

Abstract

Ortholog protein complexes are responsible for equivalent functions in different organisms. However, during evolution, each organism adapts to meet its physiological needs and the environmental challenges imposed by its niche. This selection pressure leads to structural diversity in protein complexes, which are often difficult to specify, especially in the absence of high-resolution structures. Here, we describe a multilevel experimental approach based on native mass spectrometry (MS) tools for elucidating the structural preservation and variations among highly related protein complexes. The 20S proteasome, an essential protein degradation machinery, served as our model system, wherein we examined five complexes isolated from different organisms. We show that throughout evolution, from the Thermoplasma acidophilum archaeal prokaryotic complex to the eukaryotic 20S proteasomes in yeast (Saccharomyces cerevisiae) and mammals (rat - Rattus norvegicus, rabbit - Oryctolagus cuniculus and human - HEK293 cells), the proteasome increased both in size and stability. Native MS structural signatures of the rat and rabbit 20S proteasomes, which heretofore lacked high-resolution, three-dimensional structures, highly resembled that of the human complex. Using cryoelectron microscopy single-particle analysis, we were able to obtain a high-resolution structure of the rat 20S proteasome, allowing us to validate the MS-based results. Our study also revealed that the yeast complex, and not those in mammals, was the largest in size and displayed the greatest degree of kinetic stability. Moreover, we also identified a new proteoform of the PSMA7 subunit that resides within the rat and rabbit complexes, which to our knowledge have not been previously described. Altogether, our strategy enables elucidation of the unique structural properties of protein complexes that are highly similar to one another, a framework that is valid not only to ortholog protein complexes, but also for other highly related protein assemblies.
Short abstract
Mass spectrometry approaches enabled structural elucidation of unique properties of different 20S proteasomes, a framework that is valid not only to ortholog protein complexes, but also for other highly related protein assemblies.
Introduction
Biological processes in the cell are driven by multicomponent protein complexes that form hundreds of different functional modules within the cellular environment.1 Many of these protein complexes arose from a common ancestor and are structurally and functionally conserved.2,3 Nevertheless, despite the high degree of conservation shared among ortholog protein complexes, they diverged through different evolutionary trajectories to adapt to the functional needs of present-day organisms.4 Today, valuable information on the degree of protein complex divergence is provided by bioinformatic analysis;5−8 however, in many cases, experimental characterization is lacking. To analyze the implications of evolutionary constraints on the structural features of protein complexes, we chose to apply an integrated native mass spectrometry (MS) approach, with the 20S proteasome complex serving as a model system and with a cryoelectron microscopy (EM) structure of the rat proteasome determined for validation of the MS results.
The 20S proteasome is an essential degradation machinery, designed for controlled proteolysis.9,10 It can function in its free, uncapped form and cleave proteins that contain partially unfolded regions, which can enter into its narrow aperture.11 This group of substrates includes aged, mutated, and oxidized proteins, or proteins that in their native state contain intrinsically disordered regions.11 On the other hand, to degrade structured substrates targeted for degradation by ubiquitin tagging, the 20S proteasome associates with one or two 19S regulatory complexes, forming the 26S proteasome.12,13
The architecture of the 20S proteasome particle is highly conserved, creating a 700 kDa compartment whose proteolytic active sites are restricted to its interior, so that only proteins entering this chamber are degraded.14−21 It is composed of 28 subunits, arranged in a cylindrical structure consisting of four heptameric rings: two outer α-type subunit rings embracing two central β-type subunit rings (α7β7β7α7). The two outer α-rings function as a gate that regulates the entry into the proteolytic chamber inside the two β-rings.13 The 20S proteasome can be found in all three domains of life and is ubiquitous in archaea and eukaryotes. However, a dramatic increase in complexity and diversification of the complex occurred as the organisms evolved over time. Prokaryotic 20S proteasomes (e.g., from Thermoplasma acidophilum) are generally composed of identical copies of 14 α-subunits and 14 β-subunits, while in eukaryotic proteasomes, the α- and β-subunits each differentiated into seven different subtypes, accounting for a total of 14 different subunits.22
To date, most of the structural information on 20S proteasome complexes has been driven by high-resolution structural biology methods as X-ray crystallography and cryo-EM.14−21,23−25 Nuclear magnetic resonance has also contributed to our understanding of the gating26,27 and allosteric communication within the 20S proteasome complex.28 Despite this indisputable contribution, these techniques have their own challenges and limitations, ranging from sample size and amount, up to homogeneity and rigidity restrictions. Here, we will focus on innovations in native MS that facilitate the comparative investigation of 20S proteasome complexes. Specifically, we investigated the structural properties of 20S proteasomes isolated from different organisms, i.e., archaea (T. acidophilum), yeast (Saccharomyces cerevisiae), rat (Rattus norvegicus), rabbit (Oryctolagus cuniculus), and human (HEK293 cells), species that span billions of years of evolution. Although native MS does not deliver structures at atomic resolution, its advantage lies in its rapid analysis, its low sample amount requirement, and its ability to provide insight into protein conformational dynamics and coexisting transient species in solution.29−33 We applied a set of MS-based approaches to dissect the distinct structural attributes of these highly related 20S proteasome complexes. Each method yielded a layer of information, which together provided a specific structural signature for each individual proteasome.
Our experimental analysis confirmed the subunit connectivity of the 20S proteasome α-ring and identified a proteoform for the PSMA7 subunit in glires (rat and rabbit) that has not been described before. Moreover, we show a significant increase in the collision cross-section (CCS) values of eukaryotic proteasomes compared to those of the prokaryotic archaea complex, reflecting the increased complexity of the yeast and mammalian 20S particles. Furthermore, for the eukaryotic 20S proteasome complexes, we demonstrate the diversification of individual subunits due to post-translational modifications (PTMs). In addition, we found that the yeast proteasome particle possesses the highest kinetic stability, more than the mammalian complexes, whose degree of stability is comparable. This property might confer resistance to high ethanol concentrations that are typical to yeast growth,34 ensuring a longer lifetime of the folded and active state of the 20S proteasome even under harsh physiological conditions. By solving the cryo-EM structure of the rat 20S proteasome, which was lacking an atomic structure, we validated the MS-based results. Overall, the native MS and cryo-EM results were complementary, with native MS providing relative kinetic stability and proteoform information unavailable in the cryo-EM and cryo-EM providing higher resolution structural detail. On a broader view, these results demonstrate the benefit of native MS and how it can guide structural studies of other highly related protein complexes, even those that are still lacking high-resolution structures.
Results and Discussion
Distinct Collision Cross Sections Are Exhibited by the Different Proteasome Complexes
To study the influences of evolution on the 20S proteasomes, we began by examining the overall structure of each complex from the different organisms. After establishing that all the purified proteasomes are active (Figure S1), we acquired native ion-mobility (IM) MS spectra of the simplified archaeal 20S proteasome (T. acidophilum), which contains only one type of each α- and β-subunit, and eukaryotic proteasomes isolated from yeast (S. cerevisiae), rat (R. norvegicus), rabbit (O. cuniculus), and human (HEK293 cells), each of which are composed of seven different α- and seven different β-subunits (Figure 1A,B).
Figure 1.

Native IM-MS characterization reveals differences between the archaeal, yeast, and mammalian 20S proteasome complexes. (A) Purified 20S proteasomes from five different organisms (human, rabbit, rat, yeast, and archaea) were measured on a modified Synapt G1 instrument. The three mammalian proteasomes were found to be close in mass and charge distributions. The prokaryotic archaeal 20S proteasome displays a lower mass and relatively higher charge states, while the yeast proteasome displays the highest molecular weight. Average masses for each particle are calculated from the apex of each MS peak, errors define the standard deviation of the mean. (B) IM-MS spectra of the various 20S proteasome complexes. (C) Bar plot of the experimental CCS values, which were determined from the position of the apex of the most intense mobility peak. The CCS value of the archaeal 20S proteasome is significantly lower than that of the eukaryotic complexes (p < 0.0001). Error bars represent the standard deviations of three different wave heights (14, 15, and 16 V). (D) Box plot of the width of the CCS distributions at half of the maximum height for the different 20S proteasomes. The horizontal bar indicates the median, the box shows the interquartile range (25–75%), and the whiskers extend to minimum and maximum of the result range.
The data indicate that the complex from yeast is the largest particle, with an averaged measured mass of 731 kDa, as defined from the apex of each MS peak (Figure 1A). This measurement is in agreement with the calculated mass of the particle, 730 706 Da, which was calculated from the sequence masses corrected for the detected isoforms shown in Table S1, considering the most abundant proteoform of each subunit (detailed in the Experimental Methods). The measured mass of the human 20S proteasome was 719 kDa, and those of the glires proteasome (rabbit and rat) yielded relatively similar molecular masses of 716 and 717 kDa (Figure 1A), in line with their theoretical masses of 717 257, 715 275, and 716 072 Da, respectively. The archaeal 20S proteasome displays two populations, containing a mixture of β-subunits before and after cleavage of the β-subunit propeptides. The measured mass of the mature complex is 682 kDa, which is in close agreement with its theoretical mass of 677 374 Da. Notably, the charge state series width of all eukaryotic proteasomes was wide in comparison to the archaeal proteasome peaks. This value, which is larger than the uncertainty in mass measurements (mass error), results from a combination of instrumental resolution, adduct binding and biological heterogeneity of samples (Table S1). Assuming that the contribution of instrumental resolution is similar for all proteasomes, the wider peak width of the eukaryotic particles can be attributed to more adduct binding and intrinsic heterogeneity arising from PTMs, sequence variants, and alternative splicing.
The highest charge state observed in each spectrum was then compared
to the expected theoretical maximal charge. We used the De La Mora
relationship to determine the theoretical maximum number of positive
charges that a globular spherical protein would be expected to accommodate:  , where ZR is the maximum (Rayleigh)
charge and m is the molecular weight of the protein.35 The calculated ZR for the eukaryotic 20S proteasomes is 66, and that of the archaeal
complex is 64. While there is agreement between the measured and expected
charge state values for the archaeal complex, we noticed that the
eukaryotic 20S proteasomes exhibit charge states below the ZR limit. This finding suggests that a more compact
structure was adapted upon the transition from prokaryotes to eukaryotes.
, where ZR is the maximum (Rayleigh)
charge and m is the molecular weight of the protein.35 The calculated ZR for the eukaryotic 20S proteasomes is 66, and that of the archaeal
complex is 64. While there is agreement between the measured and expected
charge state values for the archaeal complex, we noticed that the
eukaryotic 20S proteasomes exhibit charge states below the ZR limit. This finding suggests that a more compact
structure was adapted upon the transition from prokaryotes to eukaryotes.
Native IM-MS measurements enable us to separate ions not only based on their mass-to-charge ratio, but also by their shape, yielding rotationally averaged CCS values that depict the overall shapes and conformational dynamics of the various proteasome particles.36−39 We therefore continued by calculating the CCS values, using the centroid of each peak, for each of the proteasomes (Figure 1B–C). Values of 20 407 ± 30 and 20 505 ± 35 Å2 were determined for the human and yeast 20S proteasomes, respectively. On the basis of the available crystal structures, theoretical CCSs for these complexes yield values of 19 090 Å2 for the human complex and 19 045 Å2 for the yeast 20S proteasome (yeast PDB: 5CZ4, human PDB: 5LEX). These values are in agreement with the measured values (differing by about 7% from measured), given that the projection approximation algorithm used to calculate this value from known crystal structures underestimates CCSs by ∼10%.40 Likewise, we measured CCS values for the rat and rabbit 20S proteasomes, which currently do not have published crystal structures. Both IM-MS spectra yielded similar CCS values of 20 286 ± 14 and 20 209 ± 60 Å2, respectively. The CCS value calculated for the archaeal 20S was significantly smaller than those of all the other particles (19 503 ± 100 Å2), in accordance with its theoretical CCS value of 18 801 Å2 (PDB: 6BDF), and its lower molecular weight. Since the measurement is performed on a population of ions, a distribution of CCSs is obtained. This distribution is inferred by the full-width at half-maximum of the CCS peaks and conveys information on the structural heterogeneity of the proteasome complexes.41 Analysis of the CCS widths indicates that there is no significant trend among the eukaryotic proteasome species, as they all yielded similar CCS distribution values, suggesting a merely similar conformational spread (Figure 1D). However, in comparison to the eukaryotic complexes, the peak width values of the archaeal particle were smaller, suggesting a narrower conformational space.
Taken together, our measurements indicate that there is an overall agreement between the calculated CCS values and the molecular weights. The three mammalian 20S proteasomes share similar mass and CCS values, whereas the archaeal (T. acidophilum) and yeast (S. cerevisiae) 20S proteasomes have the smallest (archaeal) and largest (yeast) CCS values. The increased molecular weight of the eukaryotic proteasomes is in accordance with previous studies indicating that the additional sequence fragments that were acquired by eukaryotic proteasome subunits appear at the N- and C-terminal extensions and internal loops, which are required for determining the fixed subunit arrangement within and between the α- and β-rings.12,20,42 The increased size of the yeast complex in comparison to the mammalian 20S proteasomes, highlighted by phylogenetic analysis (Figure S2A) and multiple sequence alignment (Figure S2B) of the different eukaryotic α- and β-subunits, has been suggested to be linked to partial differences in the assembly process between yeast and mammalian complexes.43−45
CIU Unfolding Profiles Reveals the Relative Kinetic Stability of Ortholog Proteasomes
To gain information on the relative conformational resilience of the 20S species, we applied the collision-induced unfolding (CIU) approach, which couples collisional molecular perturbation with IM-MS measurements.46,47 In this type of experiment, the collision energy is elevated in a stepwise manner, causing protein activation that may consequently induce conformational change. The collision energy at which the transitions between conformations occur, the mode of the transition, and the size of the intermediates generate a characteristic unfolding trajectory of the protein complex. This information was used to define the relative kinetic stabilities of the complexes, as the activated ions are conformationally trapped in specific structures with limited reversibility.48−52Figure S3 depicts CIU fingerprints of the highest charged ion in each of the 20S proteasome spectra that allowed detection across the entire desired energy range. This charge state was chosen to record the CIU fingerprint, taking into account that high charge states can lead to numerous CIU transitions.53
Three main CIU features are observed in all 20S proteasome species (Figures 2 and S3): an initial, compact state (state 0), observed at low activation energies, and two additional unfolded states (states 1 and 2, respectively) that are generated at higher collision voltages. For the three mammalian proteasomes, the two transitions appear at similar energies. The first transition, from the initial compact state (state 0) to the first intermediate state (state 1) occurs at ∼7000 eV; an additional unfolding into state 2 occurs at an acceleration energy of ∼9700 eV. Clear CIU differences are observed, however, in the archaeal and yeast 20S proteasomes. The conformational transition from state 0 to 1 in the yeast complex is relatively delayed. Occurring at ∼7400 eV, this state does not persist over a broad collision voltage range, and the transition to state 2 occurs at ∼9100 V. The archaeal proteasome appears to be the most sensitive to elevated collision voltage. Its intermediate unfolding state (state 1) is already observed at ∼6000 eV, and the second structural transition is observed at a collision energy of ∼9000 eV. Altogether, the CIU profiles of the different 20S proteasome variants reveal differences in the transition dynamics between the three conformational states. However, all species exhibit abrupt transitions, whereby one conformer disappears and another conformer simultaneously appears, with no coexisting states, suggesting that the structural transition is highly cooperative.
Figure 2.

CIU fingerprints
of the five 20S proteasome variants indicate the
existence of two transition steps. A graphic representation of the
CIU results spanning 130 V that are converted to the relevant energy
range for each proteasome species (CIU shown in Figure S3). The data indicate shifts between three different
states. The transition of the archaeal proteasome from state 0 to
1 occurs at the lowest energy, compared to the other proteasome species.
In addition, the data reflect the apparent stability of the 0 conformer
of the yeast proteasome compared to all other forms, whereas the mammalian
proteasomes each exhibit similar transition characteristics. All experiments
started at 50 and ended at 180 V and were then converted to Elab energies using the following equation 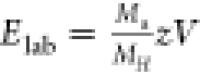 where the mass of the human
proteasome
(MH) is used as a reference for mass correction, z is charge, and V is acceleration voltage.
The numbers within the bars indicate the energy range spanned by each
conformational state.
where the mass of the human
proteasome
(MH) is used as a reference for mass correction, z is charge, and V is acceleration voltage.
The numbers within the bars indicate the energy range spanned by each
conformational state.
Similar CIU patterns in the mammalian complexes indicate their comparable topology and kinetic stability, while the ease with which the T. acidophilum archaeal 20S proteasome is structurally disrupted in comparison to the other eukaryotic particles is likely due to its reduced kinetic stability. Unexpected was the observation that the yeast 20S complex is initially resistant to gas-phase unfolding compared to mammalian proteasomes, as an higher acceleration energy was required to induce the first structural transition. Nevertheless, the second conformational transition from state 1 to state 2 was lower for the S. cerevisiae yeast complex, compared to that in the mammalian proteasomes. Overall, these results highlight the applicability of CIU patterns as specific fingerprints for distinct ortholog complexes.
SID Coupled to IM-MS Measurements Reflects 20S Proteasome Topology
An additional layer of information was obtained by fragmenting the proteasome complexes. Specifically, upon activating the complexes and inducing their dissociation, we expected to identify variations in the kinetic stability of the different proteasome species. To this end, we applied surface-induced dissociation (SID) prior to IM-MS. SID is a single-step, high-energy activation method, wherein dissociation of noncovalent interactions within the complex leads to generation of subcomplexes that form the building blocks of the complex.54−56 This method tends to favorably disrupt weaker interfaces in the protein complex.57,58 We began by examining the rabbit 20S proteasome (Figure 3). To ensure preservation of the native protein structure, we reduced the number of charges on the proteasome by utilizing triethylammonium acetate (TEAA)59 (Figure 3A, upper panel) and employed a wide isolation window, selecting the 44+ and 43+ charge states, to increase signal intensity (Figure 3A, lower panel). Upon activation, the proteasome dissociated into an array of subcomplexes with overlapping charge and oligomeric states (Figure 3B–C). We therefore harnessed the ability of IM to separate the product ions into a third dimension, based on their charge, size, and shape54 (Figure 3D–J).
Figure 3.

SID-IM-MS of the rabbit 20S proteasome complex reflects the cylindrical topology of the complex. Rabbit 20S proteasome was mixed with the charge-reducing agent TEAA and measured in a Synapt G2 instrument equipped with an SID cell (A, upper panel). The 43+ and 44+ charge states were isolated (A, lower panel) and accelerated into the surface at 150 V. (B) IM-MS plot of the SID spectrum of the rabbit 20S proteasome. The separation in drift time (vertical axis) assists in discrimination of species that are overlapping in m/z (horizontal axis) (C). The major populations of the dissociation products are designated by dashed lines (in B) and labeled with symbols that are graphically depicted in (D–J). The extracted m/z spectra from the underlined regions in the IM-MS plot (D–J), show the identified dissociation products.
Under the applied experimental conditions, a series of charge stripped peaks of the rabbit 20S proteasome precursor (α7β7β7α7) were observed (Figure 3D), in addition to various generated subcomplexes (Figure 3E–J). We identified half-proteasome species (α7β7), an α7 ring, and a β7β7α7 particle, as expected from the stacked four-ring structure of this complex. We also identified a subcomplex of the proteasome that was missing two α subunits (α7β7β7α5).60,61 Moreover, we identified a population of heterodimers corresponding in mass to PSMA2-PSMA6 (Figure 3I), discussed in detail below, and several series of α-monomers (Figure 3J). While the half-20S proteasome and the 20S proteasome missing two α-subunits have been previously described,62,60 as far as we know, this is the first time that α7 and β7β7α7 species have been identified, highlighting the advancements in technology and sample preparation methods.
We continued by comparing the SID-IM-MS spectra of the different 20S proteasome orthologs (Figure 4). The SID patterns of human and rat complexes were very similar to that of the rabbit 20S particle, in which multiple subcomplexes were produced during the dissociation process: an α7 ring, a half- proteasome (α7β7), three stacked ring assemblies (β7β7α7), a stripped complex missing two α-subunits (α7β7β7α5), the PSMA2-PSMA6 heterodimer and monomers (Figure 4A–B). For the archaeal proteasome, the most dominant population was the half-proteasome species, although the α7β7β7α5 subcomplex, 2α homodimer and monomeric α-subunits, were also detected (Figure 4D). This fragmentation pattern is consistent with the fact that the archaeal proteasome has similar α–β vs β–β interface areas but double the number of salt bridges in α–β versus β–β (Figure 4E). In contrast, clear differences were observed in the fragmentation plot of the yeast 20S proteasome (Figure 4C). Unlike the other complexes, SID at the voltage and charge state used for yeast did not dissect the yeast proteasome to an α7 ring plus three stacked ring assemblies (β7β7α7) or to two half proteasomes (α7β7) but did produce a stripped complex missing two α-subunits (α7β7β7α5) .
Figure 4.

SID-IM-MS analysis reveals a relative increase in stability of the yeast 20S proteasome. IM-MS plot of the SID spectra at 150 V of the (A) human (42+ and 43+), (B) rat (43+ and 44+), (C) yeast (41+ and 42+), and (D) archaeal (44+ and 45+) 20S proteasomes. The major dissociation products are designated by a dashed line and labeled with symbols that are graphically depicted in (F). (E) A table summarizing the interface areas, number of hydrogen bonds, and salt bridges between the α–β and β–β rings. Data were extracted from the available crystal structures of the 20S proteasomes (human 5LEX, yeast 5CZ4, and archaea 6BDF) using the PISA algorithm.
The results suggest that the yeast complex displays increased kinetic stability, in comparison to the three mammalian complexes. To further examine this assumption, we used the PISA algorithm63 to characterize the interface areas of the S. cerevisiae yeast (PDB: 5CZ4) and human (PDB: 5LEX) crystal structures (no high-resolution structures were available for rabbit and rat proteasomes) (Figure 4E). We noticed that the human 20S proteasome complex contains fewer hydrogen bonds and salt-bridges, and the total interface areas of both the α–β and β–β rings are smaller and the total number of hydrogen bonds and salt bridges is lower, in comparison to the yeast complex (Figure 4E). This observation explains the relative ease by which SID can disrupt the human complex64 and further confirms the increased kinetic stability of the yeast 20S proteasome (Figure 2).
Organisms evolve resistance to various stress factors, thus enabling higher tolerance under conditions where nonadapted organisms fail to grow. This might be the reason for the higher kinetic stability observed here for the yeast 20S proteasome; S. cerevisiae is continuously exposed to high intracellular ethanol concentrations,34,65 a condition that weakens hydrophobic interactions.66 To resist misfolding and preserve proteasome functionality under these harsh conditions, a higher unfolding barrier is probably needed.67 Thus, by increasing the number of stabilizing electrostatic interactions (hydrogen bonds and salt-bridges), in comparison to the other examined proteasome species, high kinetic stability is achieved (Figure S4). Thus, the observed higher kinetic stability of the 20S proteasome complex under harsh MS conditions likely relates to the yeast’s specific physiology and ecological niche.
Series of Heterodimers Reveals the α-Subunit Ring Order
To determine the subunit composition of the proteasome particles, we performed top-down, triple-stage MS analysis on an Orbitrap platform.68 During this analysis, the intact 20S proteasomes were initially dissociated into their constituent subunits. Following the selection and fragmentation of individual subunits, sequence analysis and PTM mapping were accomplished.68 To enhance top-down fragmentation and consequently the sequence coverage, electron capture dissociation (ECD)69 was coupled with the higher-energy collisional dissociation (HCD), giving rise to c- and z- as well as b- and y- type ions.
Data were obtained for the rat 20S proteasome by isolating ions at 12 000 m/z and applying an acceleration voltage to induce dissociation in the HCD cell (Figure 5A–B). We noticed the existence of several populations of ions in the low m/z region, around 3500–6000 m/z (Figure 5B), in addition to charge state series corresponding to the individual monomeric subunits that were stripped from the complex (1000–3500 m/z). The mass assignment process revealed that these series of peaks correspond in mass to heterodimers of α-subunits (Figure 5B–C). Under these experimental conditions, the core β-subunits were not dissociated.
Figure 5.

MS3 analysis enables elucidation of subunit organization in the α-ring. (A) Structural organization of the individual subunits in the rat 20S α-rings, as deciphered from MS3 experiments. (B–C) Analysis of the dissociation products of a MS2 experiment identifies single α-subunits, as well as α-subunit dimers of the rat 20S proteasome. Identified charge state series are labeled. The region where dimer ions are found is highlighted in gray in (B) and is enlarged in (C).
To examine the composition of the α-subunits heterodimers MS3 spectra were acquired for the 17+ charge state at 3127.2 m/z and the 20+ charge state at 2851.9 m/z (Figure S5A,C, Figure S5B,D). Following activation in the HCD cell, two series of dissociated ions were observed in the low-m/z region of each MS3 spectrum and assigned according to their measured masses to PSMA2/PSMA6 and PSMA4/PSMA7. Consequently, we were able to identify all the different pairs of heterodimers encompassing the seven different α-subunits (Figure 5B–C). On the basis of this information, we could confirm the subunit order of the rat proteasome α-ring (Figure 5A). Similar subunit arrangement information was obtained for the other 20S proteasome orthologs (Figure S6). This finding is not surprising given the available high-resolution structures of the human and yeast proteasomes20,21 and the evolutionarily conservation of the complex. Nevertheless, the results reflect the potential of the approach for determining the organization of other important cellular complexes for which very little structural data exists.
Identification of a New PSMA7 Proteoform
In characterizing the dissociated populations of the rat 20S proteasome subunits by MS2 experiments (Figure 5), we were able to accurately measure the masses and assign them to different monomeric α-subunits (Figure 6A) and dimers (Figure 5). Subunit identity was matched according to reported masses in protein databases, while considering major PTMs such as removal of the initial methionine, N-terminal acetylation, and phosphorylation. Using this approach, we assigned all the rat 20S proteasome α-subunits, except for the charge state series corresponding to PSMA7. Open source databases report the presence of three rat PSMA7 isoforms: P48004 (RC6-IL), encoding a 254 amino acid protein;70 P48004-2 (RC6-IS), a splice variant missing a six amino-acid stretch: “VVASVS” in positions 75–80;70 and isoform A0A0G2K0W9, which differs from P48004-2 by a single amino acid change of G99E.
Figure 6.

Native MS top-down analysis revealed a new rat PSMA7 proteoform. (A) MS2 activation of the rat 20S proteasome resulted in the dissociation to α-subunits. All the α-subunits could be assigned according to mass, except for PSMA7. Measured masses and type of modifications of the different subunits are indicated. Theoretical masses of each protein are shown in brackets. (B) The 21+ charge state of PSMA7 (highlighted in red in panel A) was isolated in the quadrupole and subjected to ECD and HCD fragmentation, resulting in a collection of backbone peptide fragments. Manual de novo sequencing, coupled with analysis using the LcMS-Spectator software, enabled us to reach 59% sequence coverage and confirm that the subunit is missing the initial methionine, contains an N-terminal acetylation and is missing the last two amino acids, Ala247 and Ser248. Brown brackets label b- and y-ions, orange brackets label c- and z-ions. Brackets pointing to the left and right denote the identified b- and c-ions and y- and z-ions, respectively.
The measured mass of PSMA7, 27 610 ± 0.8 Da, did not correspond to any of the reported isoforms, either with or without major PTMs. We therefore turned to MS3 analysis.68 Following isolation of a single charge state of this series, fragmentation was induced in both the ECD and HCD cells for top-down sequencing (Figure S7A). On the basis of the protein sequences of the PSMA7 isoforms, we could initially match fragments only to the N-terminal domains of the three isoforms, in which the initial methionine is removed and the N-terminus is acetylated. Because we detected no C-terminal fragments, we assumed that this protein might represent an uncharacterized isoform. Following manual de novo sequencing of the fragments, we could determine that the charge state series indeed constitutes a new proteoform, based on the protein sequence of A0A0G2K0W9, but missing the last two amino acids, Ala247 and Ser248 (Figure 6B). Inspection of the fragments using LcMS-Spectator software (PNNL, OMICS.PNL.GOV) revealed that the majority of peptide fragments in the spectrum correspond to this proteoform (Figure S7B–C), further confirming the validity of this identification.
This isoform appears as a significant population in the spectrum, and is most likely the dominant form of PSMA7 in rat liver 20S proteasomes. Bottom-up analysis of rat 20S proteasomes71 might have missed this small deletion of the two C-terminal amino acids, due to the lysine-rich KEKE motif at the C-terminus of PSMA7, that is placed immediately before the last two amino acids,70 making identification of the proteoform by trypsin-based bottom-up proteomic analysis challenging. Thus, native MS top-down analysis, which relies on accurate measurements of the intact complex and its subunits, has the potential to expose subunit isoforms that were previously unidentified.
Notably, all the identified α-subunits of the human and yeast 20S proteasomes (Figure S8A,B) could be accurately matched to protein sequences reported in open-source databases, while taking into account major PTMs, including that of PSMA7. In rabbits, on the other hand, we could not match the measured mass of PSMA7 to that of the reported isoform (GBCM01016564 https://www.ncbi.nlm.nih.gov/nuccore/610331313). However, after removal of the last two amino acids, Ala247 and Ser248, the measured mass fitted exactly to the theoretical mass of the protein missing its initial methionine and acetylated at the N-terminus, suggesting that this ragged-end C-terminal sequence might constitute a glires-specific isoform (Figure S8B).
To validate our results, we denatured the rat and rabbit 20S proteasomes and separated their constituent subunits by capillary electrophoresis (CE). Following the subunit elution from the CE device, the flow was directed straight into the mass spectrometer for intact protein mass measurements (Figures S9 and S10). Mass analysis of both the rat and rabbit 20S proteasomes confirmed that the major PSMA7 proteoform corresponds to the protein lacking the C-terminal Ala and Ser (Figures S9G and S10C, respectively), after removal of the initial methionine and acetylation. A minor subpopulation of the nonragged-end form of the PSMA7 could also be detected. Similar analysis of the 20S proteasome samples from human and yeast revealed the presence of a single full-length PSMA7 form (Figures S11 and S12, respectively).
In-depth analysis of the CE-MS results indicated that on average each subunit of the examined eukaryotic proteasomes has two coexisting variants (Figures S9–12, Table S1). Exceptions were the propeptide-containing β-subunits (PSMB1, PSMB4, PSMB5, PSMB6, and PSMB7) that displayed a single mature variant. We also noticed that the modifications were conserved among the four eukaryotic proteasome species, i.e., yeast, rat, rabbit, and human. The major variant, accounting for ∼93% of each subunit, corresponds to the removal of the first methionine residue, followed by acetylation; exceptions are PSMA1 and PSMA5, in which the initial methionine was not removed. Small subpopulations of nonacetylated proteoforms (∼7%) could also be detected for most of the subunits. The mammalian PSMA3 subunit was also singly phosphorylated, as reported earlier.72,73 However, this subunit was not detected at all for the yeast complex. Similarly, we could not detect in yeast the chromosomally tagged FLAG-His6-PSMB2 subunit.74 Lastly, in both rabbit and rat 20S proteasome complexes that were purified from liver tissue, immunoproteasome subunits were also identified, in line with the high abundance of immunoproteasome in liver samples.75 These immunoproteasome subunits were not detected in the human 20S proteasome sample originating from HEK293 cells, probably due to the overall low abundance of immunoproteasomes in these cells.76 Overall, the common modifications of the subunits among the different organisms suggest that they share a functional role.
High-Resolution Rat 20S Proteasome Structure Confirms the MS-Derived Global Structural Properties
Using cryo-EM, we determined the atomic structure of the rat 20S proteasome at 2.7 Å resolution (Figures 7 and S13). The quality of the map allowed unambiguous building of an atomic model of the complex. Unfortunately, for several of the subunits, the N- and/or C-terminus are missing due to poor electron densities that prevent reliable interpretation.
Figure 7.

cryo-EM structure of the rat 20S proteasome. (A) Cryo-EM structure of the rat 20S proteasome with a fitted atomic model. (B) Two close-up views of the structure shown in (A), which have well-resolved side chains throughout. (C) Overall view of the 20S atomic model, with each chain colored differently. (D) Close-up view of the α-ring. (E) A table summarizing the CCS value and the interface areas, number of hydrogen bonds and salt bridges between the α–β and β–β rings, as determined from cryoEM and IM-MS analyses.
The EM structure was then used for assessing the structural attributes that were revealed by native MS. The CCS value determined from the EM structure was 19 698 Å2, differing by only 4% from and in agreement with the IM-MS measured value (20 286 ± 16 Å2); this is good agreement considering the ∼10% underestimation anticipated for the projection approximation algorithm.40 As anticipated, the subunit arrangement of the α-ring is identical to that of the human and yeast 20S proteasomes, in accordance with the MS-based heterodimer analysis (Figure 5). Next, we characterized the interface areas of the rat cryo-EM structure. Fewer numbers of hydrogen bonds and salt bridges were obtained in the α–β and β–β interfaces of the rat proteasome, in comparison to the yeast crystal structure (Figures 4 and 7). In addition, the total rat proteasome interface area was smaller than that of the yeast. These observations support the different SID fragmentation patterns of the rat and yeast proteasomes (Figure 4), in which the rat complex is disrupted more easily than the yeast and further confirms the increased kinetic stability of the yeast 20S proteasome (Figure 2).
We next wished to examine whether the new PSMA7 proteoform that we identified for the glires 20S proteasomes can also be detected in the new cryo-EM structure. However, the rat structure, like the human (PDB: 5LEX) and yeast (PDB: 5CZ4) 20S proteasomes structures, was lacking the last dozen amino acids, preventing the identification of the modification. This observation highlights the complementary type of information that can be gained by native MS analysis. Although native MS cannot provide atomic resolution structures, as cryo-EM and X-ray crystallography, it allows researchers to decipher specific proteoforms that may be missing in the atomic resolution methods.
Conclusions
In this study, we presented an experimental strategy to study the structural divergence of ortholog protein complexes. We chose the 20S proteasome as our model system and applied a suite of native MS-based approaches to investigate the differences in size, kinetic stability, overall shape, organization, and subunit composition of five complexes ranging from archaea (T. acidophilum), through yeast (S. cerevisiae) to mammals (rat - R. norvegicus, rabbit - O. cuniculus and human - HEK293 cells). Although all 20S complexes are composed of 14 α- and 14 β-type subunits, which together form a ∼700 kDa assembly with a common cylindrical architecture, we could identify specific signatures for each proteasome.
We show that the mammalian 20S complexes share similar size and kinetic stability, while the archaeal and yeast complexes display smaller and larger values, respectively. Overall, we detected kinetic stabilization of the 20S proteasome throughout the course of evolution from prokaryotes to eukaryotes (Figure 8), a phenomenon that is likely linked to the divergence of the α- and β-subunits.
Figure 8.

Native mass spectrometry analysis provides input into the evolutionary trajectory of the 20S proteasome. 20S proteasomes from five species were analyzed using a collection of native mass spectrometry approaches, including MS1, MS2, MS3, IM, SID, and CIU. Our results indicated that despite the evolutionary progression, structural features of the 20S proteasome from different organisms do not change in a linear manner. Data were plotted in a three-dimensional display, showing CCS and kinetic stability values, as a function of the log of the genetic distance between the species (as calculated by Clustal Omega, EMBL-EBI). On the Y axis, the average CCS value, calculated over three wave heights of each species is shown. Error bars represent standard deviation. The width of each bar is proportional to its kinetic stability, described as the eV of the first transition state in the CIU experiments. For ease of visualization, stability values were also color-coded, as shown.
In addition, by activating the complexes within the mass spectrometer, the conserved α-subunit ring order of eukaryotic 20S proteasomes was exposed. This observation was further endorsed by solving the cryo-EM three-dimensional structure of the rat proteasome, which like the rabbit complex has been lacking a high-resolution structure. In the course of our multilevel analysis, multiple forms of the α- and β-subunits were revealed. These multiple subunit variants likely comprise compositionally distinct 20S proteasomes. This analysis also led us to the identification of a new PSMA7 proteoform in the glires proteasome. We also discovered that the yeast complex displayed the highest degree of kinetic stability (Figure 8), a property that may have arisen due to the ethanol tolerance requirement of S. cerevisiae. Overall, by integrating multiple MS-based approaches, we performed a comparative structural analysis of the 20S complexes, going from the size, shape, and kinetic stabilities of intact complexes through the variability of individual subunits to sequence analysis.
In summary, considering the breadth of information yielded, the rapid analysis, and the low sample consumption, we anticipate further applications of these still emergent MS-based approaches for assessing the structural variations of highly similar complexes. This is especially applicable given the very few experimental methods currently available for comparing kinetic stability of large protein complexes.67 We anticipate future application of this approach not only for ortholog complexes but also for many other highly similar protein assemblies. For example, this methodology may find particular use for the characterization of tissue specific protein complexes whose structures remain undefined at high resolution, for evaluating designed proteins and assembly intermediates, and for identifying ligands that induce higher kinetic stability.
Experimental Methods
Sample Preparation
20S proteasomes from rat livers, human HEK293, and yeast cells were purified as described.61 Rabbit liver 20S proteasomes were purified according to the rat preparation protocol. Archaeal 20S proteasomes were purified as previously described.77 All 20S proteasomes were buffer exchanged into 150 mM ammonium acetate, pH 7, using a Biospin 6 column (Bio-Rad) prior to the measurements. Protein concentrations were adjusted to ∼3 μM.
20S Proteasome Activity Assay
To evaluate the proteolytic activity of the different 20S proteasomes, the hydrolysis of a fluorogenic substrate, Suc-LLVY-AMC (Boston Biochem) was measured. 0.8 μM of each of the 20S proteasomes were incubated with 100 μM of the substrate for 30 min at 37 °C (for the mammalian 20S proteasomes), 30 °C (for the yeast complex), or 60 °C (for the archaeal 20S proteasome), in the presence and absence of the 20S proteasome inhibitor MG132. The fluorescence of Suc-LLVY-AMC hydrolysis was measured using a microplate reader (Infinite 200, Tecan Group), using an excitation wavelength of 380 nm and an emission wavelength of 490 nm.
Proteasome Degradation Assays
To monitor the effect of ethanol on the activity of the rat and yeast 20S proteasomes, we used a reaction mixture containing 0.1 μM of the 20S proteasome in 50 mM HEPES pH 7.5 and increasing concentrations of ethanol, from 0 to 7.5%. As a substrate, α-synuclein was used (1 μM). The reaction mixtures were incubated at 37 and 30 °C for the rat and yeast 20S proteasomes, respectively. Ten microliter samples were taken at time 0 and 30 min, and quenched by the addition of reducing sample buffer and snap frozen in liquid N2. Samples were then thawed, boiled for 5 min, and loaded onto a 15% SDS-PAGE gel. Gels were stained with Coomassie brilliant blue, and changes in the level of α-synuclein were quantified by band densitometry using Image-J. The ratio between the intensity of α-synuclein after 30 min divided by the intensity at time 0 was plotted against ethanol concentrations.
Native MS Analysis
Measurements were made using three instrumental platforms modified for the measurement of large protein complexes. Most of the IM-MS measurements were performed on a modified Synapt G1 HDMS (Waters MS Technologies, Manchester, UK). Modifications were made by MS Vision (Almere, The Netherlands) and included a pump restriction sleeve that was fitted around the first section of the source ion guide for enhanced collisional cooling of the ions, promoting transmission of high mass species. The offset voltage limit of the extraction cone was increased to 200 V to allow for ion activation as the ions enter the source ion guide. Collision gas valves were added for independent control of collision gas pressures inside the trap and transfer cells. An extra Pirani gauge was fitted so both cell pressures can be monitored separately. A switching valve in the ion mobility gas supply line was installed, allowing for switching between nitrogen for ion mobility experiments, and argon for enhanced collisional cooling of activated high mass ions. In addition, data binning on the TDC can be adjusted for better signal-to-noise ratios without sacrificing mass spectral resolution. SID measurements were performed on a Synapt G2 HDMS (Waters MS Technologies, Manchester, UK) equipped with an SID device located between the transfer and the ion mobility cells.54 High resolution measurements were performed on a Q-Exactive Plus Orbitrap EMR (Thermo Fisher Scientific, Bremen, Germany) modified with a front-end interface encompassing orthogonal ion injection and installation of a front-end trap enabling a trap-and-release mode of analysis for MS3-type experiments.68
An additional modification of the Orbitrap instrument was the installation of an ECD device, directly replacing the original transfer octupole connecting the mass selection quad with the C-trap. In this configuration, without activation, ions fly along the central axis of the cell, and no significant loss of ion transmission is observed. Electrons were produced from a heated 1 mm loop of rhenium wire (0.004 in. in diameter) located in the center of the ECD device. The filament was coated with yttrium oxide to increase electron emission. Two 7 mm high-temperature samarium alloy magnets were positioned symmetrically on each side of the filament to provide magnetic field lines parallel to the central axis and to confine electrons radially. A total of eight DC voltages were applied to various lens elements that pull electrons off the filament into the regions containing the magnets. Two outer lenses were made negative with respect to the magnetic lenses to stop electrons from exiting the cell. Outer lenses were adjusted to assist in focusing ions in and out of the ECD cell.
All instruments were externally mass-calibrated using a cesium iodide solution at a concentration of 2 mg/mL. In all measurements, ∼3 μL of protein were sprayed into the instruments using a gold-coated nESI ionization capillary prepared in-house, as previously described.78
Parameters Used on the Synapt G1
Capillary voltage of 1.2–1.7 kV, sampling cone 50 V, extraction cone 10 V, source temperature 25 °C, trap and transfer collision energies 25 and 15 V, respectively, DC bias 18 V. Nitrogen was used as the IMS gas, at a flow rate of 20 mL/min. The gas flow in the trap and transfer cells were set to 8 and 2 mL/min, respectively, corresponding to a pressure of 4.35 × 10–2 mbar in the trap and 9.38 × 10–2 mbar in the transfer cells. IM wave velocity was set to 300 m/s, and wave height was set to 15 V. Backing pressure was set to 7–9 mbar.
Parameters Used on the Synapt G2
Capillary voltage of 1.2–1.7 kV, sampling cone 30 V, extraction cone 5 V, trap and transfer collision energies 20 and 5 V, respectively, DC bias 45 V. Nitrogen was used as the IM gas, at a flow rate of 60 mL/min; trap gas flow was 8 mL/min. Helium cell gas flow was set to 120 mL/min, IM wave velocity was set to 300 m/s, and wave height was set to 20 V. Backing pressure was set to 7–9 mbar.
Parameters Used on the Q-Exactive Plus Orbitrap EMR
Inlet capillary was set to a temperature of 180 °C, capillary voltage 1.7 kV, fore vacuum pressure 1.5 mbar, and trapping gas pressure 4.1, corresponding to HV pressure of 1.2 × 10–4 mbar and UHV pressure of 3.4 × 10–10 mbar. The source was operated at a constant energy of 2 V in the flatapole bias and interflatapole lens. Bent flatapole DC bias and gradient were set to 2.5 and 35 V, respectively, and the HCD cell was operated between 10–50 V. In MS3 analyses, ion trapping was performed in cycles of 10 ms, at flatapole bias and interflatapole lens voltages of −220 and 15 V, respectively. After each trapping event, a 200 μs release time was applied, during which the inject flatapole bias and interflatapole lens voltages were set to 20 and 1 V, respectively. In these experiments, instrument settings were set for the detection of small proteins, as follows: trapping gas pressure was reduced to 1.5, resulting in an HV pressure of 4.5 × 10–5 mbar and a UHV pressure of 3.4 × 10–10 mbar. Bent flatapole DC bias and gradient were reduced to 1.8 and 15 V, respectively, and no HCD energy was applied.
Ion Mobility–Mass Spectrometry Measurements and CCS Calculations
Ion mobility
measurements, CCS calculations, and CIU experiments
were performed on a modified Synapt G1 HDMS instrument. T-wave calibration
was conducted as previously described.79 CCS values were calculated using the PULSAR software.80 Theoretical CCS values for the 20S proteasome
from human (5LEX), yeast (5CZ4), rat (6TU3), and archaea (6BDF) were
calculated using the Driftscope Projection Approximation algorithm
(Waters). The Elab energies were calculated
as follows: 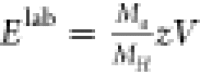 where Ma is
the mass of the different 20S proteasomes, MH is the mass of the human 20S proteasome, z is the charge, and V is the voltage applied to
the collision cell.
where Ma is
the mass of the different 20S proteasomes, MH is the mass of the human 20S proteasome, z is the charge, and V is the voltage applied to
the collision cell.
Collision-Induced Unfolding
CIU measurements were performed as described.47,80 In brief, all 20S proteasomes were gradually activated by elevating the trap collision voltage from 50 to 180 V in increments of 5 V. The highest charge state of among the different 20S proteasome particles that gave reasonable signal across the CIU voltage range was used for analysis for each. CIU fingerprint plots and data analysis were conducted using PULSAR.80
Surface-Induced Dissociation Coupled to Ion Mobility Measurements
SID-IM-MS analyses were performed on the modified Synapt G2 HDMS. All samples were preincubated with the charge-reducing agent TEAA, at a ratio of 0.1/0.9 TEAA/ammonium acetate (v/v).62 The two charge states with the highest intensity were isolated in the quadrupole mass filter, at LM and HM resolutions of 0, to maximize the isolation window. To achieve an SID of 150 V, the trap DC bias was changed to 195 V. The following voltages were applied on the SID device: entrance 1 was set to 93 V, entrance 2 at −45.5 V, front top at −145 V, front bottom at 85 V, mid bottom at −118.3 V, surface at −50 V, rear top at −182.6 V, rear bottom at −75 V, exit 1 at −77.2 V, and exit 2 at −75 V.
Capillary Electrophoresis
A CESI 8000 instrument (SCIEX, CA, USA) was interfaced with the Q Exactive Plus Orbitrap EMR mass spectrometer. Background electrolyte (BGE) was 10% acetic acid, and the same solution was used in the conductive line. For 20S proteasome subunit separation both neutral and polyethylenimine (PEI) coated capillaries were used. In order to prepare the PEI capillary, a bare fused silica (BFS) capillary was coated with polyethylenimine (Gelest Inc. SSP-060) according to the manufacturer’s instructions (Sciex CA, USA). In general, the coating procedure includes three steps: preconditioning, coating, and post coating. The preconditioning was done by rinsing the forward capillary with 0.1 M NaOH, 0.1 M HCl, and MeOH for 10 min each, at 100 PSI. The coating was done by filling the capillary with a solution containing 300 μL of PEI and 1.5 mL of anhydrous methanol (Sigma-Aldrich 322415). The capillary was left overnight in this solution and thoroughly cleaned with ethanol in the next day to remove any traces of PEI. The postcoating was done by rinsing the capillary with MeOH (5 min, 75 PSI) following by MeOH (20 min, 100 PSI). The PEI capillary was then conditioned with double distilled water (3 min, 100 PSI), 1 M NaCl (3 min, 100 PSI), and 50 mM AmAc pH 3 (6 min, 100 PSI). For spray evaluation, the capillary was rinsed with BGE (50 mM AmAc pH 3) for 2 min at 100 PSI. Then 20 kV were applied at 5 PSI for a continuous flow of a standard three protein mix (SCIEX CA, USA), dissolved in 50 mM AmAc pH 3. For the separation of samples, the capillary was first rinsed at 100 PSI with 1 M NaCl (3 min), double-distilled water (2 min), and BGE (10% acetic acid) (3 min). Injection was performed at 2 PSI for 10 s. Separation was performed for 15 min at 20 kV with a ramp time of 1 min. At the end of the run, a ramp-down from 20 to 1 kV over 3 min was performed at 25 PSI.
The neutral capillary was precoated with cross-linked neutral polyacrylamide by SCIEX. New capillaries were rehydrated overnight with a rinse of 0.1 M HCl (5 min, 100 PSI), distilled de-ionized (DDI) water (30 min, 100 PSI), and DDI at 5 PSI for the rest of the time. Following the rehydration, the capillary was conditioned at 100 PSI with 0.1 M HCl (5 min), double-distilled water (30 min), 50 mM AmAc pH 3 (3 min). Then 30 kV were applied for 30 min at 5 PSI with 50 mM AmAc pH 3 solution. For the spray evaluation, the capillary was rinsed with BGE (50 mM AmAc pH 3) for 2 min at 100 PSI. Then 30 kV were applied at 1.5 PSI for a continuous flow of the standard proteins. For the separation of samples, the capillary was first rinsed at 100 PSI with 0.1 M HCl (5 min), DDI water (5 min), followed by the BGE (10% acetic acid) for 10 min. Injection was performed at 2.5 PSI for 15 s, corresponding to approximately 6 nL of sample (0.8% of the capillary volume). Separations were performed at 30 kV, 0.5 PSI for 25 min and 2 PSI for 40 min with a ramp time of 1 min. At the end of the run, a ramp-down from 30 to 1 kV over 5 min was performed at 25 PSI.
Data Analysis
Spectra were examined and analyzed using the MassLynx software (Waters V4.2 SCN982, 2017). Minimal smoothing was applied. Spectra acquired on the Q Exactive Plus Orbitrap EMR were converted to MassLynx-compatible files using DataBridge software (Waters), and no smoothing was applied.
Calculation of the Masses of the 20S Proteasomes
The intact masses of the eukaryotic 20S proteasomes were calculated based on the sequences of the different subunit variants shown in Table S1. For each organism, the calculated mass included the sequence mass plus incorporation of PTMs found for each subunit, as shown in Figures S8–S11. These include the removal of initial methionines in PSMA2, PSMA3, PSMA4, PSMA6, PSMA7, PSMB2, PSMB3, addition of acetylation to PSMA1, PSMA2, PSMA3, PSMA4, PSMA5, PSMA6, PSMA7, PSMB2, PSMB3, and a phosphorylation to PSMA3. For the human 20S proteasome, the calculated mass also included one FLAG tag (DYKDDDDK) which was fused to the C-terminus of the PSMB2 subunits. For the yeast 20S proteasome, the calculated mass also included two FLAG-His6 tags (DYKDDDDKHHHHHH), which were fused to the C-terminus of the PSMB2 subunits.74 The mass of the archaeal 20S proteasome was calculated according the protein sequence of the α- and β-subunits, after removal of the TEV-cleavable His tag.77
Top-Down Proteomic Analysis
De novo top-down protein sequencing was performed by both manual analysis and by using LcMS-Spectator software (PNNL, OMICS.PNL.GOV).
Cryo-EM Sample Preparation and Data Collection
Purified endogenous rat liver 20S proteasome (R. norvegicus) was concentrated to ∼14 mg/mL. A 2.5 μL sample was applied to C-Flat 2/2 300 mesh holey carbon grids (Protochips), blotted for 3 s at 4 °C and 100% humidity, and plunge frozen into liquid ethane cooled by liquid nitrogen using a Vitrobot automated plunger (Thermo Fisher Scientific). Applying the sample 30 min after glow discharge improved the percentage of side views. Imaging was done using a Titan Krios G3i electron microscope (Thermo Fisher Scientific) operated at 300 kV, at a nominal magnification of 105000×, corresponding to a pixel size of 0.86 Å. A total of 2234 movies were recorded on a K3 direct detector placed at the end of a GIF Quantum Energy Ffilter (Gatan, Inc.), using automated acquisition in EPU software (Thermo Fisher Scientific). Movies were collected with a nominal defocus range of −0.6 to −1.6 μm. Each movie was fractionated into 45 frames. The dose rate was set to ∼21 e–/pixel/s, and the total exposure time was 1.5 s, corresponding to an accumulated dose of ∼37 e–/Å2.
Cryo-EM Image Processing
Image processing was performed using RELION 3.0.81 Movie frames were motion-corrected with 7 × 5 patches and dose-weighted, followed by CTF estimation using CTFFIND4.82 Images showing well-defined particles and thin ice were selected for further processing. Initially about 2500 particles were manually picked, subjected to reference-free 2D classification, and the generated class averages were used as templates for autopicking. A total of 284 161 particles were autopicked from the selected images, extracted and binned 4 × 4 (100 pixel box size, 3.44 Å/pixel), and subjected to two rounds of 2D classification in order to clean the data set, resulting in 278 679 particles. This was followed by 3D autorefine with C2 symmetry imposed, using the cryo-EM map of the recombinant human 20S proteasome (EMD-4877),83 low-pass filtered to 40 Å, as an initial reference. Refined particles were re-extracted with 2 × 2 binning (200 pixel box size, 1.72 Å/pixel), and 3D classification was used to separate the best class which showed the highest resolution. Particles belonging to the high-resolution class (245 800) were re-extracted without binning (400 pixel box size, 0.86 Å/pixel), followed by 3D-refinement with a real-space solvent mask imposed, resulting in a map with a resolution of 3.1 Å. Subsequently, the map was refined by two rounds of per-particle CTF (including beam-tilt estimation) and per-particle motion correction (polishing). The following 3D refinement with applied solvent mask resulted in a final map with resolution of 2.7 Å using gold-standard FSC = 0.143 criteria. The final map was filtered based on local resolution estimation.
Molecular Model Building and Refinement
The recombinant human 20S proteasome (PDB: 6RGQ)83 was used as an initial model. The model was docked into the local resolution filtered map as a rigid body using UCSF Chimera,84 following by real-space refinement using Phenix.85 The model was then manually adjusted to fit the electron density map using Coot.86 Amino acids which were different between the human and rat sequences were converted. Subsequently, we used iterative real-space refinements and manual model improvements using Phenix and Coot, respectively. We did not model areas in the map where the density was not clearly interpretable, mostly at the C and N termini of the chains. The map and model were visualized using UCSF Chimera.84
Data and Code Availability
The density map of the rat 20S proteasome was deposited in the Electron Microscopy Data Bank under accession code EMD-10586, and the atomic coordinates were deposited in the Protein Data Bank under accession code 6TU3.
Acknowledgments
We thank Dan Tawfik, Weizmann Institute of Science, for helpful discussions and Dr. Ron Rotkopf, Weizmann Institute of Science, for the statistical analysis in the 20S proteasome degradation assays. We also thank MS Vision for installing the upgrades on our Synapt G1 instrument. We are grateful for the support of a Starting Grant from the (ERC) (Horizon 2020)/ERC Grant Agreement No. 636752, and for an Israel Science Foundation (ISF) Grant 300/17. M.S. is the incumbent of the Aharon and Ephraim Katzir Memorial Professorial Chair. This work was partially supported by a grant from the National Institutes of Health to V.H.W. (NIH P41GM128577).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00080.
Figure S1. Peptidase activity assays of purified 20S proteasome complexes. Figure S2. Phylogenetic and multiple alignment analysis of the different subunits of the 20S proteasome complexes from five different organisms. Figure S3. CIU fingerprints of the five 20S proteasome variants. Figure S4. Activity assays of purified rat and yeast 20S proteasome complexes in the presence of ethanol. Figure S5. Identification of dimers of α-subunits by tandem MS analysis. Figure S6. Characterization the subunit arrangement of the α-ring of the rat 20S proteasome. Figure S7. Top-down MS3 analysis of a new rat PSMA7 proteoform. Figure S8. Analysis of the rabbit α-subunits identifies a new PSMA7 rabbit proteoform. Figure S9. Denaturing CE-MS analysis of the rat 20S proteasome. Figure S10. Denaturing CE-MS analysis of the rabbit 20S proteasome. Figure S11. Denaturing CE-MS analysis of the human 20S proteasome. Figure S12. Denaturing CE-MS analysis of the yeast 20S proteasome. Figure S13. Single particle cryo-EM analysis of the rat 20S proteasome. Table S1. List of the different subunit variants composing the analyzed eukaryotic 20S proteasome complexes, as identified by CE-MS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 1998, 92 (3), 291. 10.1016/S0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Perica T.; Marsh J. A.; Sousa F. L.; Natan E.; Colwell L. J.; Ahnert S. E.; Teichmann S. A. The emergence of protein complexes: quaternary structure, dynamics and allostery. Biochem. Soc. Trans. 2012, 40 (3), 475. 10.1042/BST20120056. [DOI] [PubMed] [Google Scholar]
- Sandler I.; Medalia O.; Aharoni A. Experimental analysis of co-evolution within protein complexes: the yeast exosome as a model. Proteins: Struct., Funct., Genet. 2013, 81 (11), 1997. 10.1002/prot.24360. [DOI] [PubMed] [Google Scholar]
- Wan C.; Borgeson B.; Phanse S.; Tu F.; Drew K.; Clark G.; Xiong X.; Kagan O.; Kwan J.; Bezginov A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525 (7569), 339. 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppa E.; Zea D. J.; Marino-Buslje C. Protein-protein interactions leave evolutionary footprints: High molecular coevolution at the core of interfaces. Protein Sci. 2017, 26 (12), 2438. 10.1002/pro.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rivas J.; Marsili S.; Juan D.; Valencia A. Conservation of coevolving protein interfaces bridges prokaryote-eukaryote homologies in the twilight zone. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (52), 15018. 10.1073/pnas.1611861114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A. C.; Saeed R.; Deane C. M. Predicting protein-protein interactions in the context of protein evolution. Mol. BioSyst. 2010, 6 (1), 55. 10.1039/B916371A. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Perica T.; Teichmann S. A. Evolution of protein structures and interactions from the perspective of residue contact networks. Curr. Opin. Struct. Biol. 2013, 23 (6), 954. 10.1016/j.sbi.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Glickman M. H.; Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002, 82 (2), 373. 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Goldberg A. L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426 (6968), 895. 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G.; Sharon M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4 (3), 862. 10.3390/biom4030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunjappu M. J.; Hochstrasser M. Assembly of the 20S proteasome. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843 (1), 2. 10.1016/j.bbamcr.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Finley D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843 (1), 13. 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkowska J.; Gizynska M.; Grudnik P.; Golik P.; Karpowicz P.; Gieldon A.; Dubin G.; Jankowska E. Crystal structure of a low molecular weight activator Blm-pep with yeast 20S proteasome - insights into the enzyme activation mechanism. Sci. Rep. 2017, 7 (1), 6177. 10.1038/s41598-017-05997-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshbarger W.; Miller C.; Diedrich C.; Sacchettini J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23 (2), 418. 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Lowe J.; Stock D.; Jap B.; Zwickl P.; Baumeister W.; Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268 (5210), 533. 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Lin G.; Li D.; de Carvalho L. P.; Deng H.; Tao H.; Vogt G.; Wu K.; Schneider J.; Chidawanyika T.; Warren J. D.; et al. Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461 (7264), 621. 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M.; Bajorek M.; Kohler A.; Moroder L.; Rubin D. M.; Huber R.; Glickman M. H.; Finley D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7 (11), 1062. 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- Li H.; O’Donoghue A. J.; van der Linden W. A.; Xie S. C.; Yoo E.; Foe I. T.; Tilley L.; Craik C. S.; da Fonseca P. C.; Bogyo M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530 (7589), 233. 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno M.; Mizushima T.; Morimoto Y.; Tomisugi Y.; Tanaka K.; Yasuoka N.; Tsukihara T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002, 10 (5), 609. 10.1016/S0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- Huber E. M.; Basler M.; Schwab R.; Heinemeyer W.; Kirk C. J.; Groettrup M.; Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148 (4), 727. 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Gille C.; Goede A.; Schloetelburg C.; Preißner R.; Kloetzel P.-M.; Gobel U. B.; Frommel C. A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome. J. Mol. Biol. 2003, 326 (5), 1437. 10.1016/S0022-2836(02)01470-5. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Zhang S.; Wu Z.; Li X.; Wang W. L.; Zhu Y.; Stoilova-McPhie S.; Lu Y.; Finley D.; Mao Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565 (7737), 49. 10.1038/s41586-018-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budenholzer L.; Cheng C. L.; Li Y.; Hochstrasser M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429 (22), 3500. 10.1016/j.jmb.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G. C.; Martin A.; Nogales E. The proteasome under the microscope: the regulatory particle in focus. Curr. Opin. Struct. Biol. 2013, 23 (2), 243. 10.1016/j.sbi.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Perez F.; Kay L. E. Probing the cooperativity of Thermoplasma acidophilum proteasome core particle gating by NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (46), E9846 10.1073/pnas.1712297114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham M. P.; Sekhar A.; Kay L. E. Understanding the mechanism of proteasome 20S core particle gating. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (15), 5532. 10.1073/pnas.1322079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak A. M.; Kay L. E. Proteasome allostery as a population shift between interchanging conformers. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (50), E3454 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nissan G.; Vimer S.; Tarnavsky M.; Sharon M. Structural mass spectrometry approaches to study the 20S proteasome. Methods Enzymol. 2019, 619, 179. 10.1016/bs.mie.2018.12.029. [DOI] [PubMed] [Google Scholar]
- Chen F.; Gulbakan B.; Weidmann S.; Fagerer S. R.; Ibanez A. J.; Zenobi R. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 2016, 35 (1), 48. 10.1002/mas.21462. [DOI] [PubMed] [Google Scholar]
- Liko I.; Allison T. M.; Hopper J. T.; Robinson C. V. Mass spectrometry guided structural biology. Curr. Opin. Struct. Biol. 2016, 40, 136. 10.1016/j.sbi.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Lossl P.; van de Waterbeemd M.; Heck A. J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35 (24), 2634. 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon M. Biochemistry. Structural MS pulls its weight. Science 2013, 340 (6136), 1059. 10.1126/science.1236303. [DOI] [PubMed] [Google Scholar]
- Ghareib M.; Youssef K. A.; Khalil A. A. Ethanol tolerance of Saccharomyces cerevisiae and its relationship to lipid content and composition. Folia Microbiol. (Dordrecht, Neth.) 1988, 33 (6), 447. 10.1007/BF02925769. [DOI] [PubMed] [Google Scholar]
- de la Mora J. F. Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal. Chim. Acta 2000, 406 (1), 93. 10.1016/S0003-2670(99)00601-7. [DOI] [Google Scholar]
- Ben-Nissan G.; Sharon M. The application of ion-mobility mass spectrometry for structure/function investigation of protein complexes. Curr. Opin. Chem. Biol. 2018, 42, 25. 10.1016/j.cbpa.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrer B. C.; Merenbloom S. I.; Koeniger S. L.; Hilderbrand A. E.; Clemmer D. E. Biomolecule analysis by ion mobility spectrometry. Annu. Rev. Anal. Chem. 2008, 1, 293. 10.1146/annurev.anchem.1.031207.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanucara F.; Holman S. W.; Gray C. J.; Eyers C. E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6 (4), 281. 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Wojcik R.; Zhang X.; Ibrahim Y. M.; Burnum-Johnson K. E.; Orton D. J.; Monroe M. E.; Moore R. J.; Smith R. D.; Baker E. S. Coupling front-end separations, ion mobility spectrometry, and mass spectrometry for enhanced multidimensional biological and environmental analyses. Annu. Rev. Anal. Chem. 2017, 10 (1), 71. 10.1146/annurev-anchem-061516-045212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Atri V.; Porrini M.; Rosu F.; Gabelica V. Linking molecular models with ion mobility experiments. Illustration with a rigid nucleic acid structure. J. Mass Spectrom. 2015, 50 (5), 711. 10.1002/jms.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelica V.; Shvartsburg A. A.; Afonso C.; Barran P.; Benesch J. L. P.; Bleiholder C.; Bowers M. T.; Bilbao A.; Bush M. F.; Campbell J. L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38 (3), 291. 10.1002/mas.21585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer W.; Ramos P. C.; Dohmen R. J. The ultimate nanoscale mincer: assembly, structure and active sites of the 20S proteasome core. Cell. Mol. Life Sci. 2004, 61 (13), 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y.; Kaneko T.; Okamoto K.; Bai M.; Yashiroda H.; Furuyama K.; Kato K.; Tanaka K.; Murata S. Dissecting beta-ring assembly pathway of the mammalian 20S proteasome. EMBO J. 2008, 27 (16), 2204. 10.1038/emboj.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques A. J.; Glanemann C.; Ramos P. C.; Dohmen R. J. The C-terminal extension of the beta7 subunit and activator complexes stabilize nascent 20 S proteasomes and promote their maturation. J. Biol. Chem. 2007, 282 (48), 34869. 10.1074/jbc.M705836200. [DOI] [PubMed] [Google Scholar]
- Ramos P. C.; Marques A. J.; London M. K.; Dohmen R. J. Role of C-terminal extensions of subunits beta2 and beta7 in assembly and activity of eukaryotic proteasomes. J. Biol. Chem. 2004, 279 (14), 14323. 10.1074/jbc.M308757200. [DOI] [PubMed] [Google Scholar]
- Niu S.; Rabuck J. N.; Ruotolo B. T. Ion mobility-mass spectrometry of intact protein--ligand complexes for pharmaceutical drug discovery and development. Curr. Opin. Chem. Biol. 2013, 17 (5), 809. 10.1016/j.cbpa.2013.06.019. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Han L.; Ruotolo B. T. Collisional and Coulombic unfolding of gas-phase proteins: high correlation to their domain structures in solution. Angew. Chem., Int. Ed. 2014, 53 (35), 9209. 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- Deng L.; Broom A.; Kitova E. N.; Richards M. R.; Zheng R. B.; Shoemaker G. K.; Meiering E. M.; Klassen J. S. Kinetic stability of the streptavidin-biotin interaction enhanced in the gas phase. J. Am. Chem. Soc. 2012, 134 (40), 16586. 10.1021/ja305213z. [DOI] [PubMed] [Google Scholar]
- Steinberg M. Z.; Breuker K.; Elber R.; Gerber R. B. The dynamics of water evaporation from partially solvated cytochrome c in the gas phase. Phys. Chem. Chem. Phys. 2007, 9 (33), 4690. 10.1039/b705905a. [DOI] [PubMed] [Google Scholar]
- Meyer T.; de la Cruz X.; Orozco M. An atomistic view to the gas phase proteome. Structure 2009, 17 (1), 88. 10.1016/j.str.2008.11.006. [DOI] [PubMed] [Google Scholar]
- van der Spoel D.; Marklund E. G.; Larsson D. S.; Caleman C. Proteins, lipids, and water in the gas phase. Macromol. Biosci. 2011, 11 (1), 50. 10.1002/mabi.201000291. [DOI] [PubMed] [Google Scholar]
- Servage K. A.; Silveira J. A.; Fort K. L.; Russell D. H. Cryogenic Ion Mobility-Mass Spectrometry: Tracking Ion Structure from Solution to the Gas Phase. Acc. Chem. Res. 2016, 49 (7), 1421. 10.1021/acs.accounts.6b00177. [DOI] [PubMed] [Google Scholar]
- Jhingree J. R.; Bellina B.; Pacholarz K. J.; Barran P. E. Charge mediated compaction and rearrangement of gas-phase proteins: A case study considering two proteins at opposing ends of the structure-disorder continuum. J. Am. Soc. Mass Spectrom. 2017, 28 (7), 1450. 10.1007/s13361-017-1692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Huang C.; Wysocki V. H. Surface-induced dissociation of ion mobility-separated noncovalent complexes in a quadrupole/time-of-flight mass spectrometer. Anal. Chem. 2012, 84 (14), 6016. 10.1021/ac300810u. [DOI] [PubMed] [Google Scholar]
- Song Y.; Nelp M. T.; Bandarian V.; Wysocki V. H. Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent. Sci. 2015, 1 (9), 477. 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe A.; Hsia Y.; Busch F.; Sheffler W.; King N. P.; Baker D.; Wysocki V. H. Confirmation of intersubunit connectivity and topology of designed protein complexes by native MS. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (6), 1268. 10.1073/pnas.1713646115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Wysocki V. H. Surface induced dissociation: dissecting noncovalent protein complexes in the gas phase. Acc. Chem. Res. 2014, 47 (4), 1010. 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- Harvey S. R.; Seffernick J. T.; Quintyn R. S.; Song Y.; Ju Y.; Yan J.; Sahasrabuddhe A. N.; Norris A.; Zhou M.; Behrman E. J.; et al. Relative interfacial cleavage energetics of protein complexes revealed by surface collisions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (17), 8143. 10.1073/pnas.1817632116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. H. Impact of charge state on gas-phase behaviors of noncovalent protein complexes in collision induced dissociation and surface induced dissociation. Analyst 2013, 138 (5), 1353. 10.1039/c2an36525a. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Witt S.; Felderer K.; Rockel B.; Baumeister W.; Robinson C. V. 20S proteasomes have the potential to keep substrates in store for continual degradation. J. Biol. Chem. 2006, 281 (14), 9569. 10.1074/jbc.M511951200. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G.; Vimer S.; Tarnavsky M.; Sharon M.. Structural mass spectrometry approaches to study the 20S proteasome. Methods Enzymol. 2019, In press. [DOI] [PubMed] [Google Scholar]
- Ma X.; Loo J. A.; Wysocki V. H. Surface induced dissociation yields substructure of Methanosarcina thermophila 20S proteasome complexes. Int. J. Mass Spectrom. 2015, 377, 201. 10.1016/j.ijms.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372 (3), 774. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Quintyn R. S.; Yan J.; Wysocki V. H. Surface-induced dissociation of homotetramers with d2 symmetry yields their assembly pathways and characterizes the effect of ligand binding. Chem. Biol. 2015, 22 (5), 583. 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- Voordeckers K.; Kominek J.; Das A.; Espinosa-Cantu A.; De Maeyer D.; Arslan A.; Van Pee M.; van der Zande E.; Meert W.; Yang Y.; et al. Adaptation to High Ethanol Reveals Complex Evolutionary Pathways. PLoS Genet. 2015, 11 (11), e1005635 10.1371/journal.pgen.1005635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandts J. F.; Hunt L. The thermodynamics of protein denaturation. 3. The denaturation of ribonuclease in water and in aqueous urea and aqueous ethanol mixtures. J. Am. Chem. Soc. 1967, 89 (19), 4826. 10.1021/ja00995a002. [DOI] [PubMed] [Google Scholar]
- Colon W.; Church J.; Sen J.; Thibeault J.; Trasatti H.; Xia K. Biological Roles of Protein Kinetic Stability. Biochemistry 2017, 56 (47), 6179. 10.1021/acs.biochem.7b00942. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G.; Belov M. E.; Morgenstern D.; Levin Y.; Dym O.; Arkind G.; Lipson C.; Makarov A. A.; Sharon M. Triple-stage mass spectrometry unravels the heterogeneity of an endogenous protein complex. Anal. Chem. 2017, 89 (8), 4708. 10.1021/acs.analchem.7b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. B.; Malhan N.; Vasil’ev Y. V.; Lopez N. I.; Makarov A.; Beckman J. S.; Voinov V. G. Sequencing grade tandem mass spectrometry for top-down proteomics using hybrid electron capture dissociation methods in a benchtop orbitrap mass spectrometer. Anal. Chem. 2018, 90 (18), 10819. 10.1021/acs.analchem.8b01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni R.; Tomita Y.; Tokunaga F.; Liang T. J.; Noda C.; Ichihara A.; Tanaka K. Molecular cloning of two types of cDNA encoding subunit RC6-I of rat proteasomes. Biochim. Biophys. Acta, Gene Struct. Expression 1995, 1264 (1), 45. 10.1016/0167-4781(95)00113-U. [DOI] [PubMed] [Google Scholar]
- Huang L.; Burlingame A. L. Comprehensive mass spectrometric analysis of the 20S proteasome complex. Methods Enzymol. 2005, 405, 187. 10.1016/S0076-6879(05)05009-3. [DOI] [PubMed] [Google Scholar]
- Bose S.; Stratford F. L.; Broadfoot K. I.; Mason G. G.; Rivett A. J. Phosphorylation of 20S proteasome alpha subunit C8 (alpha7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by gamma-interferon. Biochem. J. 2004, 378 (1), 177. 10.1042/bj20031122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerl I. E.; Caniard A.; Merl-Pham J.; Ben-Nissan G.; Mayr C. H.; Mossina A.; Geerlof A.; Eickelberg O.; Hauck S. M.; Sharon M.; et al. Dissecting the molecular effects of cigarette smoke on proteasome function. J. Proteomics 2019, 193, 1. 10.1016/j.jprot.2018.12.015. [DOI] [PubMed] [Google Scholar]
- Verma R.; Chen S.; Feldman R.; Schieltz D.; Yates J.; Dohmen J.; Deshaies R. J. Proteasomal proteomics: identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell 2000, 11 (10), 3425. 10.1091/mbc.11.10.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menneteau T.; Fabre B.; Garrigues L.; Stella A.; Zivkovic D.; Roux-Dalvai F.; Mouton-Barbosa E.; Beau M.; Renoud M. L.; Amalric F.; et al. Mass Spectrometry-based Absolute Quantification of 20S Proteasome Status for Controlled Ex-vivo Expansion of Human Adipose-derived Mesenchymal Stromal/Stem Cells. Mol. Cell. Proteomics 2019, 18 (4), 744. 10.1074/mcp.RA118.000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre B.; Lambour T.; Garrigues L.; Ducoux-Petit M.; Amalric F.; Monsarrat B.; Burlet-Schiltz O.; Bousquet-Dubouch M. P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13 (6), 3027. 10.1021/pr500193k. [DOI] [PubMed] [Google Scholar]
- Ruschak A. M.; Religa T. L.; Breuer S.; Witt S.; Kay L. E. The proteasome antechamber maintains substrates in an unfolded state. Nature 2010, 467 (7317), 868. 10.1038/nature09444. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum N.; Michaelevski I.; Sharon M. Analyzing large protein complexes by structural mass spectrometry. J. Visualized Exp. 2010, 40, e1954. 10.3791/1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal. Chem. 2010, 82 (22), 9557. 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Allison T. M.; Reading E.; Liko I.; Baldwin A. J.; Laganowsky A.; Robinson C. V. Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nat. Commun. 2015, 6, 8551. 10.1038/ncomms9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180 (3), 519. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A.; Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192 (2), 216. 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toste Rego A.; da Fonseca P. C. A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76 (1), 138. 10.1016/j.molcel.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (2), 213. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (4), 486. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The density map of the rat 20S proteasome was deposited in the Electron Microscopy Data Bank under accession code EMD-10586, and the atomic coordinates were deposited in the Protein Data Bank under accession code 6TU3.