

Abstract
In this review, we discuss selected examples from recent literature on the role of the support on directing the nanostructures of Au-based monometallic and bimetallic nanoparticles. The role of support is then discussed in relation to the catalytic properties of Au-based monometallic and bimetallic nanoparticles using different gas phase and liquid phase reactions. The reactions discussed include CO oxidation, aerobic oxidation of monohydric and polyhydric alcohols, selective hydrogenation of alkynes, hydrogenation of nitroaromatics, CO2 hydrogenation, C–C coupling, and methane oxidation. Only studies where the role of support has been explicitly studied in detail have been selected for discussion. However, the role of support is also examined using examples of reactions involving unsupported metal nanoparticles (i.e., colloidal nanoparticles). It is clear that the support functionality can play a crucial role in tuning the catalytic activity that is observed and that advanced theory and characterization add greatly to our understanding of these fascinating catalysts.
1. Introduction
Heterogeneous catalysis, where the reactants are in a different phase (typically gas or liquid) from the catalyst (typically solid), plays a central role in the modern-day production of chemicals and fuels.1 In 2016, the total catalyst market size was reported to be somewhere around $23 billion and is expected to reach $40 billion by 2022, with an annual growth rate of 4.8%.2 Supported metal catalysts are an important class of catalysts widely used in industry for several reactions including oxidation, (de)hydrogenation, hydrogenation, hydrotreating, deNOx reactions, ammonia synthesis, and Fischer–Tropsch synthesis. In these catalysts, the metal component is often expensive and is used in very small amounts (typically <5%).1 Smaller metal particles tend to have more active sites exposed compared to larger metallic particles. Hence, metal particle size is a critical structural parameter that determines the catalytic activity of supported metal catalysts.3 Common catalysts contain very small metal particles, typically in the nm range, which are dispersed onto a high surface area refractory support. Even before the explosion of nanoparticle synthesis methodologies in the recent nanotechnology era, small metal nanoparticles (NPs), dispersed on a solid support material were already being used as catalysts.4
Colloidal gold NPs are among the earliest nanomaterials to be produced and exploited in a technological application. For instance, gold colloids were used to introduce the dichroic behavior in the now famous Lycurgus Cup, which dates back to the Romans in the fourth century AD.5 Since then, gold and other metal NPs have been used to a generate range of colors in glassware and windows. Another important milestone in nanotechnology was the synthesis of stable colloidal Au NPs by Faraday in the mid-19th century.6 Despite its long history of use in metallurgy and glass technology, because gold was considered to be the archetypal unreactive noble metal, it was incorrectly assumed to be a poor candidate for a catalyst material. This perception changed dramatically after two seminal discoveries in the 1980s. Haruta found that supported gold NPs display unparalleled catalytic activity for low temperature CO oxidation.7 Hutchings predicted and demonstrated that supported gold is the catalyst of choice for producing vinyl chloride monomer via the acetylene hydrochlorination reaction.8 Since then, gold catalysis has become the subject of received intense attention from both the academic and industrial research communities.9−11 It has become clear that the identity of the support material and the gold–support interfacial sites generated often play a crucial role in determining the catalytic behavior of supported gold NPs.10,12
In the field of supported metal catalysts, support materials were often considered to be inert and their primary role was to enhance the stability of the small metal particles via anchoring. In the 1970s, Tauster introduced the term strong metal–support interaction (SMSI) to explain the unexpected H2 chemisorption properties of noble metal NPs supported on TiO2.13,14 After this report, the potential of the support playing a more active role has been investigated at a fundamental level, mostly using model catalysts. The role of support in practical catalysts is rather more complex and is still not clearly understood in many cases. Supports can play different roles during a catalytic reaction either directly or indirectly. These include providing specific defects sites onto which the metal NPs can be anchored and even stabilized in the case of metastable particles. The support can also enable electron transfer to or from the metal particles and provide additional functionality such as acidity or basicity to the overall supported metal catalyst. All of these factors can dramatically affect the catalytic properties of supported metal materials, and this is especially true for supported Au-based nanoparticle catalysts.
Because of the recent advances in spectroscopic and microscopic methods to characterize supported metal catalysts, a number of articles have now been published on the role of support on the catalytic properties of supported gold-based catalysts, including both monometallic and bimetallic catalysts. In this review, we discuss selected articles in which the role of support on the catalytic properties has been specifically explored. In the first part of this review, we discuss the role of support material in influencing the structural properties of Au-based NPs. Following that, each subsequent section reviews the role of the support on the catalytic properties of Au-based NPs for specific reactions, including CO oxidation, liquid phase alcohol oxidation, hydrogenation/hydrogenolysis, and C–C coupling reactions. The final section of this review briefly discusses the catalytic properties of unsupported gold-based colloids. We have primarily covered selected articles published over the last nine years on this topic, and we conclude the review with a summary and future outlook for this particular line of catalyst research.
2. The Role of the Support during Catalyst Synthesis
The role of support in Au catalysts can be first seen during the catalyst preparation, which has been a challenging task partly due to a relatively low melting temperature of Au (1064 °C) compared to that of other precious metals such as Pd (1555 °C) and Pt (1768 °C). Historically, the discovery that Au-based catalysts can be effective in many industrially important reactions is owed to the successful development of various preparation methods for Au catalysts pioneered by Haruta.7 The development and the choice of synthesis method are often dictated by the physical and chemical nature of the support materials, including surface area, surface acid/base properties, possible phase transformations, and the presence of defects. Because Au catalysts are well-known to be sensitive to the synthesis methods, clarifying the role played by the support materials for Au catalysts in the preparation process is the first step toward developing an overall understanding of their role in any specific catalytic processes.
This section aims to provide a brief overview of common supported catalyst preparation methods for Au catalysts. In some cases, the support materials interact with Au species during the catalyst preparation process to facilitate a high dispersion of Au. They can also serve as anchor sites to stabilize the catalytically active species. These will be mainly discussed in the section 2.1, Preparing Au Catalysts on Oxides and Other Conventional Supports. In other cases, support materials carry specific morphologies and functionalities that need to be preserved or achieved during the catalyst synthesis procedure. These will be discussed in the section 2.2, Preparing Au Catalysts on “Engineered” Nanostructured Support Materials. The following discussion will focus on supported monometallic Au catalysts. Publications on nanoporous Au and Au alloys will be excluded from this section.
2.1. Preparing Au Catalysts on Oxides and Other Conventional Supports
Early attempts at preparing Au catalysts on refractory supports was achieved via conventional impregnation methods using chloroauric acid (HAuCl4) as the gold precursor. Relatively large Au particles (i.e., >30 nm) were usually generated after the calcination step, and the resultant catalysts were ultimately not active.15 Later research concluded that the ineffectiveness of the impregnation method was due to (i) the weak interaction between the AuCl4– ion with the oxide surface in an acidic (pH ∼ 1) environment, (ii) particle growth during the calcination step when the chlorine content is high in the catalyst16 possibly due to a weak bonding between the cationic Au (a soft acid) and the chlorine ion (a hard base), and (iii) chlorine poisoning of the active sites.16,17 Therefore, alternative methods for preparing Au catalysts were developed to counter these adverse effects, and these are described in this section. An alternative method using sol-immobilization is discussed subsequently in the review as this is a means of fine-tuning the nature of the nanoparticle prior to being supported.
Deposition–precipitation (DP) is one of the main methods for preparing supported Au NPs. Haruta and co-workers18,19 first succeeded in making active Au catalysts using this method in the early 1990s. In a typical synthesis procedure for Au/TiO2, preformed TiO2 particles are first mixed with an aqueous solution of HAuCl4. The pH of the mixture is then adjusted using a base (e.g., NaOH) to a pH between 6 and 10 and aged at 70 °C for an hour. The suspension is then washed, dried, and calcined at 400 °C for 4 h. Similar methods have been used for depositing Au on other oxide supports such as Fe2O3,20 Al2O3,17 and MgO.19 A variant of the DP method utilizing urea instead of a strong base like NaOH for preparing Au catalysts was first attempted by Dekker et al.21 and then later by Zanella et al.22 In this method, the hydrolysis of urea (CO(NH2)2) permits gradual and homogeneous control of the OH– concentration and avoids local increases in pH and subsequent precipitation of Au(OH)3 away from the support. The DP method is also effective on other types of supports, such as activated carbon,23 nanodiamond,24 phosphates,25,26 hydrotalcite type layered double hydroxides,27,28 g-C3N4,29 and hydroxyapatite (HAP).30 The DP method can even be used to prepare single-atom dispersed Au catalysts.31 By using UV irradiation instead of a heat treatment, Flytzani-Stephanopoulos and co-workers32 prepared a Au/TiO2 material with atomically dispersed Au species that was shown to be active for the low-temperature water–gas shift (LT-WGS) reaction.
During DP synthesis, a key parameter is the surface charge state of the support material, which depends on the pH value and the isoelectric point (IEP) of the oxide.33,34 For TiO2 (IEP ∼ 4.5–6.3), the surface will be positively charged (terminated by –OH2+) at pH values lower than the IEP and negatively charged (terminated by O–) at higher pH values. In conjunction with this, the nature of the Au species generated also depends on the pH value, as well as the concentration of gold and chlorine present, the ionic strength of the solution, and the reaction temperature. According to Moreau et al.,35 the hydrolysis reactions of AuCl4– gives rise to a complex equilibrium of different gold chloro-hydroxy species at a given pH value (Figure 1a). At pH < 2, AuCl4– is the dominant Au containing species; at pH > 8, the dominant species will be Au(OH)4–; for pH values between 2 and 8, the major species present are charged AuClx(OH)4–x– anions or neutral AuClx(OH)3-x(H2O) species. Therefore, in the DP process, electrostatic interaction between the oxide surface and Au species occurs at lower pH levels, which explains a higher Au uptake under those conditions. However, the Cl content remains high, which could result in large Au particles during the calcination step. The optimum pH for TiO2 usually lies around 6–8, where electrostatic adsorption should not take place. Using X-ray absorption spectroscopy, Louis et al.36 proposed that in a DP method utilizing NaOH, Au species were grafted onto the OH– groups associated with the TiO2 support, forming Ti–O–Au(OH)3– metal complexes. This explains the relatively low Au uptake (i.e., 60% at pH = 6)37 from the Au precursor solution, which is one of the main limitations of the DP method. The DP method utilizing urea allows a higher Au loading be achieved (i.e., 8%) through the precipitation of nitrogen-containing amorphous compounds or the adsorption of an ammino-hydroxo-aquo cationic gold complex.38,39 At pH values >9, the Au loading is limited by the increasing solubility of the Au hydroxide species.38
Figure 1.

Relative calculated equilibrium concentration of gold complexes ([Cl–] = 2.5 × 10–3 M) as a function of the pH of the solution. Reproduced with permission from ref (35). Copyright 2005 Elsevier.
Another significant limitation of the DP method is that it does not work well for oxides having a low IEP, such as SiO2 (IEP < 2–4).34 Anion adsorption (AA) methods have also been attempted by many researchers. In an AA process, the surface of the support is tuned to be positively charged, on which the negatively charged gold chloro-hydroxy species can gradually electrostatically adsorb. The AA process usually takes a long time to complete (typically ∼16 h). This AA method was used by Zenella et al.,22 and Au particles smaller than 4 nm supported on TiO2 were achieved. Lessard et al.40 used this approach and made active gold catalysts for low-temperature water–gas shift reactions on La2O3 and La2O2SO4. Because of the high IEPs of these supports, the pH of the mixture was tuned so that the main species adsorbed was Cl-free (i.e., Au(OH)4–).
The Au uptake and highest Au loading attainable depends on the nature of the support. By carefully tuning the HAuCl4 concentration and the pH of the solution, Pitchon, Petit, and co-workers41,42 achieved 100% Au uptake and a 2% final loading on an Al2O3 support. Furthermore, no gold was lost during the filtration and washing steps. It was also suggested that there is some kind of anion exchange process taking place between the Au species with the surface hydroxyl groups associated with the Al2O3 support. A similar method was later used by the same group to disperse Au onto layered double hydroxides.43 Nguyen et al.44 reported a better thermal stability of the Au/γ-Al2O3 catalysts prepared by the AA method compared to those prepared by conventional deposition precipitation (DP).
Single-atom Au catalysts can be also prepared using the AA route. Wang et al.45,46 reported atomically dispersed gold on ZnZrOx, while Qiao et al. prepared single-Au atom catalysts on Co3O4,47 CeO2,48 and FeOx.49 The metal loading of these catalysts were usually kept at a very low level of around 0.05 at %.
For a SiO2 support that has a low IEP, cationic adsorption is one possible strategy to prepare active Au catalysts. Trichlorobis(ethylenediamine)gold(III) (Au(en)2Cl3) can serve as the cationic precursor, and Au/SiO2 catalysts have been prepared by Zanella et al.36 and later by Dai and Overbury50−52 using this cationic absorption approach. As mentioned earlier, in the DP urea or ammonia process, the cationic adsorption of amino-hydroxo-aquo cationic Au complexes were thought to be taking place.39,53
Preparing Au/SiO2 with the more readily available HAuCl4 precursor can be done by adjusting the surface functionality of the SiO2. Amine functional groups can provide positively charged aminium ions in an acidic solution, and therefore AuCl4– anions can be electrostatically adsorbed. In a 2009 study by Liu et al.,54 commercial SiO2 supports were refluxed with APTES (H2N(CH2)3Si(OEt)3) in ethanol for 24 h, so that the amine functional group can be grafted onto the support before adding HAuCl4. This effectively shifts the IEP of the SiO2 to a higher value.55 In a more recent study, branched polyethylenimine was used to functionalize the SiO2 to anchor glutathione-protected Au clusters.56 It should also be noted that Au catalysts on silica supports can also be prepared using a double-support strategy, which can in some sense be considered as functionalizing the SiO2 surface with another oxide. For instance, Au catalysts on TiO2,57 CoOx,58 and FeOx-modified59 SiO2 supports have been reported.
Functionalizing activated carbon surfaces by acid washing has been studied by Willock and co-workers.60 It was found that washing the carbon support with nitric or hydrochloric acid can almost exclusively generate surface hydroxyl groups, which can better assist the nucleation of Au particles compared to a carbon surface covered with ketone groups.
Another way of achieving high dispersion of Au and an intimate contact with the support is to prepare both components cooperatively. The most common approach is the coprecipitation (CP) method, developed by Haruta and co-workers18,61 for Au catalysts. They reported that certain oxide supports (e.g., α-Fe2O3, Co3O4, and NiO) can be precipitated out from the solution largely simultaneously with Au, therefore ensuring good mixing. Haruta’s original method involves quickly pouring an aqueous solution of HAuCl4 and the corresponding metal nitrate precursors into an aqueous solution of sodium carbonate (Na2CO3). The resultant precipitates (usually hydroxides) will then be subjected to washing, vacuum drying, and calcination, typically at 400 °C to form the final catalyst. In this CP process, through a quick mixing of the acidified precursor solution into the basic Na2CO3 solution (pH = 8), the AuCl4– will undergo the hydrolysis process described earlier and release Cl–, which can be further removed during the washing step. The final Au/metal oxide catalyst is generated during the calcination step. As confirmed by XRD and TEM characterization, at Au loadings of 5–10 at % with respect to the support transition metal, Au NPs below 10 nm in size are generally formed.18
Andreeva et al.62 and Hutchings and co-workers63,64 later reported slightly different CP methods for preparing such metal oxide supported Au catalysts. The modification comes from varying the sequence of mixing the acid and base precursors as compared to Haruta’s original method.18,61 In this case, the Na2CO3 solution was added gradually into an aqueous solution of metal nitrates and HAuCl4 until a pH of 8–9 was attained, followed by the usual washing, drying, and calcination steps. The resultant catalysts were shown to be active for low-temperature water–gas shift,62 CO oxidation at ambient temperature,63 and the direct synthesis of hydrogen peroxide.64
With the advances of aberration-corrected scanning transmission electron microscopy (AC-STEM), Herzing et al.65 reported that Au subnanometer clusters and isolated atomic Au species also exist in the catalysts prepared by the CP method. Furthermore, it was proposed that the subnanometer clusters might be responsible for the high activities observed in CO oxidation reactions rather than the Au NPs. Using cyanide leaching and in situ electron microscopy, Allard et al.66 demonstrated that a significant amount of atomically dispersed Au can be trapped inside the CP generated support materials, which can subsequently diffuse outward to the surface during subsequent heat treatment (Figure 2). This is not too surprising considering the nature of the CP method. He et al.67 later studied Au on Fe2O3e prepared by the above two CP methods and found that a larger fraction of Au can be trapped into the support when the acidic and basic solutions are mixed quickly (i.e., via Haruta’s original method). The dynamic evolution of Au species during the heat treatment (e.g., via diffusion and aggregation of Au on the surface and via outward diffusion of trapped internal Au species) determines the final population distribution of Au species on the oxide surface as well as the catalytic activity of the material in the CO oxidation reaction.
Figure 2.

High angle annular dark field images of the leached catalyst showing changes as a result of in situ heating. Very little change was seen with time at 250 °C. (a) Starting image recorded after a few minutes at temperature. Only slight changes were seen after 2 min at 500 °C (b), but an additional 5 min at 500 °C showed void shrinkage, coalescence and growth of Au NPs, and the diffusion of Au species to the surface to form discrete nanocrystals (arrowed). Reproduced with permission from ref (66). Copyright 2009 Oxford Academic.
Kudo et al.68 further modified Haruta’s CP method by adding HAuCl4 approximately 1.5 h after mixing the metal nitrates with Na2CO3. They found that this procedure significantly increased the available Au sites for CO adsorption, probably because the support and Au no longer precipitate simultaneously, meaning that much less Au will be trapped inside the support. The method then becomes very similar to a deposition–precipitation method. Another modification of the conventional CP method was reported later by Zhang et al.69 Here, an electrochemical approach was used to monitor the concentration of Cl– ions in the solution mixture after coprecipitation, which is thought to affect the precipitation of the support and gold hydroxides. The most active catalysts can then be reliably reproduced when the [Cl–] concentration lies in the 1–3 ppm range and the Au NPs are thought to be mainly sitting on the edge of nanosized Fe2O3 particles after calcination.
One recent example of Au catalysts prepared by coprecipitation is the Au/α-MoC catalysts reported by Ma and co-workers.70 The catalysts were prepared by mixing aqueous solutions of (NH4)6Mo7O24·4H2O and HAuCl4, followed by washing, drying, and a 500 °C calcination step. AC-STEM characterization of their materials confirmed the formation of epitaxial Au rafts, 1–2 nm in diameter and 2–4 atomic layers thick, grown on an α-MoC support, which are highly active for the water–gas shift reaction even at room temperature.
Finally, heat treatment is usually needed when preparing Au catalysts in order to convert the precursors of Au and support oxides into their active forms. One of the key roles of the support is to stabilize the Au species and maintain their high dispersion. Recently, de Jongh and co-workers71 showed that on a TiO2 support, Au particle agglomeration can be accelerated by the presence of water and/or the presence of Cl–, and it is more pronounced in an oxidizing atmosphere. In contrast, Au on nonreducible supports such as SiO2 and Al2O3 are remarkably stable in a nonoxidizing atmosphere. In another study by Zhang and co-workers,72 2–5 nm Au particles epitaxially supported on MgGa2O4 spinel were shown to retain their original particle size even after heating above the melting temperature of bulk gold (1064 °C), demonstrating the potential efficacy of particle stabilization effects from the support lattice.
2.2. Preparing Au Catalysts on “Engineered” Nanostructured Support Materials
In many cases when synthesizing Au catalysts, efficiently and homogeneously dispersing the Au is the main concern during the preparation, especially when commercial metal oxide support materials are used. In other cases, however, creating and/or maintaining the special nanostructure of the support material can be equally important. Support materials with well-designed nanostructures and architectures can not only bring additional functionalities to the Au catalysts, but it also can serve as model catalysts for mechanistic investigations.73 In this section, we will discuss the preparation of Au catalysts on a variety of support materials having specially designed nanostructures. These include (i) ordered porous materials, such as zeolites, mesoporous silica, and metal–organic frameworks (MOFs), (ii) Au catalysts with iron oxide heterostructures that allow magnetic separation, and (iii) Au catalysts with yolk–shell or core–shell nanostructures and so-called “inverse” oxide/metal catalysts.
2.2.1. Au on Mesoporous Supports
Mesoporous silica, zeolites, and other materials with controlled pore structures are widely used as catalysts and support materials.74 Preparing Au catalysts on such mesoporous materials is potentially attractive for at least two reasons: First, it is harder for Au species in the mesoporous materials to migrate and agglomerate due to the geometric constraints imposed by the structure, thus resulting in Au catalysts having superior stability compared to those prepared on conventional high surface area metal oxide supports. For example, Dayte and co-workers75 showed that the sintering of Au particles is dependent on pore size, pore wall thickness (which determines the pore wall strength), and pore connectivity. For instance, Au supported on materials with two-dimensional pore structures and lower connectivity (e.g., SBA-15) showed better stability compared to those supported on materials with 3-D pore structures and high interconnectivity (e.g., SBA-12), in which Au can migrate more easily between pores and even to the outer surface (Figure 3).
Figure 3.

HAADF-STEM images of Au catalysts samples after reduction at 200 °C for 2 h in flowing hydrogen supported by (a) MCM-41, (b) SBA-15 with 4.8 nm thick pore walls, (c) SBA-15 with 2.6 nm-thick pore walls, (d) HMM-2, (e) SBA-12, and (f) SBA-11. Reproduced with permission from ref (75). Copyright 2005 American Chemical Society.
Second, it is often desirable to combine some catalytic functionality imparted by the mesoporous support with that of gold to generate “multifunctional” catalysts. One good example is Au supported on titanium-silica 1 (TS-1), which itself is an important catalyst for the selective oxidation or epoxidation of olefins. The combination of TS-1 with Au can create a bifunctional catalyst, whereby olefins can be epoxidized over TS-1 with H2O2 formed in situ from H2 and O2 produced over Au.76−78
For mesoporous silica, the challenges in preparation are similar to those described for the regular silica supports which were discussed earlier. Mesoporous silica materials can also be deliberately decorated with functional groups, such as amine,79−83 thiol,84,85 and pyrrolidone,86,87 to increase the interaction of the support with Au. Functionalizing the preformed Au particles/clusters before deposition on the mesoporous silica support is another approach which has been tried. For example, Tsukuda et al.88 have reported an active alcohol oxidation catalyst in which Au particles supported on mesoporous silica were prepared using triphenylphosphine functionalized Au11 clusters. Secondary support layers/particles comprised of, for example, TiO2,89−92 CeO2,93−95 and BaSO4,96 within the mesoporous silica structures have also been commonly used to enhance Au attachment. Conventional preparation methods for dispersing Au on those materials can be directly applied when such a double-support configuration is employed.
To ensure most of the Au is located within the pore structure of the support, it is often desirable to have a one-pot synthesis of the mesoporous support and Au NPs. Overbury et al.97 developed a coassembly technique using a bifunctional organosilane ligand in which the amine group complexes with Au(III) and the siloxane group interacts with the silica matrix during a sol–gel template synthesis. Uniform 2–5 nm Au NPs with up to 7 wt % loading were prepared within the pores of an MCM-41 material by this method. A similar approach using thiol-containing organosilane ligands was subsequently reported by Wu et al.81,98 and Chen et al.99,100 Budroni and Corma101 have reported preparing a Au–organic–silica catalyst that was synthesized from Au NPs capped with both 1-dodecanethiol (DT) and 3-mercaptopropyltrimethoxysilane and dispersed in ethanol containing tetraethyl orthosilicate (TEOS), followed by the hydrolysis of the TEOS.
In addition to mesoporous silica, other mesoporous oxides have been also explored as support materials for Au catalysts such as mesoporous titania,102−104 mesoporous ceria,105−108 and mesoporous iron oxide.109 More traditional preparation methods, such as incipient wetness, deposition precipitation,110 ion exchange,111,112 chemical vapor deposition,113 or sol-immobilization, can be directly used to make Au catalysts on these nonsilica-based mesoporous supports.114
Au single-atom catalysts have also been successfully prepared using mesoporous supports. For example, Flytzani-Stephanopoulos and co-workers115 reported making low loading (i.e., 0.25 at %) Au catalysts on mesoporous MCM-41 and KLTL-zeolite materials doped with alkali ions (i.e., Na+ and K+) using simple incipient wetness impregnation methods. It was found that the alkali ions are responsible for stabilizing the atomically dispersed Au in the form of Au–O(OH)x, which is found to be highly active in the low-temperature water–gas shift reaction.
Porous coordination polymers, especially metal–organic-frameworks (MOF),112 are emerging as important catalysts and/or catalyst support materials. Hermes et al.116 have reported Au/MOF-5 materials prepared by a CVD method using a (CH3)Au(PMe3) precursor. Haruta and co-workers117 later prepared Au catalysts on several porous coordination polymers with a solid grinding method using Me2Au(acac), which is also a convenient source of Au in CVD-based preparation methods.113 A typical preparation procedure involves first grinding the coordination polymers with Me2Au(acac), a slightly volatile organogold complex, using an agate mortar and pestle, in air for 20 min without the need of organic solvent, and then a heat treatment at relatively low temperature (e.g., 120 °C). The resultant catalysts were found to be active for benzyl alcohol oxidation. Additional work has been reported more recently using a similar method for producing ultrafine Au NPs within MOF structures.118,119 Impregnation in organic media using Me2Au(acac) is also a possible route to make Au catalysts supported in MOFs, as reported by Epron and co-workers.120 Providing that the MOF is properly functionalized, e.g., amino-functionalized MIL-53(NH2)121 or UiO-66(NH2),122 HAuCl4 can be used as a precursor in an in situ reduction type method using NaBH4 as the reductant in the solution. Dai et al.123 have recently reported a charge modulation approach, in which charged species were created within the framework of a pyridine-functionalized conjugated organic network (P-CON) by introducing fluoride anions, which serves as a stabilization center for Au NPs.
2.2.2. Au on Magnetic Supports
Magnetically recyclable catalysts are of considerable interest to the research community as they potentially enable separation steps in various reaction process and can serve as a bridge between homogeneous and heterogeneous catalysis.124,125 For Au catalysts, there are a reasonable number of publications that focus specifically on preparing composite materials between Au NPs and magnetic NPs (e.g., iron oxide), the latter of which serves the dual purpose of acting as a “support” for the gold and providing the magnetic functionality to the catalyst.126
Sun and co-workers127 reported the synthesis of Au/Fe3O4 “dumbbell” NPs, a few nanometers in dimension on each side, using a bottom-up approach. The synthesis involves the decomposition of Fe(CO)5 on the surface of preformed Au NPs followed by oxidation in a 1-octadecene solvent. Additional control over the size, structure, and chemical nature of such composite materials are also possible by regulating the reaction conditions. By depositing such particles onto more conventional catalyst supports, such as metal oxide or carbon, highly active and magnetically recoverable catalysts can be made (Figure 4).128,129 It has also been reported that the magnetic properties of the iron oxide can be enhanced due to an interfacial interaction with the metal particle.130 A slightly different preparation method was reported by Lin et al.,131 whereby decomposition of an iron–oleate complex was carried out at high temperature (i.e., 310 °C) to produce very similar dumbbell morphologies.
Figure 4.

(a) A high resolution TEM image of a Au–Fe3O4 dumbbell nanoparticle. (b) Schematic diagram showing the concept of double support strategy and three possible interactions between Au–Fe3O4 dumbbell NPs and supports. (c,d) TEM images of Au–Fe3O4 supported on SiO2: (c) bright field, (d) dark field. The dumbbell-like NPs were highly dispersed on the support. Reproduced with permission from ref (128). Copyright 2008 The Royal Society of Chemistry.
Spivey and co-workers132 reported making particles having an Fe3O4 core of ∼5 nm diameter coated with a thin (i.e., 0.5 nm) Au shell, which were themselves supported on TiO2. These were prepared by reducing Au precursors in the presence of iron oxide NPs. Meng et al.133 have also reported a one-pot solvothermal synthesis method for making Au–Fe3O4 nanocomposites, in which the resultant iron oxide particles are usually in the 10–100 nm size range. Kong et al.134 reported a one-pot hydrothermal synthesis method, which incorporates magnetic γ-Fe2O3 and Au particles into the wall of mesoporous silica materials. The resultant material is both catalytically active and magnetically recoverable. Magnetic materials other than iron oxides have also been explored, including Ni/SiO2@Au microspheres,135 and Au supported on CoFe2O4 nanotubes.136
2.2.3. Au Encapsulated in Supports
Advances in nanotechnology have allowed novel types of support structure to be developed, opening new possibilities for supporting Au catalysts. For instance, to remediate the relatively poor thermal stability of Au nanocatalysts, nanomaterials have been engineered not only to support the Au particles but also to encapsulate them. For instance, Zhan et al.137,138 demonstrated porous carbon as having potential as a covering layer for Au NPs, which impart exceptional thermal stability to the Au NPs while still allowing catalysis to take place on the Au surface. Such a porous carbon layer was introduced through the decomposition of amine-containing carbonaceous surfactants attached to the Au NPs. Similarly, Zhang et al.139 demonstrated that a porous TiOx overlayer can be intentionally introduced to pin down Au NPs through a wet chemistry approach. Such a layer not only discourages Au from sintering but also enhances CO oxidation through a strong metal–support interaction (SMSI).
The encapsulation of Au NPs can also be achieved through a colloidal synthesis approach, forming a Au-core/porous shell structure. Gao et al.140 reported small (<3 nm) and highly stable Au NPs encapsulated by SiO2, prepared from ultrasmall gold hydroxide NPs, followed by SiO2 coating within reverse micelles, then finally converting Au hydroxide to Au through thermal annealing. Amine functionality can also be introduced during the SiO2 coating procedure in order to achieve a better affinity for the metal.141 Bai et al.142 have reported a method for preparing mesoporous aluminosilicate encapsulated Au@SiO2 multilayer core–shell catalysts. Au core–shell catalysts with TiO2143 and titanium silicate142 shells have also been reported recently.
Another type of Au catalyst with an encapsulating support was first introduced by Schüth and co-workers in 2006.144 As shown in Figure 5, the synthesis begins with Au NPs, upon which a SiO2 shell was then deposited, followed by deposition of another thin layer of porous ZrO2 with a typical pore size of 3–4 nm. The SiO2 then can be etched away, leaving Au encapsulated by a hollow ZrO2 shell, in a so-called “yolk–shell” configuration. Compared to the “core–shell” nanostructure, the “yolk–shell” architecture obviously allows more Au surface sites to be exposed for participation in catalytic reactions. The shell not only serves as the support but can also provide mass-transfer controls between the reactant molecules with the gold. The immediate benefit is a significantly improved thermal stability: the catalyst retains the same activity in CO oxidation even after a high-temperature calcination treatment at 800 °C. The same group later reported a carbon-based yolk–shell structure145 and demonstrated the possibility of using cyanide leaching as a method for fine-tuning the size of the Au particles after the yolk–shell structure had been synthesized.146,147
Figure 5.

General scheme of the yolk–shell synthetic process and TEM images (left, dark field; middle and right, bright field) of the products obtained after each step. From left to right: in the first step, colloidal gold particles with sizes of approximately 15 nm are synthesized. These colloids are covered with a dense silica layer by a modified Stöber process to form monodisperse silica spheres with one gold particle in the center of each sphere. The spheres are then covered by a thin layer of zirconia particles with pores 3–4 nm in diameter between the individual zirconia particles. Finally, the silica is leached out and loose gold particles, now no longer in the center of the spheres, are obtained. (The formula “Au, @ZrO2” indicates the presence of free space between the encapsulated gold particle and the mesoporous ZrO2 shell.) Reproduced with permission from reference (144). Copyright 2006 John Wiley and Sons.
Zaera and co-workers148,149 have prepared Au@TiO2 yolk–shell structures using a similar approach. To demonstrate that Au@SiO2 yolk–shell structures are also possible, Song and co-workers150 used a much larger Au particle (∼120 nm) as the original core, followed by SiO2 coating, and then employed cyanide to leach away some of the internal Au material. Zhang and co-workers151 used polystyrene-co-poly(4-vinylpyridine) microspheres as both templates to fabricate a porous silica shell via a sol–gel process which acts as a scaffold to immobilize the Au NPs. A yolk–shell structure was then formed after calcination, during which the polymer microsphere is burnt away. Au-based yolk–shell catalysts incorporating TiO2,152 CeO2,153,154 and ZrO2155 shells have now also been reported.
Finally, because the interface between Au and the support are often considered catalytically important,156 an inverse catalyst concept has also been investigated for Au catalysts, whereby the original support is now created in the Au NPs. Dumesic and co-workers157 demonstrated such a case in 2015, in which MoOx moieties are dispersed onto Au NPs using a controlled surface reaction approach. The interface between the MoOx species and the Au are considered to be the active site for the reverse water–gas shift reaction in this catalyst. In this case, the concept of what is actually acting as the support material is getting blurred.
In this section, we have surveyed the plethora of preparation methods for synthesizing Au catalysts on different types of support materials. One challenge in determining the full role of the support during the synthesis is that advanced techniques for characterizing the material, such as synchrotron-based X-ray absorption spectroscopy (XAS) and AC-STEM, are not always readily available to the materials chemist. Given the fact that Au catalysis is very sensitive to nanostructure, there is still much scope for understanding the details of these synthesis methods and the crucial role played by the support in determining the final catalyst nanostructure. Understanding the impact of the support on Au dispersion, the choice of Au precursors and the required treatments (e.g., heat treatment, leaching, surface functionalization) is an important goal in order to reveal the precise role of the support in the actual catalytic reactions, a topic which will be covered in more detail in subsequent sections of this review article.
3. The Role of the Support during Reactions on Au Nanostructures
3.1. CO Oxidation and Related Reactions
The CO oxidation reaction is one of the most widely studied processes in the area of Au catalysis. It also is important for our understanding of a range of related reactions, such as preferential oxidation of CO in the presence of H2 (PROX) and the water–gas shift (WGS) reaction, which will only touch on in this review. The resilience of bulk gold to oxidation has led to much discussion on the mechanisms for oxygen adsorption and activation over supported NPs. Clearly, O2 must be activated for CO to be transformed to CO2, but the observed high activity of gold NPs is in stark contrast to the well-known inert behavior of single-crystal surfaces of Au.158 The field of CO oxidation by supported Au particles has been the subject of a number of reviews.159 These have highlighted the major aspects of this chemistry, including the support influence on reactivity, discussion of the active oxidation state of gold, the importance of water in the oxidation reaction, and particle size and morphology effects. These continue to be hot topics of discussion and are also now enhanced by new approaches to catalyst synthesis, new insights from characterization methods and kinetics, and the advent of well-defined single-metal site catalysts. The area has also benefitted from close interactions between specialists in materials synthesis, characterization,160 catalytic testing, surface science, and applied theoretical chemistry.161
Theoretical studies have shown that isolated Au particles are capable of oxidizing CO.162 However, in the most widely accepted mechanism for practical rates of oxidation, oxygen adsorption is believed to occur on the support or at the metal–support interface. In particular, oxygen adsorption is thought to be favored by the presence of oxygen vacancies that would be expected on supports that are semiconductor materials (e.g., TiO2 and ZnO) as a consequence of the Schottky junction at the metal/support interface.163 Oxide supports have been broadly divided into “active” semiconducting, which can sustain oxygen vacancy creation by reduction of metal centers (i.e., reducible supports), and wide band gap materials, which tend to be irreducible and are thought to be “inactive” in oxidation reactions.164 The influence of the point of zero charge on the Au/support effects are also reported to have important implications for CO oxidation catalysis.165 For large Au NPs, (i) CO reversibly adsorbs at low temperatures (T < 150 K),166 or is in dynamic equilibrium at sufficiently high partial pressures, which is also the case at around room temperature and above,167 (ii) CO oxidation takes place readily only when oxygen is provided in atomic form,168−170 and (iii) dissociative adsorption of oxygen is strongly hindered by a high dissociation barrier due to a weak coupling to the Au substrate due to the filled d-states,165,171,172 and (iv) adsorbed CO may be directly oxidized to CO2 or transformed into carbonates or formates, which then decompose to CO2 and water or surface hydroxyl groups.159 Which route dominates depends on catalyst composition, preparation method, and reaction conditions.
Oxide supports with irreducible character, such as Al2O3, SiO2, and MgO, generally show a low ability to adsorb or store oxygen and yet CO oxidation is observed for Au NPs on these supports.173 This indicates that oxygen adsorption and dissociation must be possible on the gold clusters themselves, although DFT calculations suggest there is only a weak interaction162 except for particular facets of small NPs. For these irreducible supports, activity for CO oxidation critically depends on the diameter of the gold particles, and usually only very small particles (<2 nm) yield highly active catalysts.164,174,175 The increasing activity with smaller particle size was explained by an enhanced dissociative adsorption of oxygen on small gold particles due to a higher density of reactive defect sites (edge, kink, or step sites)176,177 or a gradual change in the electronic structure at decreasing size.174,178,179 On the other side are catalysts which are supported on reducible transition metal oxides, such as Fe2O3, CeO2, NiOx, CoOx, and TiO2. These exhibit a superior activity for oxidation reactions, with activities higher than those of irreducible supports by up to 1 order of magnitude. On these catalysts, the size of the gold particles also seems to play a more secondary role, with even large Au particles up to 30 nm in size demonstrating activity,164 although in some cases deactivation has been noted as particles sinter and so grow in size.179,169 This observation may also be explained by the metal loading, where the metal covers enough area of the support to mitigate its role providing atomic oxygen.169 One widely discussed explanation for the higher CO oxidation activity of Au NPs supported on reducible supports is the supply of oxygen from the lattice of the support in a Mars van Krevelen (MvK) process. Scheme 1 shows one reaction sequence that includes a MvK style supply of oxygen. First CO adsorbs close to the interface between metal particle and support. Then lattice oxygen forms a new bond with carbon, forming an oxygen anion vacancy. The vacancy can be filled in a second step through the dissociation of molecular oxygen at the defect site. When lattice oxygen (O2–) is removed during this oxidation route, the two electrons from the ion can be accommodated by the reduction of support cations (e.g., Ti4+ τo Ti3+) which is not possible on an irreducible support. This also means that the activation of O2 must take place at the Au/oxide interface as molecular oxygen dissociates to heal the anion defects that are formed.
Scheme 1. MvK Scheme for Oxidation of CO at the Au Nanoparticle Interface with an Oxide Support.

Note: the square symbol is used to indicate a lattice vacancy.
On the practical assessment of oxidation catalysts, Kipnis et al. has analyzed work on CO oxidation and preferential oxidation of CO in the presence of H2 (PROX) to emphasize the importance of the exothermic nature of CO oxidation reactions.180 By measuring the temperature at the inlet, middle and outlet of the catalyst bed in a plug flow reactor, the temperature gradient can be used to highlight the exotherm effect. Figure 6 shows this measurement for a PROX reaction in which a 1:1 mixture of CO was used in a large excess of H2. Not all of the CO is consumed as the oxygen will also react with H2. At steady state, there is a 20 °C difference between the inlet and the outlet temperatures as the exotherm has caused light-off at the front of the bed and the oxidation reaction becomes diffusion limited there. The rest of the catalyst has no oxygen supply, and so oxidation cannot take place in these regions. This makes it difficult to compare catalysts under high CO conversion conditions, and reliable kinetic parameters are only obtained from experiments at low CO conversion. Many researchers simply compare catalysts based on light-off curves generated by monitoring CO conversion as the temperature of the catalyst bed is increased.
Figure 6.

Preferential oxidation of CO over 1%Au/Al2O3, bed height 1.6 cm. Gas composition: vol %, O2 0.9%, CO 0.9%, H2 60% balance N2. Reaction nominally at room temperature, GHSV 41 N l gcat–1 h–1. Reproduced with permission from ref (180). Copyright 2014 Elsevier.
3.1.1. Irreducible Oxides As Supports
One of the features worthy of discussion for Au catalysts supported on reducible supports is their oxygen storage capacity. This is usually thought of as the ability of the catalyst to use up lattice oxygen from the support for CO oxidation, which can be replenished by O2. However, irreducible supports can also show some oxygen storage ability. Gleaves and co-workers have used a temporal analysis of products (TAP) reactor to study CO oxidation over a Au/SiO2 catalyst prepared by magnetron sputtering.181 TEM analysis shows that materials prepared in this way have a mean particle size of around 3.2 nm. In a TAP experiment, the catalyst bed is held under vacuum and exposed to short (10 s of ms duration) pulses of reactant gases with the outlet of the reactor monitored using a mass spectrometer. The pulses contain very few molecules (around 10–9 mol per pulse) so that gas molecule collisions are minimized and the temporal profile of gases exiting the reactor can be deconvoluted into surface reaction kinetics and a Knudsen diffusion component. In Figure 7, a titration experiment using the TAP reactor approach is shown. Here, the reactor bed is first exposed to a flow of O2 before the catalyst is placed under vacuum. The TAP reactor is then used to pulse CO. In the first few pulses, all CO is converted into CO2 so that the oxygen required can only be provided by that stored on the catalyst. By integrating the pulse areas, the amount of oxygen stored can be estimated. Analysis of the dependence of the oxygen reservoir formed in the pretreatment of the catalyst on the pretreatment pressure showed that oxygen is stored both on the Au surface and subsurface regions of the NPs. This study was also able to estimate the heat of adsorption of CO on a fully reduced Au/SiO2 sample to be −24.4 ± 3.7 kJ mol–1.
Figure 7.
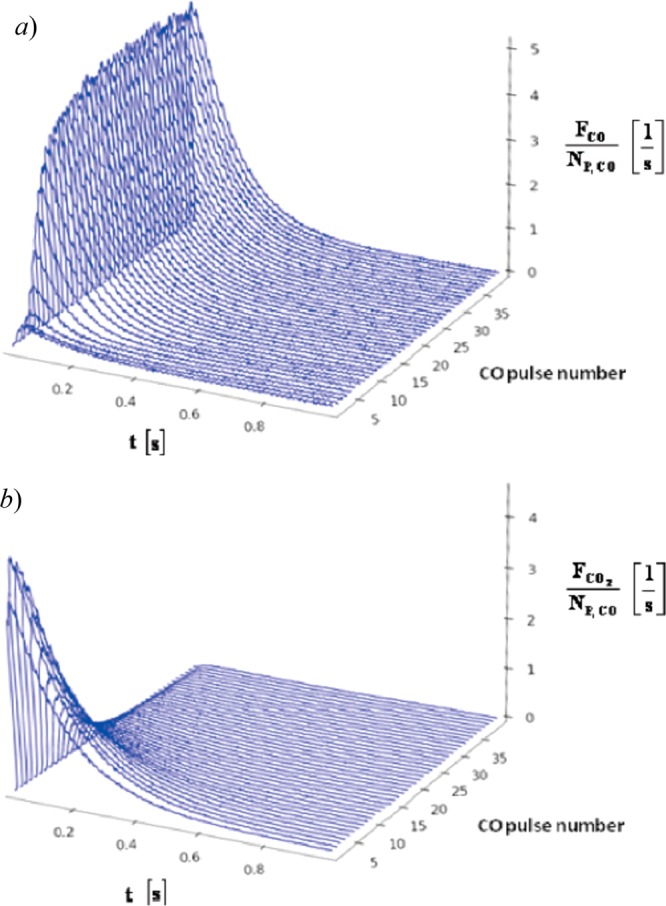
TAP CO titration experiment. Au/SiO2 catalyst exposed to a flow of 1 atm of O2 for 5 min in the TAP reactor before being placed under vacuum. Then a series of CO/Ar (50/50) pulses were used to quantify stored oxygen. (a) CO pulse intensity normalized CO exit flow. (b) CO2 exit flow normalized to CO pulse intensity. Reproduced with permission from ref (181). Copyright 2010 ACS Publications.
Schüth and co-workers have prepared Au catalysts supported on Mg(OH)2 and MgO prepared from the dehydration of the hydroxide.182 Au was deposited using a colloidal method at a loading of 0.7 wt % producing particles with a mean diameter of 3.1 nm. It was found that this produced negligible numbers of particles below 1 nm based on HAADF-STEM images. Both supports were produced from a mesoporous MgO starting point, and this turned out to be key because the high CO oxidation ability of these materials was not reproduced using conventional magnesium oxides or hydroxides. The catalysts were found to be active at temperatures as low as −85 °C (below the sublimation temperature of CO2, although the partial pressure of CO2 is too low for solid to form over the bed of the reactor).
Figure 8 shows the interesting temperature dependence observed for CO conversion. At very low temperatures and low space velocity (80 000 mL h–1 gcat–1), 100% conversion of CO is obtained, and this performance is maintained up to 30 °C, at which point the conversion dips to 73% at around 90 °C before returning to full conversion at 230 °C and above. At higher space velocity (400 000 mL h–1 gcat–1), the CO conversion is seen to rise from 35% at −85 °C until it eventually reaches 100% conversion at around −60 °C and the U-shaped behavior at higher temperatures is more pronounced. This work also used the flow reactor to titrate available oxygen by flowing CO over a preoxidized catalyst. A reservoir of active oxygen was identified, which was large enough to rule out oxygen storage by Au alone. This suggests that the mesoporous support was able to provide a reservoir of oxygen to supply the reaction even at very low temperatures. The authors also pointed out that the high activity of these materials is achieved despite their containing negligible amounts of Au particles below 1 nm. This they attribute to the support’s ability to supply active oxygen into the reaction even at very low temperatures.
Figure 8.
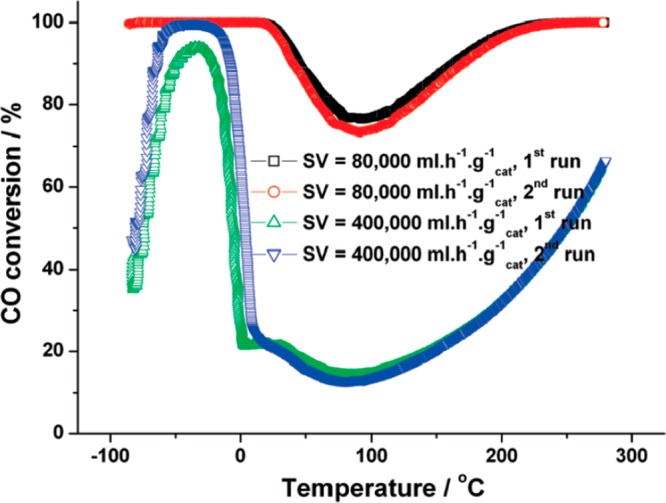
Dependence of CO conversion on temperature for Au/MgO(OH)2 prepared from mesoporous MgO. Gas composition 1% CO in 20% O2 in N2, flow rate 67 mL min–1, catalyst mass 50 mg (SV = 80 000 mL h–1 gcat–1) or 10 mg (SV = 400 000 mL h–1 gcat–1). Reproduced with permission from ref (182). Copyright 2010 American Chemistry Society.
For the Au/MgO(001) with an irreducible support, Duan and Henkelman have used DFT calculations to look at O2 adsorption and activation.183 They found that, even though the MvK process is not possible, the support still has an influence on oxygen activation. The Au work function is reduced on the MgO surface due to the interfacial dipole so that charge transfer to the O2 2π* orbital becomes easier and makes the dissociation of O2 more facile than for the Au surface away from the interface. Even so, the dissociation of O2 was found to be kinetically hindered so that the work suggests a mechanism in which O2 scission only occurs as CO2 is formed. This is similar to the Au-only mechanism originally proposed by Norskov.184
3.1.2. Reducible Oxides as Supports
Perhaps the reducible support that has been most widely studied for CO oxidation and related reactions is titania. TiO2 is a semiconducting oxide which has a number of polymorphs that differ in their chemical and electronic properties including their band gap.185 The usual forms used as catalyst supports are rutile and anatase, with the common Degussa P25 titania consisting of a mix of the two (80% anatase, 20% rutile).186 The World Gold Council (WGC) has also produced a reference standard for Au/TiO2 using P25 as the support material and depositing Au by the DP method with NaOH as the precipitating agent.
Chenakin and Kruse have synthesized pure anatase TiO2 using a titanyl oxalate complex as the titanium source.187 Gold catalysts (GC) were produced at a 1.5 wt % loading of gold using a deposition precipitation approach with NaOH (GC1) or urea (CO(NH2)2) (GC2 and GC3) as precipitating agents. The choice of urea as a precipitating agent avoids the introduction of Na+ ions into the catalyst material and other aspects of the synthesis for GC2 and GC3 were also designed to exclude Na+. Calcination of the materials was used to control mean particle size with GC3 (2.7 nm) and GC2 (2.8 nm) containing particles near to that of the WGC reference material, while the Au particles on GC1 (4.6 nm) were considerably larger. The measured CO oxidation rates (2% CO, 2% O2 in Ar; GHSV 30 000 mL gcat–1 h–1) followed an inverse power law in relation to particle size with an exponent of −2.2. This was interpreted as confirming that the periphery of the particle, and so the interface between nanoparticle and support was the important factor leading to CO oxidation activity, in line with earlier work.188 Even so, the WGC catalyst did not lie on the same trend line, showing 20% higher activity than either GC2 or GC3. The same work also reported detailed XPS analysis of the WGC catalyst (Figure 9). The Au 4f signal shows the expected characteristic shape for Au in the metallic state but shifted slightly to lower energies. This the authors interpreted as indicating negative charging of the NPs to produce Auδ-; correspondingly, the Ti 2p spectrum shows a Ti3+ feature suggesting that the oxide has been reduced through electron transfer to both Au particles and Ti cations when surface anion vacancies are formed. Water and hydroxyl groups are also evident from the O 1s spectra. A direct correlation was obtained between the fraction of Ti3+ found by XPS for the four catalyst samples and their observed reaction rates for CO oxidation. So that, besides the expected Au particle size effect, the availability of surface defects and the presence of Auδ- were found to be important factors in determining catalytic activity.
Figure 9.

XPS spectra for an Au/TiO2 catalyst (WGC) (a) Au 4f (data points) compared to a Au foil reference showing a small shift to lower binding energies in the catalyst sample. (b) Ti 2p, with an inset of the low energy shoulder on the Ti4+ peak requiring fitting with a minority Ti3+ species. (c) O 1s showing the presence of water and hydroxyl species on the catalyst surface. Reproduced with permission from ref (187). Copyright 2017 Elsevier.
DFT+U calculations have also shown that the vacancy creation energy for oxygen defects on the anatase (101) surface are affected by the presence of a Au cluster (Figure 10).189 The O vacancy defect formation energy refers to the removal of a surface oxygen anion to form 1/2O2(g), i.e.,
Figure 10.

Calculated O vacancy defect formation free energies for the (101) surface of TiO2(anatase) based on PW91 functional calculations. Interface sites refer to O removal from Ti–O–Au bonding structures, while perimeter sites are O in the normal Ti–O–Ti sites of the clean surface but close to the Au cluster. Reproduced with permission from ref (189). Copyright 2015 The Royal Society of Chemistry.
Calculations for the internal energy change for this process, based on electronic structure calculations, have been available for several years. It is now becoming more common to estimate the free energy for the reaction based on the calculated frequencies to estimate the contribution of vibrational states to the entropy and standard statistical mechanics of the ideal gas to estimate translational and rotational contributions.190,191 The resulting plot with temperature for the case of oxygen defect creation on Au/TiO2(101) anatase shows that the interface O atoms (those in Ti–O–Au bonds) have similar defect formation energies to the clean surface. For perimeter sites (Ti–O–Ti oxygens near to the Au cluster), very small clusters (Au3) have little effect, but inclusion of a Au10 cluster significantly reduces the defect formation energy, with a negative defect formation free energy occurring above around 500 K. In the same work, it is also shown that this results in an easy pathway by which CO adsorbed an Au particle can be oxidized at the interface with the TiO2 support.192
This concept of forming TiO2 with a semireduced surface has been used to rationalize the improved performance of Fe-doped TiO2 as a support compared to a reference pure TiO2 material.193 The titania support was prepared using titanium(IV) isopropoxide in a sol–gel synthesis approach with iron(III) nitrate introduced into the sol–gel to provide the desired level of doping. Au particles were added using the deposition precipitation route with urea as the reducing agent. An Fe doping level of 1 wt % was found to result in an increased Ti3+ content according to XPS and gave a conversion of 88% for CO oxidation at room temperature compared to 61% for a TiO2 catalyst prepared without any Fe-doping.
The role of the Au nanoparticle in facilitating the removal of lattice oxygen has been studied using temporal analysis of products (TAP) studies and DFT calculations by Widmann and co-workers.194 Au on P25, the most widely used commercial titania, was used in this study. As P25 TiO2 consists of ∼80% anatase, their simulation studies consider anatase (101) as the model surface. They found that, at temperatures above −20 °C, the MvK mechanism is preferred. However, below that temperature, the activation energy required to extract a lattice oxygen from the surface is too great and reaction with physisorbed molecular oxygen becomes the preferred pathway. This means that, in most practical situations, a reducible support is required to observe high activity for the CO oxidation reaction as the most efficient catalysis takes place via the MvK route. CO adsorption on the metal component, rather than support, is also preferred at these higher temperatures and the barriers to CO diffusion across the surface to the metal/oxide interface sites are comparatively small (around 0.5 eV). Also using the TAP approach, Widmann and Behm have shown that the oxygen storage capacity for Au/TiO2 shows a temperature dependence, increasing with reaction temperatures between 80 and 400 °C.195 They argue that oxygen is supplied from a highly stable active oxygen species, Oact, only formed at the perimeter of Au particles.196 The reaction of Oact with CO is activated, but the active oxygen species itself is formed in a barrierless process. The TiO2 lattice around the perimeter of the Au NPs can act as a store for Oact, which can also be replenished by adsorbed O2 under flow reactor conditions. In the TAP reactor, at higher temperatures, lattice diffusion of oxygen effectively extends the area around each particle that can be considered part of the perimeter and a greater region of the support can be used to supply Oact. In these TAP experiments, alternating CO and O2 pulses were studied. They found that CO2 is only evolved during the CO pulses, so that direct CO oxidation is preferred over any surface intermediate route under the conditions encountered in the TAP reactor.
The discussion on the relative importance of particle size effects, low coordination Au atoms and the particle interface with the oxide in the CO oxidation reaction catalyzed by Au/TiO2 has also been taken up by Lu and co-workers.197 They prepared Au/TiO2 using Degussa P25 as support and deposition–precipitation with urea to form Au NPs from the usual HAuCl4·4H2O precursor. Catalyst samples were washed to remove excess chloride once the material had been prepared. Catalysts were calcined at 523 K in 10% O2 in He, with the time of calcination used to control particle size. HRTEM was used to confirm that materials with different particle size distributions had been prepared, the samples were classified as small (av size 2.9 ± 0.6 nm), medium (av size 5.0 ± 0.8 nm), and large (av size 10.2 ± 1.6 nm). Light-off curves demonstrated the expected dependence on particle size, with the samples containing the smallest particles showing the lowest light-off temperature. The next step was to add an additional overcoat of TiO2 to the samples using atomic layer deposition (ALD) of titanium isopropoxide (TTIP); different timings of exposure to the TTIP vapor allowed control of the thickness of the overcoat. The group had already shown that the addition of TiO2 using ALD to Au catalysts prepared with irreducible supports (Au/Al2O3 and Au/SiO2) could improve the performance of these materials for CO oxidation.198 When used with Au/TiO2, HRTEM showed that the overcoat layer following the longest exposure to TTIP (50 cycles) was around 1.5 nm thick and that the ALD process did not detectably affect the Au particle size distribution. XPS also indicated that the Au NPs remained in the metallic state. CO-DRIFTS measurements showed that bands associated with low-coordination Au sites had lower intensity for the coated Au/TiO2 samples than for the materials without coating applied. This indicated that the overcoating oxide covers low coordination sites or that the ALD process causes changes in nanoparticle morphology to reduce their number. For the CO oxidation reaction, samples with 10 or 20 cycles of ALD showed small reductions in the light-off temperature but the reduction for the 50 cycle material, with the thickest coating, was significant, shifting to higher temperatures by around 40 °C. The activation energies also increased with ALD coating thickness with a measured activation energy for the uncoated small particle Au/TiO2 case of 26.5 ± 0.7 kJ mol–1 and 35.4 ± 0.9 kJ mol–1 for the 50 cycle ALD coated catalyst. Using the CO-DRIFTS data, it was found that the number of low coordination sites on the 50 cycle ALD coated small Au/TiO2 particles was more than 2 orders of magnitude lower than that on the uncoated sample and yet the CO conversion at 298 K was only halved by the addition of the coating. The small particle Au/TiO2 with this thick coating also showed higher conversions compared with the uncoated medium and large Au particle catalysts. They concluded that the coating provides additional Au···TiO2 interface sites, a factor which is able to outweigh the effect of the reduction in available low coordination Au.
Haruta has suggested that the perimeter region between Au NPs and their oxide supports is generally where the active sites for CO oxidation are located.156 Using Au supported on a wide range of oxide materials, prepared by both coprecipitation and deposition–precipitation methods, the group considered the dependence of the temperature at which CO conversion reaches 50% (T1/2) and the heat of formation of the oxides per O atom (−ΔHf0). The idea was to test if this quantity is useful as an indicator of how easily oxygen vacancies will form.199Figure 11 shows the full range of oxides considered which span oxides of Pt group metals, first row transition metal oxides, and irreducible oxides. Figure 11a illustrates a volcano type dependency with Au/Co3O4 prepared by coprecipitation giving the lowest T1/2 value at around 220 K. In fact, Co3O4 alone when prepared as nanorods has been shown to give notable low temperature CO oxidation activity.200Figure 11b highlights the change in T1/2 between the bare oxide and the Au/MOx catalyst. The value for TiO2 is lowered by nearly 600 K, which was proposed to be due to the creation of oxygen vacancies in the perimeter region around the Au clusters.
Figure 11.

(a) Correlation between T1/2 for CO oxidation and the support oxide heat of formation per atom of oxygen. (b) Difference between T1/2 for the bare oxide support and the Au/MOx catalysts. In each case, black triangles are catalysts prepared by coprecipitation (Au/M atomic ratio 1/19 in each case) and blue triangles are for deposition–precipitation with Au loadings of 1 wt %. All catalysts were calcined at 573 K for 4 h and then preoxidized in 20% O2 at 523 K for 1 h prior to use. Reaction conditions: catalyst mass, 150 mg, reactant gas 1 vol % CO in air, flow rate 50 mL min–1, SV 20 000 h–1 mL g–1, moisture content 50–200 ppm. Reproduced with permission from ref (199). Copyright 2016 Elsevier.
Neurock and Yates have also looked at the oxidation of CO over Au/TiO2 at very low temperatures (110–130 K), mainly using IR spectroscopic methods combined with DFT calculations to unravel the mechanism for CO oxidation.202 At these low temperatures, oxygen migration from the lattice is suppressed due to the barrier associated with oxygen defect creation.194 Accordingly, a model was developed in which O2 is activated at an Au–Ti4+ dual site which provides a favorable adsorption energy site (DFT calculated Eads(O2) = −1.01 eV).203 In the DFT model of Au on rutile TiO2(110), this species can oxidize CO adsorbed at the Au/TiO2 interface with a calculated barrier of only 0.1 eV. In this mechanism, the perimeter region of the catalyst is important but only to provide adsorption sites for gas phase O2 rather than supplying lattice oxygen into the reaction. Figure 12 shows how low temperature (120 K) adsorption of CO acting as an IR probe molecule can distinguish sites on the Au/TiO2 surface with distinct bands for CO adsorbed at exposed Ti4+ cations on the support as well as at cationic (Auδ+) and metallic (Au0) Au sites. Indeed, DFT calculations by Boronat et al.204 have suggested that the 2126 cm–1 feature is associated with adsorption of CO at Au in Au–O–Ti bridge structures. By pretreating catalysts in oxidizing (O2) or reducing (CO) atmospheres, the relative proportions of Auδ+ and Au0 can be controlled.201 At the low temperatures employed in these experiments, it was found that reduced catalysts are more effective at CO oxidation than preoxidized materials containing Auδ+. This is rationalized from DFT calculations which show two effects: First, low coordinated Au atoms can strongly chemisorb oxygen and are effectively locked out of taking part in CO oxidation. Second, cationic Au centers are less effective at stabilizing the surface OC···O2 transition state than are metallic Au species because the required electron transfer from Au to O2 lessened. The calculated barrier for this transition state located at the interface between a Au particle and TiO2 for Auδ+ was found to be 0.32 eV, around three times that found for metallic Au.
Figure 12.
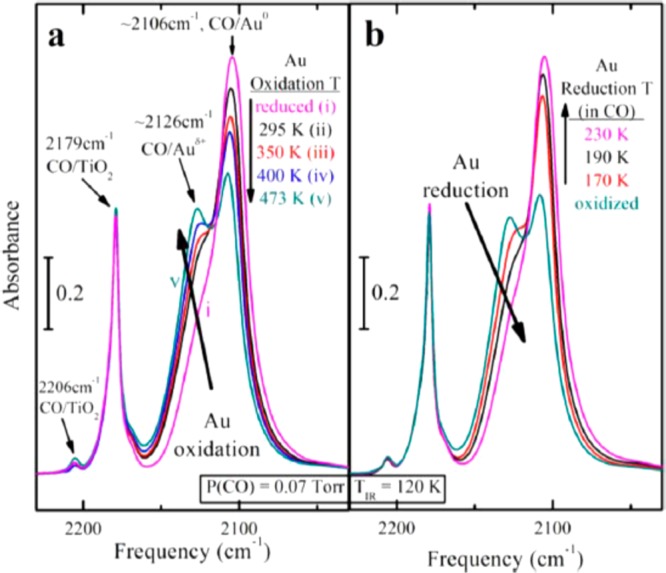
Diffuse-red spectra reflectance infrared spectra of Au/TiO2 dosed with a saturation level of CO at 120 K. (a) Catalyst premodified by oxidation in O2 at the temperatures indicated (5 Torr, min). (b) Catalyst premodified by reduction in CO at the temperatures indicated (0.07 Torr, 5 min). Reproduced with permission from ref (201). Copyright 2012 ACS Publications.
Behm and co-workers have used cyanide leaching to consider the relative importance of cationic and metallic Au in Au/TiO2 catalysts.205 Cyanide leaching can be used to preferentially remove metallic Au and so leave behind a higher proportion of cationic species than found in the as-synthesized materials. They also found that Au0 NPs are more effective for CO oxidation than is Auδ+ and used DRIFTS measurements to suggest that cationic Au can be reduced by CO under reaction conditions.
One of the most active catalysts identified so far for CO oxidation is Au supported on iron oxide. This catalyst also highlights the importance of preparation method in synthesizing the catalyst and the use of aberration corrected STEM in understanding the link between structure and activity. Hutchings and co-workers have shown that samples prepared by coprecipitation at a loading of 2.9% Au/FeOx had activities that were highly dependent on the catalyst drying procedure. Materials dried under static air in a tube furnace and heated up to 120 °C before being dried at that temperature gave virtually no activity for CO oxidation at 25 °C (0.5% vol CO, flow rate 66 000 h–1). In contrast, material prepared from the same precursor but introduced into a preheated GC oven with flowing air at 120 °C produced catalysts which showed complete conversion of CO at 25 °C using the same reaction conditions. Conventional XRD, XPS, and high resolution electron microscopy showed the particle size distribution to be the same in both samples and showed that they contained the same proportions of metallic and cationic gold.206 It was only when a higher resolution microscopy technique (namely HAADF-STEM), capable of imaging single Au atoms, became available that all Au species in these catalysts samples could be resolved.65 By taking an active Au/FeOx catalyst and then calcining it at different temperatures, the particle distribution could be altered and at the same time the FeOx support was transformed into lower surface area Fe2O3. Calcining at progressively higher temperatures led to a loss of CO oxidation activity, so that the 100% conversion found for the as-prepared and dried sample essentially became <1% conversion for a material calcined at 600 °C.
Figure 13 summarizes the resulting changes in particle size/species distributions based on a statistical analysis of HAADF-STEM imaging data. The Au species observed were classified as single atoms, subnanometer monolayer clusters, subnanometer bilayer clusters, and NPs greater than 1 nm is size. This revealed a correlation between catalyst activity and the number of 0.5 nm Au bilayer clusters present. Clearly, in these materials, Au particle size and shape are important factors in the activity for CO oxidation, but the range of particles present in the distribution produced by precipitation methods makes definitive identification of the most active form of Au difficult. Detailed electron microscopy studies have also been used to show how atomically dispersed species, subnanometer clusters and Au NPs are affected by heat treatment and cyanide leaching of metallic Au from samples.66 Colloidal deposition (also referred to as sol-immobilization) offers much narrower particle distributions, and Schüth and co-workers have used this approach to study Au/FeOx.207 By preparing materials with a mean particle size of 2.1 ± 0.54 nm and working with aberration corrected HAADF-STEM, they were able to exclude the presence of the very small particles observed on in the earlier study. Even so, these materials were highly active for CO oxidation, giving TOF values of 0.5–1 s–1, compared with the 3.5 s–1 estimated in the earlier study based only on the concentration of subnm Au bilayer clusters.
Figure 13.

Frequency with which different Au species are observed for Au/FeOx samples prepared using different drying and calcination processes. Solid bars: materials dried in flowing air at 120 °C without additional calcination. Vertical striped bars: samples dried at 120 °C and then calcined at 400 °C. White bars: samples dried at 120 °C and calcined at 550 °C. Cross-hatched bars: samples dried at 120 °C and then calcined at 600 °C. Error bars refer to standard deviation from the sampled images. Reproduced with permission from ref (65). Copyright 2008 The American Association for the Advancement of Science.
Schüth and co-workers, have also compared Au NPs supported on iron oxides (FeOx) and hydroxides (FeOH) utilizing Au NPs with a uniform size around 2 nm prepared by deposition–precipitation (DP) and colloidal deposition (CD) methods.208 The oxide materials were produced by calcination of the hydroxide (400 °C for 2 h). Electron microscopy showed that all four catalyst materials contained particles around 2 ± 0.6 nm. XRD and BET characterization also showed that the calcination step led to the formation of α-Fe2O3 for the FeOx materials with around one-third the surface area of the parent hydroxide. Opposite trends for CO oxidation were observed on the oxide and hydroxide catalysts produced by DP or CD methods, with the order of activity: DP (Au/FeOH < Au/FeOx) and CD (Au/FeOH > Au/FeOx). In this work, XANES was used to analyze the relative amounts of Auδ+ and Au0 species present in catalyst samples using features present on the Au L-III absorption edge. Both FeOx and FeOH supported Au catalysts produced by the DP method showed Auδ+ features after the drying stage of the preparation (Figure 14a), but for calcined (300 °C for 30 min) and used materials a typical Au0 absorption edge was observed. Materials produced by the CD approach showed only Au0 for the dried material (Figure 14b). This would be expected because the CD particles have been formed and reduced in the colloidal suspension. For the DP prepared samples, ionic Au species are deposited which are then reduced to form particles during calcination. In situ measurement of the XANES spectra for DP-Au/FeOx and DP-Au/FeOH confirmed that the proportion of Auδ+ remained below 10% throughout the CO oxidation reaction (1% CO/16% O2 in He, 25 °C). Even so, H2-TPR showed that the addition of Au NPs by the DP method gave rise to low temperature reduction features centered at 128 °C for DP-Au/FeOH and 232 °C for DP-Au/FeOx, with XRD showing the formation of Fe3O4 above these temperatures, indicating that Au NPs are prompting the reduction of the iron oxide and hydroxide supports. For the CD prepared catalysts, a similar effect was observed, but the reduction temperatures were higher (188 °C for CD-Au/FeOH and 257 °C for DP-Au/FeOx), suggesting that the interaction between the support and NPs is not so strong when using CD deposition. Cui et al. have also worked with Au/FeOx and Au/FeOH materials to study the effect of the strength of nanoparticle/oxide interaction, as measured by TPR, on the CO oxidation activity.209 They found that the strength of this interaction could be controlled by the pH used in the deposition–precipitation of the Au precursors and conclude that a strong interaction favors high CO conversions and imparts good catalyst stability.
Figure 14.
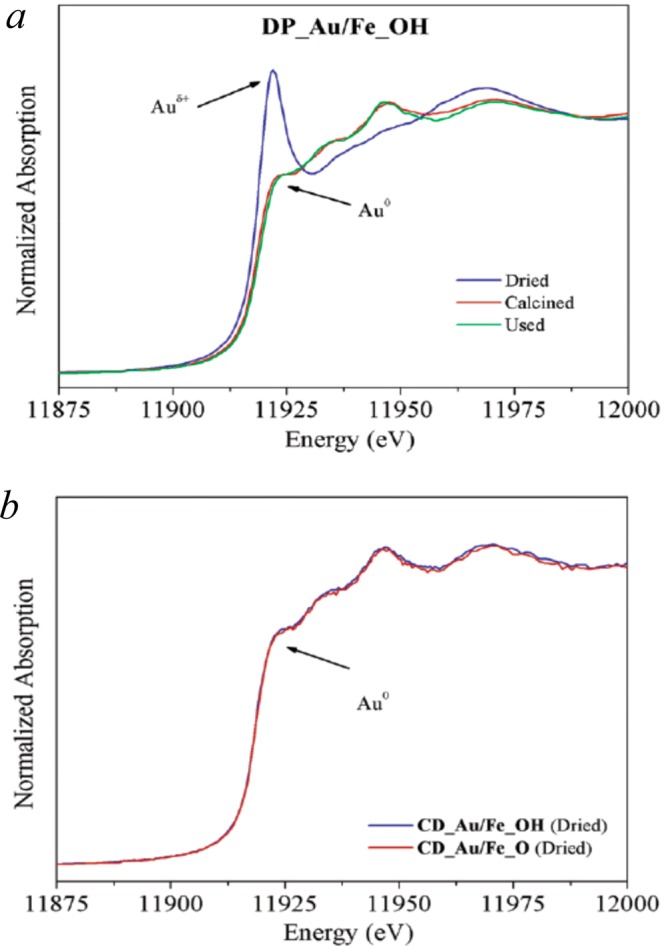
XANES spectra at the Au L-III absorption edge for (a) DP prepared Au/FeOH comparing dried, calcined (300 °C), and used catalysts, and (b) CD prepared Au/FeOx and Au/FeOH. Reproduced with permission from ref (208). Copyright 2015 The Royal Society of Chemistry.
DFT calculations using the PBE functional and DFT+U approach show that O2 will dissociate at the interface between a Au10 cluster and the clean stoichiometric (0001) surface of α-Fe2O3 more readily than on an isolated Au particle.210 In later calculations, it was also shown that the Au10 particle could become oxidized at the perimeter where the interface with the α-Fe2O3 can stabilize the oxidation of Au atoms, but this oxidation does not extend to the central region of the cluster.211 The presence of the cluster lowered the defect formation energy for oxygen vacancies in the oxide surface near to the cluster from 3.04 eV for the clean α-Fe2O3(0001) surface to 2.88 eV near to the Au10 cluster and only 0.95 eV for oxygen near to a partially oxidized (Au10O6) metal nanoparticle. Interestingly, the energy required to create an oxygen vacancy in the oxidized part of the Au cluster was calculated to be 1.96 eV, indicating that the oxygen anions in the α-Fe2O3(0001) near to the cluster are more available for a MvK oxidation of CO than are the oxygen atoms introduced into the Au cluster by O2 dissociation.
Haruta has also considered the polymorph of the iron oxide support, producing an Au catalyst supported on mesoporous maghemite, γ-Fe2O3, nanoflakes212 using a deposition–precipitation method.213 The γ-Fe2O3 appears to be a support, giving Au catalyst with higher CO oxidation activity than α-Fe2O3 due to it being more easily reduced.214 The nanoflake synthesis produces catalysts with high surface area (up to 193 m2 g–1) capable of over 90% CO conversion and a specific activity of 8.41 molCO gAu–1 h–1 at room temperature. The particle size range from electron microscopy was determined to be 2–5 nm, which, it was suggested, provides an optimal surface coverage of particle perimeter sites for catalysis.
Ceria, CeO2, is another oxide widely used in catalysis for its oxygen storage capacity, which is related to the ease of reduction of Ce4+ to Ce3+, making it possible to capitalize on the material’s redox abilities.215 Hence Au/CeO2 applied to CO oxidation is another widely studied catalyst. DRIFTS measurements have been used to identify surface species and, using the characteristic bands for key intermediates/side products, several studies have linked operando spectroscopic measurements to catalyst deactivation. For example, Chen et al. studied Au/CeO2 with Au loadings of 0.18%, 0.97%, and 5.7%, and TEM analysis confirmed that the mean particle size increases with metal loading (0.18%-Au/CeO2: 1.7 ± 0.6 nm), (0.97%-Au/CeO2: 2.6 ± 0.6 nm), and (5.7%-Au/CeO2: 3.7 ± 0.6 nm).216 Kinetic measurements in a 1% CO, 1% O2 gas mix in N2 over a temperature range of 300–360 K allowed these researchers to estimate the activation energies for CO oxidation for the three materials. The 0.97%-Au/CeO2 and 5.7%-Au/CeO2 gave similar results with estimated activation energies of 28.4 ± 1.6 and 25.7 ± 1.3 kJ mol–1, respectively, whereas the activation energy measured for the lowest Au loading (0.18%-Au/CeO2) was found to be substantially lower at 18.7 ± 3.2 kJ mol–1, in line with the expected higher activity per Au site for the smallest Au NPs. In the room temperature DRIFTS spectra obtained from these materials, bands for bidentate carbonate (CO32–), monodentate carbonate, bridged carbonate, bidentate bicarbonate (CO3H–), and carbonite (CO22–) could be detected upon the adsorption of CO. Furthermore, bands due to hydroxyl groups on the oxide surface could be classified into terminal and bridging configurations. In addition, CO(a) at Au0 and Auδ+ sites were detected, with the ratio of the two decreasing with increasing Au loading, implying that the positively charged Au centers are located at the edge of the Au NPs. By studying the DRIFTS peak as a function of time during CO oxidation (1% CO, 1% O2 in Ar), Chen et al. concluded that the oxidation activity for the direct oxidation of CO(a) does not exhibit a strong particle size dependence. By way of contrast, the production of CO2 via the decomposition of carbonates, bicarbonates, and formate species contributed to CO conversion over the larger particles studied, but these same species inhibited oxidation of CO(a) over the smaller particles contained in the 0.18%-Au/CeO2 catalysts.
El-Moemen et al. have used a similar spectroscopic approach to probe the effect of the reactant mix on the catalytic activity of 4.5 wt % Au/CeO2 catalysts for CO oxidation.217 They compared catalysts prereduced in H2 or CO with those heat treated in N2 or preoxidized at 400 °C. They find that the activation energy measured for CO oxidation is independent of the pretreatment procedure (Eact = 32 ± 2 kJ mol–1). Even so, the pretreatment does influence the initial catalytic activity and the degree of deactivation during a 17 h test (80 °C, gas mix: 1% CO, 1% O2 in N2), with preoxidized samples giving higher rates that prereduced, but the prereduced catalysts showing the greatest stability under reaction conditions. Nanoparticle sizes were not influenced by the pretreatment steps, but the surface species observed using DRIFTS experiments were affected. On the preoxidized materials signals for carbonate were detected, but these were not seen on the prereduced material. However, after around 10 min on-stream, the surface composition according to DRIFTS reached an equilibrium state that was essentially identical for all catalysts. XANES measurements have also been used by this group to show that Au rapidly reaches a metallic state during CO oxidation.218 Changing the ratio of CO to O2 in the reaction mixture to an O2-rich flow resulted in an abrupt increase in reaction rate, indicating a positive reaction order with respect to O2. Switching to a CO-rich mixture in a reaction initiated at a 1:1 ratio of CO to O2, a gradual decrease in CO conversion was observed, indicating that the oxygen reservoir on the catalyst surface was sufficient to maintain the reaction under these conditions.
The preparation method used to produce the ceria support itself can be tailored to generate cubic, polyhedral, or rod-shaped nanocrystallites of the oxide.219 The nanorod and nanocube forms show higher oxygen storage ability than the nanopolyhedra form due to the differing expression of {100}, and {110}-type facets on these structures. The nanorod form of CeO2 has a high proportion of {110} facets which are also particularly suited to providing the high dispersion of Au required for single-atom catalysts to be produced.220 Gold supported on nanorod ceria has been studied by Guo et al., who have used a deposition–precipitation approach to prepare single-Au atom catalysts and then subsequently obtained Au clusters (<2 nm in size) supported on the same material by hydrogen reduction of the corresponding single-atom samples.221 In addition, larger Au particles (3–4 nm in size) were prepared by colloidal deposition of Au onto the ceria nanorod support. XANES was used to show that the single-atom catalysts contained largely cationic Au species while the clusters and particles showed spectra typical of metallic particles. EXAFS data taken under reaction conditions also demonstrated that the three different forms of Au were stable under reaction conditions (25 °C, 1% CO, 16% O2 in He). Testing the CO oxidation activity of the three types of catalyst showed that the CO oxidation rate per unit mass of Au was higher with the 3–4 nm particles than for the metallic clusters and that the single-atom cation Au catalyst showed a much lower ability to oxidize CO. A 600 °C oxidative treatment of CeO2 nanocubes at 600 °C can produce {111} nanofacets on the {100} faces of the cubes.222 This significantly reduces the light-off temperature for CO oxidation. DRIFTS operando measurements showed that Auδ--CO species appear on these materials upon exposure to CO and can be correlated with CO oxidation activity.223 The nanofaceting appears to stabilize the Au particles and give rise to a higher proportion of Auδ--CO signal in the DRIFT spectra.
DFT ab initio molecular dynamics has also been used to study a Au20 particle supported on a CeO2(111) surface.224 The authors chose to site the particle over an oxygen defect in the surface to represent a partially reduced ceria surface. The cluster was then modeled using molecular dynamics both with and without the inclusion of adsorbed CO (Figure 15a). By plotting a distribution of the Au atom distances from the center of mass of the cluster (Figure 15b), the atomic shells of the optimized cluster can be clearly seen as separated peaks at around 2, 3.5, and 5 Å. The motion of the atoms in this MD run for the cluster without adsorbates is sufficient to smear out these peaks and gave a continuous distribution, showing that the cluster stayed connected together throughout the simulation. However, when CO was adsorbed, a single-atom can be seen to have broken away from the cluster to a distance of over 6 Å from its center of mass. Snapshots from the trajectory shows that this breakaway Au atom carries a CO molecule with it, and analysis of the electronic states shows this to be a Au+-CO complex. The electrons lost to form the Au+ cation were found to be accommodated by reduced (Ce3+) cations in the supporting ceria.225 The study went on to show that oxidation even at the periphery of the cluster is limited by the strong adsorption energy of the CO2– species that is formed. In contrast, the use of lattice oxygen to oxidize the CO in the Au+-CO complex is much more facile as the electrons generated when lattice oxygen is used to oxidize CO are accommodated by reducing the Au+ cation, which then rejoins the cluster. A similar process has been suggested based on calculations for Au/TiO2 along with microkinetic modeling, implying that this process is preferred at low temperatures (<130 °C) under oxidizing conditions but that at higher temperatures the perimeter oxidation mechanism discussed above takes over.226
Figure 15.
(a) Initial and final configurations from ab initio MD simulation of Au20/CeO2(111) with eight CO molecules adsorbed. (b) The probability distribution functions P(r) of the Au atoms relative to the center-of-mass of the Au20 based on ∼20 ps MD simulation at 700 K (Yellow spheres, Au: cyan spheres: Ce; red or blue spheres: O; green or gray spheres: C. Reproduced with permission from ref (224). Copyright 2015 Macmillan Publishers Limited.
For Au on manganese oxide supports, Schüth and co-workers have found that the preparation method for the support material can be crucial for observing high activity.227 Prior to this work, the highest activity for gold supported on manganese oxide for CO oxidation was 0.098 molCO h–1 gAu–1.228 In an earlier study, MnO2 and Mn2O3 were compared as supports with the conclusion that the redox ability of the Mn2O3 phase led to facile formation of active oxygen species on the surface of the catalyst and consequently a performance in CO oxidation as compared to Au/MnO2. Schüth and co-workers prepared Au/MnO2 using techniques designed to produce nanoscale metal oxide particles as supports. Samples of α-MnO2 nanowires (NW) were prepared using a hydrothermal method, while highly ordered mesoporous β-MnO2(meso) was produced by a nanocasting method from the aqueous manganese nitrate using SBA-15 as a hard template. In each case, PVA protected Au NPs were deposited onto the support from an aqueous colloidal solution. These catalysts exhibited CO conversion at very low temperatures. For example, the Au/α-MnO2(NW) showed a CO conversion of 37% at −90 °C flow rate; 80 000 mL h–1 gcat–1). The materials also showed some oxygen storage capacity, as CO could be used to titrate active oxygen species from the catalyst surface in the absence of gas phase O2, as shown in Figure 16.
Figure 16.
CO titration experiments for Au/MnO2 with MnO2 in the form of nanowires (NW) and nanocast mesoparticle oxide. Reaction conditions: Catalyst 50 mg, 1% CO in N2 at a flow rate of 67 mL min–1. Catalyst pretreatment gas composition: 1% CO, 20% O2, 79% He. Reproduced with permission from ref (227). Copyright 2016 American Chemical Society.
3.1.3. Non-metal-Oxide Materials as Supports
While metal oxide materials remain the most studied support materials for CO oxidation and related reactions, reports have appeared using alternative materials as supports. Haruta and co-workers have produced a Au@ZIF-8 by simple solid grinding of the Au source ((CH3)2Au(acac), acac = acetylacetonate) with the metal organic framework (MOF) material.118 ZIF-8 was chosen for its high thermal stability (over 500 °C) and large pore volume (11.6 Å pore diameter) to accommodate the Au NPs that were expected to form. No features for Au in the XRD of the resulting material were seen, but UV–vis adsorption provided some evidence that Au NPs were present. The resulting catalysts were tested for CO oxidation producing light-off curves with 50% conversion between 175 and 225 °C depending on the catalyst loading (0.5–5.0 wt % Au).
3.1.4. Ligand Protected Au NPs
Supported Au catalysts produced by conventional impregnation or deposition precipitation routes will always produce a broad range of metal particle sizes and may also result in a mix of metallic and cationic gold. Specific Au colloids in solution with protective surface ligands are one approach that has been tried to produce uniform single-size Au NPs in sufficient quantities for catalysis. For example, Au clusters protected with thiolate ligands, Aun(SR)m with R = CH2CH2Ph, have been produced displaying specific sizes (with n = 25, m = 18; n = 38, m = 24; and n = 144, m = 60), which show high thermal stability (up to 200 °C).229,230 Even though the SR ligands are tightly bound to the Au cluster, a support effect was observed with Au/CeO2 catalysts, giving superior performance to Au/TiO2. Overbury and co-workers have used this approach to synthesize Au25(SR)18 supported on CeO2 nanorods.231 They found that a heat treatment step is required to remove the SR ligands from the nanoparticle in order to observe catalytic activity, as the ligands tend to block CO adsorption. IR spectroscopy was used with CO as a probe molecule to identify a significantly greater role for Auδ+ in the CO oxidation reaction than was found for “clean” NPs deposited by more conventional deposition or impregnation methods. Removal of the thiolate ligands was determined to commence at the perimeter of the nanocluster and so the activity of Aun(SR)m after even partial removal of SR ligands emphasizes the importance of perimeter sites in CO oxidation.232
Li et al. have carried out CO oxidation experiments at 80 °C and temperature-programmed oxidation experiments to show that Au clusters stabilized with SR ligands would be expected to retain their ligands under the reaction conditions.233 Indeed, a Au144(SR)60 material pretreated at 300 °C exhibited 100% CO conversion at 40 °C, whereas a catalyst pretreated at 80 °C only achieved this level of conversion at 60 °C. This difference in performance was attributed to the thiolate ligands covering some of the catalytically active sites for the material pretreated at the lower temperature. For the ligand protected Au144(SR)60/CeO2 catalyst, an activation step was required before the catalyst showed a high level of activity. This involved holding the catalyst at 80 °C for of 10 h under the oxidative conditions (1.67% CO, 3.37% O2 in 94.96% He), and over this time period CO conversion was observed to increase from 4.5% to 75.8% and eventually reached 94.7% after 24 h.
The stabilization of Au NPs deposited on TiO2 using a colloidal deposition method with octadece-9-nyl amine as the surfactant has been studied by Dai and co-workers.138 They monitored the stability of the catalysts once the ligand shell is removed. During conventional preparation methods involving washing steps and calcination in air, the Au particles are known to migrate and sinter. In this work, an alternative approach was developed, which is illustrated schematically in Figure 17. In this process, the deposited colloidal particles were first heated to 500 °C in flowing N2, which leads to a carbonaceous layer from the decomposition products the surfactant molecules which form over the surface of the NPs. It was claimed that the formation of this layer in the inert atmosphere improves the interaction of the NPs with the support surface, possibly by reducing the oxide locally in the vicinity of the nanoparticle as the hydrocarbon chain of the surfactant is partially decomposed. The improved nanoparticle–support interaction effectively suppressed particle growth in the final calcination step. The choice of surfactant was also noted to be important with functionality in the aliphatic part of the molecule, providing cross-linking during the heat treatment. The authors used TEM, Raman spectroscopy and XRD to show that the particle size is not adversely affected by the treatment under conditions of low particle dispersion. Interestingly, 6 nm colloidal Au particles were found to have a multiply twinned icosahedral morphology which was maintained upon formation of the carbonaceous coating. Both the supported colloid and heat-treated materials exhibited a low capacity for CO adsorption. However, the materials become active for CO oxidation once a further conventional calcination step was performed to remove the carbonaceous coatings. The materials also exhibited improved thermal stability even when this second calcination step was carried out to remove the protective carbon overlayers from the Au NPs.
Figure 17.

Process for producing uniform Au particles supported on TiO2 by heating surfactant coated particles to produce a protective carbon shell compared with the conventional washing approach, which leads to particle migration and sintering. Reproduced with permission from ref (138). Copyright 2017 Wiley-VCH.
3.1.5. Particle Anchoring and Encapsulation
As has been previously discussed, the particle size effect for Au oxidation catalysis has been a matter of much discussion, with most researchers now agreeing that sizes below 5 nm are required to observe high CO oxidation activity. This roughly coincides with the length scale at which a band structure picture of the electronic states of the metal breaks down and a discrete molecular orbital picture becomes more appropriate.234 There are many applications where the catalyst needs to be robust at high temperatures, at which point, for conventionally supported Au catalysts, sintering becomes a significant route to deactivation.235 For example, in the car cold start problem, for which the activity for CO oxidation at low temperatures makes supported Au catalysts an attractive option, the catalyst will necessarily be required to spend time in the hot exhaust stream once the engine reaches its normal operating temperature. This highlights the need to devise novel approaches to firmly anchor Au NPs to the support to improve their sintering resistance and hinder coalescence. Several strategies to make support structures that hinder sintering have been developed over recent years. Even so, the deactivation of catalysts may be caused by factors other than particle agglomeration, such as surface poisoning by reaction intermediates such as carbonates or by water.217
The mobility of Au particles on conventional oxide supports has been highlighted by environmental TEM studies, where the ability to record the dynamic behavior of catalyst particles while exposed to low pressure reactant gases (100s of Pa) even at elevated temperature allow the visualization of particles in conditions approximating to their catalytically active state. Au NPs supported on the oxygen terminated surface of CeO2 have been seen to reversibly migrate and rotate even at room temperature, indicating that the interaction with the surface atoms in general is weak. However, the particles may be interacting with surface defects because they can return to their original positions/orientations rather than making further migrations on the 10 s of time scale.236
The use of microporous, mesoporous, or hierarchical materials to isolate Au NPs deposited in their channels is an attractive one. For example, Scurrell and co-workers have shown that Au deposited by incipient wetness from a KAu(CN)2 solution on to zeolite-Y shows activity for CO oxidation.231 However, on reduction to the metallic form, the initially highly dispersed Au was found to form large particles and the catalytic activity was lost. Ion exchange of zeolite-Y with transition metal cations such as Cr3+, Fe3+, Co2+, or Ni2+ was found to improve the thermal stability of the 4% Au/NaY material, with the effect inversely dependent on the reduction potentials of the exchanged metals. XRD analysis of the Au/NaY material without transition metal cation exchange shows Au particles of around 20 nm in size are obtained on reduction, whereas AuCr/NaY has an average particle size of around 5 nm and correspondingly improved CO oxidation activity. AuFe/NaY showed stable conversions for CO oxidation at 200 °C with the amount of deactivation seen to decrease with increased levels of Fe introduced by ion exchange. Even so, the level of conversion did lower from 55% for catalysts containing 2 wt % Fe to 28% at a level of 6 wt % Fe. The authors proposed that the transition metal cations can act as anchor sites for the anionic Au species deposited during the incipient wetness procedure. This then lowers the level of migration displayed by the metallic Au NPs when the catalyst is in operation.
The silica walls of the pores in mesoporous materials also provide insufficient anchoring and Au particles can be observed to migrate easily at temperatures as low as 550 °C and even make transitions between neighboring channels within the material at this temperature.237 Accordingly researchers have looked to modify mesoporous materials with additional anchor sites that can limit the migration of the NPs.
Kučerová et al. have explored a grafting approach with a Ti(OiPr)4 precursor to produce a mesoporous SBA-15 material coated with a TiOx surface layer. Earlier work had shown that impregnation of titanium(IV) isopropoxide into SBA-15 to form TiO2 in situ could improve the dispersion of Au within the mesoporous material.90 They found that the inclusion of the TiOx layer improved the catalyst activity for CO oxidation by a factor of ∼10 compared to Au/SBA-15 without this modification. IR spectroscopy of adsorbed CO was used to infer that Au deposited on the TiOx modified SBA-15 was largely associated with the TiOx coated areas of the support. For reactions carried out at 30 °C catalyst deactivation was observed but could be assigned to water inhibition and was reversible by a 400 °C calcination treatment of the used catalyst.
Pérez-Cabero et al. have selected UVM-7 as a bimodal mesoporous silica for producing supported Au catalysts using nanodomains of transition metal oxides as Au anchoring points.238 For example, the group have produced UVM-7 with TiOx nanodomains introduced into the mesopores by either (i) a one-pot cohydrolysis/condensation route in which the silica and titania regions of material are prepared together, or (ii) wet impregnation of a pure silica UVM-7 with TiO(acac)2.239 At the pH value used (7–8) for the deposition–precipitation of Au, the dominant solution phase species is anionic Au(OH)4– and, as TiO2 has a point of zero charge at ∼6, the TiOx domains deposited in the UVM-7 material provides anchor points for the deposition of Au. Calcination of these materials at 800 °C produced little growth in the Au NPs which maintained an average size of ∼3.5 nm. Materials produced in this way show high activity for CO removal from a simulated vehicle exhaust stream, even after 10 reaction cycles working at temperatures up to 600 °C in each cycle.240
TiOx modified mesoporous silica SBA-15 has been used as a substrate for supported Au catalysts by Behm and co-workers.241 They point out that to prepare a reference Au/SBA-15 via the usual deposition precipitation route from AuHCl4·3H2O is difficult as the isoelectric point for silica is quite low (∼2), so that the negatively charged Au species produced in solution will not have a high affinity for the surface at the deposition pH. Instead, they turned to using the cationic complex [Au(ethylendiamine)2]Cl3 (formed from [Au(en)2]Cl3) as the Au precursor. This was found to interact strongly with the negatively charged SBA-15 surface at pH values above 2. The SBA-15-TiOx samples were prepared using a grafting approach from a titanium isopropoxide (Ti(OiPr)4) precursor.242 Catalytic testing at reaction temperatures of 30, 80, and 180 °C showed that these materials give activity for CO oxidation, which increases with Ti loading and reaction temperature. The deactivation behavior with time-on-stream was not affected by the Ti loading with reaction rates declining by 80% or more after 1000 min even at a reaction temperature of 30 °C. However, a subsequent 400 °C heat treatment was found to completely restore the CO oxidation activity, pointing to a catalyst poisoning effect rather than particle sintering.
Creating core–shell nanostructures is another strategy that has been explored to stabilize Au NPs.244 In a two-step synthesis process, Au clusters capped with 11-mercaptoundecanoic acid (MUA) were formed and then dissolved in THF. This was then mixed with a THF solution of titanium butoxide and hydrolyzed to produce Au particles coated with a porous TiO2 shell (denoted Au@TiO2). These Au@TiO2 particles were then immobilized onto silane functionalized alumina to produce the final heterogeneous catalyst. A reference catalyst formed by impregnation of MUA-Au onto Degussa P25 TiO2 was also produced. Both materials had a nominal Au loading of 1 wt % and were calcined at 400, 600, and 700 °C. TEM was employed to show that for both catalyst variants the material prior to calcination contained uniform NPs with a mean diameter of 3 nm. The reference Au/TiO2 catalyst showed an increase of particle size even when calcined at 400 °C, with a distribution of particle sizes between 5 and 15 nm being measured. For the Au@TiO2/Si-Al2O3 material, the Au particles maintained the original 3 nm average diameter even after calcination at 600 °C. It was also suggested that the activity of these materials was improved by the highly defective TiO2 coating. Indeed, at 30 °C the Au@TiO2/Si-Al2O3 catalyst calcined at 400 °C showed a CO conversion of around 85%, while the comparable Au/TiO2 catalyst gave only 60% conversion. Light-off curves were also used to demonstrate that only calcination at 700 °C of these modified materials led to significant deactivation.
Another approach to encapsulation of Au NPs is by the synthesis of yolk–shell structures. Here Au colloids are coated with a silica shell using a method developed to produce uniform silica spheres on the 100 nm length scale. The resulting Au@SiO2 spheres are then coated by the final support material (e.g., ZrO2), and the silica is then removed by treatment with strong base (NaOH) to produce the Au yolk confined within the porous zirconia shell, as shown in Figure 5.144 This synthesis approach leads almost exclusively to one Au particle per shell and produces catalysts for CO oxidation that are resistant to sintering. The size of Au NPs and level of defects can also be controlled using post synthesis Au leaching with cyanide245 and calcination/quenching procedures.146 Detailed TEM analysis also revealed that the Au particles produced by the yolk–shell synthesis contain more twinning and stacking fault defects (Figure 18a,b) than those found for conventional colloidal synthesis methods.243 The catalytic activity measured using light-off curves could also be correlated with the measured density of these defects, with higher defect densities giving lower light-off temperatures.
Figure 18.

High resolution TEM images of Au@ZrO2 yolk–shell catalysts showing (a) multiply twinned structure of an encapsulated Au particle and (b) Au particle showing a pair of parallel stacking faults. Reproduced with permission from ref (243). Copyright 2010 American Chemical Society.
3.1.6. Effect of Water
The role of water in the oxidation of CO by supported Au NPs has been a key point of discussion for several years. Haruta246 has outlined the main ways that water may influence the low temperature CO oxidation reaction, namely: (i) water may act to maintain the cationic state of Au required by some mechanisms, (ii) water, or OH– generated from water, may be directly involved in the CO oxidation as a cocatalyst or reagent (Scheme 2), (iii) water may help to activate oxygen, or (iv) water may serve to activate intermediates or remove spectators such as carbonates. Very recently, Haruta has also reported that the influence of water on the CO oxidation rate is particularly profound for the single-atom catalyst Au1/CeO2.246
Scheme 2. Proposed Mechanism for Water as Co-catalyst in CO Oxidation.

Ojeda et al.247 showed that the yield of CO2 depends strongly on the partial pressure of water and proposed that water can act as a cocatalyst for CO oxidation via a mechanism in which it helps to activate coadsorbed O2. In this mechanism CO, O2, and H2O all adsorb reversibly to the Au or Au/oxide interface (steps 1–3). Water and oxygen can then react by proton transfer to form a surface bound hydroperoxy species (step 4). Even though kinetic fitting of equilibrium constants suggested that the hydroperoxy is a minority species, it was proposed that its oxidizing power will still facilitate the ready conversion of CO to CO2, leaving a second surface hydroxyl species (step 5). The combination of surface hydroxyls to form water and a surface oxygen atom regenerates water (closing the cycle for the cocatalyst, step 6). The resulting isolated surface oxygen atom can then readily oxidize a further CO molecule (step 7).
The effect of water on the reaction kinetics for CO oxidation has been observed for a number of catalysts. Schüth and co-workers using manganese supported Au catalysts found that the measured activation energy for CO oxidation was lower when around 3 ppm water was present in the feed gas than when the gases were carefully dried ahead of the catalyst bed. Figure 19 shows that under dry conditions the measured activation energy for Au/MnO2(nanowire-NW) catalyst and Au/MnO2(mesoporous) catalysts are both around 41 kJ mol–1. In the presence of water, the activation energy for the Au/MnO2(NW) material reduces by 13 kJ mol–1 to only 28 kJ mol–1. In situ Raman spectroscopy was used to look for evidence of superoxide or peroxide species on the catalyst surface, but only bands due to metal–oxygen modes were observed, and the researchers concluded that dioxygen anion species were not significant for this class of catalysts.
Figure 19.
Arrhenius plots for Au supported on MnO2 with and without (dry) water present in the feed stream. Reaction conditions: 5 mg catalyst, flow rate 100 mL min–1, WHSV = 1 200 000 mL h–1 gcat–1. Gas composition: 1 vol % CO, 20 vol % O2 in He. Catalysts pretreated in reactant gas at 300 °C and then cooled to required temperature and stabilized for 1 h, all CO conversions below 15%. Reproduced with permission from ref (227). Copyright American Chemical Society.
For 2 wt % Au/ZrO2, heat treatments (from 85 to 550 °C) have been used to control the hydroxylation level of the supporting oxide material and the CO oxidation activity has been systematically studied as a function of surface hydroxyl concentration.248 The degree of surface hydroxylation and the classification of the OH– groups into monodentate and bidentate was carried out using FTIR spectroscopy. CO oxidation activity was then tested using either humid or dry air as the oxygen source. The highest CO conversions were found for the catalysts that had undergone the lowest temperature heat treatment and so contained the highest surface OH– coverage. On the basis of DFT calculations, the OH– group is thought to increase the charge transfer between the Au NPs and the support, leading to a positively charged Au species to which CO can bind more strongly. The transfer of O from a coadsorbed O2 molecule is then suggested to proceed with a calculated barrier 0.2 eV lower than for the Au cluster on a clean stoichiometric oxygen terminated zirconia surface.
Saavedra et al. have studied the effect of water on a Au/TiO2 catalyst measuring a kinetic isotope effect (KIE) of kH/kD = 1.8 for CO oxidation when using 700 Pa of H2O/D2O in the feed stream of their reactor (Reaction conditions: 20 °C, 1% CO, 20% O2, space velocity (SV) = 36 l gcat–1 min–1).249 The observation of a KIE suggests that water or surface hydroxyl groups must take part in a kinetically important step in the reaction. In supporting DFT calculations, a Au10 cluster was generated on a rutile TiO2(110) surface and the exposed oxide hydroxylated. The effect of water was then considered by adding one or two water molecules to the model as required. The adsorption of water to perimeter site OH groups (Eads = −1.80 eV) was found to be significantly more favorable than adsorption of water to either the Au cluster itself (Eads = −0.85 eV) or regions of the hydroxylated surface without the Au cluster (Eads = −0.36 eV). Further calculations suggested that coadsorbed water at the Au/TiO2 interface can protonate adsorbed O2 to produce an *OOH entity capable of oxidizing CO (following step 5). However, the DFT energies and barriers suggest that *CO and *OOH interact to produce *COOH and *O rather than CO2 directly and a surface hydroxyl. This CO assisted scission of the *O–OH bond has a calculated barrier of only 0.1 eV, compared with barriers for CO oxidation by Au–O or Au–OH of 0.65 and 0.40 eV, respectively. Experimentally, they also found that for catalysts carefully dried in N2, the IR signals for molecular water decreased significantly but surface hydroxyls remain unaffected. Catalysts treated in this way showed up to an order of magnitude reduction in CO oxidation turnover frequency (TOF), supporting the idea that molecular oxygen activated by molecularly adsorbed water was the main source of oxidizing agent in these reactions. Surface hydroxyls and lattice oxygen are thought to be less significant when water is available as a cocatalyst.
Lin and co-workers reasoned that the hydrophilicity of oxide surfaces makes the direct observation of the hydroperoxy intermediate produced by the interaction of water with adsorbed O2 very difficult to detect by IR spectroscopy.250 This is the case partly because of overlapping bands from surface hydroxyl groups and water itself and partly because the behavior of the reaction as a function of water concentration shows a surface poisoning effect as the water concentration is increased. They decided that Au supported on hexagonal boron nitride should mitigate both factors, as the support has no spectroscopically competing surface species and is hydrophobic and so should not suffer from surface poisoning. Indeed, they found that Au/BN shows increasing CO conversion with H2O content with no maximum right up to a partial pressure of water of 3000 kPa. The rate of conversion of CO is low compared to oxide supported catalysts (3.5 mmol CO gAu–1 min–1), but they were able to demonstrate that the IR signature of the proposed OOH oxidant was present using DRIFTS. In the same work, oxygen labeled water H218O was used in the reaction alongside C16O and 16O2. DRIFTS measurements then found that the CO2 produced is a mix of C16O2 and C18O16O, demonstrating that the OH– produced after proton transfer from water to adsorbed O2 can also take part in the oxidation of CO. Fu et al. have used DFT calculations to look at Au on a hexagonal BN support251 and found that there is electron transfer between the BN surface and the Au9 cluster they employed. This leads to a weakening of the Au–CO bond compared to an isolated cluster, which, they suggest, explains the experimentally observed blue-shift for CO adsorbed to Au/BN reported by Lin and co-workers.250 In the DFT calculations, it was also found that water activates O2 to make HO2 and COOH in a gold-only mechanism similar to that suggested for CO oxidation on Au/TiO2 in the presence of water.249
3.2. Alcohol Oxidation
Manufacturing of bulk and fine chemicals involves functional group transformations and oxidation of alcohol to carbonyl compounds is one of the key transformations.252 Typically, these transformations are carried out using stoichiometric oxidants such as chromates, permanganates, or iodates which can result in the generation of highly hazardous waste in addition to the desired products. Hence, it is preferable to use molecular oxygen for this oxidation reaction using a catalyst.252 However, catalysts need to be able to activate molecular oxygen. Several catalysts, including supported noble metal catalysts, supported transition metal catalysts, and metal oxides, have been reported to date.253 Within the scope of this review, in this section, we discuss the role of the support during the aerobic oxidation of alcohols over supported gold nanoparticles. This section has been divided into two subsections based on the influence of the basicity or acidity of the support on the activity (conversion) and selectivity of the reaction. The acidity and basicity of the supports played a key role in the activity and selectivity for the oxidation of the alcohols.
3.2.1. Role of Support Property on the Activity of the Catalyst
Many researchers have worked extensively on the mechanism of the aerobic oxidation of alcohols over supported Au NPs and the widely accepted mechanistic steps involved are (a) step 1, deprotonation of alcohol to form an alcoholate species, (b) step 2, β-hydride elimination of the alcoholate species to form a carbonyl species, and (c) step 3, elimination of the adsorbed Hs as H2O by oxygen.253
Among these steps, the β-hydride elimination step is the rate-determining step, and this occurs mostly on the metallic site or at the metal–support interfacial site.12 When basic supports are used to support Au NPs, the basicity of the support enhances the rate of deprotonation of the alcohol to produce the alcoholate species (step 1 in Figure 20). On the basis of this strategy, several active catalysts with basic supports have been reported for the aerobic oxidation of alcohols. Kaneda and co-workers reported Au NPs supported on hydrotalcite (HT) materials as effective catalysts for the aerobic oxidation of alcohols. To demonstate the importance of the basicity of the support material, they prepared Au NPs on different supports including HT and tested them for the aerobic oxidation of phenyl ethanol to phenylacetone (Table 1).253 The catalytic data show that catalysts with MgO and HT (basic materials) gave the highest yields of the desired product. Following this strategy, several groups have reported gold NPs supported on basic materials such as MgO, ZnO, basic metal carbonates are better catalysts compared to Au NPs supported on neutral materials. Similarly, Wang et al. designed active catalysts by supporting Au NPs on Ni–Al layered double hydroxides (LDH) and exploited the basicity of the surface hydroxide groups to enhance the catalytic activity.254,255 Li et al. reported the transfer of electrons from Ni3Al-LDH as a support to Au NPs, thereby making Au negatively charged because of a strong metal–support interaction.256 Using extensive characterization, they reported that the material with the most negatively charged Au was the most active catalyst.
Figure 20.

Schematic representation of the mechanism of the aerobic oxidation of alcohols over basic metal oxide (hydrotalcite (HT)) supported gold NPs. Reproduced with permission from ref (253). Copyright 2016 Wiley-VCH.
Table 1. Role of Support on Au NPs Catalyzed Aerobic Oxidation of Alcoholsa.

| entry | catalyst | Au particle size (nm) | product yield (%)c |
|---|---|---|---|
| 1 | Au/HT | 2.7 | 99 |
| 2 | Au/MgO | 3.1 | 71 |
| 3 | Au/Al2O3 | 3.6 | 71 |
| 4 | Au/TiO2 | 3.7 | 14 |
| 5 | Au/TiO2 + Na2CO3c | 3.7 | 65 |
| 6 | Au/SiO2 | 14.0 | <1 |
Reproduced with permission from ref (253). Copyright 2016 Wiley-VCH.
Reaction conditions: Au catalyst (0.45 mol %), 1-phenylethanol (1 mmol), toluene (5 mL). Determined by GC analysis.
Reaction conditions: Au catalyst (0.45 mol %), 1-phenylethanol (1 mmol), toluene (5 mL). Na2CO3 (3 mmol).
Liu et al. reported the superior activity of Au NPs supported on Cr containing HT catalyst.257 In this catalyst, they further report that the synergistic effect between the Au NPs, and the support is because of the Cr3+/Cr6+ redox cycle at the Au–support interface. As a result of this, O2 activation takes place at the Au–Cr HT interface, which is accompanied by electron transfer from the support to the Au nanoparticle.258
Santra et al. reported Bi doped CeO2 supported Au NPs as very active catalyst for the oxidation of alcohols. The doping of Bi3+ into the CeO2 matrix increased the surface oxygen vacancy concentration of the support material and increases the basicity of the material. Consequently, Au NPs supported on Bi doped CeO2 have been found to be an effective catalyst for the aerobic alcohol oxidation reaction.259 Santra et al. also increased the surface O vacancy of the CeO2 support by introducing SnO2, which in turn resulted in the creation of Auδ±V0-Ce3+ sites (where V0 = oxygen vacancy), which are responsible for higher activity of this supported Au catalysts.260
All the examples discussed in the previous section concerned monohydric alcohols. Recently, oxidation of polyhydric alcohols such as glycerol, glucose, and hydroxymethylfurfural has received considerable attention because of the importance in the valorization of biorenewable feedstocks.261 Aerobic oxidation of these polyhydric alcohols is more challenging compared to that of monohydric alcohols, hence these oxidation reactions are typically carried out in the presence of a base (NaOH or KOH) to increase the rate of the reaction. Among all the polyhydric alcohols, glycerol is the most widely studied substrate because it is formed in large quantities as a byproduct during the biodiesel production by the transesterification of oils and fats. Hence, glycerol has been identified as one of the 12 biobased sustainable molecules that could potentially replace conventional feedstock molecules that are used in the production of chemicals and fuels.262 Oxidation of glycerol results in several products, including glyceraldehyde, glyceric acid, dihydroxy acetone, and many more (Figure 21). Villa et al. prepared Au/H-mordenite and AuPt/H-mordenite catalysts and used them for the base-free aerobic oxidation of glycerol by exploiting the acidity of the H-mordenite support.263 These are the earliest examples of the base-free oxidation of glycerol using supported metal catalysts, however, the role of acidity in enhancing the rate of the reaction has not been reported clearly. Brett et al. reported the use of bimetallic AuPd and AuPt NPs supported on Mg (OH)2 as effective catalysts for the base-free aerobic oxidation of glycerol. They utilized the basicity of Mg (OH)2 to enhance the rate of the reaction.264 Following these two initial examples, many Au NPs based catalysts have been developed for the base-free oxidation of glycerol by utilizing the acid–base properties of the support.265−267 A major problem associated with using solid base materials, such as MgO, as catalyst support is the deactivation of this catalyst during the reaction. The catalyst deactivation mainly occurs because of the leaching of the basic metal oxide upon reaction with the acidic products. Other reversible modes of deactivation are the phase transformation of MgO to Mg(OH)2 and/or MgCO3 after the reaction.268 Some other supports, e.g., hydrotalcites are found to be more stable compared to MgO-based supports for the base-free glycerol oxidation. Recently, Pan et al. reported Au/ZnO catalyst for the base-free oxidation of glycerol to dihydroxy acetone and they report the formation of Au–O–Zn sites at the Au/ZnO interface. As a result of the charge mismatch, oxygen vacancies are formed and the combination of these two sites have been found to be crucial for the oxidation of secondary alcohol group in glycerol (Figure 22). They maximized the population density of this site by preparing Au/ZnO catalyst via the deposition precipitation method followed by a high temperature thermal treatment in the presence of air. The support material plays similar roles during in the selective oxidation of other polyhydric alcohols such as ethylene glycol, propane diols, glucose, and hydroxymethyl furfural.269
Figure 21.

Schematic representation of the product formation during glycerol oxidation using supported Au catalysts. Adapted from ref (216). Copyright 2007 American Chemical Society.
Figure 22.

Role of Au-ZnO interfacial sites on the oxidation of glycerol to dihydroxy acetone. Reproduced with permission from ref (269). Copyright 2019 Elsevier.
In summary, during selective aerobic oxidation reactions, the support plays an active role via one or more of the following routes (a) by providing acidic or basic sites to generate the alcoholoate species, (b) providing electrons to generate either the Au δ- or Au0 species, and/or (c) generating oxygen vacancies that participate in the mechanism of the reaction.
3.2.2. Role of Support Property in Tuning the Selectivity
In the aerobic oxidation of benzyl alcohol, the desired product is benzaldehyde, however, other products such as toluene and benzoic acid can decrease the selectivity of this reaction. Toluene is formed by the disproportionation of two molecules of benzyl alcohol to form an equimolar mixture of toluene and benzaldehyde. In parallel, benzaldehyde is also produced by the oxidative dehydrogenation of benzyl alcohol. The benzaldehyde can be further oxidized to produce benzoic acid (Figure 23). Supported monometallic Au NPs do not catalyze the disproportionation reaction, and hence do not produce toluene. However, the activity of supported monometallic Au NPs is very low, and the addition of Pd to form bimetallic AuPd NPs increases the activity substantially. Pd, however, catalyzes the disproportionation reaction, resulting in the production of toluene. To study to the role of support, Sankar et al. prepared five supported AuPd catalysts using different supports such as TiO2, Nb2O5, C, MgO, and MgO via the sol-immobilization methodology to produce nearly uniform bimetallic NPs and tested them for the oxidation of benzyl alcohol. Bimetallic AuPd NPs supported on a basic support, e.g., ZnO or MgO, did not produce any toluene (Table 2).270
Figure 23.

Schematic representation of the reaction pathways for benzyl alcohol oxidation using supported AuPd catalysts.
Table 2. Effect of the Support on the Aerobic Oxidation of Benzyl Alcohol over Supported AuPd Catalysts270.
| selectivity (%)a |
|||
|---|---|---|---|
| catalyst | conversion (%) | benzaldehyde | toluene |
| 1%AuPd/TiO2 | 65 | 79 | 21.0 |
| 1%AuPd/Nb2O5 | 66 | 74 | 25.0 |
| 1%AuPd/C | 89 | 69 | 30.0 |
| 1%AuPd/MgO | 26 | 99 | 0.5 |
| 1%AuPd/ZnO | 39 | 99 | 0.5 |
Reaction conditions: benzyl alcohol: 18.5 mmol; substrate versus metal molar ratio: 14000; O2 pressure: 1 bar (relative); reaction time: 4 h; temperature: 120 °C. Other products include benzoic acid and benzyl benzoate.
Through careful analysis, they found that the active site for the oxidative dehydrogenation reaction is the (bi)metallic site, whereas the active site for the disproportionation reaction is the metal–support interfacial site. They tuned the metal–support interfacial site by changing the support, and through careful kinetic studies they concluded that the NPs supported on the more acidic support (TiO2, Nb2O5, activated carbon) catalyze the disproportionation reaction in addition to the oxidative dehydrogenation reaction, whereas the NPs supported on the more basic supports catalyze the oxidative dehydrogenation reaction exclusively resulting in >99% selectivity to benzaldehyde. Using detailed kinetic and in situ spectroscopic studies, they attributed this change in selectivity to different modes of adsorption of benzyl alcohol over supported Au-based catalysts. The basicity of the support promotes the adsorption of benzyl alcohol via the alcoholic oxygen atom which is crucial for the substrate to undergo the dehydrogenation pathway. Both the oxidative dehydrogenation and the disproportionation reactions do not occur without the metal, hence the cooperative role of both the metal and support is essential for these two reactions to occur.270,271
In another example, the acidity as basicity has been reported to play a crucial role in tuning the selectivity during the oxidation of glycerol using supported Au catalysts. Yuan et al. prepared a series of Au supported on MgO-Al2O3 catalysts with a systematic variation of the Mg/Al ratio to tune the acid–base properties of the support and the catalyst. All of these catalysts were tested for the base-free aerobic oxidation of glycerol at 80 °C. The Au catalyst comprising the most acidic support (Al2O3 (0.1)) resulted in the highest selectivity for dihydroxy acetone (DHA). Increasing the surface basicity and lowering the acidity, by increasing the MgO content, of the support, resulted in the selectivity to glyceric acid (GLA) being steadily increased at the expense of DHA selectivity (Figure 24).272
Figure 24.
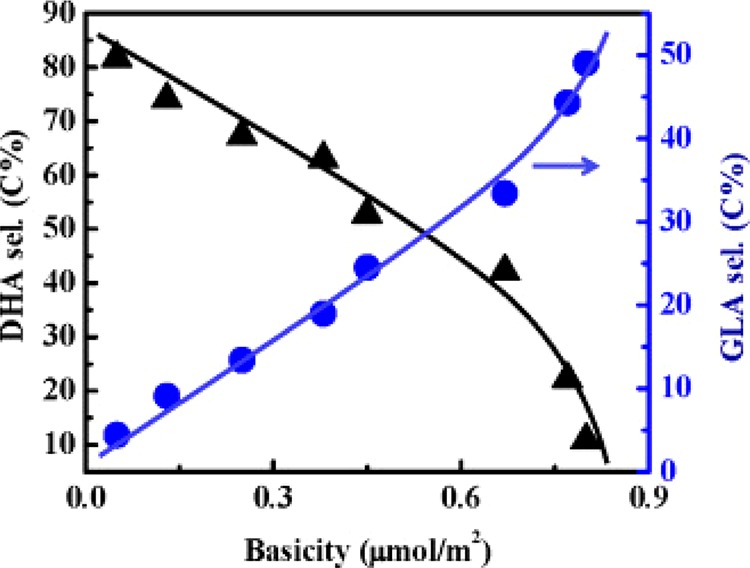
Dependencies of the DHA and GLA selectivity on the support surface basicity of the Au/MgO-Al2O3 catalysts. Reproduced with permission from ref (272). Copyright 2015 Elsevier.
Prati and co-workers studied the role of the support surface acidity on the catalytic activity and selectivity of AuPt NPs for glycerol oxidation. They found that a basic support resulted in a catalyst that was more active but less selective for the desired C3 products, producing more formic acid. However, AuPt NPs supported on acidic supports were less active but more selective toward C3 products.273 To sum up, the properties of the support material (acidity or basicity) play a key role in the resultant activity and/or selectivity of the supported metal catalysts. Basicity promotes both activity as well as selectivity, however these materials have stability issues because of support dissolution. Hence the solution is to improve the stability of the basic support materials or to improve the selectivity of the acidic support materials.
3.3. Hydrogenation Reactions
Hydrogenations are a very important class of reactions that are widely used for the production of bulk chemicals and the synthesis of fine chemicals.274,275 In general, direct hydrogenation using H2 as the reducing agent is preferred as it is the cleanest approach, and for this reaction to be viable under reasonable reaction conditions, a catalyst is required. Mostly transition metal catalysts are used to activate hydrogen, although lately nonmetal catalysts are being reported276 and especially frustrated Lewis pairs are finding growing interest.277 Traditionally, group VIII–X metals, e.g., ruthenium, palladium, and nickel, are the catalysts of choice in hydrogenation reactions. Gold was considered to be catalytically inactive. The publication of “Hydrogenation Over Supported Gold Catalyst” by Bond et al.278 in 1973 and the demonstration that gold can be a superior catalyst when applied as small NPs by Haruta et al.7 in the late 1980s were turning points for gold catalysis. Nowadays, gold is a promising catalyst with versatile application also in hydrogenation reactions.279−281 In this section, we discuss selected hydrogenation reactions in which an influence of the support material for gold-containing catalysts have been reported.
3.3.1. Selective Hydrogenation of Nitro Groups
Hydrogenation of nitro groups is an important transformation as the resulting amines are central intermediates in the chemical industry for bulk and fine chemicals alike.282 In particular, functionalized amines are of interest creating the challenging task of selectively hydrogenating the nitro group in the presence of hydrogenation-prone functionalities such as aromatic rings, carbon–carbon double bonds, or carbonyl bonds. In contrast to the selective reduction of nitro compounds with stochiometric amounts of reducing agents that lead to large amounts of waste, a catalytic process using H2 is favorable.283 However, established hydrogenation catalysts such a supported platinum catalysts usually do not differentiate between functional groups especially at longer reaction times.284−286 Thus, chemoselective catalysts for the hydrogenation of functionalized nitro compounds are needed.
Corma and Serna in 2006 showed that supported gold catalysts could be exceptional for the selective hydrogenation of nitro compounds.287 In particular, the use of Au/TiO2 led to excellent selectivity of functionalized anilines at over 95% at full or close to full conversion. This highly selective nitro group reduction was attributed to an energetically and geometrically preferential adsorption of the substrate at the interface of the NP and the TiO2 support.288 In contrast, Au/SiO2 is not a selective catalyst in this reaction, and because no preferential adsorption of the substrate was found on that catalyst, they concluded that the support plays an important role in the hydrogenation of functionalized nitroaromatics by favorably activating the nitro group.
The importance of the preferential adsorption of functionalized nitroaromatics with respect to the nature of the support for the metal catalyst was underlined in further studies on AuPt bimetallic catalysts for 3-nitrostyrene hydrogenation using both experimental and computational aproaches.289,290 Serna et al. reported that the addition of very small amounts of Pt (0.01%) to form a gold platinum bimetallic catalyst increased the TOF, significantly maintaining a very high 3-vinylaniline selectivity of 93%.289 However, an increase to 0.05% Pt decreased the selectivity to 75%. While the authors could only hypothesize on the reason for that because of difficulties in accurate characterization due to the very low Pt content, Boronat et al. gave a plausible explanation by employing DFT calculations.290 They showed that the addition of one Pt atom to a small gold cluster is sufficient to make the dissociative chemisorption of hydrogen barrierless, thereby explaining the strong increase in TOF with the very low Pt levels. With regard to the substrate adsorption, they could show that the adsorption modes are completely different on Au13/TiO2 to Pt13/TiO2 (Figures 25 and 26). The nitrostyrene molecule preferentially adsorbs on the top of the platinum NP, activating both the nitro group as well as the double bond, explaining the loss in selectivity. In contrast, the aforementioned favorable adsorption of the nitro group at the metal–support interface for Au/TiO2 catalysts is retained when the Au/Pt ratio is high enough to keep Pt atoms isolated and away from exactly that interface.
Figure 25.

Optimized geometry of nitrostyrene adsorbed (a) on the TiO2 support, (b) close to Au13 or Au12Pt NPs supported on TiO2, front view. (c) Side view for Au13/TiO2. (d) Side view for Au12Pt/TiO2. The distances are stated in Å. Reproduced with permission from ref (290). Copyright 2010 American Chemical Society.
Figure 26.
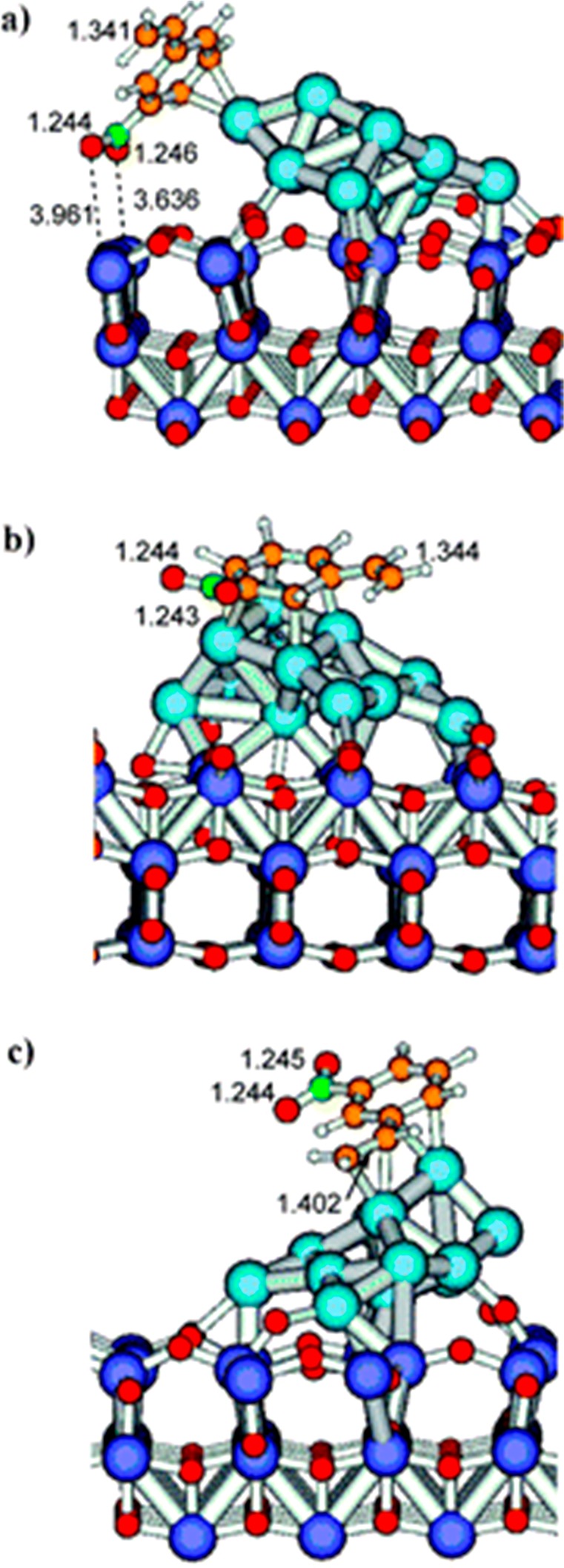
Different modes of adsorption of nitrostyrene on a Pt13 NP supported on TiO2. The distances are stated in Å. Reproduced with permission from ref (290). Copyright 2010 American Chemical Society.
Another suitable support for gold catalysts in the selective reduction of nitro compounds that aids the catalysis is Al2O3. Shimizu et al. reported a highly active Au/Al2O3 catalyst, exhibiting very good selectivity in the reduction of various functionalized nitroaromatics as well as providing a higher intrinsic activity compared to a Au/TiO2 catalyst.291 They attributed this to the acid and base properties of the support. In the reduction of 4-nitrostyrene, the use of supported gold NPs with a similar mean size resulted in a peak TOF with amphoteric Al2O3 as the support, while a strong basic support material MgO and an acidic support material SiO2 both showed very low TOFs. As gold NPs supported on carbon gave an even lower reaction rate by 2 orders of magnitude, it can be concluded that both the acidic and basic properties of the support have an influence on the observed activity. Using FTIR spectroscopy, they proposed a mechanism (Figure 27) that supports the observed structure–activity relationships. Heterolytic cleavage of H2 was proposed to occur at the gold–support interface, with the hydride located at the low coordination gold atom and the proton located at the oxygen close to the Lewis acid site. These polar hydrogen species preferentially react with the polar nitro group of the substrate enabling the chemoselectivity and preferential adsorption of the nitro group on Au/Al2O3. Such a heterolytic dissociation is in line with previous studies of H2 activation.292,293 Through DFT studies Whittaker et al. showed that H2 activation predominantly occurs across the metal–support interface for a Au/TiO2 catalyst, resulting in a proton adsorbed on a titania surface hydroxyl and a formal hydride adsorbed on the gold.293
Figure 27.

Proposed mechanism for the selective hydrogenation of nitroaromatic compounds using a Au/Al2O3 catalyst. Reproduced with permission from ref (291). Copyright 2009 American Chemical Society.
In a more recent study by Tan et al., the weak acidity and basicity provided by a zinc aluminum hydrotalcite as a support for gold nanoclusters was described to be beneficial for the selective hydrogenation of 3-nitrostyrene.294 It was shown that the support was preferentially adsorbing the nitro group of the substrate and not the carbon–carbon double bond. However, the ZnAl-hydrotalcite without any gold present was not active in the hydrogenation reaction. Thus, they considered that the small gold nanocluster (1.7–2.6 nm mean size) might be activating the hydrogen on the low coordination sites and because of the favorable adsorption of the substrate on the support the reduction could happen at the interface, resulting in the high selectivity of 3-vinylaniline. The same group investigated the influence of the divalent metal ions of hydrotalcites in 2018.295 The previous work, Au clusters supported on ZnAl-hydrotalcite was the most promising catalyst with regards to selectivity and activity. When the more basic MgAl-hydrotalcite was used as support the rate dropped considerably. Although the NiAl-hydrotalcite support led to an almost doubled activity, the selectivity to the desired product decreased substantially because of overhydrogenation and was only 9.5% after 16.5 h reaction time compared to 98.1% for the Au25-ZnAl-HT-300 catalyst. The high reactivity and overhydrogenation was attributed to the metallic nickel present in the support due to the easy reduction of nickel in close proximity of gold.
Hartfelder et al. compared Au/Al2O3 with Au/TiO2 in the hydrogenation of nitrobenzene.296 The authors showed that in the alumina-supported catalyst the gold NPs with sizes smaller than 2 nm are responsible for the observed catalytic activity, as these have the potential to activate the hydrogen. For the titania-supported catalysts, the activity was much less dependent on the particle size, which was related to the fact that titania facilitates hydrogen dissociation. As no other nitroaromatics with additional functional groups were tested, no comments on the selective nature of the catalyst can be made.
In 2017, Anandkumar et al.297 showed that the reducibility of the support is important for the hydrogenation of nitroaromatics when reducing agents such as sodium borohydride were used. Partially oxidized gold species (Auδ+) were found to be beneficial for the catalytic performance of ceria-supported gold catalysts. Through the redox cycle of Ce4+ and Ce3+, CeO2 is able to stabilize the Auδ+ on the support. Additionally, an improved adsorption of the reactants on the catalyst surface make CeO2 an excellent support. Both, the substrate 4-nitrophenol and the sodium borohydride need to adsorb on the catalyst surface. Through the hydrolysis of BH4– to B(OH)4–, hydrogen is produced and a Au–H bond is formed. Surface hydrogen can react with the 4-nitrophenolate ions to yield the product 4-aminophenolNPs (Figure 28).
Figure 28.

Schematic representation of the conversion of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP). Reproduced with permission from ref (297). Copyright 2017 The Royal Society of Chemistry.
3.3.2. Selective Hydrogenation of Alkynes and Alkadienes
Selective hydrogenation of acetylene to ethene is both academically challenging and industrially important.298−300 Cracking of naphtha results in a stream of ethene with acetylene impurities and selective hydrogenation is used to increase the purity of ethene feedstock, which is used for the production of polyethylene. Complete hydrogenation to ethane and oligomerization reaction, resulting in butadiene and higher hydrocarbons that often poison the catalyst reduce the selectivity to ethene. Most of the alkyne selective partial hydrogenation studies report the tuning of process parameters and feedstock composition to control the selectivity.301,302 Typically, Pd-based catalysts are used for this selective hydrogenation reaction; however Cu, Ni, Au, and Ag have also been reported to be active for this reaction.298−300,303 Supported Au monometallic NPs have been reported to have excellent alkene selectivity.302,304 Au has also been used as a second metal to improve the catalytic properties such as activity, selectivity, and stability of Pd-based catalysts.298,305,306
Mitsudome et al. designed a selective Au@CeO2 catalyst with a gold core and a CeO2 shell, resulting in a high number of interfacial sites for the semihydrogenation of alkynes to alkenes (Figure 29).307 When comparing this catalyst to a Au/CeO2 catalyst with gold NPs of similar size but without the core–shell structure, the core–shell structured catalyst gives 100% alkene yield even at high conversion, while the alkene yield was only 84% for the Au/CeO2 catalyst and was further decreased with increasing reaction time. The authors attribute this outstanding selectivity of the Au@CeO2 to heterolytic hydrogen dissociation occurring on the Au–ceria interfacial sites, as polar hydrogen species, especially hydrides, react more favorably with alkynes compared with alkenes, owing to the electrophilicity of alkynes. In contrast, the exposed Au sites on the unmodified Au/CeO2 catalyst induce homolytic cleavage of H2 into nonpolar hydrogen species, resulting in unselective hydrogenation of alkynes.
Figure 29.
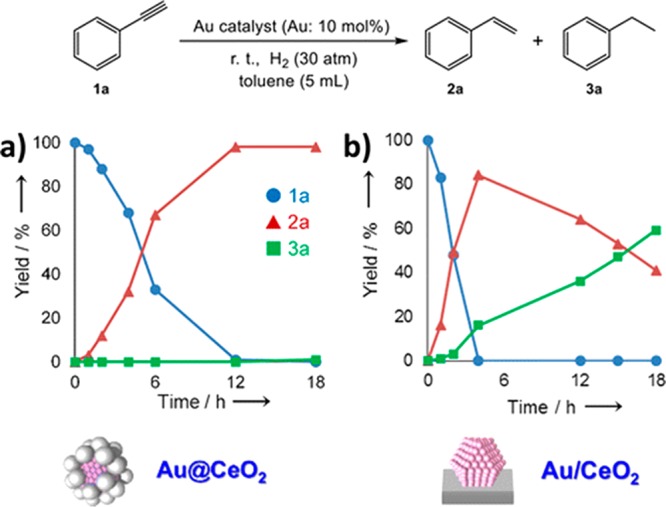
Time online study of the hydrogenation of phenylacetylene using (a) Au@CeO2 or (b) Au/CeO2. Reproduced with permission from ref (307). Copyright 2015 American Chemical Society.
Gluhoi et al.308 and Peng et al.309 reported Au/CeO2 to be a selective catalyst for the partial hydrogenation of acetylene to ethene. In both studies, the thermal pretreatment of the catalyst was varied; calcination resulted in Ce4+ and Au0, whereas reduction resulted in Ce3+ and Au0 species. The calcined sample proved more active for catalytic acetylene hydrogenation, which was ascribed to the reduced amounts of oxygen vacancies. The selectivity for both catalysts was similar.
Other impurities in alkene feedstock obtained by cracking are alkadienes. Similar to alkynes, they need to be selectively hydrogenated to the corresponding alkenes before further processing especially in the polymer production to avoid low-grade end products.310 It was shown that supported gold catalysts are active for the selective 1,3-butadiene hydrogenation, however, the metal oxide supports had little or no influence on the activity of the catalyst.311
Masoud et al. investigated Au/TiO2 and Au/SiO2 for the selective hydrogenation of 1,3-butadiene in the presence of propene.312 Au/TiO2 had a much higher initial activity, however, the catalyst deactivated faster under these reaction condition. It has been proposed that the deactivation is due to the deposition of carbonaceous species, facilitated by the surface properties of the TiO2 support. They further proposed that the active Au sites on the small clusters close to the TiO2 support are the most susceptible sites for deactivation. In contrast, on the silica support, only limited coke formation was observed, making Au/SiO2 a stable catalyst for a very long reaction time. This is an example where the role of support is not to influence the activity of the catalyst but its stability.
In contrast to metal oxides, carbon materials as supports seem to have a greater influence on the 1,3-butadiene hydrogenation as reported by Castillejos et al. in 2015.313 Au NPs supported on nitrogen-doped graphene oxides (GOs) and carbon nanotubes (CNTs) were investigated for the selective hydrogenation of 1,3-butadiene. Interestingly, the specific activities obtained with the different catalysts could neither be directly correlated to the average gold particle sizes nor the amount of smaller Au particles. They attributed this to hydrogen spillover onto the support, leading to an increased availability of the reactive hydrogen species. Hydrogen spillover, however, was not directly related to the amount of surface nitrogen groups. In the GO-supported catalysts, a continuous network of oxygen groups was proposed to facilitate the diffusion of H atoms, resulting in several strong adsorption sites. On the CNT-supported catalysts, the H atoms were suggested to spill over through defect sites where this species could also be stored. They also investigated amine-functionalized GOs and CNTs as supports for 1,3-butadiene hydrogenation.314 Interestingly, with reuse of the functionalized GO materials, the catalytic activity was enhanced although the size of the gold NP was not changed. The authors proposed that this might be due to intercalation of the substrate between the layers during restacking of the support material.
3.3.3. Hydrogenation of CO and CO2
The hydrogenation of CO and CO2 to produce methanol are reactions of great interest because methanol is not only widely used as solvent and starting material but is considered an alternative fuel and can be used for conventional energy storage for fuel cells.315 Additionally, formic acid is a promising molecule for hydrogen storage, making the hydrogenation of CO2 to yield formic acid, an important transformation that is increasingly studied.316
Hartadi et al. reported the use of TiO2, Al2O3, ZrO2, and ZnO as supports for gold NPs with very similar mean particle sizes in the hydrogenation of CO2.309 They observed the production of methanol as well as CO due to the reverse water gas shift (RWGS) reaction. The best results with regard to methanol selectivity were obtained using a Au/ZnO catalyst. Because the formation rates of methanol are in the same order of magnitude for all the catalysts evaluated, the authors attributed this to a lower activity of the Au/ZnO in the RWGS reaction. Additionally, ZnO was the only support that showed any activity for methanol production as well as in the RWGS reaction without Au present. They proposed that one reason for the improved selectivity at lower activity for Au/ZnO could be due to strong adsorption of the CO2 on the oxide support material. However, this cannot be the only reason as Au/Al2O3 shows a much lower selectivity and activity for methanol formation. Another explanation is, in an analogous manner to Cu/ZnO catalysts, in the reducing environment a (surface) reduction of the support and the formation of Zn surface species on the Au NPs can occur, which leads to stronger binding of the intermediates and a decrease in reaction barriers which ultimately results in a higher activity.
Au/ZnO was also investigated as a promising catalyst for methanol synthesis from synthesis gas as well as CO2-containing synthesis gas by Strunk et al.317 In this study, a correlation between oxygen vacancies in the ZnO support material, obtained by N2O reactive frontal chromatography, and the catalytic activity in methanol synthesis in both CO2-free and CO2-containing synthesis gas was found. Through a comparison of the N2O consumption with TEM analysis, it was concluded that the oxygen vacancies are mainly located in the perimeter of the gold NPs. These oxygen vacancies at the Au–ZnO interface were identified as the active sites for methanol synthesis.
Filonenko et al. investigated the liquid phase hydrogenation of CO2 to formates over different gold catalysts in the presence of base.318 While colloidal gold NPs were inactive, Au supported on metal oxides resulted in active catalysts for this reaction. However, the nature of support material is important, as Al2O3 led to double the TONs as compared with TiO2 as support. They also found that a high temperature pretreatment of the Au/Al2O3 had a negative influence on the catalytic activity. It was suggested to be a result of a change in the degree of hydration of the alumina, which can influence its ability to dissociate H2 heterolytically. They proposed a mechanism for CO2 hydrogenation using Au/Al2O3 (Figure 30). Starting with the heterolytic H2 dissociation at the gold and support interface into a metal hydride and surface hydroxyl, the CO2 is then adsorbed on the support, forming a bicarbonate species through reaction with the surface hydroxyl, which is subsequently attacked by the metal hydride, leading to an adsorbed formate species.
Figure 30.

Proposed reaction mechanism for the CO2 hydrogenation over an Au/Al2O3 catalyst. Reproduced with permission from ref (318). Copyright 2016 Elsevier.
In 2017, Liu et al. studied organic–inorganic hybrid silica materials in the liquid phase hydrogenation of CO2 to formate.319 It was found that through modifying the support with a Schiff base, a highly active catalyst could be obtained in the presence of a NEt3 additive in a polar solvent. Using in situ DRIFTS as well as theoretical calculations, it was found that the CO2 forms a weak carbamate zwitterionic intermediate at the Schiff base and gold interface. This activation of CO2 is important for the formation of formate. Subsequently, in the proposed mechanism (Figure 31), H2 is dissociated at the low-coordinated sites of the gold NPs and the resulting H species hydrogenate the carbamate zwitterion intermediate at the gold–Schiff base interface. They also suggest that through electron donation of the nitrogen groups the electron-rich gold surface could produce a more negative hydride, which increases the reactivity of the nucleophilic attack at the carbon atom of the CO2 molecule and therefore, is beneficial for the CO2 hydrogenation.
Figure 31.

CO2 hydrogenation over a Schiff base-modified gold nanocatalyst as proposed by Liu et al. Reproduced with permission from ref (319). Copyright 2017 Springer.
All in all, the influence of the support on the catalysis of gold-containing NPs in selective hydrogenation reactions can be versatile. For example, reduceable supports can stabilize catalytically active, partially oxidized Au species. Depending on the acid and base properties of the support, the heterolytic cleavage of hydrogen can be facilitated on the Au–support interface. Furthermore, the right support can direct the adsorption of bifunctional substrates to give high selectivities of the desired products.
3.4. C–C Coupling Reactions
A number of reviews prior to 2015 exist for the application of supported Au NPs toward C–C coupling reactions.12,320,321 More recent reviews have focused on the C–C coupling with mono- and bimetallic gold NPs in both homogeneously and heterogeneously catalyzed applications230,230,322−326 although without necessarily focusing on the role of the support. Here, we consider the specific role of the support for C–C coupling reactions, where examples exist for Glaser homocoupling (sp–sp), Ullmann homocoupling (sp2–sp2), Sonogashira cross-coupling (sp–sp2/sp3), Heck cross-coupling (sp2–sp2), Suzuki–Miyaura and Hiyama cross-coupling (sp2–sp2/sp3), as well as oxidative and reductive coupling, and three-reagent A3 cross-coupling of aldehydes, alkynes, and amines. The supporting material for the gold-based nanocatalysts used in these reactions falls into three categories: porous frameworks, photoactive oxides, and the more traditional low porosity carbons and metal oxides that are generally thought to be inactive during these reactions.
3.4.1. Porous Frameworks as Supports
Several groups have considered mesoporous or hierarchically structured silica to support gold nanocatalysts, with the advantage that these materials are easily separable from the reactants and products. Liu et al. supported AuCl on MCM-41, a hierarchical silica, identifying Au(I) active sites for oxidative cross-coupling of propargylic acetates and arylboronic acids, noting reasonable yields (71%) in only 15 min, with recyclability over seven cycles but with slower kinetics being observed after three reaction cycles.327 Sadeghzadeh et al. used a MOF-like mesoporous silica KCC-1, with phosphine linkers, to stabilize Au(III) complexes; arylation of benzoxazole with allylarene gave an 87% yield in 7 h. Similar activity was noted for a Au(III) homogeneous catalyst, although the separability of the supported catalyst makes the heterogeneous form beneficial.328 Mesoporous silica has also been used when coupled with additional materials: Movahed et al. used graphene coated with mesoporous silica, with pores of ∼2.5 nm, to support small Au NPs. The Au NPs were ∼2 nm in size and predominantly Au(0) according to XPS analysis. This catalyst was successfully applied to Suzuki–Miyaura cross-coupling for phenylboronic acid and phenyl iodides/bromides, with a 93% yield being observed with the phenyl iodide reaction after 6 h at 100 °C. The catalyst was reused six times, maintaining a 90% yield, and hot-filtration experiments showed the catalyst was working as a heterogeneous catalyst rather than as a precursor for homogeneous catalyst. Furthermore, the composite catalyst was applied to A3 coupling, with yields of 77–96% with only 4.9% leaching of the metal after six cycles, although no aggregation was noted.329 Vilhanová et al. considered a simpler silica support with amino functionalization to support Au NPs with a size of 0.8 ± 0.2 nm. When applied to Glaser-type alkyne coupling, 10 different products were successfully isolated with yields >84%; the catalysts were separable and recyclable up to five times with no changes in particle size. XPS analysis showed high quantities of Au(III), which was deemed to be part of the reaction cycle (Figure 32).330
Figure 32.

Isolated yields of diyne 2b obtained from 1b using catalyst A (Au deposited on 3-aminopropyl-functionalized silicate SBA-15, 5 mol %) and oxidant Phl(OAc)2 in DCM overnight in five cycles. (b) Z-contrast STEM images of material A before performing catalysis and (c) after five consecutive uses in the reaction outlined in (a). Particle size histograms, average particles size, and standard deviation are included, as obtained from these images. Reproduced with permission from ref (330). Copyright 2017 American Chemical Society.
Polymeric-type supports offer an alternative form of porous framework to support Au nanocatalysts, typically for larger NPs of >10 nm size. For example, Elhage et al. used nonsilanized glass wool, composed of fibers, to support Au NPs in a size range of 23 ± 10 nm. Here, photoreactivity was induced through exposure to green light to enable sp3–sp3 coupling of benzyl bromides with 80% yield in 7 h. Reusability in this case was not promising, with yields decreasing to 66% in 24 h during a second application; postapplication characterization showed larger NPs of 35 ± 12 nm had been formed.331 Poupart et al. formed supports of polystyrene-block-poly(d,l-lactide), with 1–2 μm pores, that 100 nm Au NPs were then deposited on; these proved excellent for the homocoupling of benzeneboronic acid, giving full conversion and 90% yield in 2 h.332 Liang et al. considered hydrophobic polymers of polydimethylsiloxane sponge to support Au NPs and applied them to the cross-dehydrogenative coupling of tertiary amines. A 92% yield was observed for the supported NPs, compared to 26% for unsupported NPs. The performance was observed and reusability performed over 20 cycles, with the 98% yield decreasing to 80% in 24 h. Finally, as an illustration of the potential for these porous supports, the catalyst was applied in a flow-reactor with 90% yield in 24 h.333
Thomas et al. considered a more complex composite porous system, made of self-organized strontium ion cross-linked alginate/carboxymethyl cellulose composites, then with Au NPs and graphene oxide deposited on this support. The material, which had pores of up to 100 μm, gave 98% yield in 4 h for Suzuki–Miyaura coupling and recyclability was demonstrated for up to six cycles; high yields of 85% (4 h) were also observed in the absence of graphene oxide.334 Slightly smaller pores were observed in the melamine polymer cross-linked Fe3O4 supports developed by Pourjavadi et al., being only 5–10 nm, then with small Au NPs of 6 nm in size deposited. This system was also applied to Suzuki–Miyaura coupling for aryl boronic acids and aryl halides, giving 92% yield (1.5 h), using H2O as a solvent. These catalysts offer great novelty as the magnetic core allows facile catalyst separation. However, 7% activity was lost over five reaction cycles, and the reaction rate deteriorated due to loss of catalyst.335 A magnetic-core catalyst was also created by Li et al., who supported PdAu NPs on Fe3O4@MgAl-LDH core@shell materials. This material had smaller pores of 2.1–2.7 nm, and the PdAu NPs deposited were of 1.7–2.55 nm in size. This composite material was compared to equivalent catalysts with Pd (2 nm) and Au (20 nm) particles for the Heck cross-coupling of iodobenzene with styrene, and with a PdAu ratio of 1:0.16, it was possible to achieve yields of 87.6% in 1 h; furthermore, this catalyst was recyclable over 10 experiments with >90% conversion.336
Maya et al. used bentonite as a support for 4 nm Au NPs. This support is highlighted as being inexpensive and with a readily modifiable surface; when applied for oxidative C–C coupling, the material gave up to 90% yield (12 h) in optimized conditions. The supported NPs, which had protected ligands, were compared to activated and acidified supports, as well as bare NPs, and yields were poorer. A range of ketones were investigated which, using optimized conditions, gave >80% yield, except for linear alcohols, with reusability over five reaction cycles.337 Kaur et al. have used Amberlite XAD-4 as a support for 3–8 nm Au NPs, which when applied for microwave-assisted Hiyama cross-coupling of aryl halides with phenyltrimethoxysilane, yields of up to 90% were achieved in times of only 10 min, compared to a conventional reaction time 6–12 h. In this case, the activity did, however, decrease slightly over five reaction cycles.338
3.4.2. Photoactive Supports
In contrast to porous frameworks, photoactive supports participate directly in the observed catalytic reactivity, although the reported mechanisms are contrasting. Photoactivity here refers to materials that have a chemical or physical response to illumination, rather than a direct involvement in catalysis. Clearly, there is potential for many semiconducting materials to be considered in this context, but there are few reported investigations. Crabbe et al. recently synthesized KNb3O8 supports, with a band gap of 3.7 eV, on to which they deposited Au NPs of 17.9 ± 10.1 nm. Under UVA light, Ullmann coupling of iodobenzene was achieved with 65–98% yield (2 h); this was repeated three times without significant loss of reactivity. Biaryl formation from aryl halides was also investigated, but in this case, the conversion decreased with increasing electronegativity of the halide species (Br, 20%; Cl, 0%). The mechanism for reaction was identified as a hole/electron pair forming in the photocatalyst (Figure 33), with the electron transferring to the Au nanoparticle where the reaction occurred.339
Figure 33.

Proposed pathways for the Ullmann homocoupling of aryl halides. Reproduced with permission from ref (339). Copyright 2018 The Royal Society of Chemistry.
A similar idea was proposed by Han et al. for AuPd NPs supported on TiO2 (P25) photoactive supports. The NPs were 3 nm in size, and so, under visible light, a plasmonic response was observed from the Au and “hot electrons” could transfer to the Pd, where the catalysis occurred; these electrons were postulated as being replaced by photoexcited charge carriers in the conduction band of the TiO2. When applied to the Suzuki–Miyaura cross-coupling of iodobenzene and phenylboronic acid, 98% biphenyl yield was achieved, compared to trace yield over Au Ni; in the absence of light, this dropped to 17% yield and 0% in the absence of the AuPd NPs. No change in activity or particle size was observed after four cycles. When the catalyst was investigated with a wider range of aryl halides, yields again decreased with increasing electronegativity for Br (70%) and Cl (14%).340 In contrast to these studies, Lanterna et al. report an alternative mechanism when supporting Au NPs (6 nm) on TiO2 supports for the reductive dimerization of 4-nitrobenzyl bromide, and in this case an 83% yield was achieved in 5 h, with the reaction requiring photostimulation at 532 nm. At this wavelength, this is postulated as resulting from plasmonic hot electrons being injected into the conduction band of the TiO2; these electrons then are transferred to the aryl halides from the support (Figure 34b). However, reuse experiments showed that yields decreased to ∼55% after only three cycles.341
Figure 34.

Contrasting mechanistic models for role of photocatalytic support in coupling reactions: (a) electron excited from the valence band (VB) to the conduction band (CB) of the support, creating a hole/electron pair, with electron transfer to the Fermi energy (Ef) of the nanoparticle; (b) plasmonic resonance resulting in “hot electron” injection into the support CB, where reduction can subsequently occur. Reproduced with permission from ref (339). Copyright 2018 The Royal Society of Chemistry.
3.4.3. Carbon and Metal Oxides As Supports
3.4.3.1. Carbon
On activated carbon, Parmentier et al. deposited atomically dispersed AuClx nanocatalysts, and compared these to sol-immobilized NPs of 6.6 ± 3 nm in size for the homocoupling of phenylboronic acid to form biphenyl, the AuClx nanocatalyst was shown to be active with modest yields of 25% in 6 h; in contrast, the sol-immobilized Au NPs were inactive (Figure 35). While the cationic Au(I) and Au(III) were therefore deemed reactive, it was noted that strong surface attraction of the active Au species (to the supporting carbon materials) is necessary to prevent reduction/sintering of NPs; such sintering was observed in the samples after catalytic testing (30–40 nm).342
Figure 35.

(left) Cationic Au NPs (n = 1 or 3) prove active for phenylboronic acid coupling to form biphenyl (6 h), though sintering affects reusability. (right) Neutral Au NPs are inactive.
Strong support interactions were also considered by Candu et al.336 and Primo et al.,337 who both prepared Au “platelets” with (111) facets over defective H-doped graphene. In both cases, the NPs were 3 nm deep and had surface areas of ∼20 nm. Candu et al. considered these materials for Ullmann homocoupling of iodobenzene, where good turnover numbers (TON) and selectivity to diphenyls were observed.343 Primo et al. made a comparison with 5 nm Au NPs on graphene, and a 5-fold improvement in TON was observed with 83% selectivity, although this was observed only at low conversion (0.3%).344 Graphene oxide was used to support Au NPs of 3.5 nm by Mondal et al.338 Again, a strong support–nanoparticle interaction was formed between the cationic Au and the anionic oxygen of graphene oxide. When applied to Suzuki–Miyaura cross-coupling of aryl halides and phenylboronic acid, 93% yield was observed at 70 °C. Only the combination of the support and nanoparticle was active, and hot filtration experiments showed catalyst was stable throughout, although yield did decrease by ∼10% over five cycles.345
3.4.3.2. Metal Oxides
For metal oxide supports, a stronger surface–NP interaction can occur than for the more inert carbon materials, and this can be applied in the coupling reactions. DFT calculations were used by Boronat et al. to investigate whether Au(I) or Au(III) was the active species for Sonogashira coupling reactions on Au/CeO2, the most selective of supports considered in experimental work (Figure 36). The small particles were found to spread the charge so that oxidized Auδ+ was at a particle surface, or particle–support interface, and active for deprotonation, while Au(0) was on the inside of the NPs. Interestingly, experimental testing showed homocoupling for biphenyl preferred Au(0), whereas homocoupling of alkynes was more active in NPs with mixed metal oxidation states, similar to as observed in DFT.346 Further reducible oxide supports (CeO2, TiO2, ZrO2) were tested by Liu et al. as Au nanoparticle supports, with the oxides themselves supported on SBA-15. The metal oxide quantity was 21–34% by mass, with Au NPs 1.3–2.3 nm in size. Initially, XPS measurements showed all NPs comprised some Au(III) but, after reduction, a dominance of Au(0) was observed. When tested for the base-free homocoupling of phenylboronic acid, all the reduced samples proved more active than the as-deposited catalysts. Conversion (>74%) and productivity (>94%) were high; recyclability was tested with the reduced CeO2 support, and here the yield decreased to 73% in five cycles due to leaching and aggregation, with nanoparticle sizes increasing from 1.3 to 2.3 nm.347 Albadi et al. also considered monoxide CuO and ZnO supports for Au NPs applied in Suzuki–Miyaura cross-coupling; a 90% yield was achieved in 4.5 h for coupling of aryl halides with arylboronic acid, and the catalyst was recyclable in testing over three runs.348 Gholinejad et al. also considered mixed metal oxides of CuFe2O4, supported on silica, to support Au NPs for Sonogashira coupling. The support was proposed to aid nanoparticle separation, and for the coupling of iodobenzene and phenylacetylene, this catalyst gave 95% yields and demonstrated recyclability over four reaction cycles; furthermore, the magnetic nature of the support again made it easily separable from the reaction mixture.349
Figure 36.

(a) Elementary steps in the mechanism of the Sonogashira cross-coupling reaction (blue) and the competitive iodobenzene (IB, red) and phenylacetylene (PA, green) homocoupling processes. (b) Selectivity toward the desired cross-coupling product (diphenylacetylene, DPA) obtained with different gold-based catalysts. Reproduced with permission from ref (346). Copyright 2017 Elsevier.
Several investigations have considered the use of rigorous size-selected, thiolate-protected Au NPs deposited on metal oxide supports, where here the support acts to simply stabilize the NPs. Li et al. tested small [Au25(SR)18]− [TOA]+ NPs supported on CeO2 for Ullmann heterocoupling of 4-methyl-iodobenzene with 4-nitro-iodobenzene, where TOA = tetraoctylammonium. The reactivity of the NPs is strongly affected by the type of thiolate ligand (SR) encapsulating the Au nanoparticle. Maximum conversion (91%) was observed for R = 1-naphthalenethiolate, with a selectivity of 82%; however, in recycling, this decreased to 50% and 15%, respectively, by the third reaction cycle, postulated to be due to ligand removal.350 In other similar work, Li et al. deposited Au38(SR)24 NPs (R = C2H4Ph) on TiO2, CeO2, and SiO2, and the activity on CeO2 and SiO2 was similar to the unsupported NPs for A3 coupling, being >98% in all cases. It was concluded after further analysis that both cationic “surface” and neutral “core” Au atoms play synergistic roles in the reactivity, which is aided by ligand and support interactions.351 Li et al. went further and considered the reactivity of doping Au25(SR)18 NPs (R = CH2CH2Ph) with Cu, Ag, and Pt, all supported on TiO2, all for application in the Sinogashira cross-coupling of p-iodoanisole and phenylacetylene. Overall, the pure Au nanoparticle has the highest selectivity, with the Pt-doped catalyst being equivalent; the Cu-doped catalyst in contrast preferred Ullmann homocoupling. In conclusion, the authors determined that the electronic effect (change in energy levels) from nanoparticle dopants is crucial in reactivity and is determined by the shell species (element) of the nanoparticle rather than being related to the choice of support.352
3.5. Reactions Catalyzed by Unsupported Au-Based NPs
It has been assumed that most of the catalytic activity of the supported NPs is due to the influence of the support, for example, by the creation of interfacial sites between the NP and the support or the changing in nanoparticle morphology that the support can induce on the NPs. Hence, the activity of unsupported colloidal NPs has not been considered in many studies to date. However, for the completeness of this review, we consider this topic should be addressed as there are examples where unsupported colloidal NPs can give superior performance.
Colloidal NPs have been studied to show their intrinsic catalytic activity and to elucidate the catalytic mechanism for hydrogenation, oxidation, coupling reactions, etc.353,354 These NPs mimic the metal surface activation and catalysis at the nanoscale typically observed in heterogeneous catalysis but also are dispersible in a range of solvents (unlike solid catalysts) and can be used as homogeneous catalysts. Hence, these nanoparticle catalysts can bridge both the homogeneous and heterogeneous catalysis communities, and these catalysts are sometimes referred to as semiheterogeneous.355
Nanosized gold colloids display interesting optical properties together with high catalytic activities. They are intensely colored due to the surface plasmon resonance, with the particular color of the colloid depending not only on the size and shape of the NPs but also on the properties of the solution such as the dielectric constant of the dispersion medium.353 Au and Au–Pd NPs have been shown to exhibit catalytic activities without the presence of a support for oxidation reactions for substrates as benzyl alcohol,356 glycerol,357 glucose,165,358 styrene, and CO,359,360 oxidative polymerization of aniline and pyrrole,361,362 hydrogenation of cinnamaldehyde and benzalacetone,363,364 and epoxidation and allylic oxidation.365 Monodispersed PVP stabilized Au NPs were synthesized and investigated by Tsukuda and co-workers for aerobic oxidation of benzylic alcohols and homocoupling of phenylboronic acid.356,366 The catalyst was prepared by rapid reduction using NaBH4 into an aqueous solution the [AuCl4]−–PVP complex, which yielded NPs with 1.3 nm mean diameter. The catalyst was treated with Na2SO3 to increase the particle size to study the effect of particle size on catalytic activity.
Recently, colloidal NPs were also shown to oxidize methane to methanol in aqueous solutions at mild temperatures.367,368 Compared to the same NPs supported on titanium oxide, much higher activity and selectivity was obtained with much lower consumption of H2O2.369,370 Previously, homogeneous Au and Pd have also been investigated for low temperature methane oxidation.371,372 However, without strong oxidants such as selenic acid or trifluoroacetic acid, the catalyst was not effective in activating methane to produce oxygenated products. Oxidation in aqueous conditions with homogeneous chloroauric acid led to precipitation of Au(0) and deactivation of the catalyst.373 Polymer protected bimetallic Au–Pd NPs were shown to possess much higher activity, with no deactivation observed for up to 4 h.367
This size effect on catalytic activity was further investigated under the influence of identity and concentration of the stabilizing ligand used to prepare the colloids. The role of the ligand was also studied in the enhancement or suppression of the catalytic activity while avoiding particle aggregation.374 It has been considered that strong Au–ligand interactions could decrease the particle size and avoid particle aggregation but could also decrease the catalytic activity considerably by partially or completely blocking access of the reactant molecules to the metal surface.353,374 A compromise between the activity and reusability must be established with respect to the type of stabilizing ligand used and its concentration. For example, the effect of the type of stabilizer used to prepare colloidal catalysts for glycerol oxidation has been investigated by Prati and co-workers.357 An increase in the catalytic activity was observed with decrease in the particle size when catalysts were prepared using PVA, tetrakishydroxypropylphosphonium chloride (THPC), and citrate as protecting agents (Figure 37). The catalysts showed similar selectivity, but citrate-stabilized Au NPs, with mean particle size of 9.8 nm, showed the least activity (TOF 160 h–1) compared to particles stabilized by PVA (715 h–1) and THPC (2478 h–1), which had much lower particle sizes of 2.5 and 2 nm, respectively. On aging, PVA and citrate showed much less agglomeration compared to THPC stabilized, which indicated that electrostatic stabilizers were much less effective as a protective agent compared to steric stabilizers. The bulky steric stabilizers prevented agglomeration and hence were active over longer use periods but were found to be much less active compared with THPC, probably again due to its steric congestion and limiting active site accessibility. Stabilizer-free NPs have been synthesized, but due to their instability in solution over an extended period of time, were supported onto TiO2 and tested for glycerol oxidation. Again, a decrease in activity was observed with increase in particle size and also a difference in selectivity was observed with PVP, PVA and stabilizer free catalysts.375 PVP-stabilized gold NPs were shown to display enhanced catalytic activity compared to the colloids prepared by PAA for aerobic oxidation, which was attributed due to electron donation properties observed for PVP.376
Figure 37.

(a–c) TEM images of the freshly prepared stabilized gold NPs: (a) AuPVA, (b) AuTHPC, (c) Aucitrate. Reproduced with permission from ref (357). Copyright 2009 John Wiley and Sons. (d) Comparison of selectivity for the different stabilized gold NPs, before and after aging.
Similarly, the metal:polymer ratio has been varied to probe the effect of polymer concentration on particle size and catalytic activity. Particle size was confirmed using electron microscopy, and the highest polymer to Au ratio of 50:1 gave small size particles with very narrow particle size distribution in the range of 1.8–2.8 nm.364 On the other hand, a much broader particle size distribution from 2 to 12 nm was obtained with a polymer to Au ratio of 3:1. Particles prepared with 50 times the polymer concentration showed the best activity toward the reduction of nitrophenol compared to the Au catalyst with bigger sizes. Similarly, the mean diameter of monodispersed Pd NPs was controlled from 1.7 to 3.0 nm by changing the amount of PVP added during synthesis.377
Along with the catalytic activity and selectivity, aging, and reuse of unsupported NPs has also been studied. De Vos et al. studied the stability of the Au colloids for oxidation of 1,2-propanediol.378 It was observed that significant level of activity (>50%) was maintained even after extended periods of aging (24 days). Another point noted was the recovery and reuse of the colloidal catalysts after consecutive experiments. This was addressed by using a poly(dimethylsiloxane) (PDMS) membrane to recover the sol by nanofiltration, by which over 99% of metallic colloid could be retained.379 These studies indicate the intrinsic activity of the unsupported NPs, and supporting material does not necessarily improve catalytic activity or selectivity. Colloidal catalysts can be used for a range of reactions, with stability and reuse possible for properly designed processes. However, more research is now required to understand and improve on efficiency of these colloidal catalysts as they can help to understand the role of the support and the metal–support interface in heterogeneous catalysis.
4. Future Perspectives
Since the use of gold NPs or highly dispersed gold catalysts was highlighted as effective catalysts in the 1980s, the field of gold catalysis has grown markedly and now gold and gold-containing NPs are the subject of extensive studies for a whole range of reactions in both heterogeneous and homogeneous catalysis. For supported gold and gold-containing NPs, the support is often crucial in securing the required catalytic function and this has been the focus of this review. In some cases, improved catalysis is observed in the absence of the support, but such examples are few and far between at present. However, we consider the study of unsupported colloids will provide important insights into how supported catalysts function, and so we expect such studies to continue in the future. Where there is perhaps the greatest scope for future improvement is in the field of deliberately designing supports with specific functionality. There have been some attempts at this; specifically, with the fine-tuning of acid/base properties of surfaces. However, we consider there is immense scope for further studies in this area. One approach will be to design the surfaces of the supports so they contain specific sites that can anchor gold nanoparticles. As gold is a soft metal, such an approach should be focused on adding in soft functionalities as the soft–soft interaction will necessarily lead to enhanced stability and interaction with the support. With the significant advances in materials characterization methods that we have witnessed and exploited in the past decade, coupled with advanced theoretical studies, it is clear that we will be able to probe the interaction between the support and the nanoparticle in ever more detail. Use of in situ characterization methods will be most useful, as it is known that NPs can be fluxional under reaction conditions. This we consider will be a key area for advances in future studies. However, the major problem that needs to be addressed with gold catalysis is the need to ensure that the Au NPs, clusters, or cations that constitute the active site of the catalyst, for there are examples where each of these structures is the active center and do not agglomerate as this inevitably leads to deactivation. In some cases, this may be holding up the further commercialization of Au catalysts. In this respect, attention to the design of the support so that it can inherently stabilize the active gold center is the area for the most research attention at this time. It is here that we consider that theory can help in the selection and design of novel support structures, and we envisage this being a very active research area in the future.
Acknowledgments
We thank the UK Catalysis Hub for support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grants EP/R026939/1, EP/R026815/1, EP/R026645/1, and EP/R027129/1. Qian He acknowledges the support by the National Research Foundation (NRF) Singapore, under its NRF Fellowship (NRF-NRFF11-2019-0002). Rebecca Engel acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 663830. Graham Hutchings, David Willock, and Mala Sainna thank the EPSRC for funding (EP/P033695/1).
Biographies
Meenakshisundaram Sankar is a lecturer of Physical Chemistry at Cardiff University. He obtained his Ph.D. from the National Chemical Laboratory in India. He then moved to Cardiff University as a postdoctoral research associate in the group of Prof. Graham Hutchings FRS. In 2011, he was awarded the Marie Curie Intra-European Fellowship to work at Utrecht University (Netherlands) in the group of Prof. Bert Weckhuysen. He moved back to Cardiff University as a University Research Fellow in 2014 to start his independent research group, and since 2019 he has been working as a lecturer in Physical Chemistry. His expertise lies in the area of developing heterogeneous catalysts for the valorization of renewable feedstock. He has coauthored over 60 publications in the area.
Qian He obtained his B.Eng. and M.Eng. from Tsinghua University and Ph.D. from Lehigh University in 2013 under the supervision of Professor Christopher Kiely. After his postdoctoral work in Oak Ridge National Laboratory with Dr. Albina Borisevich, he joined Cardiff University in 2016 as a University Research Fellow in the School of Chemistry. In 2019, He was awarded the NRF fellowship in Singapore and joined as an assistant professor at the Department of Materials Science and Engineering, National University of Singapore. Qian’s research focuses on developing and applying electron microscopy techniques to study catalysts and other functional nanomaterials.
Rebecca V. Engel studied chemistry at RWTH Aachen University. She received her doctoral degree from RWTH Aachen University in 2016 under the supervision of Prof. Regina Palkovits. In the same year, Rebecca moved to Cardiff to join the Group of Prof. Graham Hutchings at the Cardiff Catalysis Institute as a postdoctoral researcher. Since 2017, she is a Sêr Cymru MSCA COFUND fellow at Cardiff University. Her research interest lies in kinetic investigations in sustainable chemistry and heterogeneous catalysis.
Mala A. Sainna obtained a B.Sc. in Chemistry from the University of Maiduguri, Nigeria (2005), and an M.Sc. in Petroleum Engineering from Teesside University, United Kingdom (2010) with specialization in Catalyst and Catalysis of Fischer–Tropsch Synthesis. He then joint Compact GTL Plc at Wilton Centre as a Steam Methane Reforming and Fischer–Tropsch Scientist before moving to Manchester to pursue a Ph.D. in Computational studies of Enzymes catalysis under the supervision of Dr. Samuel De Visser at the University of Manchester. After being awarded a Ph.D. in 2015, he joint Prof. Nigel Scrutton’s research group as a Postdoc. In January 2018, he moved to Cardiff University to work in the group of Dr. David Willock as a Research Associate.
Andrew J. Logsdail received his Ph.D. in Chemistry from the University of Birmingham in 2012 under the supervision of Prof. Roy L. Johnston. He was a postdoctoral research associate in the group of Prof. C. Richard A. Catlow, FRS, in the Department of Chemistry at University College London from 2012 to 2014 and then held a Ramsay Research Fellowship at the same institution from 2014 to 2016. In 2016, he was appointed to a University Research Fellowship in the School of Chemistry at Cardiff University and was promoted in 2019 to Lecturer in Computational and Catalytic Chemistry. His research expertise is in the development and application of computational software for the modelling of catalytic materials and reactions.
Alberto Roldan graduated in 2006 at the University of Barcelona, where he pursued postgraduate studies to obtain a Master’s in electrochemistry. He received his Ph.D. with honors from the Rovira i Virgili University, and in 2010 he moved to the University College of London as a research associate. Two years later, Alberto’s research was recognized with a Ramsay Memorial Fellowship. In 2015, he was appointed a research fellowship by Cardiff University, where, in 2019, became a lecturer. Alberto’s research interest is in dynamic processes on surfaces and nanostructures, especially those relevant to heterogeneous catalysis. He employs computational simulations to link materials composition and atomic structure to resilience and reactivity with specially focused on clean energy and sustainable industry.
David J. Willock obtained his Ph.D. from Queen Mary and Westfield College, University of London, in 1991. He then moved into the area of computational chemistry in University College London before moving to the Leverhulme Centre for Catalysis in Liverpool. From 1997, he has been a member of faculty at Cardiff University and is currently a Reader in Computational Chemistry with a research group within the Cardiff Catalysis Institute. He has over 140 publications in the area focusing on materials chemistry and catalysis working together with experimentalists on structure/reactivity of metals and oxide materials.
Nishtha Agarwal obtained her M.Sc. in Chemistry from the Indian Institute of Science Education and Research Mohali (India) in 2014. She worked in Shell Technology Centre in India before moving to Cardiff. She completed her Ph.D. in nanomaterials for methane oxidation at Cardiff University within the Cardiff Catalysis Institute in collaboration with the MaxNet energy consortium under the supervision of Prof. Graham Hutchings in 2018. Currently, she is working as a Postdoc in Hutching’s group. Her research interests is in development and applications of novel catalytic materials for oxidation reactions.
Christopher J. Kiely received his Ph.D. from Bristol University in 1986. He was a faculty member at the University of Liverpool from 1989 to 2002, and then joined Lehigh University as the Harold B. Chambers Senior Professor of Materials Science and Chemical Engineering. Since 2017, he has also been Professor of Catalysis and Electron Microscopy at Cardiff University. His research expertise lies in the application and development of electron microscopy techniques for the study of catalysts and other nanomaterials.
Graham J. Hutchings is Regius Professor of Chemistry at Cardiff University. He studied chemistry at University College London. His early career was with ICI and AECI Ltd., where he became interested in gold catalysis. In 1984, he moved to academia and has held chairs at the Universities of Witwatersrand, Liverpool, and Cardiff. He was elected a Fellow of the Royal Society in 2009. He was awarded the Davy Medal of the Royal Society in 2013, the ENI Award for Advanced Environmental Solutions in 2017, the RSC Faraday Lectureship Prize, and a CBE in 2018.
Special Issue
This paper is an additional review for Chem. Rev. 2020, volume 120, issue (2), , “Nanoparticles in Catalysis”.
The authors declare no competing financial interest.
References
- Munnik P.; de Jongh P. E.; de Jong K. P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. 10.1021/cr500486u. [DOI] [PubMed] [Google Scholar]
- Catalyst Market Size, Share, Trends; Industry Growth Report 2019–2025; Grand View Research, 2019; https://www.grandviewresearch.com/industry-analysis/catalyst-market (accessed 2019-10-06).
- Liu L.; Corma A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981. 10.1021/acs.chemrev.7b00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudart M.Catalysis by Supported Metals In Adv. Catal.; Eley D. D., Pines H., Weisz P. B., Eds.; Academic Press, 1969; Vol. 20, 153–166. [Google Scholar]
- Lycurgus Cup; British Museum, 2019;https://www.britishmuseum.org/research/collection_online/collection_object_details.aspx?objectId=61219&partId=1&searchText=Lycurgus+Cup&page=1 (accessed 2019-10-06).
- Faraday M. X. The Bakerian Lecture. Experimental Relations of Gold (and Other Metals) to Light. Philos. Trans. R. Soc. London 1857, 147, 145–181. 10.1098/rstl.1857.0011. [DOI] [Google Scholar]
- Haruta M.; Kobayashi T.; Sano H.; Yamada N. Novel Gold Catalysts for the Oxidation of Carbon-Monoxide at a Temperature Far Below 0-Degree-C. Chem. Lett. 1987, 16, 405–408. 10.1246/cl.1987.405. [DOI] [Google Scholar]
- Hutchings G. J. Vapor Phase Hydrochlorination of Acetylene: Correlation of Catalytic Activity of Supported Metal Chloride Catalysts. J. Catal. 1985, 96, 292–295. 10.1016/0021-9517(85)90383-5. [DOI] [Google Scholar]
- Daniel M.-C.; Astruc D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- Liu X. Y.; Wang A.; Zhang T.; Mou C.-Y. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8, 403–416. 10.1016/j.nantod.2013.07.005. [DOI] [Google Scholar]
- Ciriminna R.; Falletta E.; Della Pina C.; Teles J. H.; Pagliaro M. Industrial Applications of Gold Catalysis. Angew. Chem., Int. Ed. 2016, 55, 14210–14217. 10.1002/anie.201604656. [DOI] [PubMed] [Google Scholar]
- Corma A.; Garcia H. Supported Gold Nanoparticles as Catalysts for Organic Reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. 10.1039/b707314n. [DOI] [PubMed] [Google Scholar]
- Tauster S. J.; Fung S. C.; Garten R. L. Strong Metal-Support Interactions. Group 8 Noble Metals Supported on Titanium Dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. 10.1021/ja00469a029. [DOI] [Google Scholar]
- Tauster S. J. Strong Metal-Support Interactions. Acc. Chem. Res. 1987, 20, 389–394. 10.1021/ar00143a001. [DOI] [Google Scholar]
- Haruta M. Catalysis of Gold Nanoparticles Deposited on Metal Oxides. CATTECH 2002, 6, 102–115. 10.1023/A:1020181423055. [DOI] [Google Scholar]
- Oh H. Selective Catalytic Oxidation of CO: Effect of Chloride on Supported Au Catalysts. J. Catal. 2002, 210, 375–386. 10.1006/jcat.2002.3710. [DOI] [Google Scholar]
- Costello C.K.; Kung M.C.; Oh H.-S.; Wang Y.; Kung H. H. Nature of the Active Site for CO Oxidation on Highly Active Au/γ-Al2O3. Appl. Catal., A 2002, 232, 159–168. 10.1016/S0926-860X(02)00092-3. [DOI] [Google Scholar]
- Haruta M.; Tsubota S.; Kobayashi T.; Kageyama H.; Genet M. J.; Delmon B. Low-Temperature Oxidation of CO Over Gold Supported ON TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. 10.1006/jcat.1993.1322. [DOI] [Google Scholar]
- Tsubota S.; Haruta M.; Kobayashi T.; Ueda A.; Nakahara Y. Preparation of Highly Dispersed Gold on Titanium and Magnesium Oxide. Stud. Surf. Sci. Catal. 1991, 63, 695–704. 10.1016/S0167-2991(08)64634-0. [DOI] [Google Scholar]
- Andreeva D.; Tabakova T.; Idakiev V.; Christov P.; Giovanoli R. Au/α-Fe2O3 Catalyst for Water–Gas Shift Reaction Prepared by Deposition–Precipitation. Appl. Catal., A 1998, 169, 9–14. 10.1016/S0926-860X(97)00302-5. [DOI] [Google Scholar]
- Dekkers M. A. P.; Lippits M. J.; Nieuwenhuys B. E. CO Adsorption and Oxidation on Au/TiO2. Catal. Lett. 1998, 56, 195–197. 10.1023/A:1019037902776. [DOI] [Google Scholar]
- Zanella R.; Giorgio S.; Henry C. R.; Louis C. Alternative Methods for the Preparation of Gold Nanoparticles Supported on TiO2. J. Phys. Chem. B 2002, 106, 7634–7642. 10.1021/jp0144810. [DOI] [Google Scholar]
- Prati L.; Rossi M. Gold on Carbon as a New Catalyst for Selective Liquid Phase Oxidation of Diols. J. Catal. 1998, 176, 552–560. 10.1006/jcat.1998.2078. [DOI] [Google Scholar]
- Navalon S.; Martin R.; Alvaro M.; Garcia H. Gold on Diamond Nanoparticles as a Highly Efficient Fenton Catalyst. Angew. Chem., Int. Ed. 2010, 49, 8403–8407. 10.1002/anie.201003216. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Yin H.; Overbury S. H.; Dai S. Metal Phosphates as a New Class of Supports for Gold Nanocatalysts. Catal. Lett. 2008, 126, 20–30. 10.1007/s10562-008-9627-x. [DOI] [Google Scholar]
- Li M. J.; Wu Z. L.; Overbury S. H. CO Oxidation on Phosphate-Supported Au Catalysts: Effect of Support Reducibility on Surface Reactions. J. Catal. 2011, 278, 133–142. 10.1016/j.jcat.2010.11.019. [DOI] [Google Scholar]
- Fang W. H.; Zhang Q. H.; Chen J.; Deng W. P.; Wang Y. Gold Nanoparticles on Hydrotalcites as Efficient Catalysts for Oxidant-Free Dehydrogenation of Alcohols. Chem. Commun. 2010, 46, 1547–1549. 10.1039/b923047e. [DOI] [PubMed] [Google Scholar]
- Fang W. H.; Chen J. S.; Zhang Q. H.; Deng W. P.; Wang Y. Hydrotalcite-Supported Gold Catalyst for the Oxidant-Free Dehydrogenation of Benzyl Alcohol: Studies on Support and Gold Size Effects. Chem. - Eur. J. 2011, 17, 1247–1256. 10.1002/chem.201002469. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Martha S.; Parida K. Facile Synthesis of Au/g-C3N4 Nanocomposites: An Inorganic/Organic Hybrid Plasmonic Photocatalyst with Enhanced Hydrogen Gas Evolution Under Visible-Light Irradiation. ChemCatChem 2014, 6, 1453–1462. 10.1002/cctc.201300949. [DOI] [Google Scholar]
- Gao J.; Fan G. L.; Yang L.; Cao X. Z.; Zhang P.; Li F. Oxidative Esterification of Methacrolein to Methyl Methacrylate over Gold Nanoparticles on Hydroxyapatite. ChemCatChem 2017, 9, 1230–1241. 10.1002/cctc.201601560. [DOI] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Yang M.; Allard L. F.; Flytzani-Stephanopoulos M. Atomically Dispersed Au-(OH)(x) Species Bound on Titania Catalyze the Low-Temperature Water-Gas Shift Reaction. J. Am. Chem. Soc. 2013, 135, 3768–3771. 10.1021/ja312646d. [DOI] [PubMed] [Google Scholar]
- Parks G. A. The Isoelectric Points of Solid Oxides, Solid Hydroxides, and Aqueous Hydroxo Complex Systems. Chem. Rev. 1965, 65, 177–198. 10.1021/cr60234a002. [DOI] [Google Scholar]
- Kosmulski M. Isoelectric Points and Points of Zero Charge of Metal (Hydr)Oxides: 50years after Parks’ Review. Adv. Colloid Interface Sci. 2016, 238, 1–61. 10.1016/j.cis.2016.10.005. [DOI] [PubMed] [Google Scholar]
- Moreau F.; Bond G. C.; Taylor A. O. Gold on Titania Catalysts for the Oxidation of Carbon Monoxide: Control of PH during Preparation with Various Gold Contents. J. Catal. 2005, 231, 105–114. 10.1016/j.jcat.2005.01.030. [DOI] [Google Scholar]
- Zanella R.; Delannoy L.; Louis C. Mechanism of Deposition of Gold Precursors onto TiO2 during the Preparation by Cation Adsorption and Deposition–Precipitation with NaOH and Urea. Appl. Catal., A 2005, 291, 62–72. 10.1016/j.apcata.2005.02.045. [DOI] [Google Scholar]
- Laguna O. H.; Romero Sarria F.; Centeno M. A.; Odriozola J. A. Gold Supported on Metal-Doped Ceria Catalysts (M = Zr, Zn and Fe) for the Preferential Oxidation of CO (PROX). J. Catal. 2010, 276, 360–370. 10.1016/j.jcat.2010.09.027. [DOI] [Google Scholar]
- Moreau F.; Bond G. C. Influence of the Surface Area of the Support on the Activity of Gold Catalysts for CO Oxidation. Catal. Today 2007, 122, 215–221. 10.1016/j.cattod.2006.12.001. [DOI] [Google Scholar]
- Delannoy L.; El Hassan N.; Musi A.; To N. N. L.; Krafft J.-M.; Louis C. Preparation of Supported Gold Nanoparticles by a Modified Incipient Wetness Impregnation Method. J. Phys. Chem. B 2006, 110, 22471–22478. 10.1021/jp062130l. [DOI] [PubMed] [Google Scholar]
- Lessard J. D.; Valsamakis I.; Flytzani-Stephanopoulos M. Novel Au/La2O3 and Au/La2O2SO4 Catalysts for the Water-Gas Shift Reaction Prepared via an Anion Adsorption Method. Chem. Commun. 2012, 48, 4857–4859. 10.1039/c2cc31105d. [DOI] [PubMed] [Google Scholar]
- Ivanova S.; Petit C.; Pitchon V. A New Preparation Method for the Formation of Gold Nanoparticles on an Oxide Support. Appl. Catal., A 2004, 267, 191–201. 10.1016/j.apcata.2004.03.004. [DOI] [Google Scholar]
- Ivanova S.; Pitchon V.; Zimmermann Y.; Petit C. Preparation of Alumina Supported Gold Catalysts: Influence of Washing Procedures, Mechanism of Particles Size Growth. Appl. Catal., A 2006, 298, 57–64. 10.1016/j.apcata.2005.09.020. [DOI] [Google Scholar]
- Arab L.; Boutahala M.; Djellouli B.; Dintzer T.; Pitchon V. Characteristics of Gold Supported on Nickel-Containing Hydrotalcite Catalysts in CO Oxidation. Appl. Catal., A 2014, 475, 446–460. 10.1016/j.apcata.2014.02.003. [DOI] [Google Scholar]
- Nguyen D. L.; Umbarkar S.; Dongare M. K.; Lancelot C.; Girardon J. S.; Dujardin C.; Granger P. Promising Stability of Gold-Based Catalysts Prepared by Direct Anionic Exchange for DeNO (x) Applications in Lean Burn Conditions. Top. Catal. 2013, 56, 157–164. 10.1007/s11244-013-9946-z. [DOI] [Google Scholar]
- Wang C.; Boucher M.; Yang M.; Saltsburg H.; Flytzani-Stephanopoulos M. ZnO-Modified Zirconia as Gold Catalyst Support for the Low-Temperature Methanol Steam Reforming Reaction. Appl. Catal., B 2014, 154–155, 142–152. 10.1016/j.apcatb.2014.02.008. [DOI] [Google Scholar]
- Wang C. Y.; Garbarino G.; Allard L. F.; Wilson F.; Busca G.; Flytzani-Stephanopoulos M. Low-Temperature Dehydrogenation of Ethanol on Atomically Dispersed Gold Supported on ZnZrOx. ACS Catal. 2016, 6, 210–218. 10.1021/acscatal.5b01593. [DOI] [Google Scholar]
- Qiao B. T.; Lin J.; Wang A. Q.; Chen Y.; Zhang T.; Liu J. Y. Highly Active Au-1/Co3O4 Single-Atom Catalyst for CO Oxidation at Room Temperature. Chin. J. Catal. 2015, 36, 1505–1511. 10.1016/S1872-2067(15)60889-0. [DOI] [Google Scholar]
- Qiao B. T.; Liu J. X.; Wang Y. G.; Lin Q. Q.; Liu X. Y.; Wang A. Q.; Li J.; Zhang T.; Liu J. Y. Highly Efficient Catalysis of Preferential Oxidation of CO in H-2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249–6254. 10.1021/acscatal.5b01114. [DOI] [Google Scholar]
- Qiao B. T.; Liang J. X.; Wang A. Q.; Xu C. Q.; Li J.; Zhang T.; Liu J. Y. Ultrastable Single-Atom Gold Catalysts with Strong Covalent Metal-Support Interaction (CMSI). Nano Res. 2015, 8, 2913–2924. 10.1007/s12274-015-0796-9. [DOI] [Google Scholar]
- Zhu H.; Liang C.; Yan W.; Overbury S. H.; Dai S. Preparation of Highly Active Silica-Supported Au Catalysts for CO Oxidation by a Solution-Based Technique. J. Phys. Chem. B 2006, 110, 10842–10848. 10.1021/jp060637q. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Ma Z.; Clark J. C.; Pan Z.; Overbury S. H.; Dai S. Low-Temperature CO Oxidation on Au/Fumed SiO2-Based Catalysts Prepared from Au(En)2Cl3 Precursor. Appl. Catal., A 2007, 326, 89–99. 10.1016/j.apcata.2007.04.004. [DOI] [Google Scholar]
- Wu Z.; Zhou S.; Zhu H.; Dai S.; Overbury S. H. DRIFTS-QMS Study of Room Temperature CO Oxidation on Au/SiO2 Catalyst: Nature and Role of Different Au Species. J. Phys. Chem. C 2009, 113, 3726–3734. 10.1021/jp809220z. [DOI] [Google Scholar]
- Somodi F.; Borbáth I.; Hegedűs M.; Tompos A.; Sajó I. E.; Szegedi Á.; Rojas S.; Fierro J. L. G.; Margitfalvi J. L. Modified Preparation Method for Highly Active Au/SiO2 Catalysts Used in CO Oxidation. Appl. Catal., A 2008, 347, 216–222. 10.1016/j.apcata.2008.06.017. [DOI] [Google Scholar]
- Liu X.; Wang A.; Yang X.; Zhang T.; Mou C.-Y.; Su D.-S.; Li J. Synthesis of Thermally Stable and Highly Active Bimetallic Au–Ag Nanoparticles on Inert Supports. Chem. Mater. 2009, 21, 410–418. 10.1021/cm8027725. [DOI] [Google Scholar]
- Phonthammachai N.; White T. J. One-Step Synthesis of Highly Dispersed Gold Nanocrystals on Silica Spheres. Langmuir 2007, 23, 11421–11424. 10.1021/la702230h. [DOI] [PubMed] [Google Scholar]
- Weng B.; Lu K. Q.; Tang Z. C.; Chen H. M.; Xu Y. J. Stabilizing Ultrasmall Au Clusters for Enhanced Photoredox Catalysis. Nat. Commun. 2018, 9, 1543. 10.1038/s41467-018-04020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia A. M.; Liotta F. L.; Pantaleo G.; Beck A.; Horváth A.; Geszti O.; Kocsonya A.; Guczi L. Effect of Ti(IV) Loading on CO Oxidation Activity of Gold on TiO2 Doped Amorphous Silica. Appl. Catal., A 2006, 310, 114–121. 10.1016/j.apcata.2006.05.027. [DOI] [Google Scholar]
- Qian K.; Huang W.; Jiang Z.; Sun H. Anchoring Highly Active Gold Nanoparticles on SiO2 by CoOx Additive. J. Catal. 2007, 248, 137–141. 10.1016/j.jcat.2007.02.010. [DOI] [Google Scholar]
- Albonetti S.; Bonelli R.; Mengou J. E.; Femoni C.; Tiozzo C.; Zacchini S.; Trifirò F. Gold/Iron Carbonyl Clusters as Precursors for TiO2 Supported Catalysts. Catal. Today 2008, 137, 483–488. 10.1016/j.cattod.2007.11.003. [DOI] [Google Scholar]
- Burgess R.; Buono C.; Davies P. R.; Davies R. J.; Legge T.; Lai A.; Lewis R.; Morgan D. J.; Robinson N.; Willock D. J. The Functionalisation of Graphite Surfaces with Nitric Acid: Identification of Functional Groups and Their Effects on Gold Deposition. J. Catal. 2015, 323, 10–18. 10.1016/j.jcat.2014.12.021. [DOI] [Google Scholar]
- Haruta M.; Yamada N.; Kobayashi T.; Iijima S. Gold Catalysts Prepared by Coprecipitation for Low-Temperature Oxidation of Hydrogen and of Carbon-monoxide. J. Catal. 1989, 115, 301–309. 10.1016/0021-9517(89)90034-1. [DOI] [Google Scholar]
- Andreeva D.; Idakiev V.; Tabakova T.; Andreev A.; Giovanoli R. Low-Temperature Water-Gas Shift Reaction on Au α-Fe2O3 Catalyst. Appl. Catal., A 1996, 134, 275–283. 10.1016/0926-860X(95)00208-1. [DOI] [Google Scholar]
- Hutchings G. J.; Rafiq H.; Siddiqui M. R. H.; Burrows A.; Kiely C. J.; Whyman R. High-Activity Au/CuO–ZnO Catalysts for the Oxidation of Carbon Monoxide at Ambient Temperature. J. Chem. Soc., Faraday Trans. 1997, 93, 187–188. 10.1039/a606482e. [DOI] [Google Scholar]
- Landon P.; Collier P. J.; Papworth A. J.; Kiely C. J.; Hutchings G. J. Direct Formation of Hydrogen Peroxide from H2/O2 Using a Gold Catalyst. Chem. Commun. 2002, 18, 2058–2059. 10.1039/b205248m. [DOI] [PubMed] [Google Scholar]
- Herzing A. A.; Kiely C. J.; Carley A. F.; Landon P.; Hutchings G. J. Identification of Active Gold Nanoclusters on Iron Oxide Supports for CO Oxidation. Science 2008, 321, 1331–1335. 10.1126/science.1159639. [DOI] [PubMed] [Google Scholar]
- Allard L. F.; Borisevich A.; Deng W.; Si R.; Flytzani-Stephanopoulos M.; Overbury S. H. Evolution of Gold Structure during Thermal Treatment of Au/FeOx Catalysts Revealed by Aberration-Corrected Electron Microscopy. J. Electron Microsc. 2009, 58, 199–212. 10.1093/jmicro/dfp016. [DOI] [PubMed] [Google Scholar]
- He Q.; Freakley S. J.; Edwards J. K.; Carley A. F.; Borisevich A. Y.; Mineo Y.; Haruta M.; Hutchings G. J.; Kiely C. J. Population and Hierarchy of Active Species in Gold Iron Oxide Catalysts for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 12905. 10.1038/ncomms12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo S.; Maki T.; Yamada M.; Mae K. A New Preparation Method of Au/Ferric Oxide Catalyst for Low Temperature CO Oxidation. Chem. Eng. Sci. 2010, 65, 214–219. 10.1016/j.ces.2009.05.044. [DOI] [Google Scholar]
- Zhang C. M.; Liu L. Q.; Cui X. J.; Zheng L. R.; Deng Y. Q.; Shi F. Chlorine as an Indicator in the Controllable Preparation of Active Nano-Gold Catalyst. Sci. Rep. 2013, 3, 1503. 10.1038/srep01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S. Y.; Zhang X.; Zhou W.; Gao R.; Xu W. Q.; Ye Y. F.; Lin L. L.; Wen X. D.; Liu P.; Chen B. B.; et al. Atomic-Layered Au Clusters on Alpha-MoC as Catalysts for the Low-Temperature Water-Gas Shift Reaction. Science 2017, 357, 389–393. 10.1126/science.aah4321. [DOI] [PubMed] [Google Scholar]
- Masoud N.; Partsch T.; de Jong K. P.; de Jongh P. E. Thermal Stability of Oxide-Supported Gold Nanoparticles. Gold Bull. 2019, 52, 105–114. 10.1007/s13404-019-00259-9. [DOI] [Google Scholar]
- Ren G. Q.; Tang Y.; Liu K. P.; Su Y.; Miao S.; Liu W.; Cong W. M.; Wang X. D.; Li W. Z.; Li J.; et al. Exceptional Antisintering Gold Nanocatalyst for Diesel Exhaust Oxidation. Nano Lett. 2018, 18, 6489–6493. 10.1021/acs.nanolett.8b03003. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Dai S. Design of Novel Structured Gold Nanocatalysts. ACS Catal. 2011, 1, 805–818. 10.1021/cs200100w. [DOI] [Google Scholar]
- Wang N.; Sun Q.; Yu J. Ultrasmall Metal Nanoparticles Confined within Crystalline Nanoporous Materials: A Fascinating Class of Nanocatalysts. Adv. Mater. 2019, 31, 1803966–23. 10.1002/adma.201803966. [DOI] [PubMed] [Google Scholar]
- Bore M. T.; Pham H. N.; Switzer E. E.; Ward T. L.; Fukuoka A.; Datye A. K. The Role of Pore Size and Structure on the Thermal Stability of Gold Nanoparticles within Mesoporous Silica. J. Phys. Chem. B 2005, 109, 2873–2880. 10.1021/jp045917p. [DOI] [PubMed] [Google Scholar]
- Russo V.; Tesser R.; Santacesaria E.; Di Serio M. Chemical and Technical Aspects of Propene Oxide Production via Hydrogen Peroxide (HPPO Process). Ind. Eng. Chem. Res. 2013, 52, 1168–1178. 10.1021/ie3023862. [DOI] [Google Scholar]
- Lewis R. J.; Ueura K.; Fukuta Y.; Freakley S. J.; Kang L.; Wang R.; He Q.; Edwards; Jennifer K.; Morgan D. J.; Yamamoto Y.; Hutchings G. J. The Direct Synthesis of H 2 O 2 Using TS-1 Supported Catalysts. ChemCatChem 2019, 11, 1673–1680. 10.1002/cctc.201900100. [DOI] [Google Scholar]
- Feng X.; Sheng N.; Liu Y. B.; Chen X. B.; Chen D.; Yang C. H.; Zhou X. G. Simultaneously Enhanced Stability and Selectivity for Propene Epoxidation with H-2 and O-2 on Au Catalysts Supported on Nano-Crystalline Mesoporous TS-1. ACS Catal. 2017, 7, 2668–2675. 10.1021/acscatal.6b03498. [DOI] [Google Scholar]
- Rombi E.; Cutrufello M. G.; Cannas C.; Casu M.; Gazzoli D.; Occhiuzzi M.; Monaci R.; Ferino I. Modifications Induced by Pretreatments on Au/SBA-15 and Their Influence on the Catalytic Activity for Low Temperature CO Oxidation. Phys. Chem. Chem. Phys. 2009, 11, 593–602. 10.1039/B813982B. [DOI] [PubMed] [Google Scholar]
- Cutrufello M. G.; Rombi E.; Cannas C.; Casu M.; Virga A.; Fiorilli S.; Onida B.; Ferino I. Synthesis, Characterization and Catalytic Activity of Au Supported on Functionalized SBA-15 for Low Temperature CO Oxidation. J. Mater. Sci. 2009, 44, 6644. 10.1007/s10853-009-3510-z. [DOI] [Google Scholar]
- Wu P.; Bai P.; Lei Z.; Loh K. P.; Zhao X. S. Gold Nanoparticles Supported on Functionalized Mesoporous Silica for Selective Oxidation of Cyclohexane. Microporous Mesoporous Mater. 2011, 141, 222–230. 10.1016/j.micromeso.2010.11.011. [DOI] [Google Scholar]
- Soni Y.; Kavya I.; Ajithkumar T. G.; Vinod C. P. One Pot Ligand Exchange Method for a Highly Stable Au-SBA-15 Catalyst and Its Room Temperature CO Oxidation. Chem. Commun. 2018, 54, 12412–12415. 10.1039/C8CC06887A. [DOI] [PubMed] [Google Scholar]
- Okumura K.; Murakami C.; Oyama T.; Sanada T.; Isoda A.; Katada N. Formation of Nanometer-Sized Au Particles on USY Zeolites under Hydrogen Atmosphere. Gold Bull. 2012, 45, 83–90. 10.1007/s13404-012-0051-z. [DOI] [Google Scholar]
- Guari Y.; Thieuleux C.; Mehdi A.; Reyé C.; Corriu R. J. P.; Gomez-Gallardo S.; Philippot K.; Chaudret B. In Situ Formation of Gold Nanoparticles within Thiol Functionalized HMS - C 16 and SBA - 15 Type Materials via an Organometallic Two-Step Approach. Chem. Mater. 2003, 15, 2017–2024. 10.1021/cm0213218. [DOI] [Google Scholar]
- Petkov N.; Stock N.; Bein T. Gold Electroless Reduction in Nanosized Channels of Thiol-Modified SBA-15 Material. J. Phys. Chem. B 2005, 109, 10737–10743. 10.1021/jp050429i. [DOI] [PubMed] [Google Scholar]
- Wang L.; Meng X. J.; Wang B.; Chi W. Y.; Xiao F. S. Pyrrolidone-Modified SBA-15 Supported Au Nanoparticles with Superior Catalytic Properties in Aerobic Oxidation of Alcohols. Chem. Commun. 2010, 46, 5003–5005. 10.1039/c000226g. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wang H.; Hapala P.; Zhu L. F.; Ren L. M.; Meng X. J.; Lewis J. P.; Xiao F. S. Superior Catalytic Properties in Aerobic Oxidation of Olefins over Au Nanoparticles on Pyrrolidone-Modified SBA-15. J. Catal. 2011, 281, 30–39. 10.1016/j.jcat.2011.03.029. [DOI] [Google Scholar]
- Liu Y.; Tsunoyama H.; Akita T.; Tsukuda T. Preparation of ∼ 1 Nm Gold Clusters Confined within Mesoporous Silica and Microwave-Assisted Catalytic Application for Alcohol Oxidation. J. Phys. Chem. C 2009, 113, 13457–13461. 10.1021/jp904700p. [DOI] [Google Scholar]
- Beck A.; Horváth A.; Stefler Gy.; Katona R.; Geszti O.; Tolnai Gy.; Liotta L. F.; Guczi L. Formation and Structure of Au/TiO2 and Au/CeO2 Nanostructures in Mesoporous SBA-15. Catal. Today 2008, 139, 180–187. 10.1016/j.cattod.2008.05.039. [DOI] [Google Scholar]
- Peza-Ledesma C. L.; Escamilla-Perea L.; Nava R.; Pawelec B.; Fierro J. L. G. Supported Gold Catalysts in SBA-15 Modified with TiO2 for Oxidation of Carbon Monoxide. Appl. Catal., A 2010, 375, 37–48. 10.1016/j.apcata.2009.12.009. [DOI] [Google Scholar]
- Liu C. H.; Guan Y. J.; Hensen E. J. M.; Lee J. F.; Yang C. M. Au/TiO2@SBA-15 Nanocomposites as Catalysts for Direct Propylene Epoxidation with O2 and H2 Mixtures. J. Catal. 2011, 282, 94–102. 10.1016/j.jcat.2011.06.001. [DOI] [Google Scholar]
- Mei B.; Wiktor C.; Turner S.; Pougin A.; van Tendeloo G.; Fischer R. A.; Muhler M.; Strunk J. Evidence for Metal-Support Interactions in Au Modified TiOx/SBA-15 Materials Prepared by Photodeposition. ACS Catal. 2013, 3, 3041–3049. 10.1021/cs400964k. [DOI] [Google Scholar]
- Hernandez J. A.; Gómez S.; Pawelec B.; Zepeda T. A. CO Oxidation on Au Nanoparticles Supported on Wormhole HMS Material: Effect of Support Modification with CeO2. Appl. Catal., B 2009, 89, 128–136. 10.1016/j.apcatb.2008.12.005. [DOI] [Google Scholar]
- Ma G. C.; Binder A.; Chi M. F.; Liu C.; Jin R. C.; Jiang D. E.; Fan J.; Dai S. Stabilizing Gold Clusters by Heterostructured Transition-Metal Oxide-Mesoporous Silica Supports for Enhanced Catalytic Activities for CO Oxidation. Chem. Commun. 2012, 48, 11413–11415. 10.1039/c2cc35787a. [DOI] [PubMed] [Google Scholar]
- Wang T.; Yuan X.; Li S. R.; Zeng L.; Gong J. L. CeO2-Modified Au@SBA-15 Nanocatalysts for Liquid-Phase Selective Oxidation of Benzyl Alcohol. Nanoscale 2015, 7, 7593–7602. 10.1039/C5NR00246J. [DOI] [PubMed] [Google Scholar]
- Tian C. C.; Chai S. H.; Mullins D. R.; Zhu X.; Binder A.; Guo Y. L.; Dai S. Heterostructured BaSO4-SiO2 Mesoporous Materials as New Supports for Gold Nanoparticles in Low-Temperature CO Oxidation. Chem. Commun. 2013, 49, 3464–3466. 10.1039/c3cc41167b. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Lee B.; Dai S.; Overbury S. H. Coassembly Synthesis of Ordered Mesoporous Silica Materials Containing Au Nanoparticles. Langmuir 2003, 19, 3974–3980. 10.1021/la027029w. [DOI] [Google Scholar]
- Wu P. P.; Bai P.; Yan Z. F.; Zhao G. X. S. Gold Nanoparticles Supported on Mesoporous Silica: Origin of High Activity and Role of Au NPs in Selective Oxidation of Cyclohexane. Sci. Rep. 2016, 6, 18817. 10.1038/srep18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Hu J.; Richards R. Intercalation of Aggregation-Free and Well-Dispersed Gold Nanoparticles into the Walls of Mesoporous Silica as a Robust “Green” Catalyst Forn-Alkane Oxidation. J. Am. Chem. Soc. 2009, 131, 914–915. 10.1021/ja808860v. [DOI] [PubMed] [Google Scholar]
- Chen L.; Hu J.; Qi Z.; Fang Y.; Richards R. Gold Nanoparticles Intercalated into the Walls of Mesoporous Silica as a Versatile Redox Catalyst. Ind. Eng. Chem. Res. 2011, 50, 13642–13649. 10.1021/ie200606t. [DOI] [Google Scholar]
- Budroni G.; Corma A. Gold–Organic–Inorganic High-Surface-Area Materials as Precursors of Highly Active Catalysts. Angew. Chem., Int. Ed. 2006, 45, 3328–3331. 10.1002/anie.200600552. [DOI] [PubMed] [Google Scholar]
- Overbury S. H.; Ortiz-Soto L.; Zhu H.; Lee B.; Amiridis M. D.; Dai S. Comparison of Au Catalysts Supported on Mesoporous Titania and Silica: Investigation of Au Particle Size Effects and Metal-Support Interactions. Catal. Lett. 2004, 95, 99–106. 10.1023/B:CATL.0000027281.96719.42. [DOI] [Google Scholar]
- Kumar A.; Kumar V. P.; Kumar B. P.; Vishwanathan V.; Chary K. V. R. Vapor Phase Oxidation of Benzyl Alcohol Over Gold Nanoparticles Supported on Mesoporous TiO2. Catal. Lett. 2014, 144, 1450–1459. 10.1007/s10562-014-1285-6. [DOI] [Google Scholar]
- Wang G. N.; Wang X. F.; Liu J. F.; Sun X. M. Mesoporous Au/TiO2 Nanocomposite Microspheres for Visible-Light Photocatalysis. Chem. - Eur. J. 2012, 18, 5361–5366. 10.1002/chem.201101410. [DOI] [PubMed] [Google Scholar]
- Violi I. L.; Zelcer A.; Bruno M. M.; Luca V.; Soler-Illia G. J. A. A. Gold Nanoparticles Supported in Zirconia-Ceria Mesoporous Thin Films: A Highly Active Reusable Heterogeneous Nanocatalyst. ACS Appl. Mater. Interfaces 2015, 7, 1114–1121. 10.1021/am5065188. [DOI] [PubMed] [Google Scholar]
- He Y. H.; Du S. Y.; Li J. W.; Zhang R. D.; Liang X.; Chen B. H. Mesoporous Ceria-Supported Gold Catalysts Self-Assembled from Monodispersed Ceria Nanoparticles and Nanocubes: A Study of the Crystal Plane Effect for the Low-Temperature Water Gas Shift Reaction. ChemCatChem 2017, 9, 4070–4082. 10.1002/cctc.201700645. [DOI] [Google Scholar]
- Lopez J. M.; Arenal R.; Puertolas B.; Mayoral A.; Taylor S. H.; Solsona B.; Garcia T. Au Deposited on CeO2 Prepared by a Nanocasting Route: A High Activity Catalyst for CO Oxidation. J. Catal. 2014, 317, 167–175. 10.1016/j.jcat.2014.06.021. [DOI] [Google Scholar]
- Cargnello M.; Gentilini C.; Montini T.; Fonda E.; Mehraeen S.; Chi M. F.; Herrera-Collado M.; Browning N. D.; Polizzi S.; Pasquato L.; et al. Active and Stable Embedded Au@CeO2 Catalysts for Preferential Oxidation of CO. Chem. Mater. 2010, 22, 4335–4345. 10.1021/cm101499x. [DOI] [Google Scholar]
- Tanaka S.; Lin J. J.; Kaneti Y. V.; Yusa S.; Jikihara Y.; Nakayama T.; Zakaria M. B.; Alshehri A. A.; You J.; Hossain M. S. A.; et al. Gold Nanoparticles Supported on Mesoporous Iron Oxide for Enhanced CO Oxidation Reaction. Nanoscale 2018, 10, 4779–4785. 10.1039/C7NR08895G. [DOI] [PubMed] [Google Scholar]
- Song H. Y.; Li G.; Wang X. S.; Xu Y. J. Characterization and Catalytic Performance of Au/Ti-HMS Catalysts on the Oxidative Desulphurization Using in Situ H2O2: Effect of Method Catalysts Preparation. Catal. Today 2010, 149, 127–131. 10.1016/j.cattod.2009.04.013. [DOI] [Google Scholar]
- Guillemot D.; Polisset-Thfoin M.; Fraissard J. Preparation of Nanometeric Gold Particles on NaHY. Catal. Lett. 1996, 41, 143–148. 10.1007/BF00811481. [DOI] [Google Scholar]
- Guillemot D.; Borovkov V. Yu.; Kazansky V. B.; Polisset-Thfoin M.; Fraissard J. Surface Characterization of Au/HY by 129 Xe NMR and Diffuse Reflectance IR Spectroscopy of Adsorbed CO. Formation of Electron-Deficient Gold Particles inside HY Cavities. J. Chem. Soc., Faraday Trans. 1997, 93, 3587–3591. 10.1039/a703431h. [DOI] [Google Scholar]
- Okumura M.; Tsubota S.; Iwamoto M.; Haruta M. Chemical Vapor Deposition of Gold Nanoparticles on MCM-41 and Their Catalytic Activities for the Low-Temperature Oxidation of CO and of H2. Chem. Lett. 1998, 27, 315–316. 10.1246/cl.1998.315. [DOI] [Google Scholar]
- Linares N.; Canlas C. P.; Garcia-Martinez J.; Pinnavaia T. J. Colloidal Gold Immobilized on Mesoporous Silica as a Highly Active and Selective Catalyst for Styrene Epoxidation with H2O2. Catal. Commun. 2014, 44, 50–53. 10.1016/j.catcom.2013.10.004. [DOI] [Google Scholar]
- Yang M.; Li S.; Wang Y.; Herron J. A.; Xu Y.; Allard L. F.; Lee S.; Huang J.; Mavrikakis M.; Flytzani-Stephanopoulos M. Catalytically Active Au-O(OH)x - Species Stabilized by Alkali Ions on Zeolites and Mesoporous Oxides. Science 2014, 346, 1498–1501. 10.1126/science.1260526. [DOI] [PubMed] [Google Scholar]
- Hermes S.; Schröter M.; Schmid R.; Khodeir L.; Muhler M.; Tissler A.; Fischer R. W.; Fischer R. A. Metal@MOF: Loading of Highly Porous Coordination Polymers Host Lattices by Metal Organic Chemical Vapor Deposition. Angew. Chem., Int. Ed. 2005, 44, 6237–6241. 10.1002/anie.200462515. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Nagaoka M.; Akita T.; Haruta M. Deposition of Gold Clusters on Porous Coordination Polymers by Solid Grinding and Their Catalytic Activity in Aerobic Oxidation of Alcohols. Chem. - Eur. J. 2008, 14, 8456–8460. 10.1002/chem.200800980. [DOI] [PubMed] [Google Scholar]
- Jiang H. L.; Liu B.; Akita T.; Haruta M.; Sakurai H.; Xu Q. Au@ZIF-8: CO Oxidation over Gold Nanoparticles Deposited to Metal-Organic Framework. J. Am. Chem. Soc. 2009, 131, 11302–11303. 10.1021/ja9047653. [DOI] [PubMed] [Google Scholar]
- Jiang H. L.; Lin Q. P.; Akita T.; Liu B.; Ohashi H.; Oji H.; Honma T.; Takei T.; Haruta M.; Xu Q. Ultrafine Gold Clusters Incorporated into a Metal-Organic Framework. Chem. - Eur. J. 2011, 17, 78–81. 10.1002/chem.201002088. [DOI] [PubMed] [Google Scholar]
- Fonseca J. da S. L.; Ferreira H. S.; Bion N.; Pirault-Roy L.; Rangel M. do C.; Duprez D.; Epron F. Cooperative Effect between Copper and Gold on Ceria for CO-PROX Reaction. Catal. Today 2012, 180, 34–41. 10.1016/j.cattod.2011.06.008. [DOI] [Google Scholar]
- Qi Y.; Luan Y.; Peng X.; Yang M.; Hou J. Y.; Wang G. Design and Synthesis of an Au@MIL-53(NH2) Catalyst for a One-Pot Aerobic Oxidation/Knoevenagel Condensation Reaction. Eur. J. Inorg. Chem. 2015, 30, 5099–5105. 10.1002/ejic.201500808. [DOI] [Google Scholar]
- Luan Y.; Qi Y.; Gao H. Y.; Zheng N. N.; Wang G. Synthesis of an Amino-Functionalized Metal-Organic Framework at a Nanoscale Level for Gold Nanoparticle Deposition and Catalysis. J. Mater. Chem. A 2014, 2, 20588–20596. 10.1039/C4TA04311A. [DOI] [Google Scholar]
- Ding S. M.; Tian C. C.; Zhu X.; Wang H. Z.; Wang H.; Abney C. W.; Zhang N.; Dai S. Engineering Nanoporous Organic Frameworks to Stabilize Naked Au Clusters: A Charge Modulation Approach. Chem. Commun. 2018, 54, 5058–5061. 10.1039/C8CC02966K. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Stubbs L. P.; Ho F.; Liu R.; Ship C. P.; Maguire J. A.; Hosmane N. S. Magnetic Nanocomposites: A New Perspective in Catalysis. ChemCatChem 2010, 2, 365–374. 10.1002/cctc.200900314. [DOI] [Google Scholar]
- Shylesh S.; Schünemann V.; Thiel W. R. Magnetically Separable Nanocatalysts: Bridges between Homogeneous and Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2010, 49, 3428–3459. 10.1002/anie.200905684. [DOI] [PubMed] [Google Scholar]
- Leung K. C. F.; Xuan S. H.; Zhu X. M.; Wang D. W.; Chak C. P.; Lee S. F.; Ho W. K. W.; Chung B. C. T. Gold and Iron Oxide Hybrid Nanocomposite Materials. Chem. Soc. Rev. 2012, 41, 1911–1928. 10.1039/C1CS15213K. [DOI] [PubMed] [Google Scholar]
- Yu H.; Chen M.; Rice P. M.; Wang S. X.; White R. L.; Sun S. Dumbbell-like Bifunctional Au–Fe3O4 Nanoparticles. Nano Lett. 2005, 5, 379–382. 10.1021/nl047955q. [DOI] [PubMed] [Google Scholar]
- Yin H.; Wang C.; Zhu H.; Overbury S. H.; Sun S.; Dai S. Colloidal Deposition Synthesis of Supported Gold Nanocatalysts Based on Au–Fe3O4 Dumbbell Nanoparticles. Chem. Commun. 2008, 0, 4357–4359. 10.1039/b807591c. [DOI] [PubMed] [Google Scholar]
- Wang C.; Yin H.; Dai S.; Sun S. A General Approach to Noble Metal–Metal Oxide Dumbbell Nanoparticles and Their Catalytic Application for CO Oxidation. Chem. Mater. 2010, 22, 3277–3282. 10.1021/cm100603r. [DOI] [Google Scholar]
- Lopes G.; Vargas J. M.; Sharma S. K.; Béron F.; Pirota K. R.; Knobel M.; Rettori C.; Zysler R. D. Ag–Fe3O4 Dimer Colloidal Nanoparticles: Synthesis and Enhancement of Magnetic Properties. J. Phys. Chem. C 2010, 114, 10148–10152. 10.1021/jp102311u. [DOI] [Google Scholar]
- Lin F. H.; Doong R. A. Bifunctional Au-Fe3O4 Heterostructures for Magnetically Recyclable Catalysis of Nitrophenol Reduction. J. Phys. Chem. C 2011, 115, 6591–6598. 10.1021/jp110956k. [DOI] [Google Scholar]
- Gaur S.; Johansson S.; Mohammad F.; Kumar C.; Spivey J. J. Catalytic Activity of Titania-Supported Core-Shell Fe3O4@Au NanoCatalysts for CO Oxidation. J. Phys. Chem. C 2012, 116, 22319–22326. 10.1021/jp3045725. [DOI] [Google Scholar]
- Meng X. W.; Li B.; Ren X. L.; Tan L. F.; Huang Z. B.; Tang F. Q. One-Pot Gradient Solvothermal Synthesis of Au-Fe3O4 Hybrid Nanoparticles for Magnetically Recyclable Catalytic Applications. J. Mater. Chem. A 2013, 1, 10513–10517. 10.1039/c3ta12141k. [DOI] [Google Scholar]
- Zhou S. J.; Jin W. M.; Ding Y.; Shao B.; Wang B. B.; Hu X.; Kong Y. In Situ Intercalation of Au Nanoparticles and Magnetic Gamma-Fe2O3 in the Walls of MCM-41 with Abundant Void Defects for Highly Efficient Reduction of 4-Nitrophenol and Organic Dyes. Dalton Trans 2018, 47, 16862–16875. 10.1039/C8DT03054E. [DOI] [PubMed] [Google Scholar]
- Zhang S. H.; Gai S. L.; He F.; Dai Y. L.; Gao P.; Li L.; Chen Y. J.; Yang P. P. Uniform Ni/SiO2@Au Magnetic Hollow Microspheres: Rational Design and Excellent Catalytic Performance in 4-Nitrophenol Reduction. Nanoscale 2014, 6, 7025–7032. 10.1039/C4NR00338A. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Jiang Y. Z.; Chi M. Q.; Yang Z. Z.; Nie G. D.; Lu X. F.; Wang C. Fabrication of Au Nanoparticles Supported on CoFe2O4 Nanotubes by Polyaniline Assisted Self-Assembly Strategy and Their Magnetically Recoverable Catalytic Properties. Appl. Surf. Sci. 2016, 363, 578–585. 10.1016/j.apsusc.2015.12.118. [DOI] [Google Scholar]
- Zhan W.; He Q.; Liu X.; Guo Y.; Wang Y.; Wang L.; Guo Y.; Borisevich A. Y.; Zhang J.; Lu G.; et al. A Sacrificial Coating Strategy Toward Enhancement of Metal-Support Interaction for Ultrastable Au Nanocatalysts. J. Am. Chem. Soc. 2016, 138, 16130–16139. 10.1021/jacs.6b10472. [DOI] [PubMed] [Google Scholar]
- Zhan W. C.; Shu Y.; Sheng Y. J.; Zhu H. Y.; Guo Y. L.; Wang L.; Guo Y.; Zhang J. S.; Lu G. Z.; Dai S. Surfactant-Assisted Stabilization of Au Colloids on Solids for Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2017, 56, 4494–4498. 10.1002/anie.201701191. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wang H.; Wang L.; Ali S.; Wang C. T.; Wang L. X.; Meng X. J.; Li B.; Su D. S.; Xiao F. S. Wet-Chemistry Strong Metal-Support Interactions in Titania-Supported Au Catalysts. J. Am. Chem. Soc. 2019, 141, 2975–2983. 10.1021/jacs.8b10864. [DOI] [PubMed] [Google Scholar]
- Zhang T. T.; Zhao H. Y.; He S. N.; Liu K.; Liu H. Y.; Yin Y. D.; Gao C. B. Unconventional Route to Encapsulated Ultrasmall Gold Nanoparticles for High-Temperature Catalysis. ACS Nano 2014, 8, 7297–7304. 10.1021/nn502349k. [DOI] [PubMed] [Google Scholar]
- Mielby J.; Kunov-Kruse A. J.; Kegnaes S. Decomposition of Formic Acid over Silica Encapsulated and Amine Functionalised Gold Nanoparticles. J. Catal. 2017, 345, 149–156. 10.1016/j.jcat.2016.11.020. [DOI] [Google Scholar]
- Bai L. L.; Yao L.; Yang Y. H.; Lee J. M. Microspheres with Au@SiO2 Core and Mesoporous Aluminosilica Shell as Superior Heterogeneous Catalysts for the Aerobic Epoxidation of Cis-Cyclooctene. Chem. Commun. 2015, 51, 4259–4262. 10.1039/C4CC09570G. [DOI] [PubMed] [Google Scholar]
- Pougin A.; Dodekatos G.; Dilla M.; Tuysuz H.; Strunk J. Au@TiO2 Core-Shell Composites for the Photocatalytic Reduction of CO2. Chem. - Eur. J. 2018, 24, 12416–12425. 10.1002/chem.201801796. [DOI] [PubMed] [Google Scholar]
- Arnal P. M.; Comotti M.; Schüth F. High-Temperature-Stable Catalysts by Hollow Sphere Encapsulation. Angew. Chem., Int. Ed. 2006, 45, 8224–8227. 10.1002/anie.200603507. [DOI] [PubMed] [Google Scholar]
- Galeano C.; Guttel R.; Paul M.; Arnal P.; Lu A. H.; Schuth F. Yolk-Shell Gold Nanoparticles as Model Materials for Support-Effect Studies in Heterogeneous Catalysis: Au@C and Au@ZrO2 for CO Oxidation as an Example. Chem. - Eur. J. 2011, 17, 8434–8439. 10.1002/chem.201100318. [DOI] [PubMed] [Google Scholar]
- Guttel R.; Paul M.; Schuth F. Ex-Post Size Control of High-Temperature-Stable Yolk-Shell Au@ZrO2 Catalysts. Chem. Commun. 2010, 46, 895–897. 10.1039/B921792D. [DOI] [PubMed] [Google Scholar]
- Guttel R.; Paul M.; Galeano C.; Schuth F. Au@ZrO2 Yolk-Shell Catalysts for CO Oxidation: Study of Particle Size Effect by Ex-Post Size Control of Au Cores. J. Catal. 2012, 289, 100–104. 10.1016/j.jcat.2012.01.021. [DOI] [Google Scholar]
- Lee I.; Joo J. B.; Yin Y. D.; Zaera F. A Yolk@Shell Nanoarchitecture for Au/TiO2 Catalysts. Angew. Chem., Int. Ed. 2011, 50, 10208–10211. 10.1002/anie.201007660. [DOI] [PubMed] [Google Scholar]
- Lee I.; Joo J. B.; Yin Y.; Zaera F. Au@Void@TiO2 Yolk-Shell Nanostructures as Catalysts for the Promotion of Oxidation Reactions at Cryogenic Temperatures. Surf. Sci. 2016, 648, 150–155. 10.1016/j.susc.2015.10.008. [DOI] [Google Scholar]
- Lee J.; Park J. C.; Song H. A Nanoreactor Framework of a Au@SiO2 Yolk/Shell Structure for Catalytic Reduction of p-Nitrophenol. Adv. Mater. 2008, 20, 1523–1528. 10.1002/adma.200702338. [DOI] [Google Scholar]
- Wang S. N.; Zhang M. C.; Zhang W. Q. Yolk-Shell Catalyst of Single Au Nanoparticle Encapsulated within Hollow Mesoporous Silica Microspheres. ACS Catal. 2011, 1, 207–211. 10.1021/cs1000762. [DOI] [Google Scholar]
- Yu Y.; Cao C. Y.; Chen Z.; Liu H.; Li P.; Dou Z. F.; Song W. G. Au Nanoparticles Embedded into the Inner Wall of TiO2 Hollow Spheres as a Nanoreactor with Superb Thermal Stability. Chem. Commun. 2013, 49, 3116–3118. 10.1039/c3cc39212k. [DOI] [PubMed] [Google Scholar]
- Li J.; Song S. Y.; Long Y.; Wu L. L.; Wang X.; Xing Y.; Jin R. C.; Liu X. G.; Zhang H. J. Investigating the Hybrid-Structure-Effect of CeO2-Encapsulated Au Nanostructures on the Transfer Coupling of Nitrobenzene. Adv. Mater. 2018, 30, 1704416. 10.1002/adma.201704416. [DOI] [PubMed] [Google Scholar]
- Liu B. C.; Yu S. L.; Wang Q.; Hu W. T.; Jing P.; Liu Y.; Jia W. J.; Liu Y. X.; Liu L. X.; Zhang J. Hollow Mesoporous Ceria Nanoreactors with Enhanced Activity and Stability for Catalytic Application. Chem. Commun. 2013, 49, 3757–3759. 10.1039/c3cc40665b. [DOI] [PubMed] [Google Scholar]
- Jin Z.; Wang F.; Wang J. X.; Yu J. C.; Wang J. F. Metal Nanocrystal-Embedded Hollow Mesoporous TiO2 and ZrO2 Microspheres Prepared with Polystyrene Nanospheres as Carriers and Templates. Adv. Funct. Mater. 2013, 23, 2137–2144. 10.1002/adfm.201202600. [DOI] [Google Scholar]
- Haruta M. Spiers Memorial Lecture Role of Perimeter Interfaces in Catalysis by Gold Nanoparticles. Faraday Discuss. 2011, 152, 11–32. 10.1039/c1fd00107h. [DOI] [PubMed] [Google Scholar]
- Carrasquillo-Flores R.; Ro I.; Kumbhalkar M. D.; Burt S.; Carrero C. A.; Alba-Rubio A. C.; Miller J. T.; Hermans I.; Huber G. W.; Dumesic J. A. Reverse Water-Gas Shift on Interfacial Sites Formed by Deposition of Oxidized Molybdenum Moieties onto Gold Nanoparticles. J. Am. Chem. Soc. 2015, 137, 10317–10325. 10.1021/jacs.5b05945. [DOI] [PubMed] [Google Scholar]
- Meyer R.; Lemire C.; Shaikhutdinov Sh. K.; Freund H.-J. Surface Chemistry of Catalysis by Gold. Gold Bull. 2004, 37, 72–124. 10.1007/BF03215519. [DOI] [Google Scholar]
- Kung M. C.; Davis R. J.; Kung H. H. Understanding Au-Catalyzed Low-Temperature CO Oxidation. J. Phys. Chem. C 2007, 111, 11767–11775. 10.1021/jp072102i. [DOI] [Google Scholar]
- Villa A.; Dimitratos N.; Chan-Thaw C. E.; Hammond C.; Veith G. M.; Wang D.; Manzoli M.; Prati L.; Hutchings G. J. Characterisation of Gold Catalysts. Chem. Soc. Rev. 2016, 45, 4953–4994. 10.1039/C5CS00350D. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Koga H.; Okumura M.; Haruta M. Advances in Gold Catalysis and Understanding the Catalytic Mechanism. Chem. Rec. 2016, 16, 2278–2293. 10.1002/tcr.201600046. [DOI] [PubMed] [Google Scholar]
- Sun K. Theoretical Investigations on CO Oxidation Reaction Catalyzed by Gold Nanoparticles. Chin. J. Catal. 2016, 37, 1608–1618. 10.1016/S1872-2067(16)62476-2. [DOI] [Google Scholar]
- Frost J. C. Junction Effect Interactions in Methanol Synthesis Catalysts. Nature 1988, 334, 577–580. 10.1038/334577a0. [DOI] [Google Scholar]
- Schubert M. M.; Hackenberg S.; van Veen A. C.; Muhler M.; Plzak V.; Behm R. J. CO Oxidation over Supported Gold Catalysts—“Inert” and “Active” Support Materials and Their Role for the Oxygen Supply during Reaction. J. Catal. 2001, 197, 113–122. 10.1006/jcat.2000.3069. [DOI] [Google Scholar]
- Comotti M.; Li W.-C.; Spliethoff B.; Schüth F. Support Effect in High Activity Gold Catalysts for CO Oxidation. J. Am. Chem. Soc. 2006, 128, 917–924. 10.1021/ja0561441. [DOI] [PubMed] [Google Scholar]
- Ruggiero C.; Hollins P. Adsorption of Carbon Monoxide on the Gold(332) Surface. J. Chem. Soc., Faraday Trans. 1996, 92, 4829–4834. 10.1039/ft9969204829. [DOI] [Google Scholar]
- Iizuka Y.; Fujiki H.; Yamauchi N.; Chijiiwa T.; Arai S.; Tsubota S.; Haruta M. Adsorption of CO on Gold Supported on TiO2. Catal. Today 1997, 36, 115–123. 10.1016/S0920-5861(96)00204-0. [DOI] [Google Scholar]
- Canning N. D. S.; Outka D.; Madix R. J. The Adsorption of Oxygen on Gold. Surf. Sci. 1984, 141, 240–254. 10.1016/0039-6028(84)90209-7. [DOI] [Google Scholar]
- Bondzie V. A.; Parker S. C.; Campbell C. T. Oxygen Adsorption on Well-Defined Gold Particles on TiO2(110). J. Vac. Sci. Technol., A 1999, 17, 1717–1720. 10.1116/1.581879. [DOI] [Google Scholar]
- Parker D. H.; Koel B. E. Chemisorption of High Coverages of Atomic Oxygen on the Pt(111), Pd(111), and Au(111) Surfaces. J. Vac. Sci. Technol., A 1990, 8, 2585–2590. 10.1116/1.576675. [DOI] [Google Scholar]
- Hammer B.; Norskov J. K. Why Gold Is the Noblest of All the Metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Roldán A.; Ricart J. M.; Illas F. Origin of the Size Dependence of Au Nanoparticles toward Molecular Oxygen Dissociation. Theor. Chem. Acc. 2011, 128, 675–681. 10.1007/s00214-010-0806-7. [DOI] [Google Scholar]
- Bond G. C.; Thompson D. T. Gold-Catalysed Oxidation of Carbon Monoxide. Gold Bull. 2000, 33, 41–50. 10.1007/BF03216579. [DOI] [Google Scholar]
- Cunningham D. A. H.; Vogel W.; Kageyama H.; Tsubota S.; Haruta M. The Relationship between the Structure and Activity of Nanometer Size Gold When Supported on Mg(OH)2. J. Catal. 1998, 177, 1–10. 10.1006/jcat.1998.2050. [DOI] [Google Scholar]
- Okumura M.; Nakamura S.; Tsubota S.; Nakamura T.; Azuma M.; Haruta M. Chemical Vapor Deposition of Gold on Al2O3, SiO2, and TiO2 for the Oxidation of CO and of H2. Catal. Lett. 1998, 51 (1), 53–58. 10.1023/A:1019020614336. [DOI] [Google Scholar]
- Grunwaldt J.-D.; Maciejewski M.; Becker O. S.; Fabrizioli P.; Baiker A. Comparative Study of Au/TiO2 and Au/ZrO2 Catalysts for Low-Temperature CO Oxidation. J. Catal. 1999, 186, 458–469. 10.1006/jcat.1999.2564. [DOI] [Google Scholar]
- Mavrikakis M.; Stoltze P.; Nørskov J. K. Making Gold Less Noble. Catal. Lett. 2000, 64, 101–106. 10.1023/A:1019028229377. [DOI] [Google Scholar]
- Roldán A.; González S.; Ricart J. M.; Illas F. Critical Size for O 2 Dissociation by Au Nanoparticles. ChemPhysChem 2009, 10, 348–351. 10.1002/cphc.200800702. [DOI] [PubMed] [Google Scholar]
- Valden M.; Pak S.; Lai X.; Goodman D. W. Structure Sensitivity of CO Oxidation over Model Au/TiO2 Catalysts. Catal. Lett. 1998, 56, 7–10. 10.1023/A:1019028205985. [DOI] [Google Scholar]
- Kipnis M. Gold in CO Oxidation and PROX: The Role of Reaction Exothermicity and Nanometer-Scale Particle Size. Appl. Catal., B 2014, 152–153, 38–45. 10.1016/j.apcatb.2014.01.030. [DOI] [Google Scholar]
- Zheng X.; Veith G. M.; Redekop E.; Lo C. S.; Yablonsky G. S.; Gleaves J. T. Oxygen and CO Adsorption on Au/SiO2 Catalysts Prepared by Magnetron Sputtering: The Role of Oxygen Storage. Ind. Eng. Chem. Res. 2010, 49, 10428–10437. 10.1021/ie100547f. [DOI] [Google Scholar]
- Jia C.-J.; Liu Y.; Bongard H.; Schüth F. Very Low Temperature CO Oxidation over Colloidally Deposited Gold Nanoparticles on Mg(OH)2and MgO. J. Am. Chem. Soc. 2010, 132, 1520–1522. 10.1021/ja909351e. [DOI] [PubMed] [Google Scholar]
- Duan Z.; Henkelman G. O2 Activation at the Au/MgO(001) Interface Boundary Facilitates CO Oxidation. Phys. Chem. Chem. Phys. 2016, 18, 5486–5490. 10.1039/C5CP05558J. [DOI] [PubMed] [Google Scholar]
- Lopez N. On the Origin of the Catalytic Activity of Gold Nanoparticles for Low-Temperature CO Oxidation. J. Catal. 2004, 223, 232–235. 10.1016/j.jcat.2004.01.001. [DOI] [Google Scholar]
- Buckeridge J.; Butler K. T.; Catlow C. R. A.; Logsdail A. J.; Scanlon D. O.; Shevlin S. A.; Woodley S. M.; Sokol A. A.; Walsh A. Polymorph Engineering of TiO2 : Demonstrating How Absolute Reference Potentials Are Determined by Local Coordination. Chem. Mater. 2015, 27, 3844–3851. 10.1021/acs.chemmater.5b00230. [DOI] [Google Scholar]
- Martra G. Lewis Acid and Base Sites at the Surface of Microcrystalline TiO2 Anatase: Relationships between Surface Morphology and Chemical Behaviour. Appl. Catal., A 2000, 200, 275–285. 10.1016/S0926-860X(00)00641-4. [DOI] [Google Scholar]
- Chenakin S.; Kruse N. Combining XPS and ToF-SIMS for Assessing the CO Oxidation Activity of Au/TiO2 Catalysts. J. Catal. 2018, 358, 224–236. 10.1016/j.jcat.2017.12.010. [DOI] [Google Scholar]
- Overbury S.; Schwartz V.; Mullins D.; Yan W.; Dai S. Evaluation of the Au Size Effect: CO Oxidation Catalyzed by Au/TiO2. J. Catal. 2006, 241, 56–65. 10.1016/j.jcat.2006.04.018. [DOI] [Google Scholar]
- Saqlain M. A.; Hussain A.; Siddiq M.; Ferreira A. R.; Leitao A. A. Thermally Activated Surface Oxygen Defects at the Perimeter of Au/TiO2: A DFT+U Study. Phys. Chem. Chem. Phys. 2015, 17, 25403–25410. 10.1039/C5CP04113A. [DOI] [PubMed] [Google Scholar]
- Tafreshi S. S.; Roldan A.; de Leeuw N. H. Micro-Kinetic Simulations of the Catalytic Decomposition of Hydrazine on the Cu(111) Surface. Faraday Discuss. 2017, 197, 41–57. 10.1039/C6FD00186F. [DOI] [PubMed] [Google Scholar]
- Roldán A.; Novell G.; Ricart J. M.; Illas F. Theoretical Simulation of Temperature Programmed Desorption of Molecular Oxygen on Isolated Au Nanoparticles from Density Functional Calculations and Microkinetics Models. J. Phys. Chem. C 2010, 114, 5101–5106. 10.1021/jp911283j. [DOI] [Google Scholar]
- Saqlain M. A.; Hussain A.; Siddiq M.; Leitão A. A. A DFT+U Study of the Mars Van Krevelen Mechanism of CO Oxidation on Au/TiO2 Catalysts. Appl. Catal., A 2016, 519, 27–33. 10.1016/j.apcata.2016.03.021. [DOI] [Google Scholar]
- Hinojosa-Reyes M.; Camposeco-Solis R.; Zanella R.; Rodríguez-González V.; Ruiz F. Gold Nanoparticle: Enhanced CO Oxidation at Low Temperatures by Using Fe-Doped TiO2 as Support. Catal. Lett. 2018, 148, 383–396. 10.1007/s10562-017-2260-9. [DOI] [Google Scholar]
- Widmann D.; Krautsieder A.; Walter P.; Brückner A.; Behm R. J. How Temperature Affects the Mechanism of CO Oxidation on Au/TiO2 : A Combined EPR and TAP Reactor Study of the Reactive Removal of TiO 2 Surface Lattice Oxygen in Au/TiO2 by CO. ACS Catal. 2016, 6, 5005–5011. 10.1021/acscatal.6b01219. [DOI] [Google Scholar]
- Widmann D.; Behm R. J. Active Oxygen on a Au/TiO2 Catalyst: Formation, Stability, and CO Oxidation Activity. Angew. Chem., Int. Ed. 2011, 50, 10241–10245. 10.1002/anie.201102062. [DOI] [PubMed] [Google Scholar]
- Widmann D.; Behm R. J. Activation of Molecular Oxygen and the Nature of the Active Oxygen Species for CO Oxidation on Oxide Supported Au Catalysts. Acc. Chem. Res. 2014, 47, 740–749. 10.1021/ar400203e. [DOI] [PubMed] [Google Scholar]
- Yao Q.; Wang C.; Wang H.; Yan H.; Lu J. Revisiting the Au Particle Size Effect on TiO 2 -Coated Au/TiO2 Catalysts in CO Oxidation Reaction. J. Phys. Chem. C 2016, 120, 9174–9183. 10.1021/acs.jpcc.5b12712. [DOI] [Google Scholar]
- Wang C.; Wang H.; Yao Q.; Yan H.; Li J.; Lu J. Precisely Applying TiO2 Overcoat on Supported Au Catalysts Using Atomic Layer Deposition for Understanding the Reaction Mechanism and Improved Activity in CO Oxidation. J. Phys. Chem. C 2016, 120, 478–486. 10.1021/acs.jpcc.5b11047. [DOI] [Google Scholar]
- Fujita T.; Horikawa M.; Takei T.; Murayama T.; Haruta M. Correlation between Catalytic Activity of Supported Gold Catalysts for Carbon Monoxide Oxidation and Metal–Oxygen Binding Energy of the Support Metal Oxides. Chin. J. Catal. 2016, 37, 1651–1655. 10.1016/S1872-2067(16)62521-4. [DOI] [Google Scholar]
- Xie X. W.; Li Y.; Liu Z. Q.; Haruta M.; Shen W. J. Low-Temperature Oxidation of CO Catalysed by Co3O4 Nanorods. Nature 2009, 458, 746–749. 10.1038/nature07877. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; McEntee M.; Neurock M.; Yates J. T. Inhibition at Perimeter Sites of Au/TiO2 Oxidation Catalyst by Reactant Oxygen. J. Am. Chem. Soc. 2012, 134, 12717–12723. 10.1021/ja304426b. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; Neurock M.; Yates J. T. Insights into Catalytic Oxidation at the Au/TiO 2 Dual Perimeter Sites. Acc. Chem. Res. 2014, 47, 805–815. 10.1021/ar400196f. [DOI] [PubMed] [Google Scholar]
- Green I. X.; Tang W.; Neurock M.; Yates J. T. Spectroscopic Observation of Dual Catalytic Sites During Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. 10.1126/science.1207272. [DOI] [PubMed] [Google Scholar]
- Boronat M.; Concepción P.; Corma A. Unravelling the Nature of Gold Surface Sites by Combining IR Spectroscopy and DFT Calculations. Implications in Catalysis. J. Phys. Chem. C 2009, 113, 16772–16784. 10.1021/jp905157r. [DOI] [Google Scholar]
- Kast P.; Kučerová G.; Behm R. J. On the Nature of the Active Au Species: CO Oxidation on Cyanide Leached Au/TiO2 Catalysts. Catal. Today 2015, 244, 146–160. 10.1016/j.cattod.2014.06.021. [DOI] [Google Scholar]
- Hutchings G.; Hall M.; Carley A.; Landon P.; Solsona B.; Kiely C.; Herzing A.; Makkee M.; Moulijn J.; Overweg A. Role of Gold Cations in the Oxidation of Carbon Monoxide Catalyzed by Iron Oxide-Supported Gold. J. Catal. 2006, 242, 71–81. 10.1016/j.jcat.2006.06.001. [DOI] [Google Scholar]
- Liu Y.; Jia C. J.; Yamasaki J.; Terasaki O.; Schuth F. Highly Active Iron Oxide Supported Gold Catalysts for CO Oxidation: How Small Must the Gold Nanoparticles Be?. Angew. Chem., Int. Ed. 2010, 49, 5771–5775. 10.1002/anie.201000452. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Gu D.; Jin Z.; Du P.-P.; Si R.; Tao J.; Xu W.-Q.; Huang Y.-Y.; Senanayake S.; Song Q.-S.; et al. Uniform 2 Nm Gold Nanoparticles Supported on Iron Oxides as Active Catalysts for CO Oxidation Reaction: Structure–Activity Relationship. Nanoscale 2015, 7, 4920–4928. 10.1039/C4NR06967F. [DOI] [PubMed] [Google Scholar]
- Cui H.-Z.; Guo Y.; Wang X.; Jia C.-J.; Si R. Gold-Iron Oxide Catalyst for CO Oxidation: Effect of Support Structure. Catalysts 2016, 6, 37–50. 10.3390/catal6030037. [DOI] [Google Scholar]
- Howard K. L.; Willock D. J. A Periodic DFT Study of the Activation of O2 by Au Nanoparticles on α-Fe2O3. Faraday Discuss. 2011, 152, 135–151. 10.1039/c1fd00026h. [DOI] [PubMed] [Google Scholar]
- Hoh S. W.; Thomas L.; Jones G.; Willock D. J. A Density Functional Study of Oxygen Vacancy Formation on α-Fe2O3(0001) Surface and the Effect of Supported Au Nanoparticles. Res. Chem. Intermed. 2015, 41, 9587–9601. 10.1007/s11164-015-1984-7. [DOI] [Google Scholar]
- Rudra S.; Nayak A. K.; Koley S.; Chakraborty R.; Maji P. K.; Pradhan M. Redox-Mediated Shape Transformation of Fe3O4 Nanoflakes to Chemically Stable Au-Fe2O3 Composite Nanorods for a High-Performance Asymmetric Solid-State Supercapacitor Device. ACS Sustainable Chem. Eng. 2019, 7, 724–733. 10.1021/acssuschemeng.8b04300. [DOI] [Google Scholar]
- Kaneti Y. V.; Tanaka S.; Jikihara Y.; Nakayama T.; Bando Y.; Haruta M.; Hossain Md. S. A.; Golberg D.; Yamauchi Y. Room Temperature Carbon Monoxide Oxidation Based on Two-Dimensional Gold-Loaded Mesoporous Iron Oxide Nanoflakes. Chem. Commun. 2018, 54, 8514–8517. 10.1039/C8CC03639J. [DOI] [PubMed] [Google Scholar]
- Zhao K.; Tang H.; Qiao B.; Li L.; Wang J. High Activity of Au/γ-Fe2O3 for CO Oxidation: Effect of Support Crystal Phase in Catalyst Design. ACS Catal. 2015, 5, 3528–3539. 10.1021/cs5020496. [DOI] [Google Scholar]
- Montini T.; Melchionna M.; Monai M.; Fornasiero P. Fundamentals and Catalytic Applications of CeO2 -Based Materials. Chem. Rev. 2016, 116, 5987–6041. 10.1021/acs.chemrev.5b00603. [DOI] [PubMed] [Google Scholar]
- Chen S.; Luo L.; Jiang Z.; Huang W. Size-Dependent Reaction Pathways of Low-Temperature CO Oxidation on Au/CeO2 Catalysts. ACS Catal. 2015, 5, 1653–1662. 10.1021/cs502067x. [DOI] [Google Scholar]
- Abd El-Moemen A.; Abdel-Mageed A. M.; Bansmann J.; Parlinska-Wojtan M.; Behm R. J.; Kučerová G. Deactivation of Au/CeO2 Catalysts during CO Oxidation: Influence of Pretreatment and Reaction Conditions. J. Catal. 2016, 341, 160–179. 10.1016/j.jcat.2016.07.005. [DOI] [Google Scholar]
- Bansmann J.; Kučerová G.; Abdel-Mageed A. M.; Abd El-Moemen A.; Behm R. J. Influence of Re-Activation and Ongoing CO Oxidation Reaction on the Chemical and Electronic Properties of Au on a Au/CeO2 Catalyst: A XANES Study at the Au L III Edge. J. Electron Spectrosc. Relat. Phenom. 2017, 220, 86–90. 10.1016/j.elspec.2017.01.002. [DOI] [Google Scholar]
- Mai H.-X.; Sun L.-D.; Zhang Y.-W.; Si R.; Feng W.; Zhang H.-P.; Liu H.-C.; Yan C.-H. Shape-Selective Synthesis and Oxygen Storage Behavior of Ceria Nanopolyhedra, Nanorods, and Nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. 10.1021/jp055584b. [DOI] [PubMed] [Google Scholar]
- Wang C. Y.; Yang M.; Flytzani-Stephanopoulos M. Single Gold Atoms Stabilized on Nanoscale Metal Oxide Supports Are Catalytic Active Centers for Various Reactions. AIChE J. 2016, 62, 429–439. 10.1002/aic.15134. [DOI] [Google Scholar]
- Guo L.-W.; Du P.-P.; Fu X.-P.; Ma C.; Zeng J.; Si R.; Huang Y.-Y.; Jia C.-J.; Zhang Y.-W.; Yan C.-H. Contributions of Distinct Gold Species to Catalytic Reactivity for Carbon Monoxide Oxidation. Nat. Commun. 2016, 7, 13481. 10.1038/ncomms13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco M.; Fernandez-Garcia S.; Lopez-Haro M.; Hungria A. B.; Chen X.; Blanco G.; Perez-Omil J. A.; Collins S. E.; Okuno H.; Calvino J. J. Critical Influence of Nanofaceting on the Preparation and Performance of Supported Gold Catalysts. ACS Catal. 2015, 5, 3504–3513. 10.1021/acscatal.5b00086. [DOI] [Google Scholar]
- Fernández-García S.; Collins S. E.; Tinoco M.; Hungría A. B.; Calvino J. J.; Cauqui M. A.; Chen X. Influence of {111} Nanofaceting on the Dynamics of CO Adsorption and Oxidation over Au Supported on CeO2 Nanocubes: An Operando DRIFT Insight. Catal. Today 2019, 336, 90–98. 10.1016/j.cattod.2019.01.078. [DOI] [Google Scholar]
- Wang Y.-G.; Mei D.; Glezakou V.-A.; Li J.; Rousseau R. Dynamic Formation of Single-Atom Catalytic Active Sites on Ceria-Supported Gold Nanoparticles. Nat. Commun. 2015, 6 (1), 6511. 10.1038/ncomms7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J.; Francis S.; Roldan A. The Influence of Support Materials on the Structural and Electronic Properties of Gold Nanoparticles – a DFT Study. Phys. Chem. Chem. Phys. 2019, 21, 19011–19025. 10.1039/C9CP03066B. [DOI] [PubMed] [Google Scholar]
- Wang Y.-G.; Cantu D. C.; Lee M.-S.; Li J.; Glezakou V.-A.; Rousseau R. CO Oxidation on Au/TiO2: Condition-Dependent Active Sites and Mechanistic Pathways. J. Am. Chem. Soc. 2016, 138, 10467–10476. 10.1021/jacs.6b04187. [DOI] [PubMed] [Google Scholar]
- Gu D.; Tseng J. C.; Weidenthaler C.; Bongard H. J.; Spliethoff B.; Schmidt W.; Soulimani F.; Weckhuysen B. M.; Schuth F. Gold on Different Manganese Oxides: Ultra-Low-Temperature CO Oxidation over Colloidal Gold Supported on Bulk-MnO2 Nanomaterials. J. Am. Chem. Soc. 2016, 138, 9572–9580. 10.1021/jacs.6b04251. [DOI] [PubMed] [Google Scholar]
- Wang L.; Huang X.; Liu Q.; Liu Y.; Cao Y.; He H.; Fan K.; Zhuang J. Gold Nanoparticles Deposited on Manganese(III) Oxide as Novel Efficient Catalyst for Low Temperature CO Oxidation. J. Catal. 2008, 259, 66–74. 10.1016/j.jcat.2008.07.010. [DOI] [Google Scholar]
- Li G.; Jin R. Atomically Precise Gold Nanoclusters as New Model Catalysts. Acc. Chem. Res. 2013, 46, 1749–1758. 10.1021/ar300213z. [DOI] [PubMed] [Google Scholar]
- Du Y.; Sheng H.; Astruc D.; Zhu M. Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 2020, 120 (2), 526–622. 10.1021/acs.chemrev.8b00726. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Jiang D.; Mann A. K. P.; Mullins D. R.; Qiao Z.-A.; Allard L. F.; Zeng C.; Jin R.; Overbury S. H. Thiolate Ligands as a Double-Edged Sword for CO Oxidation on CeO2 Supported Au25(SCH2CH2Ph)18 Nanoclusters. J. Am. Chem. Soc. 2014, 136, 6111–6122. 10.1021/ja5018706. [DOI] [PubMed] [Google Scholar]
- Nie X. T.; Qian H. F.; Ge Q. J.; Xu H. Y.; Jin R. C. CO Oxidation Catalyzed by Oxide-Supported Au-25(SR)(18) Nanoclusters and Identification of Perimeter Sites as Active Centers. ACS Nano 2012, 6, 6014–6022. 10.1021/nn301019f. [DOI] [PubMed] [Google Scholar]
- Li W.; Ge Q.; Ma X.; Chen Y.; Zhu M.; Xu H.; Jin R. Mild activation of CeO2-supported gold nanoclusters and insight into the catalytic behavior in CO oxidation. Nanoscale 2016, 8 (4), 2378–2385. 10.1039/C5NR07498C. [DOI] [PubMed] [Google Scholar]
- Taketoshi A.; Haruta M. Size-and Structure-Specificity in Catalysis by Gold Clusters. Chem. Lett. 2014, 43, 380–387. 10.1246/cl.131232. [DOI] [Google Scholar]
- Scurrell M. S. Thoughts on the Use of Gold-Based Catalysts in Environmental Protection Catalysis. Gold Bull. 2017, 50, 77–84. 10.1007/s13404-017-0194-z. [DOI] [Google Scholar]
- Kuwauchi Y.; Takeda S.; Yoshida H.; Sun K.; Haruta M.; Kohno H. Stepwise Displacement of Catalytically Active Gold Nanoparticles on Cerium Oxide. Nano Lett. 2013, 13, 3073–3077. 10.1021/nl400919c. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Che R.; Elzatahry A. A.; Zhao D. Direct Imaging Au Nanoparticle Migration Inside Mesoporous Silica Channels. ACS Nano 2014, 8, 10455–10460. 10.1021/nn503794v. [DOI] [PubMed] [Google Scholar]
- Pérez-Cabero M.; El Haskouri J.; Solsona B.; Vázquez I.; Dejoz A.; García T.; Álvarez-Rodríguez J.; Beltrán A.; Beltrán D.; Amorós P. Stable Anchoring of Dispersed Gold Nanoparticles on Hierarchic Porous Silica-Based Materials. J. Mater. Chem. 2010, 20, 6780. 10.1039/c0jm00421a. [DOI] [Google Scholar]
- Moragues A.; Puertolas B.; Mayoral A.; Arenal R.; Hungria A. B.; Murcia-Mascaros S.; Taylor S. H.; Solsona B.; Garcia T.; Amoros P. Understanding the Role of Ti-Rich Domains in the Stabilization of Gold Nanoparticles on Mesoporous Silica-Based Catalysts. J. Catal. 2018, 360, 187–200. 10.1016/j.jcat.2018.02.003. [DOI] [Google Scholar]
- Puértolas B.; Mayoral Á.; Arenal R.; Solsona B.; Moragues A.; Murcia-Mascaros S.; Amorós P.; Hungría A. B.; Taylor S. H.; García T. High-Temperature Stable Gold Nanoparticle Catalysts for Application under Severe Conditions: The Role of TiO2 Nanodomains in Structure and Activity. ACS Catal. 2015, 5, 1078–1086. 10.1021/cs501741u. [DOI] [Google Scholar]
- Kučerová G.; Strunk J.; Muhler M.; Behm R. J. Effect of Titania Surface Modification of Mesoporous Silica SBA-15 Supported Au Catalysts: Activity and Stability in the CO Oxidation Reaction. J. Catal. 2017, 356, 214–228. 10.1016/j.jcat.2017.09.017. [DOI] [Google Scholar]
- Mei B.; Becerikli A.; Pougin A.; Heeskens D.; Sinev I.; Grünert W.; Muhler M.; Strunk J. Tuning the Acid/Base and Structural Properties of Titanate-Loaded Mesoporous Silica by Grafting of Zinc Oxide. J. Phys. Chem. C 2012, 116, 14318–14327. 10.1021/jp301908c. [DOI] [Google Scholar]
- Pandey A. D.; Güttel R.; Leoni M.; Schüth F.; Weidenthaler C. Influence of the Microstructure of Gold–Zirconia Yolk–Shell Catalysts on the CO Oxidation Activity. J. Phys. Chem. C 2010, 114, 19386–19394. 10.1021/jp106436h. [DOI] [Google Scholar]
- Chen C.; Shi M.; Cargnello M.; Fornasiero P.; Murray C. B.; Gorte R. J. Au@TiO2 Core–Shell Nanostructures with High Thermal Stability. Catal. Lett. 2014, 144, 1939–1945. 10.1007/s10562-014-1351-0. [DOI] [Google Scholar]
- Raphulu M. C.; Scurrell M. S. Cyanide Leaching of Gold Catalysts. Catal. Commun. 2015, 67, 87–89. 10.1016/j.catcom.2015.04.011. [DOI] [Google Scholar]
- Fujitani T.; Nakamura I.; Haruta M. Role of Water in CO Oxidation on Gold Catalysts. Catal. Lett. 2014, 144, 1475–1486. 10.1007/s10562-014-1325-2. [DOI] [Google Scholar]
- Ojeda M.; Zhan B.-Z.; Iglesia E. Mechanistic Interpretation of CO Oxidation Turnover Rates on Supported Au Clusters. J. Catal. 2012, 285, 92–102. 10.1016/j.jcat.2011.09.015. [DOI] [Google Scholar]
- Karwacki C. J.; Ganesh P.; Kent P. R. C.; Gordon W. O.; Peterson G. W.; Niu J. J.; Gogotsi Y. Structure–Activity Relationship of Au/ZrO2 Catalyst on Formation of Hydroxyl Groups and Its Influence on CO Oxidation. J. Mater. Chem. A 2013, 1, 6051–6062. 10.1039/c3ta00081h. [DOI] [Google Scholar]
- Saavedra J.; Doan H. A.; Pursell C. J.; Grabow L. C.; Chandler B. D. The Critical Role of Water at the Gold-Titania Interface in Catalytic CO Oxidation. Science 2014, 345, 1599–1602. 10.1126/science.1256018. [DOI] [PubMed] [Google Scholar]
- Tran-Thuy T. M.; Chen C. C.; Lin S. D. Spectroscopic Studies of How Moisture Enhances CO Oxidation over Au/BN at Ambient Temperature. ACS Catal. 2017, 7, 4304–4312. 10.1021/acscatal.7b01374. [DOI] [Google Scholar]
- Fu S.-E.; Yeh C.-H.; Lin P.-J.; Nachimuthu S.; Jiang J.-C. A First Principles Study of CO Oxidation over Gold Clusters: The Catalytic Role of Boron Nitride Support and Water. Mol. Catal. 2019, 471, 44–53. 10.1016/j.mcat.2019.04.015. [DOI] [Google Scholar]
- Mallat T.; Baiker A. Oxidation of Alcohols with Molecular Oxygen on Solid Catalysts. Chem. Rev. 2004, 104, 3037–3058. 10.1021/cr0200116. [DOI] [PubMed] [Google Scholar]
- Kaneda K.; Mitsudome T. Metal–Support Cooperative Catalysts for Environmentally Benign Molecular Transformations. Chem. Rec. 2017, 17, 4–26. 10.1002/tcr.201600036. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lang X.; Zhaorigetu B.; Jia M.; Wang J.; Guo X.; Zhao J. Aerobic Oxidation of Alcohols on Au Nanocatalyst: Insight to the Roles of the Ni-Al Layered Double Hydroxides Support. ChemCatChem 2014, 6, 1737–1747. 10.1002/cctc.201400046. [DOI] [Google Scholar]
- Wang S.; Yin S.; Chen G.; Li L.; Zhang H. Nearly Atomic Precise Gold Nanoclusters on Nickel-Based Layered Double Hydroxides for Extraordinarily Efficient Aerobic Oxidation of Alcohols. Catal. Sci. Technol. 2016, 6, 4090–4104. 10.1039/C6CY00186F. [DOI] [Google Scholar]
- Li J.; Xu Y.; Wang S.; Zhang H. Ultrafine AuPd Nanoclusters on Layered Double Hydroxides by the Capt-Capped AuPd Cluster Precursor Method: Synergistic Effect for Highly Efficient Aerobic Oxidation of Alcohols. J. Phys. Chem. C 2019, 123, 15483–15494. 10.1021/acs.jpcc.9b00893. [DOI] [Google Scholar]
- Liu P.; Guan Y.; van Santen R. A.; Li C.; Hensen E. J. M. Aerobic Oxidation of Alcohols over Hydrotalcite-Supported Gold Nanoparticles: The Promotional Effect of Transition Metal Cations. Chem. Commun. 2011, 47, 11540–11542. 10.1039/c1cc15148g. [DOI] [PubMed] [Google Scholar]
- Liu P.; Degirmenci V.; Hensen E. J. M. Unraveling the Synergy between Gold Nanoparticles and Chromium-Hydrotalcites in Aerobic Oxidation of Alcohols. J. Catal. 2014, 313, 80–91. 10.1016/j.jcat.2014.03.001. [DOI] [Google Scholar]
- Santra C.; Auroux A.; Chowdhury B. Bi Doped CeO2 Oxide Supported Gold Nanoparticle Catalysts for the Aerobic Oxidation of Alcohols. RSC Adv. 2016, 6, 45330–45342. 10.1039/C6RA05216A. [DOI] [Google Scholar]
- Santra C.; Pramanik M.; Bando K. K.; Maity S.; Chowdhury B. Gold Nanoparticles on Mesoporous Cerium-Tin Mixed Oxide for Aerobic Oxidation of Benzyl Alcohol. J. Mol. Catal. A: Chem. 2016, 418–419, 41–53. 10.1016/j.molcata.2016.03.026. [DOI] [Google Scholar]
- Corma A.; Iborra S.; Velty A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. 10.1021/cr050989d. [DOI] [PubMed] [Google Scholar]
- Zhou C.-H. C.; Beltramini J. N.; Fan Y.-X.; Lu G. Q. M. Chemoselective Catalytic Conversion of Glycerol as a Biorenewable Source to Valuable Commodity Chemicals. Chem. Soc. Rev. 2008, 37, 527–549. 10.1039/B707343G. [DOI] [PubMed] [Google Scholar]
- Villa A.; Veith G. M.; Prati L. Selective Oxidation of Glycerol under Acidic Conditions Using Gold Catalysts. Angew. Chem. 2010, 122, 4601–4604. 10.1002/ange.201000762. [DOI] [PubMed] [Google Scholar]
- Brett G. L.; He Q.; Hammond C.; Miedziak P. J.; Dimitratos N.; Sankar M.; Herzing A. A.; Conte M.; Lopez-Sanchez J. A.; Kiely C. J.; et al. Selective Oxidation of Glycerol by Highly Active Bimetallic Catalysts at Ambient Temperature under Base-Free Conditions. Angew. Chem., Int. Ed. 2011, 50, 10136–10139. 10.1002/anie.201101772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N. K.; Nishimura S.; Takagaki A.; Ebitani K. Hydrotalcite-Supported Gold-Nanoparticle-Catalyzed Highly Efficient Base-Free Aqueous Oxidation of 5-Hydroxymethylfurfural into 2,5-Furandicarboxylic Acid under Atmospheric Oxygen Pressure. Green Chem. 2011, 13, 824–827. 10.1039/c0gc00911c. [DOI] [Google Scholar]
- Tongsakul D.; Nishimura S.; Ebitani K. Platinum/Gold Alloy Nanoparticles-Supported Hydrotalcite Catalyst for Selective Aerobic Oxidation of Polyols in Base-Free Aqueous Solution at Room Temperature. ACS Catal. 2013, 3, 2199–2207. 10.1021/cs400458k. [DOI] [Google Scholar]
- Takagaki A.; Tsuji A.; Nishimura S.; Ebitani K. Genesis of Catalytically Active Gold Nanoparticles Supported on Hydrotalcite for Base-Free Selective Oxidation of Glycerol in Water with Molecular Oxygen. Chem. Lett. 2011, 40, 150–152. 10.1246/cl.2011.150. [DOI] [Google Scholar]
- Guadix-Montero S.; Alshammari H.; Dalebout R.; Nowicka E.; Morgan D. J.; Shaw G.; He Q.; Sankar M. Deactivation Studies of Bimetallic AuPd Nanoparticles Supported on MgO during Selective Aerobic Oxidation of Alcohols. Appl. Catal., A 2017, 546, 58–66. 10.1016/j.apcata.2017.07.045. [DOI] [Google Scholar]
- Pan Y.; Wu G.; He Y.; Feng J.; Li D. Identification of the Au/ZnO Interface as the Specific Active Site for the Selective Oxidation of the Secondary Alcohol Group in Glycerol. J. Catal. 2019, 369, 222–232. 10.1016/j.jcat.2018.10.030. [DOI] [Google Scholar]
- Sankar M.; Nowicka E.; Tiruvalam R.; He Q.; Taylor S. H.; Kiely C. J.; Bethell D.; Knight D. W.; Hutchings G. J. Controlling the Duality of the Mechanism in Liquid-Phase Oxidation of Benzyl Alcohol Catalysed by Supported Au-Pd Nanoparticles. Chem. - Eur. J. 2011, 17, 6524–6532. 10.1002/chem.201003484. [DOI] [PubMed] [Google Scholar]
- Nowicka E.; Hofmann J. P.; Parker S. F.; Sankar M.; Lari G. M.; Kondrat S. A.; Knight D. W.; Bethell D.; Weckhuysen B. M.; Hutchings G. J. In Situ Spectroscopic Investigation of Oxidative Dehydrogenation and Disproportionation of Benzyl Alcohol. Phys. Chem. Chem. Phys. 2013, 15, 12147–12155. 10.1039/c3cp50710f. [DOI] [PubMed] [Google Scholar]
- Yuan Z.; Gao Z.; Xu B.-Q. Acid-Base Property of the Supporting Material Controls the Selectivity of Au Catalyst for Glycerol Oxidation in Base-Free Water. Chin. J. Catal. 2015, 36, 1543–1551. 10.1016/S1872-2067(15)60936-6. [DOI] [Google Scholar]
- Villa A.; Campisi S.; Mohammed K. M. H.; Dimitratos N.; Vindigni F.; Manzoli M.; Jones W.; Bowker M.; Hutchings G. J.; Prati L. Tailoring the Selectivity of Glycerol Oxidation by Tuning the Acid–Base Properties of Au Catalysts. Catal. Sci. Technol. 2015, 5, 1126–1132. 10.1039/C4CY01246A. [DOI] [Google Scholar]
- Blaser H.-U.; Malan C.; Pugin B.; Spindler F.; Steiner H.; Studer M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. 10.1002/adsc.200390000. [DOI] [Google Scholar]
- de Vries J. G.; Elsevier C. J.. The Handbook of Homogeneous Hydrogenation; Wiley: Weinheim, Germany, 2007. [Google Scholar]
- Paradies J. Metal-Free Hydrogenation of Unsaturated Hydrocarbons Employing Molecular Hydrogen. Angew. Chem., Int. Ed. 2014, 53, 3552–3557. 10.1002/anie.201309253. [DOI] [PubMed] [Google Scholar]
- Stephan D. W.; Erker G. Frustrated Lewis Pairs: Metal-Free Hydrogen Activation and More. Angew. Chem., Int. Ed. 2010, 49, 46–76. 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]
- Bond G. C.; Sermon P. A.; Webb G.; Buchanan D. A.; Wells P. B. Hydrogenation over Supported Gold Catalysts. J. Chem. Soc., Chem. Commun. 1973, 13, 444–445. 10.1039/c3973000444b. [DOI] [Google Scholar]
- Mitsudome T.; Kaneda K. Gold Nanoparticle Catalysts for Selective Hydrogenations. Green Chem. 2013, 15, 2636–2654. 10.1039/c3gc41360h. [DOI] [Google Scholar]
- Zhao J.; Ge L.; Yuan H.; Liu Y.; Gui Y.; Zhang B.; Zhou L.; Fang S. Heterogeneous Gold Catalysts for Selective Hydrogenation: From Nanoparticles to Atomically Precise Nanoclusters. Nanoscale 2019, 11, 11429–11436. 10.1039/C9NR03182K. [DOI] [PubMed] [Google Scholar]
- Hari T. K.; Yaakob Z. Recent Development of Supported Monometallic Gold as Heterogeneous Catalyst for Selective Liquid Phase Hydrogenation Reactions. Chin. J. Chem. Eng. 2015, 23, 327–336. 10.1016/j.cjche.2014.01.002. [DOI] [Google Scholar]
- Lawrence S. A.Amines: Synthesis, Properties and Application; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Song J.; Huang Z.-F.; Pan L.; Li K.; Zhang X.; Wang L.; Zou J.-J. Review on Selective Hydrogenation of Nitroarene by Catalytic, Photocatalytic and Electrocatalytic Reactions. Appl. Catal., B 2018, 227, 386–408. 10.1016/j.apcatb.2018.01.052. [DOI] [Google Scholar]
- Serna P.; Boronat M.; Corma A. Tuning the Behavior of Au and Pt Catalysts for the Chemoselective Hydrogenation of Nitroaromatic Compounds. Top. Catal. 2011, 54, 439–446. 10.1007/s11244-011-9668-z. [DOI] [Google Scholar]
- Beier M. J.; Andanson J.-M.; Baiker A. Tuning the Chemoselective Hydrogenation of Nitrostyrenes Catalyzed by Ionic Liquid-Supported Platinum Nanoparticles. ACS Catal. 2012, 2, 2587–2595. 10.1021/cs300529y. [DOI] [Google Scholar]
- Wei H.; Liu X.; Wang A.; Zhang L.; Qiao B.; Yang X.; Huang Y.; Miao S.; Liu J.; Zhang T. FeOx-Supported Platinum Single-Atom and Pseudo-Single-Atom Catalysts for Chemoselective Hydrogenation of Functionalized Nitroarenes. Nat. Commun. 2014, 5, 5634. 10.1038/ncomms6634. [DOI] [PubMed] [Google Scholar]
- Corma A.; Serna P. Chemoselective Hydrogenation of Nitro Compounds with Supported Gold Catalysts. Science 2006, 313, 332–334. 10.1126/science.1128383. [DOI] [PubMed] [Google Scholar]
- Boronat M.; Concepción P.; Corma A.; González S.; Illas F.; Serna P. A Molecular Mechanism for the Chemoselective Hydrogenation of Substituted Nitroaromatics with Nanoparticles of Gold on TiO2 Catalysts: A Cooperative Effect between Gold and the Support. J. Am. Chem. Soc. 2007, 129, 16230–16237. 10.1021/ja076721g. [DOI] [PubMed] [Google Scholar]
- Serna P.; Concepción P.; Corma A. Design of Highly Active and Chemoselective Bimetallic Gold–Platinum Hydrogenation Catalysts through Kinetic and Isotopic Studies. J. Catal. 2009, 265, 19–25. 10.1016/j.jcat.2009.04.004. [DOI] [Google Scholar]
- Boronat M.; Corma A. Origin of the Different Activity and Selectivity toward Hydrogenation of Single Metal Au and Pt on TiO2 and Bimetallic Au-Pt/TiO2 Catalysts. Langmuir 2010, 26, 16607–16614. 10.1021/la101752a. [DOI] [PubMed] [Google Scholar]
- Shimizu K.; Miyamoto Y.; Kawasaki T.; Tanji T.; Tai Y.; Satsuma A. Chemoselective Hydrogenation of Nitroaromatics by Supported Gold Catalysts: Mechanistic Reasons of Size- and Support-Dependent Activity and Selectivity. J. Phys. Chem. C 2009, 113, 17803–17810. 10.1021/jp906044t. [DOI] [Google Scholar]
- Fujitani T.; Nakamura I.; Akita T.; Okumura M.; Haruta M. Hydrogen Dissociation by Gold Clusters. Angew. Chem., Int. Ed. 2009, 48, 9515–9518. 10.1002/anie.200905380. [DOI] [PubMed] [Google Scholar]
- Whittaker T.; Kumar K. B. S.; Peterson C.; Pollock M. N.; Grabow L. C.; Chandler B. D. H2 Oxidation over Supported Au Nanoparticle Catalysts: Evidence for Heterolytic H2 Activation at the Metal–Support Interface. J. Am. Chem. Soc. 2018, 140, 16469–16487. 10.1021/jacs.8b04991. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Liu X. Y.; Zhang L.; Wang A.; Li L.; Pan X.; Miao S.; Haruta M.; Wei H.; Wang H.; et al. ZnAl-Hydrotalcite-Supported Au25Nanoclusters as Precatalysts for Chemoselective Hydrogenation of 3-Nitrostyrene. Angew. Chem., Int. Ed. 2017, 56, 2709–2713. 10.1002/anie.201610736. [DOI] [PubMed] [Google Scholar]
- Tan Y.; Liu X. Y.; Li L.; Kang L.; Wang A.; Zhang T. Effects of Divalent Metal Ions of Hydrotalcites on Catalytic Behavior of Supported Gold Nanocatalysts for Chemoselective Hydrogenation of 3-Nitrostyrene. J. Catal. 2018, 364, 174–182. 10.1016/j.jcat.2018.05.007. [DOI] [Google Scholar]
- Hartfelder U.; Kartusch C.; Makosch M.; Rovezzi M.; Sá J.; Van Bokhoven J. A. Particle Size and Support Effects in Hydrogenation over Supported Gold Catalysts. Catal. Sci. Technol. 2013, 3, 454–461. 10.1039/C2CY20485A. [DOI] [Google Scholar]
- Anandkumar M.; Vinothkumar G.; Suresh Babu K. Synergistic Effect of Gold Supported on Redox Active Cerium Oxide Nanoparticles for the Catalytic Hydrogenation of 4-Nitrophenol. New J. Chem. 2017, 41, 6720–6729. 10.1039/C7NJ01300K. [DOI] [Google Scholar]
- Nikolaev S. A.; Zanaveskin L. N.; Smirnov V. V.; Averyanov V. A.; Zanaveskin K. L. Catalytic Hydrogenation of Alkyne and Alkadiene Impurities from Alkenes. Practical and Theoretical Aspects. Russ. Chem. Rev. 2009, 78, 231. 10.1070/RC2009v078n03ABEH003893. [DOI] [Google Scholar]
- Borodziński A.; Bond G. C. Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts. Part 1. Effect of Changes to the Catalyst During Reaction. Catal. Rev.: Sci. Eng. 2006, 48, 91–144. 10.1080/01614940500364909. [DOI] [Google Scholar]
- Borodziński A.; Bond G. C. Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts, Part 2: Steady-State Kinetics and Effects of Palladium Particle Size, Carbon Monoxide, and Promoters. Catal. Rev.: Sci. Eng. 2008, 50, 379–469. 10.1080/01614940802142102. [DOI] [Google Scholar]
- Azizi Y.; Petit C.; Pitchon V. Formation of Polymer-Grade Ethylene by Selective Hydrogenation of Acetylene over Au/CeO2 Catalyst. J. Catal. 2008, 256, 338–344. 10.1016/j.jcat.2008.04.003. [DOI] [Google Scholar]
- Yan X.; Bao J.; Yuan C.; Wheeler J.; Lin W.-Y.; Li R.; Jang B. W.-L. Gold on Carbon and Titanium Oxides Composites: Highly Efficient and Stable Acetylene Hydrogenation in Large Excess of Ethylene. J. Catal. 2016, 344, 194–201. 10.1016/j.jcat.2016.09.018. [DOI] [Google Scholar]
- McCue A. J.; Anderson J. A. Recent Advances in Selective Acetylene Hydrogenation Using Palladium Containing Catalysts. Front. Chem. Sci. Eng. 2015, 9, 142–153. 10.1007/s11705-015-1516-4. [DOI] [Google Scholar]
- Jia J.; Haraki K.; Kondo J. N.; Domen K.; Tamaru K. Selective Hydrogenation of Acetylene over Au/Al2O3 Catalyst. J. Phys. Chem. B 2000, 104, 11153–11156. 10.1021/jp001213d. [DOI] [Google Scholar]
- Yang B.; Burch R.; Hardacre C.; Headdock G.; Hu P. Influence of Surface Structures, Subsurface Carbon and Hydrogen, and Surface Alloying on the Activity and Selectivity of Acetylene Hydrogenation on Pd Surfaces: A Density Functional Theory Study. J. Catal. 2013, 305, 264–276. 10.1016/j.jcat.2013.05.027. [DOI] [Google Scholar]
- Johnson M. M.; Walker D. W.; Nowack G. P.. Selective Hydrogenation Catalyst US 4,404,124, 1983.
- Mitsudome T.; Yamamoto M.; Maeno Z.; Mizugaki T.; Jitsukawa K.; Kaneda K. One-Step Synthesis of Core-Gold/Shell-Ceria Nanomaterial and Its Catalysis for Highly Selective Semihydrogenation of Alkynes. J. Am. Chem. Soc. 2015, 137, 13452–13455. 10.1021/jacs.5b07521. [DOI] [PubMed] [Google Scholar]
- Gluhoi A. C.; Bakker J. W.; Nieuwenhuys B. E. Gold, Still a Surprising Catalyst: Selective Hydrogenation of Acetylene to Ethylene over Au Nanoparticles. Catal. Today 2010, 154, 13–20. 10.1016/j.cattod.2010.02.021. [DOI] [Google Scholar]
- Peng S.; Sun X.; Sun L.; Zhang M.; Zheng Y.; Su H.; Qi C. Selective Hydrogenation of Acetylene Over Gold Nanoparticles Supported on CeO2 Pretreated Under Different Atmospheres. Catal. Lett. 2019, 149, 465–472. 10.1007/s10562-018-2628-5. [DOI] [Google Scholar]
- Derrien M. L.Selective Hydrogenation Applied to the Refining of Petrochemical Raw Materials Produced by Steam Cracking. In Studies in Surface Science and Catalysis; Cerveny L., Ed.; Elsevier, 1986; Vol. 27, Chapter 18, pp 613–666. [Google Scholar]
- Hugon A.; Delannoy L.; Louis C. Supported Gold Catalysts for Selective Hydrogenation of 1,3-Butadiene in the Presence of an Excess of Alkenes. Gold Bull. 2008, 41, 127–138. 10.1007/BF03216590. [DOI] [Google Scholar]
- Masoud N.; Delannoy L.; Schaink H.; van der Eerden A.; de Rijk J. W.; Silva T. A. G.; Banerjee D.; Meeldijk J. D.; de Jong K. P.; Louis C.; et al. Superior Stability of Au/SiO2 Compared to Au/TiO2 Catalysts for the Selective Hydrogenation of Butadiene. ACS Catal. 2017, 7, 5594–5603. 10.1021/acscatal.7b01424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillejos E.; Bachiller-Baeza B.; Asedegbega-Nieto E.; Guerrero-Ruiz A.; Rodríguez-Ramos I. Selective 1,3-Butadiene Hydrogenation by Gold Nanoparticles Deposited & Precipitated onto Nano-Carbon Materials. RSC Adv. 2015, 5, 81583–81598. 10.1039/C5RA17388D. [DOI] [Google Scholar]
- Lozano-Martín M. C.; Castillejos E.; Bachiller-Baeza B.; Rodríguez-Ramos I.; Guerrero-Ruiz A. Selective 1,3-Butadiene Hydrogenation by Gold Nanoparticles on Novel Nano-Carbon Materials. Catal. Today 2015, 249, 117–126. 10.1016/j.cattod.2014.11.023. [DOI] [Google Scholar]
- Ali K. A.; Abdullah A. Z.; Mohamed A. R. Recent Development in Catalytic Technologies for Methanol Synthesis from Renewable Sources: A Critical Review. Renewable Sustainable Energy Rev. 2015, 44, 508–518. 10.1016/j.rser.2015.01.010. [DOI] [Google Scholar]
- Moret S.; Dyson P. J.; Laurenczy G. Direct Synthesis of Formic Acid from Carbon Dioxide by Hydrogenation in Acidic Media. Nat. Commun. 2014, 5, 4017. 10.1038/ncomms5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk J.; Kähler K.; Xia X.; Comotti M.; Schüth F.; Reinecke T.; Muhler M. Au/ZnO as Catalyst for Methanol Synthesis: The Role of Oxygen Vacancies. Appl. Catal., A 2009, 359, 121–128. 10.1016/j.apcata.2009.02.030. [DOI] [Google Scholar]
- Filonenko G. A.; Vrijburg W. L.; Hensen E. J. M.; Pidko E. A. On the Activity of Supported Au Catalysts in the Liquid Phase Hydrogenation of CO2 to Formates. J. Catal. 2016, 343, 97–105. 10.1016/j.jcat.2015.10.002. [DOI] [Google Scholar]
- Liu Q.; Yang X.; Li L.; Miao S.; Li Y.; Li Y.; Wang X.; Huang Y.; Zhang T. Direct Catalytic Hydrogenation of CO2 to Formate over a Schiff-Base-Mediated Gold Nanocatalyst. Nat. Commun. 2017, 8, 1407. 10.1038/s41467-017-01673-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis M.; Garcia H. Catalysis by Supported Gold Nanoparticles: Beyond Aerobic Oxidative Processes. Chem. Rev. 2012, 112, 4469–4506. 10.1021/cr3000785. [DOI] [PubMed] [Google Scholar]
- Takale B. S.; Bao M.; Yamamoto Y. Gold Nanoparticle (AuNPs) and Gold Nanopore (AuNPore) Catalysts in Organic Synthesis. Org. Biomol. Chem. 2014, 12, 2005–2027. 10.1039/c3ob42207k. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Li G. A Critical Review on Carbon-Carbon Coupling over Ultra-Small Gold Nanoclusters. Acta. Phys. Chim. Sin. 2017, 33, 1297–1309. [Google Scholar]
- Li Z.; Abroshan H.; Liu C.; Li G. A Critical Review on the Catalytic Applications of Non-Metallic Gold Nanoclusters: Selective Oxidation, Hydrogenation, and Coupling Reactions. Curr. Org. Chem. 2017, 21, 476–488. 10.2174/1385272820666161020152707. [DOI] [Google Scholar]
- Nasrollahzadeh M.; Issaabadi Z.; Tohidi M. M.; Mohammad Sajadi S. Recent Progress in Application of Graphene Supported Metal Nanoparticles in C–C and C–X Coupling Reactions. Chem. Rec. 2018, 18, 165–229. 10.1002/tcr.201700022. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Jin R. Heterogeneous Catalysis by Gold and Gold-Based Bimetal Nanoclusters. Nano Today 2018, 18, 86–102. 10.1016/j.nantod.2017.12.009. [DOI] [Google Scholar]
- Xu B.; Madix R. J.; Friend C. M. Predicting Gold-Mediated Catalytic Oxidative-Coupling Reactions from Single Crystal Studies. Acc. Chem. Res. 2014, 47 (3), 761–772. 10.1021/ar4002476. [DOI] [PubMed] [Google Scholar]
- Liu D.; Nie Q.; Zhang R.; Cai M. Heterogeneous Gold-Catalyzed Oxidative Cross-Coupling of Propargylic Acetates with Arylboronic Acids Leading to (E)-α-Arylenones. Tetrahedron Lett. 2019, 60, 29–34. 10.1016/j.tetlet.2018.11.053. [DOI] [Google Scholar]
- Sadeghzadeh S. M.; Zhiani R.; Emrani S.; Ghabdian M. C–C Coupling Reactions Using a Gold (III) Phosphorus Complex Confined within Metal–Organic Framework Fibers in Aqueous Solution. RSC Adv. 2017, 7, 50838–50843. 10.1039/C7RA10507J. [DOI] [Google Scholar]
- Movahed S. K.; Shariatipour M.; Dabiri M. Gold Nanoparticles Decorated on a Graphene-Periodic Mesoporous Silica Sandwich Nanocomposite as a Highly Efficient and Recyclable Heterogeneous Catalyst for Catalytic Applications. RSC Adv. 2015, 5, 33423–33431. 10.1039/C5RA00062A. [DOI] [Google Scholar]
- Vilhanová B.; Václavík J.; Artiglia L.; Ranocchiari M.; Togni A.; van Bokhoven J. A. Subnanometer Gold Clusters on Amino-Functionalized Silica: An Efficient Catalyst for the Synthesis of 1,3-Diynes by Oxidative Alkyne Coupling. ACS Catal. 2017, 7 (5), 3414–3418. 10.1021/acscatal.7b00691. [DOI] [Google Scholar]
- Elhage A.; Wang B.; Marina N.; Marin M. L.; Cruz M.; Lanterna A. E.; Scaiano J. C. Glass wool: A Novel Support for Heterogeneous Catalysis. Chem. Sci. 2018, 9, 6844–6852. 10.1039/C8SC02115E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupart R.; Benlahoues A.; Le Droumaguet B.; Grande D. Porous Gold Nanoparticle-Decorated Nanoreactors Prepared from Smartly Designed Functional Polystyrene-Block-Poly(d,l-Lactide) Diblock Copolymers: Toward Efficient Systems for Catalytic Cascade Reaction Processes. ACS Appl. Mater. Interfaces 2017, 9, 31279–31290. 10.1021/acsami.6b16157. [DOI] [PubMed] [Google Scholar]
- Liang W.; Zhang T.; Liu Y.; Huang Y.; Liu Z.; Liu Y.; Yang B.; Zhou X.; Zhang J. Polydimethylsiloxane Sponge-Supported Nanometer Gold: Highly Efficient Recyclable Catalyst for Cross-Dehydrogenative Coupling in Water. ChemSusChem 2018, 11, 3586–3590. 10.1002/cssc.201801180. [DOI] [PubMed] [Google Scholar]
- Thomas M.; Sheikh M. U. D.; Ahirwar D.; Bano M.; Khan F. Gold Nanoparticle and Graphene Oxide Incorporated Strontium Crosslinked Alginate/Carboxymethyl Cellulose Composites for o-Nitroaniline Reduction and Suzuki-Miyaura Cross-Coupling Reactions. J. Colloid Interface Sci. 2017, 505, 115–129. 10.1016/j.jcis.2017.05.051. [DOI] [PubMed] [Google Scholar]
- Pourjavadi P. A.; Keshavarzi N.; Moghaddam F. M.; Hosseini S. H. Magnetic Nanocomposite of Cross-Linked Melamine Groups Decorated with Large Amounts of Gold NPs: Reduction of Nitro Compounds and Suzuki–Miyaura Coupling Reactions in Aqueous Media. ChemistrySelect 2018, 3, 2716–2722. 10.1002/slct.201702798. [DOI] [Google Scholar]
- Li J.; Wang Y.; Jiang S.; Zhang H. Facile Synthesis of Magnetic Recyclable Palladium-Gold Alloy Nanoclusters Catalysts PdAur/Fe3O4@LDH and Its Catalytic Applications in Heck Reaction. J. Organomet. Chem. 2018, 878, 84–95. 10.1016/j.jorganchem.2018.10.007. [DOI] [Google Scholar]
- Maya R. J.; Varma R. L. An Efficient and Environmentally Benign Bentonite–Gold Nanohybrid-Catalyzed Oxidative Cross-Coupling of Ketones with Benzylic Primary Alcohols. Asian J. Org. Chem. 2017, 6, 1486–1491. 10.1002/ajoc.201700301. [DOI] [Google Scholar]
- Shah D.; Kaur H. Supported Gold Nanoparticle Catalyzed Cross-Coupling of Alkoxysilanes and Aryl Halides. Curr. Catal. 2015, 4, 224–230. 10.2174/2211544704666151012192353. [DOI] [Google Scholar]
- Crabbe B. W.; Kuehm O. P.; Bennett J. C.; Hallett-Tapley G. L. Light-Activated Ullmann Homocoupling of Aryl Halides Catalyzed Using Gold Nanoparticle-Functionalized Potassium Niobium Oxides. Catal. Sci. Technol. 2018, 8, 4907–4915. 10.1039/C8CY00996A. [DOI] [Google Scholar]
- Han D.; Bao Z.; Xing H.; Yang Y.; Ren Q.; Zhang Z. Fabrication of Plasmonic Au–Pd Alloy Nanoparticles for Photocatalytic Suzuki–Miyaura Reactions under Ambient Conditions. Nanoscale 2017, 9, 6026–6032. 10.1039/C7NR01950E. [DOI] [PubMed] [Google Scholar]
- Lanterna A. E.; Elhage A.; Scaiano C. J. Heterogeneous Photocatalytic C–C Coupling: Mechanism of Plasmon-Mediated Reductive Dimerization of Benzyl Bromides by Supported Gold Nanoparticles. Catal. Sci. Technol. 2015, 5, 4336–4340. 10.1039/C5CY00655D. [DOI] [Google Scholar]
- Parmentier T. E.; Dawson S. R.; Malta G.; Lu L.; Davies T. E.; Kondrat S. A.; Freakley S. J.; Kiely C. J.; Hutchings G. J. Homocoupling of Phenylboronic Acid Using Atomically Dispersed Gold on Carbon Catalysts: Catalyst Evolution Before Reaction. ChemCatChem 2018, 10, 1853–1859. 10.1002/cctc.201701840. [DOI] [Google Scholar]
- Candu N.; Simion A.; Coman S. M.; Primo A.; Esteve-Adell I.; Parvulescu V. I.; Garcia H. Graphene Film-Supported Oriented 1.1.1 Gold(0) Versus 2.0.0 Copper(I) Nanoplatelets as Very Efficient Catalysts for Coupling Reactions. Top. Catal. 2018, 61, 1449–1457. 10.1007/s11244-018-1043-x. [DOI] [Google Scholar]
- Primo A.; Esteve-Adell I.; Coman S. N.; Candu N.; Parvulescu V. I.; Garcia H. One-Step Pyrolysis Preparation of 1.1.1 Oriented Gold Nanoplatelets Supported on Graphene and Six Orders of Magnitude Enhancement of the Resulting Catalytic Activity. Angew. Chem., Int. Ed. 2016, 55, 607–612. 10.1002/anie.201508908. [DOI] [PubMed] [Google Scholar]
- Mondal P.; Salam N.; Mondal A.; Ghosh K.; Tuhina K.; Islam Sk. M. A Highly Active Recyclable Gold–Graphene Nanocomposite Material for Oxidative Esterification and Suzuki Cross-Coupling Reactions in Green Pathway. J. Colloid Interface Sci. 2015, 459, 97–106. 10.1016/j.jcis.2015.07.072. [DOI] [PubMed] [Google Scholar]
- Boronat M.; Concepción P. Combined Theoretical and Spectroscopic Mechanistic Studies for Improving Activity and Selectivity in Heterogeneous Catalysis. Catal. Today 2017, 285, 166–178. 10.1016/j.cattod.2016.11.048. [DOI] [Google Scholar]
- Liu C.-H.; Lin C.-Y.; Chen J.-L.; Lai N.-C.; Yang C.-M.; Chen J.-M.; Lu K.-T. Metal Oxide-Containing SBA-15-Supported Gold Catalysts for Base-Free Aerobic Homocoupling of Phenylboronic Acid in Water. J. Catal. 2016, 336, 49–57. 10.1016/j.jcat.2016.01.004. [DOI] [Google Scholar]
- Albadi J.; Momeni A.; Mansournezhad A. Suzuki-Miyaura Cross-Coupling Reactions over Gold Nanoparticles Supported on CuO-ZnO Metal Oxide in Aqueous Medium. Jordan J. Chem. 2017, 12, 233–239. [Google Scholar]
- Gholinejad M.; Ahmadi J.; Nájera C. Silica Microparticles Supported Gold and Copper Ferrite Nanoparticles: A Magnetically Recyclable Bimetallic Catalyst for Sonogashira Reaction. ChemistrySelect 2016, 1, 384–390. 10.1002/slct.201600049. [DOI] [Google Scholar]
- Li G.; Abroshan H.; Liu C.; Zhuo S.; Li Z.; Xie Y.; Kim H. J.; Rosi N. L.; Jin R. Tailoring the Electronic and Catalytic Properties of Au25 Nanoclusters via Ligand Engineering. ACS Nano 2016, 10, 7998–8005. 10.1021/acsnano.6b03964. [DOI] [PubMed] [Google Scholar]
- Li Q.; Das A.; Wang S.; Chen Y.; Jin R. Highly Efficient Three-Component Coupling Reaction Catalysed by Atomically Precise Ligand-Protected Au38(SC2H4Ph)24 Nanoclusters. Chem. Commun. 2016, 52, 14298–14301. 10.1039/C6CC07825G. [DOI] [PubMed] [Google Scholar]
- Li Z.; Yang X.; Liu C.; Wang J.; Li G. Effects of Doping in 25-Atom Bimetallic Nanocluster Catalysts for Carbon–Carbon Coupling Reaction of Iodoanisole and Phenylacetylene. Prog. Nat. Sci. 2016, 26, 477–482. 10.1016/j.pnsc.2016.09.007. [DOI] [Google Scholar]
- Mikami Y.; Dhakshinamoorthy A.; Alvaro M.; García H. Catalytic Activity of Unsupported Gold Nanoparticles. Catal. Sci. Technol. 2013, 3, 58–69. 10.1039/C2CY20068F. [DOI] [Google Scholar]
- Sodium borohydride stabilizes very active gold nanoparticle catalysts. Chem. Commun., 2014, 50, 14194–14196. [DOI] [PubMed] [Google Scholar]
- Astruc D.Transition-Metal Nanoparticles in Catalysis: From Historical Background to the State-of-the Art. In Nanoparticles and Catalysis; Astruc D., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp 1–48. [Google Scholar]
- Tsunoyama H.; Sakurai H.; Negishi Y.; Tsukuda T. Size-Specific Catalytic Activity of Polymer-Stabilized Gold Nanoclusters for Aerobic Alcohol Oxidation in Water. J. Am. Chem. Soc. 2005, 127, 9374–9375. 10.1021/ja052161e. [DOI] [PubMed] [Google Scholar]
- Villa A.; Wang D.; Su D. S.; Prati L. Gold Sols as Catalysts for Glycerol Oxidation: The Role of Stabilizer. ChemCatChem 2009, 1, 510–514. 10.1002/cctc.200900118. [DOI] [Google Scholar]
- Comotti M.; Della Pina C.; Matarrese R.; Rossi M. The Catalytic Activity of ?Naked? Gold Particles. Angew. Chem., Int. Ed. 2004, 43, 5812–5815. 10.1002/anie.200460446. [DOI] [PubMed] [Google Scholar]
- Hu L.; Cao X.; Yang J.; Li M.; Hong H.; Xu Q.; Ge J.; Wang L.; Lu J.; Chen L.; et al. Oxidation of Benzylic Compounds by Gold Nanowires at 1 Atm O2. Chem. Commun. 2011, 47, 1303–1305. 10.1039/C0CC01568G. [DOI] [PubMed] [Google Scholar]
- Iizuka Y.; Tode T.; Takao T.; Yatsu K.; Takeuchi T.; Tsubota S.; Haruta M. A Kinetic and Adsorption Study of CO Oxidation over Unsupported Fine Gold Powder and over Gold Supported on Titanium Dioxide. J. Catal. 1999, 187, 50–58. 10.1006/jcat.1999.2604. [DOI] [Google Scholar]
- Chen Z.; Dellapina C.; Falletta E.; Lofaro M.; Pasta M.; Rossi M.; Santo N. Facile Synthesis of Polyaniline Using Gold Catalyst. J. Catal. 2008, 259, 1–4. 10.1016/j.jcat.2008.07.006. [DOI] [Google Scholar]
- Pina C. D.; Falletta E.; Faro M. L.; Pasta M.; Rossi M. Gold-Catalysed Synthesis of Polypyrrole. Gold Bull. 2009, 42, 27–33. 10.1007/BF03214903. [DOI] [Google Scholar]
- Zhu Y.; Qian H.; Drake B. A.; Jin R. Atomically Precise Au25(SR)18 Nanoparticles as Catalysts for the Selective Hydrogenation of α,β-Unsaturated Ketones and Aldehydes. Angew. Chem., Int. Ed. 2010, 49, 1295–1298. 10.1002/anie.200906249. [DOI] [PubMed] [Google Scholar]
- Biondi I.; Laurenczy G.; Dyson P. J. Synthesis of Gold Nanoparticle Catalysts Based on a New Water-Soluble Ionic Polymer. Inorg. Chem. 2011, 50, 8038–8045. 10.1021/ic200334m. [DOI] [PubMed] [Google Scholar]
- Boualleg M.; Guillois K.; Istria B.; Burel L.; Veyre L.; Basset J.-M.; Thieuleux C.; Caps V. Highly Efficient Aerobic Oxidation of Alkenes over Unsupported Nanogold. Chem. Commun. 2010, 46, 5361. 10.1039/c0cc00664e. [DOI] [PubMed] [Google Scholar]
- Tsunoyama H.; Sakurai H.; Ichikuni N.; Negishi Y.; Tsukuda T. Colloidal Gold Nanoparticles as Catalyst for Carbon–Carbon Bond Formation: Application to Aerobic Homocoupling of Phenylboronic Acid in Water. Langmuir 2004, 20, 11293–11296. 10.1021/la0478189. [DOI] [PubMed] [Google Scholar]
- Agarwal N.; Freakley S. J.; McVicker R. U.; Althahban S. M.; Dimitratos N.; He Q.; Morgan D. J.; Jenkins R. L.; Willock D. J.; Taylor S. H.; et al. Aqueous Au-Pd Colloids Catalyze Selective CH 4 Oxidation to CH 3 OH with O 2 under Mild Conditions. Science 2017, 358, 223–227. 10.1126/science.aan6515. [DOI] [PubMed] [Google Scholar]
- McVicker R.; Agarwal N.; Freakley S. J.; He Q.; Althahban S.; Taylor S. H.; Kiely C. J.; Hutchings G. J. Low Temperature Selective Oxidation of Methane Using Gold-Palladium Colloids. Catal. Today 2020, 342, 32–38. 10.1016/j.cattod.2018.12.017. [DOI] [Google Scholar]
- Williams C.; Carter J. H.; Dummer N. F.; Chow Y. K.; Morgan D. J.; Yacob S.; Serna P.; Willock D. J.; Meyer R. J.; Taylor S. H.; et al. Selective Oxidation of Methane to Methanol Using Supported AuPd Catalysts Prepared by Stabilizer-Free Sol-Immobilization. ACS Catal. 2018, 8, 2567–2576. 10.1021/acscatal.7b04417. [DOI] [Google Scholar]
- Ab Rahim M. H.; Forde M. M.; Jenkins R. L.; Hammond C.; He Q.; Dimitratos N.; Lopez-Sanchez J. A.; Carley A. F.; Taylor S. H.; Willock D. J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold-Palladium Alloy Nanoparticles. Angew. Chem., Int. Ed. 2013, 52, 1280–1284. 10.1002/anie.201207717. [DOI] [PubMed] [Google Scholar]
- Jones C.; Taube D.; Ziatdinov V. R.; Periana R. A.; Nielsen R. J.; Oxgaard J.; Goddard W. A. Selective Oxidation of Methane to Methanol Catalyzed, with C-H Activation, by Homogeneous, Cationic Gold. Angew. Chem. 2004, 116, 4726–4729. 10.1002/ange.200461055. [DOI] [PubMed] [Google Scholar]
- Kao L. C.; Hutson A. C.; Sen A. Low-Temperature, Palladium(II)-Catalyzed, Solution-Phase Oxidation of Methane to Methanol Derivative. J. Am. Chem. Soc. 1991, 113, 700–701. 10.1021/ja00002a063. [DOI] [Google Scholar]
- Yuan Q.; Deng W.; Zhang Q.; Wang Y. Osmium-Catalyzed Selective Oxidations of Methane and Ethane with Hydrogen Peroxide in Aqueous Medium. Adv. Synth. Catal. 2007, 349, 1199–1209. 10.1002/adsc.200600438. [DOI] [Google Scholar]
- Koczkur K. M.; Mourdikoudis S.; Polavarapu L.; Skrabalak S. E. Polyvinylpyrrolidone (PVP) in Nanoparticle Synthesis. Dalton Trans 2015, 44, 17883–17905. 10.1039/C5DT02964C. [DOI] [PubMed] [Google Scholar]
- Abis L.; Freakley S. J.; Dodekatos G.; Morgan D. J.; Sankar M.; Dimitratos N.; He Q.; Kiely C. J.; Hutchings G. J. Highly Active Gold and Gold-Palladium Catalysts Prepared by Colloidal Methods in the Absence of Polymer Stabilizers. ChemCatChem 2017, 9, 2914–2918. 10.1002/cctc.201700483. [DOI] [Google Scholar]
- Tsunoyama H.; Ichikuni N.; Sakurai H.; Tsukuda T. Effect of Electronic Structures of Au Clusters Stabilized by Poly(N -Vinyl-2-Pyrrolidone) on Aerobic Oxidation Catalysis. J. Am. Chem. Soc. 2009, 131, 7086–7093. 10.1021/ja810045y. [DOI] [PubMed] [Google Scholar]
- Teranishi T.; Miyake M. Size Control of Palladium Nanoparticles and Their Crystal Structures. Chem. Mater. 1998, 10, 594–600. 10.1021/cm9705808. [DOI] [Google Scholar]
- Mertens P. G. N.; Vankelecom I. F. J.; Jacobs P. A.; De Vos D. E. Gold Nanoclusters as Colloidal Catalysts for Oxidation of Long Chain Aliphatic 1,2-Diols in Alcohol Solvents. Gold Bull. 2005, 38, 157–162. 10.1007/BF03215255. [DOI] [Google Scholar]
- Mertens P. G. N.; Bulut M.; Gevers L. E. M.; Vankelecom I. F. J.; Jacobs P. A.; Vos D. E. D. Catalytic Oxidation of 1,2-Diols to α-Hydroxy-Carboxylates with Stabilized Gold Nanocolloids Combined with a Membrane-Based Catalyst Separation. Catal. Lett. 2005, 102, 57–61. 10.1007/s10562-005-5203-9. [DOI] [Google Scholar]