

Abstract
Trinucleotide repeats are common in the human genome and can undergo changes in repeat number and cause length-dependent chromosome fragility. Expanded CAG repeats have been linked to over 14 human diseases and are considered hotspots for breakage and genomic rearrangement. Here we describe two Saccharomyces cerevisiae based assays that evaluate the rate of chromosome breakage that occurs within a repeat tract (fragility), with variations that allow the role of transcription to be evaluated. The first fragility assay utilizes end-loss and subsequent telomere addition as the main mode of repair of a yeast artificial chromosome (YAC). The second fragility assay relies on the fact that a chromosomal break stimulates recombination-mediated repair. A PCR-based assay can be used to evaluate instability of the repeat in the same conditions used to measure repeat fragility. These assays have contributed to understanding the genetic mechanisms that cause chromosome breaks and tract-length changes at unstable trinucleotide repeats.
Keywords: Chromosome breakage, fragility, repeat instability, Yeast Artificial Chromosome (YAC), Direct duplication recombination assay (DDRA)
1. Introduction
Stretches of repetitive DNA sequence are common in eukaryotic genomes and are capable of forming alternative DNA structures, such as hairpin loops. These alternative DNA structures effectively function as a physical barrier, making it difficult for error free DNA replication and repair to occur, and can result in changes in repeat length, chromosomal breakage, and genome instability. One type of repetitive DNA sequence that is common in the human genome are CNG repeats, which when expanded, have been causally associated with over 16 human diseases including Huntington’s disease (HD), myotonic dystrophy (DM), spinocerebellar ataxias (SCAs), and fragile X syndrome (FXS) (1, 2).
Long regions of CAG/CTG and CGG/GCC trinucleotide repeats break and undergo aberrant repair at a higher frequency than non-repetitive DNA, which can result in mutation and changes in repeat copy number. Breakage of the DNA strands (referred to herein as “fragility”) and changes in repeat copy number (referred to herein as “instability”) are two sides of the same coin. For example, if normal replication or repair is impaired when encountering a repeat tract, a stalled fork, nick, gap, or double-strand break (DSB) can occur. If the fork is restarted or if there is healing within the nick, gap or break, repeat units can be lost or gained. Alternatively, the DNA lesion may be repaired by a less conservative form of repair that leads to deletion of flanking sequence, loss of a chromosome arm, or other chromosome rearrangements. This dramatic loss of genetic material can be easily monitored in S. cerevisiae by loss of a neighboring genetic marker, and is indicative that a break in the chromosome occurred. Such a marker loss assay is an underestimate of the “true” rate of breakage, as it only measures the incidents that are healed after loss of flanking DNA sequence, and not those that heal within the repeat, or those that fail to repair resulting in cell death. Nonetheless, such genetic fragility assays are very useful for comparative studies and can be used to determine if a gene or condition is involved in replication or repair of repetitive DNA.
Repeat fragility rates are generally dependent on repeat length, since longer repeats are more likely to interfere with DNA replication or repair. Therefore it is important to start any fragility assay with cells containing a known repeat size, ideally a pure population of the length of interest. This is best done using a physical PCR assay for trinucleotide repeat instability that captures all (or most) of the repeat sizes present in the starting colony. Therefore a description of PCR size analysis for CAG repeats is included in these methods (Method 1), which can be adapted for other types of repeats. A more detailed description of PCR analysis of CAG repeat instability frequencies is available in a previous issue (3). There are also genetic assay systems that can be used to select for repeat expansion or contraction events (3–5). Such selection systems differ from the PCR-based instability assay as they capture only the events that yield a phenotype in the selective system, thus they allow for the study of rare repeat length changes. However they are not appropriate for the purpose of identifying starting colonies for a fragility assay.
To determine the rate at which breakage occurs within a repeat tract, two unique yeast systems have been developed. The first is a yeast artificial chromosome (YAC) system wherein the YAC VS5 is modified such that the potential fragile sequence is integrated between a telomere seed sequence (G4T4)13 and a URA3 marker gene (6, 7) (Figure 1) (Method 2). The version of this YAC with an integrated CAG repeat tract has been termed the (CAG)n-URA3 YAC, also referred to in the literature as YAC CF1. One feature of this YAC system is that other modifications are easily made by replacing the right end of the YAC with the desired sequence (Method 3). We have used this technique to add additional genetic markers, promoters, and terminators (Figure 2). In addition the type of potential fragile element can be changed. For example, other derivatives of this YAC have been made that contain the CAG repeat in the opposite orientation (CTG)n (8), AT repeats that are present in the FRA16D common fragile site (9) (Figure 2), the expanded ATTCT repeat present in SCA10 (10), and inverted repeats (11).
Figure 1. Schematic of the YAC breakage assay.
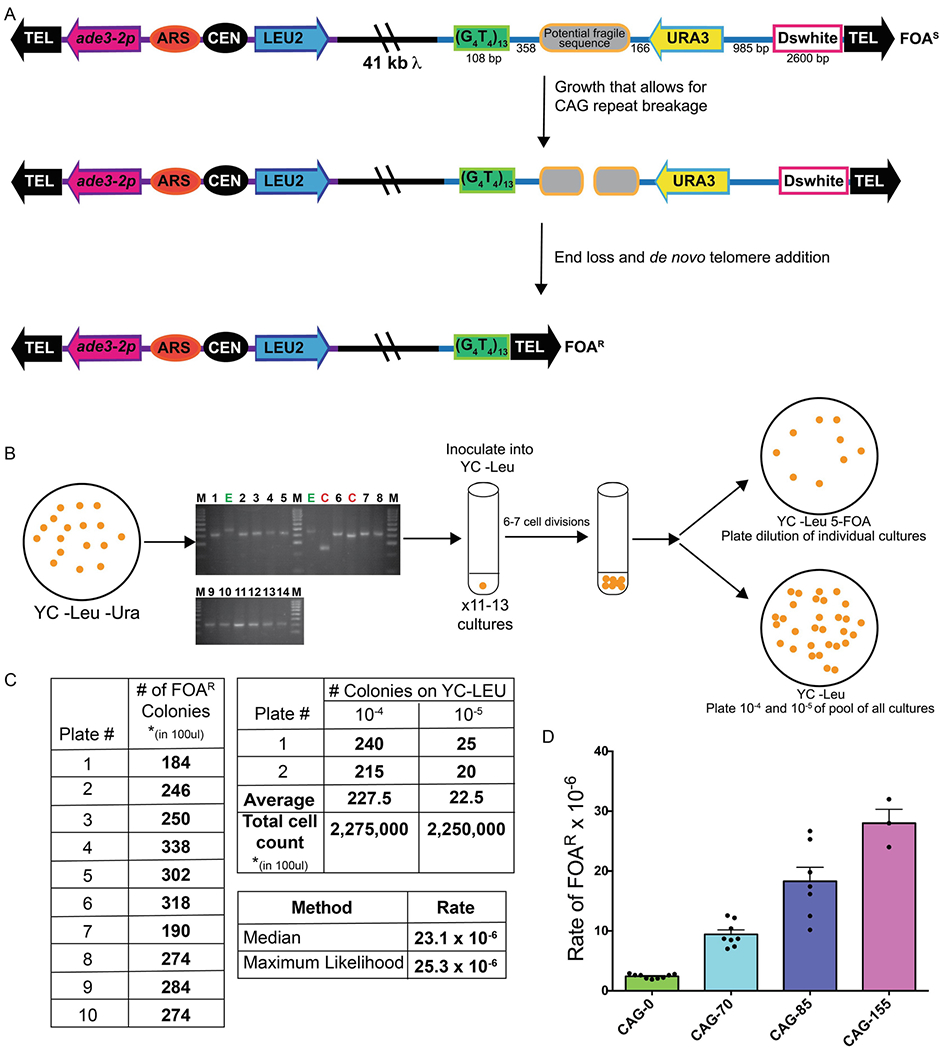
A. In this system, a potential fragile sequence (such as an expanded CAG repeat tract) is integrated onto a yeast artificial chromosome between a telomere seed sequence (G4T4)13 and the URA3 gene. Breaks that occur within the fragile sequence are subject to resection and telomere addition, which results in loss of the URA3 gene and renders cells 5-FOAR. The YAC additionally contains a LEU2 marker gene, which allows for maintenance of the YAC, a centromere (CEN4) and an origin of replication (ARS1). The blue line indicates the pYIP5 plasmid backbone, the black line indicates lambda DNA, and the purple line corresponds to pUC18 plasmid backbone. B. In the YAC end loss assay, strains are plated for single colonies on YC –Leu –Ura plates and 10 individual colonies of the correct tract length (if testing an unstable repeat sequence) are grown in YC –Leu for 6-7 cell divisions. A portion of each individual culture is plated onto YC –Leu +5-FOA plates, and a portion of the culture is pooled and serially diluted to obtain single colonies on YC –Leu, which serves as the total viable cell count. C. Example of the calculation of fragility rate for a 10-colony YAC CF1 CAG-85 fragility assay. Colony counts are used to determine the number of mutants and the number of viable cells in 100 μl of logarithmically growing cultures. D. Wild-type fragility data of S. cereviase strain BY4705 where the fragile sequence integrated between the G4T4 and URA3 marker are different tract lengths of CAG repeats. Assays done with this (CAG)n-URA3 YAC show that 5-FOA resistance increases with increasing number of CAG repeats. Data sourced from (8, 27–30).
Figure 2. Schematic of YAC modification.
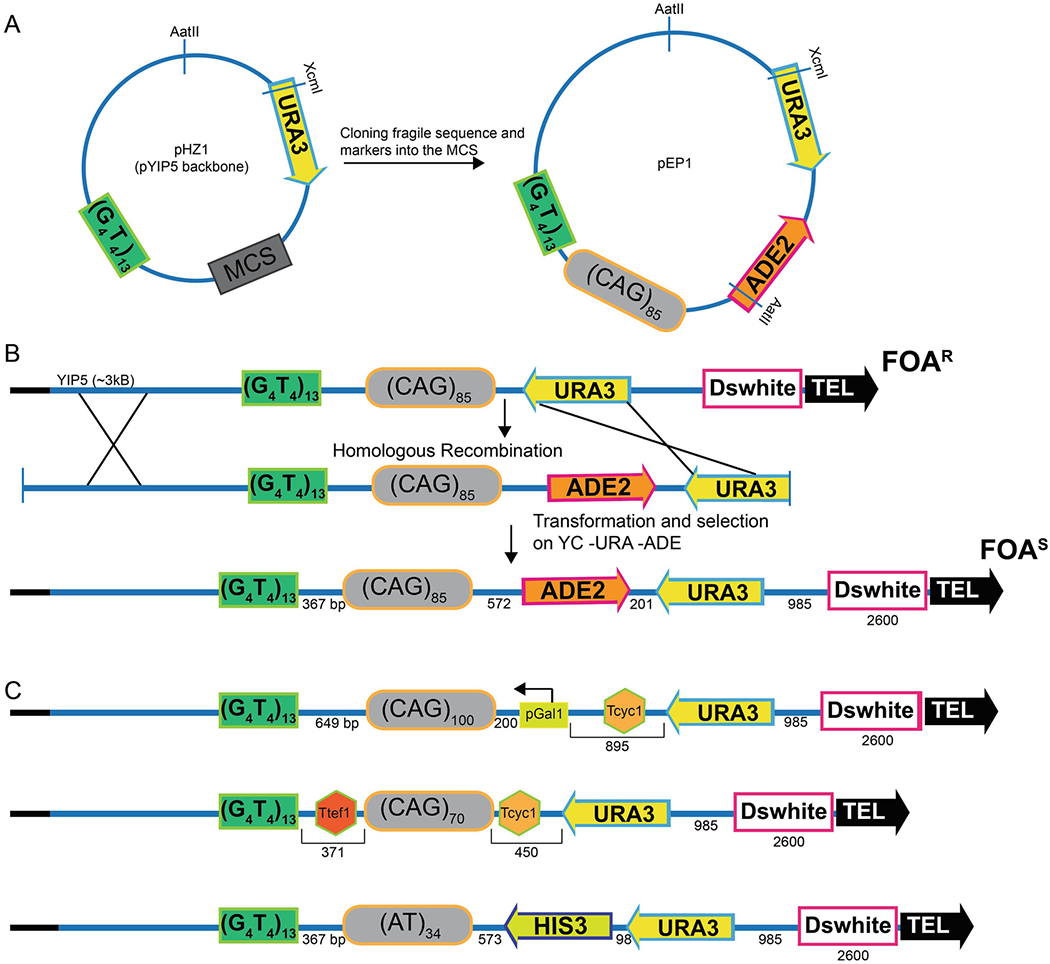
A. Schematic of pHZ1 wherein a telomere seed sequence (G4T4)13 has been inserted into the pYIP5 plasmid backbone such that G4T4 and the URA3 gene are on either side of the multiple cloning site (MCS). The AatII site has been previously used to linearize the plasmid for transformation into YAC containing strains. If AatII is not a unique restriction site after cloning (as shown for pEP1), XcmI can be also be used. B. Schematic detailing integration of a triplet repeat and a second selectable marker into the YAC construct. Transformation of the linear fragment and homologous recombination should result in a novel YAC that incorporates the ADE2 selectable marker as well as the repeat. Correct transformants can be selected for on YC -Leu -Ura -Ade plates. The resultant YAC contains the ADE2 marker gene such that transcription of ADE2 is away from the CAG repeat and Ura-colonies will be 5-FOAR and red on media that contains a limiting amount of adenine (20). Adding a second selectable marker provides an easy selection for recombinants, however other schemes are also possible. Selecting for URA3 revertants from a YAC that contains a ura3 mutation is one way to insert markerless modifications to the YAC; CRISPR-Cas9-based integration strategies could also be used. C. Examples of published CF1 YAC derivitives. In the top construct, to determine the role of transcription on CAG repeat fragility, an inducible GAL1 promoter was added adjacent to the repeat tract (20). In the middle construct, to determine the role of readthrough transcription from URA3 on CAG repeat fragility, two terminator sequences were added such that the Tcyc1 terminator sequence was added between the URA3 marker gene and the CAG repeat and the TEF1 terminator sequence was added between the G4T4 and repeat tract (19, 20). In the bottom construct, an AT34 repeat tract from the common fragile site FRA16D was integrated with the addition of the HIS3 gene (the Schizosaccharomyces pombe HIS5 gene) (9).
1.1. The YAC assay
An important feature of the YAC is that it contains little homology to any of the natural yeast chromosomes. This feature, together with the telomere seed sequence placed proximal to the potential fragile sequence, ensures that the primary mode of healing is by telomere addition rather than recombination. Healing via telomere addition is ideal as relatively few genes affect the efficiency of telomere addition. Known examples are the subunits of the enzyme telomerase, which are needed for telomere addition, and the Pif1 helicase, which inhibits promiscuous telomere addition (6). Thus, in most cases an increase in YAC end loss is caused by increased breakage rather than decreased healing. The position of the potential fragile sequence on the YAC is such that on the proximal side of the sequence there is a 108 bp (G4T4)13 telomeric seed sequence from Oxytrica, which serves as an efficient substrate for S. cerevisiae telomerase, but does not recombine with the natural yeast telomere sequence (12). Therefore, if breakage within the fragile sequence occurs, resection and degradation of the broken end exposes the G4T4 sequence as a template for telomerase (Figure 1). The G4T4 seed provides an efficient pathway for healing of breakage events, which is different from another commonly used assay for end loss of chromosome V (13), where healing can occur by a variety of mechanisms including telomere addition onto short naturally occurring GT sequences or recombination with other chromosomes.
In the YAC fragility assay, breakage that results in loss of the right arm of the YAC renders cells ura3- and resistant to 5-fluoroorotic acid (5-FOA). The number of cells that are 5-FOA resistant serves as a measure of the rate of breakage, and the effect of a potential fragile sequence can be compared to a control sequence of the same length and base composition, or a YAC that does not contain the fragile sequence. One additional design consideration of the YAC is the distance between the URA3 gene and the telomere, such that the placement of the URA3 gene promoter is far enough away from the telomere not to be subject to telomere position effect; this is the reason for the presence of filler sequence (the Drosophila white gene) next to the telomere (Figure 1). Elsewhere on the YAC, on the left arm there is a LEU2 marker gene, which allows for maintenance of the YAC, a yeast origin of replication (ARS1), and a centromere (CEN4). Finally, an ade3–2p allele can be used to screen for cells that have obtained two YACs (which occurs occasionally by nondisjunction) in strains with an ade3 background, but it is not necessary to use this feature for most applications.
For the original (CAG)n-URA3 YAC, the CAG repeat tract is integrated such that the CAG repeat is on the lagging strand template, which has been shown to be the orientation less prone to repeat tract contractions (14). For the CAG repeat, it has been established that there is a length dependent increase in breakage as measured by 5-FOA resistance (7) (Figure 1D). This YAC system can also be used to study CAG repeat stability using similar conditions used for measuring breakage (3). These instability assays revealed a length dependent increase in contractions and expansions, though contractions occur with greater frequency than expansions in a replicating yeast population (7). The CAG instability frequency measured on this YAC is similar to that measured at the chromosome II LYS2 locus for the same orientation with respect to replication (14, 15), though it is known that factors such as distance from an origin, transcription level, and chromatin structure can alter instability frequencies (see (1) for review). A useful feature of both fragility assays described herein is that both fragility and instability can be monitored under the same conditions (genomic location, strain background, transcriptional status, etc.).
1.2. The direct duplication recombination assay
The second system for measuring repeat fragility is a direct duplication recombination assay (DDRA) (Method 4). In the original version of this assay, a potential fragile sequence was integrated adjacent to a URA3 reporter and between a full length LYS2 gene and a duplication of the 3’ end of LYS2 on chromosome II (16, 17) (Figure 3A). If breaks occur within the integrated sequence, resection occurs and repair results from single strand annealing of the duplicated regions of the LYS2 gene. This results in the loss of the URA3 gene and resistance to the drug 5-FOA. In this system, it was noted that there is a length dependent increase in 5-FOA resistance which was due to breaks within or very close to an integrated CAG repeat tract (16), and expanded CGG/CCG repeats also induced length-dependent fragility (17). This system is ideal for understanding the role that recombination plays in the repair of breaks that occur within a repeat tract. Also, as it is located in the middle of a natural yeast chromosome, the replication program is known, and the location is far from elements such as the centromere and telomere that could influence chromatin structure or nuclear positioning. A modification of the DDRA utilizes the same principle, but with a duplication of the ADE2 gene, designed such that the starting strain is Ade- and recombination products will become Ade+ 5-FOAR (Brian A. Lenzmeier and Virginia A. Zakian; (18)) (Figure 3B). This modification allows for selection of recombinants, eliminating other events that can generate 5-FOAR, and the cassette can be integrated anywhere in the genome. It has been used to study increased recombination rates induced by G4 motifs (18) and AT repeats (Kaushal, Freudenreich et al, in revision). It is important to note that many genes that are involved in replication or repair are also required for efficient recombination, which makes it more difficult to determine whether a gene is required to prevent fragility or facilitate healing in this system, as compared to the YAC end loss system.
Figure 3: Schematic of the direct duplication recombination assay (DDRA).
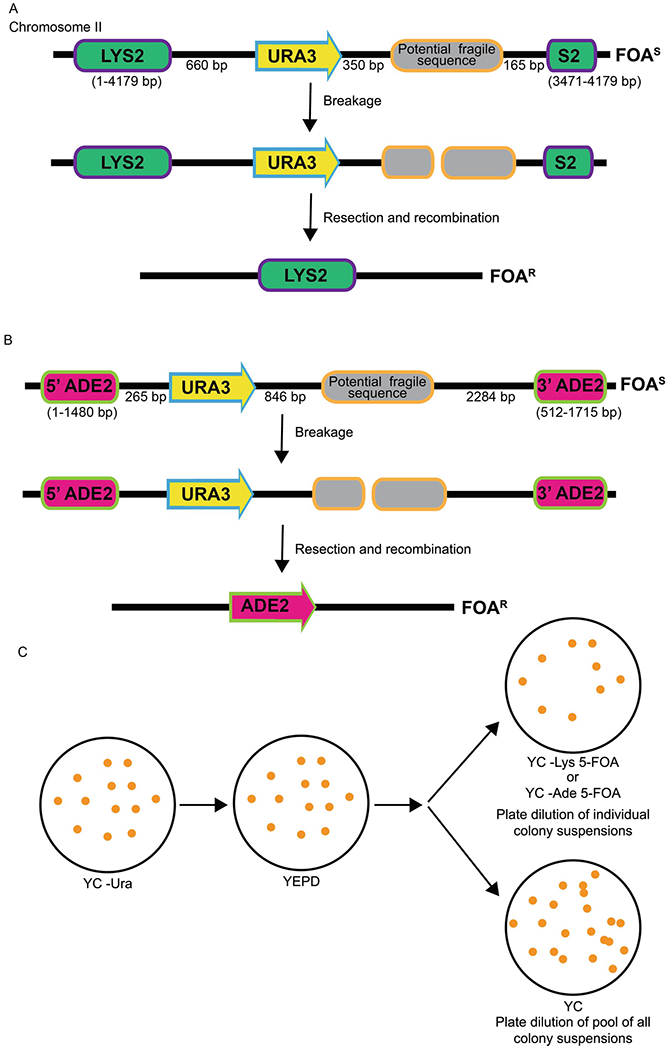
A. In this chromosomal system, a potential fragile sequence and the URA3 gene are integrated between a direct duplication of 708 bp of the 3’ end of the LYS2 gene; these cells are Lys+ and 5-FOAS. Breaks that occur within the fragile sequence are subject to resection and recombination, which renders cells Lys+ and 5-FOAR. B. In the ADE2 DDRA, a potential fragile sequence and the URA3 gene are integrated between 5’ and 3’ portions of the ADE2 gene, rendering cells Ade- and 5-FOAS. Breaks that occur within the repeat are subject to resection and repair via single strand annealing between the 968 bp duplicated region of ADE2, thus rendering cells Ade+ and 5-FOAR. C. Schematic of DDRA protocol. Cells are initially plated on YC –Ura media and then plated for single colonies on YEPD (or other nonselective media) to allow loss of URA3. Ten individual colonies are selected and each resuspended in water. A portion of each colony suspension is then plated onto 10 separate 5-FOA plates (–Lys or –Ade, depending on the assay) and 100 ul of each colony suspension is pooled, diluted and plated for single colonies onto YC plates for a total cell count.
Both assays can be modified to contain different potential fragile sequences or to modify other parameters, such as transcription level. For the YAC assay, we have made modifications to reduce transcription by flanking the CAG tract with transcriptional terminators (19) or increasing transcription by adding the inducible GAL1 promoter adjacent to the CAG tract (20) (Figure 2C). We have also added a second marker distal to the CAG tract, either ADE2 or HIS3, which allows end loss events to be verified by loss of the second marker, rather than PCR (Figure 2). For example, addition of the ADE2 marker allows identification of YAC end loss events (in an ade2 strain background) by observation of red color development of 5-FOAR colonies (20). Note that in the original (CAG)n-URA3 YAC, the lack of an efficient transcription terminator in the adjacent URA3 gene leads to read-through transcription through the CAG tract, and this is significantly reduced by introduction of either a transcription terminator or the ADE2 gene between URA3 and the CAG tract, which can effect repeat fragility and instability levels in some backgrounds (19, 20).
Taken together, both assays, the YAC end loss system and the chromosomal direct duplication recombination assay, have provided novel insight into the mechanisms of fragility and instability of repetitive DNA sequences and can be utilized to understand the breakage and repair of any sequence of interest.
2. MATERIALS
2.1. Colony PCR to determine CAG repeat tract length
Taq polymerase
10X Taq polymerase buffer
20 mM Magnesium sulfate (MgSO4)
5X CG buffer (see Note 1)
10 mM dNTPs
P20 or P200 micropipette tips
2% Metaphor agarose or other high resolution gel system
Electrophoretic gel box, running buffer (1X TBE or TAE), and power supply
10 mg/mL ethidium bromide or other DNA detection chemical
Microtubes
Table 1: Primers that are used for PCR amplification the CAG repeat tract.
These primers work for the CAG tract cloned into the YAC and DDRA constructs described herein. The forward primers anneal to sequence from the human DM1 sequence that flanks the CAG tract. The reverse primers anneal to sequence in or near the MCS that the repeat was cloned into. Primers can be used in a variety of combinations depending on the construct and repeat length.
| Direction | Primer Name | Sequence | Distance to repeat tract |
|---|---|---|---|
| Forward | CTGrev2 | 5’ CCC AGG CCT CCA GTT TGC 3’ | 74bp |
| NewCAGFor | 5’ CCT CAG CCT GGC CGA AAG AAA GAA A 3’ | 42 bp | |
| Reverse | T720 | 5’ TAA TAC GAC TCA CTA TAG GG 3’ | 80 bp |
| T720b | 5’ GAA TTC GAG CTC CAC CGC GG 3’ | 60bp | |
| NewCAGrev | 5’ CAG TCA CGA CGT TGT AAA ACG ACG G 3’ | 117bp |
2.2. Determination of chromosome fragility by a YAC end loss assay.
Starting colonies that have been checked for repeat tract length by colony PCR (see Method 3.1 for CAG repeats)
Spectrophotomer and cuvettes for measuring optical density (OD)
5 mL glass test tubes with caps
P20 or P200 micropipette tips
Roller drum or shaker at 30°C
YC –Leu liquid medium; see ref (21) for yeast media recipes; YC stands for Yeast Complete media, missing amino acids noted as “–”.
YC –Leu –Ura plates
YC –Leu plates
YC –Leu +5-FOA plates (standard 5-FOA concentration is 1.0 g/L)
Sterile glass beads or sterile spreaders
2.3: Modification of the YAC
YIP5-based plasmid with desired components added (e.g. potential fragile sequence, markers, promoters, terminators etc.)
Luria broth (LB) + 50 µg/ml ampicillin liquid media
LB + 50 µg/ml ampicillin plates
Restriction enzyme: AatII or XcmI
Water bath at 37°C
- Transformation reagents:
- 1M lithium acetate pH 7.5
- 100mM Tris-HCl, pH 7.5
- 50% polyethylene glycol (PEG) 3350; (see (22) for solution recipes)
- High molecular weight single-stranded DNA (e.g. deoxyribonucleic acid sodium salt type III from salmon testes), denatured at 100°C and cooled on ice
- Water bath at 42°C
YC –Ura –Leu yeast medium (other amino acids dropped out according to the desired marker selection; see ref (21) for recipes)
Toothpicks or sterile loops
2.4: Direct Duplication Recombination Assay (DDRA)
YC –Ura plates (see ref (21) for yeast media recipes)
YEPD plates (rich media containing yeast extract, peptone, dextrose)
YC –Lys +5-FOA plates (LYS2 assay) or YC –Ade 5-FOA plates (ADE2 assay)
YC plates
3. Methods
3.1: Colony PCR to determine CAG repeat tract length
Plate or streak for single colonies on YC –Ura, which will select against breakage events, from a master patch (it may be appropriate to drop out additional amino acids according to the fragility assay markers) (see Note 3).
Select individual colonies and clearly mark which PCR reactions will correspond to which colony.
- Combine a bulk PCR reaction mix on ice and aliquot 12.5 μl per PCR tube. The volume can be adjusted if desired. The 1X PCR reaction is:
Sterile diH2O 5.8 μl 10X buffer (w/o MgCl2) : 1.25 μl 20 mM MgSO4: 1.25 μl 6.25 pmol primer 1: 0.625 μl (of 10pmol/μl stock) 6.25 pmol primer 2: 0.625 μl (of 10pmol/μl stock) 10mM dNTPs : 0.25 μl Mix thoroughly, then add, Taq polymerase : 0.2 μl 5X CG-Rich buffer: 2.5 μl (see Note 4) Total reaction mix is 12.5 μl -
PCR cycle profile:
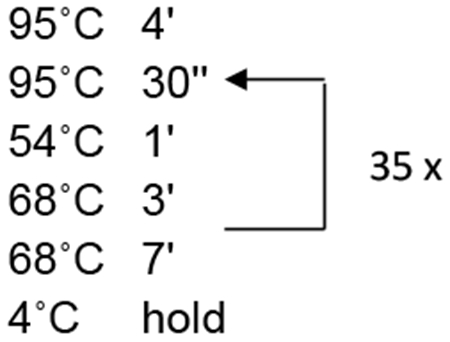
Once the PCR protocol is complete, add an appropriate amount of loading dye to the 12.5 μl reaction and run out the entire PCR reaction on a 2% Metaphor gel or other gel sizing system (see Notes 6–7). Load markers on both ends of the gel; a third marker in a middle lane is helpful for gel systems that do not run evenly across the gel box (see Notes 8–9). Run the gel in 1X TAE at 70 V for 90 min.
Post-stain with ethidium bromide or other DNA detection dye for 30 min. Transfer gel tray to diH20 and photograph the gel using a UV transilluminator (see Note 10).
3.2: Determination of chromosomal fragility by a YAC end loss assay.
Plate for single colonies on YC –Leu –Ura, which will select against breakage events, from a master patch.
If the potential fragile sequence is an unstable repeat able to be sized via PCR, do colony PCR on 10-20 colonies to check for the appropriate repeat tract length (see Method 1; Note 9).
- Inoculate colonies of correct tract length for the fluctuation assay. Colonies should be as fresh as possible; ideally less than two weeks old. Select starting colonies that have the appropriate size of repeat tract length with no visible contractions or expansions. Two methods of inoculation may be used, but whenever possible use preferentially method 1.
-
Inoculation method 1: 10 colony protocol (preferred):For this methodology there will be 10 unique colonies that are inoculated into 10 separate cultures.
- Pipet 1 mL of YC –Leu into each of ten 5 mL test tubes.
- Inoculate 1 to 3 tubes with a small number of cells and take the OD600 (using the entire 1mL culture; these are referred to as the “OD600 cultures”). The starting OD600 should be ~0.02-0.06. Once the OD600 has been taken, return the 1 mL culture to the initial test tube. Always work under sterile conditions (see Note 11)
- Inoculate the remaining tubes with approximately the same amount of cells by eye, so that they will also have a starting OD600 of ~0.02-0.06 (see Note 12)
-
Inoculation method 2: 1 colony protocol (see Note 13):For this methodology there will be 1 colony that is divided into 10 cultures.
- Suspend one entire colony with the correct tract length in 1.3 mL of YC-Leu media.
- Take 100 μl of the resuspended mix and inoculate into 0.9 mL of YC-Leu media in a 5 mL test tube. Repeat for each of the 10 cultures.
- Take 1 mL of one of the cultures out of the test tube and measure the OD600. The OD600 should be between ~0.02-0.06. Return the 1 mL culture to the initial test tube
-
Allow the cultures to grow with constant agitation or in a spinning roller drum for 6-7 doublings at 30˚C. This step allows the breakage and healing to occur (see Notes 12,14–15).
In order to determine the number of doublings, take 100 μl of the OD600 culture(s) and resuspend into 900 μl of YC –Leu media and measure the final OD. Multiply this reading by 10. If the ideal starting OD600 was ~0.02, the appropriate final OD600 range is 1.25-2.5 (see Notes 11, 12).
Spread 100 μl from each of the 10 tubes onto 10 YC –Leu 5-FOA plates using a consistent number of sterile glass beads or a sterile glass spreader. The amount plated can be adjusted up or down for strains with a higher or lower rate of 5-FOAR; just remember to adjust the number of colonies counted to the number of mutants per 100 μl in the rate calculations (see Notes 16–17).
To determine the viable total cell count, combine 100 μl from each of the 10 undiluted cell cultures and mix together. Serially dilute the mixed cultures in sterile water (usually to 10−4 and 10−5 ) and plate 100 μl on of each dilution onto YC –Leu plates. Total cell count plates should be plated in duplicate.
Incubate the plates at 30˚C for 4 days
Count colonies on the YC –Leu 5-FOA plates.
Order the plate counts from low to high to determine the median number of colonies for each set (i.e., each 10 plate assay). If necessary, adjust the number to obtain the median number of mutants in 100 μl. Average the number of colonies on the 2 “total cell count” plates to determine the number of viable cells in 100 μl. Determine the rate of 5-FOAR by using a fluctuation rate calculator such as FALCOR (http://www.keshavsingh.org/protocols/FALCOR.html) (23) or FluCalc ((24); http://flucalc.ase.tufts.edu/) using either the Maximum Likelihood Method (preferred, if the spread is not too great; this method uses the number of mutants obtained on all selective plates) or the Method of the Median (using the median number of mutants in 100 μl, as described above). See (25) for an excellent discussion of calculating fluctuation rates (see Notes 18–22).
The assay should be repeated at least three times per strain, and standard error and confidence intervals calculated.
3.3: Modification of the YAC
- Using pHZ1, a YIP5 derivitive which contains the URA3 gene and the (G4T4)13 telomeric seed sequence, clone in genetic elements and fragile sequences as desired. The MCS is between the G4T4 and URA3 marker gene. If a new fragile element is being added, it should be added between the G4T4 and URA3 marker gene (Figure 2A).
- Once all new genetic elements have been added and confirmed, linearize the plasmid such that the URA3 marker and G4T4 are on either end of the linear fragment. Previously we have used AatII or XcmI to linearize the modified pHZ1 plasmid.
Using a standard S. cerevisiae transformation protocol (26), transform the fragment into cells that contain the YAC.
Selection for proper transformants will depend on the nature of the DNA elements that are being inserted. If non-selectable elements are being added (such as a promoter or terminator sequence), transformation will need to occur in a strain that contains a URA3 point mutation so that transformation selection can occur on YC –Ura. CRISPR-Cas9 gene editing strategies can also be employed to make marker-less modifcations to the YAC.
Confirmation of integration into the YAC can be done via PCR between a new element added (e.g. ADE2 in the example) and an element not included in the integrating fragment, such as the Drosophila white gene. YAC structure can also be confirmed via Southern blot using a lambda probe to the YAC and digest with BstEII (7), or digests and probes specific to the modified area.
3.4: Direct Duplication Recombination Assay: A recombination based method to assess chromosome breakage.
Patch strain to test onto a YC –Ura plate and allow to grow for 2 days at 30°C.
Plate for single colonies on YEPD at 30°C for 3 days such that there are 50 ± 10 colonies per plate (this step allows recombination events to occur).
Randomly select 10 colonies and resuspend half of each colony in 300 μl diH20 (see Note 23).
Plate a portion of the suspension on a 5-FOA plate with the goal being to get ~100 colonies per plate to count after 4-5 days of growth at 30°C. For the LYS2 assay (Figure 3A), this should be a YC –Lys +5-FOA plate (Lysine dropout optional); For the ADE2 assay, (Figure 3B), this should be a YC –Ade +5-FOA plate (see Notes 24–25).
To determine the viable cell count combine 100 μL of each of the 10 colony suspensions. Plate 100 μl of a 10−4 dilution onto 2 YC plates. Plate a dilution that will yield ~30-300 colonies single colonies per YC plate. Total cell count plates should be plated in duplicate (see Note 26).
Count the number of colonies on the 5-FOA plates. To obtain the number of mutants and the total number of cells per 100 μl, multiply the number of colonies growing on 5-FOA and the average of the 2 total cell count plates by the dilution factors used. To determine the rate of recombination, use the method of maximum likelihood or the method of the median and a fluctuation analysis calculator (e.g. FALCOR (23) or FluCalc (24); see Method 3.2).
The assay should be repeated at least three times per strain, and standard error and confidence intervals calculated.
Acknowledgements
Work in our laboratory is currently supported by grants from the National Institute of Health (GM105473, GM122880) and National Science Foundation (MCB 1817499). We would like to thank past and present members of the C. H. Freudenreich and V.A. Zakian labs for their contributions to the development of these assays.
4. Notes
A 5X GC rich buffer may aid in amplification across the repetitive tract. One 5X GC rich buffer that has been used in amplification of the CAG repeat tract is Phusion GC Buffer (New England Biolabs, B0519S)
In order to obtain robust tract lengths PCR, primers should be designed such that there is a region of non-repetitve flanking sequence included on both sides of the repeat. Generally, we recommend between 40-80 bp of non-repetitve sequence be included on each side of the repeat. For examples of primers that span the CAG repeat for both the (CAG)n-URA3 YAC and the (CAG)n LYS2 DDRA constructs see Table 1.
This protocol is written for a CAG tract on the CF1 YAC, but can be adapted to other locations or repeats.
Taq polymerase & CG buffer should be added last to the master mix after everything else and mixed gently.
The amount of cells added to the PCR reaction should be no larger than the tip of the p200 or p20 pipette tip. Too few cells will result in a very faint PCR product, too many will result in a background smear in the lane.
The 2% Metaphor agarose gels should be made according to the manufacturers instructions.
Instead of using 2% Metaphor gels for separation of fragments, PCR reactions can be run on a denaturing PAGE gel or other high resolution gel system for size analysis, with appropriate DNA detection. Alternatively an appropriate concentration of DNA-grade agarose may be used.
The ideal molecular weight marker used should have bands that change in size every 100 bp or less. Adding an additional marker in the middle of the gel helps with sizing the CAG repeat.
If the PCR is of poor quality, then expansions may have not amplified well and could be underestimated, therefore only use good quality PCR reactions to decide on starting colonies. A 2% Metaphor gel can resolve fragments that differ by 2%, i.e. 8 bp for a fragment size of 400 bp or about 3 (CAG) repeats added or subtracted for a (CAG)85 repeat tract.
Ethidium bromide can be added directly to Metaphor gel if preferred. The ideal concentration is 0.5 μg/mL (e.g. 15 μl 10 mg/mL stock in 300 mL 1 X TAE).
There is a risk of contamination during the process of taking the OD600. If this is an issue, extra “OD600 cultures” can be started that are only used for taking the OD600 throughout the experiment and discarded before plating.
The method is designed to test for fragility occuring during log phase growth. Ensuring cells grow through several divisions in logarithmic phase is important as this growth step allows the breakage and healing to occur. The OD600 range could be altered according to the purpose of the experiment.
The 1 colony method is less preferred, but can be used when finding 10 starting colonies of the same repeat tract length is technically difficult.
For mutant strains that affect growth, discard any cultures that do not grow or grow especially poorly, and do not include them in the mix for the total cell count. For such strains, more than 10 cultures can be started to allow for this.
For wildtype, 6-7 divisions will take approximately ~16-18 hrs. For strains with poor division potential, the total number of divisions can be altered to fewer divisions, e.g. 3-5 divisions. Wildtype strains done as a comparison should be grown for the same number of divisions as mutant strains.
The amount of cells plated on 5-FOA can be adjusted for strains or YACs with higher or lower rates of breakage. For example, less than 100 μl may need to be plated for mutants showing increased end loss of the (CAG)n-URA3 YAC, and 1 mL should be plated for the (CAG)n-ADE2-URA3 YAC.
It is best to adjust the amount of cells plated on 5-FOA such that the median number of colonies is between 50-250.
Make sure you enter the number of mutants and total cells for the same volume – usually per 100 μl (per 1 mL can be used for very low rates).
For experiments with zeros or a large spread between low and high numbers of mutants (happens with lower rate assays), use the Method of the Median to calculate the rate.
Certain mutants could alter URA3 gene function or 5-FOAR by increasing the mutation rate of URA3, altering URA3 expression, or affecting the toxicity of 5-FOA, resulting in increased 5-FOAR that is not due to chromosome breakage. To confirm that there is loss of the right arm of the YAC, PCR amplify the URA3 gene (or a portion of it) from 1 colony off of each YC –Leu +5-FOA plate (10 colonies total) to confirm there is loss of the right arm of the YAC. This should be done for 3 assays (a total of 30 independent colonies checked). The expected end loss frequency for wildtype is 90-100%. Make sure to also PCR amplify from your starting strain as a positive control.
Verify the structure of the YAC in 20 independent 5-FOAR colonies (plated from independent cultures) by Southern blot, using a lambda probe to the YAC and digest with BstEII digest (see (7)).
The fragility assay can be combined with an instability assay by analyzing the repeat tract length of the daughter colonies plated for the total cell count (3). Even though these plates do not select for the presence of the repeat (e.g. the YC –Leu plates for the YAC assay), usually the rate of breakage is low enough that most colonies tested will have retained a repeat tract. One disadvantage to this method is that the starting parent colony and the daughter colonies are no longer correlated, reducing the ability to detect jackpot events.
Establish a standard size of colony to use to allow equal generations of growth between colonies and strains.
Generally, for rates in the 10−5 range, approximately 1/9th of a colony is plated, though a pilot experiment can determine the best amount to plate. Keep in mind that the more fragile the sequence, the less should be plated.
For plating onto 5-FOA, use about 10−2 cells for strains with low recombination rates, 10−3 cells for strains with high recombination rates.
The dilution plated for the viable cell count may depend on the size of the colonies that were picked for the assay. For initial assays it’s best to plate a series of dilutions, such as 10−3, 10−4 and 10-5.
References
- 1.Usdin K, House NC, Freudenreich CH. (2015) Repeat instability during DNA repair: Insights from model systems. Critical Reviews in Biochemistry and Molecular Biology 50(2):142–67. doi: 10.3109/10409238.2014.999192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMurray CT. (2010) Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11(11):786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polleys EJ, Freudenreich CH. (2018) Methods to Study Repeat Fragility and Instability in Saccharomyces cerevisiae. Methods in Molecular Biology 1672:403–19. doi: 10.1007/978-1-4939-7306-4_28. [DOI] [PubMed] [Google Scholar]
- 4.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, Narayanan V, Lobachev KS, Mirkin SM. (2009) Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Molecular Cell 35(1):82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon MJ, Bhattacharyya S, Lahue RS. (2004) Genetic assays for triplet repeat instability in yeast. Methods in Molecular Biology 277:29–45. doi: 10.1385/1-59259-804-8:029. [DOI] [PubMed] [Google Scholar]
- 6.Schulz VP, Zakian VA. (1994) The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell 76(1):145–55. [DOI] [PubMed] [Google Scholar]
- 7.Callahan JL, Andrews KJ, Zakian VA, Freudenreich CH. (2003) Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Molecular and Cellular Biology 23(21):7849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, Richard GF. (2009) SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol 16(2):159–67. doi: nsmb.1544 [pii] 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang H, Freudenreich CH. (2007) An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Molecular Cell 27(3):367–79. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherng N, Shishkin AA, Schlager LI, Tuck RH, Sloan L, Matera R, Sarkar PS, Ashizawa T, Freudenreich CH, Mirkin SM. (2011) Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proceedings of the National Academy of Sciences of the United States of America 108(7):2843–8. doi: 10.1073/pnas.1009409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu S, Wang G, Bacolla A, Zhao J, Spitser S, Vasquez KM. (2015) Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Reports doi: 10.1016/j.celrep.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pluta AF, Zakian VA. (1989) Recombination occurs during telomere formation in yeast. Nature 337(6206):429–33. doi: 10.1038/337429a0. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, Kolodner RD. (1999) Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet 23(1):81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 14.Freudenreich CH, Stavenhagen JB, Zakian VA. (1997) Stability of a CTG/CAG trinucleotide repeat in yeast is dependent on its orientation in the genome. Mol Cell Biol 17(4):2090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su XA, Dion V, Gasser SM, Freudenreich CH. (2015) Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes & Development 29(10):1006–17. doi: 10.1101/gad.256404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freudenreich CH, Kantrow SM, Zakian VA. (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279(5352):853–6. [DOI] [PubMed] [Google Scholar]
- 17.Balakumaran BS, Freudenreich CH, Zakian VA. (2000) CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum Mol Genet 9(1):93–100. [DOI] [PubMed] [Google Scholar]
- 18.Paeschke K, Capra JA, Zakian VA. (2011) DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145(5):678–91. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su XA, Freudenreich CH. (2017) Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 114(40):E8392–E401. doi: 10.1073/pnas.1711283114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch MR, House NCM, Cosetta CM, Jong RM, Salomon CG, Joyce CE, Philips EA, Su XA, Freudenreich CH. (2018) The Chromatin Remodeler Isw1 Prevents CAG Repeat Expansions During Transcription in Saccharomyces cerevisiae. Genetics 208(3):963–76. doi: 10.1534/genetics.117.300529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunham MJ, Dunham MJ, Gartenberg MR, Brown GW. Methods in yeast genetics and genomics : a Cold Spring Harbor Laboratory course manual / Maitreya J. Dunham, University of Washington, Marc R. Gartenberg, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Grant W. Brown, University of Toronto. 2015 edition / ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2015. xvii, 233 pages p. [Google Scholar]
- 22.Amberg DC, Burke D, Strathern JN, Burke D, Cold Spring Harbor Laboratory. Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual. 2005 ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2005. xvii, 230 p. p. [Google Scholar]
- 23.Hall BM, Ma CX, Liang P, Singh KK. (2009) Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25(12):1564–5. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radchenko EA, McGinty RJ, Aksenova AY, Neil AJ, Mirkin SM. (2018) Quantitative Analysis of the Rates for Repeat-Mediated Genome Instability in a Yeast Experimental System. Methods in Molecular Biology 1672:421–38. doi: 10.1007/978-1-4939-7306-4_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosche WA, Foster PL. (2000) Determining mutation rates in bacterial populations. Methods 20(1):4–17. doi: 10.1006/meth.1999.0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amberg DC, Burke DJ, Strathern JN. (2006) High-efficiency transformation of yeast. CSH protocols 2006(1). doi: 10.1101/pdb.prot4145. [DOI] [PubMed] [Google Scholar]
- 27.Sundararajan R, Gellon L, Zunder RM, Freudenreich CH. (2010) Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184(1):65–77. doi: 10.1534/genetics.109.111039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.House NC, Yang JH, Walsh SC, Moy JM, Freudenreich CH. (2014) NuA4 Initiates Dynamic Histone H4 Acetylation to Promote High-Fidelity Sister Chromatid Recombination at Postreplication Gaps. Molecular Cell 55(6):818–28. doi: 10.1016/j.molcel.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen JH, Viterbo D, Anand RP, Verra L, Sloan L, Richard GF, Freudenreich CH. (2017) Differential requirement of Srs2 helicase and Rad51 displacement activities in replication of hairpin-forming CAG/CTG repeats. Nucleic Acids Res. doi: 10.1093/nar/gkx088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang JH, Freudenreich CH. (2010) The Rtt109 histone acetyltransferase facilitates error-free replication to prevent CAG/CTG repeat contractions. DNA Repair 9(4):414–20. doi: 10.1016/j.dnarep.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]