

Abstract
Sepsis, a condition caused by severe infections, affects more than 30 million people worldwide every year and remains the leading cause of death in hospitals1,2. Moreover, antimicrobial resistance has become an additional challenge in the treatment of sepsis3, and thus, alternative therapeutic approaches are urgently needed2,3. Here, we show that adoptive transfer of macrophages containing antimicrobial peptides linked to cathepsin B in the lysosomes (MACs) can be applied for the treatment of multi-drug resistant (MDR) bacteria-induced sepsis in mice with immunosuppression. The MACs are constructed by transfection of vitamin C lipid nanoparticles (VCLNPs) that deliver antimicrobial peptide and cathepsin B (AMP-CatB) mRNA. The VCLNPs allow the specific accumulation of AMP-CatB in macrophage lysosomes, which is the key location for bactericidal activities. Our results demonstrate that adoptive MAC transfer leads to the elimination of MDR bacteria, including Staphylococcus aureus and Escherichia coli, leading to the complete recovery of immunocompromised septic mice. Our work provides an alternative strategy for overcoming MDR bacteria-induced sepsis and opens up possibilities for the development of nanoparticle-enabled cell therapy for infectious diseases.
Traditionally, sepsis has been viewed as an uncontrollable inflammatory response to pathogens2,4. However, recent clinical data reveal that more than 60% of septic patients survive through the initial inflammatory storm but rapidly progress to a longer immunosuppressive state, which is characterized by the paralysis and death of immune cells, leading to an inability to clear invading pathogens, increased susceptibility to hospital-acquired infections, and high mortality rate2,4–6. As a result, potential therapeutic targets have been extensively explored in order to treat sepsis such as removing the anaphylatoxin C5a or blocking the C5a receptor7,8. Meanwhile, approaches aiming to restore immune function have been developed and tested in patients with sepsis2,4,6. Macrophages are one of the most efficient pathogen scavengers during infections9. Indeed, in septic patients, impaired macrophages/monocytes seem to be primarily responsible for insufficient antimicrobial defense6,9–12. Several small clinical trials of immunostimulatory agents indicate benefits in reversing the deactivated macrophages/monocytes, thus enhancing infection eradication10,11. On the other hand, meta-analysis of large clinical trials did not show significant changes in reducing the patient mortality13. Differences in clinical results may be explained as follows10,14–16. First, immunostimulatory agents are not able to restore the function of impaired macrophages/monocytes to their original levels10. Second, invaded bacteria are usually trapped in macrophage phagolysosomes, where reactive oxygen species, reactive nitrogen species, and lysozymes work synergistically to clear bacteria17. However, many bacteria, such as Staphylococcus aureus and Escherichia coli, have evolved immune escape mechanisms for thwarting phagolysosomal killing, resulting in intracellular survival and recurrent infections14–16. Third, although antibiotic therapy is the standard treatment in the sepsis clinical guideline, 70%–80% of sepsis death is related to persistent infections, thus indicating the prevalence of antibiotic resistance and the paucity of effective antimicrobial agents3,6.
Based on the above research findings, we hypothesized that adoptive transfer of macrophages containing antimicrobial peptides linked to cathepsin B in the lysosomes (MACs) may enable the immunocompromised sepsis host to boost innate immunity, prevent bacterial immune evasion, and eliminate multi-drug resistant (MDR) bacteria. To test our hypothesis, we first designed and constructed the AMP-CatB mRNA encoding an antimicrobial peptide IB367 (AMP-IB367), a cathepsin B (CatB), and a CatB-sensitive linker. AMP-IB367 is a broad-spectrum AMP with rapid bactericidal activity, and has been confirmed in clinical trials18. CatB, an endogenous lysosomal protein, is first translated as an inactive precursor in the cytoplasm. After translocation into the lysosome, the precursor is processed into mature CatB19. In this study, the CatB component is incorporated to transport the AMP-IB367 into lysosomes. In order to equip macrophages with the AMP-CatB mRNA, we designed a series of vitamin-derived lipid nanomaterials. As illustrated in Fig. 1a, the mRNA is translated to functional proteins in the cytoplasm and the proteins are further translocated into lysosomes19, where the linker is cleaved by CatB proteins and thus the APM-IB367 is released20. When phagosomes encapsulating bacteria fuse with lysosomes, the ingested bacteria are exposed to both pre-stored AMP-IB367 and lysosomal antimicrobial components. Although immune evasion strategies may protect MDR bacteria from phagolysosomal killing mechanisms, the AMP-IB367 is able to kill these bacteria. Consequently, the adoptive transfer of MACs rescue MDR bacteria-induced sepsis mice with immunosuppression by restoring the innate immunity, overcoming bacterial immune evasion, and eradicating the infection.
Fig. 1 |. Schematic illustration of adoptive macrophage transfer and chemical structures of the vitamin-derived lipids.
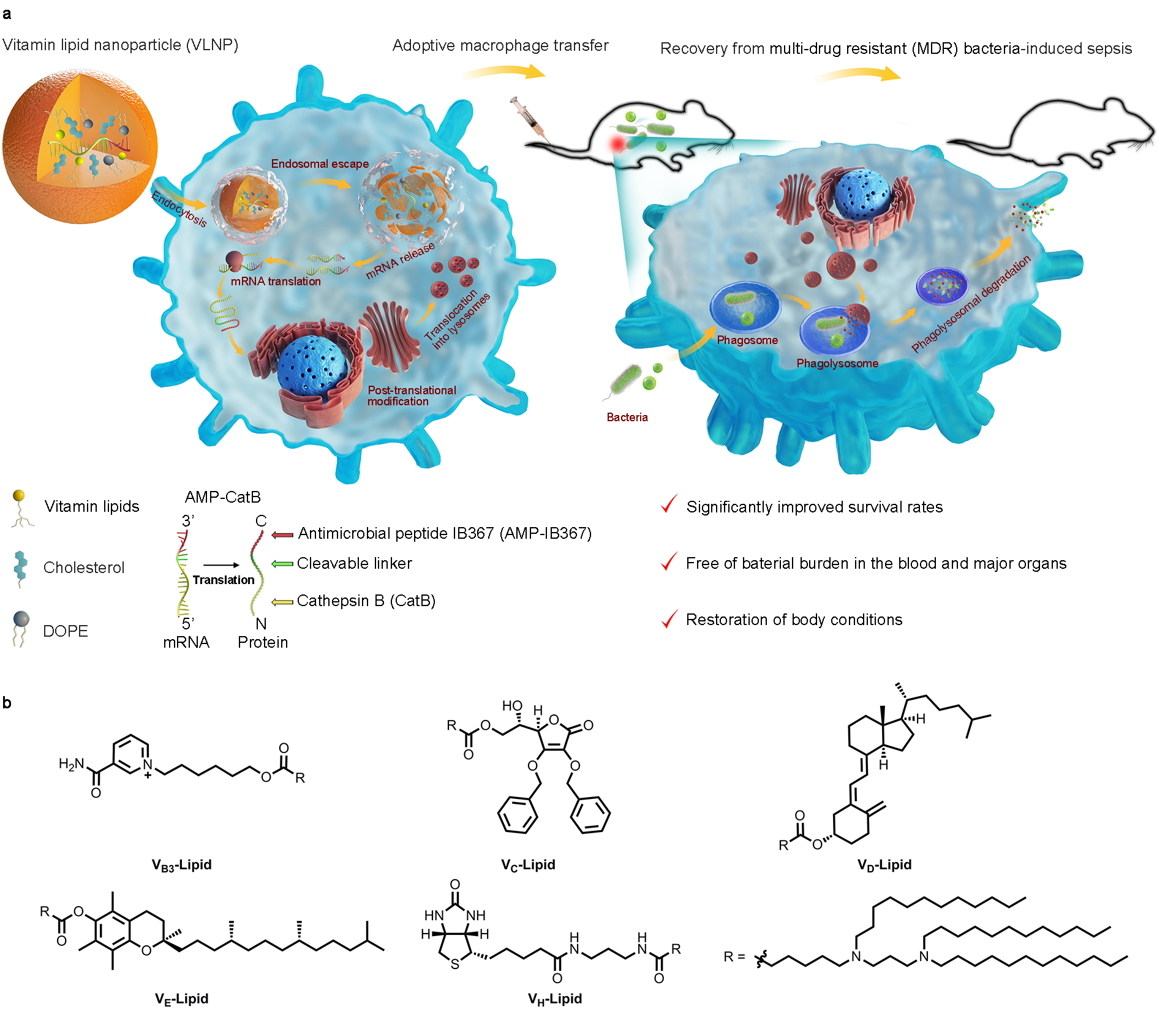
a, Construction of MACs for sepsis therapy. MACs stands for macrophages containing antimicrobial peptides linked to cathepsin B in the lysosomes (MACs). The AMP-CatB mRNA is encapsulated in the vitamin C lipid nanoparticle (VCLNP) and delivered to the macrophage where the mRNA is translated in the cytoplasm and translocated into the lysosomes. Within the lysosomes, the cleavable linker is cleaved by the lysosomal CatB, releasing the AMP-IB367. After phagosomes carrying MDR bacteria fuse with the lysosomes, the ingested MDR bacteria are eradicated by the pre-stored AMP-IB367. b, Chemical structures of vitamin-derived lipids including VB3-Lipid, VC-Lipid, VD-Lipid, VE-Lipid, and VH-Lipid.
In this study, we first synthesized five vitamin-derived lipids: VB3-Lipid, VC-Lipid, VD-Lipid, VE-Lipid, and VH-Lipid (Fig. 1b). Tertiary amines in the lipid chain can be ionized at an acidic condition and interact with mRNAs. The structures of these vitamin-derived lipids were confirmed by 1H NMR and mass spectrum (Supplementary Fig. 1–5).
Next, we formulated these vitamin-derived lipids into vitamin lipid nanoparticles (VLNPs) accordingly. Particle size of VLNPs ranged from 127 ± 1 to 174 ± 1 nm with polydispersity index (PDI) < 0.3 except VHLNPs (Supplementary Fig. 6a, b). Entrapment efficiency of mRNA was from 52% to 99% and all VLNPs were positively charged (Supplementary Fig. 6c, d). In initial screening in RAW264.7 cells using mRNA encoding firefly luciferase, we noted that VCLNPs were 20-fold more effective for mRNA delivery than other four VLNPs (Fig. 2a). Moreover, VCLNPs were 10-fold better than Lipofectamine 3000 and were 50-fold better than electroporation at the same mRNA concentration (Fig. 2a). In addition, the highest luminescence intensity of VCLNP group was observed at 12 h from the 6 to 24 h time course (Fig. 2b). To further optimize the formulation of VCLNPs, we prepared 16 different formulations based on an L16 (4)4 orthogonal table (Supplementary Fig. 6e). Formulation B2 (Lipid:DOPE:Cholesterol = 30:30:40) emerged from this study (Fig. 2c, d). Then, we validated its delivery efficiency by comparison with the top formulation A10 (Lipid:DOPE:Cholesterol = 30:30:50) in the orthogonal table. Luminescence intensity of the predicted formulation B2 was significantly higher than formulation A10 (P < 0.05, Fig. 2d, e). In the second round of optimization, we examined the mass ratio of VC-Lipid:mRNA of formulation B2 from 5:1 to 20:1 (Fig. 2d). Luminescence intensity was enhanced as we increased the mass ratio of VC-Lipid:mRNA to 15:1 (formulation C5, Fig. 2d, e). Formulation C5 of VCLNPs was in a spherical morphology from the Cryo-TEM image (Fig. 2f, Supplementary Fig. 6f). Based on these results, we chose this VCLNPs formulation with improved mRNA delivery efficiency over 7-fold than its initial formulation for further studies including cellular uptake, endocytic pathways, and endosomal escape. Using an Alexa-Fluor 647 labelled RNA, we observed more efficient cellular uptake of VCLNPs than Lipofectamine 3000 and electroporation (Supplementary Fig. 7a, b). After incubation with methyl-beta-cyclodextrin (MβCD, inhibitor of caveolae-mediated endocytosis), cellular uptake of VCLNPs was dramatically reduced by about 96% (Supplementary Fig. 7c, d), indicating a major role of caveolae-mediated endocytosis for these VCLNPs. To explore the endosomal escape mechanism, we performed a calcein assay. Calcein, a membrane-impermeable dye, is normally entrapped into cell endosomes. We treated cells with both calcein and VCLNPs, and observed diffused green fluorescence in cell cytoplasm, suggesting that endosome membranes were ruptured and consequently VCLNPs are released to the cytoplasm (Supplementary Fig. 7e, f).
Fig. 2 |. Screening, optimization, and characterization of vitamin lipid nanoparticles (VLNPs).
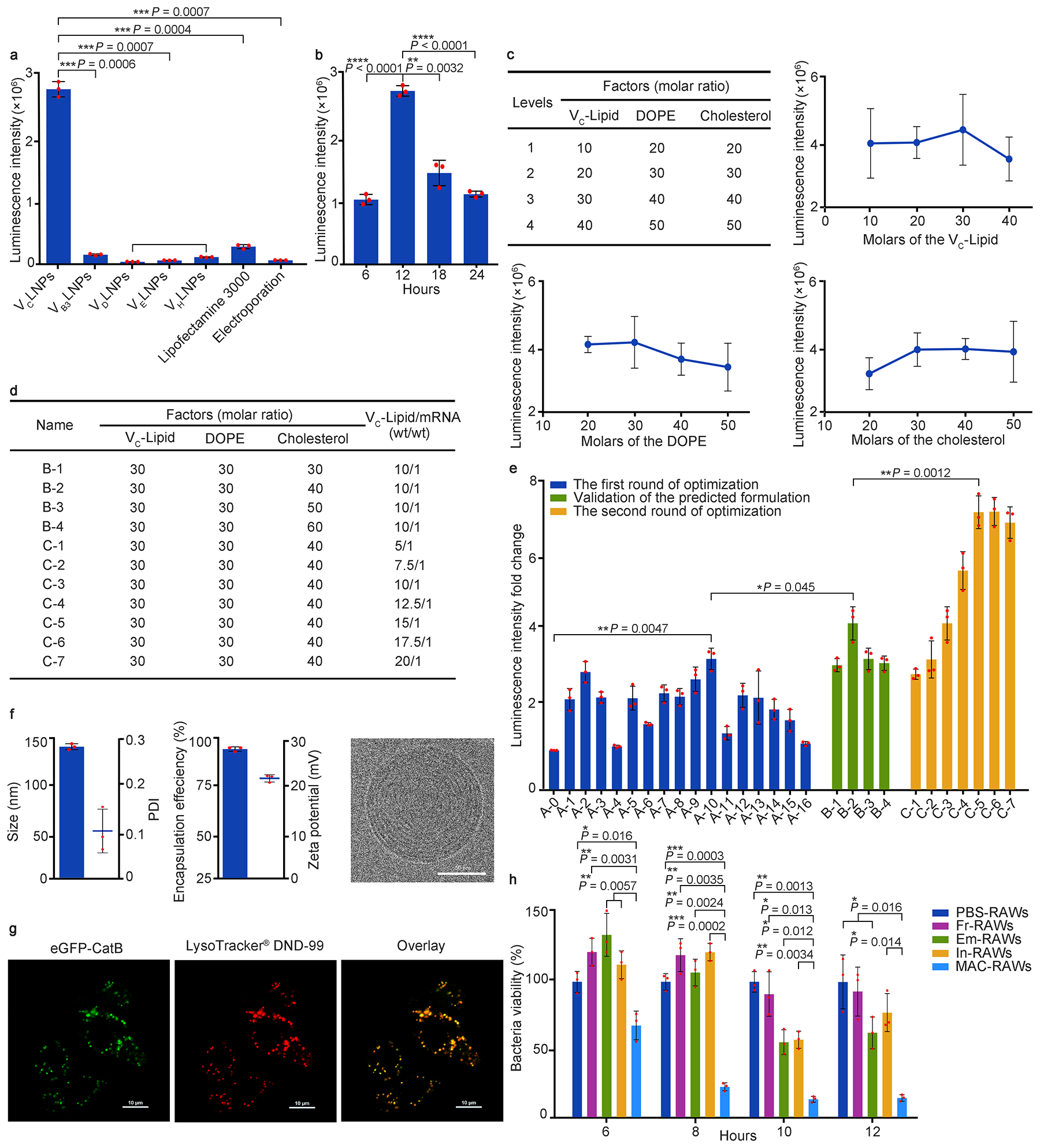
a, mRNA delivery efficiency of VLNPs in RAW264.7 cells. b, Expression kinetics of mRNA delivered by VCLNPs in RAW264.7 cells. c, The first round of optimization: four levels and impact trend of each VCLNP’s component. d, Formulation table for the validation of the predicted formulation, and the second round of optimization: the mass ratio of VC-Lipid:mRNA. e, Fold changes of luminescence intensity in the two rounds of optimization. f, Characterization of the optimal VCLNP formulation, including size, polydispersity index (PDI), encapsulation efficiency, zeta potential, and Cryo-TEM image (Scale bar = 50 nm). g, Confocal imagines of the RAW264.7 cells incubated with VCLNPs encapsulating eGFP-CatB mRNA (Scale bar = 10 μm). Data in g are representative images from n = 3 independent experiments. h, Intracellular survival of MDR Staphylococcus aureus (MDRSA) in the RAW264.7 cells. Data in a, b, c, e, and h are from n = 3 biologically independent samples. All data are presented as mean ± s.d. Statistical significance was analyzed by the two-tailed Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test whether CatB was able to transport the payload into the lysosomes, we constructed eGFP-CatB mRNA and delivered it into RAW264.7 cells using VCLNPs. Confocal images of live cells exhibited that eGFP-CatB co-localized with LysoTracker® Red DND-99 in lysosomes, with a Pearson’s correlation coefficient of 0.93 (Fig. 2g, Supplementary Fig. 7g), indicating that CatB carried its payload into lysosomes. Then, to evaluate the bactericidal activity of MACs, we quantified the intracellular survival of multi-drug resistant Staphylococcus aureus (MDRSA) in RAW264.7 cells treated by PBS (PBS-RAWs), free AMP-CatB mRNA (Fr-RAWs), empty VCLNPs (Em-RAWs), AMP-CatB mRNA VCLNPs/CatB inhibitor II (In-RAWs), and AMP-CatB mRNA VCLNPs (MAC-RAWs). Among these treatments, MAC-RAWs showed the strongest bactericidal activity at all the time points tested with the percentage inhibition from 33% to 87% (Fig. 2h). When we inhibited CatB function using the CatB inhibitor II, bactericidal activity was dramatically reduced (Fig. 2h), thus suggesting the importance of releasing AMP-IB367. No significant differences in cell numbers were observed in all five groups (Supplementary Fig. 8a).
Given the potent in vitro bactericidal activity of MAC-RAWs, we tested therapeutic effects in MDRSA-induced sepsis mice with immunosuppression. After three consecutive days of cyclophosphamide (CY) treatment, decrease of body weights (BWs), white blood cells (WBCs), and lymphocytes (LYMs) mimicked an immunocompromised state in septic patients. After intraperitoneally (i.p.) infected by MDRSA, mice were treated with PBS, PBS-RAWs, or MAC-RAWs. PBS-RAWs were injected both i.p. and intravenously (i.v.) to treat local and blood bacteria. For the MAC-RAWs, we first conducted two administration methods with the same total cell number: i.p. injection alone (MAC-RAWs (i.p.)) and i.p. + i.v. injections (MAC-RAWs (i.p. + i.v.)). Since lethality in immunosuppressed sepsis is associated with ineradicable pathogens, we measured bacterial colony forming units (CFUs) in mouse blood after 24 h of cell transfer. Similar to PBS treatment, PBS-RAWs did not reduce bacterial burden in the blood, while MAC-RAWs (i.p.) and MAC-RAWs (i.p. + i.v.) significantly reduced bacterial CFUs in blood (P < 0.01 and P < 0.001, respectively) (Fig. 3a). These results demonstrated bactericidal activity of MACs in vivo. Moreover, MAC-RAWs (i.p. + i.v.) showed much stronger ability to clear bacteria than MAC-RAWs (i.p.) (P < 0.0001, Fig. 3a). Importantly, the survival rate of MAC-RAW (i.p. + i.v., 58%) group was significantly improved compared to MAC-RAW (i.p.) group (P < 0.01, Fig. 3b). Similarly, MAC-RAWs (i.p. + i.v.) showed better therapeutic effects on survival rate (P < 0.05) than MAC-RAWs (i.v.) (Supplementary Fig. 8b). In the MAC-RAW (i.p. + i.v.) group, bacteria in blood were not detectable in 3 out of the 7 survived mice at 480 h (Fig. 3f). Then, we performed a repeat treatment for the 4 mice with persistent infections, which consequently cleared remaining bacteria in these mice (Fig. 3g). After a month, the levels of BW, WBC, and LYM of the seven survived mice fully recovered (Fig. 3c–e). Moreover, bacteria were not detectable in blood and major organs (heart, liver, spleen, lung, and kidneys) in these mice. The better therapeutic effects of MACs administrated via i.p. + i.v. injections than i.p. or i.v. injection alone may be due to the infection process of sepsis21,22. After i.p. inoculation, bacteria are transported by the lymphatic system to blood within a short period of time and then distributed to other organs via blood circulation21,22. Administration of i.p. + i.v. contributes to eliminating the bacteria in both peritoneal cavity and blood, thus leading to reduced bacterial CFUs and improved survival. When mice were i.v. infected by MDRSA, MAC-RAWs (i.v.) showed dramatically better therapeutic effects on survival rate (P < 0.01) compared to the control group (Supplementary Fig. 8c). Therefore, administration routes need to be carefully determined according to infection sites.
Fig. 3 |. Therapeutic effects of MAC-RAWs in MDRSA-induced sepsis mice with immunosuppression.
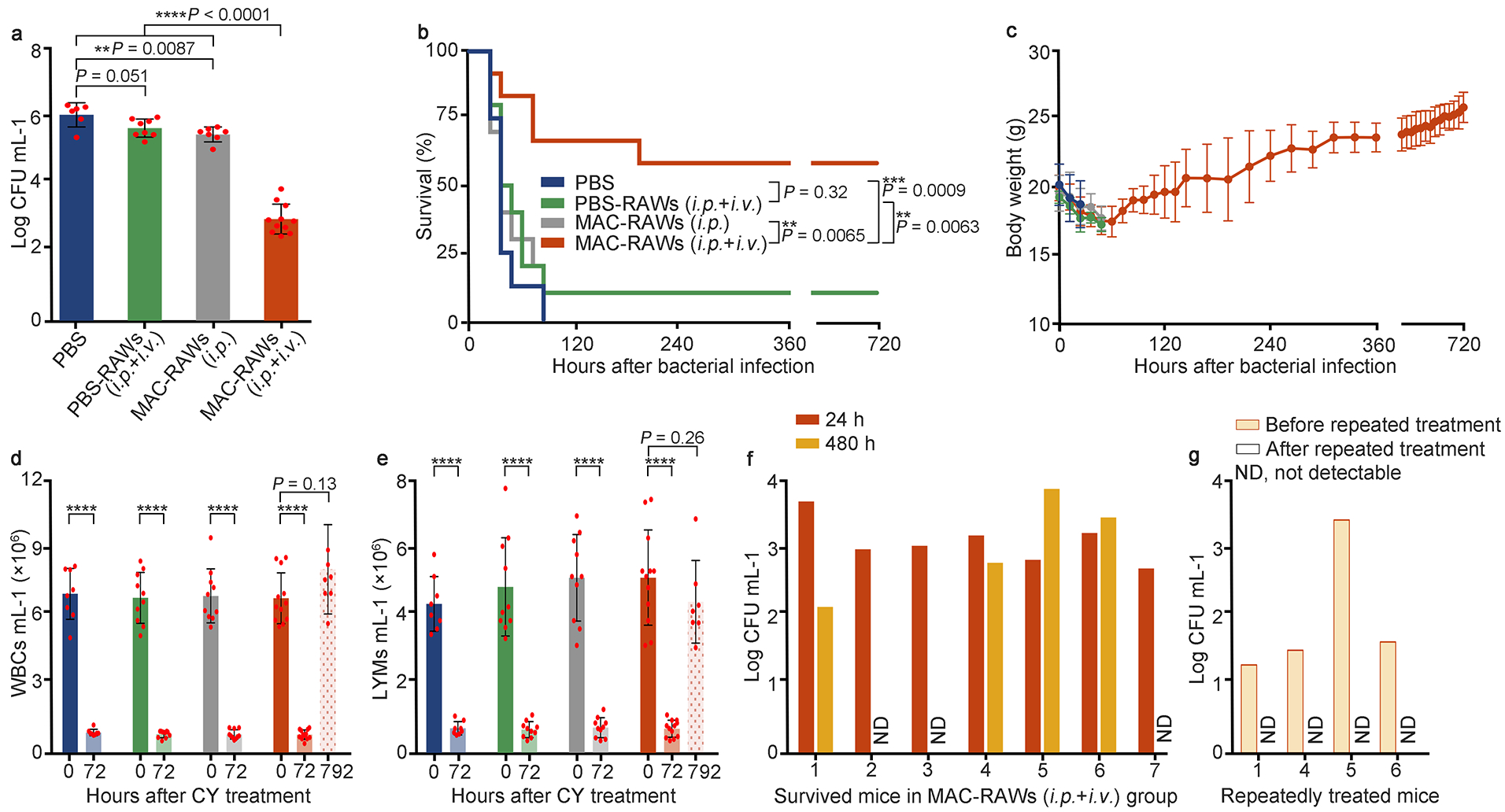
a, Bacterial burden in blood at 24 h after cell transfer. n = 6, 8, 7, and 11 live mice for PBS, PBS-RAWs (i.p. + i.v.), MAC-RAWs (i.p.), and MAC-RAWs (i.p. + i.v.) groups, respectively. b, Percentage survival of mice with sepsis induced by i.p. bacterial inoculation. c-e, The body weights (BWs), white blood cells (WBCs), and lymphocytes (LYMs) of mice. c, BWs; d, WBCs; e, LYMs. Data in b, c, d, and e (except time point 792 h), n = 8, 10, 10, and 12 for PBS, PBS-RAWs (i.p. + i.v.), MAC-RAWs (i.p.), and MAC-RAWs (i.p. + i.v.) groups, respectively. f, g, Bacterial burden in blood of each survived mouse treated by MAC-RAWs (i.p. + i.v.). Data in a, c, d, and e are the mean ± s.d. Statistical significance was analyzed by the two-tailed Student’s t-test. Data in b were analyzed by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant; ND, not detectable.
These promising results encouraged us to evaluate bactericidal activity using primary bone- marrow-derived macrophages (BMDMs), which are translatable for future clinical applications. We first generated BMDMs from mouse bone marrow and confirmed ~83.5% cells were F4/80 positive (Supplementary Fig. 9a). Then, we screened VLNPs and tested their expression profile in BMDMs. Similar to the results in RAW264.7 cells, VCLNPs were more effective in mRNA delivery than other treatments (Supplementary Fig. 9b) and the maximum luminescence intensity was observed at 12 h (Supplementary Fig. 9c). Next, we prepared MAC-BMDMs using AMP-CatB mRNA VCLNPs, and evaluated the in vitro bactericidal activity against MDRSA and multi-drug resistant Escherichia coli (MDR E.coli). Compared to BMDMs treated by PBS (PBS-BMDMs), free AMP-CatB mRNA (Fr-BMDMs), empty VCLNPs (Em-BMDMs), and AMP-CatB mRNA VCLNPs/CatB inhibitor II (In-BMDMs), AMP-CatB mRNA VCLNPs (MAC-BMDMs) showed the strongest bactericidal activity against both MDRSA and MDR E.coli (Supplementary Fig. 9d–g). In addition, AMP-CatB mRNA VCLNPs did not induce obvious cytotoxicity in BMDMs (Supplementary Fig. 9h).
Next, we applied MAC-BMDMs to treat MDRSA-induced sepsis mice with immunosuppression. Based on the data from RAW264.7 cells, MAC-BMDMs were administrated to mice via both i.p. and i.v. injections. Analysis of blood at 24 h showed that MAC-BMDMs were more effective in eliminating bacteria than PBS-BMDMs (P < 0.001, Supplementary Fig. 10a). Furthermore, MAC-BMDMs rescued 58% of mice from immunosuppressed sepsis compared with only 10% rescued by PBS-BMDMs (P < 0.01, Supplementary Fig. 10b). Except for one mouse from the MAC-BMDM group displayed persistent infection, no bacteria were found in blood in other six survived mice at 480 h (Supplementary Fig. 10f, g). Similarly, after a repeat treatment for that mouse, all the mice showed normal levels of BW, WBC, and LYM (Supplementary Fig. 10c–e), and undetectable levels of MDRSA in their blood and major organs.
In order to profile the biodistribution of these macrophages, we delivered mRNA encoding firefly luciferase into BMDMs (FLuc-BMDMs). Six hours post i.p. + i.v. injections of the FLuc-BMDMs, we measured the luminescence intensity in the following major organs usually infected by bacteria in this mouse model21,22: peritoneal cavity, spleen, liver, lung, kidneys, heart, and blood. The results showed a similar biodistribution in healthy and sepsis mice (Supplementary Fig. 10h). In the sepsis mice infected by i.p. bacterial inoculation, the rank order of luminescence intensity is as follows: peritoneal cavity, spleen, lung, liver, and blood. The BMDMs biodistribution was relatively consistent to the bacterial distribution in these sepsis mice (Supplementary Fig. 10i).
Because sepsis hosts are usually exposed to mixed bacterial infections, a formidable challenge for curing sepsis, we established a mouse sepsis model with infections by both MDRSA and MDR E.coli. Treatment with MAC-BMDMs significantly reduced bacterial burden in blood compared to the treatment with PBS and PBS-BMDMs, respectively (Fig. 4a). Therapeutic efficacy of MAC-BMDMs was also reflected in the survival rate (83%), which was much higher than that of PBS group (P < 0.001) and PBS-BMDM group (P < 0.05) (Fig. 4b). Finally, normal levels of BW, WBC, and LYM were observed in all survived mice (Fig. 4c–f), and no persistent infections were detected in blood and major organs.
Fig. 4 |. Therapeutic effects of MAC-BMDMs in mixed MDR bacteria (Staphylococcus aureus and Escherichia coli)-induced sepsis mice with immunosuppression.
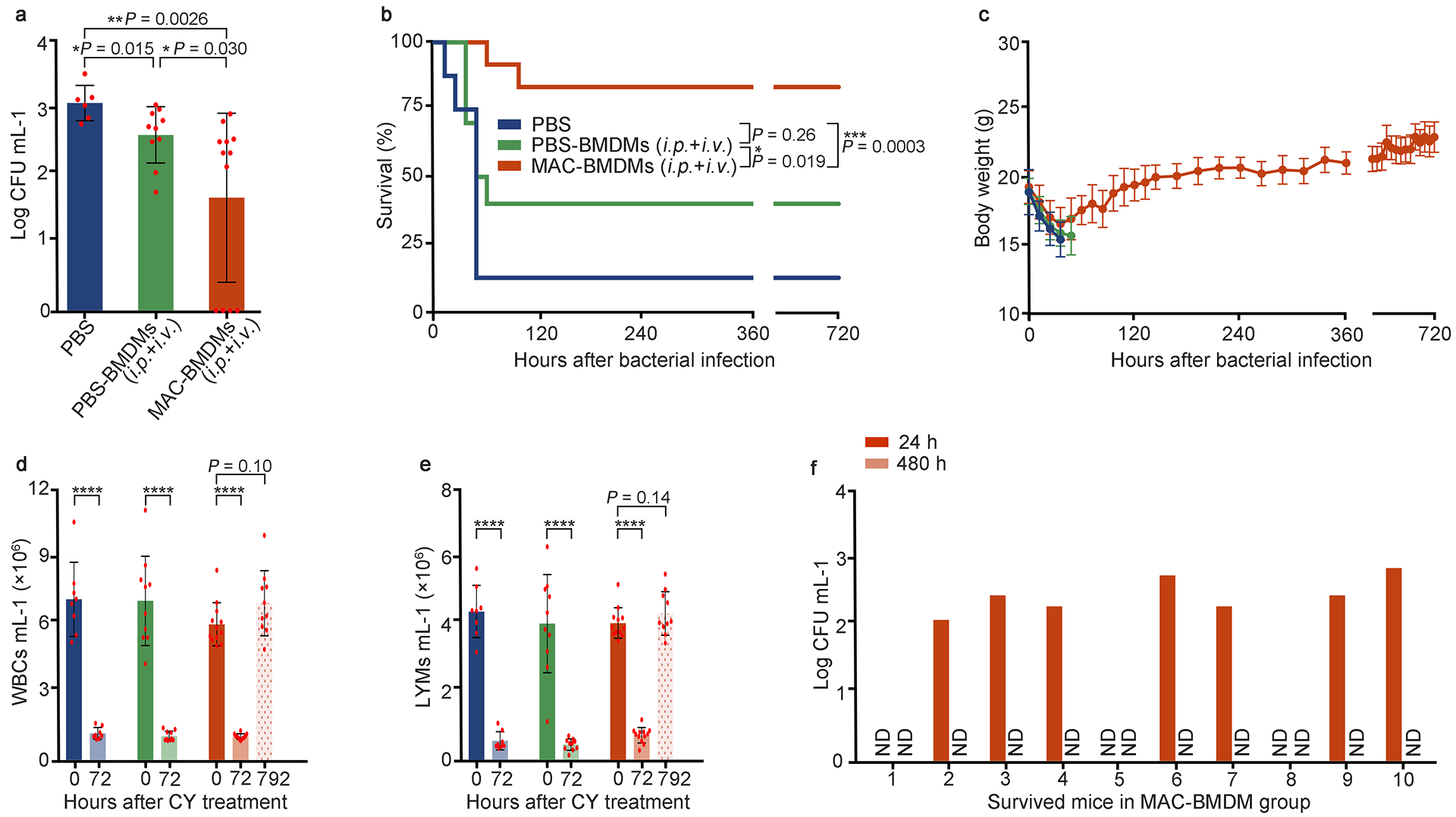
a, Bacterial burden in blood at 24 h after cell transfer. n = 6, 10, and 12 live mice for PBS, PBS-BMDMs (i.p. + i.v.), and MAC-BMDMs (i.p. + i.v.) groups, respectively. b, Percentage survival of mice with sepsis induced by i.p. bacterial inoculation. c-e, The BWs, WBCs, and LYMs of mice. c, BWs; d, WBCs; e, LYMs. Data in b, c, d, and e (except time point 792 h), n = 8, 10, and 12 for PBS, PBS-BMDMs (i.p. + i.v.), and MAC-RAWs (i.p. + i.v.) groups, respectively. f, Bacterial burden in blood of each survived mouse treated by MAC-BMDMs. Data in a, c, d, and e are the mean ± s.d. Statistical significance was analyzed by the two-tailed Student’s t-test. Data in b were analyzed by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant; ND, not detectable.
In summary, we have developed macrophages containing antimicrobial peptides linked to cathepsin B in the lysosomes (MACs) via VCLNPs-mediated delivery of mRNA. Our results prove the concept that adoptive transfer of MACs beneficially reduce bacterial burden and improve survival in multi-drug resistant (MDR) bacteria-induced sepsis mice with immunosuppression by restoring innate immune defense, preventing bacterial immune evasion, and eradicating the infection. In the future, this platform will mainly focus on treating the late phase of sepsis with immunosuppression. Autologous MACs might be prepared around one week, but the weakness of patient and emergency of sepsis may make it challenging to collect and engineer autologous macrophages. Yet, with the advance of induced pluripotent stem cells (iPSCs) technologies, allogeneic “universal” macrophages may be manufactured23, enabling iPSCs-derived MACs to become an off-the-shelf therapy for clinical applications, including sepsis. Overall, the adoptive transfer of MACs may provide a strategy for curing patients with sepsis and MDR bacterial infections.
Methods
Chemicals and reagents.
The following agents were purchased from Sigma-Aldrich, including cholesterol, gentamicin, cathepsin B (CatB) inhibitor II, cyclophosphamide (CY), 5-(N-methyl-N-isopropyl) amiloride (EIPA), methyl-beta-cyclodextrin (MβCD), and chlorpromazine hydrochloride (CPZ). The following agents were obtained from Thermo Fisher Scientific, including F4/80 monoclonal antibody, recombinant murine macrophage colony-stimulating factor (M-CSF), and LysoTracker® DND-99. 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids. Bright-Glo luciferase substrate was purchased from Promega.
Cells and bacteria.
The RAW264.7 cell line was obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, ATCC) with 10% fetal bovine serum (Gibco, Invitrogen). The cell line was not independently authenticated and mycoplasma contamination was tested by the supplier. Murine bone-marrow-derived macrophages (BMDMs) were obtained by adaptation of previous procedures24, cultured in DMEM containing 10% fetal bovine serum and 100 ng/ml M-CSF. Multi-drug resistant Staphylococcus aureus (MDRSA, ATCC BAA-44) were grown in trypticase soy agar or trypticase soy broth (BD Biosciences) at 37 °C with aeration. According to the data from ATCC, the MDRSA are resistant to the following antibiotics: amoxicillin/clavulanic acid, penicillin, ciprofloxacin, cephalothin, doxycycline, gentamicin, erythromycin, imipenem, methicillin, tetracycline, oxacillin, azithromycin, clindamycin, ceftriaxone, rifampin, amikacin, and tobramycin. Multi-drug resistant Escherichia coli (MDR E. coli, ATCC BAA-2340) was grown in nutrient agar or nutrient broth (BD Biosciences) at 37 °C with aeration. According to the data from ATCC, the MDR E. coli are resistant to the following antibiotics: amoxicillin/clavulanic acid, ticarcillin, piperacillin, ampicillin/sulbactam, cefalotin, cefuroxime, cefotetan, cefpodoxime, cefotaxime, ceftizoxime, cefazolin, cefoxitin, ceftazidime, ceftriaxone, cefepime, doripenem, meropenem, ertapenem, imipenem, nalidixic acid, moxifloxacin, norfloxacin, ciprofloxacin, levofloxacin, tobramycin, aztreonam, and trimethoprim/sulfamethoxazole.
Synthesis of vitamin-derived lipids.
Compound 1, vitamin B3 derivative, vitamin C derivative, vitamin D derivative, vitamin E derivative, and vitamin H (also called vitamin B7) derivative, were synthesized according to the methods reported previously25.
Preparation, characterization, and optimization of VLNP.
mRNAs used in this study were constructed by our mRNA platform based on the reported method25. The preparation of mRNA LNPs was reported previously26. Briefly, newly synthesized vitamin-derived lipids were formulated with DOPE, cholesterol as well as firefly luciferase (FLuc) mRNA by pipetting for in vitro screenings or by a microfluidic mixing device for ex vivo studies. The NanoZS Zetasizer (Malvern) was used to measure the size and zeta potential. The entrapment efficiency was measured by the Ribogreen assay26. The morphology of VCLNPs was examined on Thermo Scientific™ Glacios™ Cryo-TEM using the methods described before25. In order to effectively deliver mRNA into macrophages, an initial screening was performed using the five vitamin lipid nanoparticles (VLNPs), Lipofectamine 3000, and electroporation. Based on our previous method26, newly synthesized vitamin-derived lipids were formulated with DOPE, cholesterol (Lipid:DOPE:Cholesterol = 20:30:40, mole ratio) as well as FLuc mRNA (Lipid:mRNA = 10:1, mass ratio) in the initial screening. Delivery efficiency of mRNA was determined by the luciferase expression assay26. Next, the dynamics of FLuc expression followed by two rounds of optimization were carried out after the initial screening. Briefly, VcLNPs from A-1 to A-16 were prepared based on the orthogonal array design table L16 (4)4 and the top formulation was predicted by the FLuc expression data. After the top formulation was validated, the second round of optimization focused on fine tuning the mass ratio of VC-Lipid:mRNA. Electroporation for macrophages was performed using Nucleofector kit (Lonza) and the suggested protocols by the Nucleofector 2b Device.
Cellular uptake and endosomal escape.
RAW264.7 cells were seeded in a 24-well plate at 105 cells/well and cultured for 24 h. Then, the cells were treated by FLuc mRNA and Alexa-Fluor 647-labeled RNA (1:1, weight ratio) using Lipofectamine 3000, VcLNPs, or electroporation. After 3 h incubation, cellular uptake was quantified by a flow cytometer (LSRII, BD). To study endocytic pathways of VcLNPs, cellular uptake assay was performed in the presence of different endocytotic inhibitors, 5-(N-methyl-N-isopropyl)amiloride (EIPA), methyl-beta-cyclodextrin (MβCD), and chlorpromazine hydrochloride (CPZ), which inhibit macropinocytosis-, caveolae-, and clathrin-mediated endocytosis, respectively. For the endosomal escape assay, 2 × 104 cells were plated in an imaging dish (Ibidi) for 24 h, and then 150 μg/mL of calcein was added to the cells with or without VcLNPs containing Alexa-Fluor 647-labeled RNA for 6 h at 37 °C27. After washing with PBS to remove extracellular calcein and VcLNPs, cells were lively imaged under the Nikon A1R Live Cell Imaging Confocal Microscope. The images were analysed by NIS-Elements AR 5.20.00.
Analysis of lysosome co-localization.
To test whether AMP-CatB can specificly accumulate in the lysosomes, we prepared the eGFP-CatB mRNA and delivered it into RAW264.7 cells using VCLNPs. Then, the lysosomes were stained with LysoTracker® Red DND-99, a well-established lysosome probe. The co-localization of eGFP-CatB and LysoTracker® Red DND-99 was visualized under the Nikon A1R Live Cell Imaging Confocal Microscope. The images were analysed by NIS-Elements AR 5.20.00.
Cytotoxicity of vitamin C lipid nanoparticles in BMDMs.
Cytotoxicity of VCLNPs in BMDMs was examined by adaptation of the previous MTT assay25. 2 × 104 BMDMs were seeded into each well of a 96-well plate in 100 μL of growth medium. After 12 h of incubation with PBS, free AMP-CatB mRNA, empty VCLNPs, AMP-CatB mRNA VCLNPs/CatB inhibitor II, and AMP-CatB mRNA VCLNPs, MTT solution was added. After additional 4 h incubation, 100 uL of 10% SDS-HCl was added into each well. The purple formazan was dissolved overnight and the absorbance was recorded at 570 nm by the plate reader (Molecular Devices Spectramax M5).
In vitro antimicrobial assay.
The intracellular antimicrobial assay was conducted according to the reported method15. Briefly, after treated by PBS, free AMP-CatB mRNA, empty VCLNPs, AMP-CatB mRNA VCLNPs/CatB inhibitor II, and AMP-CatB mRNA VCLNPs, the RAW264.7 cells or BMDMs were incubated with MDRSA or MDR E.coli at multiplicity of infection of 25 for 120 min. After washing by PBS, medium containing gentamicin (100 μg/mL) was supplemented and the cells were incubated for additional 1 h to clear extracellular bacteria. At different time points, cells were washed by PBS and lysed with 0.1% Triton-X 100. Lastly, the lysates were cultured on the nutrient agar or trypticase soy agar for counting bacterial colony forming units (CFUs).
In vivo treatment of MDR-induced sepsis mice with immunosuppression.
All mouse experiments were performed under the protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Ohio State University and were also complied with all relevant ethical regulations as applicable. C57BL/6 mice (6–7 weeks) were purchased from the Jackson Lab. The immunosuppressed model of sepsis was conducted as the methods in the literature21,22,28. Briefly, C57BL/6 mice were i.p. injected CY at a dosage of 100 mg/kg for 3 consecutive days before bacterial infection. Their immunocompromised states were assessed by monitoring the body weights (BWs), counting white blood cells (WBCs) by a hemocytometer, and counting lymphocytes (LYMs) by the Kwik-Diff stain. For i.p. bacterial inoculation, mice were infected with 0.1 ml of bacterial suspension (5×108 CFUs/mouse for MDRSA infection and 2×108 CFUs/mouse for mixed MDRSA and MDR E. coli infections). For, i.v. bacterial inoculation, mice were infected with 0.1 ml of bacterial suspension (1×108 CFUs/mouse). Afterwards, the mice were administrated by 0.2 mL PBS or 0.2 mL cell suspension (total 2 million cells/mouse). After 24 h infection, the blood was collected from the facial vein of live mice to quantify bacterial CFUs. The survival of mice were assessed every 12 h within the first 6 days and then every 24 h for the following 24 days based on 20% loss of BW and the early removal criteria. After 30 days, the mice were euthanized and major organs (heart, liver, spleen, lung, and kidneys) were aseptically homogenized to quantify the bacterial CFUs.
Biodistribution of macrophages and bacteria.
The biodistribution of macrophages was performed on both healthy and sepsis mice. In this experiment, we first delivered FLuc mRNA into BMDMs (FLuc-BMDMs) for 12 h. Next, each mouse was administered 0.2 mL PBS or 0.2 mL cell suspension (total 4 million cells) via i.p. + i.v. injections. After 6 h, bioluminescence signals in the blood, peritoneal fluid, and major organs were measured using a Xenogen IVIS imaging system as reported in the literature26. The bacterial CFUs in the blood, peritoneal fluid, and major organs of sepsis mice induced by i.p. bacterial inoculation were quantified 6 h after infection.
Supplementary Material
Acknowledgements
We acknowledge the use of the core facility provided by the Campus Microscopy & Imaging Facility at the Ohio State University. X.Z. acknowledges the support from Fundamental Research Funds for the Central Universities (No. DUT18RC(3)027). Y.D. acknowledges the support from National Institutes of Health (NIH) through the Maximizing Investigators’ Research Award R35GM119679 of the National Institute of General Medical Sciences as well as the start-up fund from the College of Pharmacy at The Ohio State University.
Footnotes
Competing interests
The authors have no competing interests to declare.
Supplementary information is available in the online version of the paper.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Reinhart K, et al. Recognizing sepsis as a global health priority—a WHO resolution. New England Journal of Medicine 377, 414–417 (2017). [DOI] [PubMed] [Google Scholar]
- 2.van der Poll T Immunotherapy of sepsis. The Lancet infectious diseases 1, 165–174 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Huttunen R & Aittoniemi J New concepts in the pathogenesis, diagnosis and treatment of bacteremia and sepsis. Journal of Infection 63, 407–419 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS & Karl IE The pathophysiology and treatment of sepsis. New England Journal of Medicine 348, 138–150 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Otto GP, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Critical care 15, R183 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotchkiss RS, Monneret G & Payen D Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. The Lancet infectious diseases 13, 260–268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Czermak BJ, et al. Protective effects of C5a blockade in sepsis. Nature medicine 5, 788 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Ward PA & Fattahi F New strategies for treatment of infectious sepsis. Journal of leukocyte biology (2019). [DOI] [PubMed] [Google Scholar]
- 9.Huang X, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proceedings of the National Academy of Sciences of the United States of America 106, 6303–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Docke WD, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nature medicine 3, 678–681 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Presneill JJ, Harris T, Stewart AG, Cade JF & Wilson JW A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. American journal of respiratory and critical care medicine 166, 138–143 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Galbraith N, Walker S, Galandiuk S, Gardner S & Polk HC Jr. The Significance and Challenges of Monocyte Impairment: For the Ill Patient and the Surgeon. Surgical infections 17, 303–312 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Bo L, Wang F, Zhu J, Li J & Deng X Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Critical care (London, England) 15, R58 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster TJ Immune evasion by staphylococci. Nature reviews. Microbiology 3, 948–958 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Garzoni C & Kelley WL Staphylococcus aureus: new evidence for intracellular persistence. Trends in microbiology 17, 59–65 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Lewis AJ, Richards AC & Mulvey MA Invasion of Host Cells and Tissues by Uropathogenic Bacteria. Microbiology spectrum 4(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pauwels AM, Trost M, Beyaert R & Hoffmann E Patterns, Receptors, and Signals: Regulation of Phagosome Maturation. Trends in immunology 38, 407–422 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giles FJ, Redman R, Yazji S & Bellm L Iseganan HCl: a novel antimicrobial agent. Expert opinion on investigational drugs 11, 1161–1170 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Linke M, Herzog V & Brix K Trafficking of lysosomal cathepsin B-green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. Journal of cell science 115, 4877–4889 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Vasey PA, et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clinical cancer research : an official journal of the American Association for Cancer Research 5, 83–94 (1999). [PubMed] [Google Scholar]
- 21.Frimodt-Møller N, Knudsen J & Espersen F The mouse peritonitis/sepsis model in Handbook of animal models of infection 127–136 (Elsevier, 1999). [Google Scholar]
- 22.McVicker G, et al. Clonal expansion during Staphylococcus aureus infection dynamics reveals the effect of antibiotic intervention. PLoS pathogens 10, e1003959 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crow D Could iPSCs Enable “Off-the-Shelf” Cell Therapy? Cell 177, 1667–1669 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Ying W, Cheruku PS, Bazer FW, Safe SH & Zhou B Investigation of macrophage polarization using bone marrow derived macrophages. JoVE (Journal of Visualized Experiments), (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, et al. Chemotherapy drugs derived nanoparticles encapsulating mRNA encoding tumor suppressor proteins to treat triple-negative breast cancer. Nano Research, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano letters 15, 8099–8107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su X, Fricke J, Kavanagh DG & Irvine DJ In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Molecular pharmaceutics 8, 774–787 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, et al. High-throughput synergy screening identifies microbial metabolites as combination agents for the treatment of fungal infections. Proceedings of the National Academy of Sciences of the United States of America 104, 4606–4611 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.