

SUMMARY
Many eukaryotic cells distribute their intracellular components asymmetrically through regulated active transport driven by molecular motors along microtubule tracks. While intrinsic and extrinsic regulation of motor activity exists, what governs the overall distribution of activated motor-cargo complexes within cells remains unclear. Here, we utilize in vitro reconstitution of purified motor proteins and non-enzymatic microtubule-associated proteins (MAPs) to demonstrate that MAPs exhibit distinct influences on the motility of the three main classes of transport motors: kinesin-1, kinesin-3, and cytoplasmic dynein. Further, we dissect how combinations of MAPs affect motors, and unveil MAP9 as a positive modulator of kinesin-3 motility. From these data, we propose a general “MAP code” that has the capacity to strongly bias directed movement along microtubules and helps elucidate the intricate intracellular sorting observed in highly polarized cells such as neurons.
eTOC Blurb
Monroy et al. analyze the effects of six microtubule-associated proteins (MAPs) on three transport motors to understand how MAPs contribute to polarized transport. They find that MAPs differentially gate motor access to the microtubule, providing a template for how motor activity is spatially regulated within cells.
INTRODUCTION
Within cells, nothing works in isolation. Therefore, in order to dissect the complexity of intracellular processes, it is essential to study the behaviors of molecules both individually and collectively. One intricate process is the polarized active transport along microtubules that is required within neurons for the establishment and maintenance of distinct dendritic and axonal compartments (Kapitein and Hoogenraad, 2011; Stiess and Bradke, 2010). This transport system is driven by kinesin motors and cytoplasmic dynein, which travel towards the microtubule plus and minus ends, respectively (Paschal and Vallee, 1987; Vale et al., 1985). Of the large kinesin family, the kinesin-1, -2, and -3 classes are thought to act as the predominant long-distance transport motors, while other kinesins serve more specialized cellular roles (Hirokawa et al., 2010; Lipka et al., 2016). In neurons, kinesin-3 and dynein drive various cargoes within both axons and dendrites, while kinesin-1 carries cargoes into the axon, but is largely excluded from the dendrites (Ayloo et al., 2017; Jenkins et al., 2012; Kapitein et al., 2010; Karasmanis et al., 2018; Lipka et al., 2016; Maday et al., 2014; Tas et al., 2017). How cells compartmentalize molecular motor activity remains unclear.
Post-translational modifications of tubulin have been proposed to act as a “tubulin-code” that can be read out by activated motor proteins to direct their movement to specific cellular compartments (Gadadhar et al., 2017; Janke, 2014; Park and Roll-Mecak, 2018; Yu et al., 2015). Although detyrosination decreases the motility of the activated dynein-dynactin-BicD (DDB) complex four-fold (McKenney et al., 2016), the reported biophysical effects of certain tubulin modifications on kinesin-1 are relatively modest. Acetylation increases kinesin-1 velocity 1.2-fold (Reed et al., 2006) and detyrosination decreases kinesin-1 processivity 1.4-fold, with no effect on velocity (Sirajuddin et al., 2014), raising questions about how such effects could directly, substantially impact kinesin-1 transport in vivo. Within cells, transport motors must encounter a large variety of non-enzymatic microtubule-associated proteins (MAPs) that decorate the microtubule cytoskeleton (Ramkumar et al., 2018). Disruption of this bidirectional transport system due to mutations in motor complexes or MAPs leads to a wide range of neurodevelopmental and neurodegenerative disorders (Chevalier-Larsen and Holzbaur, 2006; Gleeson et al., 1998; Harms et al., 2012), highlighting the importance of the interplay between these classes of proteins.
Since the identification of “structural” MAPs that co-purified with polymerized brain tubulin (Borisy et al., 1975), MAPs have been described as stabilizers, nucleation-promoting factors, and bundlers of microtubules (Bechstedt and Brouhard, 2012; Bulinski and Borisy, 1979; Bulinski and Bossler, 1994; Chen et al., 1992; Moores et al., 2006; Wieczorek et al., 2015). However, recent work suggests that these MAPs may also function to direct motor transport (Ecklund et al., 2017; Lipka et al., 2016; Monroy et al., 2018; Tan et al., 2019). Perhaps the most well-studied MAP with regards to its effects on motors is the Alzheimer’s disease-associated MAP, tau, which was originally thought to be axon-specific, but can also be observed in mature dendrites (Figure S1) (Dotti et al., 1987; Tan et al., 2019). Tau inhibits the processive motility of kinesin-1 and kinesin-3 (Chaudhary et al., 2018; Dixit et al., 2008; McVicker et al., 2011; Monroy et al., 2018; Vershinin et al., 2007), but does not strongly impede the movement of activated dynein-dynactin complexes (Tan et al., 2019). These differential effects are due to a steric clash between tau and the relatively large kinesin motor domain, which does not exist for the smaller dynein microtubule-binding domain (Kellogg et al., 2018; Shigematsu et al., 2018; Tan et al., 2019). MAP2 is localized to dendrites and the axon initial segment and has been shown to inhibit kinesin-1 in vivo (Gumy et al., 2017). Based on the similarities in their microtubule-binding sites (Al-Bassam et al., 2002; Kellogg et al., 2018), it is likely that MAP2 affects kinesins and dynein akin to tau. MAP7 is important for a range of kinesin-1 functions in vivo (Barlan et al., 2013; Metivier et al., 2019; Metzger et al., 2012; Sung et al., 2008; Tymanskyj et al., 2018), and has been shown to directly recruit kinesin-1 to the microtubule lattice (Chaudhary et al., 2019; Hooikaas et al., 2019; Monroy et al., 2018). Kinesin-1 is most likely able to navigate the tau-rich axon in part due to the presence of MAP7, which displaces tau from the microtubule (Monroy et al., 2018). Interestingly, MAP7 inhibits kinesin-3, but does not substantially affect dynein motility (Monroy et al., 2018). DCX and its paralogue, doublecortin-like kinase-1 (DCLK1) robustly stimulate microtubule polymerization (Bechstedt and Brouhard, 2012; Fourniol et al., 2013; Patel et al., 2016), but are restricted to distal dendrites and axonal growth cones (Lipka et al., 2016; Liu et al., 2012; Tint et al., 2009), indicating they may have specific roles commensurate with their localization patterns. Both MAPs have been reported to interact with the kinesin-3 motor domain and promote kinesin-3 cargo transport within dendrites (Lipka et al., 2016; Liu et al., 2012). An understudied MAP is MAP9/ASAP, which plays a role in organizing the mitotic spindle in cultured cells (Saffin et al., 2005; Venoux et al., 2008), and is associated with cell degeneration and cancer (Forman et al., 2016; Rouquier et al., 2014). Although MAP9 is highly expressed in the vertebrate nervous system throughout development (Fontenille et al., 2014), its molecular functions remain unknown. Why there are distinct, yet overlapping localization patterns for these MAPs within neurons and how these MAPs may contribute, individually and collectively, to sorting motors into specified compartments remain outstanding questions.
Here, we present a comprehensive analysis of the effect of six MAPs on three classes of transport motors in an effort to elucidate a “MAP code” that could underlie polarized transport. We find that tau and MAP2C act as general inhibitors of kinesin-1 and kinesin-3, preventing these motors from accessing the lattice, while three MAPs that localize within dendrites, DCX, DCLK1, and MAP9 differentially gate access to the microtubule by inhibiting kinesin-1, but not kinesin-3, providing a molecular system by which these motors are spatially regulated in neurons. We dissect the mechanism by which kinesin-3 is able to progress through these MAPs, highlighting a key role for MAP9 in specifically promoting kinesin-3 translocation. Furthermore, MAP9 is the only neuronal MAP examined thus far that substantially inhibits the processive DDB complex. Overall, our study provides general mechanistic principles for how MAPs help to orchestrate the distribution of specific motors within the crowded intracellular environment by gating access to the microtubule lattice.
RESULTS
Compartmentally distinct MAPs differentially affect kinesin-1 and kinesin-3
In order to understand how motors are differentially directed into dendritic or axonal compartments, we first sought to determine the localization patterns of six MAPs within neurons at the same developmental time points. We performed immunocytochemistry on DIV4, DIV7, and DIV14 primary mouse hippocampal cultures with antibodies against tau, MAP2, MAP7, DCX, DCLK1, and MAP9 (Figure S1). We found that three of these MAPs, tau, MAP7, and MAP9, localized throughout both dendrites and axons, while the other three, MAP2, DCLK1 and DCX, were predominantly restricted to dendrites, consistent with prior localization studies (Figure S1–S2) (Dotti et al., 1987; Lipka et al., 2016; Monroy et al., 2018; Tan et al., 2019; Tint et al., 2009).
Because of the previously reported spatial distributions of the major transport kinesin family motors (Ayloo et al., 2017; Jenkins et al., 2012; Kapitein et al., 2010; Karasmanis et al., 2018; Lipka et al., 2016; Maday et al., 2014; Tas et al., 2017), we set out to investigate how compartmentally distinct MAPs may affect kinesin-1 and kinesin-3 motility along microtubules in an effort to understand how MAPs, in general, could contribute to polarized motor transport in vivo. Towards this goal, we utilized a molecular reconstitution system of purified proteins to probe for direct effects of MAPs on motor motility. Using multi-color total internal reflection fluorescence microscopy (TIRF-M), we imaged the progression of purified, fluorescently labeled truncated motors, kinesin-1 (KIF5B1-560, hereafter referred to as KIF5B in the figures) and kinesin-3 (KIF1A1-393, hereafter referred to as KIF1A in the figures) in the absence or presence of six fluorescently labeled full-length MAPs (Figures 1–2 and S3A–B). Strikingly, we observed that other than MAP7, which increased kinesin-1 landing rate 25-fold, as previously reported (Monroy et al., 2018), all other MAPs significantly decreased the landing rate of kinesin-1 on the microtubule lattice, with the greatest effect being a 15-fold reduction by MAP2C (Figure 1A–B, S4A). This inhibition was consistent even at a 10-fold higher concentration of kinesin-1 (Figure 1B). It was especially surprising that DCX and DCLK1 both inhibited kinesin-1, considering they do not share overlapping binding sites with the kinesin-1 motor domain (Fourniol et al., 2013), suggesting this effect may be due to steric interference away from the surface of the microtubule. With the exception of MAP7, all other MAPs present in the dendrites blocked kinesin-1 from landing on the microtubule, suggesting that the presence of MAP7 may not be sufficient to facilitate kinesin-1 transport in all cellular compartments. To test this idea, we asked if MAP7 could support kinesin-1 motility in the presence of an inhibitory MAP (iMAP). Both DCX and MAP9 bound simultaneously with MAP7 on microtubules, suggesting their binding sites do not overlap (Figure 1C). In the presence of either of these iMAPs, we found that although kinesin-1 was recruited to the microtubule by MAP7, its movement along the lattice was still largely impeded (Figure 1B–D). Thus, diverse iMAPs have dominant effects on kinesin-1 movement even after the motor has been recruited to the microtubule surface by MAP7.
Figure 1. Kinesin-1 is inhibited from landing and progressing on the microtubule lattice by tau, MAP2C, DCX, DCLK1, and MAP9.
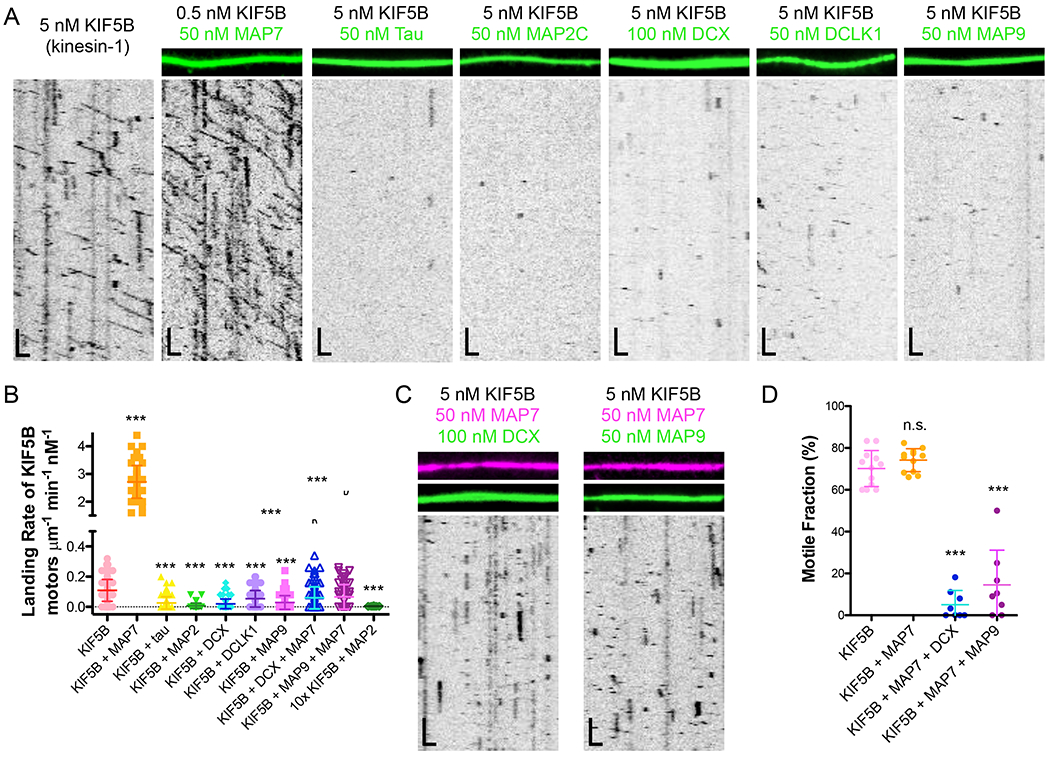
(A) TIRF-M images and kymographs of KIF5B-mScarlet (kinesin-1) at indicated concentrations + 1 mM ATP in the absence and presence of 50 nM sfGFP-MAP7, 50 nM sfGFP-tau, 50 nM sfGFP-MAP2C, 100 nM DCX-sfGFP, 50 nM sfGFP-DCLK1, or 50 nM sfGFP-MAP9. Scale bars: 1 μm (x), 10 sec (y). (B) Quantification of the landing rates of KIF5B-mScarlet + 1 mM ATP in the absence and presence of each MAP or MAP combination (means ± s.d. in motors μm−1min−1nM−1 are: 0.11 ± 0.07 for KIF5B alone (n=134 kymographs from 3 independent trials), 2.72 ± 0.59 for KIF5B + MAP7 (n=83 kymographs from 2 independent trials), 0.03 ± 0.04 for KIF5B + tau (n=100 kymographs from 2 independent trials), 0.01 ± 0.02 for KIF5B + MAP2C (n=93 kymographs from 2 independent trials), 0.02 ± 0.03 for KIF5B + DCX (n=94 kymographs from 2 independent trials), 0.05 ± 0.05 for KIF5B + DCLK1 (n=92 kymographs from 2 independent trials), 0.03 ± 0.05 for KIF5B + MAP9 (n=96 kymographs from 2 independent trials), 0.06 ± 0.07 for KIF5B + MAP7 + DCX (n=114 kymographs from 2 independent trials), 0.07 ± 0.08 for KIF5B + MAP7 + MAP9 (n=70 kymographs from 2 independent trials), and 0.005 ± 0.001 for 10x the concentration of KIF5B (50 pM) + MAP2C (n=20 kymographs from 2 independent trials). All datapoints are plotted with lines indicating means ± s.d. P < 0.0001 (***) calculated by one-way ANOVA with Bonferroni correction. (C) TIRF-M images and kymographs of 5 nM KIF5B-mScarlet + 1 mM ATP in the presence of 50 nM BFP-MAP7 (pink) with 100 nM DCX-sfGFP (green), or 50 nM BFP-MAP7 (pink) with 50 nM sfGFP-MAP9 (green). Scale bars: 1 μm (x), 10 sec (y). (D) Quantification of the percentage of motile KIF5B motors in the absence (70.2 ± 8.6 %; n=169 molecules from 12 kymographs from 3 independent trials) or presence of 50nM MAP7 (76.2 ± 5.5; n=160 molecules from 12 kymographs from 2 independent trials), 50 nM MAP7 with 100 nM DCX (5.1 ± 6.8; n=160 molecules from 8 kymographs from 2 independent trials), or 50 nM MAP7 with 50 nM MAP9 (14.5 ± 16.6; n=111 molecules from 8 kymographs from 2 independent trials). P < 0.0001 (***) and P = 0.0540 (n.s.) calculated by one-way ANOVA with Bonferroni correction.
Figure 2. Kinesin-3 access to the microtubule lattice is differentially gated by MAP7, tau, MAP2C, DCX, DCLK1, and MAP9.
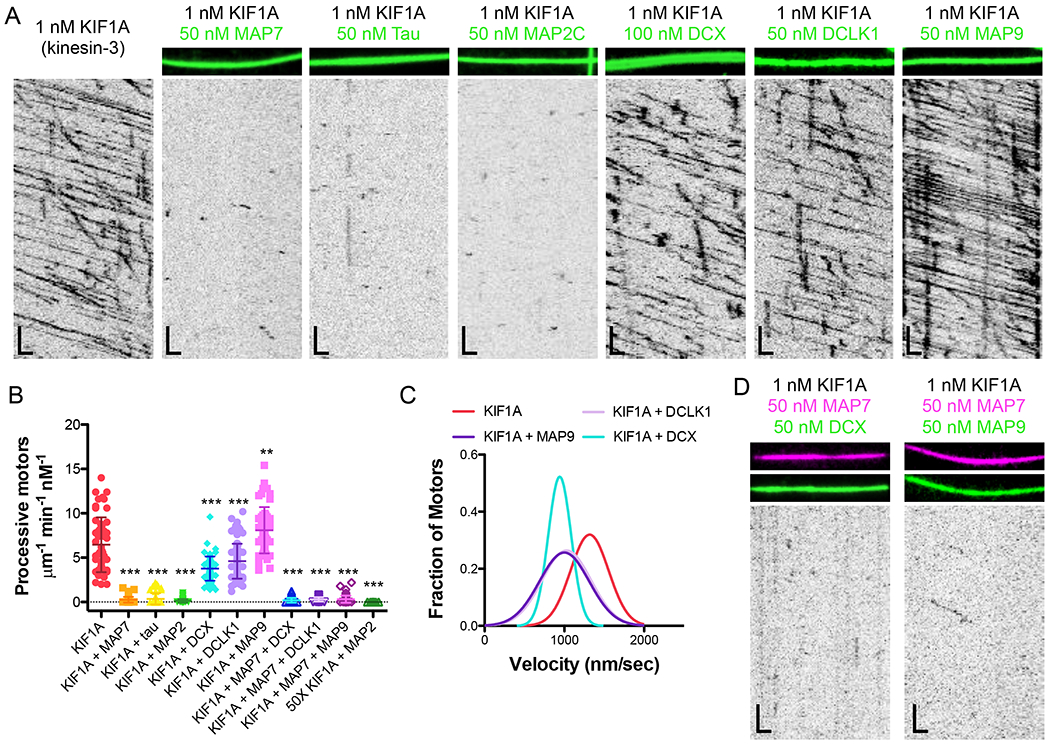
(A) TIRF-M images and kymographs of 1 nM KIF1A-mScarlet (kinesin-3) + 1 mM ATP in the absence and presence of 50 nM sfGFP-MAP7, 50 nM sfGFP-tau, 50 nM sfGFP-MAP2C, 100 nM DCX-sfGFP, 50 nM sfGFP-DCLK1, or 50 nM sfGFP-MAP9. Scale bars: 1 μm (x), 5 sec (y). (B) Quantification of the number of processive KIF1A-mScarlet motors + 1 mM ATP in the absence and presence of each MAP or MAP combination (means ± s.d. in motors μm−1min−1nM−1 are: 6.46 ± 3.09 for KIF1A alone (n=54 kymographs from 3 independent trials), 0.26 ± 0.33 for KIF1A + MAP7 (n=75 kymographs from 3 independent trials), 0.36 ± 0.64 for KIF1A + tau (n=71 kymographs from 3 independent trials), 0.13 ± 0.21 for KIF1A + MAP2C (n=69 kymographs from 3 independent trials), 3.77 ± 1.36 for KIF1A + DCX (n=65 kymographs from 3 independent trials), 4.61 ± 1.97 for KIF1A + DCLK1 (n=83 kymographs from 3 independent trials), 8.10 ± 2.61 for KIF1A + MAP9 (n=55 kymographs from 3 independent trials), 0.19 ± 0.26 for KIF1A + MAP7 + DCX (n=66 kymographs from 2 independent trials), 0.30 ± 0.25 for KIF1A + MAP7 + DCLK1 (n=46 kymographs from 2 independent trials), 0.33 ± 0.40 for KIF1A + MAP7 + MAP9 (n=76 kymographs from 2 independent trials), and 0.006 ± 0.004 for 50x the concentration of KIF1A (50 μM) + MAP2C (n=19 kymographs from 2 independent trials). All datapoints are plotted with lines indicating means ± s.d. P < 0.0001 (***) and P = 0.0034 (**) calculated by one-way ANOVA with Bonferroni correction. (C) Velocity histograms of KIF1A + 1 mM ATP in the absence and presence of DCX, DCLK1, and MAP9 with Gaussian fits. Means ± s.d. are 1252.5 ± 279.7, 931.0 ± 177.0, 1070.6 ± 280.6, and 1023.8 ± 293.6 nm/sec for KIF1A alone, KIF1A + DCX, KIF1A + DCLK1, and KIF1A + MAP9, respectively. P < 0.0001 for KIF1A vs. KIF1A + each MAP calculated by one-way ANOVA with Bonferroni correction. n=179, 77, 80 and 86 KIF1A motors for KIF1A alone, KIF1A + DCX, KIF1A + DCLK1, and KIF1A + MAP9, respectively from 2 independent trials each. (D) TIRF-M images and kymographs of 1 nM KIF1A-mScarlet + 1 mM ATP in the presence of 50 nM BFP-MAP7 (pink) with 50 nM DCX-sfGFP (green), or 50 nM BFP-MAP7 (pink) with 50 nM sfGFP-MAP9 (green). Scale bars: 1 μm (x), 5 sec (y).
Considering the similarity in motor domains and binding footprints on the microtubule lattice between kinesin-1 and kinesin-3 (Gigant et al., 2013; Kikkawa et al., 2001), we were curious if we would observe the same global inhibition of kinesin-3 by these six MAPs. Indeed, we observed that MAP7, tau, and MAP2C largely inhibited kinesin-3 from accessing the microtubule (Figure 2A–B). The inhibition by MAP2C was consistent even at a 50-fold higher concentration of kinesin-3 (Figure 2B). Strikingly, saturating amounts of DCX, DCLK1, and MAP9 were permissive for kinesin-3 motility (Figure 2A–B). Prior studies have reported that DCX and DCLK1 interact with the motor domain of kinesin-3 and are important for kinesin-3 transport of cargo within dendrites in vivo (Lipka et al., 2016; Liu et al., 2012). However, we did not observe an increase in the number of processive kinesin-3 motors on the microtubule or motor velocity in the presence of DCX or DCLK1 (Figure 2A–C, S4B), indicating that these MAPs do not directly recruit kinesin-3 or allosterically enhance its motor activity. Interestingly, MAP9 was the only MAP to significantly increase the number of processive kinesin-3 motors on the lattice (Figure 2A–B). We next wanted to test if DCX, DCLK1, and MAP9 could recruit kinesin-3 or promote kinesin-3 motility in the presence of an iMAP. On microtubules co-decorated with saturating amounts of MAP7 and either DCX, DCLK1, or MAP9, we still observed a significant inhibition of kinesin-3 landing events (Figure 2B,D). Thus, unlike MAP7, which can recruit kinesin-1 in the presence of an iMAP, we do not observe a similar effect for DCX, DCLK1, or MAP9 on kinesin-3. However, our data reveal that MAP9, which is present in both dendrites and axons (Figure S1), is the only MAP that positively modulates kinesin-3 motility by increasing the number of moving motors on the microtubule.
The K-loop of kinesin-3 is necessary for MAP9 to exert its effects on the motor
It is striking that three MAPs present in dendrites, DCX, DCLK1, and MAP9, impede kinesin-1, but not kinesin-3. We therefore wanted to investigate the differential effects of these three MAPs on kinesin-1 and kinesin-3. At sub-saturating concentrations of DCX (5 nM), where we observe cooperative clusters of DCX molecules on the microtubule lattice (Bechstedt and Brouhard, 2012), substantially more kinesin-3 motors entered and passed through these clusters than kinesin-1 motors, the majority of which detached upon encountering DCX assemblies (Figure S4C–D). Similar to kinesin-3, the activated DDB complex, which also transports cargo within dendrites, largely moved through cooperative DCX clusters unimpeded (Fig S4C–D).
We, and others, have previously shown that MAP7 directly interacts with kinesin-1 to recruit kinesin-1 to the microtubule (Hooikaas et al., 2019; Monroy et al., 2018). We therefore asked whether a motor must directly interact with a MAP in order to progress through a MAP-decorated lattice. Solution-based pull-down assays using purified proteins revealed that neither DCLK1 nor MAP9 stably interacted with our kinesin-3 construct (KIF1A1-393) (Figure S4E). We cannot rule out a possible interaction between these MAPs and the kinesin-3 tail domain; however, previous data suggested that the motor domain directly interacts with DCX and DCLK1 (Lipka et al., 2016; Liu et al., 2012). Although our result is in contrast to these studies, it is consistent with our observations that DCX, DCLK1, and MAP9 are unable to recruit kinesin-3 in the presence of an iMAP, unlike the ability of MAP7 to recruit kinesin-1 in a similar assay (Figure 1B–C, 2B,D).
A primary difference between the kinesin-1 and kinesin-3 constructs used in this study is the presence of six lysines, known as the K-loop, within loop 12 of the kinesin-3 motor domain (Figure S4F–G)(Soppina and Verhey, 2014). We mutated two of these lysines to alanines (KIF1AA) and observed the effects on motility in the absence and presence of DCX, DCLK1, and MAP9 (Figure 3A–C, S4G). Similar to prior studies of this region (Okada and Hirokawa, 2000; Soppina and Verhey, 2014), we observed a significant reduction in the number of processive motors on the microtubule, without a decrease in velocity (Figure 3A–C; means ± s.d.: 6.46 ± 3.09 vs. 0.03 ± 0.4 motors μm−1 min−1 nM−1, and 1252.5 ± 279.7 vs. 1503.7 ± 641.9 nm/sec for KIF1A vs. KIF1AA). The presence of DCX or DCLK1 further decreased the number of processive KIF1AA motors. Conversely, the presence of MAP9 on the microtubule increased the number of KIF1AA motors ~3-fold (Figure 3A–C), revealing that MAP9 may directly contact kinesin-3 on the microtubule surface, despite our inability to detect a stable interaction between the two molecules in solution (Fig. S4E). Mutation of all six lysines to alanines abolished the ability of MAP9 to stimulate KIF1AA-loop landing and progression on the microtubule (Figure S5A–B), indicating that the remaining four lysines in the K-loop of KIF1AA may be sufficient to interact with MAP9. From these data, we suggest that MAP9 could transiently contact the positively charged K-loop as kinesin-3 steps along the lattice.
Figure 3. Mechanistic dissection of kinesin-3 progression through MAP-decorated microtubule lattices.
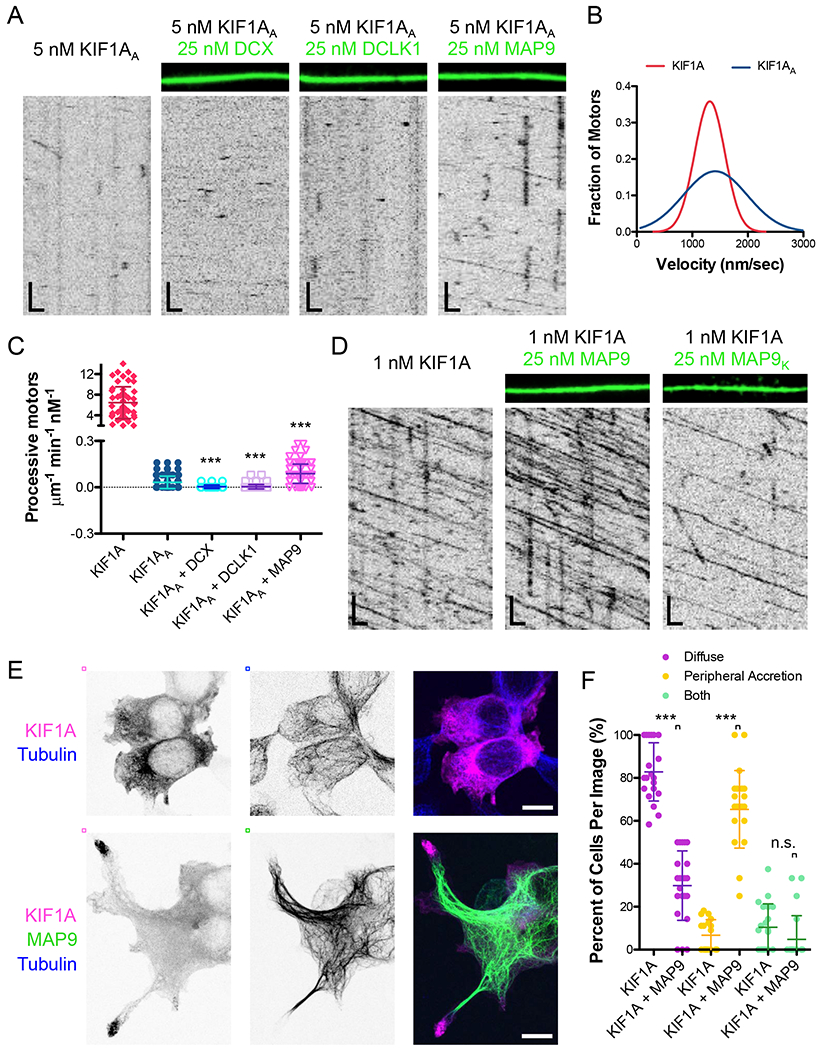
(A) TIRF-M images and kymographs of 5 nM KIF1AA-mScarlet (kinesin-3A) + 1 mM ATP in the absence and presence of 50 nM DCX-sfGFP, 50 nM sfGFP-DCLK1, or 50 nM sfGFP-MAP9. Scale bars: 1 μm (x), 5 sec (y). (B) Velocity histograms of KIF1A and KIF1AA (in the presence of MAP9) + 1 mM ATP with Gaussian fits. KIF1A data are reproduced from Figure 2C. There were too few KIF1AA motors alone to analyze, thus we quantified motor velocity in the presence of MAP9 due to the increased motor density. Mean ± s.d velocity for KIF1AA + MAP9 was 1503.7 ± 642.0 nm/sec (n=102 motors from 3 independent trials). P < 0.0001 using a student’s t-test for KIF1A vs. KIFIAA. (C) Quantification of the number of processive KIFlA-mScarlet motors + 1 mM ATP in the absence and presence of DCX, DCLK1, and MAP9. KIF1A data are reproduced from Figure 2B. Means ± s.d. in motors μm−1min−1nM−1 are: 6.46 ± 3.09 for KIF1A, 0.03 ± 0.04 for KIF1AA alone (n=143 kymographs from 3 independent trials), 0.003 ± 0.01 for KIF1AA + DCX (n=92 kymographs from 2 independent trials), 0.004 ± 0.01 for KIF1AA + DCLK1 (n=94 kymographs from 2 independent trials), 0.09 ± 0.06 for KIF1AA + MAP9 (n=135 kymographs from 3 independent trials). All datapoints are plotted with lines indicating means ± s.d. P < 0.0001 (***) calculated by one-way ANOVA with Bonferroni correction. (D) TIRF-M images and kymographs of 1 nM KIF1A-mScarlet + 1 mM ATP in the absence and presence of 25 nM sfGFP-MAP9 or 25 nM sfGFP-MAP9K. Scale bars: 1 μm (x), 5 sec (y). Mean ± s.d for KIF1A + MAP9K was 1.22 ± 0.68 μm−1min−1nM−1 (n=94 kymographs from 2 independent trials). P < 0.0001 using a student’s t-test. (E) Confocal images of HEK cells expressing KIF1A alone (top) or KIF1A + MAP9 (bottom) and immunostained for tubulin. Scale bars: 10 μm. (F) Quantification of the cellular localization of KIF1A in the absence or presence of MAP9. Datapoints represent individual images with lines indicating means ± s.d. P < 0.0001 (***) for KIF1A vs. KIF1A + MAP9 for diffuse (82.8 ± 13.6 and 29.8 ± 16.2 % of cells for KIF1A and KIF1A + MAP9, respectively) and peripheral accretion (6.8 ± 7.2 and 65.4 ± 18.0 % of cells for KIF1A and KIF1A + MAP9, respectively) phenotypes and P = 0.1032 for the presence of both phenotypes (10.5 ± 10.9 and 4.8 ± 11.0 % of cells for KIF1A and KIF1A + MAP9, respectively) using a student’s t-test. n=108 and 157 cells for KIF1A and KIF1A + MAP9, respectively, from 2 separate cultures.
In order to examine a potential interaction between MAP9 and kinesin-3 on the microtubule, we examined the MAP9 sequence for a conserved acidic region that could transiently interact with the K-loop of kinesin-3 (Figure S5C–E). Mutation of a conserved stretch of three glutamic acids (aa 502-504) to lysines (Figure S5D–E) strongly perturbed the ability of kinesin-3 motors to bind and move along microtubules saturated with MAP9K (Figure 3D and S5F). These mutations did not substantially affect the microtubule binding affinity of MAP9K (Figure S5F), indicating that the inhibition of kinesin-3 is not due to an altered affinity of MAP9K for the microtubule. These data indicate that MAP9 may enable kinesin-3 progression via direct contact with the motor on the microtubule surface. The presence of MAP9 modestly decreased kinesin-3 velocity (Figure 2C), which could also be due to a transient interaction between these proteins, similar to the effect of MAP7 on kinesin-1 (Monroy et al., 2018). Collectively, these data reveal a mechanism by which MAP9 enables kinesin-3 motility along microtubules.
We next examined if MAP9 could affect the motility of full-length kinesin-3 in vivo. Prior studies have demonstrated that wild type KIF1A transfected into cultured cells exhibits a diffuse cytoplasmic localization, likely due to autoinhibition of the motor (Hammond et al., 2009). However, certain mutations cause the motor to relocalize to microtubules or accumulate at the periphery of the cells due to release of motor autoinhibition (Chiba et al., 2019; Niwa et al., 2016). We observed a similar pattern for KIF1A transfected into HEK293 cells (82.8 ± 13.6 % of cells exhibited diffuse localization of KIF1A), but upon transfection of both KIF1A and MAP9, KIF1A localization was strongly redistributed to the tips of processes extending from these cells (Figure 4E–F; 65.4 ± 18.0 % of cells exhibited peripheral accretion of KIF1A). These data suggest that MAP9 enhances the motility of this motor in vivo and may play a role in releasing the autoinhibition of full-length KIF1A, which will be a fascinating future direction to explore.
Figure 4. Addition of the kinesin-3 K-loop into loop 12 of kinesin-1 enhances on-rate and enables motility through a lattice saturated with MAP9.
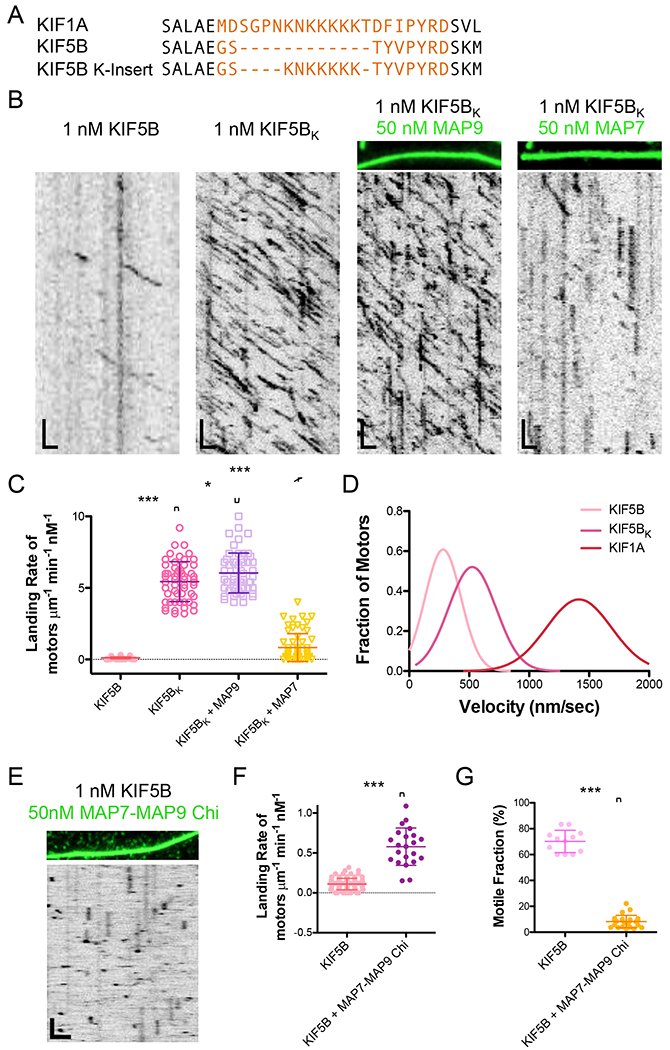
(A) Sequence alignment comparing loop 12 of kinesin-3 (KIF1A) and kinesin-1 (KIF5B) and the kinesin-1 chimera construct with the K-loop residues inserted into loop 12 of KIF5B for the studies in (B). (B) TIRF-M images and kymographs of 1 nM KIF5B-mScarlet compared with 1 nM KIF5BK (kinesin-1 chimera with the K-loop insertion from kinesin-3) + 1 mM ATP in the absence and presence of 50 nM sfGFP-MAP9 or 50 nM sfGFP-MAP7. Scale bars: 1 μm (x), 5 sec (y). (C) Quantification of the landing rates of KIF5B-mScarlet compared with KIF5BK-mScarlet + 1 mM ATP in the absence and presence of MAP9 or MAP7. KIF5B data are reproduced from Figure 2B. Means ± s.d. in motors μm−1min−1nM−1 are: 5.44 ± 1.39 for KIF5BK alone (n=55 kymographs from 2 independent trials), 6.04 ± 1.39 for KIF5BK + MAP9 (n=56 kymographs from 2 independent trials), and 0.83 ± 0.98 for KIF5BK + MAP7 (n=63 kymographs from 2 independent trials). All datapoints are plotted with lines indicating means ± s.d. P < 0.0001 (***) and P = 0.025 (*) using a student’s t-test. (D) Velocity histograms of KIF5B, KIF5BK, and KIF1A + 1 mM ATP with Gaussian fits. KIF1A data are reproduced from Figure 3B. Mean ± s.d velocities for KIF5B and KIF5BK are 358.2 ± 295.7 nm/sec (n=188 motors from 3 independent trials) and 489.0 ± 164.5 nm/sec (n=241 motors from 2 independent trials), respectively. P < 0.0001 using a student’s t-test for KIF5B vs. KIF5BK. (E) TIRF-M image and kymograph of 1 nM KIF5B-mScarlet (kinesin-1) + 1 mM ATP in the presence of 50 nM sfGFP-MAP7(aa565-610)-MAP9(aa302-647)-chimera. Scale bars: 2 μm (x), 10 sec (y). (F) Quantification of the landing rates of KIF5B-mScarlet + 1 mM ATP in the absence and presence of the MAP7-MAP9-chimera. Means ± s.d. in motors μm−1min−1nM−1 are: 0.11 ± 0.07 for KIF5B alone (reproduced from Figure 1B) and 0.58 ± 0.23 for KIF5B + MAP7-MAP9-chimera (n=22 kymographs from 2 independent trials). (G) Quantification of the percentage of motile KIF5B motors in the absence (70.2 ± 8.6 %; reproduced from Figure 1D) or presence of 50nM MAP7-MAP9-chimera (8.2 ± 4.8; n=1173 molecules from 24 kymographs from 2 independent trials). For (F) and (G), all datapoints are plotted with lines indicating means ± s.d. P < 0.0001 (***) using a student’s t-test.
If the K-loop of kinesin-3 is the defining feature that allows for its movement on a microtubule crowded with MAPs that inhibit kinesin-1, then the transposition of this region into kinesin-1 should confer resistance to the strong effects of its iMAPs. In support of this idea, when the kinesin-3 K-loop was inserted into loop 12 of kinesin-1 (Gigant et al., 2013; Kikkawa et al., 2001), the motor was able to transport cargoes into dendrites from which it is normally excluded (Karasmanis et al., 2018). Based on prior studies, we engineered a chimeric kinesin-1 with the K-loop of kinesin-3 inserted into loop 12 of kinesin-1 (Figure 4A), and found that the kinesin-1 chimera (KIF5BK) exhibited a 40-fold increase in landing rate compared to the wild type motor, with a small increase in velocity (Figure 4B–D; means ± s.d.: 0.11 ± 0.07 vs. 5.44 ± 1.39 motors μm−1 min−1 nM−1, and 358.2 ± 295.7 nm/sec vs. 489.0 ± 164.5 nm/sec for KIF5B vs. KIF5BK). In contrast to wild type KTF5B, KTF5BK was able to land and translocate on a MAP9-decorated microtubule, but was impeded by the presence of MAP7, which normally inhibits kinesin-3 (Figure 4B–C). This result suggests that the K-loop is the structural element responsible for the inhibition of kinesin-3 by MAP7, because although KTF5BK includes the MAP7 interaction region, it is still significantly inhibited on a lattice saturated with MAP7. We next wanted to test if an increased on-rate was sufficient to enable kinesin-1 to progress through a MAP9-saturated lattice. We engineered a chimeric MAP combining the microtubule-binding domain of MAP9 with the kinesin-1 binding domain of MAP7. The MAP7 region of this chimeric protein recruited wild type kinesin-1 to the microtubule, increasing its landing rate 5-fold, but kinesin-1 was unable to move on the lattice (Figure 4E–G). Thus, these data indicate the K-loop of kinesin-3 confers its ability to move through MAP9 independently of its ability to increase the motor’s on-rate. In summary, the presence of MAP9 on the microtubule is incompatible with the movement of kinesin-1, and we hypothesize this could be due to structural interference between the two molecules on the surface of the microtubule.
MAP9 inhibits the dynein-dynactin-BicD complex by blocking p150 binding
Next, we wanted to examine the effect of MAP9 on dynein, because we observed that DCX did not significantly impair DDB translocation along the microtubule (Figure S4C–D), and in prior studies, we found that tau and MAP7 did not dramatically impede DDB motility either (Monroy et al., 2018; Tan et al., 2019). In contrast to these other MAPs, MAP9 significantly inhibited processive DDB from accessing the lattice as evidenced by the 4-fold reduction in the number of processive motors on the microtubule in the presence of MAP9 (Figure 5A–B). DDB complexes contain two distinct microtubule-binding domains (MTBD): the small MTBD on the dynein motor domain (Carter et al., 2008) that directly contacts the tubulin surface, and the CAP-Gly/basic domain of the p150Glued subunit of dynactin that binds to the unstructured tubulin tails (King and Schroer, 2000; McKenney et al., 2016). We assayed each MTBD independently to determine how MAP9 inhibits the fully assembled DDB complex, and found that MAP9 dramatically decreased the amount of p150 on the microtubule 57-fold, but had little effect on the amount of dynein motor domain on the lattice (Figure 5C–F). These data indicate that MAP9 does not share overlapping binding sites on the microtubule surface with the dynein motor domain, which we hypothesize is the mechanism of its inhibition of kinesin-1 (Figures 1 and 4). Instead, our data support a model whereby MAP9 likely inhibits DDB from initially landing on the microtubule by blocking p150 binding to the tubulin tail domains. Overall, these data reveal that MAP9 acts as an inhibitor for kinesin-1 and dynein, but enables kinesin-3 to land and progress on the microtubule.
Figure 5: MAP9 inhibits the processive dynein-dynactin-BicD complex by blocking p150 binding.
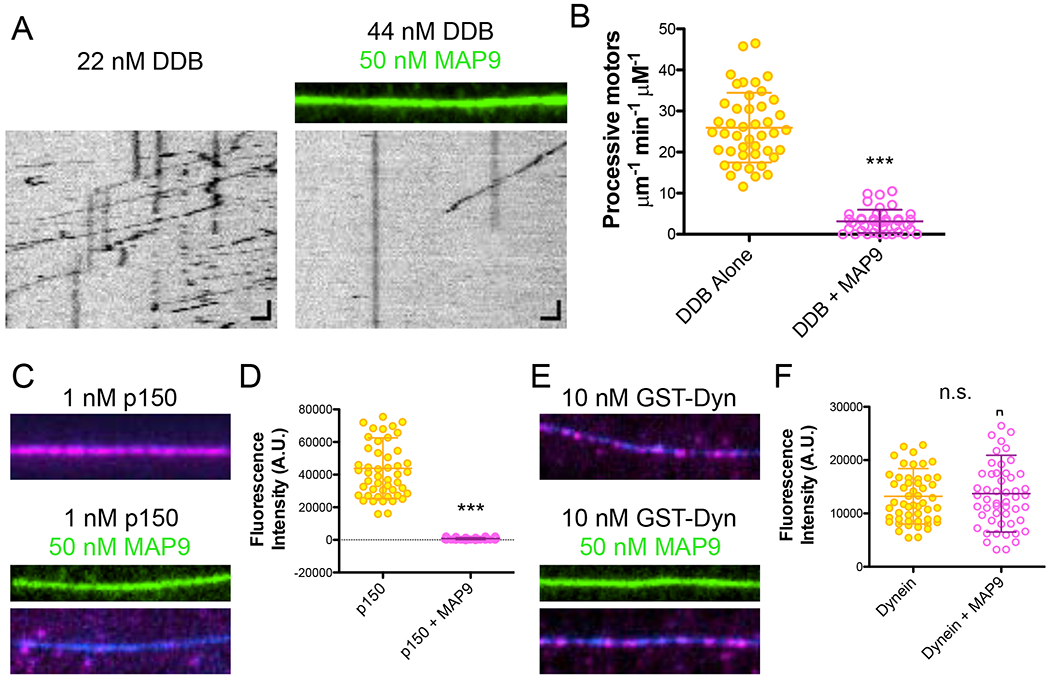
(A) TIRF-M images and kymographs of dynein-dynactin-BicD2 (DDB)-TMR at indicated concentrations + 1 mM ATP in the absence and presence of 50 nM sfGFP-MAP9. Scale bars: 1 μm (x), 5 sec (y). (B) Quantification of the number of processive DDB complexes + 1 mM ATP in the absence and presence of MAP9. Means ± s.d. in motors μm−1min−1nM−1 are: 26.0 ± 8.5 for DDB alone (n=44 kymographs from 2 independent trials) and 3.2 ± 2.8 for DDB + MAP9 (n=41 kymographs from 2 independent trials). P < 0.0001 (***) using a student’s t-test. (C) TIRF-M images of 1 nM p150-mScarlet in the absence and presence of 50 nM sfGFP-MAP9. Image width: 10.8 μm. (D) Quantification of p150-mScarlet fluorescence intensity in the absence and presence of MAP9 (means ± s.d. are 44239.7 ± 18637.0 and 764.5 ± 12.0 A.U. and n=50 and 50 microtubules for p150 and p150 + MAP9, respectively from 2 independent trials; P < 0.0001 (***)). (E) TIRF-M images of 10 nM TMR-GST-dynein motor domain in the absence and presence of 50 nM sfGFP-MAP9. Image width: 10.8 μm. (F) Quantification of TMR-GST-dynein fluorescence intensity in the absence and presence of MAP9 (means ± s.d. are 13208.3 ± 5202.0 and 13719.4 ± 7205.4 A.U. and n=53 and 52 microtubules for dynein and dynein + MAP9, respectively from 2 independent trials; P = 0.6773 (n.s.)).
MAP9 competes for binding on the microtubule surface with DCX and DCLK1
Very little is known about the mechanism of microtubule binding by MAP9, but we found it intriguing that, similar to DCX and DCLK1, it inhibited kinesin-1, but allowed for kinesin-3 movement. However, unlike DCX, MAP9 inhibits the association of the processive dynein complex with the microtubule. We were therefore curious about the binding site of MAP9 on the lattice. First, we analyzed whether MAP9 and DCX or DCLK1 could bind simultaneously to individual microtubules. We mixed equimolar concentrations of MAP9 with DCX or DCLK1 and observed that MAP9 was excluded from sites of DCX or DCLK1 enrichment, and vice versa (Figure 6A). Interestingly, this anti-correlation is similar to what we observed for MAP9 and DCLK1 within distal dendrites (Figure S6). Conversely, at equimolar concentrations, MAP9 simultaneously bound microtubules coated in tau or MAP7 (Figure 6B). Our prior study on MAP7 and tau led us to hypothesize that MAP7 binds along the protofilament “ridge” of the microtubule similar to tau (Monroy et al., 2018). DCX and DCLK1 bind at the vertex of four tubulin heterodimers, occupying the “valley” between two adjacent protofilaments (Fourniol et al., 2013; Kim et al., 2003). Taken together, these data suggest that MAP9 also binds within the interprotofilament valley, similar to DCX and DCLK1 (Figure 6C), but could potentially make contacts with the ridge of the protofilament and even the tubulin C-terminal tails to obstruct the binding domains of kinesin-1 and p150, respectively. In addition, it is noteworthy that MAP9 can bind the microtubule simultaneously with MAP7 and tau, all three of which are present in the axon and play distinct roles in allowing kinesin-1, kinesin-3, and dynein to access the lattice.
Figure 6: MAP9 competes for binding on the microtubule with DCX and DCLK1, but not with tau and MAP7.
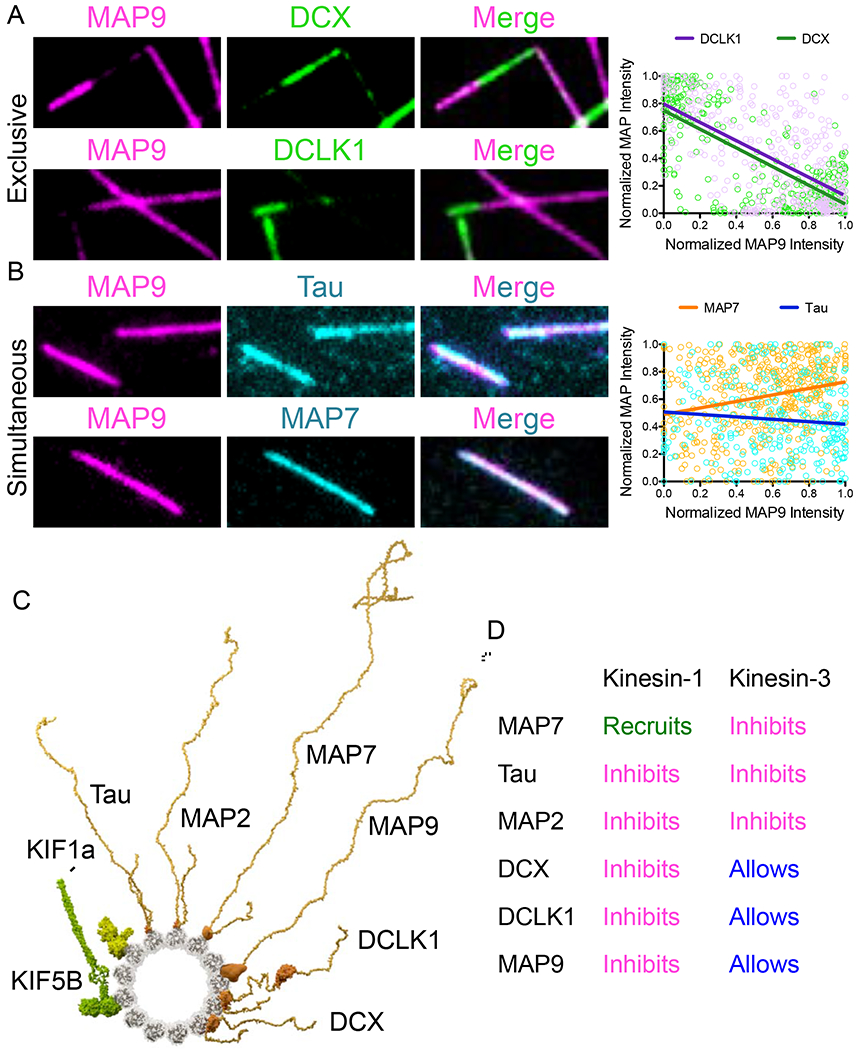
(A) TIRF-M images of 50 nM sfGFP-MAP9 and 50 nM mTagBFP-DCX or 50 nM sfGFP-MAP9 and 50 nM TagRFP-DCLK1 show these MAPs exclude each other into homotypic patches on microtubules. Right: corresponding graph displaying individual XY pairs per pixel for MAP9 intensity vs. DCLK1 or DCX intensity on the microtubule, fit with a linear regression. Pearson’s correlation coefficients: −0.609 for MAP9 vs. DCLK1 (n=403 XY pairs from n=10 microtubules, P < 0.0001) and −0.731 for MAP9 vs. DCX (n=323 XY pairs from n=10 microtubules, P < 0.0001). (B) TIRF-M images of 50 nM sfGFP-MAP9 and 50 nM mScarlet-tau or 50 nM sfGFP-MAP9 and 50 nM mTagBFP-MAP7 show these MAPs bind to microtubules simultaneously. Right: corresponding graph displaying individual XY pairs per pixel for MAP9 intensity vs. MAP7 or tau intensity on the microtubule, fit with a linear regression. Pearson’s correlation coefficients: 0.234 for MAP9 vs. MAP7 (n=460 XY pairs from n=10 microtubules, P < 0.0001) and −0.095 for MAP9 vs. tau (n=293 XY pairs from n=10 microtubules, P = 0.106). Image width: 7.0 μm. (C) Minus-end on view of modeled MAPs and kinesin motors bound to the microtubule. KIF5B1-560: motor domains (4hna), dimeric kinesin (3kin), coiled-coil (1d7m) as in Zhang et al. (2012). KIF1A1-393: motor domains (1ia0), dimer based on conventional kinesin (3kin) as in Huo et al. (2012). Tau2N4R and MAP2C: microtubule-binding region based on tau (6cvn). MAP7 and MAP9: plausible microtubule-binding regions via de novo and homology modeling (Buchan et al., 2010). DCLK1: microtubule-binding N-DC1 domain (1mg4), kinase domain (5jzj). DCX: microtubule-binding N-DC1 domain (2xrp). MAPs are shown as full-length pseudo-models, with projection domains and domain linkers modeled as unfolded to convey intrinsic disorder predicted for those regions. (D) Summary table of results: MAPs have distinct effects on different motors.
DISCUSSION
Overall, we have found that for each of the major cargo transport motors, kinesin-1, kinesin-3, and dynein, there is at least one MAP that inhibits each motor and at least one MAP that allows for each motor to progress unimpeded along the microtubule (Figure 6D). In addition, the effects of MAPs on motor landing and motility we report here are more dramatic than those previously reported for tubulin post-translational modifications, suggesting a strong potential for the MAP code in directing motor transport in vivo.
It is an outstanding question how motors are spatially distributed into specific neuronal compartments. Three MAPs that localize to the dendrites all inhibit kinesin-1, but allow for kinesin-3 motility on the microtubule, suggesting neurons employ multiple modalities to specifically allow kinesin-3, but prevent kinesin-1 transport within dendrites. In vivo, loss of DCX or DCLK1 impedes the ability of kinesin-3 to transport cargo within dendrites (Lipka et al., 2016; Liu et al., 2012). Our results suggest this effect is not due to direct recruitment of kinesin-3 by these MAPs. Instead, removal of DCX or DCLK1 could affect the distribution of other MAPs that inhibit kinesin-3, or these MAPs could alter the accumulation of posttranslational modifications that could cause a cascade of effects on motor transport. The distinct distal localization patterns of DCX and DCLK1 suggest that they are unlikely candidates for the spatial regulation of kinesin-1 and kinesin-3 movement along the entire length of neuronal processes. Our results, combined with other studies, indicate that MAP9 and non-canonical MAPs, such as septins (Karasmanis et al., 2018), may be more important for facilitating kinesin-3 transport in dendrites and excluding kinesin-1. Our in vitro and in vivo data support a role for MAP9 in promoting kinesin-3 motility on the lattice; however, further studies will be necessary to demonstrate the functional importance of this mechanism in vivo, and whether MAP9 is required for kinesin-3 cargo transport in cells.
Co-binding analyses between MAPs led us to hypothesize that MAP9 binds within the interprotofilament valley, but based on our experiments dissecting its inhibition of kinesin-1, it may also contact the protofilament ridge, overlapping with kinesin motor domain binding sites. Interestingly, MAP9 blocks p150 from binding the microtubule, but not the dynein motor domain, indicating that MAP9 does not share a common binding site with the dynein motor domain, which has a smaller binding footprint than that of kinesin-1 (Carter et al., 2008; Gigant et al., 2013). With regards to its inhibition of p150, MAP9 could potentially contact and affect the C-terminal tails of tubulin, which p150 directly binds (Wang et al., 2014), or the MAP9 projection domain may sterically impede the p150-tubulin interaction. High-resolution structural analysis of MAP9 bound to microtubules will be needed to fully answer these questions.
It is tempting to speculate that MAP binding could designate individual microtubules, or even subsets of protofilaments, as specific tracks for anterograde or retrograde transport. This type of organization, analogous to emerging results in intraflagellar transport (Bertiaux et al., 2018), would prevent collisions between motors and their cargoes and could conceivably allow for MAPs or other factors to independently modulate transport in either direction. Further, while MAP9 can facilitate kinesin-3 and MAP7 can recruit kinesin-1, our data show that under saturating conditions with a simultaneously bound iMAP, these motors are still inhibited. Thus, we speculate that subsets of microtubules or protofilaments must be devoid of iMAPs to enable motor transport. The cooperative binding exhibited by tau and DCX could be necessary to establish and maintain homotypic MAP zones on the lattice. However, for MAPs that do not exhibit cooperative binding behaviors, such as MAP7 and MAP9, other mechanisms may regulate their binding. Tubulin isotypes or tubulin posttranslational modifications could dictate exclusive MAP7 or MAP9 regions of the lattice to prevent simultaneous binding and enable efficient transport of kinesin-1 or kinesin-3. Future work will be necessary to determine how tubulin modifications affect MAP binding, and contribute to gating motor access to the microtubule.
MAP7 also has kinesin-independent roles in regulating microtubule dynamics (Gallaud et al., 2014; Metivier et al., 2019; Tymanskyj et al., 2017), and is important to promote axonal branch regeneration after nerve injury (Tymanskyj and Ma, 2019). Whether MAP9 has roles in regulating the microtubule cytoskeleton that are independent of its roles in directing motors, or whether MAP7 and MAP9 coordinate their binding activities to regulate microtubule dynamics remains to be determined.
Finally, this study has implications beyond polarized neuronal transport to any cell or process that relies on MAPs and molecular motors. Many of these MAPs are found in other cell types, such as muscles (tau, MAP7) (Metzger et al., 2012; Murakami et al., 1995), or in specialized cellular processes, such as mitosis (MAP7, MAP9, DCLK1) (Gallaud et al., 2014; Metivier et al., 2019; Qu et al., 2019; Saffin et al., 2005; Venoux et al., 2008). It is therefore important to consider how MAPs affect different types of motors when analyzing any system in which they must share a common microtubule lattice. Further work will be necessary to elucidate how MAPs co-decorate the microtubule in vivo to dictate access by specific motors.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kassandra Ori-McKenney (kmorimckenney@ucdavis.edu). Cell lines and plasmids are available upon request from the authors.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Protein Expression and Purification Systems
Tubulin was isolated from porcine brain using the high-molarity PIPES procedure as previously described (Castoldi and Popov, 2003). For bacterial expression of sfGFP-tau, sfGFP-MAP2, sfGFP-MAP7, mTagBFP-MAP7, sfGFP-MAP9, mTag-BFP-MAP9, sfGFP-MAP9EEE>KKK, DCX-sfGFP, sfGFP-DCLK, K560-mScarlet, K420-mScarlet, KIF1A-mScarlet, KIF1A KK>AA-mscarlet, KIF1AK-loop>A-loop-mscarlet, KIF5BK-insert-mscarlet, and mScarlet-p150, BL21-RIPL cells were grown at 37°C until ~O.D. 0.6 and protein expression was induced with 0.1 mM IPTG. Cells were grown overnight at 18°C, harvested, and frozen. Cell pellets were resuspended in lysis buffer (50 mM Tris pH 8, 150 mM K-acetate, 2 mM Mg-acetate, 1 mM EGTA, 10% glycerol) with protease inhibitor cocktail (Roche), 1 mM DTT, 1 mM PMSF, and DNAseI. Cells were then passed through an Emulsiflex press and cleared by centrifugation at 23,000 x g for 20 mins. Clarified lysate from bacterial expression was passed over a column with Streptactin Superflow resin or Streptactin XT Superflow resin (Qiagen/IBA). After incubation, the column was washed with four column volumes of lysis buffer, then bound proteins were eluted with 3 mM desthiobiotin (Sigma) or 50 mM D-biotin (CHEM-IMPEX) in lysis buffer. Eluted proteins were concentrated on Amicon concentrators and passed through a superose-6 10/300 (GE Healthcare) gel-filtration column in lysis buffer using a Bio-Rad NGC system. Peak fractions were collected, concentrated, and flash frozen in LN2. Protein concentration was determined by measuring the absorbance of the fluorescent protein tag and calculated using the molar extinction coefficient of the tag. Recombinant SNAPf-GST-hDyn protein was prepared using the Bac-to-Bac baculovirus system (Invitrogen) as previously described (McKenney et al., 2014; McKenney et al., 2016). The purified protein was labeled with 10 μM SNAP-Cell TMR-Star (NEB) while bound to the StrepTactin resin during purification. The protein was subjected to a cycle of MT binding and release by ATP to select for active motors. Briefly, motors were bound to an excess of stabilized porcine MTs in BRB80 buffer with 10 μM taxol. MTs were pelleted at 60,000 g for 10 min at room temperature. Bound motors were released by re-suspension of the MT pellet in BRB80 with 10 μM taxol and 10 mM ATP. MTs were pelleted again as before, and the eluted motors were frozen in LN2 after the addition of 20% sucrose and 1 mg/ml BSA as cryoprotectants. The resulting preparations were analyzed by SDS polyacrylamide gel electrophoresis (SDS-PAGE).
Cell Lines
For primary hippocampal neuronal cultures, all animals were used with approval from the University of California Davis Institutional Animal Care and Use Committees (IACUC #20293 to S.S.) and housed and cared for in accordance with the guidelines provided by the National Institutes of Health. CD-1 mice (Charles Rivers) were used for neuronal cultures. Females were mated and the morning a vaginal plug was observed was considered embryonic day 0.5 (E0.5). Hippocampi were dissected out from E16.5-18.5 mouse embryonic brains and hippocampal neurons were isolated using Worthington Papain Dissociation System (Worthington Biochemical Corporation) following manufacturer’s protocol. Gender was not considered as a factor. Primary neurons were plated at density of 100k neurons per glass coverslips coated with 0.05 mg/ml Poly-D-Lysine (mol wt 70,000-150,000, Millipore-Sigma) and cultured in Neurobasal media supplemented with glucose, GlutaMAX (ThermoFisher), B27 and P/S. Cells were cultured for the indicated amount of time in a cell culture incubator at 37°C and 5% CO2. Media was changed every two days until neurons were fixed at DIV4, DIV7, or DIV14. For Flp-In T-Rex 293 cells (Thermofisher #R78007, an authenticated cell line), cells were cultured in standard DMEM media supplemented with 10% FBS and 10% P/S in a cell culture incubator at 37°C and 5% CO2 until they reached ~80-90% confluency. Cells were seeded at 100,000 cells per coverslip and transfected with pEGFP-N1-HsKIF1A full-length and/or pMSCARLET-N1-HsMAP9 (see molecular biology) using the FuGENE 6 (Promega E2691) transfection reagent. Cells were maintained in the above conditions until fixation 24 hrs after transfection.
METHOD DETAILS
Molecular Biology and Biochemistry
The cDNAs for protein expression in this study were as follows: human Tau-2N4R (Addgene #16316), human MAP7 (GE Dharmacon MGC Collection #BC025777), human MAP2 (Transomics #BC172263), human MAP9 (Transomics # BC146864), human DCX (Addgene #83928), mouse DCLK1 (Transomics #BC133685), human KIF5B (aa 1-560; a gift from R. Vale), and human KIF1A (aa 1-393; Addgene # 61665). Tau-2N4R, MAP2 MAP7, MAP9, and DCLK proteins were cloned in frame using Gibson cloning into a pET28 vector with an N-terminal strepII-Tag, mTagBFP or a superfolder GFP (sfGFP) cassette. DCX proteins were cloned in frame using Gibson cloning into a pET28 vector with a C-terminal sfGFP cassette. K560 and K420 were cloned in frame using Gibson cloning into pET28 vector with a C-terminal mScarlet-strepII cassette. KIF1A cloned in frame using Gibson cloning into pET28 vector with a C-terminal leucine-zipper-mScarlet-strepII cassette. All modified KIF1A constructs also contain the leucine zipper. KIF1AKK>AA, KIF1AK-loop>A-loop, KIF5BK-insert, and MAP9EEE>KKK mutations were introduced by overlapping PCR and Gibson cloning into pET28 vector with a C-terminal mScarlet-strepII cassette. The MAP7-MAP9 chimera was made by overlapping PCR and Gibson cloning to prepare a pET28 vector with a strepII tag and sfGFP tag at the N-terminus, followed by amino acids 565-610 of MAP7 then amino acids 302-647 of MAP9. pEGFP-N1-KIF1A was a gift from Shinsuke Niwa. For pMSCARLET-N1-HsMAP9, mscarlet-HsMAP9 was cloned in frame using Gibson cloning into the pEGFP-N1 vector, swapping out EGFP for mScarlet.
Pull-down assays were performed with either sfGFP-DCLK or sfGFP-MAP9 tagged at the C-terminal end with a FLAG epitope. FLAG beads (Thermofisher) were washed into assay buffer (100 mM Tris, pH 7.4, 150 mM NaCl, 4 mM MgCl, 1 mM EGTA, 0.5% TX-100, supplemented with 1 mM DTT and 4 mM ATP), then incubated with 500 nM DCLK, 500 nM MAP9, or buffer (beads alone control) for 1 hour rotating at 4°C. FLAG beads were then washed in assay buffer five times, then resuspended in assay buffer and 500 nM for KIF1A was added to the beads alone control and the experimental conditions. The 350 μL final volume solutions were incubated for 1 hour rotating at 4°C. The supernatants were collected, then the bead pellets were washed five times in assay buffer and resuspended in one bead bed volume. Gel samples of the supernatants and pellets were analyzed by SDS-PAGE. The supernatant samples that were run on SDS-PAGE were 15% of the pellet samples.
Microscopy
For TIRF microscopy experiments, imaging was performed on a Nikon Eclipse TE200-E microscope equipped with an Andor iXon EM CCD camera, a 100X, 1.49 NA objective, four laser lines (405, 491, 568, and 647 nm) and Micro-Manager software (Edelstein et al., 2010). Experiments were conducted at room temperature. For TIRF-M experiments, a mixture of native tubulin, biotin-tubulin, and fluorescent-tubulin purified from porcine brain (~10:1:1 ratio) was assembled in BRB80 buffer (80mM PIPES, 1mM MgCl2, 1mM EGTA, pH 6.8 with KOH) with 1mM GTP for 15 min at 37°C, then polymerized MTs were stabilized with 20 μM taxol. Microtubules were pelleted over a 25% sucrose cushion in BRB80 buffer to remove unpolymerized tubulin. Flow chambers containing immobilized microtubules were assembled as described (McKenney et al., 2014). All experiments were performed in assay buffer (60 mM Hepes pH 7.4, 50 mM K-acetate, 2 mM Mg-acetate, 1 mM EGTA, and 10% glycerol) supplemented with 0.1mg/mL biotin-BSA, 0.5% Pluronic F-168, and 0.2 mg/mL κ-casein (Sigma) and 10 uM taxol.
For all MAP plus motor experiments, saturating concentrations for the MAPs and single molecule concentrations for the motors were premixed and flowed into the chamber at the same time. For the MAP competition plus motor experiments, saturating concentrations of the MAPs and single molecule concentrations for the motors were premixed and flowed into the chamber at the same time. For live imaging, images were taken every 0.5 seconds for a total of 4 minutes for K560 (K420) and every 0.24 seconds for a total of 2 minutes for KIF1A (KIF1A KK>AA, KIF1AK-loop>A-loop, KIF5BK-insert). Kymographs were made from movies of K560, K420, KIF1A, KIF1AKK>AA, KIF1AK-loop>A-loop, and KIF5BK-insert in the absence or presence of MAPs and landing rates and velocity parameters were measured for individual events and runs, respectively. For KIF1A and DDB experiments where the motors were highly processive and most runs began before the movies were started, we quantified the number of processive motors per μm per min instead of the number of landing events to provide the most accurate numbers for these motors. Velocity data were fit with a Gaussian equation.
For all competition experiments, proteins were premixed at equimolar concentrations and flowed into the chamber at the same time. Competition experiments were performed with a mixture of either mTagBFP-MAP9 and sfGFP-DCX or sfGFP-DCLK or mTagBFP-MAP9 and sfGFP-tau or sfGFP-MAP7. For analysis of the competition experiments, line scans were taken of individual microtubules to determine intensity values for each MAP at each pixel along the length of the microtubule. The intensities were normalized as a fraction of the highest intensity for the particular MAP on that particular microtubule. The resulting XY pair per pixel was plotted on a XY graph and fit with a linear regression equation. Pearson’s correlation coefficient was determined for each MAP pair to determine if there was a negative, positive or lack of correlation between the MAPs on the microtubule.
For all saturation curves, a concentration series was performed for each protein. For fluorescence intensity analysis, ImageJ was used to draw a line across the microtubule of the MAP channel and the integrated density was measured. The line was then moved adjacent to the microtubule of interest and the local background was recorded. The background value was then subtracted from the value of interest to give a corrected intensity measurement. The fluorescence intensity data were fit with a one site binding hyperbola equation to derive the KD for each MAP. For certain cooperative MAPs, such as DCX, we decided to use a higher concentration (100 nM) for Figures 1 and 2, because we would occasionally see a very small region of the lattice that was devoid of MAP, though DCX reached saturation by overall intensity measurements at lower concentrations. This may be due to the heterogeneity of the tubulin, which we purify from brain lysate, or a specific preference of MAPs for particular lattice conformations.
Neuronal cultures were fixed in 4% paraformaldehyde for 20 minutes at room temperature, washed several times with PBS, permeabilized in PBS with 0.3% Triton X-100 (PBS-TX), and blocked with 5% BSA in PBS for 1 hour at room temperature. Cultures were then incubated overnight at 4°C with primary antibodies at a concentration of 1:500 for rabbit anti-MAP7 (Thermofisher PA5-31782), 1:500 for mouse monoclonal anti-Tau-1 (Millipore MAB3420), 1:500 anti-chicken tau (Genetex GTX49353), 1:500 anti-rabbit MAP2 (Sigma M3696), 1:500 anti-rabbit MAP9 (Invitrogen PA5-58145), 1:500 anti-mouse DCLK (Invitrogen MA5-26800), 1:500 anti-mouse DCX (Invitrogen MA5-17066), 1:1000 for mouse anti-alpha Tubulin (Sigma Clone DM1A T9026), or 1:1000 for rabbit anti-beta Tubulin (Abcam ab6046). Secondary antibodies were used at 1:1000 for Cy3 donkey anti-rabbit, Cy5 donkey anti-mouse, or Cy3 goat anti-chicken and incubated for 1 hour at room temperature. Cells were then rinsed several times with PBS and mounted using VectaShield mounting medium (Vector Laboratories).
Flp-In T-Rex 293 cells were fixed 24 hours later using 4% paraformaldehyde for 20 minutes at room temperature, washed several times with PBS, permeabilized in PBS with 0.3% Triton X-100 (PBS-TX), and blocked with 5% BSA in PBS for 1 hour at room temperature. Cells were then incubated overnight at 4°C with primary antibody at a concentration of 1:1000 for mouse anti-alpha Tubulin (Sigma Clone DM1A T9026). The secondary antibody Cy5 donkey anti-mouse was used at 1:1000. Coverslips were imaged on a Leica SPE laser scanning confocal microscope using an oil immersion 60x or 100x objective.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed with a two-tailed unpaired Student’s t-test, or a one-way ANOVA with Bonferroni post-hoc correction. Unless otherwise stated, all data was analyzed manually using ImageJ (FIJI). Graphs were created using Graphpad Prism and statistical tests were performed using this program. All variances given represent standard deviation. The statistical details of each experiment can be found in the figure legends, including statistical tests used, exact value of “n”, what “n” represents, definition of how data are plotted in each graph, and the dispersion and precision measures. Significance is defined in the figure legends: *** indicates P < 0.0001, ** indicates a P-value between 0.001 and 0.0001, * indicates a P-value between 0.05 and 0.001, and P > 0.05 indicates there is no significant difference.
DATA AND CODE AVAILABLITY
This study did not generate or analyze datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-MAP7 | Thermofisher | PA5-31782 |
| Mouse monoclonal anti-Tau-1 | Millipore | MAB3420 |
| Anti-chicken tau | Genetex | GTX49353 |
| Anti-mouse MAP2 | Sigma | M3696 |
| Anti-rabbit MAP9 | Invitrogen | PA5-58145 |
| Anti-mouse DCLK | Invitrogen | MA5-26800 |
| Anti-mouse DCX | Invitrogen | MA5-17066 |
| Mouse anti-alpha tubulin | Sigma Clone DM1A | T9026 |
| Rabbit anti-beta tubulin | Abcam | Ab6046 |
| Rabbit anti-MAP7 | Genetex | GTX120907 |
| Anti-rabbit MAP2 | Millipore | #AB5622 |
| Anti-mouse MAP7 | Sigma | SAB1409973 |
| Cy3 donkey anti-rabbit | Invitrogen | #A27034 |
| Cy5 donkey anti-mouse | Invitrogen | #A28180 |
| Cy3 goat anti-chicken | Invitrogen | A11039 |
| Vectashield no DAPI | Vector Laboratories | H-1000 |
| Bacterial and Virus Strains | ||
| Escherichia coli (BL21DE3) | Agilent | 200131 |
| Escherichia Coli (XL10Gold) | Agilent | 200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Biotinylated poly(L-lysine)-[g]-poly(ethylene-glycol) (PLL-PEG-Biotin | SuSoS AG | PLL(20)-G[3.5]-PEG(2)/PEG(3.4)-biotin(50%) |
| Streptavidin | ThermoFischer | 21135 |
| Trolox (6-hydroxy-2,5,6,7,8-tetramethylchroman-2-carbonsaure, 97%) | Acros | AC218940050 |
| 3,4-Dihydroxybenzoic Acid (Protocatechuic acid) | Sigma-Aldrich | 37580 |
| Protocatachuate 3,4-Dioxygenase from Pseudomonas sp. | Sigma-Aldrich | P8279 |
| κ-caesin from bovine milk | Sigma-Aldrich | C0406 |
| Pierce Bovine Serum Albumin, Biotinylated | Thermo-Fischer | 209130 |
| Paclitaxel | Sigma-Aldrich | T7402 |
| Pluronic F-157 | Sigma-Aldrich | P2443 |
| Glass cover slides (18x18-1.5) | Fischer | 12-541A |
| Superfrost Microscope slides | Fisher | 12-550-143 |
| Adenosine 5’-triphosphate disodium salt hydrate | Sigma-Aldrich | A2383 |
| Guanosine 5’-triphosphate sodium salt hydrate | Sigma-Aldrich | G8877 |
| Bovine Serum Albumin | Sigma-Aldrich | A2058 |
| Casein | Sigma-Aldrich | C7078 |
| Nonidet P 40 substitute (NP-40) | Sigma-Aldrich | 74385 |
| PDMS (polydimethylsiloxane, sylgard 184) | Sigma-Aldrich | 761036 |
| DNAseI | NEB | M0303L |
| Streptactin Superflow resin | Qiagen | 30002 |
| Streptactin XT Superflow resin | IBA | 2-4010-025 |
| d-Desthiobiotin | Sigma | D1411 |
| D-biotin | CHEM-IMPEX | #00033 |
| Anti-FLAG M2 Affinity gel beads | Thermofisher | A2220 |
| FuGENE 6 transfection reagent | Promega | E2691 |
| Experimental Models: Cell Lines | ||
| Primary mouse hippocampus neurons | S. Simo | |
| Flp-In T-Rex 293 Cell Line | Thermofisher | #R78007 |
| Recombinant DNA | ||
| Human Tau-2N4R | Addgene | #16316 |
| Human MAP7 | GE Dharmacon MGC Collection | #BC025777 |
| Human MAP2 | Transomics | #BC172263 |
| Human MAP9 | Transomics | #BC146864 |
| Human DCX | Addgene | #83928 |
| Mouse DCLK | Transomics | #BC133685 |
| Human KIF5B (aa 1-560) | R. Vale | |
| Human KIF1A (aa 1-393) | Addgene | #61665 |
| pEGFP-N1-HsKIF1A full-length | S. Niwa | |
| Software and Algorithms | ||
| FIJI | Schindelin et al., 2012 | https://Fiji.sc/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| μManager | Edelstein et al., 2010 | https://micro-manager.org/ |
Highlights.
Microtubule-associated proteins (MAPs) act as modulators of motor movement
Dendrite-localized DCX, DCLK1, and MAP9 inhibit kinesin-1, but not kinesin-3
MAP9 enhances kinesin-3 motility in vitro and in vivo via the motor’s K-loop
MAP9 inhibits the dynein-dynactin complex by blocking the p150-tubulin interaction
ACKNOWLEDGEMENTS
This work was supported by the March of Dimes Basil O’Connor Award, NIH grants 1R00HD080981 and 1R35GM133688, Pew Scholar Research Award, and Simons Foundation SFARI Award to K.M.O.M, March of Dimes Basil O’Connor Award and NIH grants 5R00NS089428 and 1R35GM124889 to R.J.M, NIH grant R01NS109176 to S.S, JSPS Kakenhi #17KK0139 and #17H05010 to S.N, and NSFGRF grant 1650042 to B.Y.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Al-Bassam J, Ozer RS, Safer D, Halpain S, and Milligan RA (2002). MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol 157, 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayloo S, Guedes-Dias P, Ghiretti AE, and Holzbaur ELF (2017). Dynein efficiently navigates the dendritic cytoskeleton to drive the retrograde trafficking of BDNF/TrkB signaling endosomes. Mol Biol Cell 28, 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlan K, Lu W, and Gelfand VI (2013). The microtubule-binding protein ensconsin is an essential cofactor of kinesin-1. Curr Biol 23, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechstedt S, and Brouhard GJ (2012). Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev Cell 23, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertiaux E, Mallet A, Fort C, Blisnick T, Bonnefoy S, Jung J, Lemos M, Marco S, Vaughan S, Trepout S , et al. (2018). Bidirectional intraflagellar transport is restricted to two sets of microtubule doublets in the trypanosome flagellum. J Cell Biol 217, 4284–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisy GG, Marcum JM, Olmsted JB, Murphy DB, and Johnson KA (1975). Purification of tubulin and associated high molecular weight proteins from porcine brain and characterization of microtubule assembly in vitro. Ann NY Acad Sci 253, 107–132. [DOI] [PubMed] [Google Scholar]
- Bulinski JC, and Borisy GG (1979). Self-assembly of microtubules in extracts of cultured HeLa cells and the identification of HeLa microtubule-associated proteins. Proc Natl Acad Sci U S A 76, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulinski JC, and Bossler A (1994). Purification and characterization of ensconsin, a novel microtubule stabilizing protein. J Cell Sci 107 (Pt 10), 2839–2849. [DOI] [PubMed] [Google Scholar]
- Carter AP, Garbarino JE, Wilson-Kubalek EM, Shipley WE, Cho C, Milligan RA, Vale RD, and Gibbons IR (2008). Structure and functional role of dynein’s microtubule-binding domain. Science 322, 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi M, and Popov AV (2003). Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr Purif 32, 83–88. [DOI] [PubMed] [Google Scholar]
- Chaudhary AR, Berger F, Berger CL, and Hendricks AG (2018). Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic 19, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary AR, Lu H, Krementsova EB, Bookwalter CS, Trybus KM, and Hendricks AG (2019). MAP7 regulates organelle transport by recruiting kinesin-1 to microtubules. J Biol Chem 294, 10160–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, and Hirokawa N (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E, and Holzbaur EL (2006). Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Chiba K, Takahashi H, Chen M, Obinata H, Arai S, Hashimoto K, Oda T, McKenney RJ, and Niwa S (2019). Disease-associated mutations hyperactivate KIF1A motility and anterograde axonal transport of synaptic vesicle precursors. Proc Natl Acad Sci U S A 116, 18429–18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, and Holzbaur EL (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Banker GA, and Binder LI (1987). The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience 23, 121–130. [DOI] [PubMed] [Google Scholar]
- Ecklund KH, Morisaki T, Lammers LG, Marzo MG, Stasevich TJ, and Markus SM (2017). She1 affects dynein through direct interactions with the microtubule and the dynein microtubule-binding domain. Nat Commun 8, 2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2010). Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14, Unit14 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenille L, Rouquier S, Lutfalla G, and Giorgi D (2014). Microtubule-associated protein 9 (Map9/Asap) is required for the early steps of zebrafish development. Cell Cycle 13, 1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman OP, Hitti RJ, Boursnell M, Miyadera K, Sargan D, and Mellersh C (2016). Canine genome assembly correction facilitates identification of a MAP9 deletion as a potential age of onset modifier for RPGRIP1-associated canine retinal degeneration. Mamm Genome 27, 237–245. [DOI] [PubMed] [Google Scholar]
- Fourniol F, Perderiset M, Houdusse A, and Moores C (2013). Structural studies of the doublecortin family of MAPs. Methods Cell Biol 115, 27–48. [DOI] [PubMed] [Google Scholar]
- Gadadhar S, Bodakuntla S, Natarajan K, and Janke C (2017). The tubulin code at a glance. J Cell Sci 130, 1347–1353. [DOI] [PubMed] [Google Scholar]
- Gallaud E, Caous R, Pascal A, Bazile F, Gagne JP, Huet S, Poirier GG, Chretien D, Richard-Parpaillon L, and Giet R (2014). Ensconsin/Map7 promotes microtubule growth and entrosome separation in Drosophila neural stem cells. J Cell Biol 204, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Wang W, Dreier B, Jiang Q, Pecqueur L, Pluckthun A, Wang C, and Knossow M (2013). Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat Struct Mol Biol 20, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME , et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Katrukha EA, Grigoriev I, Jaarsma D, Kapitein LC, Akhmanova A, and Hoogenraad CC (2017). MAP2 Defines a Pre-axonal Filtering Zone to Regulate KIF1- versus KIF5-Dependent Cargo Transport in Sensory Neurons. Neuron 94, 347–362 e347. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Cai D, Blasius TL, Li Z, Jiang Y, Jih GT, Meyhofer E, and Verhey KJ (2009). Mammalian Kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol 7, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM , et al. (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78, 1714–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, and Tanaka Y (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. [DOI] [PubMed] [Google Scholar]
- Hooikaas PJ, Martin M, Muhlethaler T, Kuijntjes GJ, Peeters CAE, Katrukha EA, Ferrari L, Stucchi R, Verhagen DGF, van Riel WE , et al. (2019). MAP7 family proteins regulate kinesin-1 recruitment and activation. J Cell Biol 218, 1298–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C (2014). The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol 206, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins B, Decker H, Bentley M, Luisi J, and Banker G (2012). A novel split kinesin assay identifies motor proteins that interact with distinct vesicle populations. J Cell Biol 198, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, and Hoogenraad CC (2011). Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol Cell Neurosci 46, 9–20. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, and Hoogenraad CC (2010). Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol 20, 290–299. [DOI] [PubMed] [Google Scholar]
- Karasmanis EP, Phan CT, Angelis D, Kesisova IA, Hoogenraad CC, McKenney RJ, and Spiliotis ET (2018). Polarity of Neuronal Membrane Traffic Requires Sorting of Kinesin Motor Cargo during Entry into Dendrites by a Microtubule-Associated Septin. Dev Cell 46, 204–218 e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, and Nogales E (2018). Near-atomic model of microtubule-tau interactions. Science 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, and Hirokawa N (2001). Switch-based mechanism of kinesin motors. Nature 411, 439–445. [DOI] [PubMed] [Google Scholar]
- Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, Walsh CA, Otlewski J, Bushweller JH , et al. (2003). The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol 10, 324–333. [DOI] [PubMed] [Google Scholar]
- King SJ, and Schroer TA (2000). Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol 2, 20–24. [DOI] [PubMed] [Google Scholar]
- Lipka J, Kapitein LC, Jaworski J, and Hoogenraad CC (2016). Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J 35, 302–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JS, Schubert CR, Fu X, Fourniol FJ, Jaiswal JK, Houdusse A, Stultz CM, Moores CA, and Walsh CA (2012). Molecular basis for specific regulation of neuronal kinesin-3 motors by doublecortin family proteins. Mol Cell 47, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, and Holzbaur EL (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney RJ, Huynh W, Tanenbaum ME, Bhabha G, and Vale RD (2014). Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 345, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney RJ, Huynh W, Vale RD, and Sirajuddin M (2016). Tyrosination of alphatubulin controls the initiation of processive dynein-dynactin motility. EMBO J 35, 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker DP, Chrin LR, and Berger CL (2011). The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of Tau to inhibit kinesin-mediated transport. J Biol Chem 286, 42873–42880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metivier M, Monroy BY, Gallaud E, Caous R, Pascal A, Richard-Parpaillon L, Guichet A, Ori-McKenney KM, and Giet R (2019). Dual control of Kinesin-1 recruitment to microtubules by Ensconsin in Drosophila neuroblasts and oocytes. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger T, Gache V, Xu M, Cadot B, Folker ES, Richardson BE, Gomes ER, and Baylies MK (2012). MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature 484, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy BY, Sawyer DL, Ackermann BE, Borden MM, Tan TC, and Ori-McKenney KM (2018). Competition between microtubule-associated proteins directs motor transport. Nat Commun 9, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores CA, Perderiset M, Kappeler C, Kain S, Drummond D, Perkins SJ, Chelly J, Cross R, Houdusse A, and Francis F (2006). Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J 25, 4448–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami N, Ishiguro K, Ihara Y, Nonaka I, Sugita H, and Imahori K (1995). Tau protein immunoreactivity in muscle fibers with rimmed vacuoles differs from that in regenerating muscle fibers. Acta Neuropathol 90, 467–471. [DOI] [PubMed] [Google Scholar]
- Niwa S, Lipton DM, Morikawa M, Zhao C, Hirokawa N, Lu H, and Shen K (2016). Autoinhibition of a Neuronal Kinesin UNC-104/KIF1A Regulates the Size and Density of Synapses. Cell Rep 16, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, and Hirokawa N (2000). Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc Natl Acad Sci U S A 97, 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, and Roll-Mecak A (2018). The tubulin code in neuronal polarity. Curr Opin Neurobiol 51, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, and Vallee RB (1987). Retrograde transport by the microtubule associated protein MAP 1C. Nature 330, 181–183. [DOI] [PubMed] [Google Scholar]
- Patel O, Dai W, Mentzel M, Griffin MD, Serindoux J, Gay Y, Fischer S, Sterle S, Kropp A, Burns CJ , et al. (2016). Biochemical and Structural Insights into Doublecortin-like Kinase Domain 1. Structure 24, 1550–1561. [DOI] [PubMed] [Google Scholar]
- Qu D, Weygant N, Yao J, Chandrakesan P, Berry WL, May R, Pitts K, Husain S, Lightfoot S, Li M , et al. (2019). Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer. J Oncol 2019, 6402925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkumar A, Jong BY, and Ori-McKenney KM (2018). ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins. Dev Dyn 247, 138–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, and Verhey KJ (2006). Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16, 2166–2172. [DOI] [PubMed] [Google Scholar]
- Rouquier S, Pillaire MJ, Cazaux C, and Giorgi D (2014). Expression of the microtubule-associated protein MAP9/ASAP and its partners AURKA and PLK1 in colorectal and breast cancers. Dis Markers 2014, 798170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffin JM, Venoux M, Prigent C, Espeut J, Poulat F, Giorgi D, Abrieu A, and Rouquier S (2005). ASAP, a human microtubule-associated protein required for bipolar spindle assembly and cytokinesis. Proc Natl Acad Sci U S A 102, 11302–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H, Imasaki T, Doki C, Sumi T, Aoki M, Uchikubo-Kamo T, Sakamoto A, Tokuraku K, Shirouzu M, and Nitta R (2018). Structural insight into microtubule stabilization and kinesin inhibition by Tau family MAPs. J Cell Biol 217, 4155–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM, and Vale RD (2014). Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol 16, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppina V, and Verhey KJ (2014). The family-specific K-loop influences the microtubule on-rate but not the superprocessivity of kinesin-3 motors. Mol Biol Cell 25, 2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiess M, and Bradke F (2010). Neuronal polarization: The cytoskeleton leads the way. Dev Neurobiol. [DOI] [PubMed] [Google Scholar]
- Sung HH, Telley IA, Papadaki P, Ephrussi A, Surrey T, and Rorth P (2008). Drosophila ensconsin promotes productive recruitment of Kinesin-1 to microtubules. Dev Cell 15, 866–876. [DOI] [PubMed] [Google Scholar]
- Tan R, Lam AJ, Tan T, Han J, Nowakowski DW, Vershinin M, Simo S, Ori-McKenney KM, and McKenney RJ (2019). Microtubules gate tau condensation to spatially regulate microtubule functions. Nat Cell Biol 21, 1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas RP, Chazeau A, Cloin BMC, Lambers MLA, Hoogenraad CC, and Kapitein LC (2017). Differentiation between Oppositely Oriented Microtubules Controls Polarized Neuronal Transport. Neuron 96, 1264–1271 e1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint I, Jean D, Baas PW, and Black MM (2009). Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J Neurosci 29, 10995–11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymanskyj SR, and Ma L (2019). MAP7 Prevents Axonal Branch Retraction by Creating a Stable Microtubule Boundary to Rescue Polymerization. J Neurosci 39, 7118–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymanskyj SR, Yang B, Falnikar A, Lepore AC, and Ma L (2017). MAP7 Regulates Axon Collateral Branch Development in Dorsal Root Ganglion Neurons. J Neurosci 37, 1648–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymanskyj SR, Yang BH, Verhey KJ, and Ma L (2018). MAP7 regulates axon morphogenesis by recruiting kinesin-1 to microtubules and modulating organelle transport. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS, and Sheetz MP (1985). Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venoux M, Basbous J, Berthenet C, Prigent C, Fernandez A, Lamb NJ, and Rouquier S (2008). ASAP is a novel substrate of the oncogenic mitotic kinase Aurora-A: phosphorylation on Ser625 is essential to spindle formation and mitosis. Hum Mol Genet 17, 215–224. [DOI] [PubMed] [Google Scholar]
- Vershinin M, Carter BC, Razafsky DS, King SJ, and Gross SP (2007). Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci U S A 104, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Crevenna AH, Kunze I, and Mizuno N (2014). Structural basis for the extended CAP-Gly domains of p150(glued) binding to microtubules and the implication for tubulin dynamics. Proc Natl Acad Sci U S A 111, 11347–11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek M, Bechstedt S, Chaaban S, and Brouhard GJ (2015). Microtubule-associated proteins control the kinetics of microtubule nucleation. Nat Cell Biol 17, 907–916. [DOI] [PubMed] [Google Scholar]
- Yu I, Garnham CP, and Roll-Mecak A (2015). Writing and Reading the Tubulin Code. J Biol Chem 290, 17163–17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.