

Abstract
Protein adsorption to the surface of a nanoparticle can fundamentally alter the character, behavior, and fate of a nanoparticle in vivo. Current methods to capture the protein corona rely on physical separation techniques and are unable to resolve key, individual protein–nanoparticle interactions. As a result, the precise link between the “synthetic” and the “biological” identity of a nanoparticle remains unclear. Herein, we report an unbiased photoaffinity-based approach to capture, characterize, and quantify the protein corona of liposomes in their native state. Compared to conventional methods, our photoaffinity approach reveals markedly different interacting proteins as well as reduced total protein binding to liposome surfaces. Identified proteins do not follow protein abundancy patterns of human serum, as has been generally reported, but are instead dominated by soluble apolipoproteins–endogenous serum proteins that have evolved to recognize the lipidic surface of circulating lipoproteins. We believe our findings are the most accurate characterization of a liposome’s biological identity but, more fundamentally, reveal liposome–protein binding is, in many cases, significantly less complex than previously thought.
Short abstract
Serum protein adsorption can drastically affect the in vivo performance of a liposome. A method to capture, characterize, and quantify the protein corona of a liposome in its native state is presented.
Introduction
The protein corona of a nanoparticle describes a subset of proteins that preferentially adsorb to the surface of a nanoparticle upon administration in vivo. Formation of the protein corona creates the “biological identity” of a nanoparticle.1−3 To some extent, it is the protein corona, not the underlying synthetic surface of a nanoparticle, which the body “sees” and interacts with (Figure 1a). The adsorbed protein corona can, therefore, significantly influence the in vivo fate of a nanoparticle,4,5 for instance, by promoting bodily clearance and/or shielding active targeting ligands displayed from a nanoparticle surface.6−8 The composition and extent of the protein corona is dependent on the “synthetic identity” of a nanoparticle (e.g., size, surface charge, chemical composition),9−11 the biological media to which the nanoparticle is exposed,12,13 and the kinetics of protein binding;14−17 however, the general formation of a protein corona is believed omnipresent for all types of nanoparticles.4,18
Figure 1.

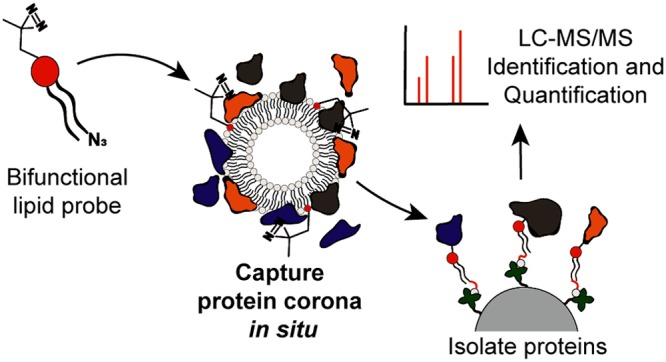
Protein corona identification using photoaffinity-based chemoproteomics. (a) Liposomes exposed to biological medium are confronted by a wide variety of endogenous proteins, a subset of which preferentially bind to the synthetic surface of the liposome to create the biological identity of the liposome. (b) Centrifugation protocols to isolate nanoparticle protein complexes rely on efficient sedimentation and can disrupt weak protein–nanoparticle interactions, induce protein aggregation, and/or lead to the capture of large, unbound proteins. (c) Schematic representation of a PAL approach for the capture and identification of a liposome protein corona. (d) Bifunctional PAL probe, IKS02, structurally similar to common PC phospholipids.
The most common method to isolate and identify the protein corona of a nanoparticle involves sedimentation of nanoparticle–protein complexes following incubation in biological fluids, such as (human) serum or blood (Figure 1b).19 Depending on the density of a nanoparticle, this requires centrifugation speeds high enough (typically >14 000g) to ensure enough pelleted material for subsequent characterization. However, subjecting nanoparticle–protein complexes to significant centrifugal force runs the risk of disrupting native and weak protein–nanoparticle interactions and can induce protein aggregation and/or sedimentation of large, unbound proteins.20 This, in turn, leads to the capture and inclusion of false positive proteins and a biased profile of protein binding to a nanoparticle surface, often mirroring serum protein abundancy.21,22 As such, while reported protein corona data sets have highlighted important general differences in serum protein binding based on, for example, nanoparticle size or surface charge,23 it is not yet possible to identify key individual nanoparticle–protein interactions from the long, empirical lists of proteins typically reported. Characterizing the protein corona of liposomes and other lipid nanoparticles is especially problematic, given the low density of these lipidic particles requires higher centrifugal forces than dense (e.g., inorganic) nanoparticles. In studies involving lipid nanoparticles, the identified protein corona is typically dominated by highly abundant serum albumin and high molecular weight proteins (e.g., complement C3 and α-2-macroglobulin).10,12,15,24,25
Herein, an unbiased photoaffinity labeling (PAL) method to capture the protein corona of liposomes, in their native state, is described (Figure 1c). Photoaffinity labeling has been successfully applied in chemoproteomic strategies to study lipid metabolism,26 identify inhibitor off-targets, and discover new small-molecule therapeutics.27,28 Here, it is introduced to the field of nanotechnology and nanomedicine for the first time. Of the myriad nanoparticle-based drug delivery systems reported, liposomes are the most widely investigated and approved for clinical use.29,30 In this study, we apply our photoaffinity method to three clinically relevant liposome formulations—AmBisome (anionic), EndoTAG-1 (cationic), and Myocet (neutral)—to assess the influence of liposome surface charge on protein binding. We recently described, in mechanistic detail, the biodistribution and bodily clearance of these same three liposome formulations in vivo (embryonic zebrafish).31 Following photoaffinity capture and purification of the protein corona, label-free quantitative mass spectrometry revealed distinct and highly reproducible protein corona fingerprints for all three liposome formulations. In contrast to centrifugation protocols, our photoaffinity method identified only a small subset of bound serum proteins, devoid of abundant serum albumin and dominated by apolipoproteins. The dominance of apolipoproteins, adsorbed to the surface of liposomes, over more abundant serum has not before been reported.15,32,33
Results
Probes for PAL require two key features:27 (1) photoactivatable chemical functionality that, upon in situ sample irradiation, can covalently cross-link to any molecule/protein in close proximity, and (2) a bioorthogonal handle for conjugation of a reporter molecule or selective pull-down of the probe–protein complex from the biological environment. Both functionalities should be small to avoid significant disruption of the native liposomal system and the potential capture of proteins that would not otherwise bind to the liposome surface. Accordingly, a PAL probe, IKS02, structurally similar to endogenous phosphatidylcholine (PC) phospholipids, was designed and synthesized via robust phosphoramidite synthetic protocols (Figure 1d and Figure S1). PC lipids are present in virtually all clinically approved liposomal formulations.34 Within the PAL probe design, the zwitterionic PC lipid headgroup was maintained so as not to alter the surface charge or surface charge density of liposomes containing IKS02. Likewise, the incorporation of long-chain fatty acids (>C12) not only mirrors chain lengths typical of most reported (clinical and experimental) liposome formulations but, given the extreme water insolubility of long chain PC lipids, largely excludes any possibility of incorporated IKS02 dissociating from a liposome membrane under physiological conditions.35 Diazirine functionality was chosen as photoactivatable group given its small size and high photoefficiency.36 Irradiation of diazirines with UV-A (∼350 nm) light generates a highly reactive carbene intermediate that can spontaneously react with all residues, as well as the backbone, of a surface-bound protein. We chose to install diazirine functionality at the phosphatidylcholine headgroup of the lipid probe to maximize the capture of proteins directly interacting with the liposome surface. As a bioorthogonal ligation handle, azide functionality was incorporated at the terminus of one fatty acid chain of the PAL probe. In this position, it is most likely buried within the liposome lipid bilayer, minimizing potential unwanted interactions with surface-bound proteins. Following photo-crosslinking and liposome solubilization, azide functionality was used to selectively couple the protein–lipid conjugate to either a fluorescent alkyne-Cy5 probe or an alkyne-biotin label. The latter could be used to selectively pull down and isolate the protein–lipid conjugate from the biological media. In both cases, conjugation reactions were performed using standard bioorthogonal click chemistry protocols.28,37
Three liposome formulations, either approved for clinical use or under development (Myocet, AmBisome, and EndoTAG-1) were selected to test our photoaffinity method, as well as investigate qualitative and quantitative differences in the adsorbed protein corona as a function of liposome surface charge. Myocet (lipid composition: POPC/cholesterol −55:45, reported size (clinical): 150–200 nm) is a zwitterionic, neutral liposomal-doxorubicin formulation used in breast cancer therapy.38 AmBisome (lipid composition: DSPC/DSPG/cholesterol −53:21:26; size: 78 nm) is a negatively charged liposomal-amphotericin B formulation used to treat fungal infections.39 EndoTAG-1 (lipid composition: DOTAP/DOPC – 51.5:48.5; size: 200 nm) is a positively charged liposomal-paclitaxel formulation that targets the tumor vasculature.40 Liposomes, formulated without encapsulated drugs and based on the lipid composition of these three formulations, were prepared by thin-film hydration and extrusion. For the photoaffinity method, IKS02 was incorporated within liposome formulations at 5 mol % (∼1 probe per 10 nm2 liposome surface). Dynamic light scattering and zeta potential measurements revealed all liposomes were ∼100 nm in diameter (polydispersity index (PDI) < 0.1) and that surface charges were not significantly affected by incorporation of IKS02 (Table S1).
Next, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to resolve the protein corona fingerprint of all three liposome formulations obtained either via our photoaffinity method or via a conventional centrifugation protocol (Figure 2).10,12,15,24,25,32 In all cases, liposomes were incubated in human serum at 37 °C for 1 h prior to the capture/isolation of the liposome–protein complex. For the centrifugation protocol, liposome–protein complexes were sedimented (15 min, 17 500g) and carefully washed to remove unbound proteins present in the supernatant. Pelleted liposome–protein complexes were dissolved in denaturing buffer, resolved by SDS-PAGE (10 μg/well), and visualized by Coomassie Blue staining (Figure 2a,d). As a negative control, human serum, diluted with buffer containing no liposomes, was subjected to the same centrifugation protocol (Figure 2c). From the obtained gels, only cationic EndoTAG-1 liposomes displayed a distinct and unique protein binding profile. In contrast, AmBisome (negative) and Myocet (neutral) liposomes showed very similar protein fingerprints to the serum-only control, with the clearest visible band, at ∼70 kDa, corresponding to serum albumin. These results highlight two inseparable and competing flaws of using centrifugation to isolate liposome–protein complexes from serum protein mixtures. On the one hand, efficient sedimentation of nanoparticle–protein complexes relies on a threshold particle density, either of the nanoparticle or the formed nanoparticle–protein complex. In this case, using fluorescently labeled liposomes (DOPE-LR, 1 mol %), a clearly significant fraction of liposomes remained in suspension following centrifugation (15 min, 17 500g) and was, therefore, excluded from subsequent characterization (Figure S2a,b). Furthermore, in all cases, the measured size of the pelleted liposomes was significantly larger than that of the original formulation (Figure S2c–e). Whether, or not, aggregation adversely affects protein corona formation is, however, unclear given the significant background of large, unbound serum proteins in the pelleted fraction. Alternatively, by increasing centrifugal speeds to achieve greater nanoparticle–protein complex sedimentation, the risk of sedimenting (large), nonadsorbed serum proteins is also increased. This is exemplified by the significant amount of resolved proteins present in the SDS-PAGE of the serum-only control (Figure 2d, far right lane).
Figure 2.

Liposome protein corona fingerprints resolved by gel electrophoresis. (a) Schematic representation of the centrifugation protocol. (b) Schematic representation of the photoaffinity protocol. (c) SDS-PAGE of human serum, stained with Coomassie Blue. (d) SDS-PAGE of the liposome protein coronas, as well as captured proteins of a liposome free control sample, isolated by centrifugation and stained with Coomassie Blue. (e) SDS-PAGE of the liposome protein coronas, isolated by photoaffinity method, visualizing Cy5 labeled lipid–protein conjugates by in-gel fluorescence. Unique bands highlighted with red arrows. (below) Coomassie Blue loading controls displayed as cropped images. Complete gels displaying all controls and complete Coomassie Blue stained gels displayed in Figure S3.
For the photoaffinity method, liposomes containing IKS02 were incubated in serum and subsequently irradiated (15 min, 350 nm, 15 mW cm–2) in situ. After covalent capture of the protein corona, liposomes were solubilized with detergent (Triton X-100). The cross-linked lipid–protein complexes were then conjugated to a fluorescent, alkyne-Cy5 probe, and the fluorescent lipid–protein complex was resolved by SDS-PAGE. In-gel fluorescence was used to reveal the subset of proteins that successfully cross-linked to the lipid probe (Figure 2b,e). As negative control (“-UV”), liposomes containing IKS02 were not irradiated but otherwise processed identically. In this case, the resolved protein corona fingerprints showed distinct and unique protein profiles, both between formulations as well as compared to the resolved protein corona of the same liposomes isolated via centrifugation. In the case of cationic EndoTAG-1 liposomes, multiple unique protein bands, notably at ∼10, 28, 40, 45, and 150 kDa, were detected in the “+UV” sample exclusively. In addition, proteins, notably at ∼28 and 80 kDa, appear significantly enriched over the “–UV” background. Similarly, unique protein bands, notably at ∼20, 28, and 35 kDa, were present for the +UV AmBisome formulation. In the case of Myocet, no unique protein labeling over “–UV” background was observed, indicating a possible lack of significant protein binding. Interestingly, the band intensity for serum albumin, at ∼70 kDa, for both AmBisome and Myocet formulations, was similar for both the +UV sample and −UV control, indicating albumin may not constitute a significant proportion of the protein corona of either of these liposome formulations. Background proteins resolved in all three −UV control samples broadly followed protein abundancy patterns of human serum. This suggests background labeling is due to nonselective protein binding and can be attributed to relatively low amounts of cross-linked lipid–protein complexes compared to the total amount of protein in the sample. In addition, background labeling appeared selective for the presence of the copper click catalyst (Figure S3). This labeling was minimized by reducing the copper sulfate concentration and increasing chelating agent concentration, as described previously.41,42 Although complete elimination of background labeling was not achieved, the resolved protein corona of the three liposome formulations isolated via our photoaffinity approach clearly show significant differences in both distribution and abundancy of proteins compared to both the negative (−UV) control and compared to the resolved protein corona of the same liposome formulations isolated via centrifugation.
To characterize the specific composition of the protein coronas visualized by gel electrophoresis, we performed label-free, quantitative mass spectrometry on the photoaffinity captured protein corona of EndoTAG-1, AmBisome, and Myocet liposomes. Over the past decade, label-free quantification (LFQ) has emerged as a straightforward and accurate method to quantify relative protein amounts within complex proteomic samples that do not allow for metabolic labeling, such as human blood or serum.43 Recently, this method has been used to determine the abundance of proteins within the protein corona of nanoparticles isolated via centrifugation.44 For each liposome formulation, six separate samples were incubated in serum and subsequently irradiated (+UV). Alongside, six control samples were incubated in serum but not irradiated (−UV). The liposomes were solubilized, and the captured lipid–protein complexes were conjugated to alkyne-functionalized biotin, followed by streptavidin-agarose bead enrichment and on-bead digestion (Figure 3a). In all cases, enolase digest (50 fmol) was added to the enriched samples as an internal standard. The samples were resolved using nano ultra performance liquid chromatography mass spectrometry (nanoUPLC-MS/MS), and peptide fragments were identified and quantified based on the LFQ TOP3 approach using the ISOQuant software.45,46 To gain a high accuracy for the label-free quantification, strict processing parameters were selected. These included a total of six replicates, a minimum peptide score of 6.0, as well as a minimum of three unique identified peptides per protein (Table S2). In addition and to ensure that identified proteins were consistently bound to the liposome surface, only proteins that were present in six of six (+UV) samples were considered for further analysis. All selection criteria can, of course, be modified retrospectively to meet any desired output (see Supporting Information files for fully editable raw and processed proteomic data sets). To correct for the background labeling observed in the gel electrophoresis experiments, volcano plots were constructed to identify enriched and statistically significant proteins of the protein corona (Figure 3b–d). To ensure statistical significance, a ratio comparing the average relative protein quantities (expressed in ppm) of +UV versus −UV samples was plotted against its p-value, determined by a t test followed by a Benjamini-Hochberg correction. Proteins that were significantly enriched (twofold and p < 0.05) over the background were selected as a true part of the protein corona. Proteins completely absent in the background were listed as “exclusive for +UV” and added to the selection. Finally, selected proteins were ranked based on abundance (Figures 3b–d and S4). In addition, LFQ allows for the absolute quantification of a proteomic sample, in which the amount of the protein is calculated (in fmol) as compared to an internal standard (50 fmol enolase digest). In this case, the sum of the absolute amounts of individual proteins, background corrected and meeting the strict selection criteria, was significantly higher for cationic EndoTAG-1 liposomes than anionic AmBisome or neutral Myocet liposomes (Figure 3e). Indeed, the amount of protein adsorbed to the surface of Myocet liposomes was vanishingly small. These results broadly mirror total protein abundancies observed in gel electrophoresis experiments and confirm that serum protein adsorption is most prolific on cationic liposome surfaces. In contrast, precise quantitative analysis is not possible for protein coronas isolated via centrifugation methods due to the variability in sedimentation efficiency between different liposome formulations.
Figure 3.

LFQ identification of the liposome protein corona, isolated via photoaffinity method. (a) Schematic representation of photoaffinity labeling and enrichment for MS/MS identification of the liposome protein corona. (b–d) LFQ MS for EndoTAG-1, AmBisome, and Myocet liposomes. Volcano plots of enrichment over background (log2(+UV/–UV)) plotted against the statistical significance of this comparison (−log10(p-value)). Proteins meeting all selection criteria labeled in green. Proteins without background labeling are listed as exclusive for +UV. Abundance plots displaying the replicate abundancies of the top 10 proteins (ppm) within the +UV samples. Complete abundance plots containing all proteins, including −UV abundancy values and tables, can be found in Supporting Information Figure 5. (e) Absolute quantification of protein binding to EndoTAG-1, AmBisome, and Myocet liposomes. Values calculated as the average absolute amount of protein of the +UV replicates corrected for the average absolute amount of protein of the −UV replicates.
To verify the accuracy of our photoaffinity approach, we next performed a competition assay in which AmBisome liposomes, containing IKS02, were incubated in predefined mixtures of purified serum proteins together with increasing concentrations of unlabeled AmBisome liposomes (Figure 4a). As a defined protein mixture, apolipoprotein E (APOE, 2 μgmL–1), apolipoprotein A-I (APOA1, 2 μgmL–1)—both of which apparently bound to the surface of AmBisome—were combined with abundant but apparently nonbinding serum albumin (ALBU, 25 μgmL–1), transferrin (TRFE, 10 μgmL–1), and prothrombin (THRB, 2 μgmL–1). The relative concentrations of individual proteins was chosen to approximate endogenous serum protein abundance (Table S3). In the absence of any competing and unlabeled AmBisome liposomes, our photoaffiinity approach again revealed the selective binding of apoE and apoA1 to the anionic surface of AmBisome liposomes (Figure 4b). The relative abundancy of apoE and apoA1 on the surface of the liposomes was comparable to that observed for experiments using human serum with slight variation in absolute values likely reflecting small differences in relative protein concentrations compared to endogenous human serum. Furthermore, this experiment confirmed the complete absence of binding of more abundant serum proteins (e.g., ALBU and TRFE) to the surface of AmBisome liposomes (Figure 4b).
Figure 4.

Validation of apolipoprotein E and A1 binding to AmBisome liposomes. (a) Liposomes were incubated in a mixture of purified human serum proteins consisting of apolipoprotein E (APOE, 2 μgmL–1), serum albumin (ALBU, 25 μgmL–1), apolipoprotein A-I (APOA1, 2 μgmL–1), transferrin (TRFE, 10 μgmL–1), and prothrombin (THRB, 2 μgmL–1). (b) Volcano plot of protein enrichment over background (log2(+UV/–UV)) plotted against the statistical significance of this comparison (−log10(p-value)). Proteins meeting all selection criteria labeled in green. Abundance plot displaying the abundancies of apoE and apoA1 within the +UV samples. (c) Competition assay of apolipoprotein E and A1 binding. Increasing concentrations (1:1 to 1:9 molar ratios) of unlabeled AmBisome liposomes were incubated, together with AmBisome liposomes, containing IKS02, in the above predefined mixture of human serum proteins. Captured apoE and apoA1 on the surface of IKS02-labeled AmBisome liposomes were separated by SDS-PAGE and visualized by in-gel fluorescence (Cy5). Protein loading determined by Coomassie Blue (coom.). Protein structures were obtained from the protein data bank (PDB): (APOE: 2L7B, APOA1: 1AV1, ALBU: 1E78, THRB: 6C2W, TRFE: 1D3K). Illustrations were generated using Illustrate.48
In the presence of increasing concentrations of unlabeled but otherwise compositionally identical liposomes, our photoaffinity probe captured decreasing amounts of apoE and apoA1 bound to the surface of IKS02-labeled AmBisome liposomes (Figure 4c). Importantly, this result confirmed that unlabeled AmBisome liposomes also bind, and compete for, apoE and apoA1, indicating that the incorporation of our photoaffinity probe (5 mol %) within a liposome membrane does not signifcantly affect specific serum protein binding. Again, this experiment confirmed no enrichment of ALBU, THRB, or TRFE on the surface of AmBisome (Figure S5). Analogous experiments using Myocet confirmed the complete absence of serum proteins at the surface of these liposomes, whereas for EndoTAG-1, selective binding of apoE and apoA1 was again observed (Figure S6). However, significant amounts of surface-bound THRB were not captured on the surface of EndoTAG-1, as was the case in human serum. Again, this may be due to an underestimation of the relative concentration of THRB in the predefined protein mixture, but it is noteworthy that THRB was detected here with high statistical significance (+UV vs −UV), albeit low enrichment. While we have used six biological replicates to reliably determine enrichment at a minimum of twofold over the background throughout this study,47 it may be the case that proteins with high statistical significance (e.g., p < 0.01) but low enrichment (e.g., ∼1.5×) should still be considered important components of the liposome protein corona.
To compare liposome protein coronas isolated via centrifugation, six replicates of each liposome formulation were incubated in human serum, centrifuged, washed, and resolved with SDS-PAGE, followed by in-gel digestion (Figure S7).49,50 Following digestion, the same nanoUPLC-MS/MS methods and LFQ criteria, as for the photoaffinity labeling, were applied to identify specific, isolated proteins. Given the variability in sedimentation efficiency, isolated proteins from the centrifugation method were ranked on abundancy without background correction (Figure S8 and Tables S4–S7). For all three liposome formulations, the number of individually identified proteins present in the protein corona isolated via centrifugation were higher than those identified via the photoaffinity method (Figure 4a). This was most evident for EndoTAG-1 liposomes, where our photoaffinity method identified a total of 20 unique proteins compared to 100+ for centrifugation, and for Myocet liposomes, where our photoaffinity method identified just two proteins compared to 24 for centrifugation.
To further correlate identified proteins to their natural abundance, complete human serum was digested in solution, followed by identification and quantification (Table S3). With these data, the distribution of protein molecular weights and isoelectric points for both methods, and all three liposome formulations, could be compared to the protein composition of the serum sample used in this study (Figure 5b,c). In the case of protein molecular weight, protein coronas isolated via centrifugation methods showed similar size distributions to that of native serum. In contrast, coronas isolated via photoaffinity methods contained no proteins with a molecular weight of more than 150 kDa, with AmBisome and Myocet exclusively binding proteins less than 60 kDa molecular weight. This disparity is most likely due to the sedimentation of large, unbound proteins during centrifugation. In the case of protein isoelectric point (pI), both EndoTAG-1 and AmBisome predominantly bound acidic serum proteins (pI < 7), irrespective of isolation method. Interestingly, there was no significant enrichment of basic serum proteins (pI > 7) on the surface of anionic, AmBisome liposomes. Accordingly, protein pI distributions on the surface of AmBisome and EndoTAG-1 liposomes broadly follow the pI distribution of proteins in human serum, in which the majority of proteins are acidic. This also explains the high amount of total protein binding to cationic, EndoTAG-1 liposomes.
Figure 5.

Comparison of liposome protein coronas isolated via centrifugation or photoaffinity method. (a) Number of distinct serum proteins adsorbed to the surface of EndoTAG-1, AmBisome, and Myocet liposomes. (b) Molecular weight (in kDa) distributions of identified proteins for each liposome formulation and complete human serum. Photoaffinity samples are labeled “p” (e.g., pEndoTag), and centrifugation samples are labeled “c” (e.g., cEndoTag). (c) pI distributions of identified proteins for each liposome formulation and complete human serum. (d) Heat map displaying the abundancy of proteins associated with individual liposome formulations. For the centrifugation method, protein abundance was calculated as the average abundance (ppm) for every protein over the average total amount of protein in the sample. For the photoaffinity method, protein abundance was calculated as the average abundance (ppm) for every protein over the average total amount of protein (meeting the selection criteria).
Finally, a heat map was constructed to compare individual proteins present on the surface of each of the three liposome formulations, isolated via either centrifugation or photoaffinity methods (Figure 5d). Here, the relative abundance of a protein within a sample is displayed for the top 10 most abundant proteins in human serum, as well as the top 10 most abundant apolipoproteins. From this heat map, it is clear that protein coronas isolated via centrifugation closely follow native protein abundancies in human serum, and human serum albumin (ALBU) and complement component 3 (CO3) are abundantly present in all samples, as well as in the control sample (i.e., no liposomes). In contrast, our photoaffinity method reveals the most abundant serum proteins do not constitute a significant component of the protein corona of any of the three liposomal formulations tested. Instead, isolated protein coronas are dominated by apolipoproteins. These results show that photoaffinity labeling can be used to selectively determine the protein corona of liposomes without a bias toward large abundant proteins.
Discussion
The dominance of apolipoproteins on the surface of all three liposome formulations can be rationally explained in terms of endogenous protein function. The evolved function of soluble apolipoproteins is to bind secreted lipoproteins (e.g., HDL, LDL, VLDL, and chylomicrons), to coordinate the transport and metabolism of endogenous and exogenous (dietary) fats throughout the body.51 The general structure of a lipoprotein consists of a phospholipid monolayer surrounding a solid lipid core, rich in triglycerides and cholesteryl esters. Following secretion into the bloodstream, lipoproteins can associate with various exchangeable and soluble apolipoproteins (apo), the most abundant being apoA (I, II, and IV), apoC (I, II, and III), and apoE.51 Specific apolipoprotein binding to the surface of a lipoprotein is determined by the physicochemical properties of a lipoprotein, in particular, its size and curvature, as well as local environmental factors (e.g., local apolipoprotein concentrations). The changing apolipoprotein “signature” on the surface of a lipoprotein, throughout its lifecycle, dictates a lipoprotein’s fate in the body. Given the natural affinity of soluble apolipoproteins for the surface of endogenous and circulating lipid nanoparticles (i.e., lipoproteins), it is perhaps unsurprising that these serum proteins also dominate the protein corona of liposomes.
At a fundamental level, our finding that virtually no serum proteins, including highly abundant serum albumin, bind to the surface of Myocet liposomes suggests that the general formation of a protein corona on a nanoparticle may not always be relevant. Likewise, the enrichment and high abundance of acidic apoE (pI 5.5) on the surface of anionic AmBisome liposomes is unexpected, although it can be rationalized by the presence of a cationic heparin binding site on the surface of apoE.52−54 Overall, while the implications of these findings on in vivo liposome fate will require comprehensive mechanistic studies in animal models,55 the ability to accurately characterize and quantify the protein corona of a liposome in complex biological mixtures, prior to first injections in animals, provides a strong rationale for further in vivo experiments.
Finally, it is important to recognize the limitations of our photoaffinity method as described. Given its chemical structure, the IKS02 photoaffinity probe can only be reasonably applied to lipidic (nano)materials (e.g., liposomes, micelles, solid lipid particles, lipid-coated particles, etc.). Assuming synthetic accessibility, however, there is no reason why a bifunctional probe with separate photoaffinity and conjugation handles could not be designed for other self-assembled, organic materials (e.g., polymersomes, hydrogels, etc.). More fundamentally, however, our photoaffinity approach can only capture the hard protein corona of a liposome (i.e., proteins directly adsorbed to the nanoparticle surface) and will not resolve potentially important proteins of any (outer) “soft” corona that may form.4,56,57 It is worth noting, however, that in the case of lipoprotein-bound apolipoproteins, biological function relies on direct binding of apolipoprotein to a target receptor/enzyme (e.g., apoE-LDLr mediated uptake of low-density lipoprotein (LDL) particles in hepatocytes).51
In conclusion, our photoaffinity-based chemoproteomics approach enables the capture, identification, and quantification of the protein corona of a liposome in its native state. Through this approach, we have revealed liposome protein coronas that are quantitatively and qualitatively different from each other but also significantly less complex than those previously reported. While we have focused on human serum solutions in this study, the ability to capture proteins in situ provides a unique opportunity to isolate and characterize the adsorbed protein corona of a liposome, in its native state, in any ex vivo or in vitro protein sample, such as human blood or plasma, and even in vivo (e.g., using light transparent zebrafish embryos). Furthermore, light activation can be applied with high spatiotemporal resolution, offering the chance to resolve evolving nanoparticle–protein interactions in both time and space. These features represent a significant technological advance over current methods and, going forward, may enrich our fundamental understanding of the protein corona as well as its impact on nanoparticle behavior and performance in vitro and in vivo.
Acknowledgments
The research was financially supported via a NWO VICI grant (724.014.001) and an M-ERA grant awarded to A.K. for R.P. and F.C. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE58 partner repository with the dataset identifier PXD016229.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b01222.
Figures and tables, materials and methods (chemical synthesis), detailed experimental chemical procedures, materials and methods (biological and proteomics), detailed procedures (biological and proteomics), NMR and HRMS spectra of chemical compounds (PDF)
Proteomic data: quantified protein lists generated using ISOQuant (XLSX)
Proteomic data: identified and quantified peptide fragments lists generated using ISOQuant (XLSX)
Author Contributions
R.P. and F.C. conceived the research. R.P. designed the experiments. R.P., S.C., and I.K.S. carried out all the experiments and analyzed the data. B.I.F. designed and performed the nanoUPLC-MS/MS measurements and assisted in the analysis. A.K. directed the project. R.P and F.C. wrote the manuscript with feedback from all the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Lazarovits J.; Chen Y. Y.; Sykes E. A.; Chan W. C. W. Nanoparticle-Blood Interactions: The Implications on Solid Tumour Targeting. Chem. Commun. 2015, 51, 2756–2767. 10.1039/C4CC07644C. [DOI] [PubMed] [Google Scholar]
- Cai R.; Chen C. The Crown and the Scepter: Roles of the Protein Corona in Nanomedicine. Adv. Mater. 2019, 31, 1–13. 10.1002/adma.201805740. [DOI] [PubMed] [Google Scholar]
- Nguyen V. H.; Lee B.-J. Protein Corona: A New Approach for Nanomedicine Design. Int. J. Nanomed. 2017, 12, 3137–3151. 10.2147/IJN.S129300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkey C. D.; Chan W. C. W. Understanding and Controlling the Interaction of Nanomaterials with Proteins in a Physiological Environment. Chem. Soc. Rev. 2012, 41, 2780–2799. 10.1039/C1CS15233E. [DOI] [PubMed] [Google Scholar]
- Nel A. E.; Mädler L.; Velegol D.; Xia T.; Hoek E. M. V; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano-Bio Interface. Nat. Mater. 2009, 8, 543–557. 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Bertrand N.; Grenier P.; Mahmoudi M.; Lima E. M.; Appel E. A.; Dormont F.; Lim J. M.; Karnik R.; Langer R.; Farokhzad O. C.. Mechanistic Understanding of in Vivo Protein Corona Formation on Polymeric Nanoparticles and Impact on Pharmacokinetics. Nat. Commun. 2017, 8. 10.1038/s41467-017-00600-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. Y.; Kim H. S.; Palanikumar L.; Go E. M.; Jana B.; Park S. A.; Kim H. Y.; Kim K.; Seo J. K.; Kwak S. K.; Kim C.; Kang S.; Ryu J. H. Cloaking Nanoparticles with Protein Corona Shield for Targeted Drug Delivery. Nat. Commun. 2018, 9, 1–9. 10.1038/s41467-018-06979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvati A.; Pitek A. S.; Monopoli M. P.; Prapainop K.; Bombelli F. B.; Hristov D. R.; Kelly P. M.; Åberg C.; Mahon E.; Dawson K. A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez R.; Hadjidemetriou M.; Sanchez-Iglesias A.; Liz-Marzan L. M.; Kostarelos K. In Vivo Formation of Protein Corona on Gold Nanoparticles. The Effect of Their Size and Shape. Nanoscale 2018, 10, 1256–1264. 10.1039/C7NR08322J. [DOI] [PubMed] [Google Scholar]
- Corbo C.; Molinaro R.; Taraballi F.; Toledano Furman N. E.; Hartman K. A.; Sherman M. B.; De Rosa E.; Kirui D. K.; Salvatore F.; Tasciotti E. Unveiling the in Vivo Protein Corona of Circulating Leukocyte-like Carriers. ACS Nano 2017, 11, 3262–3273. 10.1021/acsnano.7b00376. [DOI] [PubMed] [Google Scholar]
- Grenier P.; Viana I. M. de O.; Lima E. M.; Bertrand N. Anti-Polyethylene Glycol Antibodies Alter the Protein Corona Deposited on Nanoparticles and the Physiological Pathways Regulating Their Fate in Vivo. J. Controlled Release 2018, 287, 121–131. 10.1016/j.jconrel.2018.08.022. [DOI] [PubMed] [Google Scholar]
- Amici A.; Caracciolo G.; Digiacomo L.; Gambini V.; Marchini C.; Tilio M.; Capriotti A. L.; Colapicchioni V.; Matassa R.; Familiari G.; Palchetti S.; Pozzi D.; Mahmoudi M.; Laganà A. In Vivo Protein Corona Patterns of Lipid Nanoparticles. RSC Adv. 2017, 7, 1137–1145. 10.1039/C6RA25493D. [DOI] [Google Scholar]
- Wang M.; Gustafsson O. J. R.; Pilkington E. H.; Kakinen A.; Javed I.; Faridi A.; Davis T. P.; Ke P. C. Nanoparticle–Proteome in Vitro and in Vivo. J. Mater. Chem. B 2018, 6, 6026–6041. 10.1039/C8TB01634H. [DOI] [PubMed] [Google Scholar]
- Casals E.; Pfaller T.; Duschl A.; Oostingh G. J.; Puntes V. Time Evolution of the Nanoparticle Protein Corona. ACS Nano 2010, 4, 3623–3632. [DOI] [PubMed] [Google Scholar]
- Barrán-Berdón A. L.; Pozzi D.; Caracciolo G.; Capriotti A. L.; Caruso G.; Cavaliere C.; Riccioli A.; Palchetti S.; Laganá A. Time Evolution of Nanoparticle-Protein Corona in Human Plasma: Relevance for Targeted Drug Delivery. Langmuir 2013, 29, 6485–6494. 10.1021/la401192x. [DOI] [PubMed] [Google Scholar]
- Hadjidemetriou M.; Al-Ahmady Z.; Kostarelos K. Time-Evolution of in Vivo Protein Corona onto Blood-Circulating PEGylated Liposomal Doxorubicin (DOXIL) Nanoparticles. Nanoscale 2016, 8, 6948–6957. 10.1039/C5NR09158F. [DOI] [PubMed] [Google Scholar]
- Cedervall T.; Lynch I.; Lindman S.; Berggard T.; Thulin E.; Nilsson H.; Dawson K. A.; Linse S. Understanding the Nanoparticle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2050–2055. 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docter D.; Westmeier D.; Markiewicz M.; Stolte S.; Knauer S. K.; Stauber R. H. The Nanoparticle Biomolecule Corona: Lessons Learned – Challenge Accepted?. Chem. Soc. Rev. 2015, 44, 6094–6121. 10.1039/C5CS00217F. [DOI] [PubMed] [Google Scholar]
- Carrillo-Carrion C.; Carril M.; Parak W. J. Techniques for the Experimental Investigation of the Protein Corona. Curr. Opin. Biotechnol. 2017, 46, 106–113. 10.1016/j.copbio.2017.02.009. [DOI] [PubMed] [Google Scholar]
- Weber C.; Morsbach S.; Landfester K. Possibilities and Limitations of Different Separation Techniques for the Analysis of the Protein Corona. Angew. Chem., Int. Ed. 2019, 58, 12787–12794. 10.1002/anie.201902323. [DOI] [PubMed] [Google Scholar]
- Lundqvist M.; Augustsson C.; Lilja M.; Lundkvist K.; Dahlbäck B.; Linse S.; Cedervall T. The Nanoparticle Protein Corona Formed in Human Blood or Human Blood Fractions. PLoS One 2017, 12, 1–15. 10.1371/journal.pone.0175871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal P.; Hall J. B.; McLeland C. B.; Dobrovolskaia M. A.; McNeil S. E. Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv. Drug Delivery Rev. 2009, 61, 428–437. 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundqvist M.; Stigler J.; Elia G.; Lynch I.; Cedervall T.; Dawson K. A. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 14265. 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi D.; Caracciolo G.; Digiacomo L.; Colapicchioni V.; Palchetti S.; Capriotti A. L.; Cavaliere C.; Zenezini Chiozzi R.; Puglisi A.; Laganà A. The Biomolecular Corona of Nanoparticles in Circulating Biological Media. Nanoscale 2015, 7, 13958–13966. 10.1039/C5NR03701H. [DOI] [PubMed] [Google Scholar]
- Hadjidemetriou M.; Al-Ahmady Z.; Mazza M.; Collins R. F.; Dawson K.; Kostarelos K. In Vivo Biomolecule Corona around Blood-Circulating, Clinically Used and Antibody-Targeted Lipid Bilayer Nanoscale Vesicles. ACS Nano 2015, 9, 8142–8156. 10.1021/acsnano.5b03300. [DOI] [PubMed] [Google Scholar]
- Gubbens J.; Ruijter E.; de Fays L. E. V; Damen J. M. A.; de Kruijff B.; Slijper M.; Rijkers D. T. S.; Liskamp R. M. J.; de Kroon A. I. P. M. Photocrosslinking and Click Chemistry Enable the Specific Detection of Proteins Interacting with Phospholipids at the Membrane Interface. Chem. Biol. 2009, 16, 3–14. 10.1016/j.chembiol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Murale D. P.; Hong S. C.; Haque M. M.; Lee J. S. Photo-Affinity Labeling (PAL) in Chemical Proteomics: A Handy Tool to Investigate Protein-Protein Interactions (PPIs). Proteome Sci. 2016, 15, 1–34. 10.1186/s12953-017-0123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M.; Stolze S. C.; Westphal M. V.; van Stralen L.; Martella A.; van Rooden E. J.; Guba W.; Varga Z. V.; Deng H.; van Kasteren S. I.; Grether U.; Ijzerman A. P.; Pacher P.; Carreira E. M.; Overkleeft H. S.; Ioan-Facsinay A.; Heitman L. H.; van der Stelt M. Selective Photoaffinity Probe That Enables Assessment of Cannabinoid CB2 Receptor Expression and Ligand Engagement in Human Cells. J. Am. Chem. Soc. 2018, 140, 6067–6075. 10.1021/jacs.7b11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T. M.; Cullis P. R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Delivery Rev. 2013, 65, 36–48. 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- Bulbake U.; Doppalapudi S.; Kommineni N.; Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. 10.3390/pharmaceutics9020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell F.; Bos F. L.; Sieber S.; Arias-Alpizar G.; Koch B. E.; Huwyler J.; Kros A.; Bussmann J. Directing Nanoparticle Biodistribution through Evasion and Exploitation of Stab2-Dependent Nanoparticle Uptake. ACS Nano 2018, 12, 2138–2150. 10.1021/acsnano.7b06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S.; Docter D.; Kuharev J.; Musyanovych A.; Fetz V.; Hecht R.; Schlenk F.; Fischer D.; Kiouptsi K.; Reinhardt C.; Landfester K.; Schild H.; Maskos M.; Knauer S. K.; Stauber R. H. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772. 10.1038/nnano.2013.181. [DOI] [PubMed] [Google Scholar]
- Capriotti A. L.; Caracciolo G.; Caruso G.; Cavaliere C.; Pozzi D.; Samperi R.; Laganà A. Analysis of Plasma Protein Adsorption onto DC-Chol-DOPE Cationic Liposomes by HPLC-CHIP Coupled to a Q-TOF Mass Spectrometer. Anal. Bioanal. Chem. 2010, 398, 2895–2903. 10.1007/s00216-010-4104-y. [DOI] [PubMed] [Google Scholar]
- Chang H.-I.; Cheng M.-Y. Clinically-Proven Liposome-Based Drug Delivery: Formulation, Characterization and Therapeutic Efficacy. J. Nanomed. Biother. Discovery 2012, 01, 1–8. 10.4172/scientificreports.195. [DOI] [Google Scholar]
- Marrink S.-J.; Berger O.; Tieleman P.; Jähnig F. Adhesion Forces of Lipids in a Phospholipid Membrane Studied by Molecular Dynamics Simulations. Biophys. J. 1998, 74, 931–943. 10.1016/S0006-3495(98)74016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Jang S. Y.; Wang D.; Liew S. S.; Li Z.; Lee J. S.; Yao S. Q. A Suite of “Minimalist” Photo-Crosslinkers for Live-Cell Imaging and Chemical Proteomics: Case Study with BRD4 Inhibitors. Angew. Chem., Int. Ed. 2017, 56, 11816–11821. 10.1002/anie.201706076. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Baskin J. M.; Prescher J. A.; Lo A.; Bertozzi C. R. A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol. 2006, 1, 644–648. 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- Batist G.; Barton J.; Chaikin P.; Swenson C.; Welles L. Myocet (Liposome-Encapsulated Doxorubicin Citrate): A New Approach in Breast Cancer Therapy. Expert Opin. Pharmacother. 2002, 3, 1739–1751. 10.1517/14656566.3.12.1739. [DOI] [PubMed] [Google Scholar]
- Cornely O. A.; Maertens J.; Bresnik M.; Ebrahimi R.; Ullmann A. J.; Bouza E.; Heussel C. P.; Lortholary O.; Rieger C.; Boehme A.; Aoun M.; Horst H.; Thiebaut A.; Ruhnke M.; Reichert D.; Vianelli N.; Krause S. W.; Olavarria E.; Herbrecht R. Liposomal Amphotericin B as Initial Therapy for Invasive Mold Infection: A Randomized Trial Comparing a High – Loading Dose Regimen with Standard Dosing (AmBiLoad Trial). Clin. Infect. Dis. 2007, 44, 1289–1297. 10.1086/514341. [DOI] [PubMed] [Google Scholar]
- Schmitt-Sody M.; Strieth S.; Krasnici S.; Sauer B.; Schulze B.; Teifel M.; Michaelis U.; Naujoks K.; Dellian M. Neovascular Targeting Therapy: Paclitaxel Encapsulated in Cationic Liposomes Improves Antitumoral Efficacy. Clin. Cancer Res. 2003, 9, 2335–2341. (https://clincancerres.aacrjournals.org/content/9/6/2335). [PubMed] [Google Scholar]
- Yang Y.; Yang X.; Verhelst S. H. L. Comparative Analysis of Click Chemistry Mediated Activity-Based Protein Profiling in Cell Lysates. Molecules 2013, 18, 12599–12608. 10.3390/molecules181012599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens C. J.; Johnson S. N.; Pressnall M. M.; Leon M. A.; Berkland C. J. Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjugate Chem. 2018, 29, 686–701. 10.1021/acs.bioconjchem.7b00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Shweiki M. H. D. R.; Mönchgesang S.; Majovsky P.; Thieme D.; Trutschel D.; Hoehenwarter W. Assessment of Label-Free Quantification in Discovery Proteomics and Impact of Technological Factors and Natural Variability of Protein Abundance. J. Proteome Res. 2017, 16, 1410–1424. 10.1021/acs.jproteome.6b00645. [DOI] [PubMed] [Google Scholar]
- Docter D.; Distler U.; Storck W.; Kuharev J.; Wünsch D.; Hahlbrock A.; Knauer S. K.; Tenzer S.; Stauber R. H. Quantitative Profiling of the Protein Coronas That Form around Nanoparticles. Nat. Protoc. 2014, 9, 2030–2044. 10.1038/nprot.2014.139. [DOI] [PubMed] [Google Scholar]
- Silva J. C.; Gorenstein M. V.; Li G.; Vissers J. P. C.; Geromanos S. J. Absolute Quantification of Proteins by LCMS E. Mol. Cell. Proteomics 2006, 5, 144–156. 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- Distler U.; Kuharev J.; Navarro P.; Levin Y.; Schild H.; Tenzer S. Drift Time-Specific Collision Energies Enable Deep-Coverage Data-Independent Acquisition Proteomics. Nat. Methods 2014, 11, 167–170. 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- Old W. M.; Meyer-Arendt K.; Aveline-Wolf L.; Pierce K. G.; Mendoza A.; Sevinsky J. R.; Resing K. A.; Ahn N. G. Comparison of Label-Free Methods for Quantifying Human Proteins by Shotgun Proteomics. Mol. Cell. Proteomics 2005, 4, 1487–1502. 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Autin L.; Olson A. J. Illustrate: Software for Biomolecular Illustration. Structure 2019, 27, 1716–1720. 10.1016/j.str.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A.; Tomas H.; Havli J.; Olsen J. V.; Mann M. In-Gel Digestion for Mass Spectrometric Characterization of Proteins and Proteomes. Nat. Protoc. 2006, 1, 2856–2860. 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P.; Aberg C.; Salvati A.; Dawson K. A. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7, 779–786. 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- Mahley R. W.; Innerarity T. L.; Rall S. C. J.; Weisgraber K. H. Plasma Lipoproteins: Apolipoprotein Structure and Function. J. Lipid Res. 1984, 25, 1277–1294. [PubMed] [Google Scholar]
- Saito H.; Dhanasekaran P.; Nguyen D.; Baldwin F.; Weisgraber K. H.; Wehrli S.; Phillips M. C.; Lund-Katz S. Characterization of the Heparin Binding Sites in Human Apolipoprotein E. J. Biol. Chem. 2003, 278, 14782–14787. 10.1074/jbc.M213207200. [DOI] [PubMed] [Google Scholar]
- Dong J.; Peters-Libeu C. A.; Weisgraber K. H.; Segelke B. W.; Rupp B.; Capila I.; Hernaiz M. J.; LeBrun L. A.; Linhardt R. J. Interaction of the N-Terminal Domain of Apolipoprotein E4 with Heparin. Biochemistry 2001, 40, 2826–2834. 10.1021/bi002417n. [DOI] [PubMed] [Google Scholar]
- Chen J.; Li Q.; Wang J. Topology of Human Apolipoprotein E3 Uniquely Regulates Its Diverse Biological Functions. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14813–14818. 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarovits J.; Sindhwani S.; Tavares A. J.; Zhang Y.; Song F.; Audet J.; Krieger J. R.; Syed A. M.; Stordy B.; Chan W. C. W. Supervised Learning and Mass Spectrometry Predicts the in Vivo Fate of Nanomaterials. ACS Nano 2019, 13, 8023–8034. 10.1021/acsnano.9b02774. [DOI] [PubMed] [Google Scholar]
- Weber C.; Simon J.; Mailänder V.; Morsbach S.; Landfester K. Preservation of the Soft Protein Corona in Distinct Flow Allows Identification of Weakly Bound Proteins. Acta Biomater. 2018, 76, 217–224. 10.1016/j.actbio.2018.05.057. [DOI] [PubMed] [Google Scholar]
- Kari O. K.; Ndika J.; Parkkila P.; Louna A.; Lajunen T.; Puustinen A.; Viitala T.; Alenius H.; Urtti A. In Situ Analysis of Liposome Hard and Soft Protein Corona Structure and Composition in a Single Label-Free Workflow. Nanoscale 2020, 12, 1728–1741. 10.1039/C9NR08186K. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Pérez-Riverol Y.; Reisinger F.; Ríos D.; Wang R.; Hermjakob H. The Proteomics Identifications (PRIDE) Database and Associated Tools: Status in 2013. Nucleic Acids Res. 2012, 41, D1063–D1069. 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.