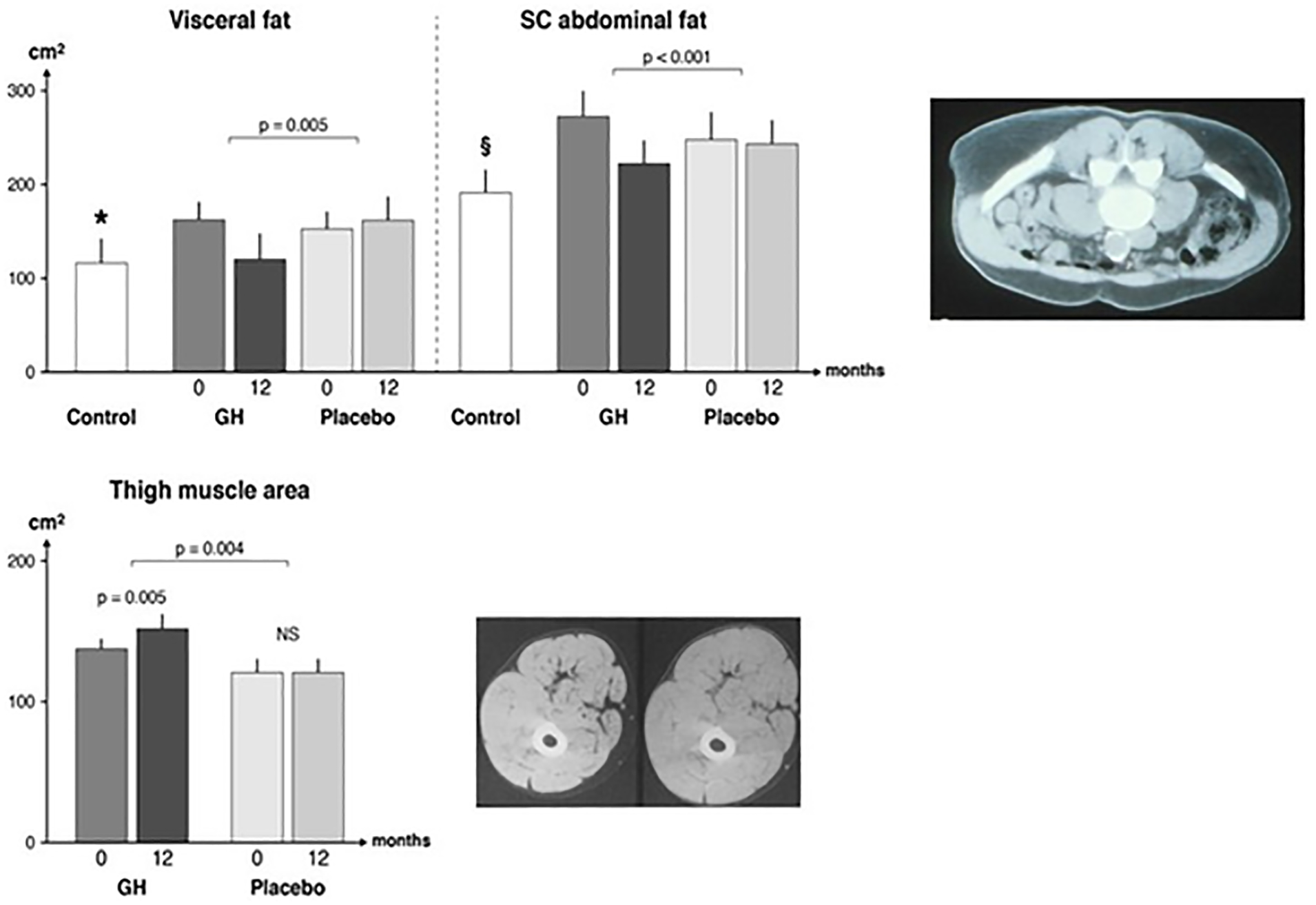
Patients with GH-deficiency participated in a double-blind parallel study receiving either GH or placebo. The patients were studied at baseline (0) and after 12 months treatment (12). The results were compared with a reference population (n = 29) matched for age and sex (control). The P values denote a comparison of changes between the GH and placebo groups, respectively. Visceral fat lower in the reference group as compared to all patients at baseline, as shown by * (P = 0.02). Subcutaneous fat was lower in the reference group as compared to all patients a baseline, as shown by § (P = 0.03). The data were originally presented in42. The CT images are representative scans from the mid-umbilical region before (upper) and after (lower) GH treatment (upper panel). This scan derives from a separate trial125 and is reproduced with the permission from the principal investigator. The lower panel includes a representative CT scan from the first study42 before (right) and after (left) GH treatment. Upper CT image panel reproduced with permission125. Lower CT image panel reproduced with permission from REF42.
By contrast, long-term and unregulated GH excess might cause glucose intolerance, as seen in patients with active acromegaly43. Interestingly, active acromegaly is also associated with reduced fat mass, increased lean body mass and increased serum FFA levels. Disease control reverses insulin resistance43 and this effect occurs together with an increase in fat mass and a decrease in lean body mass44. The insulin antagonistic effect of GH exposure is rapidly reversible and preceded by elevations in serum FFA levels, as demonstrated in a study in adult patients with GHD where GH was infused at different time points relative to the assessment of insulin-stimulated glucose uptake (Figure 3)45. Thus, a causal link between the lipolytic effects of GH and the antagonistic effects GH has on insulin-stimulated glucose uptake is undisputed, but the underlying molecular mechanisms are not yet fully settled.
Figure 3: Effects of GH in humans on FFA levels and insulin sensitivity.
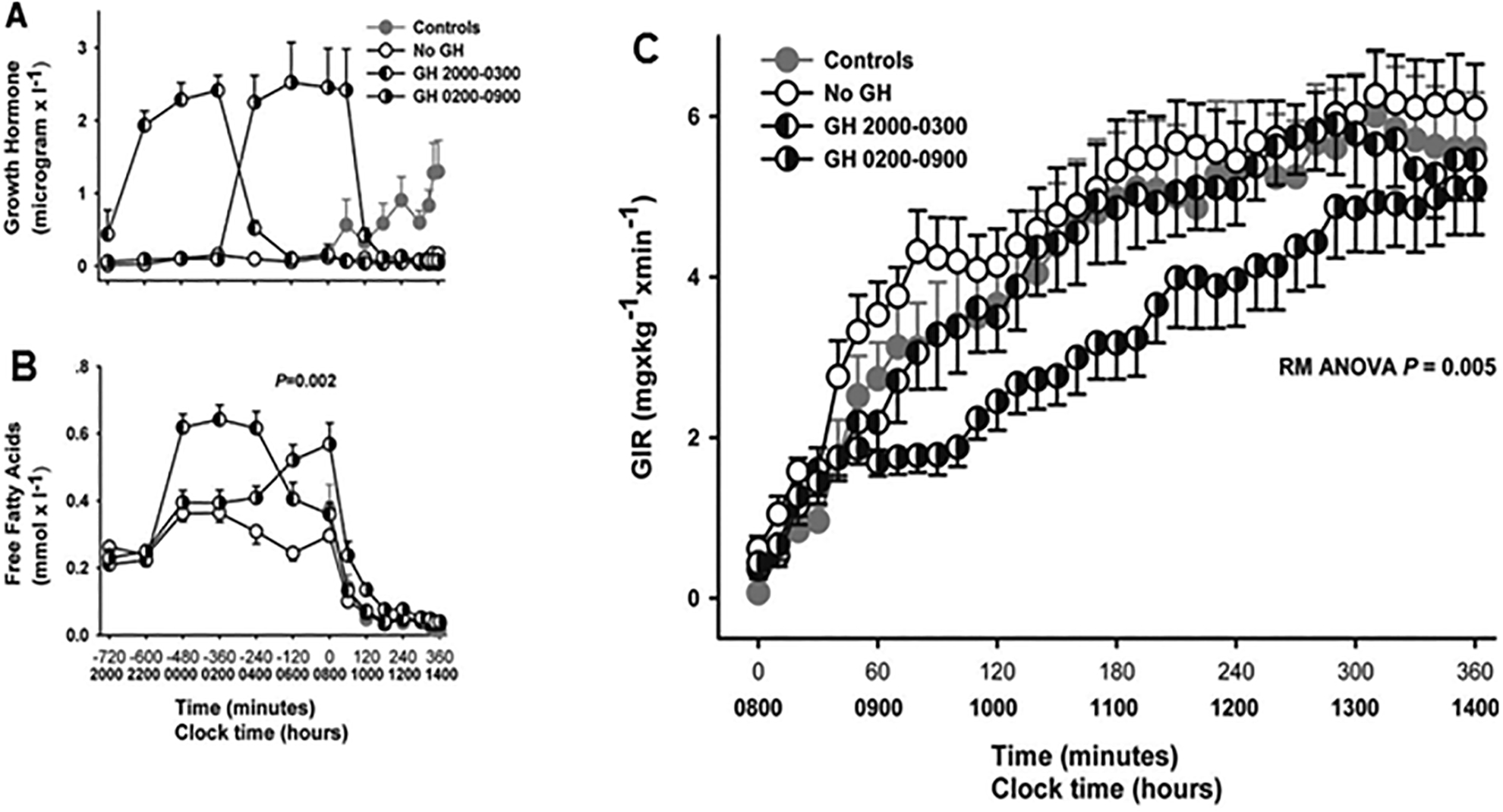
Effects of a 7-h GH infusion terminated at either 03h or 09h as compared with no GH in adult hypopituitary patients with GH-deficiency. A reference group (Control) of healthy, age-matched and sex-matched volunteers was studied once without receiving any treatment. Panel a: GH levels. Panel b: FFA levels. Panel c: glucose infusion rate (GIR) as assessed by a euglycemic clamp starting at 08 h. The figure provides evidence that ongoing GH infusion (GH from 02:00 – 09:00) induces insulin resistance as demonstrated by the reduced GIR, and this is temporally associated with elevated FFA levels as shown in panel b. Adapted from a previously published study45 and used with permission.
In contrast to what has been reported in humans with insulin resistance induced by lipid infusion46, no evidence has been obtained from human studies that GH inhibits insulin signalling through insulin receptor substrate 1 (IRS1)-associated phosphatidylinositol 3-kinase (PI3K) and AKT47,48. Of note, this mechanism of action for insulin resistance has been reported in GH-treated rodents49,50. However, a study in healthy human volunteers has shown that GH infusion is accompanied by elevated serum FFA levels and suppresses pyruvate dehydrogenase activity, which indicates that substrate competition between glucose and lipid intermediates at the entry point of the citric acid cycle could be one causative mechanism of GH-mediated insulin resistance51.
These metabolic effects of GH also raise the question whether targeting the GH receptor (GHR) with either exogenous GH or the GH antagonist pegvisomant would prove useful for the treatment of insulin resistance and/or obesity. Indeed, a meta-analysis of studies of GH treatment for obesity in adults demonstrate a moderate but statistically significant reduction in adipose tissue mass, in particular visceral fat, together with a more favorable lipid profile and an increase in lean body mass as compared with placebo52. However, the analysis also shows that GH treatment concomitantly increases fasting levels of plasma glucose and insulin52. Conversely, pegvisomant treatment might be hypothesized to improve insulin sensitivity, which was reported in young patients with type 1 diabetes mellitus treated with pegvisomant for 4 weeks53. However, the treatment was associated with a pronounced reduction in serum IGFI levels towards the range seen in hypopituitarism, which is not surprising given that GH stimulates IGF1 production53. In patients with type 2 diabetes mellitus (T2DM) (without acromegaly), no experimental data are available, but pegvisomant therapy is likely to increase fat mass, which is not favorable in the context of T2DM. As such, it might be premature to conclude a role for either GH or pegvisomant beyond the licensed indications.
Taken together, over 50 years of research in healthy human volunteers and patients with GHD or acromegaly document a potent lipolytic effect of GH, which predominates in the fasting state and constitutes a physiologic mechanism to provide energy from fat depots. However, unregulated and sustained GH excess might cause elevated circulating FFA levels, resulting in overt glucose intolerance. The molecular mechanisms responsible for the lipolytic effects of GH remain unresolved although, as described below, current studies provide evidence of relevant pathways54,55.
GH-induced intracellular signalling
GH exerts its intracellular effects by binding to preformed, single-pass transmembrane receptor dimers on the cellular surface. Within adipose tissue, GHRs are expressed on preadipocytes, mature adipocytes and the assorted cells (including, fibroblasts, immune cells, and endothelial cells) that comprise the stromal vascular fraction56. The GHR does not have kinase activity; however, GHR is constitutively associated with two non-receptor tyrosine kinases, Janus kinase 2 (JAK2) and SRC kinase57. An important 2008 study showed that an agonist-induced conformational change in the GHR can determine the choice of the intracellular signalling pathway58. For example, GH binding to the GHR leads to a conformational change of the receptor, which unmasks the catalytic domains of JAK2 and enables transphosphorylation of adjacent JAK2 molecules59. Independent of JAK2, GH binding to the GHR also leads to autophosphorylation of SRC kinase58,60. Of note, the profound effects of GH on all tissues, including adipose tissue, is largely dependent on GH-induced activation of JAK2 and SRC kinase pathways. As discussed later, JAK2 and SRC signalling downstream of GH activate different signalling pathways and differentially regulate physiological processes within adipose tissue. Therefore, the ability of GH to activate specific pathways downstream of GHR binding holds interesting therapeutic potential.
Researchers investigating GH signalling pathways have extensively studied phosphorylation and activation of the signal transducer and activator of transcription (STAT) family of transcription factors downstream of JAK261–64. After GH binding to GHR and JAK2 transphosphorylation, JAK2 phosphorylates the GHR at several tyrosine residues leading to the recruitment and phosphorylation of several STAT proteins, including STAT1, STAT3, and STAT565,66. The phosphorylated and dimerized STAT proteins then translocate to the nucleus, where they bind to gamma-activated site (GAS) DNA motifs to regulate the expression of GH target genes. Of the various STAT proteins, two isoforms of STAT5 exist and appear to mediate the majority of the biological effects of GH65. These isoforms, STAT5A and STAT5B, have over 90% sequence homology67. In humans, STAT5B is the major mediator of GH action. Moreover, in both humans and mice, mutation or ablation of STAT5B leads to GH insensitivity68–71.
As mentioned earlier, phosphorylation and activation of SRC kinase through the GHR is independent of JAK260,72. Activated SRC kinase phosphorylates Protein Kinase C (PKC) and activates the extracellular signal regulated kinases (ERK1 and ERK2) and MAPK signalling pathways72. Through the MAPK–ERK pathway, GHR can also activate the PI3K–AKT pathway via phosphorylation of IRS1 and/or IRS2, as evidenced by studies in mice57. Of note, AKT-mediated activation of PDE3B would be expected to inhibit GH-induced lipolysis. However, our new observations show that a single bolus of GH is sufficient to repress PDE3B mRNA levels in subcutaneous adipose tissue of human volunteers (unpublished observation). Interestingly, bovine GH (bGH) transgenic mice have increased adipose tissue expression of the p85α regulatory subunit of PI3K, whereas the opposite is found in GH-deficient mice49. In bGH-expressing mice, the increased expression of p85α regulatory subunit homodimers can enable them to bind and sequester IRS173, thereby preventing the activation of PI3K and resulting in insulin resistance. However, as stated earlier, studies in humans have demonstrated that GH-induced insulin resistance is independent of effects on MAPK–ERK and PI3K signalling pathways45,47,48,74,75. Thus, GH-induced intracellular signalling substantially alters the physiology of adipose tissue through its actions on various metabolic pathways (Figure 1).
When evaluating the role of GH in any metabolic pathway, the interplay between GH and IGF1 must also be considered. In 1985, Howard Green and colleagues proposed “a dual effector theory of GH action”, in which GH induces preadipocyte differentiation, whereas IGF1 stimulates clonal expansion of the differentiated adipocytes76. Thus, GH and IGF1 have well established and distinct functions in adipose tissue. Importantly, there are other well-known activities that clearly differentiate GH versus IGF1 actions: first, GH is diabetogenic, whereas IGF1 is not6; second, GH stimulates glomerulosclerosis[G], whereas IGF1 does not77; and third, growth in mice is differentially influenced by the two hormones. For example, one report shows that GH contributes 14%, IGF1 contributes 35%, GH and IGF1 together contribute 34% and other factors contribute 17% to total mouse growth78. In the context of this Review, it is important to note that GH is lipolytic, whereas IGF1 is not. Thus, the role of IGF1 in lipolysis is not discussed; however, an indirect role of IGF1 on lipolysis is possible via an influence on adipose tissue structure and function (for example, adipokine expression, senescence, fibrosis and depot differences). Future studies are necessary to delineate the specific molecular targets specifically effected by either GH or IGF1 actions in adipose tissue.
Mechanisms of GH-induced lipolysis
Studies in humans, mice and cellular models have elucidated some of the molecular mechanisms of GH-induced lipolysis. For example, GH treatment in cultured adipocytes or patients with obesity has been shown to increase phosphorylation and activation of HSL to stimulate lipolysis79,80. During weight loss in patients with obesity, GH administration regulates HSL activity in adipose and muscle tissue80. In rat adipocytes, GH treatment decreases the levels of inhibitory G protein coupled receptor alpha subunits, resulting in a decrease in sensitivity to anti-lipolytic agents81,82. Also, GH treatment of human adipose tissue represses lipoprotein lipase activity both in vivo80 and in vitro38,83 (not shown in Figure 1). Interestingly, in adults with GHD, 1 month of GH treatment down-regulates mRNA expression of CIDEA84, a lipid droplet protein that protects against lipolysis84,85. Furthermore, G0S2 expression in adipose tissue is suppressed during fasting, where both GH secretion and lipolysis are elevated86, but it is unclear if this effect is directly mediated by GH84,86,87. Finally, either STAT5 or JAK2 ablation specifically in adipose tissue of mice decreases lipolysis88–90; however, this effect is not specific to GH-induced lipolysis, and the detailed molecular mechanisms underlying this reduction are unknown. The net effect is that GH strongly induces lipolysis in adipose tissue (Figure 1). Importantly, the above studies warrant further identification of the precise molecular pathways and lipolytic signals that are triggered by GH.
Studies from our research groups have identified a molecular mechanism that links GH-mediated lipolysis and insulin resistance in human adipocytes. In this work, we carried out experiments in human volunteers, mouse models and cultured cells to demonstrate that GH suppresses the expression of FSP27 at both the mRNA and protein level54,55 (Figure 1). Of note, FSP27 levels are associated with insulin sensitivity in humans with obesity26,85,91, and nonsense mutation of FSP27 in humans leads to increased lipolysis, hypertriglyceridemia and insulin resistant diabetes mellitus.92. In addition, adipose tissue-specific disruption of FSP27 causes insulin resistance in high fat diet-fed mice93.
The expression of FSP27 is regulated by peroxisome proliferator-activated receptor gamma (PPARγ)28,85,94. Our studies54,55 tested the hypothesis that GH transcriptionally controls the lipolytic flux of FFAs by affecting PPARγ activity to downregulate FSP27 in adipocytes. Indeed, GH-induced modulation of FSP27 expression is mediated through activation of both MEK–ERK and STAT5-dependent intracellular signalling pathways, which interact to differentially manipulate the activity of PPARγ on the FSP27 promoter54. For example, GH induces MEK–ERK activation, which causes PPARγ inactivation. As a counter-regulatory mechanism, GH induces phosphorylation of STAT5, which directly activates PPARγ. However, the GH-induced MEK–ERK pathway is the dominant pathway, leading to PPARγ inactivation, decreased FSP27 expression and thereby increasing lipolysis and insulin resistance55 (Figure 1). More studies are required to dissect the molecular interaction between FSP27 and PPARγ and to identify any additional mechanistic pathways that are regulated by GH to induce lipolysis.
Previous in vitro studies in human HEK293 cells have demonstrated that MAPK pathway activation decreases PPARγ transcriptional activity via MEK–ERK activation95, which leads to PPARγ downregulation95. Furthermore, many studies have linked PPARγ Ser273 phosphorylation with the development of insulin resistance in mice96–98. Interestingly, overexpression of FSP27 in cultured primary human adipocytes as well as exposure to a GHR antagonist, pegvisomant, can block MEK–ERK-mediated phosphorylation of PPARγ at Ser273, which stabilizes PPARγ in the nucleus and prevents GH-induced lipolysis and insulin resistance54,55. Thus, these studies identify a novel molecular MEK–ERK–PPARγ–FSP27 pathway that regulates GH-induced lipolysis and insulin resistance. In addition, the findings suggest that pegvisomant treatment maintains insulin sensitivity in patients with acromegaly, at least in part, through its regulation of FSP27 expression. Furthermore, the MEK–ERK and PPARγ-dependent mechanism suggests several molecular targets of intervention to reduce GH-dependent lipolysis. Many of these targets already have pharmacological agents in clinical use, including: MEK inhibitors (Trametinib), PPARγ agonists (Thiazolidinediones) and anti-lipolytic agents (Acipimox)8.
Insights from mouse models
Extremes in the GH–IGF1 axis in both humans and animal models provide valuable insight as to the actions of GH on adipose tissue in vivo. In particular, mouse models, due to their genetic tractability, have been instrumental in deciphering the physiological and metabolic effects of GH action.
Mouse models with altered GH activity.
Different mouse models have been generated with altered GH action (for select examples, see Table 1). For example, mice engineered to express bGH have elevated GH action. For a model of decreased GH action, researchers can utilize GHR antagonist (GHA) mice, which is similar to human congenital GH deficiency, and inducible adult GH deficiency models (aiGHRKO and AOiGHD mice), which are similar to adult human GHD. Finally, mouse models with no GH action are available, either with complete GH-resistance found in GHR gene disrupted (GHR–/–) mice or with GH-deficiency found in Ames and Snell dwarf mice (Table 1).
Table 1:
Characteristics of mouse lines with altered GH action
| Name of mouse line | bGH | LiGHRKO | FaGHRKO | AdGHRKO | AOiGHD | aiGHRKO | GHA | Ames and Snell dwarf mice | GHR–/– |
|---|---|---|---|---|---|---|---|---|---|
| GH alteration | Transgenic for bovine GH | Liver specific disruption of GHR | Fat specific disruption of GHR using ap2 promoter driving cre expression | Adipocyte-specific disruption of GHR using adipQ promoter driving cre expression | Adult GH deficiency via somatotroph destruction | Adult GH deficiency via adult inducible deletion of GHR | GHR antagonist gene | Homozygous recessive mutation in Pit-1 (Snell) or prop1 (Ames) | Disruption of GHR gene |
| General features | |||||||||
| GH action | ↑↑ | ↓ liver; ↑ all extrahepatic tissues | ↓ in AT only | ↓ in adipocytes only | ↓ (10–12 weeks) | ↓ (6 weeks) | ↓ | ↓ | Absent |
| GH | ↑↑ | ↑ | ↔ | ↔ | ↓ | ↑ | ↑ | ↓↓ | ↑ |
| IGF1 | ↑↑ | ↓ | ↑ male | ↔ | ↓ | ↓↓ | ↓ | ↓↓ | ↓↓ |
| Body weight | ↑↑ | ↓ | ↑ | ↔ | ↔ | ↓ | ↓ | ↓↓ | ↓↓ |
| Insulin sensitivity | ↓ | ↓ | ↔ | ↑ | ↑ | ↓ | ↓ ↔ | ↑ | ↑ |
| Lifespan | ↓ | ↔ | ↓ | N.d. | N.d. | ↔ male; ↑ female |
↔ | ↑↑ | ↑↑ |
| White adipose tissue features | |||||||||
| Mass | ↑ young ↓ old |
↑ young ↓ old |
↑↑ | ↑↑ | ↑ (after induction) | ↑ (after induction) | ↑↑ | ↑ | ↑↑ |
| Depot differences | All depots impacted | All depots impacted | All depots impacted | All depots impacted | Sc and Retro | sc | sc | sc | sc |
| Leptin | ↓ | ↑ female | ↑ female ↔ male |
↔ | ↑ | ↔ | ↑↑ | ↑ ↔ |
↑ |
| Adiponectin | ↓ | ↑ | ↓ male ↔ female |
↓ | ↔ | N.d. | ↑ | ↑ | ↑↑ |
| Resistin | ↓ | ↑ | ↔ | ↓ | N.d. | ↑ female | ↑ | ↔ | ↑ |
| Senescence | ↑ | N.d. | N.d. | N.d. | N.d. | N.d. | ↔ | N.d. | ↓ |
| Fibrosis | ↑ | ↑ | ↓ | ↓ | N.d. | N.d. | ↓ | N.d. | ↓ |
| Immune Cells** | |||||||||
| M1 | ↔ | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | ↑ |
| M2 | ↑ | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | ↑ |
| T cells | ↑ | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | N.d. | ↑ |
↑, increase; ↔, no change; ↓, decrease; GHR, growth hormone receptor, N.d., no data.
Sc, subcutaneous white adipose tissue; retro, retroperitoneal white adipose tissue; M1, classically activated macrophages; M2, alternatively activated macrophages
Data obtained from flow cytometry only
Although comparable human clinical conditions do not exist, tissue-specific mouse lines also allow one to explore the role of GH in selected tissues and/or cell types. Relevant to adipose tissue, two separate adipose tissue-specific GHR gene disrupted or knockout (FaGHRKO and AdGHRKO) mouse lines have been characterized99,100. In addition, as the majority of circulating IGF1 is produced by hepatocytes, liver-specific GHR knockout (LiGHRKO) mice allows the evaluation of other tissues, including adipose tissue, under conditions of elevated GH and low IGF-1. This condition resembles a fasting state and enables the in vivo molecular dissection of IGF1 independent and dependent roles of the GH signalling axis101 (Table 1). Of note, several of these mouse lines share features with clinical conditions (for example, bGH mice with acromegalic gigantism and GHR–/– mice with Laron Syndrome. However, in addition to size and species differences, most of the genetically engineered mice have life-long disruption of the GH–IGF1 axis and/or perturbations in the normal pulsatile action of GH.
In these mouse models, excess GH reduces overall adiposity whereas the opposite condition increases adiposity. For example, adult bGH mice and LiGHRKO mice are leaner than littermate controls, having notably less fat mass for most of their adult lives101–103. Conversely, adult mice with decreased GH action (that is, Ames, Snell, GHA, FaGHRKO, AdGHRKO, GHR–/–, AOiGHD and aiGHRKO) have increased fat mass99,102,104–108. Importantly, body composition trends are age-dependent and sex-dependent, highlighting the need to consider these factors in experimental design. For example, longitudinal body composition analyses in bGH mice show resistance to midlife gains in adipose tissue, but these mice have greater fat mass at younger ages (<3 months of age for males and <4 months of age in females), and bGH female mice have a delayed and less exaggerated difference in body composition as compared with bGH males103. In addition, LiGHRKO mice have a very similar trend to bGH mice in fat mass gains101. In contrast, Ames and Snell mice have increased adiposity104,109; however, this trend is attenuated in Ames mice at older ages106, whereas GHA mice continue to see fat mass gains with advancing age110.
Longitudinal body composition data are available for a variety of other lines including GHR–/–, FaGHRKO, AdGHRKO and aiGHRKO mice; all lines with reduced GH action in adipose tissue have increased fat mass throughout life, albeit the increase in adipose tissue mass is less exaggerated for females105,107,110. Upon challenge with a high fat diet, bGH mice are resistant to obesity, with preferential accumulation of lean tissue instead of adipose tissue111,112, whereas GHA, Ames and GHR–/– mice are more susceptible to gaining additional fat mass when compared with wild type mice109,111,113,114. Interestingly, despite increased obesity, Ames dwarf, GHA and GHR–/– mice are resistant to the detrimental effects of high fat feeding on glucose homeostasis and insulin sensitivity109,111,113,114, which is consistent with the idea of GH-induced lipolysis as a causative factor for insulin resistance.
Adipokine expression.
Adipokine secretion is altered by GH in both mice and humans, although the molecular pathways have yet to be fully elucidated. Interestingly, GHR–/– mice and patients with Laron Syndrome exhibit increased leptin levels, which mirrors the increase in adipose tissue mass. Similarly, reduced circulating leptin (which inhibits appetite) is observed in bGH transgenic mice and patients with acromegaly115,116. Likewise, acute GH stimulation of adipocytes in vitro leads to increased leptin secretion in a STAT5-independent manner117.
Similar to leptin, levels of circulating adiponectin (which regulates glucose and lipid metabolism) are increased in GHR–/– mice and patients with Laron Syndrome99,118. Although differences in the literature exist regarding adiponectin levels in patients with acromegaly, GH treatment in human adipose tissue in vitro and in rodents in vivo leads to reduced adiponectin expression and secretion119. This reduced expression of adiponectin can be attributed to a STAT5-mediated transcriptional repression on the adiponectin promoter120. By contrast, GH levels are inversely correlated to ghrelin (which increases appetite) levels; that is, GH treatment acutely decreases circulating ghrelin levels in children with GHD, and remission of acromegaly after surgical therapy increases total ghrelin levels121,122. Other adipokines have been evaluated in several of these mouse lines (reviewed in56).
Differences in adipose tissue depots.
Adipose tissue is a heterogeneous tissue with well-documented depot differences in cellular developmental origin, proliferative capacity, glucose and lipid metabolism, insulin sensitivity, fatty acid composition, cytokine pattern, thermogenic ability and vascularization. Importantly, key proteins in lipolysis, such PLIN1 and HSL, have expression levels that have been reported to vary not only by depot but also by obesity status123. Thus, it might not be surprising that the effect of GH on adipose tissue differs depending on the depot in both humans and mice. Interestingly, the depot most affected seems to vary by species. In humans, the visceral depot is the most affected by alterations in the action of GH, and the greatest reduction of white adipose tissue mass occurs in the visceral depot in patients with active acromegaly124. In addition, in humans with GHD, GH treatment decreases total body fat, with the largest decrease observed in the visceral depots125,126.
Much of our understanding of how GH affects adipose tissue in a depot-specific manner comes from studies done in mice, where multiple depots can be compared in a single animal. Overall, a reduction in GH action, as seen in GHR–/– and GHA mice, causes a striking and specific enlargement of the subcutaneous fat depot, whereas an excess in GH action appears to decrease the mass of all depots similarly102,107,110. Importantly, despite similar reductions in the mass of adipose tissue in these studies102,107,110, molecular signatures of the adipose tissue depots reveal a more significant genotype effect in subcutaneous depots as compared to other depots. For example, compared with wild type mice, the subcutaneous adipose tissue of bGH mice shows a striking increase in gene expression pathways related to T cell infiltration and activation, but only modest changes are seen in epididymal adipose tissue127. Furthermore, GHR–/– mice show subtle but distinctive expression signatures between epididymal and subcutaneous adipose tissue depots128. These data confirm that adipose tissue mass alone might not be a sufficient readout of the effect of GH on a specific adipose tissue depot.
Many examples in the literature support depot-specific differences at the cellular or molecular level (reviewed in ref.56,129). For example, histological analysis of adipose tissue sections from mouse lines with extremes in GH action show substantial alterations between the different models in morphology and adipocyte size in subcutaneous adipose tissue; however, the epididymal adipose tissue depot is fairly uniform among mouse lines56. Furthermore, the capacity of preadipocytes for proliferation and differentiation is dependent on the depot of origin of the isolated cells130. Of note, adipose tissue depot differences in insulin receptor, IGF1 receptor and GHR have also been reported, with highest IGF1 receptor (IGF1R) expression in epididymal and mesenteric depots, higher insulin receptor expression in retroperitoneal and mesenteric depots and highest GHR expression in the retroperitoneal depot131. This finding is important for the hormone responsiveness of specific depots. In addition, the levels of pregnancy-associated plasma protein-A (PAPPA), which might increase local IGF1 action through its proteolytic activity, are statistically significantly higher in visceral adipose tissue from adipose tissue explants from patients with obesity and mouse mesenteric adipose tissue samples from wild type, bGH and GHR–/– mice, which could also contribute to depot-specific responses to the GH–IGF1 axis132,133. Importantly, adipocyte-specific deletion of GHR versus IGF1R in mice134 suggests that the depot differences are mainly GH-dependent; that is, GHR deletion specifically in adipocytes using an adiponectin Cre promoter results in an increase in adipose tissue mass with the subcutaneous depot most effected. By contrast, IGF1R deletion in adipocytes is associated with a 25% reduction in adipose tissue mass that is uniform among the depots134. Collectively, these observations underscore the need to study more than one adipose tissue depot and other cell types in addition to adipocytes to fully understand the role that GH has on adipose tissue metabolism.
Emerging areas of research
Cellular senescence in adipose tissue.
Senescent cells are cells that undergo irreversible cell-cycle arrest, and the tissue accumulation of senescent cells is common during normal aging and in response to various metabolic stressors such as oncogene activation, DNA and oxidative damage, or metabolic insults like high glucose135. In addition to growth arrest, senescent cells produce a senescence-associated secretary phenotype, which is a complex mixture of secreted factors that include proinflammatory cytokines, chemokines, growth factors and proteases136. Collectively, the accumulated senescent cells and their secretory products induce a potent proinflammatory state that alters tissue inflammation, angiogenesis and fibrosis135. The cellular growth arrest occurring in senescence is seemingly regulated through two main pathways, p16INK4a–Rb and p53–p21CIP1 (REF137). Importantly, administration of senolytic agents, which selectively eliminate senescence cells, have been shown to improve physical function and increase longevity in mice138.
Increases in cellular senescence are commonly associated with obesity and type 2 diabetes mellitus139. Furthermore, the accumulation of these cells can have a major effect on age-related adipose tissue dysfunction, leading to lipotoxicity and chronic inflammation, irrespective of adipose tissue mass140,141. Interestingly, GH action is positively correlated with the accumulation of senescent cells in adipose tissue of mice. More specifically, bGH mice (10 months of age) and GH-injected (19 months of age) mice accumulate more senescent cells and have higher p16INK4a expression in adipose tissue than their littermate controls142. Conversely, primary subcutaneous preadipocytes from 18-month old GHR–/–, Ames and Snell dwarf mice, exhibit decreased burden of senescent cells and lower p16 INK4a expression (a marker of senescence) in most adipose tissue depots compared with control littermates142. In contrast to 18-month old GH deficient Ames dwarf mice and GHR–/–mice, 18 month old GHA mice do not have any change in adipose tissue senescent cell burden despite considerable obesity143. This finding suggests that senescent cell burden is more related to aging than to the level of GH action. Alternatively, the protection against generating senescent cells afforded by decreased GH action, low IGF1 or improved insulin sensitivity in the GHA mice is counterbalanced by the extreme obesity of GHA mice with advancing age.
Of note, the p53 tumour suppressor pathway is also upregulated by GH in adipose tissue, and this pathway has been suggested to mediate the insulin resistance seen with acromegaly in bGH mice144; however, the influence of p53 on adipose tissue senescence is not established in these studies. Relevant to this Review, an increase in the number of senescent preadipocytes contributes to a decline in insulin responsiveness and increase the overall lipotoxicity in the tissue145,146. Taken together, GH-induced alteration in senescent cells probably influences the lipolytic potential of adipose tissue, although this hypothesis has yet to be thoroughly studied.
Adipose tissue fibrosis.
An important non-cellular component of white adipose tissue is the extracellular matrix (ECM), which surrounds individual adipocytes and provides structural support for the tissue. Of note, adipose tissue is one of the few tissues in the body that undergoes constant alterations in cell size, which requires a flexible, accommodating ECM. Therefore, when an excessive accumulation of ECM proteins or fibrosis occurs, the rigid ECM impedes adipocyte growth and promotes local and systemic pathologies, including chronic inflammation, immune cell recruitment, insulin resistance and cell death147,148. Interestingly, a 2018 study from our research group showed that GH action is positively correlated with ECM deposition. For example, bGH mice and LiGHRKO have excess fibrosis and collagen deposition in adipose tissue, whereas GHA, AdGHRKO and FaGHRKO mice (Table 1) have reduced collagen content100,149. This phenomenon is most prominent in the subcutaneous fat depot and correlated better with GH action rather than IGF1 action. Whether the increase in fibrosis is a direct effect of GH or an indirect effect of the hormone’s potent lipolytic action remains to be determined. Regardless, the marked fibrosis in bGH mice probably contributes to the limited ability of these mice to store lipids in adipose tissue and might influence the capacity of a depot for lipolysis.
Intra-depot adipocyte heterogeneity.
In addition to depot specific differences found in adipose tissue, individual white adipocytes within a single depot might differ in insulin-stimulated glucose uptake, maximal lipogenic rate, response to catecholamines, uptake of free fatty acids, as well as regulation of oxidative phosphorylation and glycolysis149–155. Indeed, lineage tracing analyses have demonstrated that adipocytes, even within a single adipose tissue, develop from different developmental origins156,157. These studies are further supported by genetic mouse models, including ablation of HSL and fat-specific knockout of the insulin receptor, which both lead to a bimodal distribution of adipocyte cell size158,159. Similarly, GH excess in bGH mice alters adipocyte size distribution in subcutaneous adipose tissue, yielding a subset of very small adipocytes149. Thus, the genetic changes in these mouse models unmask an intrinsic heterogeneity within white adipocytes of a single adipose tissue depot.
In a 2019 study, we identified three distinct subpopulations of white adipocytes that have clear phenotypic and functional differences, which we termed Type 1, Type 2 and Type 3 adipocytes160. In comparison to Type 1 cells, Type 2 and 3 adipocytes display increased insulin-mediated glucose uptake and tend to have increased de novo lipogenesis. By contrast, stimulation of Stat5-Tyr694 phosphorylation by GH is highest in Type 2 adipocytes160. As insulin and GH positively and negatively regulate adipocyte size, respectively, we hypothesize that the differential responses of these adipocyte subpopulations might, at least in part, contribute to the changes in size distribution observed upon adipose tissue-specific ablation of insulin receptor or in bGH mice. Thus, in addition to depot-specific effects, distinct responses of adipocyte subpopulations might mediate GH action in adipose tissue. Furthermore, a 2019 study utilizing transcriptomic profiling of clonally grown mesenchymal progenitor cells identified four adipocyte subtypes in humans, suggesting that adipocyte subpopulations might also mediate physiological responses in humans161.
Conclusions
From the historic work in humans showing GH activation of FFA flux, to mechanistic studies that show the inhibiting effect of GH on insulin-induced lipogenesis, to the most recent work depicting the effect of GH effect on FSP27 and PPARγ, we now have a clearer understanding of additional molecular ‘players’ in GH-induced lipolysis. To complement this work, data derived from mice with altered GH action has led to the discovery of differential effects of GH on adipose tissue depots as well as the participation of GH in mediating collagen deposition and senescence, primarily in subcutaneous fat depots. New and exciting data that reveal different populations of adipocytes in white adipose tissue depots and perhaps the ability of GH to target its lipolytic effect to a subpopulation of adipocytes in a given depot are indeed intriguing.
GH is well known to strongly induce whole body lipolysis and the molecular mechanisms responsible for the lipolytic action of GH within the adipocyte are now better understood. However, future studies that consider the surrounding tissue milieu and inherent subcellular differences on these molecular processes should be evaluated. For example, researchers could evaluate the influence of the unique adipokine profile induced by GH on lipolysis, or the responsiveness of distinct adipocyte subpopulations to GH as well as the variation in lipolytic and lipogenic machinery among these subpopulations. Also of interest are GH-induced alterations in senescent cells, the senescent associated secretory proteins, and depot dependent differences in adipose tissue fibrosis. All of these factors probably directly or indirectly influence the balance of adipogenic, lipogenic and lipolytic capacity of adipose tissue. As the pharmacological inhibition of lipolysis restores insulin sensitivity during GH exposure in humans8 and FSP27 overexpression inhibits GH-induced insulin resistance in human adipocytes, the regulation of GH action in adipose tissue might be a valuable therapeutic target to improve metabolic health in patients with acromegaly.
In this Review, we have updated the decades’ old observation of the effects of GH on adipose tissue and described new molecular and cellular mechanisms. A common denominator of the metabolic effects of GH on adipose tissue lipolysis appears to be insulin antagonism leading to fat loss as well as reduced glucose uptake. Such actions are also potential targets for treating our major health threats of obesity and T2DM.
Key points.
GH exposure in humans potently stimulates the release of free fatty acids (FFAs) from adipose tissue into the circulation after a lag phase of 1–2 hours and with a peak effect after 3–4 hrs.
This GH-induced increase in circulating FFAs is causally linked to the antagonistic effects of GH on basal and insulin-stimulated glucose uptake.
Overexpression of FSP27 or exposure to a GHR antagonist, pegvisomant, can block the diabetogenic effects of GH.
GH-induced activation of the MEK–ERK pathway has a key role in PPARγ inactivation and FSP27 downregulaton, thus increasing lipolysis and insulin resistance.
GH impacts adipose tissue in a depot-specific manner and influences other features of adipose tissue (for example, senescence, adipocyte subpopulations and fibrosis) all of which could influence lipolysis.
Acknowledgements
J.J.K. acknowledges the support of the state of Ohio’s Eminent Scholar Program that includes a gift from Milton and Lawrence Goll and AMVETS. J.J.K. and D.E.B. acknowledge the support of NIH/NIA AG059779, The Edison Biotechnology Institute and Diabetes Institute at Ohio University. V.P. acknowledges the support of NIH/NIDDK grant DK10171, NIH/NHLBI HL139049 and funds from Osteopathic Heritage Foundation’s Vision 2020 to Heritage College of Osteopathic Medicine at Ohio University. K.Y.L. acknowledges the support of start-up funds from Ohio University Heritage College of Osteopathic Medicine, the Ohio University Diabetes Institute and the American Diabetes Association Junior Faculty Development Award 1-17-JDF-055.
GLOSSARY
- GH-deficiency
a rare disorder characterized by the inadequate secretion of GH and can be categorized into congenital or acquired, and/or childhood or adult-onset
- Acromegaly
a condition caused by hypersecretion of GH from a pituitary tumor that is managed by surgical tumor removal or medical control
- Glomerulosclerosis
a degenerative process that involves scarring within the renal glomeruli of the kidney
- Laron Syndrome
a rare condition of GH resistance characterized by short stature, which is often caused by mutations in the GHR gene and is inherited in an autosomal recessive manner
- Panhypopituitarism
Inadequate production or absence of anterior pituitary hormones. Also known as hypopituitarism
- Senolytic agents
small molecules that can selectively target and kill senescent cells
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brockmann GA & Bevova MR Using mouse models to dissect the genetics of obesity. Trends Genet 18, 367–376, (2002). [DOI] [PubMed] [Google Scholar]
- 2.Friedman JM A war on obesity, not the obese. Science 299, 856–858, (2003). [DOI] [PubMed] [Google Scholar]
- 3.Moller N & Jorgensen JO Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 30, 152–177, (2009). [DOI] [PubMed] [Google Scholar]
- 4.Luft R, Ikkos D, Gemzell CA & Olivecrona H Effect of human growth hormone in hypophysectomised diabetic subjects. Lancet 1, 721–722, (1958). [DOI] [PubMed] [Google Scholar]
- 5.Davidson MB Effect of growth hormone on carbohydrate and lipid metabolism. Endocr Rev 8, 115–131, (1987). [DOI] [PubMed] [Google Scholar]; This classic review provides a historical perspective and a thorough understanding of early studies on the role of GH on carbohydrate and lipid metabolism.
- 6.Rabinowitz D & Zierler KL A Metabolic Regulating Device Based on the Actions of Human Growth Hormone and of Insulin, Singly and Together, on the Human Forearm. Nature 199, 913–915, (1963). [DOI] [PubMed] [Google Scholar]
- 7.Tunaru S et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9, 352–355, (2003). [DOI] [PubMed] [Google Scholar]
- 8.Nielsen S, Moller N, Christiansen JS & Jorgensen JO Pharmacological antilipolysis restores insulin sensitivity during growth hormone exposure. Diabetes 50, 2301–2308, (2001). [DOI] [PubMed] [Google Scholar]; The lipolytic action of GH is responsible for its reduction of insulin sensitivity in human volunteers.
- 9.Unger RH, Clark GO, Scherer PE & Orci L Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta 1801, 209–214, (2010). [DOI] [PubMed] [Google Scholar]
- 10.Boden G Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 46, 3–10, (1997). [PubMed] [Google Scholar]
- 11.Boden G, Chen X, Ruiz J, White JV & Rossetti L Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93, 2438–2446, (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dresner A et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103, 253–259, (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrannini E, Barrett EJ, Bevilacqua S & DeFronzo RA Effect of fatty acids on glucose production and utilization in man. J Clin Invest 72, 1737–1747, (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajala MW & Scherer PE Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144, 3765–3773, (2003). [DOI] [PubMed] [Google Scholar]
- 15.Ahmadian M, Wang Y & Sul HS Lipolysis in adipocytes. Int J Biochem Cell Biol 42, 555–559, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkins CM et al. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279, 48968–48975, (2004). [DOI] [PubMed] [Google Scholar]
- 17.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH & Sul HS Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279, 47066–47075, (2004). [DOI] [PubMed] [Google Scholar]
- 18.Zechner R et al. FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15, 279–291, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmermann R et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386, (2004). [DOI] [PubMed] [Google Scholar]
- 20.Granneman JG, Moore HP, Krishnamoorthy R & Rathod M Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem 284, 34538–34544, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lass A et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 3, 309–319, (2006). [DOI] [PubMed] [Google Scholar]
- 22.Subramanian V et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem 279, 42062–42071, (2004). [DOI] [PubMed] [Google Scholar]
- 23.Miyoshi H et al. Control of adipose triglyceride lipase action by serine 517 of perilipin A globally regulates protein kinase A-stimulated lipolysis in adipocytes. J Biol Chem 282, 996–1002, (2007). [DOI] [PubMed] [Google Scholar]
- 24.Yang X et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 11, 194–205, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong J et al. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol 195, 953–963, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller P et al. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem 283, 14355–14365, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JY et al. Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am J Physiol Endocrinol Metab 294, E654–667, (2008). [DOI] [PubMed] [Google Scholar]
- 28.Puri V et al. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem 282, 34213–34218, (2007). [DOI] [PubMed] [Google Scholar]
- 29.Nishino N et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest 118, 2808–2821, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grahn TH et al. Fat-specific protein 27 (FSP27) interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J Biol Chem 289, 12029–12039, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh M et al. Fat-specific protein 27 inhibits lipolysis by facilitating the inhibitory effect of transcription factor Egr1 on transcription of adipose triglyceride lipase. J Biol Chem 289, 14481–14487, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raben MS Growth hormone. 1. Physiologic aspects. N Engl J Med 266, 31–35, (1962). [DOI] [PubMed] [Google Scholar]
- 33.Raben MS & Hollenberg CH Effect of growth hormone on plasma fatty acids. J Clin Invest 38, 484–488, (1959). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glick SM, Roth J, Yalow RS & Berson SA Immunoassay of Human Growth Hormone in Plasma. Nature 199, 784–787, (1963). [DOI] [PubMed] [Google Scholar]
- 35.Giustina A & Veldhuis JD Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 19, 717–797, (1998). [DOI] [PubMed] [Google Scholar]
- 36.Roth J, Glick SM, Yalow RS & Bersonsa. Hypoglycemia: a potent stimulus to secretion of growth hormone. Science 140, 987–988, (1963). [DOI] [PubMed] [Google Scholar]
- 37.Zierler KL & Rabinowitz D Roles of Insulin and Growth Hormone, Based on Studies of Forearm Metabolism in Man. Medicine (Baltimore) 42, 385–402, (1963). [DOI] [PubMed] [Google Scholar]
- 38.Richelsen B et al. Growth hormone treatment of obese women for 5 wk: effect on body composition and adipose tissue LPL activity. Am J Physiol 266, E211–216, (1994). [DOI] [PubMed] [Google Scholar]
- 39.Krag MB et al. Growth hormone-induced insulin resistance is associated with increased intramyocellular triglyceride content but unaltered VLDL-triglyceride kinetics. Am J Physiol Endocrinol Metab 292, E920–927, (2007). [DOI] [PubMed] [Google Scholar]
- 40.Norrelund H et al. Effects of GH on urea, glucose and lipid metabolism, and insulin sensitivity during fasting in GH-deficient patients. Am J Physiol Endocrinol Metab 285, E737–743, (2003). [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen JO et al. Marked effects of sustained low growth hormone (GH) levels on day-to-day fuel metabolism: studies in GH-deficient patients and healthy untreated subjects. J Clin Endocrinol Metab 77, 1589–1596, (1993). [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen JO et al. Growth hormone versus placebo treatment for one year in growth hormone deficient adults: increase in exercise capacity and normalization of body composition. Clin Endocrinol (Oxf) 45, 681–688, (1996). [DOI] [PubMed] [Google Scholar]
- 43.Moller N et al. Basal- and insulin-stimulated substrate metabolism in patients with active acromegaly before and after adenomectomy. J Clin Endocrinol Metab 74, 1012–1019, (1992). [DOI] [PubMed] [Google Scholar]
- 44.Bredella MA et al. Body Composition and Ectopic Lipid Changes With Biochemical Control of Acromegaly. J Clin Endocrinol Metab 102, 4218–4225, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krusenstjerna-Hafstrom T et al. Growth hormone (GH)-induced insulin resistance is rapidly reversible: an experimental study in GH-deficient adults. J Clin Endocrinol Metab 96, 2548–2557, (2011). [DOI] [PubMed] [Google Scholar]
- 46.Shulman GI Unraveling the cellular mechanism of insulin resistance in humans: new insights from magnetic resonance spectroscopy. Physiology (Bethesda) 19, 183–190, (2004). [DOI] [PubMed] [Google Scholar]
- 47.Nielsen C et al. Growth hormone signaling in vivo in human muscle and adipose tissue: impact of insulin, substrate background, and growth hormone receptor blockade. J Clin Endocrinol Metab 93, 2842–2850, (2008). [DOI] [PubMed] [Google Scholar]
- 48.Jessen N et al. Evidence against a role for insulin-signaling proteins PI 3-kinase and Akt in insulin resistance in human skeletal muscle induced by short-term GH infusion. Am J Physiol Endocrinol Metab 288, E194–199, (2005). [DOI] [PubMed] [Google Scholar]
- 49.del Rincon JP et al. Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes 56, 1638–1646, (2007). [DOI] [PubMed] [Google Scholar]
- 50.Dominici FP et al. Influence of the crosstalk between growth hormone and insulin signalling on the modulation of insulin sensitivity. Growth Horm IGF Res 15, 324–336, (2005). [DOI] [PubMed] [Google Scholar]
- 51.Nellemann B et al. Growth hormone-induced insulin resistance in human subjects involves reduced pyruvate dehydrogenase activity. Acta Physiol (Oxf) 210, 392–402, (2014). [DOI] [PubMed] [Google Scholar]
- 52.Mekala KC & Tritos NA Effects of recombinant human growth hormone therapy in obesity in adults: a meta analysis. J Clin Endocrinol Metab 94, 130–137, (2009). [DOI] [PubMed] [Google Scholar]
- 53.Thankamony A et al. Short-term administration of pegvisomant improves hepatic insulin sensitivity and reduces soleus muscle intramyocellular lipid content in young adults with type 1 diabetes. J Clin Endocrinol Metab 99, 639–647, (2014). [DOI] [PubMed] [Google Scholar]
- 54.Sharma R et al. Growth hormone controls lipolysis by regulation of FSP27 expression. J Endocrinol 239, 289–301, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; In both mice and cell culture models, GH regulates lipolysis and insulin sensitivity employing ERK and STAT5 dependent mechanisms to control PPARγ mediated transcription of FSP27.
- 55.Sharma VM et al. Growth hormone acts along the PPARgamma-FSP27 axis to stimulate lipolysis in human adipocytes. Am J Physiol Endocrinol Metab 316, E34–E42, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; In human adipocytes, GH regulates lipolysis through ERK-dependent phosphorylation of PPARγ and transcriptional regulation of FSP27.
- 56.Troike KM et al. Impact of Growth Hormone on Regulation of Adipose Tissue. Compr Physiol 7, 819–840, (2017). [DOI] [PubMed] [Google Scholar]
- 57.Brooks AJ & Waters MJ The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol 6, 515–525, (2010). [DOI] [PubMed] [Google Scholar]; This excellent article provides a thorough understanding of the GH–GHR interaction as a function of downstream intracellular signalling.
- 58.Rowlinson SW et al. An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat Cell Biol 10, 740–747, (2008). [DOI] [PubMed] [Google Scholar]; The GH–GHR interaction can activate both STAT5 and ERK dependent intracellular signalling pathways. In light of findings that GH-lipolysis and insulin resistance is primarily dependent on ERK-dependent signaling, specific and yet to be discovered ERK-dependent GH analogs might have strong therapeutic and clinical significance.
- 59.Waters MJ The growth hormone receptor. Growth Horm IGF Res 28, 6–10, (2016). [DOI] [PubMed] [Google Scholar]
- 60.Lanning NJ & Carter-Su C Recent advances in growth hormone signaling. Rev Endocr Metab Disord 7, 225–235, (2006). [DOI] [PubMed] [Google Scholar]
- 61.Herrington J, Smit LS, Schwartz J & Carter-Su C The role of STAT proteins in growth hormone signaling. Oncogene 19, 2585–2597, (2000). [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Darus CJ, Xu BC & Kopchick JJ Identification of growth hormone receptor (GHR) tyrosine residues required for GHR phosphorylation and JAK2 and STAT5 activation. Mol Endocrinol 10, 1249–1260, (1996). [DOI] [PubMed] [Google Scholar]
- 63.Xu BC, Wang X, Darus CJ & Kopchick JJ Growth hormone promotes the association of transcription factor STAT5 with the growth hormone receptor. J Biol Chem 271, 19768–19773, (1996). [PubMed] [Google Scholar]
- 64.Hansen LH et al. Identification of tyrosine residues in the intracellular domain of the growth hormone receptor required for transcriptional signaling and Stat5 activation. J Biol Chem 271, 12669–12673, (1996). [DOI] [PubMed] [Google Scholar]
- 65.Ram PA, Park SH, Choi HK & Waxman DJ Growth hormone activation of Stat 1, Stat 3, and Stat 5 in rat liver. Differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phosphorylation. J Biol Chem 271, 5929–5940, (1996). [DOI] [PubMed] [Google Scholar]
- 66.Smit LS et al. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol Endocrinol 10, 519–533, (1996). [DOI] [PubMed] [Google Scholar]
- 67.Moriggl R et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol 16, 5691–5700, (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davey HW, Park SH, Grattan DR, McLachlan MJ & Waxman DJ STAT5b-deficient mice are growth hormone pulse-resistant. Role of STAT5b in sex-specific liver p450 expression. J Biol Chem 274, 35331–35336, (1999). [DOI] [PubMed] [Google Scholar]
- 69.Davey HW et al. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology 142, 3836–3841, (2001). [DOI] [PubMed] [Google Scholar]
- 70.Kofoed EM et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 349, 1139–1147, (2003). [DOI] [PubMed] [Google Scholar]
- 71.Scalco RC et al. Growth hormone insensitivity with immune dysfunction caused by a STAT5B mutation in the south of Brazil: evidence for a founder effect. Genet Mol Biol 40, 436–441, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu T, Ling L & Lobie PE Identification of a JAK2-independent pathway regulating growth hormone (GH)-stimulated p44/42 mitogen-activated protein kinase activity. GH activation of Ral and phospholipase D is Src-dependent. J Biol Chem 277, 45592–45603, (2002). [DOI] [PubMed] [Google Scholar]
- 73.Ueki K et al. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol 22, 965–977, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorgensen JO et al. GH receptor signaling in skeletal muscle and adipose tissue in human subjects following exposure to an intravenous GH bolus. Am J Physiol Endocrinol Metab 291, E899–905, (2006). [DOI] [PubMed] [Google Scholar]
- 75.Krusenstjerna-Hafstrom T et al. Insulin and GH signaling in human skeletal muscle in vivo following exogenous GH exposure: impact of an oral glucose load. PLoS One 6, e19392, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Green H, Morikawa M & Nixon T A dual effector theory of growth-hormone action. Differentiation 29, 195–198, (1985). [DOI] [PubMed] [Google Scholar]
- 77.Doi T et al. Glomerular lesions in mice transgenic for growth hormone and insulinlike growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am J Pathol 137, 541–552, (1990). [PMC free article] [PubMed] [Google Scholar]
- 78.Lupu F, Terwilliger JD, Lee K, Segre GV & Efstratiadis A Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol 229, 141–162, (2001). [DOI] [PubMed] [Google Scholar]; These studies systematically address the specific contribution of the IGF1 versus GH on linear growth
- 79.Dietz J & Schwartz J Growth hormone alters lipolysis and hormone-sensitive lipase activity in 3T3-F442A adipocytes. Metabolism 40, 800–806, (1991). [DOI] [PubMed] [Google Scholar]
- 80.Richelsen B et al. Regulation of lipoprotein lipase and hormone-sensitive lipase activity and gene expression in adipose and muscle tissue by growth hormone treatment during weight loss in obese patients. Metabolism 49, 906–911, (2000). [DOI] [PubMed] [Google Scholar]
- 81.Doris R, Vernon RG, Houslay MD & Kilgour E Growth hormone decreases the response to anti-lipolytic agonists and decreases the levels of Gi2 in rat adipocytes. Biochem J 297 (Pt 1), 41–45, (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yip RG & Goodman HM Growth hormone and dexamethasone stimulate lipolysis and activate adenylyl cyclase in rat adipocytes by selectively shifting Gi alpha2 to lower density membrane fractions. Endocrinology 140, 1219–1227, (1999). [DOI] [PubMed] [Google Scholar]
- 83.Ottosson M et al. Growth hormone inhibits lipoprotein lipase activity in human adipose tissue. J Clin Endocrinol Metab 80, 936–941, (1995). [DOI] [PubMed] [Google Scholar]
- 84.Zhao JT et al. Identification of novel GH-regulated pathway of lipid metabolism in adipose tissue: a gene expression study in hypopituitary men. J Clin Endocrinol Metab 96, E1188–1196, (2011). [DOI] [PubMed] [Google Scholar]
- 85.Puri V et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci U S A 105, 7833–7838, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nielsen TS et al. Fasting, but not exercise, increases adipose triglyceride lipase (ATGL) protein and reduces G(0)/G(1) switch gene 2 (G0S2) protein and mRNA content in human adipose tissue. J Clin Endocrinol Metab 96, E1293–1297, (2011). [DOI] [PubMed] [Google Scholar]
- 87.Pedersen MH et al. Substrate Metabolism and Insulin Sensitivity During Fasting in Obese Human Subjects: Impact of GH Blockade. J Clin Endocrinol Metab 102, 1340–1349, (2017). [DOI] [PubMed] [Google Scholar]
- 88.Kaltenecker D et al. Adipocyte STAT5 deficiency promotes adiposity and impairs lipid mobilisation in mice. Diabetologia 60, 296–305, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nordstrom SM, Tran JL, Sos BC, Wagner KU & Weiss EJ Disruption of JAK2 in adipocytes impairs lipolysis and improves fatty liver in mice with elevated GH. Mol Endocrinol 27, 1333–1342, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi SY et al. Adipocyte-specific deficiency of Janus kinase (JAK) 2 in mice impairs lipolysis and increases body weight, and leads to insulin resistance with ageing. Diabetologia 57, 1016–1026, (2014). [DOI] [PubMed] [Google Scholar]
- 91.Slayton M, Gupta A, Balakrishnan B & Puri V CIDE Proteins in Human Health and Disease. Cells 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rubio-Cabezas O et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med 1, 280–287, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanaka N et al. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J Biol Chem 290, 3092–3105, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zandbergen F et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem J 392, 313–324, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burgermeister E et al. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol 27, 803–817, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banks AS et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi JH et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li P et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 147, 815–826, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.List EO et al. The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Molecular Endocrinology 27, 524–535, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.List EO et al. Adipocyte-Specific GH Receptor-Null (AdGHRKO) Mice Have Enhanced Insulin Sensitivity With Reduced Liver Triglycerides. Endocrinology 160, 68–80, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.List EO et al. Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology 155, 1793–1805, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berryman DE et al. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res 14, 309–318, (2004). [DOI] [PubMed] [Google Scholar]
- 103.Palmer AJ et al. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology 150, 1353–1360, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bartke A Impact of reduced insulin-like growth factor-1/insulin signaling on aging in mammals: novel findings. Aging Cell 7, 285–290, (2008). [DOI] [PubMed] [Google Scholar]
- 105.Berryman DE et al. Two-year body composition analyses of long-lived GHR null mice. J Gerontol A Biol Sci Med Sci 65, 31–40, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heiman ML, Tinsley FC, Mattison JA, Hauck S & Bartke A Body composition of prolactin-, growth hormone, and thyrotropin-deficient Ames dwarf mice. Endocrine 20, 149–154, (2003). [DOI] [PubMed] [Google Scholar]
- 107.Junnila RK et al. Disruption of the GH Receptor Gene in Adult Mice Increases Maximal Lifespan in Females. Endocrinology 157, 4502–4513, (2016). [DOI] [PubMed] [Google Scholar]
- 108.Luque RM et al. Metabolic Impact of Adult-Onset, Isolated, Growth Hormone Deficiency (AOiGHD) Due to Destruction of Pituitary Somatotropes. PLoS One 6, e15767, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hill CM et al. Long-lived hypopituitary Ames dwarf mice are resistant to the detrimental effects of high-fat diet on metabolic function and energy expenditure. Aging Cell 15, 509–521, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berryman DE, Lubbers ER, Magon V, List EO & Kopchick JJ A dwarf mouse model with decreased GH/IGF-1 activity that does not experience life-span extension: potential impact of increased adiposity, leptin, and insulin with advancing age. Journals of Gerontology. Series A, Biological Sciences and Medical Sciences 69, 131–141, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berryman DE et al. Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology 147, 2801–2808, (2006). [DOI] [PubMed] [Google Scholar]
- 112.Olsson B et al. Bovine growth hormone transgenic mice are resistant to diet-induced obesity but develop hyperphagia, dyslipidemia, and diabetes on a high-fat diet. Endocrinology 146, 920–930, (2005). [DOI] [PubMed] [Google Scholar]
- 113.Robertson K, Kopchick JJ & Liu JL Growth hormone receptor gene deficiency causes delayed insulin responsiveness in skeletal muscles without affecting compensatory islet cell overgrowth in obese mice. Am J Physiol Endocrinol Metab 291, E491–498, (2006). [DOI] [PubMed] [Google Scholar]
- 114.Yang T et al. Growth hormone receptor antagonist transgenic mice are protected from hyperinsulinemia and glucose intolerance despite obesity when placed on a HF diet. Endocrinology 156, 555–564, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Silha JV et al. Perturbations in adiponectin, leptin and resistin levels in acromegaly: lack of correlation with insulin resistance. Clin Endocrinol (Oxf) 58, 736–742, (2003). [DOI] [PubMed] [Google Scholar]
- 116.Ueland T et al. Associations between body composition, circulating interleukin-1 receptor antagonist, osteocalcin, and insulin metabolism in active acromegaly. J Clin Endocrinol Metab 95, 361–368, (2010). [DOI] [PubMed] [Google Scholar]
- 117.Fain JN, Ihle JH & Bahouth SW Stimulation of lipolysis but not of leptin release by growth hormone is abolished in adipose tissue from Stat5a and b knockout mice. Biochem Biophys Res Commun 263, 201–205, (1999). [DOI] [PubMed] [Google Scholar]
- 118.Kanety H et al. Total and high molecular weight adiponectin are elevated in patients with Laron syndrome despite marked obesity. Eur J Endocrinol 161, 837–844, (2009). [DOI] [PubMed] [Google Scholar]
- 119.Nilsson L et al. Prolactin and growth hormone regulate adiponectin secretion and receptor expression in adipose tissue. Biochem Biophys Res Commun 331, 1120–1126, (2005). [DOI] [PubMed] [Google Scholar]
- 120.White UA, Maier J, Zhao P, Richard AJ & Stephens JM The modulation of adiponectin by STAT5-activating hormones. Am J Physiol Endocrinol Metab 310, E129–136, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eden Engstrom B, Burman P, Holdstock C & Karlsson FA Effects of growth hormone (GH) on ghrelin, leptin, and adiponectin in GH-deficient patients. J Clin Endocrinol Metab 88, 5193–5198, (2003). [DOI] [PubMed] [Google Scholar]
- 122.Reyes-Vidal C et al. Prospective study of surgical treatment of acromegaly: effects on ghrelin, weight, adiposity, and markers of CV risk. J Clin Endocrinol Metab 99, 4124–4132, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ray H, Pinteur C, Frering V, Beylot M & Large V Depot-specific differences in perilipin and hormone-sensitive lipase expression in lean and obese. Lipids Health Dis 8, 58, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Freda PU et al. Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J Clin Endocrinol Metab 93, 2334–2343, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bengtsson BA et al. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J Clin Endocrinol Metab 76, 309–317., (1993). [DOI] [PubMed] [Google Scholar]
- 126.Johannsson G et al. Growth hormone treatment of abdominally obese men reduces abdominal fat mass, improves glucose and lipoprotein metabolism, and reduces diastolic blood pressure. J Clin Endocrinol Metab 82, 727–734, (1997). [DOI] [PubMed] [Google Scholar]
- 127.Benencia F et al. Male Bovine GH Transgenic Mice Have Decreased Adiposity With an Adipose Depot-Specific Increase in Immune Cell Populations. Endocrinology 156, 1794–1803, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stout MB et al. Transcriptome profiling reveals divergent expression shifts in brown and white adipose tissue from long-lived GHRKO mice. Oncotarget 6, 26702–26715, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Berryman DE & List EO Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. Int J Mol Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Flint DJ, Binart N, Kopchick J & Kelly P Effects of growth hormone and prolactin on adipose tissue development and function. Pituitary 6, 97–102, (2003). [DOI] [PubMed] [Google Scholar]
- 131.Hjortebjerg R et al. Insulin, IGF-1, and GH Receptors are Altered in an Adipose Tissue Depot-Specific Manner in Male Mice with Modified GH Action. Endocrinology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gude MF et al. PAPP-A, IGFBP-4 and IGF-II are secreted by human adipose tissue cultures in a depot-specific manner. Eur J Endocrinol 175, 509–519, (2016). [DOI] [PubMed] [Google Scholar]
- 133.Hjortebjerg R et al. Depot-specific and GH-dependent regulation of IGF binding protein-4, pregnancy-associated plasma protein-A, and stanniocalcin-2 in murine adipose tissue. Growth Horm IGF Res 39, 54–61, (2018). [DOI] [PubMed] [Google Scholar]
- 134.Boucher J et al. Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function. Diabetes 65, 2201–2213, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kirkland JL & Tchkonia T Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kuilman T & Peeper DS Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9, 81–94, (2009). [DOI] [PubMed] [Google Scholar]
- 137.McHugh D & Gil J Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 217, 65–77, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu L et al. Improves the In Vitro Developmental Competence and Reprogramming Efficiency of Cloned Bovine Embryos by Additional Complimentary Cytoplasm. Cell Reprogram 21, 51–60, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Villaret A et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 59, 2755–2763, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Crewe C, An YA & Scherer PE The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127, 74–82, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stout MB, Justice JN, Nicklas BJ & Kirkland JL Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology (Bethesda) 32, 9–19, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stout MB et al. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging (Albany NY) 6, 575–586, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Comisford R et al. Growth Hormone Receptor Antagonist Transgenic Mice Have Increased Subcutaneous Adipose Tissue Mass, Altered Glucose Homeostasis and No Change in White Adipose Tissue Cellular Senescence. Gerontology 62, 163–172, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bogazzi F et al. Growth hormone is necessary for the p53-mediated, obesity-induced insulin resistance in male C57BL/6J x CBA mice. Endocrinology 154, 4226–4236, (2013). [DOI] [PubMed] [Google Scholar]
- 145.Tchkonia T et al. Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Palmer AK et al. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 64, 2289–2298, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Khan T et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29, 1575–1591, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pasarica M et al. Adipose tissue collagen VI in obesity. The Journal of clinical endocrinology and metabolism 94, 5155–5162, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Householder LA et al. Increased fibrosis: A novel means by which GH influences white adipose tissue function. Growth Horm IGF Res 39, 45–53, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hagberg CE et al. Flow Cytometry of Mouse and Human Adipocytes for the Analysis of Browning and Cellular Heterogeneity. Cell Rep 24, 2746–2756 e2745, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gao H et al. CD36 Is a Marker of Human Adipocyte Progenitors with Pronounced Adipogenic and Triglyceride Accumulation Potential. Stem cells 35, 1799–1814, (2017). [DOI] [PubMed] [Google Scholar]
- 152.Varlamov O, Chu M, Cornea A, Sampath H & Roberts CT Jr. Cell-autonomous heterogeneity of nutrient uptake in white adipose tissue of rhesus macaques. Endocrinology 156, 80–89, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Seydoux J et al. Adrenoceptor heterogeneity in human white adipocytes differentiated in culture as assessed by cytosolic free calcium measurements. Cellular signalling 8, 117–122, (1996). [DOI] [PubMed] [Google Scholar]
- 154.Gliemann J & Vinten J Lipogenesis and insulin sensitivity of single fat cells. The Journal of physiology 236, 499–516, (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Salans LB & Dougherty JW The effect of insulin upon glucose metabolism by adipose cells of different size. Influence of cell lipid and protein content, age, and nutritional state. J Clin Invest 50, 1399–1410, (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sanchez-Gurmaches J & Guertin DA Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nature communications 5, 4099, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chau YY et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat Cell Biol 16, 367–375, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bluher M, Patti ME, Gesta S, Kahn BB & Kahn CR Intrinsic heterogeneity in adipose tissue of fat-specific insulin receptor knock-out mice is associated with differences in patterns of gene expression. J Biol Chem 279, 31891–31901, (2004). [DOI] [PubMed] [Google Scholar]
- 159.Xia B et al. Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice. PLoS Genet 13, e1007110, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee KY et al. Developmental and functional heterogeneity of white adipocytes within a single fat depot. EMBO J 38, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Min SY et al. Multiple human adipocyte subtypes and mechanisms of their development. bioRxiv, (2019). [Google Scholar]