

Abstract
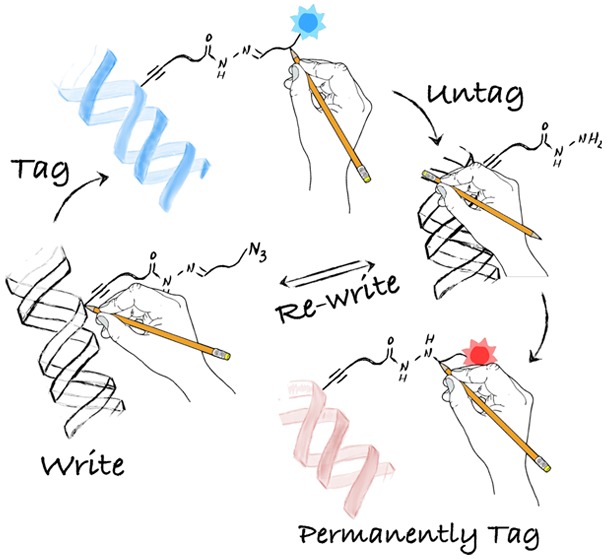
Current methods for bioconjugation rely on the introduction of stable linkers that lack the required versatility to perform sequential functionalizations. However, sequential manipulations are an increasing requirement in chemical biology because they can underpin multiple analyses of the same sample to provide a wider understanding of cell behavior. Here, we present a new method to site-selectively write, remove, and rewrite chemical functionality to a biomolecule, DNA in this case. Our method combines the precision and robustness of methyltransferase-directed labeling with the reversibility of acyl hydrazones and the efficiency of click chemistry. Underpinning the method is a new S-adenosyl-l-methionine derivative to site-selectively label DNA with a bifunctional chemical handle containing an acyl hydrazone-linker and a terminal azide. Functional tags are conjugated via the azide and can be removed (i.e., untagged) when needed at the acyl hydrazone via exchange with hydroxyl amine. The formed hydrazide-labeled DNA is a versatile intermediate that can be either rewritten to reset the original chemical handle or covalently reacted with a permanent tag. This ability to write, tag, untag, and permanently tag DNA is exploited to sequentially introduce two fluorescent dyes on DNA. Finally, we demonstrate the potential of the method by developing a protocol to sort labeled DNA using magnetic beads, with subsequent amplification of the sorted DNA sample for further analysis. The presented method opens new avenues for site-selective bioconjugation and should underpin integrative approaches in chemical biology where sequential functionalizations of the same sample are required.
Short abstract
We present a method using MTases, acyl hydrazone, and click chemistry to site-selectively tag, untag, and rewrite on DNA. Sequential labeling of DNA or sorting PCR DNA demonstrate its versatility.
Introduction
Established and emerging approaches for studying biomolecules rely on their conjugation with chemical groups or functional tags (Figure 1A).1−4 The introduced functionalities enable manipulations that go from simple extraction and purification from complex mixtures to advanced analytical studies. This way bioconjugation has enabled research into post-translational modifications and the biological processes where biomolecules are involved and, more recently, the exploitation of these molecules for application.5−10 Critically, being able to introduce these chemical handles in a precise location of the biomolecule minimizes the impact that bioconjugation has on secondary structure and activity and can throw light on modifications and regulation of biomolecules. Common methods for site-selective functionalization of these biomolecules include introducing site-specific mutations or the development of selective chemical handles that discriminate similar reacting groups within these molecules.11−17
Figure 1.

Schematic representation of methods for labeling of biomolecules. (A) Current methodologies are based on the introduction, via one or multiple steps, of stable chemical linkers. Common challenges include site-selectivity, tolerance to functional groups, or reversibility. (B) Our method achieves site-selective MTase-directed labeling of DNA ① to then introduce chemical tags via azide–alkyne cycloaddition ②. This introduced functionality can then be removed (i.e., untagged) via exchange at the Schiff-base ③, to give an intermediate hydrazide-labeled DNA that can be rewritten via Schiff-base formation ④ to give the original functionality introduced via MTase-labeling. Alternatively, this hydrazide-labeled DNA can be functionalized via covalent chemistry to introduce a permanent tag if needed ⑤.
In recent years, there has been a growing interest in the use of chemoenzymatic methods to label biomolecules.18−20 These methods not only introduce site-selective chemical modifications but can label biomolecules in complex mixtures, such as living cells or their lysates. Methyltransferases (MTases) are emerging as a key class of enzymes for the site-selective functionalization of biomolecules because of their versatility in terms of targets and functional groups they can accommodate.21−25 Most MTases are responsible for transferring a methyl group from the naturally occurring cofactor S-adenosyl-l-methionine (AdoMet) and have been identified with targets as diverse as small molecules, carbohydrates, proteins, and nucleic acids. Most importantly, a wide range of MTases can accommodate larger groups in their binding pocket, so that by manipulating the chemical structure of AdoMet it is possible to hijack MTase machinery to introduce functional groups to biomolecules.21−28 Commonly, MTase-directed ligation is used to introduce “clickable” groups that are used to label the biomolecules with fluorescent and other functional tags (Figure 1A).26−31 MTase-directed ligation is now finding application in imaging and in genomic and metabolomic analysis.
Invariably, chemical and enzymatic strategies to label biomolecules have been focused on developing stable linkages that rely on the covalent attachment of functional tags. However, this approach often results in the modification of the biomolecule with bulky functional groups that can compromise the chemical and physical properties of the targeted biomolecule. In recent years, efforts have been made to develop reversible chemistries for bioconjugation that let scientists remove the introduced chemical functionality once it has served its purpose.5,16,17 For instance, recent examples in the field of MTase-directed labeling have explored the use of disulfide linkers32 or of light-cleavable moieties,33,34 that can be cleaved under the right circumstances. Invariably, however, current bioconjugation techniques lack the versatility to be rewritten if needed and limit the study of these biomolecules to a single analytical process. This approach is at odds with the current trends in cell biology, where integrative approaches that combine multiple analytical methods are needed to provide a holistic understanding of cell behavior.35−38 This need is particularly true in single-cell analyses, where material is limited.
In order to address these limitations, here we present a new method for the functionalization of nucleic acids. The selectivity and robustness of MTase-directed labeling is combined with the reversibility of Schiff-bases and the efficiency of click chemistry, to introduce a bifunctional chemical handle on DNA that allows us to perform multiple functionalizations (Figure 1B). To this end, we have developed a new class of AdoMet derivatives, containing an acyl hydrazone or oxime linker and a terminal azide at the sulfonium center. First, we demonstrate that representative MTases are able to accommodate these bifunctional cofactors to site-selectively label DNA, including oligonucleotide and plasmid DNA. Then, we identify that the acyl hydrazone-linker is amenable to cleavage via exchange with H2NOH·HCl, providing a route to untag the chemical functionality introduced. Furthermore, we demonstrate that the formed hydrazide-labeled DNA can be rewritten to recover the original chemical handle. Alternatively, this hydrazide-labeled DNA can be covalently functionalized using nonreversible chemistries to introduce permanent tags if required. The potential of the method to underpin new discoveries at the interface between chemistry and biology is demonstrated through the sequential labeling of DNA with two fluorescent dyes and the development of a new protocol to sort fragments of DNA using magnetic beads. Labeled DNA fragments are sorted under mild conditions with high yields, remaining functional to then be amplified by real time polymerase chain reaction (real time PCR).
Results and Discussion
Cofactor Synthesis—AdoMet Derivatives Carrying Schiff-Base Linkers and a Terminal Azide
Our initial aim was to develop an AdoMet analog that carried a reactive and rewritable (or reversible) chemical handle. While several strategies are available to introduce reactive moieties to AdoMet derivatives, few of these are reversible,32−34 and none allow repeated functionalization of the target site. An ideal reactive linker should carry a chemical functionality orthogonal to common biological moieties (e.g. hydroxyl, amino, and carboxyl). In nature, Schiff-bases are commonly used to this end and chemists have now exploited the versatility of the C=N bond to develop a plethora of applications for this dynamic chemistry.39−44 The stability of the C=N bond can be tailored as a function of the “amine” used, with hydrazides and alkoxyamines demonstrating the largest stability ranges. In particular, Dawson et al. have exploited this versatility to reversibly tag biotin onto proteins through an acyl hydrazone linker.45 The biotin-modified protein could be captured via affinity chromatography to then be released under mild conditions via competitive exchange. While this work clearly demonstrates the potential of Schiff-bases to reversibly label biomolecules, the site-selective modification of biomolecules with these chemistries still remains a challenge.
With these principles in mind, we prepared linkers 1a and 1b, carrying the desired hydrazide or alkoxyamine moieties protected as tert-butylcarbamates (see sections 3.1–3.7 in the Supporting Information for details). These linkers were then reacted with S-adenosyl-l-homocysteine (AdoHcy) under standard conditions to give Boc-protected AdoMet derivatives 2 (Scheme 1). Although MTases are not particularly affected by impurities in the cofactor mixture, the excess of linker 1 was removed via extraction with diethyl ether because the presence of this linker could result in nonspecific alkylation of DNA in subsequent assays. The crude AdoMet derivatives 2 obtained this way were freeze-dried and immediately deprotected under acidic conditions. Purification of the hydrazide- and alkoxyamine-AdoMet derivatives 3 was performed by HPLC, to separate both diastereomers of each cofactor 3 (Figure S20 and Figure S23), a separation that was not possible at later stages. Our initial intention was to use these cofactors to introduce the reactive moiety in DNA but these deprotected AdoMet derivatives 3 slowly degraded (Figures S26 and S27), in particular following freeze-drying (Figure S28). For instance, HPLC analysis of cofactor 3b after freeze-drying revealed the presence of an additional peak at higher retention times (∼31 min). MS analysis of this peak suggested a mass of 536.61 Da, very close to that of the alkoxy AdoMet derivative (537.62 Da). This difference in molecular mass, together with the increase in retention time, suggested degradation of 3b was also occurring via intramolecular nucleophilic attack of the terminal amine of the linker to the sulfonium center (Figure S28).
Scheme 1. Synthesis of AdoMet Derivatives 4.

Conditions: (i) HCO2H, AcOH, 35 °C; (ii) TFA; (iii) (1) TFA, (2) 5, 20 mM ammonium acetate pH 5.5 water/MeOH. Full details can be found in the Supporting Information.
Degradation of AdoMet and derivatives is common but does not compromise their application. As just mentioned, MTases can promote transalkylation even with the complex mixture obtained upon degradation. Moreover, degradation is normally suppressed upon storage of these cofactors at low temperature and in a mildly acidic buffer (in our case 0.1% acetic acid 15–20 mM in cofactor).46,47 However, we decided to try to minimize the degradation observed via this intramolecular rearrangement (Figure S28). To this end, AdoMet derivatives 3 were reacted immediately after purification by HPLC with commercially available benzaldehyde 5 (Scheme 1). A slight excess of aldehyde 5 (1.2 equiv) was employed to ensure full functionalization of the intermediate 3, and the obtained cofactors 4 freeze-dried and stored in 0.1% acetic acid solution without further purification. The cofactor analogues 4 that were formed now contained a reactive terminal azide, suitable for tagging via standard azide–alkyne cycloaddition and with the additional advantage of expanding the range of functional molecules that we had access to in the tagging step of our cycle (Figure 1). Additionally, cofactors 4 carried a Schiff-base linker that maintained the reversible functionality, giving us a route to remove and rewrite chemical functionality. The chemical identity of these cofactors was confirmed via HRMS (section 3.10 in the Supporting Information). No degradation of AdoMet derivatives 4 was observed following this protocol (Figure S31, 4b as a representative example), and these cofactors could be used in the reversible labeling of DNA without further purification.
Figure 4.

Rewriting oligo DNA. (A) Schematic representation of untagging and rewriting of chemical functionality on oligo DNA using the AdoMet derivative 4a and aldehyde 5. Analytical UPLC chromatograms of oligo DNA following incubation with M.TaqI and 4a, followed by incubation with 10 equiv of H2NOH·HCl in 10 mM ammonium acetate pH 4 (B), and oligo DNA from B followed by incubation with aldehyde 5 (C). Chromatogram of oligo DNA following incubation with M.TaqI and 4a (D) and of aldehyde 5 incubated with 10 equiv of H2NOH·HCl in 10 mM ammonium acetate pH 4 (E) shown for comparison. UPLC conditions: 0.5% triethyl amine, water/MeCN gradient at 60 °C. Under these conditions, oligo DNA melts and both strands of DNA can be observed independently.
Writing Chemical Functionality—MTase-Directed Labeling
Following successful synthesis of AdoMet derivatives 4, a restriction assay was used to demonstrate that representative MTases could accommodate cofactors 4 to efficiently and site-selectively label DNA.31 In this assay, M.TaqI, an N6-adenine DNA MTase, was used to label pUC19 DNA, which has four recognition sites (TCGA) for this enzyme (Figure 2).48 Successful alkylation of DNA by M.TaqI results in protection of the plasmid from restriction digestion by R.TaqI, an endonuclease with the same target site as M.TaqI.49 Our intention was to demonstrate not only that cofactors 4 could be employed by a relevant MTase but also that they could modify DNA with a complex topology (Figure 2B).
Figure 2.

MTase-directed labeling of plasmid DNA. (A) Schematic representation of restriction assay and (B) gel electrophoresis of pUC19 following enzymatic treatment with M.TaqI and/or R.TaqI in the presence and absence of AdoMet (375 μM) or AdoMet derivative 4b. In the absence of M.TaqI-mediated alkylation (lanes 4, 8, and 12), pUC19 is cut into fragments, of which the largest three can be identified by gel electrophoresis. M.TaqI-mediated alkylation with AdoMet (line 10) or derivative 4b (lanes 1–3 and 5–6) results in partial to full protection from restriction by R.TaqI, with mainly open circular and supercoiled plasmid DNA being observed by gel electrophoresis. Controls in the absence of AdoMet derivatives (lanes 11 and 12), in the absence of M.TaqI (lanes 4, 8, and 12) and in the absence of R.TaqI (lane 9) are included.
The native M.TaqI substrate, AdoMet, was used as a positive control for these experiments. In the absence of cofactor, M.TaqI is unable to alkylate pUC19, and bands corresponding to the three biggest DNA fragments formed upon digestion with R.TaqI were observed (Figure 2, lane 11). A similar effect is observed in the absence of M.TaqI (Figure 2, lane 12), demonstrating that neither isomer of AdoMet derivative 4b interferes with the ability of R.TaqI to digest plasmid DNA (Figure 2, lane 4 and 8). More importantly, in the presence of both diastereomers of 4b, M.TaqI was able to functionalize pUC19, although with different efficiencies, and with limited evidence of DNA digestion. In this case, only bands corresponding to open circular or supercoiled DNA were observed (Figure 2, lanes 1–3 and 5–7), similar to those observed when AdoMet was used (Figure 2, lane 10) or when no digestion was performed (Figure 2, lane 9). Dilution of the amount of cofactor used revealed that the second fraction had a higher activity. A similar effect was observed for the acyl hydrazone derivative 4a (Figure S32), demonstrating that both analogues had the potential to be employed for the labeling of biomolecules. The reactivity of both diastereomers was unexpected, as it is commonly assumed that MTases are stereoselective,27 and thus we considered that isomerization around the sulfonium center was occurring in the assay conditions. However, no isomerization was observed by HPLC following incubation at 50 °C of the second isomer of cofactor 4b (Figure S31). Thus, we can not determine whether the activity of M.TaqI with both isomers of 4b is due to a lack of specificity (for this stereocenter) of this MTase,22,50 isomerization in the presence of the MTase, or the presence of small amounts of the active stereoisomer.
Having demonstrated that M.TaqI was able to catalyze transalkylation of plasmid DNA with cofactors 4, we then decided to test whether other relevant DNA MTases could use these AdoMet derivatives as cofactors. M.MpeI is a cytosine-C5 MTase. Much like human DNMT1, this bacterial enzyme targets the CpG dinucleotide, whose methylation is involved in the mechanism of gene regulation in vertebrates.32,51,52 Thus, pUC19 was incubated with mutant M.MpeI (Q136A, N347A) and 4a, and then challenged with R.HaeII, a restriction enzyme that targets a subset of the CpG dinucleotides.53 To our delight, we also observed efficient transalkylation of plasmid DNA using this MTase (Figure S33), although only isomer II seemed to show significant activity with this enzyme.
Further evidence of the ability of MTases to alkylate DNA with AdoMet derivatives 4 was carried out using a 14-base pair (bp) oligonucleotide with one copy of the M.TaqI sequence (TCGA) in the center. MTase-directed labeling was achieved in the same way as the protection assay described above; however, restriction digestion was not carried out. Our intention here was to monitor the MTase-directed labeling of the oligo DNA via HPLC, so that further details about the labeling process could be observed. Following incubation of the oligo DNA with M.TaqI in the presence of AdoMet or AdoMet derivatives 4, samples were analyzed by HPLC and MS. HPLC analysis was performed above the melting temperature of the DNA so that both strands could be clearly identified in the chromatogram (Figure 3). A clear shift in the retention time was seen upon labeling with AdoMet (Figure S36), 4a, or 4b (Figure 3, middle) when compared to the retention times of the unmodified DNA (Figure 3, top and Figure S35). This shift was observed for both peaks, demonstrating that M.TaqI was able to label both strands as a consequence of the palindromic nature of the sequence this MTase recognizes. Moreover, the shift was proportional to the size and nature of the linker transferred, with the AdoMet methylation resulting in a small shift in retention time (Figure S36) and the oxime derivate 4b giving the biggest shift (Figure 3). In these chromatograms, we could also see what we expected to be, and later confirmed, free hydrazide (Figure 3, left, peaks at ∼13 min) and hydroxylamine (Figure 3, right, peaks at ∼19 min). The presence of this small amount of untagged oligo DNA is likely due to hydrolysis under the HPLC conditions. Analysis of the individual peaks was carried out using MS, which confirmed labeling was successful and that only one linker per DNA chain had been conjugated. MS also confirmed the nature of the side chain functionality introduced following incubation (Figures S35–S44).
Figure 3.

Writing and untagging chemical functionality on oligo DNA. Analytical HPLC chromatograms of oligo DNA (top) and oligo DNA following incubation with M.TaqI and 4 (middle), and with M.TaqI and 4, followed by incubation with 10 equiv of H2NOH·HCl in 10 mM ammonium acetate pH 4.0 (bottom). HPLC conditions: 0.1 M triethylammonium acetate buffer, pH 7.0 (A)/MeCN (B) gradient at 60 °C. Under these conditions, oligo DNA melts and both strands of DNA can be observed independently.
Untagging the Introduced Functionality—Competitive Exchange of the Schiff-Base
Following successful labeling of the oligo DNA, these samples were then used to demonstrate the reversible nature of the Schiff-base introduced and its potential to be efficiently cleaved. Aliquots of labeled oligo DNA were incubated at 50 °C for 1.5 h with 10 equiv of H2NOH·HCl. This competing reagent was introduced to facilitate exchange with the Schiff-base and the pH of the samples was adjusted to pH 4.0.54,55 HPLC analysis showed a clear shift in the retention time of the acyl hydrazone-labeled DNA following treatment with the competing reagent (Figure 3, left). Over 85% of the functional linker was cleaved under these conditions. The new peak shifted to lower retention time, as expected following the loss of the potentially hydrophobic aldehyde 5. MS analysis confirmed that these peaks (∼13 min) corresponded to the hydrazide-labeled oligo DNA (Figures S46 and S47). Conversely, under these conditions the oxime-labeled DNA remained intact (Figure 3, right and Figure S48), consistent with the higher stability of this type of Schiff-base.56
Incubation of DNA with hydroxylamine has been associated with the induction of mutations as a result of a nucleophilic attack to pyrimidine bases to give 6-hydroxylamino-5,6-dihydroxycytidine and N4-dihydroxycytidine among others.57,58 Moreover, loss of purine bases has been reported at pH 4.59 To evaluate whether these side-reactions were being observed under the conditions used for cleavage of the Schiff-base, analysis of the HRMS for each of the oligo DNAs was performed. To our delight, no evidence of these side-reactions was observed. We believe the absence of DNA damage is the result of the low concentration of H2NOH·HCl used (typically 20 mM) and the short incubation times (typically 1.5 h).
Rewriting the Original Functionality via Schiff-Base Formation
The next stage was to demonstrate that the labeled DNA could be rewritten to recover the original functionality introduced during the MTase-directed labeling with cofactor 4a (Figure 4A). To this end, the hydrazide-functionalized oligo DNA (Figure 4B) was incubated in the presence of an excess of aldehyde 5. As predicted, a clear shift in the retention time of the main peaks associated with oligo DNA was observed (Figure 4C). Comparison of this chromatogram to that obtained following incubation of oligo DNA with M.TaqI and cofactor 4a (Figure 4D) showed good overlap of the peaks associated with azide-functionalized DNA at ∼6.2 and 6.4 min and a similar ratio of this peak to that of the free hydrazide (at 3.8 and 4.0 min). The presence of a small amount of hydrazide-labeled oligo DNA is likely due to hydrolysis of the Schiff-base under the UPLC conditions. Three additional peaks were observed following rewriting with aldehyde 5, which overlapped with those observed when this aldehyde was incubated with H2NOH·HCl (Figure 4E). Analysis of the individual peaks was carried out by UPLC-MS, which confirmed rewriting was successful, that only one aldehyde had been conjugated, and the nature of the chemical functionality on the oligo DNA (Figures S20–S50).
Write, Untag, and Permanently Tag
One common limitation of the current chemistries used for site-selective functionalization of biomolecules is their lack of versatility. Once a chemical moiety is introduced to, for instance, facilitate the purification of the biomolecule (e.g. biotin), these moieties remain attached to the targeted biomolecule. More importantly, these moieties can not be further functionalized under mild and straightforward conditions, to introduce new functionality (e.g. fluorophores, targeting ligands) often required for further research. Our strategy for MTase-directed labeling of DNA using AdoMet derivative 4a results in DNA that now carries a Schiff-base that can be efficiently cleaved. Removing the chemical functionality via exchange with H2NOH·HCl results in a hydrazide-labeled DNA that should be easily functionalized using standard covalent bioconjugation techniques. To this end, fragments of DNA generated by PCR, containing 17 CpG sites,60 were site-selectively labeled with M.MpeI. Labeling was followed by incubation with H2NOH·HCl to remove the azide moiety and then reacted with a commercially available NHS-activated fluorophore ATTO 647N (7) to introduce a permanent fluorescent tag (Figure S34A). The reaction was monitored via gel electrophoresis (Figure S34B) and showed specific conjugation of ATTO 647N (7) to hydrazide-labeled DNA. While no red-fluorescence was observed in the absence of ATTO 647N (7) (Figure S34B, lanes 1, 3, and 5), this dye is positively charged and was able to nonspecifically associate with the DNA in all samples (Figure S34B, lanes 2, 4, and 6). When we compared the intensities of the red and green channels (SYBR Green), to evaluate the degree of labeling with ATTO 647N (7) per unit of DNA, we observed that the hydrazide-labeled PCR fragments were giving the highest ratio (Figure S34B, lanes 6), 4.8 times higher for the DNA than in the absence of the untag step (H2NOH·HCl treatment, Figure S34B, lanes 4), and over 23 times higher than in the absence of MTase-directed labeling (Figure S34B, lanes 2).
Sequential Functionalization with Complementary Fluorescent Dyes
The final stage was to demonstrate the utility of our strategy and the potential to perform multiple functionalizations on the same sample of DNA. To this end, we decided to explore the consecutive labeling of short DNA fragments with two different fluorescent dyes. DNA fragments were first incubated with M.MpeI and cofactor 4a, to yield azide-functionalized DNA (Figure 5A, step ①). This functionalization resulted in a small shift in the migration of the DNA on the gel (Figure 5B, step ①) but, as expected, no fluorescence was observed (Figure 5C, step ①). A further shift in the migration was observed when the azide-functionalized DNA was reacted with TAMRA-DBCO 6 (Figure 5B, step ②), but more importantly, emission from DNA-associated TAMRA fluorophore was clearly observed (Figure 5C, step ②). Removal of the TAMRA tag was achieved by incubation with an excess of H2NOH·HCl. No fluorescence was observed from the resulting DNA fragments (Figure 5C, step ③), and a shift back to the original migration was observed (Figure 5B, step ③), suggesting that this hydrazide linker had little impact on the physical properties of the DNA. Incubation of this hydrazide-labeled DNA with NHS-activated ATTO 647N (7) resulted in a new shift in migration (Figure 5B, step ④) and the corresponding appearance of fluorescence, now visible under red illumination (Figure 5C, step ④).
Figure 5.

Sequential functionalization. (A) Schematic representation of the sequential functionalization of DNA: DNA was first written via site-selective MTase-directed labeling using cofactor 4a ①. The obtained azide-functionalized DNA fragments were then reacted with TAMRA-DBCO tag (6) via azide–alkyne cycloaddition ②. The introduced TAMRA was then removed via exchange with H2NOH·HCl ③, to give hydrazide-labeled DNA fragments that were covalently functionalized with NHS-activated ATTO 647N (7) as a permanent tag ④. Functionalization was monitored using gel electrophoresis. Conditions: DNA concentration; 7 ng/μL, release buffer; 10 mM ammonium acetate, pH 6.8, 1 M NaCl, 0.01% SDS. DNA stained with GelRed. Gel was visualized using a Bio-Rad Pharos FX (GelRed: excitation, trans-UV; emission filter, 590/110 nm; TAMRA: excitation, epi-green illumination; emission filter: 602/50 nm; ATTO 647N (7): excitation, epi-red illumination; emission filter: 700/50 nm). TAMRA channel was colored yellow and ATTO 647N (7) was colored red for visualization. (B) GelRed channel and (C) composite image of TAMRA and ATTO 647N channels. Full chemical structures of the fluorescent dyes are available in the Supporting Information.
Application—DNA Sorting via Reversible Capture and Further Amplification via PCR
Having demonstrated that each of the steps of our method were feasible and that sequential functionalization of DNA was possible, we decided to test the potential of this method to underpin sequential manipulations of DNA that go beyond fluorescent tagging. A common application of MTase-directed labeling of DNA is the manipulation of DNA for sequencing.23,26,27 In this application, DNA carrying sequences of interest is normally sorted through site-selective labeling with affinity tags such as biotin, followed by capture with magnetic beads or via affinity chromatography. Release of the captured nucleic acid is commonly achieved using denaturing conditions that disrupt the biotin-(strept)avidin binding. However, denaturing conditions for release can result in low percentages of labeled-DNA isolated (see Figure 6B for an example). We anticipated that an alternative strategy could be employed using the AdoMet derivative 4a, but the presence of the acyl hydrazone linker would allow us to efficiently release the captured DNA under mild conditions. A similar concept has been employed for the development of photoreleasable and reducible linkers that avoid the use of denaturing conditions.
Figure 6.

Capture and release of DNA using MTase-directed labeling and competitive exchange. (A) Schematic representation of functionalization of DNA fragments with a biotin tag using both MTase-directed labeling and subsequent DBCO conjugation. (B) Schematic representation of the capture and release of DNA fragments using the AdoMet derivative 4a. (C) Percentage of DNA remaining following capture with magnetic beads and (D) percentage DNA released from the magnetic beads following treatment with H2NOH·HCl (cycles 1–3) and denaturing conditions (reflux in denaturing buffer, cycles 4 and 5). DNA was either unlabeled (■), incubated with M.MpeI and 4a (●), or incubated with M.MpeI and azide containing cofactor 8 (○). Conditions: DNA concentration; 7 ng/μL; capture buffer: 10 mM Tris, 1 M NaCl, pH 8.5; release buffer: 20 mM H2NOH·HCl (22624 equiv per labeling site), 10 mM ammonium acetate, pH 6.8, 1 M NaCl, 0.01% SDS; denaturing buffer: 0.1% SDS. N = 3. Error bars indicate range. The amount of DNA was quantified using a Qubit fluorometer and normalized to the starting amount of DNA (C) or the amount of DNA captured (D).
Our studies thus focused on the capture and release of the 203 bp DNA fragment containing 17 CpG sites targeted for alkylation by M.MpeI.60 After labeling with cofactor 4a, the fragments were further modified by reaction with a bifunctional linker molecule, carrying biotin and dibenzocyclooctyne (DBCO) groups (Figure 6A). MTase-directed labeling was also performed with AdoMet derivative 8 that, while including an azide moiety for further functionalization using DBCO chemistry, did not contain the reversible acyl hydrazone linker. Capture experiments, using streptavidin-coated beads, were performed using a high salt tris buffer. We consistently captured in excess of 80% of the DNA (Figure 6C, Figures S51 and S52), while almost no unlabeled DNA was adsorbed onto the magnetic bead under these conditions (Figure 6C, ■).
Following capture, the DNA-coated beads were washed and then suspended in an ammonium acetate buffer solution with H2NOH·HCl at 50 °C, to release the captured DNA via cleavage of the acyl hydrazone linker. Initial experiments with 3 equiv of H2NOH·HCl per CpG site on the DNA failed to release significant amounts of captured DNA (Figure S51). Increasing the amount of this competing agent significantly increased the amount of released DNA up to ∼50% of the original DNA captured. We believe the need to increase the amount of H2NOH·HCl is probably due to a combination of factors including moving from solution to solid phase, the relative high number and density of labeling sites present in the captured DNA, and the length of the DNA (203 bp in PCR fragments vs 14 bp for oligo DNA). The effect of pH on the release efficiency was also evaluated (Figure S52). While the optimal pH for acyl hydrazone exchange is around pH 4.5,54,55 the degree of ionization of the captured DNA would be higher at higher pH, minimizing nonspecific interactions between the bead and the DNA. In our case, no significant effect of pH was observed on the amount of DNA released, with similar levels obtained for all three pH values tested (4.1, 4.9, and 7) (Figure S52). Further optimization of this protocol included reducing the concentration of DNA used during the capture experiments, with almost quantitative release obtained when 7 ng/μL instead of >22 ng/μL was used (Figure 6D, ●).
To demonstrate that release was the result of the cleavage of the acyl hydrazone linker and not the result of nonspecific interactions of the excess of H2NOH·HCl, a sample of DNA was modified using M.MpeI and azide-containing AdoMet derivative 8. This cofactor lacks the required chemistry for reversible capture of the DNA (Figure 6B) but reacts in a similar fashion to 4a in the capture of MTase-labeled DNA (Figure 6A). In fact, similar levels of DNA capture were obtained using either of the cofactor analogues, 4a or 8. Incubating the captured DNA with an excess of H2NOH·HCl gave no observable release of DNA modified with cofactor 8, even after four cycles of incubation (Figure 6D, ○). This lack of release was in sharp contrast to the almost quantitative release observed for DNA labeled with AdoMet derivative 4a (Figure 6D, ●). DNA labeled with AdoMet derivative 8 was only released from the magnetic beads using standard denaturing conditions (boiling in 0.1% SDS), although only 20% of the captured DNA was recovered, as opposed to the nearly quantitative release with cofactor 4a. Moreover, no additional release of DNA was observed from those beads used to capture the DNA modified with cofactor 4a (Figure 6D, ○).
Finally, and to show that the presence of the hydrazide linker did not affect the functionality of the released DNA, we tested the ability of a polymerase enzyme (Sso7d, Bio-Rad) to further amplify DNA that was captured and released from the beads. Polymerase chain reaction (PCR) is a common tool in molecular biology that underpins applications ranging from quantification of DNA to genetic fingerprinting and diagnosis.61,62 Amplification by PCR can be affected by the presence of bulky groups such as DBCO or biotin, and by DNA damage induced by incubation with hydroxyl amine.32,63,64 In our case, following capture and release, real time PCR (SsoAdvanced Universal SYBR Green Supermix, Biorad) showed an amplification curve that evolved in a similar fashion for both the modified and control DNA samples (Figure S53), indicating that the introduction of the acyl hydrazone-linker, and the incubation with 20 mM H2NOH·HCl, had no impact on the ability of the DNA to be amplified via PCR. Although we can not rule out that the polymerase used in this assay can bypass damaged sites effectively, we anticipate side-reactions will be minimized in our conditions by the low concentration of hydroxylamine used, the short incubation times used, and running the experiments at pH 6.8 or above (Figure 6). Similar side-reactions could be expected upon prolonged storage of the untagged DNA through nucleophilic attack of the hydrazide moiety to cytosine residues, leading to inter- and intrastrand cross-links. However, we believe that this potential side-reaction can be minimized by immediately reacting the purified DNA fragments, following steps ④ or ⑤ in our reported methodology (Figure 1B), much like cofactors 4 were prepared to minimize the intramolecular nucleophilic attack observed for cofactors 3. Finally, the concentration determined using real time PCR (35 ± 11 ng/μL) was in good agreement with that obtained using a complementary, fluorescence-based measurement (Qubit fluorometer) of the eluted DNA (concentration for the DNA 38 ng/μL). All together, these results demonstrate the potential of our approach, and of AdoMet derivative 4a to underpin the development of a mild and selective method to sort labeled DNA with high recovery efficiencies and functionality.
Conclusion
Here, we have presented a new versatile method to site-selectively write, remove, and rewrite functional tags on DNA. To this end, two new S-adenosyl-l-methionine (AdoMet) derivatives incorporating a bifunctional chemical handle, carrying both a Schiff-base linker and a terminal azide, were prepared. The ability of two relevant methyltransferases (MTases), including M.MpeI a bacterial analog of human DNMT1, to label DNA using these cofactors was demonstrated, and the versatility of this methodology was explored by labeling DNA across a range of sizes, including oligo and plasmid DNA. The combination of MTase-directed labeling and reversible acyl hydrazone chemistry allowed us to site-selectively label DNA with a bifunctional chemical handle, to enable tagging and untagging of chemical functionality. Each of the steps has been demonstrated using a range of analytical and bioanalytical tools. This combination of known chemistries has not been described before, and we have demonstrated the potential of this methodology to underpin new biological applications through the sequential labeling of DNA with two complementary fluorescent dyes, via writing → tagging → untagging and permanent tagging. Moreover, a new protocol was developed to sort labeled fragments of plasmid DNA, fragments that could be quantitatively recovered under mild conditions. The recovered DNA fragments retained the functionality of an unmodified DNA for PCR amplification, a critical step in molecular biology and the application of functional DNA.
We think the versatility of our method will have an impact beyond the research presented here. The combination of azide–alkyne cycloaddition and acyl hydrazone exchange should enable the introduction of a broad range of chemical moieties and functional tags. Also, a range of MTases that target other biomolecules (e.g. RNA, proteins) have been reported, thus making the presented method adaptable for bioconjugation to a broad range of targets. Moreover, the principles behind the presented method (i.e. write → tag → untag ⇄ rewrite...) should be readily expanded to other methods for site-selective functionalization of these biomolecules, including other chemical and chemoenzymatic methods. Ultimately, these methodologies should enable sequential analyses on the same target biomolecule (e.g. imaging → capture → sequence), expanding the analytical methods available to understand the biological role of these biomolecules and our ability to exploit them. Our efforts to explore some of these directions will be reported in due course.
Acknowledgments
The authors thank the University of Birmingham for the John Evans Fellowship to P.F.-T. and scholarships to A.W., K.U., A.E.R., and J.K. This work was supported by the European Union 634890, “BeyondSeq”, Royal Society U.K. (RG140613), the EPSRC (EP/N020901/1, EP/M506461/1), the Wellcome Trust (177ISSFPP), and the Birmingham Science City and the European Regional Development Fund. The authors thank Rachel O’Reilly for access to the ChemiDoc MP Imager and Tom Wilks for help with gel electrophoresis imaging. Finally, we thank both colleagues and Andrew Dove for critical reading of the manuscript and feedback.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b01023.
Full experimental details, including materials and methods, safety statement, structure of the reactive dyes used, and experimental details for the synthesis and characterization of cofactors 4 and precursors, for MTase expression and purification, for gel electrophoresis, for oligonucleotide HPLC and UPLC-MS analysis, for PCR amplification, for PCR product labeling, and for DNA sorting. (PDF)
Author Contributions
A.W., E.J., R.K.N., and P.F.-T. conceived and designed the experiments. A.W., S.C., K.U., and E.J. synthesized and purified cofactors. A.E.R. and J.K. produced and confirmed the activity of the M.TaqI and M.MpeI enzymes. A.W. and E.J. performed the experiments demonstrating rewritability. Q.S. performed real time PCR. A.W. performed all other experiments. P.F.-T. and R.K.N. secured funding. A.W., E.J., R.K.N., and P.F.-T. analyzed the data and wrote the paper, with all other authors contributing to the final version of the manuscript.
The authors declare the following competing financial interest(s): A.W., E.J., R.K.N., and P.F.-T. are named on a patent application (GB1913598.7) related to this work.
Supplementary Material
References
- Hermanson G. T.Bioconjugate Techniques; 2013. [Google Scholar]
- Cobo I.; Li M.; Sumerlin B. S.; Perrier S. Smart Hybrid Materials by Conjugation of Responsive Polymers to Biomacromolecules. Nat. Mater. 2015, 14, 143–159. 10.1038/nmat4106. [DOI] [PubMed] [Google Scholar]
- Koniev O.; Wagner A. Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation. Chem. Soc. Rev. 2015, 44 (15), 5495–5551. 10.1039/C5CS00048C. [DOI] [PubMed] [Google Scholar]
- Benizri S.; Gissot A.; Martin A.; Vialet B.; Grinstaff M. W.; Barthélémy P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjugate Chem. 2019, 30 (2), 366–383. 10.1021/acs.bioconjchem.8b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-J.; Groves B.; Muscat R. A.; Seelig G. DNA Nanotechnology from the Test Tube to the Cell. Nat. Nanotechnol. 2015, 10, 748–760. 10.1038/nnano.2015.195. [DOI] [PubMed] [Google Scholar]
- Lapa S. A.; Chudinov A. V.; Timofeev E. N. The Toolbox for Modified Aptamers. Mol. Biotechnol. 2016, 58 (2), 79–92. 10.1007/s12033-015-9907-9. [DOI] [PubMed] [Google Scholar]
- Li J.; Green A. A.; Yan H.; Fan C. Engineering Nucleic Acid Structures for Programmable Molecular Circuitry and Intracellular Biocomputation. Nat. Chem. 2017, 9, 1056–1067. 10.1038/nchem.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiber E.-A.; Hardisty R.; van Delft P.; Balasubramanian S. Mapping and Elucidating the Function of Modified Bases in DNA. Nat. Rev. Chem. 2017, 1, 0069. 10.1038/s41570-017-0069. [DOI] [Google Scholar]
- Wang L. Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc. Chem. Res. 2017, 50 (11), 2767–2775. 10.1021/acs.accounts.7b00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Schultz P. G. Playing with the Molecules of Life. ACS Chem. Biol. 2018, 13 (4), 854–870. 10.1021/acschembio.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-Selective Protein-Modification Chemistry for Basic Biology and Drug Development. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Hu Q.-Y.; Berti F.; Adamo R. Towards the next Generation of Biomedicines by Site-Selective Conjugation. Chem. Soc. Rev. 2016, 45 (6), 1691–1719. 10.1039/C4CS00388H. [DOI] [PubMed] [Google Scholar]
- Haimovich A. D.; Muir P.; Isaacs F. J. Genomes by Design. Nat. Rev. Genet. 2015, 16, 501–516. 10.1038/nrg3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright T. H.; Vallée M. R. J.; Davis B. G. From Chemical Mutagenesis to Post-Expression Mutagenesis: A 50 Year Odyssey. Angew. Chem., Int. Ed. 2016, 55 (20), 5896–5903. 10.1002/anie.201509310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh G. C.; Burns J. R.; Howorka S. Comparing Proteins and Nucleic Acids for Next-Generation Biomolecular Engineering. Nat. Rev. Chem. 2018, 2 (7), 113–130. 10.1038/s41570-018-0015-9. [DOI] [Google Scholar]
- Spicer C. D.; Pashuck E. T.; Stevens M. M. Achieving Controlled Biomolecule-Biomaterial Conjugation. Chem. Rev. 2018, 118 (16), 7702–7743. 10.1021/acs.chemrev.8b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt E. A.; Cal P. M. S. D.; Oliveira B. L.; Bernardes G. J. L. Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem. 2019, 3 (3), 1. 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- Anhäuser L.; Rentmeister A. Enzyme-Mediated Tagging of RNA. Curr. Opin. Biotechnol. 2017, 48, 69–76. 10.1016/j.copbio.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Li C.; Wang L.-X. Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem. Rev. 2018, 118 (17), 8359–8413. 10.1021/acs.chemrev.8b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Park K.-Y.; Suazo K. F.; Distefano M. D. Recent Progress in Enzymatic Protein Labelling Techniques and Their Applications. Chem. Soc. Rev. 2018, 47 (24), 9106–9136. 10.1039/C8CS00537K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Zheng Y. G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem. Biol. 2016, 11 (3), 583–597. 10.1021/acschembio.5b00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen J.; Vranken C.; Leen V.; Neely R. K.; Janssen K. P. F.; Hofkens J. Methyltransferase-Directed Labeling of Biomolecules and Its Applications. Angew. Chem., Int. Ed. 2017, 56 (19), 5182–5200. 10.1002/anie.201608625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat. Rev. Genet. 2018, 19, 81–92. 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- Luo M. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev. 2018, 118 (14), 6656–6705. 10.1021/acs.chemrev.8b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkuvienė M.; Mickutė M.; Vilkaitis G.; Klimašauskas S. Repurposing Enzymatic Transferase Reactions for Targeted Labeling and Analysis of DNA and RNA. Curr. Opin. Biotechnol. 2019, 55, 114–123. 10.1016/j.copbio.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff C.; Lukinavičius G.; Klimas̆auskas S.; Weinhold E. Direct Transfer of Extended Groups from Synthetic Cofactors by DNA Methyltransferases. Nat. Chem. Biol. 2006, 2, 31–32. 10.1038/nchembio754. [DOI] [PubMed] [Google Scholar]
- Dalhoff C.; Lukinavičius G.; Klimašauskas S.; Weinhold E. Synthesis of S-Adenosyl-L-Methionine Analogs and Their Use for Sequence-Specific Transalkylation of DNA by Methyltransferases. Nat. Protoc. 2006, 1, 1879–1886. 10.1038/nprot.2006.253. [DOI] [PubMed] [Google Scholar]
- Wang R.; Zheng W.; Yu H.; Deng H.; Luo M. Labeling Substrates of Protein Arginine Methyltransferase with Engineered Enzymes and Matched S-Adenosyl-l-Methionine Analogues. J. Am. Chem. Soc. 2011, 133 (20), 7648–7651. 10.1021/ja2006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K.; Chen Y.; Wu H.; Bothwell I. R.; Blum G. J.; Zeng H.; Dong A.; Zheng W.; Min J.; Deng H.; et al. Defining Efficient Enzyme-Cofactor Pairs for Bioorthogonal Profiling of Protein Methylation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (42), 16778–16783. 10.1073/pnas.1216365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken C.; Deen J.; Dirix L.; Stakenborg T.; Dehaen W.; Leen V.; Hofkens J.; Neely R. K. Super-Resolution Optical DNA Mapping via DNA Methyltransferase-Directed Click Chemistry. Nucleic Acids Res. 2014, 42 (7), e50-e50 10.1093/nar/gkt1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer M. H.; Vranken C.; Deen J.; Frederickx W.; Vanderlinden W.; Wand N.; Leen V.; Gehlen M. H.; Hofkens J.; Neely R. K. Methyltransferase-Directed Covalent Coupling of Fluorophores to DNA. Chem. Sci. 2017, 8 (5), 3804–3811. 10.1039/C6SC04229E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriukienė E.; Labrie V.; Khare T.; Urbanavičiu̅tė G.; Lapinaitė A.; Koncevičius K.; Li D.; Wang T.; Pai S.; Ptak C.; et al. DNA Unmethylome Profiling by Covalent Capture of CpG Sites. Nat. Commun. 2013, 4, 2190. 10.1038/ncomms3190. [DOI] [PubMed] [Google Scholar]
- Anhäuser L.; Muttach F.; Rentmeister A. Reversible Modification of DNA by Methyltransferase-Catalyzed Transfer and Light-Triggered Removal of Photo-Caging Groups. Chem. Commun. 2018, 54 (5), 449–451. 10.1039/C7CC08300A. [DOI] [PubMed] [Google Scholar]
- Heimes M.; Kolmar L.; Brieke C. Efficient Cosubstrate Enzyme Pairs for Sequence-Specific Methyltransferase-Directed Photolabile Caging of DNA. Chem. Commun. 2018, 54 (90), 12718–12721. 10.1039/C8CC05913F. [DOI] [PubMed] [Google Scholar]
- Crosetto N.; Bienko M.; van Oudenaarden A. Spatially Resolved Transcriptomics and Beyond. Nat. Rev. Genet. 2015, 16, 57. 10.1038/nrg3832. [DOI] [PubMed] [Google Scholar]
- Dean K. M.; Palmer A. E. Advances in Fluorescence Labeling Strategies for Dynamic Cellular Imaging. Nat. Chem. Biol. 2014, 10, 512. 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser M.; Wojcik M.; Kim D.; Mahmoudi M.; Li W.; Xu K. Correlative Super-Resolution Microscopy: New Dimensions and New Opportunities. Chem. Rev. 2017, 117 (11), 7428–7456. 10.1021/acs.chemrev.6b00604. [DOI] [PubMed] [Google Scholar]
- Stuart T.; Satija R. Integrative Single-Cell Analysis. Nat. Rev. Genet. 2019, 20 (5), 257–272. 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- Adams J. P. Imines, Enamines and Oximes. J. Chem. Soc. Perkin 1 2000, (2), 125–139. 10.1039/a808142e. [DOI] [Google Scholar]
- Ramström O.; Lehn J.-M. Drug Discovery by Dynamic Combinatorial Libraries. Nat. Rev. Drug Discovery 2002, 1, 26–36. 10.1038/nrd704. [DOI] [PubMed] [Google Scholar]
- Wojtecki R. J.; Meador M. A.; Rowan S. J. Using the Dynamic Bond to Access Macroscopically Responsive Structurally Dynamic Polymers. Nat. Mater. 2011, 10, 14–27. 10.1038/nmat2891. [DOI] [PubMed] [Google Scholar]
- Belowich M. E.; Stoddart J. F. Dynamic Imine Chemistry. Chem. Soc. Rev. 2012, 41 (6), 2003–2024. 10.1039/c2cs15305j. [DOI] [PubMed] [Google Scholar]
- Rosales A. M.; Anseth K. S. The Design of Reversible Hydrogels to Capture Extracellular Matrix Dynamics. Nat. Rev. Mater. 2016, 1, 15012. 10.1038/natrevmats.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Jin Y.. Dynamic Covalent Chemistry: Principles, Reactions, and Applications; John Wiley & Sons, 2018. [Google Scholar]
- Dirksen A.; Yegneswaran S.; Dawson P. E. Bisaryl Hydrazones as Exchangeable Biocompatible Linkers. Angew. Chem., Int. Ed. 2010, 49 (11), 2023–2027. 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavičius G.; Tomkuvienė M.; Masevičius V.; Klimašauskas S. Enhanced Chemical Stability of AdoMet Analogues for Improved Methyltransferase-Directed Labeling of DNA. ACS Chem. Biol. 2013, 8 (6), 1134–1139. 10.1021/cb300669x. [DOI] [PubMed] [Google Scholar]
- Huber T. D.; Wang F.; Singh S.; Johnson B. R.; Zhang J.; Sunkara M.; Van Lanen S. G.; Morris A. J.; Phillips G. N.; Thorson J. S. Functional AdoMet Isosteres Resistant to Classical AdoMet Degradation Pathways. ACS Chem. Biol. 2016, 11 (9), 2484–2491. 10.1021/acschembio.6b00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedecke K.; Pignot M.; Goody R. S.; Scheidig A. J.; Weinhold E. Structure of the N6-Adenine DNA Methyltransferase M•TaqI in Complex with DNA and a Cofactor Analog. Nat. Struct. Biol. 2001, 8, 121–125. 10.1038/84104. [DOI] [PubMed] [Google Scholar]
- Sato S.; Hutchinson C. A.; Harris J. I. A Thermostable Sequence-Specific Endonuclease from Thermus Aquaticus. Proc. Natl. Acad. Sci. U. S. A. 1977, 74 (2), 542. 10.1073/pnas.74.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pljevaljčić G.; Schmidt F.; Weinhold E. Sequence-Specific Methyltransferase-Induced Labeling of DNA (SMILing DNA). ChemBioChem 2004, 5 (3), 265–269. 10.1002/cbic.200300739. [DOI] [PubMed] [Google Scholar]
- Lister R.; Pelizzola M.; Dowen R. H.; Hawkins R. D.; Hon G.; Tonti-Filippini J.; Nery J. R.; Lee L.; Ye Z.; Ngo Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch A. Phylogeny of Methylomes. Science 2010, 328 (5980), 837–838. 10.1126/science.1190738. [DOI] [PubMed] [Google Scholar]
- Slatko B. E.; Croft R.; Moran L. S.; Wilson G. G. Cloning and Analysis of the HaeIII and HaeII Methyltransferase Genes. Gene 1988, 74 (1), 45–50. 10.1016/0378-1119(88)90248-X. [DOI] [PubMed] [Google Scholar]
- Dirksen A.; Dirksen S.; Hackeng T. M.; Dawson P. E. Nucleophilic Catalysis of Hydrazone Formation and Transimination: Implications for Dynamic Covalent Chemistry. J. Am. Chem. Soc. 2006, 128 (49), 15602–15603. 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- Mahon C. S.; Jackson A. W.; Murray B. S.; Fulton D. A. Templating a Polymer-Scaffolded Dynamic Combinatorial Library. Chem. Commun. 2011, 47 (25), 7209–7211. 10.1039/c1cc11998b. [DOI] [PubMed] [Google Scholar]
- Kalia J.; Raines R. T. Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem., Int. Ed. 2008, 47 (39), 7523–7526. 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese E. B.; Freese E. Two Separable Effects of Hydroxylamine on Transforming DNA. Proc. Natl. Acad. Sci. U. S. A. 1964, 52 (5), 1289–1297. 10.1073/pnas.52.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P.; Smith R. P. Biologic Activity of Hydroxylamine: A Review. CRC Crit. Rev. Toxicol. 1985, 14 (1), 87–99. 10.3109/10408448509023765. [DOI] [PubMed] [Google Scholar]
- An R.; Jia Y.; Wan B.; Zhang Y.; Dong P.; Li J.; Liang X. Non-Enzymatic Depurination of Nucleic Acids: Factors and Mechanisms. PLoS One 2014, 9 (12), 1–17. 10.1371/journal.pone.0115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski M.; Czapinska H.; Bochtler M. CpG Underrepresentation and the Bacterial CpG-Specific DNA Methyltransferase M. MpeI. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (1), 105–110. 10.1073/pnas.1207986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis K. B.; Faloona F. A. [21] Specific Synthesis of DNA in Vitro via a Polymerase-Catalyzed Chain Reaction. Methods Enzymol. 1987, 155, 335–350. 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- Kramer M. F.; Coen D. M. Enzymatic Amplification of DNA by PCR: Standard Procedures and Optimization. In Current Protocols in Molecular Biology 2001, 15.1.1–15.1.14. 10.1002/0471142727.mb1501s56. [DOI] [PubMed] [Google Scholar]
- Ren X.; El-Sagheer A. H.; Brown T. Azide and Trans-Cyclooctene DUTPs: Incorporation into DNA Probes and Fluorescent Click-Labelling. Analyst 2015, 140 (8), 2671–2678. 10.1039/C5AN00158G. [DOI] [PubMed] [Google Scholar]
- Aslanzadeh J. Application of Hydroxylamine Hydrochloride for Post-PCR Sterilization. Mol. Cell. Probes 1993, 7 (2), 145–150. 10.1006/mcpr.1993.1020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.