

Abstract
Overexpression and activation of c-Jun N-terminal kinases (JNKs) have been observed in multiple cancer cell lines and tumor samples. Various JNK isoforms have been reported to promote lung and liver cancer, as well as keratinocyte transformation, suggesting an important role of JNK signaling in promoting tumor development. However, there are three JNK isoforms, and it is unclear how each individual isoform, especially the ubiquitously expressed JNK1 and JNK2, functions in melanoma. Our previous study found that C116S mutations in both JNK1 and JNK2 rendered them insensitive to the covalent pan-JNK inhibitor JNK-IN-8 while retaining kinase activity. To delineate the specific roles of JNK1 and JNK2 in melanoma cell proliferation and invasiveness, we expressed the wild type (WT) and C116S mutants in melanoma cell lines and used JNK-IN-8 to enable chemical–genetic dissection of JNK1 and JNK2 activity. We found that the JNK2C116S allele consistently enhanced colony proliferation and cell invasiveness in the presence of JNK-IN-8. When cells individually expressing WT or C116S JNK1/2 were subcutaneously implanted into immunodeficient mice, we again found that bypass of JNK-IN-8-mediated inhibition of JNK signaling by expression of JNK2C116S specifically resulted in enhanced tumor growth in vivo. In addition, we observed a high level of JNK pathway activation in some human BRAF inhibitor (BRAFi) resistant melanoma cell lines relative to their BRAFi sensitive isogenic counterparts. JNK-IN-8 significantly enhanced the response to dabrafenib in resistant cells overexpressing JNK1WT, JNK2WT, and JNK1C116S but had no effect on cells expressing JNK2C116S, suggesting that JNK2 signaling is also crucial for BRAFi resistance in a subset of melanomas. Collectively, our data show that JNK2 activity is specifically required for melanoma cell proliferation, invasiveness, and BRAFi resistance and that this activity is most important in the context of JNK1 suppression, thus providing a compelling rationale for the development of JNK2 selective inhibitors as a potential therapy for the treatment of melanoma.
Graphical Abstract
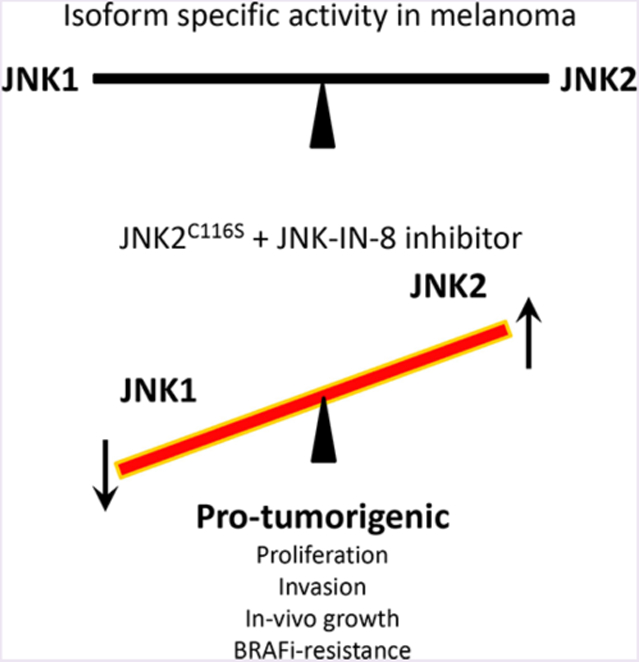
c-Jun N-terminal kinases (JNKs) collectively form one of the canonical mitogen-activated protein kinase (MAPK) families and are activated by stress, growth factors, and cytokines. In humans, JNK1, JNK2, and JNK3 are encoded by three separate genes, MAPK8–10, respectively, and are spliced alternatively to yield 10 isoforms with molecular weights ranging from 46 to 54 kDa.1 JNK1 and JNK2 are expressed ubiquitously, while JNK3 expression is believed to be more limited to neuronal tissue, testis, and cardiomyocytes. By activating c-Jun and other transcription factors, JNKs can regulate apoptosis, cellular proliferation, tumor cell migration and motility, and immune responses. Activation of JNK has been associated with both oncogenic and tumor-suppressive functions, suggesting that its signaling functions are likely to be context specific.2–4 For example, we have reported that suppression of JNK signaling is important for the induction of cutaneous squamous cell carcinoma (cuSCC) in the context of BRAFi treatment.5 Other evidence supports the suggestion that JNK1 and JNK2 might have opposing activities in tumorigenesis due to selective interactions with different transcription factors.6,7
Despite significant advances in immunotherapy and targeted therapy, metastatic melanoma continues to represent a major area of continuing medical need, particularly in the setting of resistance to therapy. As such, the search for additional vulnerabilities and co-dependencies in melanoma is necessary but made complex by the multifaceted mechanisms of resistance. Although distinct drivers such as mutant BRAF and NRAS have now been established for melanoma, one unifying theme is that melanomas typically exhibit high ERK activity. In this context, ERK and JNK pathway crosstalk has long been the subject of investigation.8 Although initial work linked the two signaling modules tightly,8 more recent studies suggest that JNK activity is required for both optimal proliferation and inducing apoptosis.9,10 Furthermore, JNK activation has been implicated in BRAF inhibitor resistance.11,12 Exome sequencing has recently identified hypomorphic alleles of kinases upstream of JNK with some data suggesting these alleles render melanoma cells resistant to apoptosis.13
However, there has been little exploration of whether specific JNK isoforms contribute to these phenotypes. JNKs often compensate for each other, especially in genetic deletion models, due to the high degree of similarity among the isoforms.2,14–16 Indeed, there may also be conflation of data obtained in some experiments especially with the assumed equivalence of siRNA-based knockdown of protein and chemical inhibition of kinase activity.17
To clarify this issue, we developed a chemical genetic method to dissect the functions of JNK1 and JNK2 and used a combination of in vitro and in vivo approaches to investigate the roles of individual JNK isoforms in melanoma tumori-genesis and acquired BRAFi resistance. We found that JNK2, but not JNK1, is required for the invasiveness and long-term proliferation of melanomas. Furthermore, activation of JNK2 signaling is specifically involved in acquired resistance to BRAF inhibitors in a subset of melanoma cells for which c-Jun is activated. Our finding supports the development of JNK2 selective inhibitors to achieve more effective combination therapy to protect late stage melanoma patients from BRAFi resistance. The results of this study also help to establish new guidance for the selection and stratification of late stage melanoma patients in clinical trials.
We further hypothesized that JNK1 and JNK2 exhibit potentially opposing activities in melanoma tumorigenesis and adaptive BRAF inhibitor resistance and that these distinct activities could be exploited for therapeutic gain.
RESULTS
JNK2 Is Significantly More Highly Expressed Than JNK1 in Melanoma.
RNA sequencing data derived from cutaneous melanoma clinical data sets (PanCancer Atlas; n = 448) and deposited in cBioPortal demonstrate that both JNK1 (MAPK8) and JNK2 (MAPK9) are expressed in melanomas. JNK2 is significantly more highly expressed by almost 4.5-fold (Figure 1A). Furthermore, JNK1 appears to be more frequently deleted and JNK2 more frequently amplified in multiple data sets, although these are rare events overall (Figure 1B).
Figure 1.
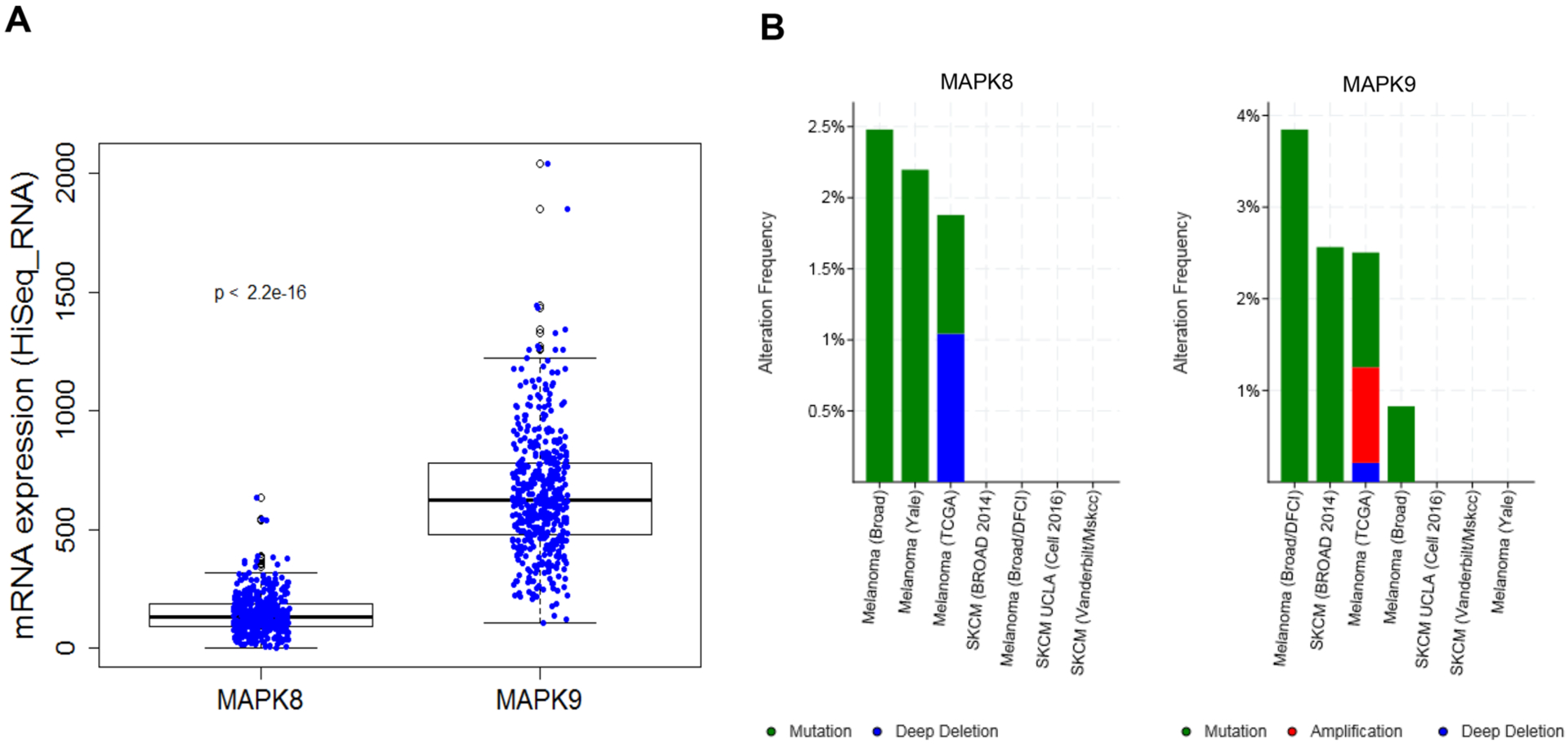
cBioportal data analysis of MAPK8 and MAPK9 genes. (A) Aggregate RNA expression levels of MAPK8 and MAPK9 genes in multiple cutaneous melanoma clinical cohorts show that MAPK9 is significantly more highly expressed (4.5-fold; p < 2.2 × 10−16). (B) Somatic variants in MAPK8 and MAPK9 are rare, with MAPK8 exhibiting deep deletion more frequently and MAPK9 exhibiting amplification more frequently.
JNK2 Activity, but Not JNK1 Activity, Is Required for Cell Proliferation in Melanoma Cells.
To delineate the specific roles of JNK1 and JNK2 on melanoma cell proliferation and invasiveness, we employed a chemical–genetic approach. The basis of our approach is the potent, selective, and irreversible pan-JNK inhibitor, JNK-IN-8, which covalently binds the cysteine 116 (C116) residue that is conserved in JNK1, JNK2, and JNK3.18 We used C116S mutant alleles of JNK1 (JNK1C116S) and JNK2 (JNK2C116S), which confer resistance to JNK-IN-8 without affecting the kinase activity of JNKs, to dissect the individual contribution of each kinase. For our experiments, we expressed WT and mutant JNK1C116S and JNK2C116S alleles using lentiviral transduction in 501MEL, WM239A, and HMEL-T1 cell lines and used JNK-IN-8 to enable chemical–genetic dissection of JNK1 and JNK2 activity (Figure 2A,B). Importantly, we confirmed that these mutant alleles retain signaling to c-Jun by exposing the lines to an established activator of JNKs, ultraviolet radiation (Newport, SOL-UV), and showing that c-JUN is still phosphorylated in cells expressing the C116S mutant alleles even in the presence of JNK-IN-8 (Supplementary Figure 1).
Figure 2.
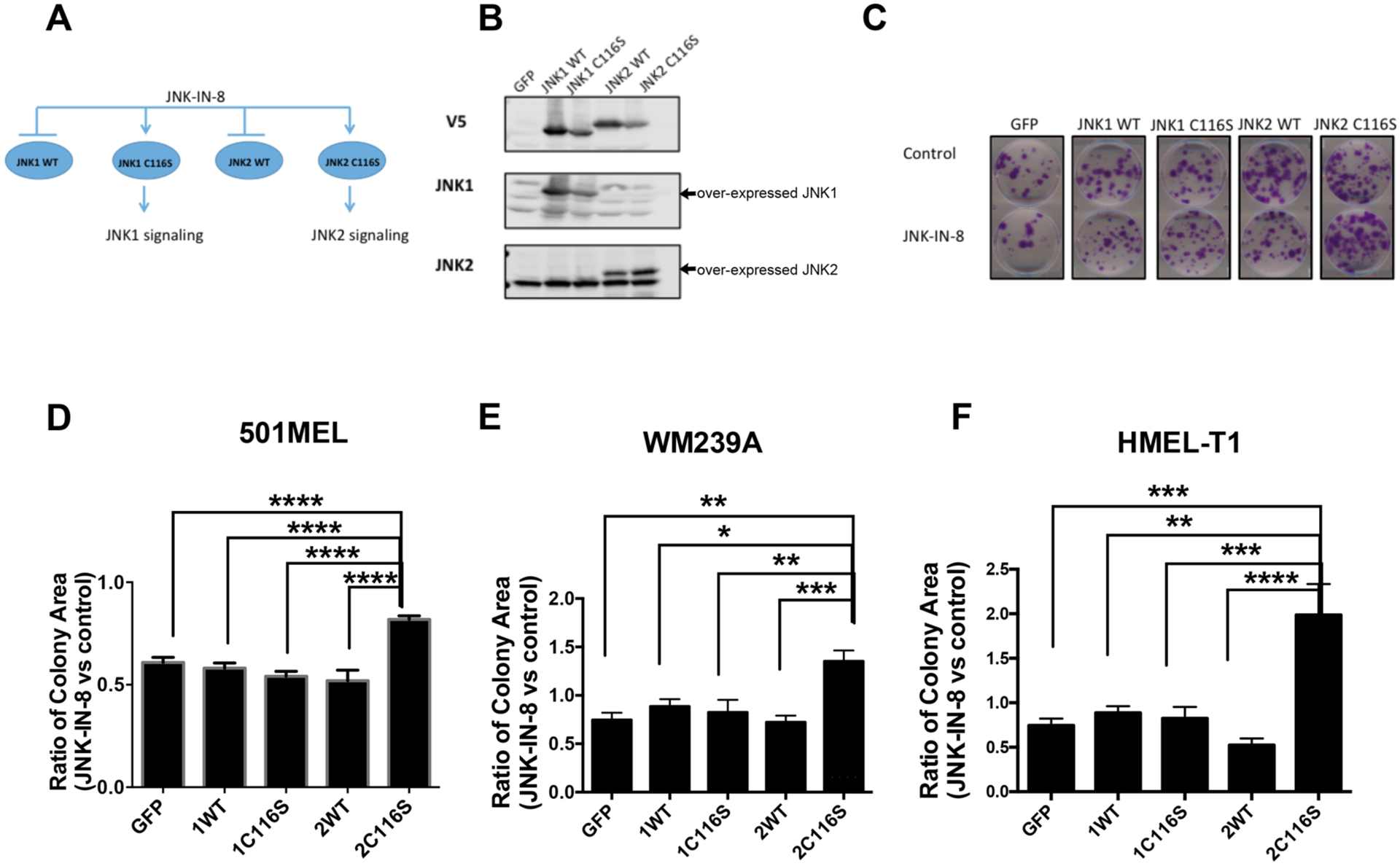
JNK2 activity promotes long-term cell proliferation. (A) Schematic of chemical genetic methods for delineating the specific roles of JNK1 and JNK2 signaling. GFP, JNK1WT, JNK1C116S, JNK2WT, and JNK2C116S were overexpressed in 501MEL, WM239A, and HMEL-T1 cell lines. Administration of JNK-IN-8 can distinguish the signaling of JNK1 and JNK2. (B) Representative Western blots of 501MEL cell lines indicated overexpression of JNK1WT, JNK1C116S, JNK2WT, and JNK2C116S. (C) Representative colony formation assay after continual treatment with JNK-IN-8 in a 501MEL cell line for 14 days, quantified by total colony area (ImageJ), showing a strong growth advantage for the cells overexpressing JNK2C116S vs cells overexpressing JNK1WT, JNK1C116S, and JNK2WT. The scale bar on the representative images is 5 cm. (D–F) Comparison of the colony area treated with JNK-IN-8 normalized to control among 501MEL, WM239A, and HMEL-T1 cell lines, respectively, overexpressing GFP, JNK1WT, JNK1C116S, JNK2WT, and JNK2C116S (*p < 0.05; **p < 0.001; ***p < 0.0001; n = 7).
To test the effect on long-term proliferation on melanoma cells, colony formation was assessed following JNK-IN-8 treatment in 501MEL, WM239A, and HMEL-T1 BRAF mutant/NRAS wild-type melanoma cell lines. The transduction of the individual JNK1/2WT and JNK1/2C116S mutants had a small effect on total colony numbers without much effect of JNK-IN-8 (Supplementary Figure 2). However, we found that JNK-IN-8 dramatically inhibited long-term cell proliferation in terms of the average colony area. In comparing the individual transduced cells with and without an inhibitor (Supplementary Figure 2), we sought to assess whether any of the JNK1/2C116S mutant-expressing lines would exhibit a unique response to the inhibitor suggestive of a selective requirement for the activity of one of the isoforms specifically. Indeed, by then using the ratio of responses of inhibitor-treated cells over control-treated cells, we found that JNK-IN-8 inhibited individual colony expansion by approximately 40% in 501MEL cells over-expressing GFP, JNK1WT, JNK2WT, or JNK1C116S compared to vehicle control but did so by only 19% in cells overexpressing JNK2C116S. Similarly, we observed 35% and 99% increased levels of colony formation in WM239A and HMEL-T1 cells, respectively, that overexpressed JNK2C116S and 12–28% growth inhibition in JNK-IN-8-treated cells overexpressing JNK1WT, JNK2WT, or JNK1C116S. Cumulatively, these results indicate JNK2 activity is more important for cell proliferation than JNK1 activity [n = 7 (Figure 2C–F)].
JNK2 Activity, but Not JNK1 Activity, Is Required for Invasion in Melanoma Cells.
We next asked whether JNK2 might also be required for other cancer-related properties of cells. JNKs have been reported to promote epithelial–mesenchymal transition (EMT) and invasiveness in carcinoma cells.19–21 To investigate the role of JNK1 and JNK2 in promoting the invasiveness and motility of melanoma cells, we also conducted invasion assays in 501MEL and HMEL-T1 melanoma cells. Here, as described above, we first assessed the effect of transduction of the individual JNK1/2WT and JNK1/2C116S mutants showing somewhat varied responses (Supplementary Figure 3). Again, we sought to assess whether any of the JNK1/2C116S mutant-expressing lines would exhibit a unique response to the inhibitor suggestive of a selective requirement for the activity of one of the isoforms specifically. Indeed, by then using the ratio of responses of inhibitor-treated cells to control-treated cells, we found that 501MEL and HMEL-T1 cells expressing JNK2C116S were alone able to significantly augment cell invasion by 84% and 416%, respectively, as compared to GFP control cells [n = 10 (Figure 3A,B)]. We also observed a 67 ± 3.1% reduction in 501MEL cells overexpressing the C116S JNK1 mutant but not in HMEL-T1 cells. However, there was no significant difference in cell migration among cells overexpressing JNK1/2WT or JNK1/2C116S. Taken together, these findings indicated that JNK2 is required for melanoma invasiveness but JNK1 is not.
Figure 3.
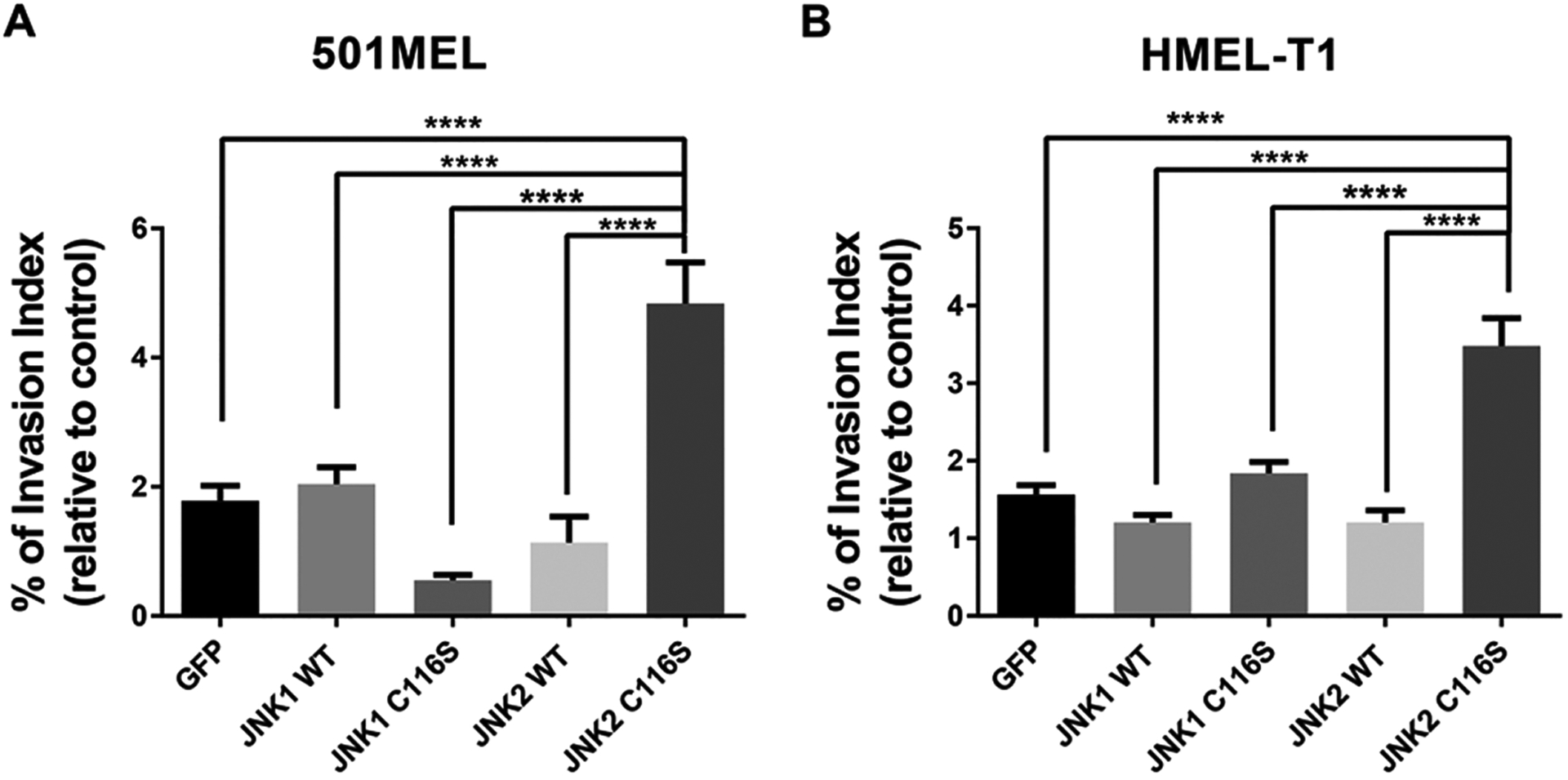
JNK2 significantly promotes melanoma cell invasion in vitro. Dramatic increase in the level of cell invasion in (A) 501MEL and (B) HMEL-T1 cell lines overexpressing JNK2C116S compared to cells overexpressing GFP, JNK1WT, JNK1C116S, JNK2WT, and JNK2C116S (*p < 0.05; ****p < 0.0001; n = 10).
Activation of JNK2 Significantly Promotes Tumori-genesis in a Xenograft Mouse Model.
Given the role of JNK2 in cell proliferation and invasiveness in vitro, we next focused on addressing whether specific activation of JNK2 is tumorigenic in vivo. When cells stably expressing JNK1/2WT or JNK1/2C116S were subcutaneously implanted into opposite flanks of immunodeficient NCr nude mice, we again found that only JNK2C116S-mediated bypass of JNK inhibition by JNK-IN-8 resulted in the most robust tumor growth (Figure 4A–E). Interestingly JNK1C116S-expressing tumors exhibited a slight decrement in tumor growth following treatment with JNK-IN-8 (Figure 4C), also consistent with the notion that JNK2 is the more important driver of tumorigenesis relative to JNK1. Nevertheless, it is clear here, as in the in vitro experiments, that the retention of JNK2 activity in the context of simultaneous inhibition of JNK1 results in the most tumorigenic phenotypes. No apparent toxicity was observed in JNK-IN-8-treated mice.
Figure 4.
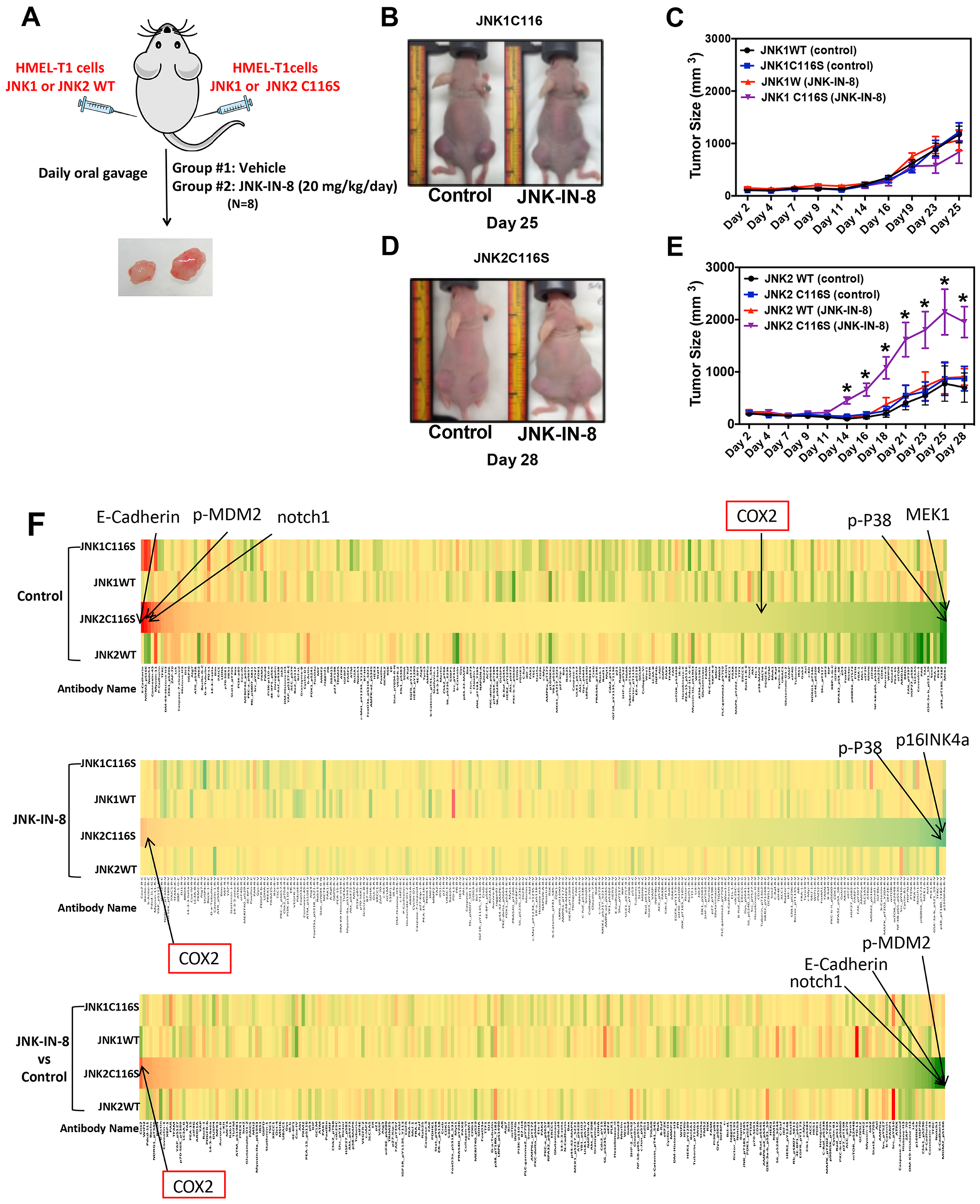
Activation of JNK2 significantly promotes tumorigenesis in vivo. (A) Experimental design of a xenograft mouse model. (B and C) Role of JNK1 signaling in tumor formation (n = 8 for each group). (D and E) Role of JNK2 signaling in tumor formation (n = 8 for each group). (F) RPPA heat map of significant changes of potential downstream target genes of activation of the JNK2 signaling pathway. Red indicates upregulated genes. Green indicates downregulated genes. Yellow indicates no significant changes.
Proteomics Analysis Identified Downstream Target Pathways of JNK2 Signaling.
To investigate the potential downstream targets of JNK2 signaling in tumorigenesis in vivo, we collected five tumors from each of the four cell lines implanted for each drug condition (in Figure 4, n = 40) and performed reverse-phase protein arrays (RPPA) to interrogate the status of 243 proteins across multiple cancer-related pathways. We generated two types of comparisons.
First, we wanted to assess whether a specific set of proteins was significantly more or less expressed in samples with intact JNK2 signaling in the presence of JNK-IN-8. Notably, in the tumors expressing JNK2C116S, COX-2 levels were higher following JNK-IN-8 treatment than those of tumor samples overexpressing JNK1WT, JNK2WT, and JNK1C116S. On the other hand, NOTCH1, E-Cadherin, and phosphorylated MDM2 (S166) levels were dramatically decreased in these cells in the presence of JNK-IN-8 (in Figure 4F, n = 5).
Second, we wanted to see whether intact JNK1 and JNK2 activity was individually associated with expression of distinct sets of proteins and phosphoproteins within tumors. For this analysis, we identified proteins that were up- or downregulated in both WT and C116S mutants of JNK1/2 in the absence of JNK-IN-8. Proteins that changed in common between JNK1 and JNK2 were then removed from further consideration. We then focused on proteins for which levels changed in control-treated tumors in a manner that could be suppressed by JNK-IN-8 administration but retained in JNK1/2C116S-expressing tumors in the context of JNK-IN-8. This is a significantly smaller set of proteins; however, JNK1-specific changes were observed in 22 proteins, and JNK2-specific changes in 29 proteins (Supplementary Table 1).
Interestingly, multiple JNK2-specific proteins in the tumors, as judged by this assay, are known to be involved in cellular motility and invasiveness. These include ARAF, ATM, BIRC3, MMP2, NOTCH3, and TSC1.22–27 Determining whether these are direct kinase targets of JNK2 will require further investigation.
The JNK2 Signaling Pathway Contributes to BRAFi Resistance in a Subset of Melanoma Cells.
Given the ability of JNK2 to drive tumorigenic properties of melanoma cells, including the apparent activation of RAS signaling in the proteomic pathway analysis described above, we then asked whether JNK2 could impact resistance to targeted therapies. Using phospho-c-Jun as a reporter of JNK pathway activity, we observed a high level of expression of phosphorylated JNK, phosphorylated c-Jun, and total c-Jun in m229R and SK-MEL28R human melanoma cells resistant to the BRAF inhibitor (BRAFi) compared to paired isogenic m229S and SK-MEL28S sensitive cells (Figure 5A).
Figure 5.
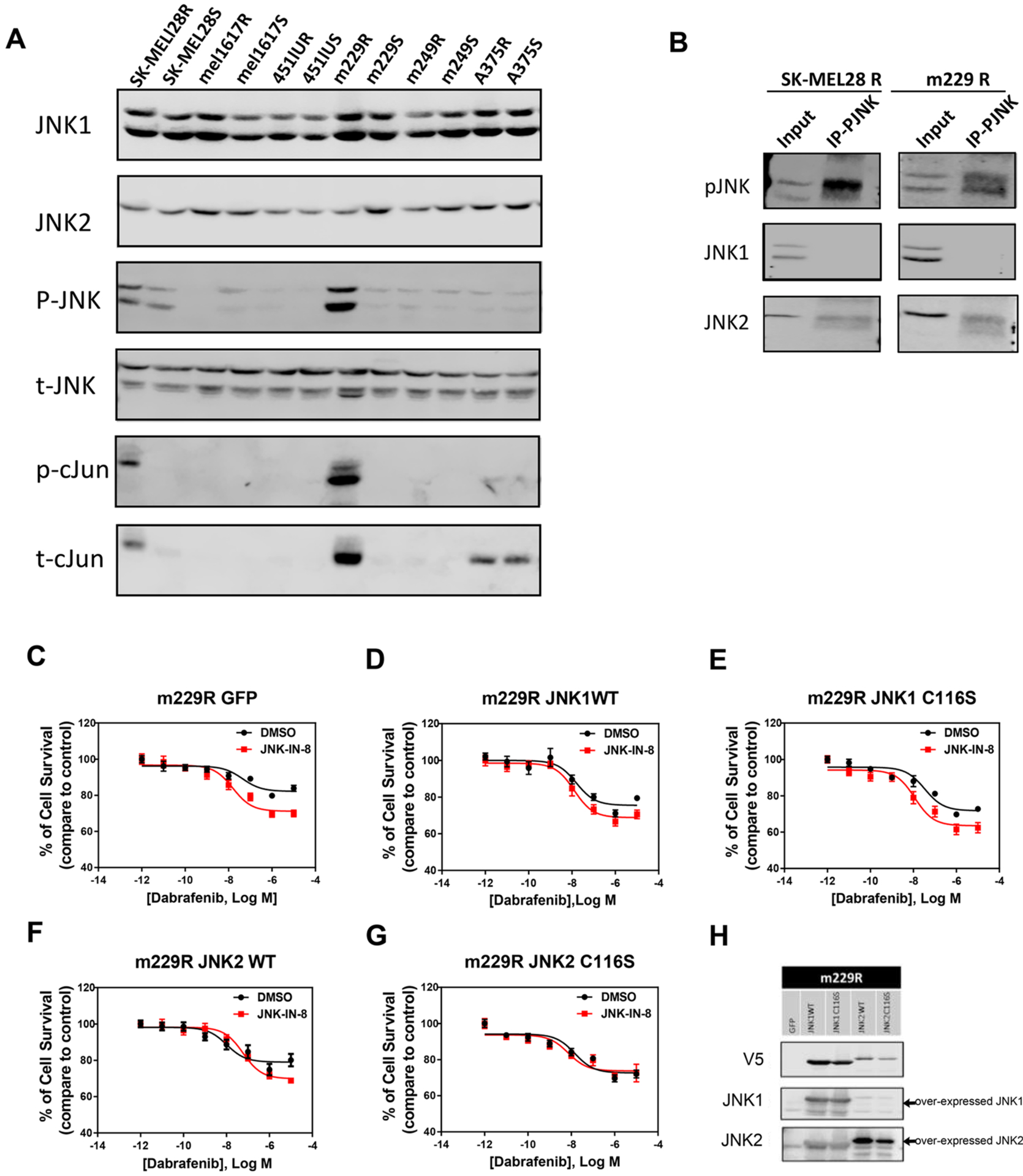
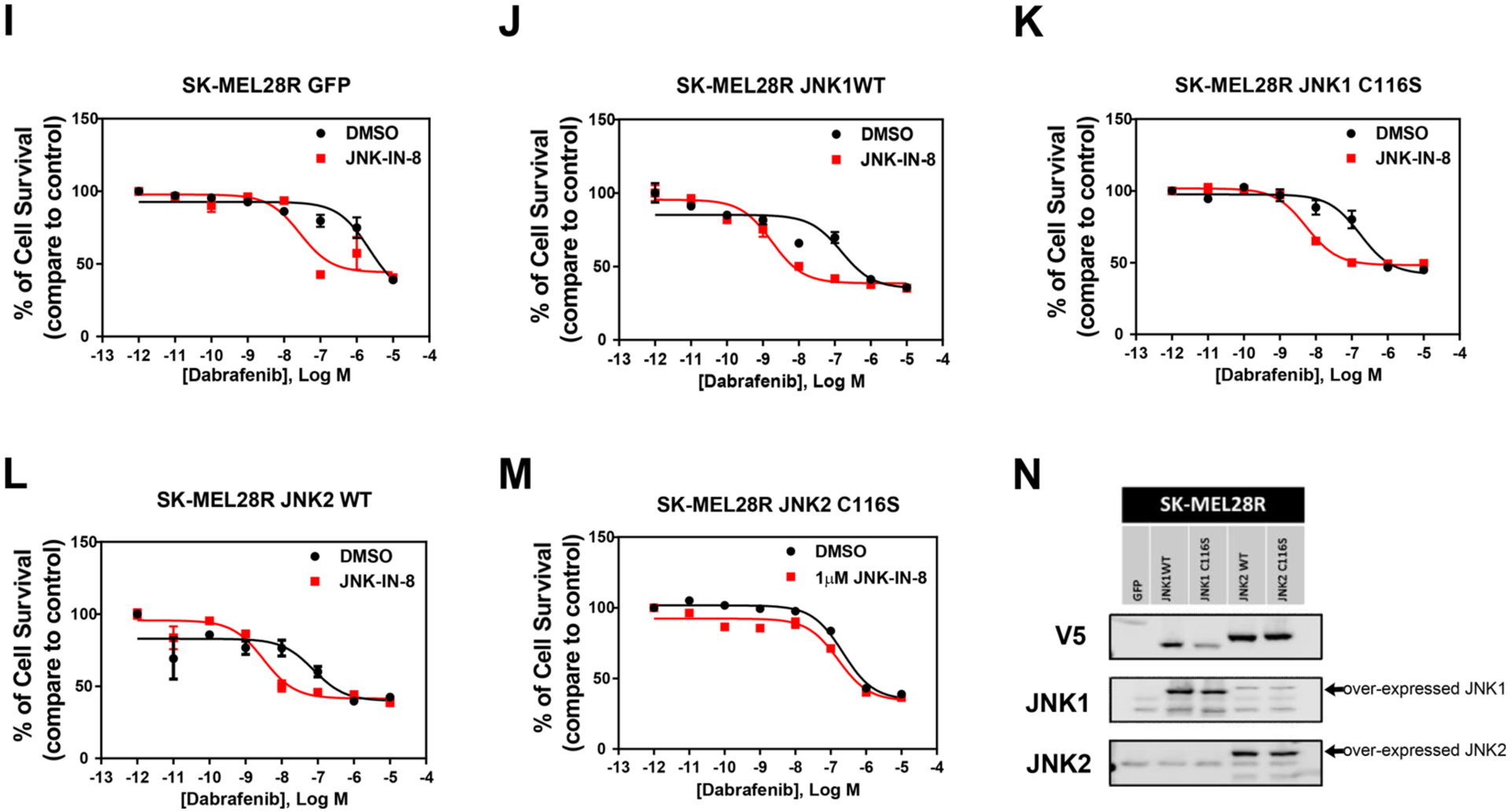
Roles of JNK1 and JNK2 in adaptive BRAFi resistance in a subset of melanoma cell lines. (A and B) Comparison of activation of JNKs and c-Jun between melanoma paired resistant and sensitive cells. (H and N) Western blots of m229R and SK-MEL28R resistant cells, respectively, indicated overexpression of JNK1WT, JNK1C116S, JNK2WT, and JNK2C116S. (C–G and I–M) Sensitivity to BRAFi was measured by the dose–response curve of cell survival to dabrafenib. JNK-IN-8 significantly increased the inhibition effect of dabrafenib in cells overexpressing GFP, JNK1WT, JNK1C116S, and JNK2WT but had no effect on cells overexpressing JNK2C116S.
To further investigate the potential roles of an individual JNK in BRAFi resistance, we examined whether JNK1, JNK2, or both contribute to the high activity of the JNK signaling pathway. Our results demonstrated that only JNK2 protein was detectable in immunoprecipitated complexes following pull-down with phospho-JNK in both SK-MEL28R and m229R resistant cells (Figure 5B). This finding indicated that mainly JNK2 is activated in the BRAFi resistant cells. Here again, we individually transduced these cell lines with JNK1/2WT and JNK1/2C116S. In the absence of JNK-IN-8, the responses are not substantially altered in the lines overexpressing the wild-type and mutant JNKs (black curves, Figure 5C–G,I–M). In addition, to confirm the effect of JNK2 in the adaptive BRAFi resistance, we compared the dose–response curve upon administration of the pan-JNK inhibitor among cells over-expressing WT and mutated JNK1 and JNK2. We found that JNK-IN-8 significantly enhanced the effect of dabrafenib on resistant cells expressing JNK1WT, JNK1C116S, and JNK2WT but had no such effect on cells expressing JNK2C116S (Figure 5C–G). Similar results were observed in SK-MEL28R cells (Figure 5I–M). Interestingly, the inhibition of JNK2 effects in the cell lines appears to be somewhat different in that the maximum effect of dabrafenib is augmented in m229R cells and the IC50 is decreased in SK-MEL28R cells (Supplementary Table 2). These data suggest that activation of JNK2 is sufficient for inducing the BRAFi resistance in this subset of melanoma cell lines.
DISCUSSION
The individual contributions of JNK isoforms to tumorigenesis and specific tumorigenic phenotypes have been difficult to discern because of a lack of appropriate approaches. Through a chemical genetics approach using a covalent, irreversible inhibitor of JNK, JNK-IN-8, we have implicated JNK2 as an important driver of invasiveness, proliferation, and BRAFi resistance in melanoma, and our results additionally suggest that JNK1 exhibits opposing tumor-suppressive activity. To further confirm the role of JNK2 in tumor growth, we evaluated the effect of individual JNKs on tumor growth in a xenograft mouse model.
Although resistance to BRAFi is now well-documented, the impact of other pathways in dictating the response to therapy is not entirely clear because there has been a primary focus on how to better target ERK signaling. c-Jun has emerged recently as a mediator of phenotype switching and BRAFi resistance in melanoma.11 Besides supporting the contributions of the stress-activated JNK kinase signaling pathway to adaptive BRAFi resistance in melanoma cells, we further uncovered that this central role of JNK signaling pathway is due to activation of JNK2 and not JNK1.
Importantly, our data strongly suggest that concomitant inhibition of JNK1 by JNK-IN-8 in the context of JNK2C116S expression (and intact JNK2 signaling) contributes to tumor progression. If this were the case, one would expect that JNK1C116S-expressing cells and tumors might suffer a decrement in invasiveness or progression. This is observed in the invasiveness of 501MEL (Figure 3A) and to a less significant degree in JNK1C116S-expressing HMEL-T1 tumors (Figure 4C).
Inhibitors of JNK2, by preventing both its activation and cellular activity, could potentially be effective inhibitors of BRAF mutant melanoma. Due to the high degree of structural and regulatory similarity between JNK1 and JNK2, there are challenges in developing specific inhibitors for individual JNKs. JNK-interacting peptide (JIP)-based peptides with selectivity against JNK2 have been shown to inhibit breast cancer cell migration.,28 suggesting a potential structural basis for isoform-specific inhibitors. Other JNK inhibitors based upon simultaneous targeting of ATP- and substrate-binding sites suggest an additional strategy by which JNK2 selective inhibitors may potentially be made.29
In conclusion, our novel chemical genetic approach allows the discernment of the roles of individual JNKs in melanoma. We have shown that JNK2 signaling plays a critical role in tumor maintenance and adaptive BRAFi drug resistance in melanoma, supporting the idea that developing compounds that specifically inhibit JNK2 may be therapeutically useful, particularly in a context in which JNK1 activity is augmented.
METHODS
Animals and Cell Lines.
Eight-week-old female athymic nude mice were used for all in vivo experiments. For in vitro experiments, cells were cultured in RPMI 1640 (Sigma) supplemented with 10% fetal bovine serum (FBS) (Sigma), glutamine, and penicillin/streptomycin (Invivogen). Cells were treated with PLX4720, vemurafenib (Selleck Chemicals), or DMSO (1:2000). Cells were maintained at 37 °C in atmospheric oxygen and 5% CO2.
Data Analysis Using cBioPortal.
We conducted an integrative analysis of complex cancer genomics and clinical profiles using cBioPortal data, an open access resource (http://www.cbioportal.org/). This web-based tool was used to query the specific gene expression of JNK1 (MAPK8) and JNK2 (MAPK9) in four cohorts of melanoma patient samples: skin cutaneous melanoma (Broad, Cell 2012, 121 samples), melanoma (Broad/Dana Farber, Nature 2012, 25 samples), skin cutaneous melanoma (TCGA, Provisional, 479 samples), and skin cutaneous melanoma (Yale, Nature Genetics 2012, 91 samples).
Lentivirus Production and Transduction.
Standard protocols for lentivirus packaging and transduction were used as described previously.5 In brief, 10 μg of lentiviral plasmids (pLenti6/GFP, pLenti6/V5-JNK1WT, pLenti6/V5-JNK1C116S, pLenti6/V5-JNK2WT, and pLenti6/V5-JNK2C116S) and 0.3 μg of VSVG/2.7 μg of Δ8.9 Packaging Mix were mixed in 1.5 mL of Opti-MEM medium. The diluted Lipofectamine 3000 reagent was combined with the plasmid mixture and incubated for 30 min at room temperature. HEK293T cells were then transfected with the plasmids and Lipofectamine 3000 mixture and incubated for 72 h at 37 °C and 5% CO2 following the manufacturer’s instructions. Lentiviral supernatants were collected after transfection for 48 and 72 h and centrifuged at 3000 rpm and 4 °C for 10 min. For lentiviral transduction, melanoma cells were transduced using lentiviral supernatants for 48 h. GFP expression was observed under an inverted fluorescence microscope.
Colony Formation Assay.
A total of 250 cells per well were inoculated in a six-well plate in 3 mL of RPMI 1640 medium supplemented with 10% FBS. Two weeks after being plated, the cells were fixed with 100% cold methanol and stained with 1% crystal violet (Sigma) in PBS to visualize the colonies. The number of colonies that were larger than 50 mm (approximately 100 cells) in diameter in each well was counted.
Matrigel Invasion Assay.
A BD BioCoat Matrigel Invasion Chamber was used to measure cell invasion according to the manufacturer’s instructions. Cells (1 × 105 cells/well) suspended in 0.3 mL of RPMI 1640 medium were added to the upper compartment of a 24-well Matrigel-coated or noncoated 8 mm membrane, and RPMI 1640 medium supplemented with 30% FBS was added to the lower compartment. After being incubated for 22 h at 37 °C and 5% CO2, the cells were fixed with 100% methanol and stained with 1% crystal violet in PBS. The number of cells that migrated across the control membrane or penetrated the Matrigel-coated membrane was determined in 10 fields across the center and the periphery of the membrane.
Western Blotting.
Standard protocols for Western blotting were used as described previously.5 Briefly, cells were lysed in standard buffers with protease inhibitors (Roche) and phosphatase inhibitors (Santa Cruz) with extracts run on sodium dodecyl sulfate (SDS)– polyacrylamide gels and transferred to an Immobilon-P transfer membrane (Millipore). Blots were blocked in Odyssey blocking buffer. Proteins were detected using the following commercially available primary antibodies. The primary antibodies against JNK1 2C6 (catalog no. 3708), JNK2 (catalog no. 4672), V5-Tag (catalog no. 13202), SAPK/JNK (catalog no. 9252), phospho-SAPK/JNK Thr183/Tyr185 (catalog no. 4668), c-Jun 60A8 (catalog no. 9165), and phospho-c-Jun Ser63 (catalog no. 9261) were purchased from Cell Signal (Beverly, MA) and used at the indicated concentrations as described by the manufacturer. Fluorescently labeled DyLight800 and DyLight680 secondary antibodies (catalog nos. 5257, 5366, 5470, and 5151; 1:5000; Cell Signaling Technology) were used, and signals were detected using the Li-COR Odyssey Fc imager (LI-COR Biosciences).
Immunoprecipitation.
Dynabeads protein A (50 μL) was coated with 10 μg of targeted antibody p-JNK (Cell Signaling) overnight at 4 °C according to the manufacturer’s instructions (Life Technology). Before being washed and incubated with the cell extract, the antibody was covalently coupled to the beads using BS3 (Thermo Fisher Scientific) as cross-linkers. Dynabeads with the antibody were incubated with 50 μg of the cell extract under gentle rotation at 4 °C overnight. Beads were then washed three times with washing buffer [PBS (pH 7.4) with 0.02% Tween 20] and eluted with 20 μL of elution buffer and 10 μL of 6× SDS loading buffer. Subsequently, beads were heated for 10 min at 70 °C, and the eluates were subjected to Western blotting to detect the amount of activated JNK1 or JNK2 bound to the beads.
Cell Viability.
Cells were plated in a 96-well clear bottom black microplate (Corning) with complete culture medium for 18 h. Cells were treated with increasing concentrations of dabrafenib in the presence or absence of JNK-IN-8 for 72 h. Cell viability was assessed with Cell Titer Glo (Promega) according to the manufacturer’s instructions. Sigmoid curve fitting was performed with a four-parameter logistic equation implemented with Prism (GraphPad Inc.).
In Vivo Tumorigenicity Assay.
Animal studies were conducted according to protocols approved by the M. D. Anderson Cancer Center Institutional Animal Care and Use Committee. A total of 5 × 106 HMEL-t1 cells in PBS (pH 7.3) per mouse were injected subcutaneously into the flank of 8-week-old female nude (NCr, Taconic) athymic mice. Each mouse was implanted with two different tumors (JNKWT and matching JNKC116S) and treated with either the drug or the vehicle. Eight mice were used per condition: JNK1WT/JNK1C116S vehicle, JNK1WT/JNK1C116S JNK-IN-8, JNK2WT/JNK2C116S vehicle, and JNK2WT/JNK2C116S JNK-IN-8 (for a total of 32 mice). Tumors were allowed to grow for 3–4 weeks to a diameter of 150 mm, at which point drug treatment commenced. Mice were treated with either vehicle or JNK-IN-8 (20 mg/kg orally, once daily) beginning on the same day as inoculation. Tumors were completely excised, weighed, and measured with calipers. The width and length of the tumor were measured, and the tumor volume (cubic millimeters) was calculated using the formula tumor volume (mm3) = length (mm) × width (mm2) × 0.52. Mice were weighed biweekly. Final weights increased by an average of 11.2% across all control mice and 11.0% in JNK-IN-8-treated mice.
Reverse-Phase Protein Array (RPPA).
RPPA analysis was performed in the University of Texas M. D. Anderson Cancer Center RPPA/Functional Proteomics core facility. In general, cell lysates generated from the in vivo tumorigenicity assay were serially diluted 2-fold for five dilutions (from undiluted to a 1:16 dilution) and arrayed on nitrocellulose-coated slides in an 11 × 11 format. Samples were probed with 243 antibodies by a tyramide-based signal amplification approach and visualized by the DAB colorimetric reaction. Slides were scanned on a flatbed scanner to produce 16-bit tiff images. The density of each spot from tiff images was identified and quantified by Array-Pro Analyzer. Relative protein levels for each sample were determined and analyzed using bioinformatics methods.
Statistical Analysis.
The results are presented as means ± the standard error of the mean from at least three independent experiments, unless noted otherwise. Asterisks indicate a statistical comparison between the control and experimental group by a Student’s t test, by two-way analysis of variance (ANOVA) with secondary Bonferroni multiple-comparison test, or by one-way or repeated-measures ANOVA with a Dunnett multiple-comparison test as indicated in the figure legends. p values of <0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the animal support services at the South Campus Vivarium (M. D. Anderson Cancer Center) and RPPA Core Facility (M. D. Anderson Cancer Center) (National Cancer Institute Grant CA16672). K.Y.T. acknowledges support from the Elsa U. Pardee Foundation and M. D. Anderson Cancer Center IRG Program. K.N.D. acknowledges support from the National Institutes of Health (GM123252), the Cancer Research and Prevention Institute of Texas (RP180880, RP160657, and RP140108), and the Welch Foundation (F-1390). The authors gratefully acknowledge receiving cell lines from Lynda Chin, MD (MD Anderson Cancer Center), Roger Lo, MD, PhD (UCLA) and Meenhard Herlyn, DVM, DSc (Wistar Institute).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.9b00083.
Supplementary Figures 1 –3 and Supplementary Tables 1 and 2 (PDF)
The authors declare the following competing financial interest(s): N.S.G. is a founder, science advisory board member (SAB), and equity holder in Gatekeeper, Syros, Petra, C4, B2S, and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Her2llc, Deerfield, and Sanofi. N.S.G. and T.Z. are inventors of a patent covering JNK-IN-8 owned by DFCI.
REFERENCES
- (1).Weston CR, and Davis RJ (2007) The JNK signal transduction pathway. Curr. Opin. Cell Biol 19, 142–149. [DOI] [PubMed] [Google Scholar]
- (2).Girnius N, Edwards YJ, Garlick DS, and Davis RJ (2018) The cJUN NH2-terminal kinase (JNK) signaling pathway promotes genome stability and prevents tumor initiation. eLife 7, e36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kennedy NJ, Sluss HK, Jones SN, Bar-Sagi D, Flavell RA, and Davis RJ (2003) Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes Dev. 17, 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P, Jacks T, and Davis RJ (2011) Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol. Cell. Biol 31, 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vin H, Ojeda SS, Ching G, Leung ML, Chitsazzadeh V, Dwyer DW, Adelmann CH, Restrepo M, Richards KN, Stewart LR, Du L, Ferguson SB, Chakravarti D, Ehrenreiter K, Baccarini M, Ruggieri R, Curry JL, Kim KB, Ciurea AM, Duvic M, Prieto VG, Ullrich SE, Dalby KN, Flores ER, and Tsai KY (2013) BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. eLife 2, No. e00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pietkiewicz S, Sohn D, Piekorz RP, Grether-Beck S, Budach W, Sabapathy K, and Janicke RU (2013) Oppositional regulation of Noxa by JNK1 and JNK2 during apoptosis induced by proteasomal inhibitors. PLoS One 8, No. e61438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ke H, Harris R, Coloff JL, Jin JY, Leshin B, Miliani de Marval P, Tao S, Rathmell JC, Hall RP, and Zhang JY (2010) The c-Jun NH2-terminal kinase 2 plays a dominant role in human epidermal neoplasia. Cancer Res. 70, 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson S, and Ronai Z (2007) Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 11, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pathria G, Garg B, Garg K, Wagner C, and Wagner SN (2016) Dual c-Jun N-terminal kinase-cyclin D1 and extracellular signal-related kinase-c-Jun disjunction in human melanoma. Br. J. Dermatol 175, 1221–1231. [DOI] [PubMed] [Google Scholar]
- (10).Haass NK (2016) JNK-ERK signalling in melanoma: rewired or entangled? Br. J. Dermatol 175, 1139–1140. [DOI] [PubMed] [Google Scholar]
- (11).Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M, Bollag G, Long GV, Tulchinsky E, Rizos H, Pearson RB, McArthur GA, Dhillon AS, and Ferrao PT (2015) The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signaling l8, No. ra82. [DOI] [PubMed] [Google Scholar]
- (12).Fallahi-Sichani M, Moerke NJ, Niepel M, Zhang T, Gray NS, and Sorger PK (2015) Systematic analysis of BRAF(V600E) melanomas reveals a role for JNK/c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol. Syst. Biol 11, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Stark MS, Woods SL, Gartside MG, Bonazzi VF, Dutton-Regester K, Aoude LG, Chow D, Sereduk C, Niemi NM, Tang N, Ellis JJ, Reid J, Zismann V, Tyagi S, Muzny D, Newsham I, Wu Y, Palmer JM, Pollak T, Youngkin D, Brooks BR, Lanagan C, Schmidt CW, Kobe B, MacKeigan JP, Yin H, Brown KM, Gibbs R, Trent J, and Hayward NK (2012) Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet 44, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cellurale C, Girnius N, Jiang F, Cavanagh-Kyros J, Lu S, Garlick DS, Mercurio AM, and Davis RJ (2012) Role of JNK in mammary gland development and breast cancer. Cancer Res. 72, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cellurale C, Weston CR, Reilly J, Garlick DS, Jerry DJ, Sluss HK, and Davis RJ (2010) Role of JNK in a Trp53- dependent mouse model of breast cancer. PLoS One 5, No. e12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Anbalagan M, and Sabapathy K (2012) JNK1 and JNK2 play redundant functions in Myc-induced B cell lymphoma formation. Int. J. Cancer 130, 1967–1969. [DOI] [PubMed] [Google Scholar]
- (17).Lu H, Liu S, Zhang G, Wu B, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, Zhang W, Chen G, Cheng C, Zeng J, Wu LW, Zhang J, Liu X, Xu W, Krepler C, Sproesser K, Xiao M, Miao B, Liu J, Song CD, Liu JY, Karakousis GC, Schuchter LM, Lu Y, Mills G, Cong Y, Chernoff J, Guo J, Boland GM, Sullivan RJ, Wei Z, Field J, Amaravadi RK, Flaherty KT, Herlyn M, Xu X, and Guo W (2017) PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, and Gray NS (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang J, Kuiatse I, Lee AV, Pan J, Giuliano A, and Cui X (2010) Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol. Cancer Res 8, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Santibanez JF (2006) JNK mediates TGF-beta1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett. 580, 5385–5391. [DOI] [PubMed] [Google Scholar]
- (21).Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, and Xu J (2011) Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 71, 3980–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Van Nes J, Chan A, Van Groningen T, Van Sluis P, Koster J, and Versteeg R (2013) A NOTCH3 transcriptional module induces cell motility in neuroblastoma. Clin. Cancer Res 19, 3485–3494. [DOI] [PubMed] [Google Scholar]
- (23).Seidelin JB, Larsen S, Linnemann D, Vainer B, Coskun M, Troelsen JT, and Nielsen OH (2015) Cellular inhibitor of apoptosis protein 2 controls human colonic epithelial restitution, migration, and Rac1 activation. Am. J. Physiol Gastrointest Liver Physio l308, G92–99. [DOI] [PubMed] [Google Scholar]
- (24).Jacob A, and Prekeris R (2015) The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Costa AM, Pinto F, Martinho O, Oliveira MJ, Jordan P, and Reis RM (2015) Silencing of the tumor suppressor gene WNK2 is associated with upregulation of MMP2 and JNK in gliomas. Oncotarget 6, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen WT, Ebelt ND, Stracker TH, Xhemalce B, Van Den Berg CL, and Miller KM (2015) ATM regulation of IL-8 links oxidative stress to cancer cell migration and invasion. eLife 4, e07270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mooz J, Oberoi-Khanuja TK, Harms GS, Wang W, Jaiswal BS, Seshagiri S, Tikkanen R, and Rajalingam K (2014) Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci. Signaling l7, No. ra73. [DOI] [PubMed] [Google Scholar]
- (28).Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, Van Den Berg CL, and Dalby KN (2011) Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chem. Biol 6, 658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stebbins JL, De SK, Pavlickova P, Chen V, Machleidt T, Chen LH, Kuntzen C, Kitada S, Karin M, and Pellecchia M (2011) Design and characterization of a potent and selective dual ATP- and substrate-competitive subnanomolar bidentate c-Jun N-terminal kinase (JNK) inhibitor. J. Med. Chem 54, 6206–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.