

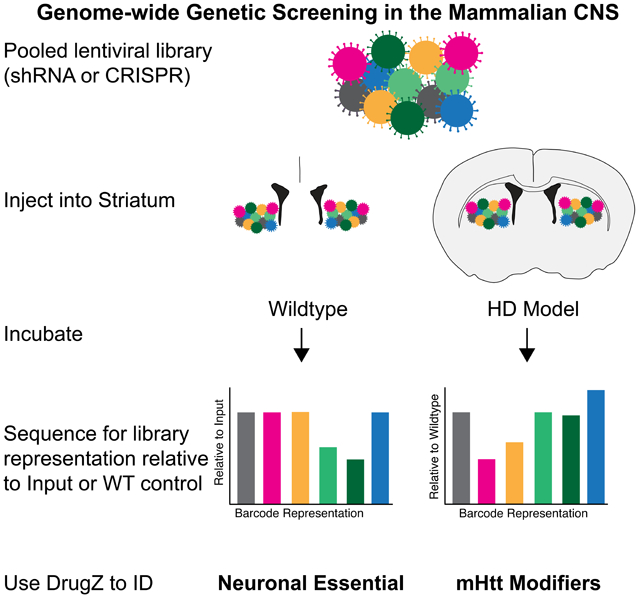
Summary
Unbiased in vivo genome-wide genetic screening is a powerful approach to elucidate new molecular mechanisms, but such screening has not been possible to perform in the mammalian central nervous system (CNS). Here we report the results of the first genome-wide genetic screens in the CNS using both shRNA and CRISPR libraries. Our screens identify many classes of CNS neuronal essential genes, and demonstrate that CNS neurons are particularly sensitive not only to perturbations to synaptic processes, but also autophagy, proteostasis, mRNA processing, and mitochondrial function. These results reveal a molecular logic for the common implication of these pathways across multiple neurodegenerative diseases. To further identify disease-relevant genetic modifiers, we applied our screening approach to two mouse models of Huntington’s disease (HD). Top mutant Huntingtin toxicity modifier genes included several Nme genes and several genes involved in methylation-dependent chromatin silencing and dopamine signaling, results that reveal new HD therapeutic target pathways.
Keywords: Huntington’s disease, genome-wide screening, neuronal essential genes, Nme1
Graphical Abstract
eTOC Blurb
Wertz et al. report a method for unbiased genome-wide genetic screening in the CNS to identify neuronal essential genes in vivo. They also apply this approach to uncover modifiers of mutant Huntingtin toxicity in vivo and show that Nme1 expression can modulate Huntington’s disease model phenotypes.
Introduction
Neurons are among the longest-lived and highest energy consuming cells of the body. As such, neurons possess cell biological properties that likely impart unique vulnerabilities that are not possessed by other cell types in the body. Knowledge of such unique neuronal genetic vulnerabilities may aid in the design of new therapeutic strategies that are effective for the treatment of multiple neurodegenerative diseases. For example, all of the most common neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, are associated with mutated proteins that aggregate (Mattson and Magnus, 2006), implying that neurons may be especially vulnerable to proteostasis perturbations. Although unbiased genetic screening has identified essential genes across many cell types, no such screening has yet been conducted in vivo in the mammalian central nervous system (CNS) for any neuronal type, due to the difficulty of delivering genome-wide manipulations to a non-dividing and anatomically protected cell population. However, such screening could identify unique neuronal genetic vulnerabilities.
Among neuronal cell types, spiny projection neurons (SPNs) of the caudate/putamen (striatum) show the most pronounced loss in Huntington’s disease (HD) (Vonsattel et al., 1985), which is caused by CAG trinucleotide repeat expansions in the huntingtin gene (Group, 1993). Human genome-wide association studies have identified activity of the DNA mismatch repair pathway as an important modifier of HD age-of-onset (Genetic Modifiers of Huntington's Disease, 2015), while previous HD model studies have linked the polyglutamine mutant Huntingtin protein (mHTT) to dysregulation of many cellular processes, including gene transcription and proteostasis (Finkbeiner, 2011). However, no study has systematically compared, in the mammalian CNS, the HD-modifying potential of all genes in the genome in a controlled, isogenic genetic background. These data could point to genetic modifiers of and therapeutic targets for HD that are currently unrecognized.
In order to find in vivo CNS neuronal essential genes, we developed an approach to apply pooled shRNA or CRISPR genome-wide genetic screening to the adult CNS of wild type mice. We also performed genome-wide genetic screening for modifiers of mHTT toxicity in two extensively characterized mouse models of HD: the R6/2 transgenic exon 1 HTT model (Mangiarini et al., 1996) and the zQ175 knock-in full length HTT model (Heikkinen et al., 2012; Menalled et al., 2012). These HD models display several pathological HD hallmarks, but only a small amount of cell death in the striatum, a characteristic that can be exploited for phenotypic enhancer screening. Our data reveal that CNS neurons are not only vulnerable to many perturbations of synaptic function, but that they also are very vulnerable to perturbations to autophagy, proteostasis, mRNA processing, and mitochondrial function. While perturbations to these same pathways also sensitized striatal SPNs to mHTT toxicity, our HD model studies reveal several classes of genetic modifiers of mHTT toxicity. These included genes involved in methylation-dependent chromatin silencing, dopamine signaling, and genes belonging to the Nme gene family. In particular, we were able to validate Nme1 as a modifier of mHTT toxicity and ameliorate HD model phenotypes by over-expression of Nme1 in the striatum. Our study reveals the power of unbiased genetic screening in the CNS to uncover genetic modifiers of neuronal viability in normal and diseased contexts, and points to new therapeutic targets for HD.
Results
An In Vivo Method for Unbiased Screening in the Mammalian CNS
For our genome-wide CNS screening, we employed two well-characterized high-titer VSV-G pseudotyped lentiviral genome-wide murine pooled libraries: one for short hairpin RNA (shRNA), RNAi-based screening and one for single guide RNA (sgRNA) CRISPR-based screening (Doench et al., 2016; Moffat et al., 2006) (STAR Methods). Both libraries were ultra-concentrated and introduced into adult mouse striata by means of double bilateral intracranial stereotaxic injections. Lentiviral transduction results in the integration of shRNA or sgRNA elements into the host cell DNA and thus permanently marks each infected cell. After 4 weeks or 7 months of injection in WT animals, the striata were dissected and genomic (gDNA) sequenced for library representation. shRNAs or sgRNAs that target genes for which knockdown or knockout leads to striatal toxicity are expected to be underrepresented in striatal tissue relative to the input shRNA/sgRNA library since they lead to cell death. Thus, the output of our pooled screening approach was differential representation of genome-integrated library elements upon infected cell loss, as revealed by high throughput DNA sequencing (Figure 1A). Since the striatal cell population is composed of a finite number of terminally differentiated non-dividing neurons, we first determined parameters needed to ensure enough cells were sampled per library element, and that the sequencing depth was sufficient to allow differential analysis. We utilized VSV-G coated lentivirus for our screening studies due to its high neuronal tropism in the striatum (Blomer et al., 1997) and low toxicity in the striatum (Blomer et al., 1997; Mazarakis et al., 2001). We employed a control GFP-encoding VSV-G lentivirus to assess viral spread (average area of 26.41 ± 3.78%, ~120,000 GFP-positive cells per striatal hemisphere; Figure S1A-S1E), and multiplicity of infection (Figure S1F-S1G) across the injection site. There is a high probability that very near to the injection site some cells might be transduced by more than one genetic perturbation, however the likelihood that the synergistic effect of two gene perturbations would generate synthetic lethality is very small due to the nature of the pooled screening approach (STAR Methods and Figure S1H) (Costanzo et al., 2016; Shearer and Saunders, 2015). Injection of lentivirus in the CNS could elicit a small, localized inflammatory response; to address this possibility, we stained viral-injected tissue with markers of inflammation and observed a very small increase in Iba1 and GFAP immunostaining directly adjacent to the needle track, but no evidence of broad inflammation (Figure S1I-L). Thus with our VSV-G lentivirus pooled screening experimental approach, we can achieve a bias to high neuronal tropism, single gene effects, as well as low inflammatory effects.
Figure 1. An In Vivo Method for Unbiased Screening in the Mammalian CNS.

(A) Schematic of pooled screening via stereotaxic intracranial injection into the mouse dorsal striatum. (B) Plot of Pearson correlation values vs. replicate numbers pooled per sample for libraries of increasing complexity (1K, 3.1K, 20K, and 92K). As more replicates are added per sample, correlation values increase. (C) Heatmap of Pearson correlation values as replicates were pooled per sample in the 92K library. (D) ROC-AUC analysis of increasing library coverage with 1 (blue), 5 (yellow), 20 (green) and 74 (red) samples per replicate. AUC values are indicated in parentheses. (E-G) Scatterplots of the log2 normalized library representation in genomic DNA from WT mice vs. the input shRNA library after 7 months (E), 4 weeks (F), and CRISPR library (G) after 4 weeks of in vivo incubation. Blue points indicate library elements with > 1 log2 fold depletion in the WT gDNA as compared to the input library. Pearson correlation r = 0.87 (E), r = 0.82 (F), and r = 0.95 (G). Insets represent full library coverage; dotted red line indicates region expanded in panels D-F for visualization of differentially recovered elements.
Our quantification of viral spread and MOI indicated that for a 1,000-element library, each element would be represented in ~120 cells per sample; in a more complex genome-scale library, each element would be represented in ~1 cell per sample, requiring multiple samples to be pooled to enable robust statistical analyses. To validate this for a genome-wide library, we injected sgRNA sub-libraries of increasing complexity into wild-type (WT) animals. We then analyzed the correlation of sgRNA representation between replicates with increasing numbers of combined samples for each of the sub-libraries (Figure 1B, Figure S2A) and used this to estimate the number of samples we would need for genome-scale coverage. Combinations of 1, 5, 20 or 74 samples per replicate of the genome-wide shRNA library injected into wildtype mice were examined by Receiver Operator Characteristic-Area Under the Curve (ROC-AUC) (Doench et al., 2016) analysis and Pearson correlation of the log2 normalized read counts showed improved genome-wide library performance with increasing sample number (Figure 1C-D). For both the shRNA and sgRNA libraries, upon pooling samples into a single replicate, representation was highly correlated with the input at genome-scale (Figure 1E-G), which would allow identification of genetic perturbations that lead to cell death and depletion of elements from the library. Further, the cumulative frequency and average log2 normalized shRNA or sgRNA representation were consistent across groups (Figure S2B-S2E), confirming sufficient coverage for in vivo genome-wide screening.
shRNA Screening in Wild Type Mice to Identify in vivo CNS Neuronal Essential Genes
We next sought to identify CNS in vivo neuronal essential genes relevant for both short-term and long-term neuronal survival by examining screening data for evidence of neuronal death at 4 weeks and 7 months after genetic manipulation. The magnitude of the relative library element depletion was larger and more consistent in the shRNA screens (Figure 1E-F) than the CRISPR screen (Figure 1G). This could be a consequence of low efficiency of CRISPR gene editing in non-dividing cells in the brain (Nami et al., 2018), or due to the use of a Cas9 transgenic mouse (instead of Cas9 co-delivery in virus with gRNA). We used DrugZ, a sensitive algorithm optimized for identification of genetic modifiers in negative selection screens (Colic et al., 2019), to map differential representation of individual shRNA/sgRNAs onto their gene targets to identify neuronal essential genes at the per-gene level. We took the DrugZ normalized Z score (normZ) from the shRNA and CRISPR screens and compared these scores to log2 normalized gene expression data we collected from WT mouse striatum (Figure 2A-B, Table S1, and STAR Methods). Genes with lower normZ scores represented those that were most depleted in the screens, and these were expressed at high levels in the WT striatum, which indicated that we recovered genes relevant to striatal biology.
Figure 2. Identification of Neuronal Essential Genes by Pooled Genome-wide in vivo Screening.

Contour plots of normZ scores vs. log2 WT striatal gene expression for the 7-month (A) and 4-week (B) shRNA screens. (C) Scatterplot of the log2 normalized fold-change in WT compared to input library at 4 weeks vs. 7 months after in vivo incubation with the genome-wide shRNA library. Green points represent individual shRNA hairpins with an average of > 1 log2 fold depletion in shRNA representation at 4 weeks and 7 months. Pearson correlation r = 0.78. (D) Density plot of the sum normZ scores for the WT shRNA screens shows a bimodal distribution overlaid with two Gaussians to highlight the depleted essential genes (green) as compared to the non-essential genes (black). Genes were identified as candidate neuronal essential genes below the threshold of the intersection of the two Gaussians (red dotted line). (E) Plot of normZ values vs. rank of candidate neuronal essential genes. Top candidate essential genes in relevant biological pathways are highlighted in color as marked. (F) Plots of top GO terms and (G) KEGG pathways significantly associated with candidate neuronal essential genes identified by in vivo screening from the current study (green), C. elegans (purple) and D. melanogaster (blue), as well as human core essential genes in the CEG2 list (grey) represented with Fisher’s exact test −log10 p-value.
First focusing on the shRNA screen data, we sought to identify factors necessary for both short- and long-term neuronal survival and thus compared the average log2 normalized foldchange in shRNA element representation directly in the WT samples at 4 weeks and 7 months vs. the input shRNA library. This comparison revealed a subset of shRNAs that were depleted at both timepoints (Figure 2C). The sum of the 4 week and 7 month normZ scores for these WT data showed a bimodal distribution and, when modeled with two Gaussian distributions, essential genes (green, Figure 2D) and non-essential genes (black, Figure 2D) intersected at a normZ score of ~0.005 and corresponded with DrugZ FDR of 0.038 (red dotted line, Figure 2D). DrugZ analysis identified 3,875 candidate neuronal essential genes with normZ scores below this empirically determined threshold (Figure 2E, and Table S2). Unbiased pathway analyses revealed that these candidates were enriched in genes involved in many basic neuronal KEGG pathways and gene ontology biological process (GO) terms, including synaptic vesicle function, proteasome function, mitochondrial function, and neurotrophin signaling (Figure 2F-2G, and Table S2).
Previous in vivo screens in D. melanogaster (Yamamoto et al., 2014) and C. elegans (Firnhaber and Hammarlund, 2013) have been used to identify neuronal essential genes in nonmammalian metazoans. We compared our neuronal essential genes (green) to orthologs from these screens and observed overlap of many genes, and analysis of these overlapping genes from D. melanogaster (blue) and C. elegans (purple) revealed broad conservation of GO terms and KEGG pathways (Figure 2F-G). We next hypothesized that comparison of our neuronal essential gene list to other essential gene sets from non-neuronal mammalian cell type screens would reveal both common biology and pathways unique to mammalian neuronal cells. Thus, we compared our neuronal essential gene to a widely used reference set of core essential genes [‘CEG2’ from (Hart et al., 2017)], which was derived from aggregate CRISPR screen data from a panel of different dividing human cancer cell lines. We predicted that these cells would be, like neurons, highly metabolic and that this biology would be reflected in overlap of KEGG and GO terms related to basic cellular processes of these cells (grey dots Figure 2F-G). This overlap indeed revealed shared enrichment of genes related to mRNA processing, proteostasis, and oxidative phosphorylation. These finding suggest that several essential gene pathway members for mammalian neurons (as opposed to metazoan neurons) are essential because of mammalian neurons’ high-energy consumption and metabolism. Several of our neuronal essential genes related to neurotrophin signaling and synaptic function have been previously demonstrated to be essential for neuronal viability in mammals [e.g. (Baquet et al., 2004; Verhage et al., 2000)], confirming that our screen also recovered previously identified individual neuronal essential genes from mouse CNS studies.
We observed that several neuronal essential genes in the striatum are themselves striatal-enriched [>1.5 log2 fold-change enrichment in striatum vs. the rest of the brain, from (Kasukawa et al., 2011)] (Figure S3A). Chromatin Enrichment Analysis (Lachmann et al., 2010) and previous striatal ChIP-Seq studies (Niewiadomska-Cimicka et al., 2017) revealed that these striatal-enriched genes are transcriptional targets of the retinoic acid receptor beta (Rarb) (Figure S3B and Table S2) an important regulator of striatal development (Rataj-Baniowska et al., 2015). This suggests that striatal-enriched genes regulated by post-developmental retinoic acid-dependent signaling are essential for the viability of striatal neurons. Given previous reports of loss of striatal-enriched gene expression in response to mHTT (Luthi-Carter et al., 2000), we reasoned that several of these striatal-enriched genes may themselves be downregulated in response to mHTT. Thus, we examined their expression level in the striatum of the R6/2 (Table S1 and STAR Methods) and zQ175 [previously published data: (Langfelder et al., 2016)] mouse models of HD at timepoints age-matched to our screen. This analysis revealed decreased expression of striatal-enriched candidate essential genes in the striatum of HD models (Figure S3C and Table S2), suggesting that mHTT toxicity is in part due to transcriptional downregulation of these striatal neuronal essential genes.
As there is normally not much cell turnover the adult striatum, we did not expect to find genes in our screen that would be enriched in library representation (i.e. that would prevent cell death in wild type striatum). Indeed, in contrast to our identification of several thousand candidate neuronal essential genes, only a single gene, RNA Guanylyltransferase and 5’-Phosphatase (Rngtt; sumZ score = 4.02) displayed enriched in library representation in this wild type screen. This single gene identification also confirms that neither a local neurotoxic inflammatory response at the injection site nor potential glia proliferation effects exist, that otherwise could have a confounding effect on the DrugZ analysis and identification of neuronal essential genes.
CRISPR sgRNA Screening in Wild Type Mice to Identify in vivo CNS Neuronal Essential Genes
While the magnitude of library element depletion was lower in the CRISPR screen, DrugZ analysis of the sgRNA representation for neuronal essential genes identified 668 candidate genes (Figure 3A-B and Figure S3D). Of these, 214 hits were common with the shRNA neuronal essential screen, hypergeometric p-value = 0.023 (Figure S3E). Candidate neuronal essential genes identified by the CRISPR screen were enriched in many of the same KEGG pathway and GO terms identified by the shRNA screen, including: proteostasis, spliceosome, endocytosis, and synaptic vesicle function (Figure 3C-F and Table S2). As expected, these genes also overlapped at the pathway level with the CEG2 list and the previously identified D. melanogaster and C. elegans neuronal essential genes (Figure 3E-F). These results are complementary to our primary shRNA screen results and confirm that, despite lower sensitivity, pooled genome-wide CRISPR libraries can be used for in vivo screening in CNS neurons.
Figure 3. In vivo CRISPR Screening Validates Neuronal Essential Genes and Pathways.

(A) Contour plot of normZ scores vs. log2 WT striatal gene expression for the CRISPR 4-week screen. (B) Plot of normZ values vs. rank of candidate neuronal essential genes by DrugZ analysis of CRISPR sgRNA representation. Top candidate essential genes in relevant biological pathways are highlighted in color as marked. (C-D) Venn diagrams of the overlap of significant KEGG pathways and GO terms from the CRISPR and shRNA screens. Top (E) GO terms and (F) KEGG pathways significantly associated with neuronal essential genes identified by the CRISPR screen (yellow), C. elegans (purple) and D. melanogaster (blue), as well as human core essential genes in the CEG2 list (grey) represented with Fisher’s exact test −log10 p-value. (G) Schematic of significant pathways and biological processes enriched in neuronal essential genes.
Together, our genome-wide screens have uncovered genes involved in the maintenance of neuron viability in vivo in the adult CNS. These genes belong to many cellular pathways, including: core cellular pathways of highly metabolic cells; protein phosphorylation, endocytosis, and autophagy pathways; and many neuronal pathways, including those related to neurotrophin signaling and synaptic transmission (schematic Figure 3G). We have also identified among these essential genes a set of striatal-enriched genes, and make the prediction that suppression of the transcriptional dysregulation induced by mHTT of these particular striatal-enriched genes would help to suppress mHTT toxicity.
Identification of genetic modifiers of mHTT toxicity in two models of HD
Next, we sought to identify genes that become essential for neuronal viability in the context of mHTT (modifiers of mHTT toxicity). To do this we utilized two HD models, the R6/2 (exon 1 fragment mHTT transgenic model) and zQ175 (full length mHTT knockin model), and we compared the shRNA representation in HD model striatum vs. age-matched isogenic WT control striatum dissected at phenotypically relevant time points (Figure 4A). In both model screens, overall shRNA representation was highly correlated at a genome-wide level (Figure S4A-B) and we identified a population of shRNAs with > 1 log2 fold depletion in the mutant as compared to WT (blue) that is distinct from the population of shRNAs that contribute to WT neuronal viability (green) (Figure 4B, 4D). DrugZ analysis revealed 564 candidate mHTT protective genes (factors that promote survival and for which knockdown is toxic), in the R6/2 vs. isogenic control comparison (Figure 4C and Table S3), and overlap of these hits with the neuronal essential genes (identified in Figure 2) revealed 263 genes that are unique to the R6/2 model and have an effect only in the presence of mHTT (Figure 4F). Some of these 263 genes that were essential only in the R6/2 mHTT context belonged to KEGG and GO terms similar to those seen as essential for neuronal cell survival in a WT context (i.e. different genes, but similar KEGG and GO terms), including: transcription, proteostasis, as well as MAPK and neurotrophin signaling terms (Figure 4C, 4G, Figure S4C, and Table S3). However, the majority of gene terms belonged to new KEGG and GO terms that included: dopaminergic synapse, integrin-mediated signaling, methylation-dependent chromatin silencing, fatty acid degradation, and circadian rhythm terms (Figure 4C, 4G, Figure S4C, and Table S3). Analysis of the zQ175 vs. isogenic control comparison revealed 436 candidate mHTT protective genes (Figure 4E and Table S3), of which 253 are unique to the zQ175 model and have an effect only in the presence of mHTT. These also included several genes in KEGG and GO terms similar to those essential for neuronal cell survival in a WT context, including transcription and cell cycle associated terms (Figure 4E, 4H, Figure S4D, and Table S3). However, in the zQ175 model screen as well, the majority of gene terms belonged to unique KEGG and GO terms that included mitochondrial respiratory chain complex I and methylation-dependent chromatin silencing terms (Figure 4E, 4H, Figure S4D, and Table S3). The fact that several top mHTT toxicity modifiers in both HD models belonged to KEGG and GO terms similar to those seen in the WT essential context implies that the presence of either full length or N-terminal fragment mHTT sensitizes neurons to make them even more highly vulnerable to perturbations in neuronal essential functions. In addition to these essential functions, however, our screening implicated several other cellular functions as uniquely modifying survival in the presence of mHTT in both HD models, including methylation-dependent chromatin silencing. This latter finding reinforces the notion that the extensive changes in DNA methylation that have been associated with mHTT and HD are causal in neuronal death in HD (Horvath et al., 2016; Ng et al., 2013). Finally, inspection of individual gene hits in both screens for those of highest magnitude effect revealed that genes linked to dopamine signaling (Drd2 in the zQ175 model) and that Nme gene family members (Nme1 and Nme4 in the R6/2 model; and Nme1 and Nme3 in the zQ175 model) promote survival in the context of either form of mHTT (Figure 4C and E, and Table S3). Although several Nme gene family members were recovered as neuronal essential genes with small effects, they became much more essential in the context of either form of mHTT (Figure 4C, 4E, Table S2, and Table S3).
Figure 4. Identification of in vivo Modifiers of Neuronal Cell Death in the R6/2 and zQ175 HD Mouse Models.

(A) Schematic of genome-wide shRNA screening in HD model and isogenic control mice at disease-relevant time points. (B and D) Scatterplots of the log2 normalized fold-change in shRNA representation in R6/2 or zQ175 mice vs. isogenic WT controls. Blue points represent individual hairpins with > 1 log2 fold differential depletion in mutant vs. WT. Green points represent individual shRNA hairpins with an average of > 1 log2 fold depletion in average shRNA representation in WT compared to input library (Figure 2C). Pearson correlation r = 0.94 (R6/2) and r = 0.81 (zQ175). (C and E) Plots of normZ scores vs. rank of candidate protective factors identified by DrugZ analysis in R6/2 and zQ175 models. Top genes in relevant biological pathways are highlighted in color as marked. (F) Venn diagram of neuronal essential genes vs. HD genetic modifiers. 41 (28+13) candidate protective factors are common to both the R6/2 and zQ175 screens, hypergeometric p-value = 1.30e-4. (G and H) Top GO terms significantly associated with candidate protective factors unique to the R6/2 (red) and zQ175 (orange) models respectively, as well as essential genes identified in Figure 3 (green) represented with Fisher’s exact test –log10 p-value < 0.05.
Previous RNAi in vitro screens in HD models have identified modifiers of mHtt toxicity. Two such in vitro studies used exogenous overexpression of fluorescent protein-tagged mHtt exon 1 fragments with expanded CAG repeats and screened for enhanced aggregation of mHtt after RNAi knockdown in D. melanogaster (Doumanis et al., 2009) or mouse neuro2a cells (Yamanaka et al., 2014), while an in vivo screen in an htt57-128Q-CFP C. elegans model (Lejeune et al., 2012) relied on the enhancement of a motor behavior to uncover genetic mHtt modifiers. As expected, few of the HD modifiers identified in the current study had previously been uncovered in these types of RNAi screens (Table S4). The presence of these genes in previous screens validates our findings, and underscores their relevance to mHTT affects across cell types. However, screening directly in the CNS of HD models increases our sensitivity to uncover those genes that directly influence SPN cell death, and has enabled us to identify many modifiers previously unrecognized in other model contexts.
Recent genome-wide association (GWA) studies by the Genetic Modifiers of Huntington’s Disease (GeM-HD) consortium and the TRACK-HD consortium have identified a number of genetic variants that are associated with HD age-of-onset or progression in human patients (Consortium., 2019; Genetic Modifiers of Huntington's Disease, 2015; Moss et al., 2017). Two hits from our screen, transcription elongation regulator 1 (Tcerg1; R6/2 model data) and transforming acidic coiled-coil containing protein 3 (Tacc3; zQ175 model data), reached genome-wide significance in the GeM-HD age-of-onset modifier study. Tcerg1, which had also been previously implicated as an HD age-of-onset modifier by an earlier independent study (Holbert et al., 2001), is a glutamine-rich protein that influences transcription and pre-mRNA splicing, while Tacc3 encodes a motor spindle protein that has been shown to play a role in HD-related pathways including DNA damage. FAN1 and other members of DNA-damage repair pathways have been implicated as strong genetic modifiers of HD onset by the GeM-HD study (Consortium., 2019; Genetic Modifiers of Huntington's Disease, 2015). A member of the DNA repair complex, Fanconi Anemia Complementation Group E (FANCE), has not previously been identified from these GWA studies, but was a hit in both our R6/2 and zQ175 screen data. Further, we identified an additional overlap of our screen hits to 75 genes from the GeM-HD study and 49 genes from the TRACK-HD study that did not reach genome-wide significance in those studies (Table S4). Finally we identified in our zQ175 screen as protective of mHTT toxicity the geneubiquitin carboxy-terminal hydrolase L1 (Uchl1), which was previously identified as an HD age-of-onset modifier by other human genetic studies (Naze et al., 2002). Thus even though all of these human genetic studies relied on clinical HD measures, while we were assessing mHTT toxicity in our screen, we were still able to identify direct overlaps and novel DNA-damage pathways members that likely play an important role in SPN loss in the caudate and putamen of human HD patients. Since these human genetic studies detect common variants that impart largely small effects from non-developmentally essential genes, our screening data provide an important resource of a large number of likely higher magnitude effect modifier genes that may not be recoverable from human GWA studies.
Finally, our screens also uncovered genes for which knockdown is protective in an HD model (candidate mHTT vulnerability genes). Using the DrugZ analysis as described above, we identified 426 genes in the R6/2 model and 494 genes in the zQ175 model with p-value < 0.05 (Figure S4E-J and Table S3). However, since both HD models used in our screens display only a small amount of striatal cell loss, these candidates require additional validation. Of particular interest is the fact that we recovered complement component 3 (C3), for which knockdown has previously been shown to protect against neurodegeneration in Alzheimer’s disease (Shi et al., 2017) as a top candidate mHTT vulnerability gene in both HD models.
Nme1 modulates HD model phenotypes in the R6/2 mouse striatum
As each HD mouse model has its own particular disease course and may model different aspects of HD biology, of most interest are those 41 protective genes identified in both the R6/2 and zQ175 HD model screens (Figure 4F, Table S3). Of these genes we focused on Nme1, nucleoside diphosphate kinase 1, as it has been shown to display decreased expression in the caudate (−0.46, p-value = 2.38E-07) and cerebellum (−0.31, p-value = 0.00013) of HD patient brains (Hodges et al., 2006). Nme1 was the first metastasis suppressor to be identified (Rosengard et al., 1989), and numerous functions subsequently have been ascribed to it, including modulation of cytosolic chaperone function (Leung and Hightower, 1997) as well as nucleoside diphosphate and histidine kinase activity, and in the nervous system, synaptic vesicle recycling (Krishnan et al., 2001; Marshall et al., 2010). Nme1 also has links to degenerative disease and alpha-synuclein protein aggregation, as it was recently identified as overlapping with a deletion locus that displayed significant genome-wide association to dementia with Lewy bodies (DLB) (Kun-Rodrigues et al., 2019). Nme1 is expressed throughout the CNS, but it has lower expression in the striatum in both mouse and human brain tissue samples (Figure S5A-C), which may lead to increased intrinsic vulnerability of striatal cells to mHTT toxicity.
In order to validate Nme1 knockdown as enhancing mHTT toxicity in vivo, we tested the post-developmental effects of lowering Nme1 protein levels (Nme1 knockdown, KD) in wild-type adult and R6/2 HD model striatum by adeno-associated virus serotype 9 (AAV9)-mediated transduction of an shRNA construct targeting Nme1 (identified in our primary screen) under the U6 promoter. Nme1 shRNA injections were performed into the right striatal hemisphere, with the left striatal hemisphere receiving a matched injection of a control scramble shRNA construct (Control KD) (Schematic: Figure 5A, top panel). Both KD viruses contained a GFP cDNA that was used as a marker of viral spread (Figure S5D). Striatal Nme1 mRNA levels were reduced in each case by approximately half by the Nme1 KD virus (Figure 5B). To assess neuronal toxicity induced upon Nme1 KD, we first tested for the spontaneous rotational motor behavior that is observed upon unilateral striatal lesion (Dunnett and Iversen, 1982). We anticipated that if Nme1 KD led to a high level of striatal SPN death in the HD model, mice would exhibit asymmetrical turning behavior. These experiments revealed an HD-model specific induction of contralateral turning upon unilateral Nme1 KD (Figure 5C). Stereological assessment of NeuN+ neurons in Nme1 KD vs. Control KD striatal areas revealed loss of striatal neurons due to the Nme1 KD only in the mHTT background (Figure 5D), results that were confirmed with a second, independent shRNA Nme1 KD2 (Figure S5E-F). Together these data confirm the genome-wide screen result that reduction of Nme1 expression enhanced the lethality of mHTT toxicity.
Figure 5. Nme1 Knockdown Increases Neuronal Cell Death and mHTT Aggregation, while Nme1 Overexpression Improves Behavioral Phenotypes and Decreases mHTT Aggregation in vivo in the R6/2 HD Mouse Model.

(A) Schematic of injections of injections into R6/2 and isogenic WT control animals. Unilateral knockdown experiments were started at 6-weeks of age [Nme1 knockdown 1 experiment (KD), n = 15 per group; Nme1 knockdown 2 experiment (KD2), n = 10 per group], while bilateral overexpression (OX) experiments were started at 5-weeks of age (n = 10 per group). (B) qPCR quantification of the relative fold-change of Nme1 mRNA levels in Nme1 KD as compared to Control KD striatum for R6/2 and isogenic controls (n = 4 per group). Nme1 levels are normalized to housekeeping gene Eif4a2. One-tailed, paired t-test of deltaCt values, * p-value = 0.034 WT and * p-value = 0.029 R6/2. (C) Rotational behavior analysis (at 9 weeks of age, 3 weeks after viral transduction) to assess the effect of unilateral striatal neuron loss due to injection of Nme1 KD. WT n = 14, R6/2 n = 12; Ordinary one-way ANOVA p-value = 0.0002 with Bonferroni’s multiple comparisons test, * p-value = 0.03, **** p-value < 0.0001. (D) Quantification of NeuN-positive cells in the Control KD vs. Nme1 KD injected striatal hemispheres of WT (n = 4) and R6/2 (n = 4) mice. Points are an average of three images per section from 8 or 16 sections; one-tailed, paired t-test, * p-value = 0.01. (E) qPCR quantification of the relative foldchange of Nme1 mRNA levels in Nme1 OX as compared to Control striatum (n = 5 per group). Nme1 levels are normalized to housekeeping gene Eif4a2; one-tailed, unpaired student’s t-test of DeltaCt values, **** p-value <0.0001. (F) Open field distance horizontal travelled (at 9 weeks of age, 4 weeks after viral transduction) in a 60 min testing window, n = 9 for all groups except WT Nme1 OX n = 8; one-tailed, unpaired student’s t-test, ns p-value = 0.082. (G) Open field resting time (at 9 weeks of age, 4 weeks after viral transduction) in a 60 min testing window, n = 9 for all groups except WT Nme1 OX n = 8; one-tailed, unpaired student’s t-test, * p-value = 0.049. (H) Stereotypic counts (at 9 weeks of age, 4 weeks after viral transduction) in a 60 min testing window, n = 9 for all groups except WT_Nme1-OX n = 8, one-tailed, unpaired t-test, * p-value = 0.024. (I) Stereotypic time (at 9 weeks of age, 4 weeks after viral transduction) in a 60 min testing window, n = 9 for all groups except WT Nme1 OX n = 8, one-tailed, unpaired student’s t-test, * p-value = 0.019. (J) Representative 40x Z-stack indirect immunofluorescence images demonstrating mHTT aggregation in Control KD vs. Nme1 KD injected striatal hemispheres of R6/2 mice as revealed by EM48 antibody reactivity, as well as merged signal with nuclear DAPI stain. Scale bar = 50μm. (K) Quantification of mHTT aggregates as EM48+ puncta per image in Control KD vs. Nme1 KD injected hemispheres of R6/2 mice (n = 4 per group). Points are an average of three images per section from 16 sections, * p-value = 0.011, one-tailed paired student’s t-test. (L) Representative 40x Z-stack indirect immunofluorescence images demonstrating mHTT aggregation in Control OX vs. Nme1 OX striatum of R6/2 mice as revealed by EM48 antibody reactivity, as well as merged signal with nuclear DAPI stain. Scale bar = 50μm. (M) Quantification of mHTT aggregates as EM48+ puncta per image in Control OX vs. Nme1 OX striatum of R6/2 mice (n = 4 per group). Points are an average of three images per section from 8 sections, * p-value = 0.014, one-tailed unpaired student’s t-test. EM48 images have been modified to increase contrast for visualization purposes. All images were modified identically and quantification was performed on unmodified images as described in the methods. All error bars represent mean and SEM.
To test whether Nme1 overexpression (OX) was sufficient to suppress mHTT toxicity, we overexpressed Nme1 under the Eif2a promoter bilaterally into the striatum of 5-week old R6/2 HD model or isogenic control mice by AAV9-mediated transduction (Schematic: Figure 5A, bottom panel). This transduction resulted in Nme1 OX that was approximately 5-fold in the R6/2 model, and approximately 15-fold in the isogenic control mice (Figure 5E). Although the R6/2 HD model phenotypes progress very rapidly, even this brief OX of Nme1 resulted in an amelioration of several HD model phenotypes, including open field locomotion (Figure 5F-G), stereotypic movements (Figure 5H-I), as well as body weight and arousability (Figure S5G-I).
To investigate the mechanistic basis for the genetic effect of Nme1 on mHTT toxicity, we performed single nuclear RNA-Sequencing (snRNA-Seq, n = 5 per group; STAR Methods) studies on the striatal tissue from the Nme1 OX study. Differential gene expression analysis of identified cell types (Figure S5J and STAR Methods) revealed that, in both dSPN and iSPNs, Nme1 OX resulted in the increased expression of several E3 ubiquitin ligases, including Ube3a (Table S5). As levels of Ube3a have previously been implicated as modulating mHTT aggregates in HD mouse model brain (Maheshwari et al., 2014), it is possible that the effects of Nme1 on mHTT toxicity are linked to mHTT aggregation. If this were correct, we expected to observe a change in mHTT aggregation upon either Nme1 KD or OX. We next assessed mHTT aggregation via EM48 aggregation-specific antibody immunoreactivity in both a Nme1 KD and Nme1 OX context. We observed an approximate 13% increase in EM48 puncta number and a 24% increase in mHTT aggregate size in the Nme1 KD vs. Control KD striata (Figure 5J-K and S5K). These results were also confirmed with a second, independent shRNA Nme1 KD2 (Figure S5L-M). We observed an approximate 20% decrease in EM48 puncta number (but no significant decrease in mHTT aggregate size) in Nme1 OX vs. Control OX striata (Figure 5L-M and S5N). These data suggest that the ability of Nme1 expression to affect mHTT toxicity is due, at least in part, to alterations in proteostasis and hence modulation of mHTT clearance or aggregation.
Discussion
In this study we have performed the first genome-wide knockdown screens in vivo in the mammalian CNS (Figure 1). These screen data afford us unique insights into the genes and biological processes necessary for the survival of non-dividing neurons integrated into their native microenvironment over long time-scales. Our WT screening data have identified genes that are required for neuronal cell viability (Figures 2 and 3), and reveal that neurons are highly dependent upon processes that support high cellular metabolic activity, including: proteostasis, mRNA processing, and mitochondrial function. These findings explain why all of the most common neurodegenerative diseases have been linked to some facet of these processes. Our data also reveal several other pathways that are necessary for neuronal viability, and that certain striatal-enriched genes are essential to maintain neuron viability in the normal post-developmental context. Some of these candidate neuronal essential genes in the striatum were transcriptionally downregulated in both HD models used in this study, suggesting that mHTT toxicity is in part due to the decreased expression of certain SPN-enriched genes that occurs in these models.
Further, our screening data have identified modifiers of mHTT-induced toxicity directly in both the R6/2 and zQ175 models of HD (Figure 4). Many of these HD-relevant pathways overlap with pathways that are essential in a non-HD (WT) context, indicating that mHTT sensitizes neurons to dysfunction in these core neuronal essential pathways. While we identified a number of genes previously linked to modifying mHTT toxicity in other disease models as well as HD age-of-onset from human GWA studies (Table S3-S4), our study has also importantly revealed several novel in vivo modifiers of mHTT toxicity, including genes linked to methylation-dependent chromatin silencing, dopamine signaling, and Nme family gene function. These are genes that likely cannot be recovered from screens in simpler model organisms or from analysis of common genetic variants in non-essential genes in the human population.
We have further directly validated Nme1 as a mHTT protective factor in vivo, and demonstrate that it displays low basal expression in WT striatum (Figure S5). Nme1 protein interacts with related family member, Nme3 (protective in zQ175 model), which regulates mitochondrial dynamics and is important for neuronal survival (Chen et al., 2019). Therefore, aside from effects on mHTT aggregation (Figure 5), deficits to mitochondrial function upon loss of Nme1 may also contribute to cellular toxicity.
Together the data presented in this study establish that genome-wide genetic screening in the CNS is a robust method to identify in vivo disease modifiers, which include both neuronal essential genes and mHTT-specific modulators. More broadly, our screen has revealed that C3 and Nme1, which affect Alzheimer’s disease and dementia with Lewy bodies (DLB) risk, respectively (Kun-Rodrigues et al., 2019; Seshadri et al., 2010), are among the top enhancers and suppressors, respectively, of mHTT toxicity in vivo. These findings further support the notion that there exist classes of genes that modify disease risk or disease progression across various neurodegenerative diseases. Future genome-wide CNS genetic screens in mouse models of other neurodegenerative diseases promise to reveal other such genes. Finally, future applications of this screening in a cell type-specific manner and around phenotypes related to striatal function, not only survival, hold great promise for uncovering new disease-relevant mechanisms.
STAR Methods
Lead Contact and Materials Availability
• Further information and requests for resources and reagents should be directed to the Lead Contact, Myriam Heiman (mheiman@mit.edu).
• This study did not generate new unique reagents.
Experimental Models and Subject Details
Animals:
All mouse husbandry and experimental procedures were conducted with the approval of the Massachusetts Institute of Technology Animal Care and Use Committee. Mice were housed under pathogen-free conditions, with food and water provided ad libitum on a standard 12h light/12h dark cycle. No procedures were performed on the mice prior to the outlined experiments. For all studies, littermate mice were group-housed and male littermates were randomly assigned to experimental groups and used at ages described in the Method Details and figure legends. Only male mice were used given HD model differences in phenotype progression between male and female mice. Behavioral experiments were performed on mice at the time points outlined in the Method Details, and mice were naïve to the tests unless otherwise stipulated. Symptomatic HD model mice were provided with DietGel76A (ClearH2O, Portland, ME) for additional nutritional support. All mice were obtained from the Jackson Laboratory (Bar Harbor, ME). C57BL/6J wild-type mice (Jackson Laboratory stock # 000664, RRID:IMSR_JAX:000664) were used between 8-12 weeks of age for the optimization of screening parameters. R6/2 HD model and isogenic control mice (B6CBA-Tg(HDexon1)62Gpb/1J, Jackson Laboratory stock #002810, RRID:IMSR_JAX:002810) were used between 6-10 weeks of age. zQ175 HD model and isogenic control mice (B6J.zQ175DN KI, Jackson Laboratory stock #029928, RRID:IMSR_JAX:029928) were used between 3-10 months of age. Rosa26 Cas9 knockin mice (Platt et al., 2014) (Jackson Laboratory stock #026179, RRID:IMSR_JAX:026179) were used between 6-10 weeks of age. Group samples sizes for genome-wide genetic screening were determined empirically, as described below in the In vivo Genetic Screening: Library scaling and coverage estimation Method Detail. Group samples sizes for Nme1 knockdown and overexpression experiments were based on results from previous studies that allowed for significant differences among HD R6/2 model genotypes (Hachigian et al., 2017) and by the maximum number allowed by surgical handling and behavioral testing capacity.
Human samples:
Human TissueScan cDNA arrays (Origene, Rockville, MD) composed of 24 different brain regions from normal, de-identified post mortem human brain tissue were used for quantification of relative gene expression across brain regions as described in the qRT-PCR Method Detail. Comparisons were made only across brain regions of the same TissueScan array.
Method Details:
In vivo lentiviral injection parameters:
Multiplicity of infection (MOI):
MOI was estimated by measurement of direct green fluorescent protein (GFP) fluorescence intensity, which correlates linearly with the number of viral integration events (Tsai et al., 2015). The distribution of direct GFP fluorescence intensity was measured in the striatal tissue of a young adult C57BL/6J animal that had been injected with a GFP cDNA lentivirus using the same titer and injection parameters as used for the shRNA lentiviral library (see below). Striatal tissue sections were washed 3 × 5 min in phosphate buffered saline (PBS), incubated with 4',6-diamidino-2-phenylindole (DAPI, 0.1μg/mL, ThermoScientific, Rockford IL) for 10 min, mounted with ProLong Gold Antifade Mountant (ThermoScientific, Rockford IL), and imaged on an LSM 710 microscope (Carl Zeiss AG, Oberkochen, Germany). Sections closest to the injection site were identified by the needle track and coordinates relative to Bregma. 40x magnification tiled Z-stack images were collected across the width of the striatum closest to the injection site and stitched with high confidence (0.9) using the Zenn Black software (Carl Zeiss AG, Oberkochen, Germany). Sum projections of the Z-stacks were created using the FIJI software (Schindelin et al., 2012), cells were identified by DAPI, and endogenous GFP signal intensity was measured for each transduced cell and normalized by subtracting local background adjacent to the cell body. The highest MOI and therefore GFP intensity would occur directly at the injection site, while GFP-positive cells further from the injection would contain only a single viral integration event. Endogenous GFP-intensity measurements were binned in 100μm increments from the site of the injection. The median endogenous fluorescence intensity at the periphery of the injection site (greater than 300μm from the needle) is 65.32 while at the center of the injection site the median intensity is 371, indicative of a highest average possible 5.68 MOI immediately around the injection site.
To model the effect of this higher MOI on our screening data, we relied on the fact that the likelihood of a given pair of genes being targeted in the same cell is proportional to the product of the relative abundance of all shRNA targeting either gene. Therefore, we took the shRNA-level abundance pool as measured by input plasmid read count and collapsed it to a gene-level relative abundance by summing the readcounts for each hairpin targeting the gene. We then converted this to a gene-frequency score by dividing by the total of all gene abundance counts. The relative likelihood of any given gene pair being targeted in the same cell is therefore the product of their gene frequency scores (related to Figure S1H). At our estimated MOI the likelihood of any given cell receiving multiple library elements is high near the injection site. However, since each gene-level score is aggregated across multiple hairpin-level observations, and each hairpin-level observation is aggregated across as many cells as possible, the effect of any synergistic or synthetic lethal effect of multiple reagents per cell would be washed out by the other cells in the population. If Gene A is synthetic lethal with Gene B, and Gene A is perturbed in 100 cells, the synergistic effect of GeneA/GeneB dual targeting in one cell is washed out by the other 99 cells. The likelihood that A/B dual targeting occurs in a significant fraction of cells is very small, given the abundance profile in Figure S1, and the likelihood that A/C or other pairs also generate synthetic lethality is also very small, based on extensive measurement of genetic interactions in yeast [background probability ~2.5%; (Costanzo et al., 2016)]. Therefore synergistic effects are unlikely to be present in pooled library screens.
Lentiviral spread:
Viral spread was estimated by measurement of amplified GFP signal (Hachigian et al., 2017) from a young adult C57BL/6J animal injected with a GFP cDNA lentivirus using the same titer and injection parameters as used for the shRNA lentiviral library (see below). Serial 20μm sections through the striatum were cut and mounted on slides and stained for GFP and DAPI (Hachigian et al., 2017). Tiled images of the striata were acquired at 20x magnification and stitched with high confidence (0.9) using the Zenn Black software (Carl Zeiss AG, Oberkochen, Germany), and maximum intensity projections were created using FIJI software (Schindelin et al., 2012). Relative total-striatal area and GFP-positive injection area was measured by creating a region of interest selection based on the greatest extent of the amplified GFP signal. Quantification demonstrated widespread viral infection across the injection site. GFP-positive cells were manually quantified in maximum intensity projections of 10μm Z-stacks for each section across the whole of the injection site to estimate the total number of GFP-positive cells. An adult mouse striatal hemisphere is estimated to contain approximately 700,000 neurons, of which, 95% are striatal projection neurons. The injection site covers an area of 26.41 ± 3.78% of the striatum. Cell counting revealed 970 ± 367 GFP positive cells per 10μm volume of the section, the total volume of the injection covered 60 20μm sections therefore we estimate ~120,000 cells per striatal hemisphere were infected.
In vivo Genetic Screening:
Lentiviral library concentration:
All lentiviral libraries used in the study, including the VSV-G pseudotyped lentiviral murine genome-wide shRNA library (Moffat et al., 2006), CRISPR murine Asiago sgRNA library (Doench et al., 2016), and the GFP cDNA control virus, were provided as cell culture supernatants by the Broad Institute Genetic Perturbation Platform (GPP) (Root et al., 2006). The shRNA library is composed of 92,426 unique elements with an average of 4-6 shRNAs targeting each gene, while the ‘Asiago’ sgRNA library consists of 80,000 elements with 4 sgRNAs targeting each gene. Both libraries contain genetic perturbations under the control of a U6 promoter element. Libraries were ultra-concentrated by filtration through a 0.45μM polyethersulfone membrane (Nalgene Nunc, Penfield NY), 10-fold concentration using the Lenti-X concentrator precipitation solution (Clontech, Mountain View, CA), resuspension in PBS, and final pelleting through a sucrose cushion solution [20% sucrose, 100 mM NaCl, 1 mM EDTA, and 25 mM HEPES (pH 7.4)] by centrifugation at 46,698 X average relative centrifugal force for 2 h and 15 min at 4°C in a SW32 Ti rotor (Beckman Coulter, Brea CA). The final lentiviral pellet was gently resuspended overnight at 4°C in 1/1,000 the original starting volume in freezing solution [PBS, 10% sucrose, and 25 mM HEPES (pH 7.3)], frozen in single-use aliquots at a rate of −1°C /min in a cell freezing chamber (Nalgene Nunc, Penfield NY), and stored at −80°C until use. Final viral titers were determined using the p24 ELISA kit (Clontech, Mountain View, CA) to be approximately 5.0 x 109 viral particles/mL for the shRNA library and 6.08 x 109 viral particles/mL for the sgRNA library.
Pooled lentiviral injection:
The concentrated shRNA lentiviral library was intracranially injected by stereotaxic surgery at 6 weeks of age (R6/2 model mice n=43 and isogenic controls, n= 46) or 3 months of age (zQ175 model mice and isogenic controls, n=40 per group). Concentrated sgRNA library was injected by stereotaxic surgery at 6 weeks of age in WT Cas9+/− mice (n=35). Mice were anesthetized with inhaled isofluorane, placed in a flat skull position in a stereotaxic frame (Stoelting, Wood Dale IL) and injected using a cemented needle (Hamilton Company, Reno NV). 1000 nl of virus was injected at (3nl/s) at each of the following 4 coordinates per mouse (in mm relative to Bregma ML = Medial-lateral; AP = Anterior-posterior; DV=Dorsal-ventral): ML= +2.0mm, AP=0.30mm, DV= −3.70mm; ML= −4.0mm, AP=0.30mm, DV= −3.70mm; ML= −3.4mm, AP=0.90mm, DV= −3.30mm; and ML= +1.7mm, AP=0.90mm, DV= −3.30mm. The needle was left in place for 1-minute post-injection prior to withdrawal. After injection and incubation of the sgRNA or shRNA libraries in the HD model animals and their relevant isogenic controls, striatal cell genomic DNA (gDNA) was harvested at the phenotypically appropriate time points (4 weeks post-injection for the sgRNA and shRNA R6/2 screens and 7 months post-injection for the zQ175 screen) using the Gentra Puregene Genomic DNA purification kit (Qiagen, Hilden, Germany). For gDNA processing, sequencing, and downstream analysis, each striatal hemisphere was treated as an independent experimental sample. Genomic DNA samples were subjected to quality control analysis and samples with either low purity (260nm/280nm <1.6) or near 0 concentration were excluded.
Illumina Sequencing:
5-10 μg of gDNA per sample was PCR amplified, a process that incorporated P5/P7 sequencing adaptors and barcodes for individual samples. PCR products were purified using the AMPure XP-PCR kit according to the manufacture’s protocol (Beckman Coulter, Brea CA). Samples were then pooled and sequenced on a single flow lane of an Illumina HiSeq 2000 (Illumina, San Diego CA) sequencer at the MIT BioMicro Center as previously described (Shema et al., 2015). Reads were identified by the ACCGG (shRNA) or CACCG (sgRNA) sequence in the library vector preceding the shRNA or sgRNA insert sequence. Reads then underwent deconvolution using PoolQ (Broad Institute Genomics Perturbation Platform) which involved mapping of individual shRNA or sgRNA inserts to a file containing all possible elements in the library, and assigned to the correct barcoded sample. After Illumina sequencing and deconvolution of library elements, no samples were excluded from downstream analysis. Read counts for individual library elements were converted to reads per million by dividing the reads for a single shRNA by the total reads in a sample multiplied by 1 million. This was followed by addition of 1 and log2 transformation for downstream analysis. For analyses where samples were pooled into larger replicates, raw reads were summed prior to log2 transformation.
Library scaling and coverage estimation:
Sampling parameters needed to obtain genome-wide screening coverage were determined empirically using CRISPR sgRNA (inert as they were injected into WT mouse striata) libraries of intermediate sub-genome size to infer the replicate number required for sufficient in vivo integrated lentiviral recovery and coverage of a genome-scale 80,000 or 92,000 element library. Inert libraries of increasing complexity (1,000; 3,100; and 20,000 elements) were stereotaxically injected as described above into the dorsal striata of 20 young adult C57BL/6J animals and library representation was determined after 4 weeks of incubation. For sample-size estimation for the genome-scale screen, samples were combined semi-randomly into replicates of 1, 5, 10 or 20 samples and the Pearson correlation of the replicates determined. The 1,000, 3,100, and 20,000 elements libraries were recovered at a threshold of greater than 500 reads/element at one, one, and five pooled striatal samples per replicate, respectively, at a Pearson r value of > 0.80. Using these data, we estimated that at least 40 replicates would be required for genome-wide coverage of the 80,000 and 92,000-element libraries in the adult dorsal striatum of HD model and control animals. For the genome-scale shRNA library, 1, 5, 20, 37, and 74 samples were semi-randomly combined into replicates and the Pearson correlation determined to confirm that a single pooled replicate of WT vs. Input or WT vs. Mutant should be used. Therefore pooling was non-random and while each screen was not replicated, replication of hits was seen across the 4 week and 7 month screens. Further, the CRISPR screen and shRNA screen in the WT context provided replication of each other’s results. The following number of striatal samples were harvested and pooled into a single replicate for final genome-wide shRNA screening after 4 weeks (R6/2 screen) or 7 months (zQ175 screen) of incubation: 80 R6/2 samples; 87 R6/2 non-carrier isogenic control samples; 74 zQ175 samples; and 74 zQ175 non-carrier isogenic control samples. For sgRNA screening after 4 weeks, 52 WT control samples were used. High pooled sample numbers gave us library coverage equivalent to the input plasmid libraries packaged to make the lentivirus, with ~100 million mapped reads per replicate for each screen.
Screen Analysis:
As the genome-wide library contains on average 4-6 shRNAs targeting each gene, and the Asiago library contains 4 sgRNAs per gene, examining the combined effect of more than one genetic perturbation per gene assists in assessing the possibility off-target or seed-based effects. Relative library representation was determined as described above, and the DrugZ algorithm (Colic et al., 2019) was used to rank each gene’s relative depletion in the screen based upon the relative recovery of the shRNAs or sgRNAs. Briefly, DrugZ determines the fold change of each shRNA or sgRNA reagent relative to a user-specified control sample, in this case the initial plasmid pool. The variance for each fold change is estimated based on the distribution of fold changes for the 1,000 reagents with most similar abundance in the control sample. Using this variance estimate, a Z-score is calculated for each reagent, and a gene-level Z-score is determined by summing the reagent-level Z-scores and normalizing by the square root of the number of reagents, yielding the final normZ score. P-values are calculated based on the standard normal distribution and false discovery rates are estimated using the method of Benjamini & Hochberg. We used a log2 gene expression > −1 in WT striatum cutoff to identify genes expressed in the striatum. In the WT shRNA screen, sum normZ scores showed a bimodal distribution. We fit the data with a two-component Gaussian mixture model (using the mixtools package in R) approximating hits (essential genes) and non-hits (genes with no knockdown phenotype). False discovery rate at a given normZ score was calculated as the ratio of the areas under the Gaussian models for all values less than normZ. At the intersection of the Gaussian models at normZ score 0.005, FDR was estimated as 0.038; this cutoff was used as the empirical threshold to determine neuronal essential candidate genes. In all other screens a threshold of normZ p-value < 0.05 was used to determine candidate ‘hits’. Data analysis revealed a number of targets that replicated between screen modalities (shRNA and sgRNA) and mutant models (R6/2 and zQ175).
EnrichR Pathway Analysis:
Analysis of gene enrichment in Gene Ontology Biological Process 2018, KEGG Mouse 2019, and ChEA 2016 terms were performed using Enrichr (using the Gsea.py package in Python) (Chen et al., 2013; Kuleshov et al., 2016). Significant pathways were identified by Fisher’s exact test with nominal p-value < 0.05. For HD modifier analyses we used a background list of the 10,306 genes in the population as determined by representation of the shRNA library in DrugZ analysis filtered by log2 gene expression > −1 in WT mouse striatal samples.
In vivo Nme1 Knockdown:
Nme1 KD (shRNA sequences from the genome-wide screen hit) and Control KD shRNA sequences were packaged into AAV serotype 9 vectors for neuronal targeting (SignaGen, Rockville MD). Viral vectors all contained the following elements: AAV-U6-shRNA(shRNA)-EF1α-GFP, with sequences for a generic shRNA control (5’ – AGTACTGCTTACGATACGG-3’), Nme1 KD (TRCN0000360451, 5‘–ACGTGCCACTGTTAGATTAAA-3’), and Nme1 KD2 (TRCN0000360522, 5‘–TCAGGACCAGTGGTTGCTATG-3’). High titer AAV9 virus (~1x1013 viral genomes/ml) was stereotaxically injected in R6/2 or isogenic control mice at 6-weeks of age as described previously (Hachigian et al., 2017) (Nme1 KD, n = 15; Nme1 KD2, n = 10). Briefly, mice were anesthetized with inhaled isofluorane, placed in a flat skull position in a stereotaxic frame (Stoelting, Wood Dale IL), and injected with 1μl of Nme1 KD or Control KD virus at a rate of 3nl/s. Unilateral injections were used, with control virus in the mouse’s left hemisphere, and Nme1 KD virus in the mouse’s right hemisphere such that each animal could be used as its own control for immunostaining and behavioral analyses. Injection coordinates (in mm relative to Bregma) were as follows: Anterior-posterior = 0.6mm, Medial-lateral = +/− 1.85mm, Dorsal-ventral = −3.5mm.
In vivo Nme1 Overexpression:
Nme1 OX and Control GFP sequences were packaged into AAV serotype 9 vectors for neuronal targeting (SignaGen, Rockville MD). The Nme1 sequence used for overexpression was NM_008704.2. Viral vectors contained the following elements: AAV-Eif2a-Nme1-EF2a-GFP or AAV-Eif2a-Null-EF2a-GFP. High titer AAV9 virus (~1x1013 viral genomes/mL) was stereotaxically injected in R6/2 or isogenic control mice at 5-weeks of age as described for Nme1 KD experiments. Bilateral injections were used, and mice were chosen at random for Nme1 OX or Control GFP injection and housed with mice injected with the same virus, with group sizes of n = 10. Injection coordinates (in mm relative to Bregma) were as follows: Anterior-posterior = 0.6mm, Medial-lateral = +/− 1.85mm, Dorsal-ventral = −3.5mm.
Behavioral Testing:
Behavioral analyses were performed on R6/2 and isogenic wild-type controls as previously described (Hachigian et al., 2017). Behavioral testing was conducted by an investigator blinded to genotype, and scored by a second blinded investigator. Mice were identified as outliers and excluded from further analysis if they scored greater than two standard deviations from the mean on multiple behavioral tests.
Arousability:
Arousability was measured in the Nme1 OX or Control OX animals at 9 weeks of age. Briefly, the state of responsive arousal was measured as reactivity to removal of the top of the home cage. Scores were recorded as the fraction of animals per cage that were showing arousal after 15, 20, and 45 seconds after cage opening.
Open Field Analysis:
Open field analysis was completed on Nme1 OX or Control OX animals at 9 weeks of age as previously described (Hachigian et al., 2017). Total horizontal activity, resting time, and stereotypic movements were measured over 60 min in Med Associates Inc. (Fairfax, VT) open field chamber equipped with infrared break-beam sensors.
Rotational asymmetry measurement:
Animals were placed under an overturned metal mesh pencil cup [diameter 4.375 inches, height 5.5 inches (Rolodex #82406)] that was placed in front of two mirrors, each at a 45° angle, to allow visualization of the mouse from all sides. Mice were allowed to freely explore the environment while video was recorded for a single 5-min trial. Rotational behavior was quantified during the 5-min trial to identify the effect of unilateral Nme1 KD. A fixed starting point was chosen based on the orientation of the mouse at the beginning of the video. A rotation was counted when the mouse completed a 180° turn from the starting point in the clockwise (CW) or counter clockwise (CCW) direction. As all mice were injected with the Control KD virus on the left and Nme1 KD virus on the right hemisphere, a CCW turn is described as contralateral to the lesion, while a CW turn is ipsilateral to the lesion.
Indirect Immunofluorescence:
For Nme1 KD studies, tissue samples were prepared for immunofluorescence by transcardial perfusion followed by tissue processing and staining as previously described (Heiman et al., 2008). Serial 20μm-thick sections through the striatum covering the entire injection site were co-stained with a GFP antibody (Ab6556, 1:500, Abcam, Cambridge MA), and either the EM48 antibody (MAB5374, 1:100, Millipore Sigma, St. Louis, MO) for visualizing mHTT aggregates, or else a NeuN antibody (MAB377, 1:100, Millipore Sigma, St. Louis, MO) for visualizing neuronal nuclei. Every 6th section through the striatum was sampled at 3 fields per hemisphere in the GFP-positive injection site using 40x Z-stacks of 6μm in depth by an investigator blinded to genotype. For Nme1 OX studies, fresh frozen brains were sectioned sagittally across the entire injection site in 10uM thick sections through the striatum and co-stained as described above. Image analysis was completed using FIJI (Schindelin et al., 2012).
qRT-PCR:
Mouse brain tissue was dissected, flash frozen in liquid nitrogen, and disrupted using the TissueLyser (Qiagen, Hilden, Germany) for 2 x 2min at 25 Hz, and RNA was purified using the RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany). cDNA was prepared from 20ng of RNA using the Superscript III first strand synthesis system (ThermoScientific, Rockford IL). TaqMan Universal Master Mix (ThermoScientific, Rockford IL) was used for qRT-PCR, and 20μL reactions were run on a StepOnePlus system (ThermoScientific, Rockford IL). The following TaqMan probes were used (ThermoScientific, Rockford IL): Mm02619580_g1 for mouse Actb; Hs01060665_g1 (Pimer Limited VIC) for human ACTB; Mm00834357_g1 for mouse Eif4a2; Mm01612215_m1 for mouse Nme1; Hs02621161_s1 for human NME1; and Hs00259967_m1 for human PPP1R1B (DARPP-32). Human TissueScan cDNA arrays (Origene, Rockville, MD) were used for quantification of relative NME1 expression across brain regions. Multiplexed qPCR with TaqMan Gene Expression Master Mix was used according to the manufacturer’s protocol (ThermoScientific, Rockford IL). Primer-limited Taqman probes for Human ACTB and Mouse Actb were used for in-well normalization. As a control, the striatal enriched gene PPP1R1B (DARPP-32) was used to confirm array specificity.
Single Nuclear (snRNA) RNA Sequencing and Analysis:
Nuclei isolation protocol was adapted from (Mathys et al., 2019). All procedures were performed on ice. Striata were dissected and tissue was homogenized in 700 μL of homogenization buffer (320 mM sucrose, 5 mM CaCl2, 3 mM Mg(CH3COO)2, 10 mM Tris HCl [pH 7.8], 0.1 mM EDTA [pH 8.0], 0.1% NP-40, 1 mM β-mercaptoethanol, and 0.4 U/μL SUPERaseIn RNase Inhibitor (ThermoFisher Scientific, Waltham MA) with a 2 mL KIMBLE Dounce tissue grinder (MilliporeSigma, Burlington MA) using 10 strokes with loose pestle followed by 10 strokes with tight pestle. Homogenized tissue was filtered through a 40 μm cell strainer and mixed with 450 μL of working solution (50% OptiPrep density gradient medium (MilliporeSigma, Burlington MA), 5 mM CaCl2, 3 mM Mg(CH3COO)2, 10 mM Tris HCl [pH 7.8], 0.1 mM EDTA [pH 8.0], and 1 mM β-mercaptoethanol). The mixture was then slowly pipetted onto the top of an OptiPrep density gradient containing 750 μL of 30% OptiPrep Solution (134 mM sucrose, 5 mM CaCl2, 3 mM Mg(CH3COO)2, 10 mM Tris HCl [pH 7.8], 0.1 mM EDTA [pH 8.0], 1 mM β-mercaptoethanol, 0.04% NP-40, and 0.17 U/μL SUPERase In RNase Inhibitor) on top of 300 μL of 40% OptiPrep Solution (96 mM sucrose, 5 mM CaCl2, 3 mM Mg(CH3COO)2, 10 mM Tris HCl [pH 7.8], 0.1 mM EDTA [pH 8.0], 1 mM β-mercaptoethanol, 0.03% NP-40, and 0.12 U/μL SUPERase In RNase Inhibitor) inside a Sorenson Dolphin microcentrifuge tube (MilliporeSigma, Burlington MA). Nuclei were pelleted at the interface of the OptiPrep density gradient by centrifugation at 10,000 x g for 5 min at 4°C using a fixed angle rotor (FA-45-24-11-Kit). The nuclear pellet was collected by aspirating ~100μL from the interface and transferring to a 2.5 mL Eppendorf tube. The pellet was washed with 2% BSA (in 1x PBS) containing 0.12 U/μL SUPERase In RNase Inhibitor. The nuclei were pelleted by centrifugation at 300 x g for 3 min at 4°C using a swing-bucket rotor (S-24-11-AT). Nuclei were washed three times with 2% BSA and centrifuged under the same conditions. The nuclear pellet was re-suspended in 100 μL of 2% BSA.
Droplet-based snRNA sequencing libraries were prepared using the Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics, Pleasanton CA) according to the manufacturer’s protocol and sequenced on a NovaSeq 6000 at the Broad Institute Genomics Platform. FASTQ files were aligned to the pre-mRNA annotated Mus musculus reference genome version GRCm38 that included EGFP. The resulting feature-barcode count matrices contained 177,874 cells with a median of 1,441 unique genes and 2,669 unique molecular identifiers (UMIs) per nucleus. The distributions of total UMIs per cell were plotted and nuclei that fell in the upper tail of this distribution (> 20,000 UMIs) were removed, as these were most often multiple nuclei captured by a single droplet.
The batchelor package (Haghverdi et al., 2018) was used to remove the batch effect observed across biological replicates within each experimental group from the count matrix. Batch-corrected data was used as input to the archetypal analysis for cell type identification (ACTION) algorithm (Mohammadi et al., 2018) to identify a set of identify landmark cells or ‘archetypes’, each representing a potential underlying cell state. Using ACTION-decompositions with varying numbers of archetypes, we employed our recently developed ACTION-based network (ACTIONet) framework (Mohammadi et al., 2019) to create a multi-resolution nearest neighbor graph that linked cells by their transcriptomic file. A modified version of the stochastic gradient descent-based layout method was used in the uniform manifold approximation and projection (UMAP) algorithm, to visualize the ACTIONet graph. A curated set of known cell type-specific markers was used to annotate individual cells with their expected cell type and assign a confidence score to each annotation. Multiple iterations of this process were performed to further sub-cluster and distinguish cells with similar transcriptional profiles. At each iteration, the network was visualized to assess clustering and annotation accuracy and a filtering step was performed to remove cells annotated with low-confidence, cells with ambiguous profiles resembling dissimilar cell types (generally corresponding to doublet nuclei), cells corresponding to nodes with low coreness in the network (generally corresponding to high ambient RNA content or doublet nuclei), and those corresponding to cell types with counts too low for differential expression analysis. At each iteration genes that had a total of less than 10 counts in any experimental group and nuclei with less than 500 unique genes were also removed.
Confidently annotated cells were extracted and gene counts were log-normalized using the R package scran with cell type, genotype, and transgene as normalization factors. Differential expression analysis of the log-normalized counts was performed using Wilcoxon rank-sum test on a by-cell-type basis for sufficiently abundant cell types. Genes were considered differentially expressed if they had an absolute log-fold change > 0.1 with FDR < 0.001 vs. the respective control in each statistical test.
Bulk RNA Sequencing and Analysis:
Striatal RNA samples (R6/2 model, 11 weeks of age; 5 biological replicates each for R6/2 and non-carrier isogenic controls) were prepared for RNA-Seq using the Ovation RNA-Seq System v2 (NuGEN, San Carlos CA) and the Nextera DNA Library Preparation Kit (Illumina, San Diego CA). The quality of prepared bar-coded libraries was assessed using an Advanced Analytical-fragment Analyzer (Advanced Analytical, Ankeny IA) before mixing for sequencing at approximately 40 million reads/sample on the Illumina HiSeq 2000 (Illumina, San Diego CA) platform at the MIT BioMicro Center. The raw fastq data of 50-bp single-end sequencing reads were aligned to the mouse mm9 reference genome using the STAR 2.4.0 RNA-Seq aligner (Dobin et al., 2013). The mapped reads were processed by htseq-count of HTSeq software (Anders et al., 2015) with mm9 gene annotation to count the number of reads mapped to each gene. Gene differential expression testing between the R6/2 and isogenic control experimental groups was performed using the TopHat-Cufflinks package (Trapnell et al., 2012). Genes with adjusted p-value <0.05 were chosen as differentially expressed genes. Previously published striatal gene differential expression from zQ175 and isogenic control age matched to the cohort used for the zQ175 arm of the screen was used (Langfelder et al., 2016).
Quantification and Statistical Analysis:
Genetic Screening and Transcriptional Profiling:
Use of the relevant pipelines for quantification, determination of normality of data, and appropriate statistical analysis of the genetic screening experiments (DrugZ), bulk RNA-Seq (Tophat Cufflinks) and snRNAseq data (ACTION) are described in the figures and figure legends as well as the Methods Detail sections above.
Gene overlap analysis:
The significance of the overlap when comparing two gene lists was determined using the hypergeometric p-value test in Python. We used 10,306 as the total number of genes in the population as determined by representation of the shRNA library in DrugZ analysis filtered by log2 gene expression > −1 in WT mouse striatal samples. The overlap was considered significant with p-value < 0.05.
Nme1 Experiments:
Analysis of NeuN-positive nuclei and EM48 positive aggregates in Nme1 KD and Nme1 OX injected brains was automated using a Macro in FIJI (Schindelin et al., 2012). Briefly, Z-stacks were flattened into SUM projections, background corrected (rolling ball = 50), filtered (Mean = 1.5), thresholded (DAPI = ‘Triangle’, or EM48 = ‘Moments’), and positive puncta counted by ‘analyze particles’ built-in function (size = 0.001-50.00, circularity = 0.00-1.00) to determine average size and number of EM48-positive structures per image. NeuN positive cells were counted manually by an investigator blinded to genotype using the same imaging and sampling parameters as described above. We used GraphPad Prism 8 to perform the statistical analysis of the Nme1 KD and Nme1 OX experiments. Specific information on the number (n) of values used as well as the statistical tests applied to the data can be found in the figures and/or figure legends. We considered results statistically significant with p < 0.05.
Data and Software Availability:
All sequencing datasets generated as part of this study are publicly available in NCBI GEO under accession #GSE109416 (R6/2 model gene expression data), #GSE141856 (snRNA-Seq Nme1 OX expression data), and SRA project number SRP189737, Bioproject PRNJA529414 (screening raw reads).
Supplementary Material
Table S1 RNA-Seq analysis of total RNA from R6/2 and WT striatum at 11 weeks of age for WT striatal gene expression and DEG identification, Related to Figure 2 and 3.
Table S2 WT neuronal essential gene analysis from the shRNA and CRISPR screens; DrugZ candidate genes, GOBP, and KEGG pathways p-value < 0.05, core essential gene (CEG2 list) overlap, and transcriptional (Brainstars study striatal enriched) DEG overlaps, Related to Figures 2 and 3.
Table S3 R6/2 and zQ175 screens; DrugZ candidate HD modifier genes, GOBP and KEGG pathways (p-value < 0.05), Related to Figure 4.
Table S4 Analysis of candidate modifier genes overlapping with RNAi screens for mHtt toxicity in D. melanogaster and C. elegans and HD patient GWA studies, Related to Figure 4.
Table S5 Single nuclear RNA-Sequencing (snRNA-Seq) of cell type-specific changes to gene expression (p-value adj. < 0.001 and log2 fold change >0.1 or < −0.1) in direct and indirect spiny projection neurons of the R6/2 model or control mice upon Nme1 overexpression, Related to Figure 5.
Key Resources Table:
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| GFP | Abcam | ab6556, RRID:AB_305564 |
| Nme1 | Abcam | ab154547, RRID:AB_2818979 |
| Eif4a2 | Abcam | ab31218, RRID:AB_732123 |
| Secondary anti-Rabbit HRP | Jackson Immunoresearch | 111-035-003, RRID:AB_2313567 |
| NeuN | Millipore Sigma | MAB377, RRID:AB_2298772 |
| EM48 | Millipore Sigma | MAB5374, RRID:AB_177645 |
| Alexafluor Anti-rabbit 488 | ThermoFisher | A21206, RRID:AB_141708 |
| Alexafluor Anti-mouse 647 | ThermoFisher | A31571, RRID:AB_162542 |
| Bacterial and Virus Strains | ||
| Murine genome-wide shRNA library | Broad Institute of MIT GPP | CP0007 |
| Murine genome-wide Asiago sgRNA library | Broad Institute of MIT GPP | CP0085 |
| Murine 1000 sgRNA library | Broad Institute of MIT GPP | CP0014 |
| Murine Asiago Pool 1 20,000 sgRNA library | Broad Institute of MIT GPP | CP0016 |
| Human 3,100 sgRNA library | Broad Institute of MIT GPP | CP0056 |
| U6-shRNA(shRNA)-EF1α-GFP Scramble Control 5’ – AGTACTGCTTACGATACGG-3’ | SignaGen | SL100894 |
| U6-shRNA(shRNA)-EF1α-GFP Nme1 KD 5’–ACGTGCCACTGTTAGATTAAA-3’ | SignaGen | TRCN0000360451 |
| U6-shRNA(shRNA)-EF1α-GFP Nme1 KD2 (, 5‘–TCAGGACCAGTGGTTGCTATG-3’ | SignaGen | TRCN0000360522 |
| AAV-Eif2a-Nme1-EF2a-GFP Nme1 OX sequence NM_008704.2 | SignaGen | N/A |
| AAV-Eif2a-Null-EF2a-GFP GFP Control | SignaGen | N/A |
| Biological Samples | ||
| TissueScan cDNA - Mouse Developmental Array | Origene | CAT#MDRT301 |
| TissueScan cDNA - Human Brain Array | Origene | CAT#HBRT501 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Gentra Puregene Tissue Kit (4g) | Qiagen | CAT#158667 |
| Lenti-X Concentrator Solution | Clontech | CAT#631232 |
| Lenti-X™ p24 Rapid Titer Kit | TaKaRa | CAT#632200 |
| TaqMan® Gene Expression Master Mix | ThermoFisher | CAT#4369016 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | CAT#74804 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v3 | 10x Genomics, Pleasanton CA | CAT#1000075 |
| Chromium Chip B Single Cell Kit | 10x Genomics, Pleasanton CA | CAT#1000074 |
| Chromium i7 Multiplex Kit | 10x Genomics, Pleasanton CA | CAT#120262 |
| Deposited Data | ||
| Raw screening data | This Paper | SRA project number SRP189737, Bioproject PRNJA529414 |
| RNAseq Data R6/2 | This Paper | NCBI GEO #GSE109416 |
| snRNAseq Data Nme1OX | This Paper | NCBI GEO #GSE141856 |
| RNAseq Data zQ175 | Langfelder et al., 2016 | NCBI GEO #GSE65776 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J hemi | The Jackson Laboratory | JAX: 002810, RRID:IMSR_JAX:002810 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J non carrier controls | The Jackson Laboratory | JAX: 002810, RRID:IMSR_JAX:002810 |
| Mouse: B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J | The Jackson Laboratory | JAX:026179, RRID:IMSR_JAX:026179 |
| Mouse: C57BL/6J WT controls | The Jackson Laboratory | JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: B6J.zQ175DN KI Heterozygous | The Jackson Laboratory | JAX:029928 RRID:IMSR_JAX:029928 |
| Mouse: B6J.zQ175DN KI Wild type | The Jackson Laboratory | JAX:029928 RRID:IMSR_JAX:029928 |
| Oligonucleotides | ||
| Taqman gene expression Human NME1 Hs02621161_s1 | ThermoFisher | CAT#4331182 |
| Taqman gene expression Mouse Nme1 Mm01612215_m1 | ThermoFisher | CAT#4331182 |
| Taqman gene expression Human PPP1R1B (DARPP-32) Hs00259967_m1 | ThermoFisher | CAT#4331182 |
| Taqman gene expression Mouse Actb Mm02619580_g1 (VIC-MGB_PL) | ThermoFisher | CAT#4448892 |
| Taqman gene expression Mouse Eif4a2 Mm00834357_g1 (VIC-MGB_PL) | ThermoFisher | CAT#4448892 |
| Taqman gene expression Human ACTB Hs01060665_g1 (VIC-MGB_PL) | ThermoFisher | CAT#4448892 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| FIJI | Schneider et al., 2012 | https://imagej.net/Fiji, RRID:SCR_002285 |
| DrugZ | Colic et. al. 2019 | https://github.com/hart-lab/drugz |
| EnrichR | Chen et. al. 2013 | https://amp.pharm.mssm.edu/Enrichr/, RRID:SCR_001575 |
| Gsea.py | Mootha et. al. 2003 | https://github.com/zqfang/GSEApy |
| Batchelor | Haghverdi et al., 2018 | https://bioconductor.org/packages/release/bioc/html/batchelor.html |
| ACTIONet | Mohammadi et al., 2019 | https://github.com/shmohammadi86/ACTIONet |
| TopHat-Cufflinks | Trapnell et al., 2012) | https://github.com/cole-trapnell-lab |
Highlights:
Unbiased genome-wide genetic screening in the mouse brain
Identification of in vivo neuronal essential genes
Identification of genetic modifiers of mutant Huntingtin toxicity in vivo
Validation of Nme1 as a suppressor of mutant Huntingtin toxicity
Acknowledgements
The authors thank the CHDI Foundation for providing the zQ175 mice used in this study. This work was supported by NIH/NINDS award 1 R01 NS085880 (M. Heiman), an award from the JPB Foundation (M. Heiman), an award from the Bev Hartig Huntington’s Disease Foundation (M. Heiman), a Fay/Frank Seed Award from the Brain Research Foundation (M. Heiman), the Jeptha H. and Emily V. Wade Award (M. Heiman), and fellowships from the JPB Foundation and the Hereditary Disease Foundation (M.H.W.), the NIH (5T32EB019940-05, S.S.P.), and the JPB Foundation (H.L.). T.H. and M.C. were supported by NIH/NIGMS award R35GM130119 (T.H.), CPRIT grant RR160032 (T.H.), and the NIH/NCI Cancer Center Support Grant P30 CA016672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, and Jones KR (2004). Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 4250–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomer U, Naldini L, Kafri T, Trono D, Verma IM, and Gage FH (1997). Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. Journal of virology 71, 6641–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CW, Wang HL, Huang CW, Huang CY, Lim WK, Tu IC, Koorapati A, Hsieh ST, Kan HW, Tzeng SR, et al. (2019). Two separate functions of NME3 critical for cell survival underlie a neurodegenerative disorder. Proceedings of the National Academy of Sciences of the United States of America 116, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma'ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D, et al. (2019). Identifying chemogenetic interactions from CRISPR knockout screens with drugZ. bioRxiv 232736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium., G.M.o.H.s.D. (2019). CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset. Cell 178, 887–900 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumanis J, Wada K, Kino Y, Moore AW, and Nukina N (2009). RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PloS one 4, e7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnett SB, and Iversen SD (1982). Spontaneous and drug-induced rotation following localized 6-hydroxydopamine and kainic acid-induced lesions of the neostriatum. Neuropharmacology 21, 899–908. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S (2011). Huntington's Disease. Cold Spring Harbor perspectives in biology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnhaber C, and Hammarlund M (2013). Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. PLoS genetics 9, e1003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington's Disease, C. (2015). Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group, T.H.s.D.C.R. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Hachigian LJ, Carmona V, Fenster RJ, Kulicke R, Heilbut A, Sittler A, Pereira de Almeida L, Mesirov JP, Gao F, Kolaczyk ED, et al. (2017). Control of Huntington's Disease-Associated Phenotypes by the Striatum-Enriched Transcription Factor Foxp2. Cell reports 21, 2688–2695. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, and Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nature biotechnology 36, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A, et al. (2017). Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 7, 2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimaki K, Vartiainen N, Puolivali J, Hendricks SJ, Glaser JR, Bradaia A, Wadel K, Touller C, Kontkanen O, et al. (2012). Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PloS one 7, e50717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. (2006). Regional and cellular gene expression changes in human Huntington's disease brain. Human molecular genetics 15, 965–977. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, et al. (2001). The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proceedings of the National Academy of Sciences of the United States of America 98, 1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, Eszes M, Faull RL, Curtis MA, Waldvogel HJ, et al. (2016). Huntington's disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging 8, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasukawa T, Masumoto KH, Nikaido I, Nagano M, Uno KD, Tsujino K, Hanashima C, Shigeyoshi Y, and Ueda HR (2011). Quantitative expression profile of distinct functional regions in the adult mouse brain. PloS one 6, e23228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KS, Rikhy R, Rao S, Shivalkar M, Mosko M, Narayanan R, Etter P, Estes PS, and Ramaswami M (2001). Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron 30, 197–210. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun-Rodrigues C, Orme T, Carmona S, Hernandez DG, Ross OA, Eicher JD, Shepherd C, Parkkinen L, Darwent L, Heckman MG, et al. (2019). A comprehensive screening of copy number variability in dementia with Lewy bodies. Neurobiology of aging 75, 223 e221–223 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, and Ma'ayan A (2010). ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nature neuroscience 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune FX, Mesrob L, Parmentier F, Bicep C, Vazquez-Manrique RP, Parker JA, Vert JP, Tourette C, and Neri C (2012). Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC genomics 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SM, and Hightower LE (1997). A 16-kDa protein functions as a new regulatory protein for Hsc70 molecular chaperone and is identified as a member of the Nm23/nucleoside diphosphate kinase family. The Journal of biological chemistry 272, 2607–2614. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Human molecular genetics 9, 1259–1271. [DOI] [PubMed] [Google Scholar]
- Maheshwari M, Shekhar S, Singh BK, Jamal I, Vatsa N, Kumar V, Sharma A, and Jana NR (2014). Deficiency of Ube3a in Huntington's disease mice brain increases aggregate load and accelerates disease pathology. Human molecular genetics 23, 6235–6245. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506. [DOI] [PubMed] [Google Scholar]
- Marshall JC, Collins J, Marino N, and Steeg P (2010). The Nm23-H1 metastasis suppressor as a translational target. European journal of cancer 46, 1278–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer's disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, and Magnus T (2006). Ageing and neuronal vulnerability. Nature reviews Neuroscience 7, 278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, Carter EE, Barber RD, Baban DF, Kingsman SM, et al. (2001). Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Human molecular genetics 10, 2109–2121. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, et al. (2012). Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PloS one 7, e49838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Mohammadi S, Davila-Velderrain J, and Kellis M (2019). Multi-resolution single-cell state characterization via joint archetypal/network analysis. bioRxiv. [Google Scholar]
- Mohammadi S, Ravindra V, Gleich DF, and Grama A (2018). A geometric approach to characterize the functional identity of single cells. Nature communications 9, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DJH, Pardinas AF, Langbehn D, Lo K, Leavitt BR, Roos R, Durr A, Mead S, investigators, T.-H., investigators, R., et al. (2017). Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. The Lancet Neurology 16, 701–711. [DOI] [PubMed] [Google Scholar]
- Nami F, Basiri M, Satarian L, Curtiss C, Baharvand H, and Verfaillie C (2018). Strategies for In Vivo Genome Editing in Nondividing Cells. Trends in biotechnology 36, 770–786. [DOI] [PubMed] [Google Scholar]
- Naze P, Vuillaume I, Destee A, Pasquier F, and Sablonniere B (2002). Mutation analysis and association studies of the ubiquitin carboxy-terminal hydrolase L1 gene in Huntington's disease. Neuroscience letters 328, 1–4. [DOI] [PubMed] [Google Scholar]
- Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, and Fraenkel E (2013). Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proceedings of the National Academy of Sciences of the United States of America 110, 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska-Cimicka A, Krzyzosiak A, Ye T, Podlesny-Drabiniok A, Dembele D, Dolle P, and Krezel W (2017). Genome-wide Analysis of RARbeta Transcriptional Targets in Mouse Striatum Links Retinoic Acid Signaling with Huntington's Disease and Other Neurodegenerative Disorders. Molecular neurobiology 54, 3859–3878. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rataj-Baniowska M, Niewiadomska-Cimicka A, Paschaki M, Szyszka-Niagolov M, Carramolino L, Torres M, Dolle P, and Krezel W (2015). Retinoic Acid Receptor beta Controls Development of Striatonigral Projection Neurons through FGF-Dependent and Meis1-Dependent Mechanisms. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 14467–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DE, Hacohen N, Hahn WC, Lander ES, and Sabatini DM (2006). Genome-scale loss-of-function screening with a lentiviral RNAi library. Nature methods 3, 715–719. [DOI] [PubMed] [Google Scholar]
- Rosengard AM, Krutzsch HC, Shearn A, Biggs JR, Barker E, Margulies IM, King CR, Liotta LA, and Steeg PS (1989). Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature 342, 177–180. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, et al. (2010). Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama 303, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer RF, and Saunders DN (2015). Experimental design for stable genetic manipulation in mammalian cell lines: lentivirus and alternatives. Genes to cells : devoted to molecular & cellular mechanisms 20, 1–10. [DOI] [PubMed] [Google Scholar]
- Shema R, Kulicke R, Cowley GS, Stein R, Root DE, and Heiman M (2015). Synthetic lethal screening in the mammalian central nervous system identifies Gpx6 as a modulator of Huntington's disease. Proceedings of the National Academy of Sciences of the United States of America 112, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, and Lemere CA (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Tsai TH, Chang CP, Chen SF, Lee YM, and Shyue SK (2015). Linear correlation between average fluorescence intensity of green fluorescent protein and the multiplicity of infection of recombinant adenovirus. Journal of biomedical science 22, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, et al. (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, and Richardson EP Jr. (1985). Neuropathological classification of Huntington's disease. Journal of neuropathology and experimental neurology 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Jaiswal M, Charng WL, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H, Haelterman NA, Xiong B, et al. (2014). A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159, 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka T, Wong HK, Tosaki A, Bauer PO, Wada K, Kurosawa M, Shimogori T, Hattori N, and Nukina N (2014). Large-scale RNA interference screening in mammalian cells identifies novel regulators of mutant huntingtin aggregation. PloS one 9, e93891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 RNA-Seq analysis of total RNA from R6/2 and WT striatum at 11 weeks of age for WT striatal gene expression and DEG identification, Related to Figure 2 and 3.
Table S2 WT neuronal essential gene analysis from the shRNA and CRISPR screens; DrugZ candidate genes, GOBP, and KEGG pathways p-value < 0.05, core essential gene (CEG2 list) overlap, and transcriptional (Brainstars study striatal enriched) DEG overlaps, Related to Figures 2 and 3.
Table S3 R6/2 and zQ175 screens; DrugZ candidate HD modifier genes, GOBP and KEGG pathways (p-value < 0.05), Related to Figure 4.
Table S4 Analysis of candidate modifier genes overlapping with RNAi screens for mHtt toxicity in D. melanogaster and C. elegans and HD patient GWA studies, Related to Figure 4.
Table S5 Single nuclear RNA-Sequencing (snRNA-Seq) of cell type-specific changes to gene expression (p-value adj. < 0.001 and log2 fold change >0.1 or < −0.1) in direct and indirect spiny projection neurons of the R6/2 model or control mice upon Nme1 overexpression, Related to Figure 5.