

Abstract
Orai and Stim proteins are the mediators of calcium release-activated calcium signaling and are important in the regulation of bone homeostasis and disease. This includes separate regulatory systems controlling mesenchymal stem cell differentiation to form osteoblasts, which make bone, and differentiation and regulation of osteoclasts, which resorb bone. These systems will be described separately, and their integration and relation to other systems, including Orai and Stim in teeth, will be briefly discussed at the end of this review.
Keywords: Osteoporosis, Osteogenesis, Mineralization, Bone remodeling
Graphical Abstract
Introduction
Despite the apparently static function of the skeleton as a scaffold for the body, bone is actually a highly dynamic tissue that undergoes continuous cycles of formation and degradation in response to a complex interplay of local and systemic signals. The continuous remodeling of the skeleton not only allows for adaptation to changing mechanical stresses but is critical for systemic calcium as well as phosphate homeostasis, since bone represents the principal store for calcium and phosphate in the human body. Particularly important are the signals that coordinate the activity of two specialized cell types: the osteoblasts that produce and mineralize bone matrix and the osteoclasts that break it down. Dysregulation of bone turnover is central to the pathogenesis of many common skeletal disorders such as osteoporosis, which can result from either excess resorption by osteoclasts or insufficient bone formation by osteoblasts. Because of the clinical importance of bone loss disorders, a major focus of bone research has been the identification and characterization of the signaling systems that regulate bone cells, including the intracellular signal transduction mechanisms that mediate these signals, as these represent potential targets for therapeutic intervention [1, 2].
In addition to maintenance of skeletal integrity and regulation of calcium homeostasis, other functions for bone cells are now recognized. Numerous interactions between the bone cells and the immune system have been identified, leading to the recognition of the sub-discipline of osteoimmunology [3, 4]. Bone also provides a unique environment for hematopoiesis, highlighted by the prominence of hematologic symptoms in disorders of impaired bone resorption [5, 6]. The role of bone more broadly as an endocrine organ is emphasized by the recognition that FGF23, which is primarily produced by bone cells, is a key factor in several systemic disorders [7, 8]. These bone cell functions are also subject to complex regulation, and their characterization another focus in research on bone and disease.
Calcium signaling pathways are now recognized as mediators in the regulation of bone cell function, but much about the specific roles and mediators remains controversial. This review will focus specifically on the significance of store-operated Ca2+ entry (SOCE); briefly, the endoplasmic reticulum (ER) is the major Ca2+ store found in cells. The activation of phospholipase C-coupled receptors leads to the production of inositol (1,4,5) trisphosphate (IP3), a soluble second messenger that binds to receptors located on ER membrane, leading to ER Ca2+ depletion. Loss of ER Ca2+ content is sensed by Stim1 and Stim2, two type 1A transmembrane proteins found on the ER membrane. Upon activation, Stim proteins translocate within the ER towards the plasma membrane (PM), where they physically associate with and activate members of the Orai family of channels. Here, we will examine recent evidence defining roles for Orai and Stim in bone cells, considering potential roles for these proteins in the pathogenesis and treatment of bone disorders.
Osteoclasts, Osteoclastogenesis, and Calcium Signaling
Osteoclasts are specialized bone resorption cells, required for skeletal remodeling, repair, and the release of systemic calcium stores. Briefly summarizing established biology, osteoclasts derive from hematopoietic stem cells; they belong to the monocytic lineage which also produces macrophages and dendritic cells. Osteoclastic differentiation (osteoclastogenesis) requires expression of specialized genes including a vacuolar ATPase for acid secretion (to dissolve bone mineral) as well as proteolytic enzymes such as cathepsins K and B and matrix metalloproteinase 9, to break down the collagenous matrix [9]. For efficient bone resorption, osteoclast precursors also must fuse to form giant multinucleated cells. The mature osteoclasts attach tightly to the bone surface through integrins attached to a peripheral actin ring. Acid and proteases are then released on the bone in the sealed space beneath the osteoclast leading to resorption [9].
A key stimulus for osteoclast formation is the TNF family member Receptor Activator of NF-kappaB ligand (RANKL) [10, 11]. In vitro, addition of RANKL to osteoclast precursors grown with monocyte colony-stimulating factor (MCSF) is sufficient to induce formation of resorption competent osteoclasts. Mice deficient in either RANKL or its receptor RANK, lack osteoclasts and bone resorption resulting in osteopetrosis with absence of the marrow space [10-13]. Investigation of RANKL signal transduction revealed that RANKL like other TNF family members acts through Traf proteins, in particular Traf6, to stimulate the phosphorylation and degradation of IkappaB, allowing nuclear translocation and activation of NF-kappaB, which in turn regulates expression of osteoclast-specific genes [14-16]. Both Traf6 deficient mice and mice doubly deficient in the NF-kB subunits p50 and p52 lack osteoclasts and exhibit osteopetrosis [15, 17, 18]. However, activation of the Traf6-NF-kB pathway in osteoclast precursors is not sufficient for osteoclastogenesis as osteoclast formation cannot be rescued by a TRAF6 mutant sufficient for NF-kB activation [15], implying the existence of other necessary signaling pathways.
Calcium signaling is now recognized as critical for osteoclastogenic differentiation [19-22]. In 2002, two groups performing gene expression profiling to identify transcripts upregulated by RANKL during osteoclastogenesis noted a marked increase in expression of the calcium-dependent transcription factor NFATc1 (also called NFAT2) [23, 24]. Osteoclast precursors deficient in NFATc1 could not form osteoclasts in response to RANKL in vitro, but precursors transduced with constitutively active NFATc1 could form multinucleated osteoclasts even in the absence of RANKL [23], indicating that this pathway was both necessary and sufficient. Furthermore, blastocyst complementation studies confirmed the importance of NFATc1 in vivo [25]. As in other cell types, NFATc1 activation in osteoclast precursors followed increases in intracellular calcium that stimulated its dephosphorylation by the calcium-dependent phosphatase calcineurin [26, 27]. Calcium signaling has also been linked to activation of other important transcriptional regulators of osteoclastogenesis. Elevation of intracellular calcium [Ca2+]i appears to accelerate NFkappaB nuclear translocation [28], while the cAMP response element-binding protein (CREB) is activated through phosphorylation by a calcium/calmodulin-dependent kinase [29, 30]. Complete osteoclastic differentiation required sustained NFATc1 upregulation with autoamplification, which appeared to depend on the calcium oscillations that are observed in osteoclast precursors following 24-48 hours treatment with RANKL [23, 31]. Yet, at that time no pathway from RANK to calcium channels had been established. In 2004, Koga et al. identified costimulatory receptors such as TREM-2 and PIR-A that are linked to the ITAM containing adaptor proteins DAP12 or FcRgamma that lead to activation of phospholipase C gamma (PLCgamma) in osteoclast precursors. Knock-out studies showed that loss of these adaptor proteins impaired osteoclastogenesis while having no effect on the activation of IkappaB or other known RANK signal transducers such as the c- Jun N- terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38) [31]. Moreover, PLCgamma inhibitors or genetic knockout of PLCgamma2 impaired NFATc1 upregulation and osteoclastic differentiation [32, 33]. PLC activation was also shown to depend on Syk, tec-family tyrosine kinases (Btk, Tec), the B-cell linker protein (BLNK) and the adaptor SH2-containing leukocyte protein of 76 kDa (SLP76) [19, 31, 34-36]. Activation of PLC suggested the involvement of its product inositol 1,4,5-trisphosphate (IP3) which can stimulate IP3 receptors (IP3R) in bone to release ER Ca2+ stores [37]. While it appears that osteoclast precursors express all three IP3 receptors, gene knockout studies have shown that it is IP3R2 that is critical for calcium oscillations during osteoclastogenesis [38, 39]. But identification of the PLC-IP3-calcium pathway did not fully explain these phenomena as the mechanism for the influx of intracellular calcium following store depletion remained unclear. Subsequent work has suggested that Orai1 and Stim1 mediate this effect.
Osteoclasts and Orai/Stim
Osteoclast precursors, both from human peripheral blood and murine bone marrow, have been shown to express Orai1 and Stim 1, with detection of Stim2 also reported [40-42]. Several groups investigated whether RANKL-induced osteoclastic differentiation alters Orai or Stim expression, with varying results. We found that when peripheral blood mononuclear cells are treated with RANKL, both Orai1 and Stim1 protein levels gradually decline, with a parallel decrease in store-operated calcium entry noted during osteoclastogenic differentiation [40]. Using a RANKL-responsive murine monocytic cell line, RAW264.7, other groups found an early increase in Stim1, which then declined with either a similar pattern for Orai1 or no significant change [41, 42]. These groups investigated the possibility that transient receptor potential subfamily vanilloid (TRPV) channels might also be involved, particularly after osteoclastic differentiation is complete. Li et al. found evidence that in mature osteoclasts, calcium entry in response to fluid flow is mediated by TRPV4 channels rather than Orai1/Stim1 [42]. Functional studies of the effects of Orai1/Stim1 inhibition, however, demonstrated a clear role for this pathway in transducing differentiation signals from the RANKL receptor. Using the RAW264.7 cells line, Hwang and Putney demonstrated that shRNA knockdown of Orai1 inhibited RANKL-stimulated formation of multinucleated osteoclasts; this effect was confirmed in primary human osteoclast precursors transiently transfected with Orai1 siRNA [43]. Functional assays in vitro confirmed that the reduction of mature osteoclast formation after Orai1 knockdown significantly impaired bone mineral resorption. Orai1 knockout mice were also evaluated, but unexpectedly these showed evidence of an osteoblast defect as well (see below) [44, 45]. Nevertheless, histologic sections of bone from the Orai1 knockouts showed a marked reduction in tartrate-resistant acid phosphatase (TRAP) positive osteoclasts that were predominantly mononuclear, consistent with the defect in osteoclastogenesis found in vitro in the absence of Orai1 [44]. However, it is possible that in vivo, as in cocultures of osteoclast precursors with osteoblasts, a calcium independent pathway to NFATc1 activation linked to the cancer Osaka thyroid (Cot) kinase may, at least partially, compensate for the absence of Orai1/Stim1-mediated signaling [38, 46].
Orai/Stim and bone loss disorders
Studies to date point most strongly to a role for Orai1/Stim1 in RANKL differentiation signaling, though a few observations raise the possibility of effects on mature osteoclast function. The ability of Orai/Stim inhibition to impair the formation of mature multinucleated osteoclasts and thus the efficient resorption of bone suggests that their inhibitors might be used to treat bone loss disorders associated with osteoclast excess. One such disorder of high prevalence is post-menopausal estrogen deficiency induced osteoporosis [47, 48]. Kertesz et al. noted that while phospholipase C-gamma, which acts upstream from Orai/1/Stim1, is significant for basal osteoclastic bone resorption, it is not required for bone loss stimulated by estrogen deficiency [49]. However, since Orai1 can be activated through other pathways, it may still be relevant to assess the dependence of estrogen-deficiency osteoporosis on this calcium pathway. Unfortunately, the limited survival of Orai1-null mice hampered assessment of ovariectomy effects on bone in these animals as such changes take months to develop [44, 45, 50, 51]. As new Orai/Stim inhibitors become available, however, their ability to reduce bone loss may be tested in wild-type mouse models of estrogen-deficiency osteoporosis. Of course, since Orai1 and Stim1 are widely expressed, inhibitors may have effects on multiple tissues, in particular the immune system [52-55]. But notably we observed substantial inhibition of osteoclast formation by the inhibitor 3,4-dichloropropioaniline (DCPA) at concentrations which blocked SOC by only 50% [40]. Hence osteoclastogenesis may be sufficiently sensitive to permit selective inhibition at safe concentrations. Alternatively, it may be possible to use liposomes or nanoparticle carriers to target osteoclasts or osteoclast precursors specifically [56-60]. Although osteoporosis is the most common bone-loss disorder, targeting the bone loss associated with immune-mediated inflammatory arthritides (e.g. rheumatoid arthritis) using Orai1/Stim1 inhibitors may be more straightforward, since in this context inhibiting both immune function and bone resorption would be beneficial. The potential for a variety of calcium channels to serve as therapeutic targets in rheumatoid arthritis has recently been reviewed [61]. We have tested DCPA as a novel inhibitor of Orai1/Stim1-mediated store operated calcium entry that impairs the formation of multinucleated osteoclasts in vitro [40] as a therapy for inflammatory arthritis. Using the collagen-induced arthritis (CIA) mouse model of rheumatoid arthritis [62-64], we determined the effects of DCPA treatment on disease development [65]. DCPA significantly reduced disease as reflected by the low arthritic index in the DCPA-treated compared to the placebo-treated animals. Histologic studies showed a reduction in joint inflammation and tissue destruction, while microcomputed tomography confirmed a reduction in bone loss in the paws. Liu et al. applied an Orai1 shRNA knockdown approach to the same mouse model of rheumatoid arthritis [66]. Initial studies evaluating tolerance of systemic gene silencing by lentiviral shRNA constructs showed reduced survival with knockdown of Orai1 [66], but in subsequent studies, intra-articular injection of Orai1 shRNA lentiviral particles was tolerated and produced a marked amelioration of arthritic symptoms even when treatment was begun after symptoms arose [67]. Histologic evaluation and microcomputed tomography studies confirmed a reduction in bone damage by this treatment, with few osteoclasts identified by TRAP staining in joint sections. Indirect effects on osteoclastogenesis were also possible, since osteoclastogenic RANKL as well as inflammatory cytokines such as TNF-alpha, which enhances osteoclastogenesis, were reduced in joint extracts from Orai1 shRNA-treated mice. To clarify the relative contribution of a reduction in RANKL concentration versus a reduced response of osteoclast precursors to RANKL, cell-type specific inhibition of Orai1 will be required. Additional studies using a xenograft model of human rheumatoid arthritis, examined the therapeutic effects of YM-58483 (also known as BTP2), a small molecule inhibitor of CRAC, and of a neutralizing antibody to Orai1 [68] [69]. Production of IL-6 by the xenograft cells and serum Rheumatoid factor were both decreased by these treatments, and histologic sections showed reduced staining for the osteoclast-associated enzyme tartrate resistant acid phosphatase. However, both inhibitors were also associated with development of hyperglycemia, and YM-58483 showed hepatic and renal toxicity, again suggesting that targeting Orai1 inhibitors to specific cell types or tissues may be necessary for their therapeutic application.
The investigation of Orai proteins in osteoclasts has focused almost entirely on Orai1. However, the role of Orai3 in inflammatory arthritis has also been examined. Again, using the CIA mouse model to investigate therapeutic effects in inflammatory arthritis, Orai3 shRNA lentivirus was applied systemically; in contrast to lentivirus with Orai1 shRNA, this was well tolerated [66]. The consequent knockdown of Orai3 significantly reduced clinical symptoms as reflected by a reduced arthritis score and paw swelling, while histologic sections showed decreased bone erosion as well as reduced inflammation. To assess whether the effects on bone reflected intrinsic changes in osteoclasts or their precursors, marrow cells were isolated from Orai3 shRNA-treated mice or controls, and their responses to RANKL treatment determined. Testing these cells in resorption pit assays, revealed a substantial reduction in resorptive capacity by Orai3 knockdown, but only a mild and statistically insignificant decrease in TRAP positive cells. These results appear to favor a role for Orai3 in mature osteoclasts rather than in osteoclastogenesis. However, whether multinucleation of the TRAP positive cells was impaired by Orai3 inhibition, as it is by Orai1 inhibition, is not reported. It is possible that only TRAP positive mononuclear cells are present when Orai3 is knocked down as seen with inhibition of Orai1; such a lack of osteoclast precursor fusion with giant cell formation could in itself explained the impaired bone resorption. Interestingly, an association between susceptibility to rheumatoid arthritis and a polymorphism in the Orai1 gene (rs7135617) has been reported [70]; this is an intronic polymorphism that may affect gene splicing, but its specific implications for Orai1 function or expression and its effects in osteoclasts or precursors remain to be determined.
Osteoblasts, Bone Formation and Calcium Signaling
Osteoblasts are specialized bone cells responsible for the production of the mature mineralized skeleton. During prenatal development, a cartilaginous skeleton first forms that, over time, is degraded by osteoclasts and progressively replaced by calcified bone through the activity of osteoblasts. Outside the growth plates (primarily in the long bones of the axial skeleton), skeletal mineralization is largely complete by the early postnatal period. Thus, one indication of osteoblast impairment is delayed bone mineralization with persistence of fetal cartilage in the postnatal skeleton. In addition to a critical role during growth and development, osteoblasts act throughout life in concert with osteoclasts to remodel the skeleton. If osteoclastic resorption during bone turnover is not matched by commensurate bone production, that is, if osteoblast number or function is insufficient, bone loss results.
Osteoblasts are derived from bone marrow mesenchymal stem cells. Osteoblastic differentiation results in a capacity to synthesize the components of osteoid, the non-mineralized matrix of bone, of which the major element is type I collagen, and to mineralize the osteoid through the secretion of calcium and phosphate with concomitant removal of hydrogen ions [71]. As bone is laid down, the osteoblasts, formerly on the surface of the bone, become encased within the mineralized matrix and further differentiate into osteocytes. Although apparently isolated in small lacunae within the bone, osteocytes have long cellular process that extend through small channels (canaliculi) in the bone through which they maintain contact with other osteocytes and cells still at the bone surface. The osteocyte processes contain gap junctions through which materials may pass from cell to cell within osteocytes of a single bone forming unit, called an osteon. These cellular processes also allow for the rapid spread of signals amongst osteocytes, including calcium signals, for coordinating their function, although receptor mechanisms as well as gap junction transport are involved in this regulation [72, 73]. It is now recognized that signals from osteocytes to the bone surface play a key role in the regulation of bone turnover [74] [75]. Both osteoblasts and osteocytes regulate osteoclastogenesis through the production of RANKL and osteoprotegerin, while another product of these cells, FGF23, acts systemically, affecting heart and kidney as well as bone [74-76].
Numerous factors have been identified that regulate the survival, proliferation, differentiation and function of osteoblasts, their precursors, and osteocytes [77, 78]. These include hormones such as parathyroid hormone (PTH), thyroid stimulating hormone, cortisol, estrogens and androgens, as well as growth factors, including IGF-1, FGF2, and multiple members of the transforming growth factor family (e.g. TGF-beta, bone morphogenetic protein 2 (BMP2) and BMP4) [79-88]. Wnt and Notch signals are also critical regulators of osteoblasts, and a role for the sympathetic nervous system has also been identified [89-91]. The signal transduction mechanisms that mediate these signals in osteoblasts are also diverse [92, 93] but while calcium signaling has been identified in osteoblasts, its role has not been as extensively studied as it has for osteoclasts. Osteoblasts are known to express a number of G-protein coupled receptors that can activate phospholipase C and release ER calcium stores, thereby potentially stimulating store operated calcium entry. The parathyroid hormone receptor PTHR1 has been linked to phospholipase C activation in osteoblasts; however, the major effects of PTH on osteoblasts seem to be mediated by the adenylate cyclase-cAMP-protein kinase A pathway rather than calcium [83, 88]. PTHR1 activation does cause significant calcium influx in osteocytes, but this appears to occur via L-type calcium channels [94]. ATP stimulation of osteoblast purinergic receptors also increases [Ca2+]I. While this is due at least in part to calcium entry through P2X7, P2Y receptors that activate phospholipase C are also present. These include P2Y1, which, in mesenchymal stem cells, induces calcium oscillations dependent on calcium influx through Orai1/Stim1 [95-97]. Osteoblasts also express the calcium sensing receptor (CaSR) by which elevated extracellular calcium concentrations can stimulate osteoblast proliferation, differentiation and matrix mineralization. Signaling by the CaSR is known to be mediated by phospholipase C activation, IP3 production and elevation of intracellular calcium [98]. Recently it was shown that elevation of osteoblast [Ca2+]i following CaSR stimulation could be inhibited by the CRAC inhibitors 2-APB and YM-58483. Furthermore, osteoblast proliferation stimulated by increased extracellular calcium could be inhibited not only by phospholipase C and CaSR inhibitors but also by 2-APB and YM-58483 (though not by verapamil or nifedipine) strongly implicating Orai1/Stim1 in receptor mediated osteoblast regulation [99].
Osteoblasts and Orai/Stim
The first evidence of the importance of Orai1 for osteoblastic bone formation, however, came from analyses of Orai1 knock-out mice. Although such knock-outs were first generated in 2008, initial studies, though noting reduced size at birth, did not perform detailed skeletal studies [54, 100, 101]. In 2012, two groups, using separately derived Orai1 knockout mice, identified significant skeletal abnormalities in these mice that were inconsistent with its known role in osteoclastogenesis [44] [45]. Vertebral sections from 3-week-old mice revealed retained cartilage in knockout animals at sites where mineralized bone was already mature in wild-type controls. Whole skeletal staining with Alcian blue for cartilage and Alizarin red for bone confirmed that the reduction in mineralized bone and persistence of cartilage was systemic [44]. When analyzed by microcomputed tomography set for detection of mineralized bone, the cartilaginous areas appeared as multiple defects in the cortical bone [44]. In addition, histomorphometric analysis of trabecular bone showed fewer trabeculae and significant trabecular thinning, with an overall reduction in bone reflected by a decreased ratio of bone volume to tissue volume (BV/TV) in the Orai1 knockout [44]. These findings were recapitulated in older animals lacking Orai1: microcomputed tomography of femora from 6-week-old, 8-week-old and 12-week-old Orai1 knockout mice, respectively, demonstrated reduced trabecular thickness and increased trabecular separation compared to controls, as well as reduced bone mineral density compared to age-matched controls [45, 102]. One study further showed that the reduced bone mass in the knock-outs was progressive with age [102]. However, it should be noted that the femur is not optimal for such studies since analysis can be affected by differences in animal size, and trabecular bone mass naturally decreases with age. Overall, these data support the hypothesis that Orai1 deficiency during development significantly impairs production of mineralized bone and suggest that further investigation into the roles of Orai in osteoblasts is warranted.
One potential mechanism for reduced bone mass in the knockout mice is a reduction in osteoblast numbers possibly reflected reduced proliferation or survival in the absence of Orai1. Vertebral and femoral bone sections from knockout mice showed decreased staining for the osteoblast marker enzyme alkaline phosphatase (ALP) [44, 103] indicating a lack of mature osteoblasts, potentially reflecting reduced osteoblast numbers. However, Choi et al., 2018, estimated that osteoblasts, as a fraction of bone surface area, were increased in Orai1 knockout animals relative to controls [102]. Moreover, in cultures of osteoblasts from wild-type and Orai1 knockout mice, they found that cell numbers were greater in its absence [102]. However, Hwang et al. found no differences in cell numbers in osteoblast cultures from Orai1 knockout versus wild-type mice, and transduction of an osteoblastic cell line with the dominant-negative Orai1 E106Q mutant had no observable effect on cell numbers [45]. It is possible that these contradictory findings simply reflect differences in specific culture conditions used. As noted above, in vitro studies have suggested that some proliferative signals, such as increased extracellular calcium, may depend on store operated calcium entry. It has since been shown that osteoblasts express all three Orai and both Stim proteins [103, 104]. The potential for functional redundancy or a compensatory activity by other family members can make it difficult to ascertain which specific functions can be ascribed to which genes
It is notable that skeletal defects have not been described for humans with mutations in Orai1 (or Stim1), though some do show abnormalities of dentition. Species differences in Orai expression or function may underlie this discrepancy. Differences in phenotype are also seen amongst patients with different mutations – and none are recapitulated by the gene disruption in the mice. The limited lifespan of patient’s with Orai mutations might also be a factor: it is also possible that skeletal abnormalities might have become manifest had the patients survived longer.
Osteoblast differentiation and function are affected by deficiency of Orai1
An alternative mechanism for the skeletal abnormalities in the Orai1 knockout mice is that osteoblast differentiation, matrix production and/or mineralization are impaired in the absence of Orai1. Several studies used cultures of osteoblasts or osteoblast precursors from Orai1 knockout mice to investigate how the differentiation or functioning of osteoblasts might be altered. Reduced expression of osteoblast marker genes such as alkaline phosphatase, type I collagen, and osteocalcin, was reported by several groups when Orai is absent or inhibited, though results are not fully concordant [44, 45, 102, 103]. In particular, Hwang et al. found little difference for early markers of differentiation, with alkaline phosphatase activity appearing equivalent in cultures from wild-type and knock-out mice; they suggest that only the later stages of osteoblast differentiation depend significantly on Orai1 [45]. However, most studies found alkaline phosphatase reduced in the absence of Orai1 both in vitro and in vivo. Osteocyte marker gene expression was also tested and showed reductions in the cultures from the Orai1 knockout mice compared to controls [102, 103]. Additionally, Lee et al. found evidence of Orai1 involvement in the osteogenic effects of BMP2, a major regulator of osteoblast differentiation. Bone marrow stromal cells, precursors to osteoblasts, obtained from Orai1 knockout mice not only failed to differentiate in response to osteogenic conditions, but did not upregulate downstream targets of BMP signaling, indicating a link between Orai1 and BMP signals that are critical for proper osteoblast maturation [103]. A consistent finding across multiple studies was a reduction in matrix mineralization in the absence of Orai1, both in vitro and in vivo [44, 45, 102, 103]. The possibility that it is primarily osteoblast function and specifically mineralization that is dependent on Orai1 will require further investigation.
Regulatory functions of osteoblasts and osteocytes were also examined. Osteoblast production of the osteoclastogenic factor RANKL, and its antagonist osteoprotegerin, was tested for dependence on Orai1, with variable results [45, 103]. In contrast, a clear effect was observed for FGF23 expression: deletion or knockdown of Orai1 significantly reduced osteoblast FGF23 production. Choi et al. [102] found that FGF23 is reduced in cells lacking Orai1, and found associated changes in osteocyte morphology (shape and border changes). Zhang et al. [104] showed that both Stim1 and Orai1 expression was under the control of NF-kappaB in osteoblasts: treatment with NF-kappaB inhibitors attenuated store operated calcium entry. Further, they demonstrated that 1,25 dihydroxyvitamin D-induced FGF23 expression was blocked by either pharmacological or genetic inhibition of Orai or by NF-kappaB inhibition. Overall, these investigations reveal a critical role for NF-KappaB-induced Stim1 and Orai1 expression in vitamin D-induced FGF23 expression. Because of the involvement of FGF23 not only in bone health and disease but also in renal and cardiovascular disease, the identification of Orai1 as a regulator of FGF23 is potentially of broad clinical interest.
Expression of Stim in osteoblast function and bone tumor progression
Osteoblasts express all three Orai and both Stim (stromal interaction molecule) genes [92]. At the level of osteoblast precursor mesenchymal stem cells, Orai1 facilitates bone morphogenetic protein signaling required for normal osteoblast differentiation, ultimately leading to higher levels of expression of members of the Stim and Orai families [103]. This has been further demonstrated at the protein level in osteoblasts or osteoblastic cell lines [105]. Interestingly, Stim1 overexpression in MC3T3-E1 osteoblastlike cells caused changes consistent with increased osteoblast function; this was reflected in calcium deposition activity as well as increased Runx2, BMP4, and type 1 collagen production relative to control cells. Considered together, these observations indicate that SOCE promotes osteoblast differentiation [105].
In clinically related work, Sun et al. [106] noted that SOCE and its major mediator Stim1 are implicated in a number of pathological processes associated with cancer progression. Hence, Stim1 expression was increased in chemotherapy-resistant osteosarcoma, and that patients with Stim1 expression had poor overall survival relative to Stim1-negative osteosarcoma patients [106]. Further, Stim1 overexpression in osteosarcoma cells conferred cisplatin resistance [106]. ER Ca2+ depletion is a major cause of ER stress; as an ER Ca2+ sensor, Stim1 responds to loss of ER Ca2+ content by facilitating the entry of Ca2+, which would be capable of reversing this effect. Although cisplatin primarily kills cells through formation of DNA lesions, it also causes ER stress which can be reversed via upregulation of Stim1 [106]. Hence, at least within this context, increased expression of Stim and associated SOCE confers increased survival on osteosarcoma, thereby increasing disease severity.
Orai and tooth differentiation
Tooth development is a separate and specialized topic closely related to bone formation. LaCruz et al. [107] reviewed the topic and discussed multiple reports of abnormal enamel phenotypes being a result of Stim or Orai mutations. Screening of genes involved in formation of dental enamel identified Stim1 as a key gene in ameloblasts, the cell type that controls enamel production [107]. Similarly, to transcriptional profiles observed in osteoblasts [103], mRNAs for all three Orai genes and both Stim genes were expressed in the ameloblast cell line LS8 [108]. This observation was confirmed in primary ameloblasts by Nurbaeva et al. [109]; the ability of ameloblastic cells to exhibit store operated calcium entry was also confirmed in this study. A murine line was generated in 2017 which exhibited ameloblast-specific deletion of Stim1 and Stim2 [110]. Ameloblasts from these mice exhibited impairment of SOCE along with substantial hypomineralization, thinning of enamel and mechanical weakness of the teeth. Pathological differences in the formation of the ruffled border were noted and attributed to ER stress and mitochondrial dysfunction. Overall, these studies reveal a fundamental role for SOCE in enamel formation. However, it should be noted that the factors responsible for initiating SOCE were not determined in these studies. Interestingly, the small peptide hormone cholecystokinin was shown to be upregulated during ameloblast maturation; Ca2+ imaging revealed that the presence of cholecystokinin increased cytoplasmic Ca2+ levels which could then be blocked with pharmacological CRAC-channel inhibitors [111]. Similar observations were made using either acetylcholine or ATP [111]. Considered in the context of the studies described above, these observations reveal that loss of Stim1/2 interferes with receptor-mediated Ca2+ responses in ameloblasts. Future studies may reveal the relative contributions of SOCE to physiological signals in ameloblasts regulated cholecystokinin, acetylcholine, ATP and/or other agonists.
Conclusion
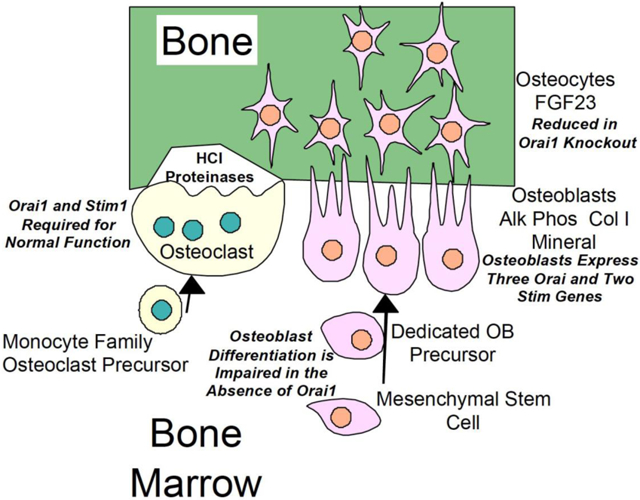
Orai and Stim proteins are critical players for numerous processes in cells that produce mineral, as well as in mineral degrading cells. The contributions of Stim and Orai to these respective cell types are summarized in Figure 1. Normal osteoclast differentiation requires expression of both Orai1 and Stim1; when Orai1 is absent, only rudimentary bone degradation is seen. Differentiation and function of bone forming osteoblasts is also impaired with Orai1 deletion, although through fundamentally different mechanisms. These fundamental differences are important, since they may provide a window to specifically target one cell type over another. Hence, although osteoclasts are critical for normal bone function, over-activity of osteoclasts is associated with both osteoporosis and bone erosion associated with arthritis. Differences in Stim/Orai subtype-dependence and/or relative dependence on Orai1, as well, may reveal a promising therapeutic window to target one cell type over another. The extent to which this is possible, however, will depend on detailed future analyses of Stim/Orai expression and function, as well as a greater understanding of their role(s) in osteoclasts and osteoblast both in physiological and pathophysiological contexts.
Figure 1.

Diagram showing expression of Stim and Orai1 proteins in osteoblasts and osteoclasts, and functional consequences of this expression.
Acknowledgments
This work was supported by the Department of Veteran’s Affairs grant BX002490, by NIH grants AR055208 to HB, AR065407 to HB, JS, and LR, GM117907 to JS and ES023845 to JB, as well as GM104942 (LR). Opinions expressed are not those of the Department of Veteran’s Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Park-Min KH, Ji JD, Antoniv T, Reid AC, Silver RB, Humphrey MB, Nakamura M, Ivashkiv LB, IL-10 suppresses calcium-mediated costimulation of receptor activator NF-kappa B signaling during human osteoclast differentiation by inhibiting TREM-2 expression, Journal of immunology (Baltimore, Md. : 1950), 183 (2009) 2444–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Boyce BF, Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts, Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 28 (2013) 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T, Takayanagi H, Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems, Physiological reviews, 97 (2017) 1295–1349. [DOI] [PubMed] [Google Scholar]
- [4].Humphrey MB, Nakamura MC, A Comprehensive Review of Immunoreceptor Regulation of Osteoclasts, Clinical reviews in allergy & immunology, 51 (2016) 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miyamoto T, Role of osteoclasts in regulating hematopoietic stem and progenitor cells, World journal of orthopedics, 4 (2013) 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Galan-Diez M, Kousteni S, The osteoblastic niche in hematopoiesis and hematological myeloid malignancies, Current molecular biology reports, 3 (2017) 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fukumoto S, Martin TJ, Bone as an endocrine organ, Trends in endocrinology and metabolism: TEM, 20 (2009) 230–236. [DOI] [PubMed] [Google Scholar]
- [8].Han Y, You X, Xing W, Zhang Z, Zou W, Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts, Bone research, 6 (2018) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boyle WJ, Simonet WS, Lacey DL, Osteoclast differentiation and activation, Nature, 423 (2003) 337–342. [DOI] [PubMed] [Google Scholar]
- [10].Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ, Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation, Cell, 93 (1998) 165–176. [DOI] [PubMed] [Google Scholar]
- [11].Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T, Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J, RANK is essential for osteoclast and lymph node development, Genes & development, 13 (1999) 2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan SC, Sun Y, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ, RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism, Proceedings of the National Academy of Sciences of the United States of America, 97 (2000) 1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y, The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor, The Journal of biological chemistry, 273(1998)28355–28359. [DOI] [PubMed] [Google Scholar]
- [15].Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J, Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis, The EMBO journal, 20 (2001) 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J, Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice, Genes to cells : devoted to molecular & cellular mechanisms, 4 (1999) 353–362. [DOI] [PubMed] [Google Scholar]
- [17].Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R, Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2, Nature medicine, 3 (1997) 1285–1289. [DOI] [PubMed] [Google Scholar]
- [18].Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U, Requirement for NF-kappaB in osteoclast and B-cell development, Genes & development, 11 (1997) 3482–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Negishi-Koga T, Takayanagi H, Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation, Immunological reviews, 231 (2009) 241–256. [DOI] [PubMed] [Google Scholar]
- [20].Robinson LJ, Blair HC, Barnett JB, Zaidi M, Huang CL, Regulation of bone turnover by calcium-regulated calcium channels, Annals of the New York Academy of Sciences, 1192 (2010) 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hwang SY, Putney JW Jr., Calcium signaling in osteoclasts, Biochimica et biophysica acta, 1813 (2011) 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kajiya H, Calcium signaling in osteoclast differentiation and bone resorption, Advances in experimental medicine and biology, 740 (2012) 917–932. [DOI] [PubMed] [Google Scholar]
- [23].Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T, Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts, Developmental cell, 3 (2002) 889–901. [DOI] [PubMed] [Google Scholar]
- [24].Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T, Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator, The Journal of biological chemistry, 277 (2002) 41147–41156. [DOI] [PubMed] [Google Scholar]
- [25].Asagiri M, Takayanagi H, The molecular understanding of osteoclast differentiation, Bone, 40 (2007) 251–264. [DOI] [PubMed] [Google Scholar]
- [26].Im SH, Rao A, Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling, Molecules and cells, 18 (2004) 1–9. [PubMed] [Google Scholar]
- [27].Takayanagi H, The role of NFAT in osteoclast formation, Annals of the New York Academy of Sciences, 1116 (2007) 227–237. [DOI] [PubMed] [Google Scholar]
- [28].Komarova SV, Pilkington MF, Weidema AF, Dixon SJ, Sims SM, RANK ligand-induced elevation of cytosolic Ca2+ accelerates nuclear translocation of nuclear factor kappa B in osteoclasts, The Journal of biological chemistry, 278 (2003) 8286–8293. [DOI] [PubMed] [Google Scholar]
- [29].Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H, Regulation of osteoclast differentiation and function by the CaMK-CREB pathway, Nature medicine, 12 (2006) 1410–1416. [DOI] [PubMed] [Google Scholar]
- [30].Ang ES, Zhang P, Steer JH, Tan JW, Yip K, Zheng MH, Joyce DA, Xu J, Calcium/calmodulin-dependent kinase activity is required for efficient induction of osteoclast differentiation and bone resorption by receptor activator of nuclear factor kappa B ligand (RANKL), Journal of cellular physiology, 212 (2007) 787–795. [DOI] [PubMed] [Google Scholar]
- [31].Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T, Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis, Nature, 428 (2004) 758–763. [DOI] [PubMed] [Google Scholar]
- [32].Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R, PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2, The Journal of clinical investigation, 116 (2006) 2869–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen Y, Wang X, Di L, Fu G, Chen Y, Bai L, Liu J, Feng X, McDonald JM, Michalek S, He Y, Yu M, Fu YX, Wen R, Wu H, Wang D, Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis, The Journal of biological chemistry, 283 (2008) 29593–29601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee SH, Kim T, Jeong D, Kim N, Choi Y, The tec family tyrosine kinase Btk Regulates RANKL-induced osteoclast maturation, The Journal of biological chemistry, 283 (2008) 11526–11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, Takai T, Kodama T, Morio T, Geha RS, Kitamura D, Kurosaki T, Ellmeier W, Takayanagi H, Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals, Cell, 132 (2008) 794–806. [DOI] [PubMed] [Google Scholar]
- [36].Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC, The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 6158–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Falsafi R, Tatakis DN, Hagel-Bradway S, Dziak R, Effects of inositol trisphosphate on calcium mobilization in bone cells, Calcified tissue international, 49 (1991) 333–339. [DOI] [PubMed] [Google Scholar]
- [38].Kuroda Y, Hisatsune C, Nakamura T, Matsuo K, Mikoshiba K, Osteoblasts induce Ca2+ oscillation-independent NFATc1 activation during osteoclastogenesis, Proceedings of the National Academy of Sciences of the United States of America, 105 (2008) 8643–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Morikawa K, Goto T, Tanimura A, Kobayashi S, Maki K, Distribution of inositol 1,4,5-trisphosphate receptors in rat osteoclasts, Acta histochemica et cytochemica, 41 (2008) 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhou Y, Lewis TL, Robinson LJ, Brundage KM, Schafer R, Martin KH, Blair HC, Soboloff J, Barnett JB, The role of calcium release activated calcium channels in osteoclast differentiation, Journal of cellular physiology, 226 (2011) 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kajiya H, Okamoto F, Nemoto T, Kimachi K, Toh-Goto K, Nakayana S, Okabe K, RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations, Cell calcium, 48 (2010) 260–269. [DOI] [PubMed] [Google Scholar]
- [42].Li P, Bian X, Liu C, Wang S, Guo M, Tao Y, Huo B, STIM1 and TRPV4 regulate fluid flow-induced calcium oscillation at early and late stages of osteoclast differentiation, Cell calcium, 71 (2018) 45–52. [DOI] [PubMed] [Google Scholar]
- [43].Hwang SY, Putney JW, Orai1-mediated calcium entry plays a critical role in osteoclast differentiation and function by regulating activation of the transcription factor NFATc1, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 26 (2012) 1484–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Robinson LJ, Mancarella S, Songsawad D, Tourkova IL, Barnett JB, Gill DL, Soboloff J, Blair HC, Gene disruption of the calcium channel Orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development, Laboratory investigation; a journal of technical methods and pathology, 92 (2012) 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hwang SY, Foley J, Numaga-Tomita T, Petranka JG, Bird GS, Putney JW Jr., Deletion of Orai1 alters expression of multiple genes during osteoclast and osteoblast maturation, Cell calcium, 52 (2012) 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kuroda Y, Hisatsune C, Mizutani A, Ogawa N, Matsuo K, Mikoshiba K, Cot kinase promotes Ca2+ oscillation/calcineurin-independent osteoclastogenesis by stabilizing NFATc1 protein, Molecular and cellular biology, 32 (2012) 2954–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Manolagas SC, Kousteni S, Jilka RL, Sex steroids and bone, Recent progress in hormone research, 57 (2002) 385–409. [DOI] [PubMed] [Google Scholar]
- [48].Sipos W, Pietschmann P, Rauner M, Kerschan-Schindl K, Patsch J, Pathophysiology of osteoporosis, Wiener medizinische Wochenschrift (1946), 159 (2009) 230–234. [DOI] [PubMed] [Google Scholar]
- [49].Kertesz Z, Gyori D, Kormendi S, Fekete T, Kis-Toth K, Jakus Z, Schett G, Rajnavolgyi E, Dobo-Nagy C, Mocsai A, Phospholipase Cgamma2 is required for basal but not oestrogen deficiency-induced bone resorption, European journal of clinical investigation, 42 (2012) 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jee WS, Yao W, Overview: animal models of osteopenia and osteoporosis, Journal of musculoskeletal & neuronal interactions, 1 (2001) 193–207. [PubMed] [Google Scholar]
- [51].Komori T, Animal models for osteoporosis, European journal of pharmacology, 759 (2015) 287–294. [DOI] [PubMed] [Google Scholar]
- [52].Feske S, CRAC channelopathies, Pflugers Archiv : European journal of physiology, 460 (2010) 417–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lian J, Cuk M, Kahlfuss S, Kozhaya L, Vaeth M, Rieux-Laucat F, Picard C, Benson MJ, Jakovcevic A, Bilic K, Martinac I, Stathopulos P, Kacskovics I, Vraetz T, Speckmann C, Ehl S, Issekutz T, Unutmaz D, Feske S, ORAI1 mutations abolishing store-operated Ca(2+) entry cause anhidrotic ectodermal dysplasia with immunodeficiency, The Journal of allergy and clinical immunology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A, Hair loss and defective T- and B-cell function in mice lacking ORAI1, Molecular and cellular biology, 28 (2008) 5209–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Parekh AB, Store-operated CRAC channels: function in health and disease, Nature reviews. Drug discovery, 9 (2010) 399–410. [DOI] [PubMed] [Google Scholar]
- [56].Teong B, Kuo SM, Tsai WH, Ho ML, Chen CH, Huang HH, Liposomal Encapsulation for Systemic Delivery of Propranolol via Transdermal Iontophoresis Improves Bone Microarchitecture in Ovariectomized Rats, International journal of molecular sciences, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ramos AP, Cruz MAE, Tovani CB, Ciancaglini P, Biomedical applications of nanotechnology, Biophysical reviews, 9 (2017) 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen S, Zheng L, Zhang J, Wu H, Wang N, Tong W, Xu J, Huang L, Zhang Y, Yang Z, Lin G, Wang X, Qin L, A novel bone targeting delivery system carrying phytomolecule icaritin for prevention of steroid-associated osteonecrosis in rats, Bone, 106 (2018) 52–60. [DOI] [PubMed] [Google Scholar]
- [59].Carbone EJ, Rajpura K, Allen BN, Cheng E, Ulery BD, Lo KW, Osteotropic nanoscale drug delivery systems based on small molecule bone-targeting moieties, Nanomedicine : nanotechnology, biology, and medicine, 13 (2017) 37–47. [DOI] [PubMed] [Google Scholar]
- [60].Cai M, Yang L, Zhang S, Liu J, Sun Y, Wang X, A bone-resorption surface-targeting nanoparticle to deliver anti-miR214 for osteoporosis therapy, International journal of nanomedicine, 12 (2017) 7469–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Park R, Ji JD, Calcium channels: the potential therapeutic targets for inflammatory bone destruction of rheumatoid arthritis, Inflammation research : official journal of the European Histamine Research Society … [et al.], 65 (2016) 347–354. [DOI] [PubMed] [Google Scholar]
- [62].Caplazi P, Baca M, Barck K, Carano RA, DeVoss J, Lee WP, Bolon B, Diehl L, Mouse Models of Rheumatoid Arthritis, Veterinary pathology, 52 (2015) 819–826. [DOI] [PubMed] [Google Scholar]
- [63].Clutter SD, Wilson DC, Marinov AD, Hirsch R, Follistatin-like protein 1 promotes arthritis by up-regulating IFN-gamma, Journal of immunology (Baltimore, Md. : 1950), 182 (2009) 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Brand DD, Latham KA, Rosloniec EF, Collagen-induced arthritis, Nature protocols, 2 (2007) 1269–1275. [DOI] [PubMed] [Google Scholar]
- [65].Blair HC, Soboloff J, Robinson LJ, Tourkova IL, Larrouture QC, Witt MR, Holaskova I, Schafer R, Elliott M, Hirsch R, Barnett JB, Suppression of arthritis-induced bone erosion by a CRAC channel antagonist, RMD open, 2 (2016) e000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu S, Kiyoi T, Takemasa E, Maeyama K, Systemic lentivirus-mediated delivery of short hairpin RNA targeting calcium release-activated calcium channel 3 as gene therapy for collagen-induced arthritis, Journal of immunology (Baltimore, Md. : 1950), 194 (2015) 76–83. [DOI] [PubMed] [Google Scholar]
- [67].Liu S, Kiyoi T, Takemasa E, Maeyama K, Intra-articular lentivirus-mediated gene therapy targeting CRACM1 for the treatment of collagen-induced arthritis, Journal of pharmacological sciences, 133 (2017) 130–138. [DOI] [PubMed] [Google Scholar]
- [68].Liu S, Hasegawa H, Takemasa E, Suzuki Y, Oka K, Kiyoi T, Takeda H, Ogasawara T, Sawasaki T, Yasukawa M, Maeyama K, Efficiency and Safety of CRAC Inhibitors in Human Rheumatoid Arthritis Xenograft Models, Journal of immunology (Baltimore, Md. : 1950), 199 (2017) 1584–1595. [DOI] [PubMed] [Google Scholar]
- [69].Ohga K, Takezawa R, Arakida Y, Shimizu Y, Ishikawa J, Characterization of YM-58485/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo, International immunopharmacology, 8 (2008) 1787–1792. [DOI] [PubMed] [Google Scholar]
- [70].Yen JH, Chang CM, Hsu YW, Lee CH, Wu MS, Hwang DY, Chen BK, Liao HT, Wu MT, Chang WC, A polymorphism of ORAI1 rs7135617, is associated with susceptibility to rheumatoid arthritis, Mediators of inflammation, 2014 (2014) 834831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Blair HC, Larrouture QC, Tourkova IL, Liu L, Bian JH, Stolz DB, Nelson DJ, Schlesinger PH, Support of bone mineral deposition by regulation of pH, American journal of physiology. Cell physiology, 315 (2018) C587–c597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Loiselle AE, Jiang JX, Donahue HJ, Gap junction and hemichannel functions in osteocytes, Bone, 54 (2013) 205–212. [DOI] [PubMed] [Google Scholar]
- [73].Batra N, Kar R, Jiang JX, Gap junctions and hemichannels in signal transmission, function and development of bone, Biochimica et biophysica acta, 1818 (2012) 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bonewald LF, The Role of the Osteocyte in Bone and Nonbone Disease, Endocrinology and metabolism clinics of North America, 46 (2017) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Plotkin LI, Bellido T, Osteocytic signalling pathways as therapeutic targets for bone fragility, Nature reviews. Endocrinology, 12 (2016) 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Erben RG, Physiological Actions of Fibroblast Growth Factor-23, Frontiers in endocrinology, 9 (2018) 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Marie PJ, Signaling pathways affecting skeletal health, Current osteoporosis reports, 10 (2012) 190–198. [DOI] [PubMed] [Google Scholar]
- [78].Zuo C, Huang Y, Bajis R, Sahih M, Li YP, Dai K, Zhang X, Osteoblastogenesis regulation signals in bone remodeling, Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA, 23 (2012) 1653–1663. [DOI] [PubMed] [Google Scholar]
- [79].Ornitz DM, Marie PJ, Fibroblast growth factor signaling in skeletal development and disease, Genes & development, 29 (2015) 1463–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sanchez-Duffhues G, Hiepen C, Knaus P, Ten Dijke P, Bone morphogenetic protein signaling in bone homeostasis, Bone, 80 (2015) 43–59. [DOI] [PubMed] [Google Scholar]
- [81].Wu M, Chen G, Li YP, TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease, Bone research, 4 (2016) 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yakar S, Werner H, Rosen CJ, Insulin-like growth factors: actions on the skeleton, Journal of molecular endocrinology, 61 (2018) T115–t137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yavropoulou MP, Michopoulos A, Yovos JG, PTH and PTHR1 in osteocytes. New insights into old partners, Hormones (Athens, Greece), 16 (2017) 150–160. [DOI] [PubMed] [Google Scholar]
- [84].Yuen T, Sun L, Liu P, Blair HC, New M, Zallone A, Zaidi M, Beyond Reproduction: Pituitary Hormone Actions on Bone, Progress in molecular biology and translational science, 143 (2016) 175–185. [DOI] [PubMed] [Google Scholar]
- [85].Zhou H, Cooper MS, Seibel MJ, Endogenous Glucocorticoids and Bone, Bone research, 1 (2013) 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Komori T, Glucocorticoid Signaling and Bone Biology, Hormone and metabolic research = Hormonund Stoffwechselforschung = Hormones et metabolisme, 48 (2016) 755–763. [DOI] [PubMed] [Google Scholar]
- [87].Hachemi Y, Rapp AE, Picke AK, Weidinger G, Ignatius A, Tuckermann J, Molecular mechanisms of glucocorticoids on skeleton and bone regeneration after fracture, Journal of molecular endocrinology, 61 (2018) R75–r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Datta NS, Abou-Samra AB, PTH and PTHrP signaling in osteoblasts, Cellular signalling, 21 (2009) 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Moorer MC, Riddle RC, Regulation of Osteoblast Metabolism by Wnt Signaling, Endocrinology and metabolism (Seoul, Korea), 33 (2018) 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Canalis E, Notch in skeletal physiology and disease, Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA, 29 (2018) 2611–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Corr A, Smith J, Baldock P, Neuronal Control of Bone Remodeling, Toxicologic pathology, 45 (2017) 894–903. [DOI] [PubMed] [Google Scholar]
- [92].Abdallah BM, Jafari A, Zaher W, Qiu W, Kassem M, Skeletal (stromal) stem cells: an update on intracellular signaling pathways controlling osteoblast differentiation, Bone, 70 (2015) 28–36. [DOI] [PubMed] [Google Scholar]
- [93].Hankenson KD, Gagne K, Shaughnessy M, Extracellular signaling molecules to promote fracture healing and bone regeneration, Advanced drug delivery reviews, 94 (2015) 3–12. [DOI] [PubMed] [Google Scholar]
- [94].Prideaux M, Dallas SL, Zhao N, Johnsrud ED, Veno PA, Guo D, Mishina Y, Harris SE, Bonewald LF, Parathyroid Hormone Induces Bone Cell Motility and Loss of Mature Osteocyte Phenotype through L-Calcium Channel Dependent and Independent Mechanisms, PloS one, 10 (2015) e0125731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lenertz LY, Baughman CJ, Waldschmidt NV, Thaler R, van Wijnen AJ, Control of bone development by P2X and P2Y receptors expressed in mesenchymal and hematopoietic cells, Gene, 570 (2015) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kawano S, Otsu K, Kuruma A, Shoji S, Yanagida E, Muto Y, Yoshikawa F, Hirayama Y, Mikoshiba K, Furuichi T, ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells, Cell calcium, 39 (2006) 313–324. [DOI] [PubMed] [Google Scholar]
- [97].Peng H, Hao Y, Mousawi F, Roger S, Li J, Sim JA, Ponnambalam S, Yang X, Jiang LH, Purinergic and Store-Operated Ca(2+) Signaling Mechanisms in Mesenchymal Stem Cells and Their Roles in ATP-Induced Stimulation of Cell Migration, Stem cells (Dayton, Ohio), 34 (2016) 2102–2114. [DOI] [PubMed] [Google Scholar]
- [98].Goltzman D, Hendy GN, The calcium-sensing receptor in bone--mechanistic and therapeutic insights, Nature reviews. Endocrinology, 11 (2015) 298–307. [DOI] [PubMed] [Google Scholar]
- [99].Hu F, Pan L, Zhang K, Xing F, Wang X, Lee I, Zhang X, Xu J, Elevation of extracellular Ca2+ induces store-operated calcium entry via calcium-sensing receptors: a pathway contributes to the proliferation of osteoblasts, PloS one, 9 (2014) e107217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP, Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels, Nature immunology, 9 (2008) 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Bosl M, Stoll G, Nieswandt B, Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation, Blood, 113 (2009) 2056–2063. [DOI] [PubMed] [Google Scholar]
- [102].Choi H, Srikanth S, Atti E, Pirih FQ, Nervina JM, Gwack Y, Tetradis S, Deletion of Orai1 leads to bone loss aggravated with aging and impairs function of osteoblast lineage cells, Bone reports, 8 (2018) 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lee SH, Park Y, Song M, Srikanth S, Kim S, Kang MK, Gwack Y, Park NH, Kim RH, Shin KH, Orai1 mediates osteogenic differentiation via BMP signaling pathway in bone marrow mesenchymal stem cells, Biochemical and biophysical research communications, 473 (2016) 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zhang B, Yan J, Umbach AT, Fakhri H, Fajol A, Schmidt S, Salker MS, Chen H, Alexander D, Spichtig D, Daryadel A, Wagner CA, Foller M, Lang F, NFkappaB-sensitive Orai1 expression in the regulation of FGF23 release, Journal of molecular medicine (Berlin, Germany), 94 (2016) 557–566. [DOI] [PubMed] [Google Scholar]
- [105].Chen Y, Ramachandran A, Zhang Y, Koshy R, George A, The ER Ca(2+) sensor STIM1 can activate osteoblast and odontoblast differentiation in mineralized tissues, Connective tissue research, 59 (2018) 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Sun X, Wei Q, Cheng J, Bian Y, Tian C, Hu Y, Li H, Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells, Human cell, 30 (2017) 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lacruz RS, Smith CE, Bringas P Jr., Chen YB, Smith SM, Snead ML, Kurtz I, Hacia JG, Hubbard MJ, Paine ML, Identification of novel candidate genes involved in mineralization of dental enamel by genome-wide transcript profiling, Journal of cellular physiology, 227 (2012) 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nurbaeva MK, Eckstein M, Snead ML, Feske S, Lacruz RS, Store-operated Ca2+ Entry Modulates the Expression of Enamel Genes, Journal of dental research, 94 (2015) 1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Nurbaeva MK, Eckstein M, Concepcion AR, Smith CE, Srikanth S, Paine ML, Gwack Y, Hubbard MJ, Feske S, Lacruz RS, Dental enamel cells express functional SOCE channels, Scientific reports, 5 (2015) 15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Eckstein M, Vaeth M, Fornai C, Vinu M, Bromage TG, Nurbaeva MK, Sorge JL, Coelho PG, Idaghdour Y, Feske S, Lacruz RS, Store-operated Ca(2+) entry controls ameloblast cell function and enamel development, JCI insight, 2 (2017) e91166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Nurbaeva MK, Eckstein M, Devotta A, Saint-Jeannet JP, Yule DI, Hubbard MJ, Lacruz RS, Evidence That Calcium Entry Into Calcium-Transporting Dental Enamel Cells Is Regulated by Cholecystokinin, Acetylcholine and ATP, Frontiers in physiology, 9 (2018) 801. [DOI] [PMC free article] [PubMed] [Google Scholar]